

ZYVOX
LINEZOLID
Solución inyectable
300 ml Inyectable I.V. , 600 Miligramos
1 Caja , 10 Tabletas , 600 Miligramos
Bolsa, 1 Inyectable, 300 Mililitros
FORMA FARMACÉUTICA Y FORMULACIÓN
Tabletas: 600 mg de Linezolid; excipientes c.s.p.* 1 tableta.
*Almidón de maíz, celulosa microcristalina, hidroxipropilcelulosa, almidón glicolato de sodio, estearato de magnesio, agua purificada, opadry blanco YS-1-18202-A, opacode rojo S-1-15-118 y cera carnauba.
Solución inyectable:
Cada 100 mL contienen: 200 mg de Linezolid; vehículo c.s.p.** 100 mL.
**Citrato de sodio dihidratado, ácido cítrico anhidro, glucosa monohidratada, solución de ácido clorhídrico 10%, solución de hidróxido de sodio 10% y agua para inyección en un vehículo acuoso para administración intravenosa.
INDICACIONES Y USO: Las formulaciones de ZYVOX® están indicadas para el tratamiento de las siguientes infecciones provocadas por cepas susceptibles de los microorganismos designados (ver Precauciones: Uso pediátrico, Dosificación y administración y Estudios clínicos). Linezolid no está indicado para el tratamiento de infecciones por Gram negativos. Es vital que se inicie inmediatamente una terapia específica contra Gram negativos si hay documentación o sospecha de un patógeno Gram negativo concomitante (ver Advertencias).
• Infecciones producidas por Enterococcus faecium resistente a la vancomicina, incluidos los casos con bacteriemia concurrente.
• Neumonía nosocomial causada por Staphylococcus aureus (cepas resistentes y sensibles a la meticilina) o Streptococcus pneumoniae (incluidas las cepas resistentes a multifármacos [MDRSP]).
• Infecciones complicadas de la piel y de la estructura cutánea, incluidas las infecciones de pie diabético, sin osteomielitis concomitante causadas por Staphylococcus aureus (cepas susceptibles y resistentes a la meticilina), Streptococcus pyogenes o Streptococcus agalactiae. ZYVOX® no ha sido estudiado en el tratamiento de úlceras de decúbito.
• Infecciones no complicadas de la piel y de la estructura cutánea causadas por Staphylococcus aureus (cepas susceptibles a meticilina solamente) o Streptococcus pyogenes.
• Neumonía adquirida en la comunidad causada por Streptococcus pneumoniae (incluidas las cepas resistentes a multifármacos [MDRSP]), incluidos los casos con bacteriemia concurrente o Staphylococcus aureus (cepas sensibles a la meticilina únicamente).
A fin de reducir el desarrollo de bacterias resistentes a los fármacos y mantener la eficacia de ZYVOX® y otros fármacos antibacterianos, ZYVOX® sólo debe utilizarse en el tratamiento o prevención de infecciones comprobadas o con sospechas fundadas de haber sido causadas por bacterias susceptibles. Una vez disponible la información del cultivo y de la susceptibilidad, deben utilizarse estos elementos para elegir o modificar el tratamiento antibacteriano. Si no se dispone de dichos datos, los patrones epidemiológicos locales y de susceptibilidad pueden ayudar a la selección empírica del tratamiento.
La seguridad y eficacia de las formulaciones de ZYVOX® administradas por mas de 28 días no ha sido evaluado en estudios clinicos controlados.
CONTRAINDICACIONES: El uso de las formulaciones de ZYVOX® está contraindicado en pacientes con hipersensibilidad conocida a linezolid o cualquiera de los demás componentes del producto.
Inhibidores de la monoamino oxidasa: Linezolid no debe administrarse en pacientes que estén tomando algún producto medicinal que inhiba la monoamino oxidasa A o B (por ej., fenelzina, isocarboxazida) ni tampoco dentro de las dos semanas siguientes al uso de dicho producto medicinal.
REACCIONES ADVERSAS
Pacientes adultos: La seguridad de las formulaciones de ZYVOX® fue evaluada en 2046 pacientes adultos enrolados en 7 ensayos clínicos controlados Fase III con comparador, quienes fueron tratados durante 28 días.
En el Cuadro 6 se muestra la incidencia de eventos adversos relacionados con el fármaco informado en por lo menos 1% de los pacientes adultos en estos ensayos de acuerdo a la dosis de ZYVOX®.
Cuadro 6. Incidencia (%) de eventos adversos relacionados con el fármaco producidos en >1% de pacientes adultos tratados con ZYVOX® en ensayos clínicos controlados con comparador
|
Evento adverso |
Infecciones no complicadas de la piel y de la estructura cutánea |
Todas las demás indicaciones |
||
|
ZYVOX® 400 mg PO q12h (n=548) |
Claritromicina 250 mg PO q12h (n=537) |
ZYVOX® 600 mg q12h (n=1498) |
Todos los demás comparadores* (n=1464) |
|
|
% de pacientes con 1 evento adverso relacionado con el fármaco |
25,4 |
19,6 |
20,4 |
14,3 |
|
% de pacientes que descontinuaron debido a eventos adversos relacionados con el fármaco† |
3,5 |
2,4 |
2,1 |
1,7 |
|
Diarrea |
5,3 |
4,8 |
4,0 |
2,7 |
|
Náuse |
3,5 |
3,5 |
3,3 |
1,8 |
|
Dolor de cabeza |
2,7 |
2,2 |
1,9 |
1,0 |
|
Alteración del gusto |
1,8 |
2,0 |
0,9 |
0,2 |
|
Moniliasis vaginal |
1,6 |
1,3 |
1,0 |
0,4 |
|
Infección fúngica |
1,5 |
0,2 |
0,1 |
<0,1 |
|
Pruebas anormales de la función hepática |
0,4 |
0 |
1,3 |
0,5 |
|
Vómito |
0,9 |
0,4 |
1,2 |
0,4 |
|
Decoloración de la lengua |
1,1 |
0 |
0,2 |
0 |
|
Mareo |
1,1 |
1,5 |
0,4 |
0,3 |
|
Moniliasis oral |
0,4 |
0 |
1,1 |
0,4 |
|
* Los comparadores incluyeron cefpodoxima proxetil 200 mg PO q12h; ceftriaxona 1 g IV q12h; dicloxacilina 500 mg PO q6h; oxacilina 2 g IV q6h; vancomicina 1 g IV q12h. † Los eventos adversos relacionados con el fármaco que se informaron con mayor frecuencia y que produjeron descontinuación en los pacientes tratados con ZYVOX® fueron náusea, dolor de cabeza, diarrea y vómitos. |
||||
Pacientes pediátricos: Se evaluó la seguridad de las formulaciones de ZYVOX® en 215 pacientes pediátricos con edades que oscilaban entre el nacimiento hasta los 11 años, y en 248 pacientes pediátricos con edades que oscilaban entre los 5 hasta los 17 años (146 de los 248 tenían entre 5 y 11 años, y 102 pacientes entre 12 y 17). Estos pacientes fueron enrolados en 2 estudios clínicos controlados Fase 3 con comparador, y recibieron tratamiento durante 28 días. En esos estudios, 83% y 99%, respectivamente, de los eventos adversos informados con ZYVOX® fueron descritos como de intensidad leve a moderada. En el estudio realizado en pacientes pediátricos hospitalizados (desde el nacimiento hasta los 11 años) con infecciones por bacterias Gram positivas, y que fueron aleatorizados en una proporción 2:1 (linezolid:vancomicina), la mortalidad fue 6% (13/215) en el brazo de linezolid y 3% (3/101) en el brazo de vancomicina. Sin embargo, dadas las enfermedades subyacentes severas de la población de pacientes, no se pudo establecer ninguna causalidad.
En el Cuadro 7 se muestra la incidencia de eventos adversos informados en al menos1% de pacientes pediátricos tratados con ZYVOX® en esos estudios.
Cuadro 7. Incidencia (%) de eventos adversos relacionados con el fármaco que se produjeron en >1% de pacientes pediátricos (y>1 paciente) en cualquier grupo de tratamiento en estudios clínicos controlados con comparador
|
Evento |
Infecciones no complicadas de la piel y de la estructura cutánea* |
Todas las demás indicaciones† |
||
|
ZYVOX® |
Cefadroxilo |
ZYVOX® |
Vancomicina |
|
|
% de pacientes con ≥1 evento adverso relacionado con el fármaco |
19,2 |
14,1 |
18,8 |
34,3 |
|
% de pacientes que descontinuaron debido a un evento adverso relacionado con el fármaco |
1,6 |
2,4 |
0,9 |
6,1 |
|
Diarrea |
5,7 |
5,2 |
3,8 |
6,1 |
|
Náusea |
3,3 |
2,0 |
1,4 |
0 |
|
Dolor de cabeza |
2,4 |
0,8 |
0 |
0 |
|
Heces sueltas |
1,2 |
0,8 |
1,9 |
0 |
|
Trombocitopenia |
0 |
0 |
1,9 |
0 |
|
Vómito |
1,2 |
2,4 |
1,9 |
1,0 |
|
Dolor abdominal generalizado |
1,6 |
1,2 |
0 |
0 |
|
Dolor abdominal localizado |
1,6 |
1,2 |
0 |
0 |
|
Anemia |
0 |
0 |
1,4 |
1,0 |
|
Eosinofilia |
0,4 |
0,4 |
1,4 |
0 |
|
Vértigo |
1,2 |
0,4 |
0 |
0 |
|
Prurito fuera del lugar de aplicación |
0,4 |
0 |
0 |
2,0 |
|
* Los pacientes entre 5 y 11 años recibieron ZYVOX® 10 mg/kg por mes cada 12 horas o cefadroxilo 15 mg/kg por mes cada 12h. Los pacientes de 12 años o más recibieron ZYVOX® 600 mg por mes cada 12h o cefadroxilo 500 mg PO q12h. † Los pacientes desde el nacimiento hasta los 11 años recibieron ZYVOX® 10 mg/kg IV por mes cada 8h o vancomicina 10 a 15 mg/kg IV q6-24h, según la edad y la depuración renal. |
||||
Cambios en los parámetros de laboratorio: ZYVOX® ha sido asociado con trombocitopenia cuando se utilizó en dosis de hasta 600 mg inclusive cada 12 horas durante 28 días. En los estudios clínicos controlados con comparador, de Fase 3, el porcentaje de pacientes adultos que desarrolló un recuento de plaquetas considerablemente bajo (definido como menos que el 75% del límite inferior normal y/o basal) fue 2,4% (rango entre estudios: 0,3% a 10%) con ZYVOX® y 1,5% (rango entre estudios: 0,4% a 7%) con un comparador. En un estudio realizado en pacientes pediátricos hospitalizados, con edades que van desde el nacimiento hasta los 11 años, el porcentaje de pacientes que desarrolló un recuento de plaquetas substancialmente bajo (definido como menos que el 75% del límite inferior normal y/o basal) fue 12,9% con ZYVOX® y 13,4% con vancomicina. En un estudio realizado en pacientes pediátricos ambulatorios con edades entre los 5 y 17 años, el porcentaje de pacientes que desarrolló un recuento plaquetario sustancialmente bajo fue 0% con ZYVOX® y 0,4% con cefadroxilo. La trombocitopenia asociada con el uso de ZYVOX® parece ser dependiente de la duración de la terapia (en general mayor de 2 semanas de tratamiento). Los recuentos de plaquetas en la mayoría de los pacientes retornaron al rango normal/basal durante el período de seguimiento. No se identificaron eventos adversos clínicos relacionados en los ensayos clínicos de Fase 3 en pacientes que desarrollaban trombocitopenia. Se identificaron eventos de hemorragia en pacientes con trombocitopenia en un programa de uso compasivo para ZYVOX®; la función de linezolid en estos eventos no puede determinarse (ver Advertencias).
Los cambios observados en otros parámetros de laboratorio, sin considerar la relación con el fármaco, no indicaron diferencias importantes entre ZYVOX® y los comparadores. Estos cambios en general no fueron clínicamente significativos, no indujeron a interrumpir la terapia y fueron reversibles. La incidencia de pacientes adultos y pediátricos con al menos un valor hematológico o de bioquímica en suero considerablemente anormal se presenta en los Cuadros 8, 9, 10 y 11.
Cuadro 8. Porcentaje de pacientes adultos que experimentaron por lo menos un valor hematológico de laboratorio considerablemente anormal* en estudios clínicos controlados con comparador con ZYVOX®
|
Prueba de laboratorio |
Infecciones no complicadas de la piel y de la estructura |
Todas las demás indicaciones |
||
|
ZYVOX® 400 mg q12h |
Claritromicina 250 mg q12h |
ZYVOX® 600 mg q12h |
Todos los demás comparadores† |
|
|
Hemoglobina (g/dL) |
0,9 |
0,0 |
7,1 |
6,6 |
|
Recuento de plaquetas (x 103/mm3) |
0,7 |
0,8 |
3,0 |
1,8 |
|
Leucocitos (x 103/mm3) |
0,2 |
0,6 |
2,2 |
1,3 |
|
Neutrófilos (x 103/mm3) |
0,0 |
0,2 |
1,1 |
1,2 |
|
* <75% (<50% para neutrófilos) del Límite Inferior Normal (LLN) para valores normales a nivel basal; <75% (<50% para neutrófilos) de LLN y de la basal para valores anormales a nivel basal. † Los comparadores incluyeron cefpodoxima proxetil 200 mg por mes cada 12h; ceftriaxona 1 g IV cada 12h; dicloxacilina 500 mg por mes cada 6h; oxacilina 2 g IV cada 6h; vancomicina 1 g IV cada 12h. |
||||
Cuadro 9. Porcentaje de pacientes adultos que experimentaron por lo menos un valor de bioquímica en suero de laboratorio considerablemente anormal* en estudios clínicos controlados con comparador con ZYVOX®
|
Prueba de laboratorio |
Infecciones no complicadas de la piel y de la estructura |
Todas las demás indicaciones |
||
|
ZYVOX® 400 mg q12h |
Claritromicina 250 mg q12h |
ZYVOX® 600 mg q12h |
Todos los demás comparadores† |
|
|
AST (U/L) |
1,7 |
1,3 |
5,0 |
6,8 |
|
ALT (U/L) |
1,7 |
1,7 |
9,6 |
9,3 |
|
LDH (U/L) |
0,2 |
0,2 |
1,8 |
1,5 |
|
Fosfatasa alcalina (U/L) |
0,2 |
0,2 |
3,5 |
3,1 |
|
Lipasa (U/L) |
2,8 |
2,6 |
4,3 |
4,2 |
|
Amilasa (U/L) |
0,2 |
0,2 |
2,4 |
2,0 |
|
Bilirrubina total (mg/dL) |
0,2 |
0,0 |
0,9 |
1,1 |
|
Nitrógeno ureico sanguíneo (mg/dL) |
0,2 |
0,0 |
2,1 |
1,5 |
|
Creatinina (mg/dL |
) 0,2 |
0,0 |
0,2 |
0,6 |
|
* >2 veces el límite superior normal (ULN) para valores normales en la basal; >2 veces el ULN y >2 veces el valor basal para valores anormales en la basal. † Los comparadores incluyeron cefpodoxima proxetil 200 mg por mes cada 12h; ceftriaxona 1 g IV cada 12h; dicloxacilina 500 mg por mes cada 6h; oxacilina 2 g IV cada 6h; vancomicina 1 g IV cada12h. |
||||
Cuadro 10. Porcentaje de pacientes pediátricos que experimentaron por lo menos un valor hematológico de laboratorio considerablemente anormal* en estudios clínicos controlados con comparador con ZYVOX®
|
Prueba de laboratorio |
Infecciones no complicadas de la piel y de la estructura† |
Todas las demás indicaciones‡ |
||
|
ZYVOX® |
Cefadroxilo |
ZYVOX® |
Vancomicina |
|
|
Hemoglobina (g/dL) |
0,0 |
0,0 |
15,7 |
12,4 |
|
Recuento de plaquetas (x 103/mm3) |
0,0 |
0,4 |
12,9 |
13,4 |
|
Leucocitos (x 103/mm3) |
0,8 |
0,8 |
12,4 |
10,3 |
|
Neutrófilos (x 103/mm3) |
1,2 |
0,8 |
5,9 |
4,3 |
|
* <75% (<50% para neutrófilos) del límite inferior normal (LLN) para valores normales en la basal; <75% (<50% para neutrófilos) del LLN y <75% (<50% para neutrófilos, <90% para hemoglobina si la basal <LLN) del valor basal para valores anormales en la basal. † Los pacientes entre 5 y 11 años recibieron ZYVOX® 10 mg/kg por mes cada 12h o cefadroxilo 15 mg/kg por mes cada 12h. Los pacientes de 12 años o mayores recibieron ZYVOX® 600 mg por mes cada 12h o cefadroxilo 500 mg por mes cada 12h. ‡ Los pacientes desde el nacimiento hasta los 11 años recibieron ZYVOX® 10 mg/kg IV/por mes cada 8h o vancomicina 10 a 15 mg/kg IV q6-24h, según la edad y la depuración renal. |
||||
Cuadro 11. Porcentaje de pacientes pediátricos que experimentaron por lo menos un valor de laboratorio de bioquímica en suero considerablemente anormal* en estudios clínicos controlados con comparador con ZYVOX®
|
Prueba de laboratorio |
Infecciones no complicadas de la piel y de la estructura† |
Todas las demás indicaciones‡ |
||
|
ZYVOX® |
Cefadroxilo |
ZYVOX® |
Vancomicina |
|
|
ALT (U/L) |
0,0 |
0,0 |
10,1 |
12,5 |
|
Lipasa (U/L) |
0,4 |
1,2 |
-- |
-- |
|
Amilasa (U/L) |
-- |
-- |
0,6 |
1,3 |
|
Bilirrubina total (mg/dL) |
-- |
-- |
6,3 |
5,2 |
|
Creatinina (mg/dL) |
0,4 |
0,0 |
2,4 |
1,0 |
|
* >2 veces el límite superior normal (ULN) para valores normales en la basal; >2 veces el ULN y >2 (>1.5 para bilirrubina total) veces el valor basal para valores anormales en la basal. † Los pacientes entre 5 y 11 años recibieron ZYVOX® 10 mg/kg por mes cada 12h o cefadroxilo 15 mg/kg por mes cada 12h. Los pacientes de 12 años a más recibieron ZYVOX® 600 mg por mes cada 12h o cefadroxilo 500 mg por mes cada 12h. ‡ Los pacientes desde el nacimiento hasta los 11 años recibieron ZYVOX® 10 mg/kg IV/por mes cada 8h o vancomicina 10 a 15 mg/kg IV q6-24h, según la edad y la depuración renal. |
||||
Experiencia posterior a la comercialización: Estos eventos han sido seleccionados para su inclusión debido a su gravedad, frecuencia de información, posible relación causal con ZYVOX®, o a una combinación de estos factores.
Debido a que estos eventos fueron informes voluntarios de una población de tamaño desconocido, no puede realizarse la estimación de la frecuencia ni determinarse la relación causal.
Durante el uso posterior a la comercialización de ZYVOX® se ha informado mielosupresión (incluida anemia, leucopenia, pancitopenia, y trombocitopenia) (ver Advertencias). En pacientes tratados con ZYVOX®, se han informado casos de neuropatía periférica y neuropatía óptica que algunas veces evolucionaron hacia pérdida de la visión. Se ha informado de acidosis láctica con el uso de ZYVOX® (ver Precauciones). Si bien estos informes provienen primordialmente de pacientes tratados durante períodos más largos que la duración máxima recomendada de 28 días estos eventos también han sido informados en pacientes que recibieron ciclos más cortos de terapia. Se ha informado de síndrome de serotonina en pacientes que reciben agentes serotoninérgicos concomitantes, incluidos antidepresivos como los inhibidores de la recaptación de serotonina selectiva (SSRI) y ZYVOX® (ver Precauciones). Se han informado casos de convulsiones con el uso de ZYVOX® (ver Precauciones). Se ha informado de anafilaxis, angioedema y alteraciones ampollosas de la piel, tales como los descritos en el síndrome de Stevens-Johnson. Se ha informado hipoglucemia, incluidos episodios sintomáticos (ver Precauciones).
INTERACCIONES MEDICAMENTOSAS
Inhibición de la Monoamino Oxidasa: Linezolid es un inhibidor no selectivo y reversible de la monoamino oxidasa.
Agentes serotoninérgicos y andrenérgicos: Linezolid tiene el potencial para interaccionar con agentes serotoninergicos y adrenérgicos.
MICROBIOLOGÍA
Mecanismo de acción: Linezolid es un agente antibacteriano sintético perteneciente a una nueva clase de antibióticos, las oxazolidinonas, con utilidad clínica en el tratamiento de infecciones causadas por bacterias aeróbicas Gram positivas. El espectro in vitro de la actividad de linezolid también incluye ciertas bacterias Gram negativas y bacterias anaeróbicas. Linezolid inhibe la síntesis de proteínas bacterianas por medio de un mecanismo de acción diferente al de otros agentes antibacterianos; por ello, es improbable la resistencia cruzada entre linezolid y otras clases de antibióticos. Linezolid se une a un sitio en el ARN ribosomal 23S bacteriano de la subunidad 50S y previene la formación de un complejo de iniciación 70S funcional, el cual es un componente esencial en el proceso de traducción bacteriana. Los resultados de los estudios de curvas tiempo-muerte han mostrado que linezolid es bacteriostática contra enterococos y estafilococos. Se observó que linezolid era bactericida para estreptococos en la mayoría de las cepas.
Mecanismo de resitencia: En estudios clínicos, se desarrolló resistencia a linezolid en 6 pacientes infectados con Enterococcus faecium (4 pacientes recibieron 200 mg q12h, menos que la dosis recomendada, y dos pacientes recibieron 600 mg q12h). En un programa de uso compasivo, se desarrolló resistencia a linezolid en 8 pacientes con E. faecium y en un paciente con Enterococcus faecalis. Todos los pacientes tuvieron dispositivos prostéticos no retirados o abscesos no drenados. La resistencia a linezolid se produce “in vitro” con una frecuencia de 1 x 10-9 a 1 x 10-11. Estudios “in vitro” han mostrado que las mutaciones puntuales en el ARNr 23S están relacionadas con la resistencia a linezolid. Se han publicado informes sobre E. faecium resistente a la vancomicina que ha desarrollado resistencia a linezolid durante su uso clínico. En un informe se mencionó la aparición de diseminación nosocomial de E. faecium resistente a vancomicina y a linezolid. Hubo también un informe de Staphylococcus aureus (resistente a la meticilina) que desarrolló resistencia a linezolid durante su uso clínico. La resistencia a linezolid en esos organismos se asoció con una mutación puntual en ARNr 23S (sustitución de timina por guanina en la posición 2576) del organismo. También la resistencia a linezolid en los estafilococos mediada por la enzima metiltransferasa ha sido reportada. Esta resistencia está mediada por el gen cfr (cloranfenicol-florfenicol) situado en un plásmido que es transferible entre estafilococos.
Resistencia a linezolid no ha sido resportado en Streptococcus spp., incluyendo Streptococcus pneumoniae.
Ineracciones con otros antimicrobianos: Según los estudios in vitro, existe una adición o indiferencia entre linezolid y vancomicina, gentamicina, rifampina, imipenem-cilastatina, aztreonam, ampicilina o estreptomicina.
Se ha demostrado que linezolid es activa contra la mayoría de aislados de los siguientes microorganismos, tanto in vitro como en infecciones clínicas, como se describe en la sección Indicaciones y uso.
• Microorganismos Gram positivos aeróbicos:
– Enterococcus faecium (sólo cepas resistentes a la vancomicina)
– Staphylococcus aureus (incluidas las cepas resistentes a la meticilina)
– Streptococcus agalactiae
– Streptococcus pneumoniae
– Streptococcus pyogenes
Los siguientes datos in vitro se encuentran disponibles, pero se desconoce su significado clínico. Por lo menos 90% de los siguientes microorganismos muestran una concentración inhibitoria mínima in vitro (MIC) menor o equivalente al punto de corte de susceptibilidad de linezolid. No obstante, no se ha establecido la seguridad y efectividad de linezolid para tratar infecciones clínicas debido a estos microorganismos en ensayos clínicos adecuados y bien controlados.
• Microorganismos Gram positivos:
– Enterococcus faecalis (incluidas las cepas resistentes a la vancomicina)
– Enterococcus faecium (cepas susceptibles a la vancomicina)
– Staphylococcus epidermidis (incluidas las cepas resistentes a la meticilina)
– Staphylococcus haemolyticus
– Estreptococos del grupo viridans.
• Microorganismos Gram negativos:
– Pasteurella multocida
Métodos para pruebas de susceptibilidad: Si están disponibles, los resultados de las pruebas de susceptibilidad in vitro se proporcionarán al médico como informes periódicos donde se describirá el perfil de susceptibilidad de los patógenos nosocomiales y adquiridos en la comunidad. Estos informes ayudarán al médico a elegir los antimicrobianos más eficaces.
• Técnicas de dilución: Para determinar las concentraciones inhibitorias mínimas (MIC) de antimicrobianos se emplean métodos cuantitativos. Estas MIC dan los estimados de susceptibilidad de las bacterias frente a los compuestos antimicrobianos. Las MIC deben determinarse utilizando un procedimiento estándar. Los procedimientos estándar están basados en un método de dilución (caldo o agar) o equivalente, con concentraciones de inóculos estándar y concentraciones estándar de polvo de linezolid. Los valores de MIC deben interpretarse según los criterios que se contemplan en el Cuadro 4.
• Técnicas de difusión: Los métodos cuantitativos que requieren medir los diámetros de zona también ofrecen estimados reproducibles de susceptibilidad de las bacterias a los compuestos antimicrobianos. Uno de esos procedimientos estándar requiere el uso de concentraciones de inóculos estándar. Este procedimiento emplea discos de papel impregnados con 30 µg de linezolid para probar la susceptibilidad de los microorganismos a linezolid. Los criterios de interpretación de la difusión en disco se indican en el Cuadro 4.
Cuadro 4. Criterios Interpretativos de Susceptibilidad para Linezolid
|
Patógeno |
Criterios interpretativos de susceptibilidad |
|||||
|
Concentraciones inhibitorias mínimas (MIC en µg/mL) |
Difusión en disco (diámetros de zona en mm) |
|||||
|
S |
I |
R |
S |
I |
R |
|
|
Enterococcus spp. |
≤2 |
4 |
≥8 |
≥23 |
21-22 |
≤20 |
|
Staphylococcus spp.a |
≤4 |
--- |
--- |
≥21 |
--- |
-- |
|
Streptococcus pneumoniaea |
≤2 |
--- |
--- |
≥21 |
--- |
-- |
|
Streptococcus spp. distintos de S pneumoniaea |
≤2 |
--- |
--- |
≥21 |
--- |
--- |
|
a. La falta actual de datos sobre cepas resistentes impide definir categorías distintas a “susceptibles”. Las cepas que arrojan resultados de prueba que sugieren una categoría “no susceptible” deben volverse a evaluar, y si se confirma el resultado el aislamiento debe enviarse a un laboratorio de referencia para adicional evaluación. |
||||||
Un informe de “Susceptible” indica que el patógeno probablemente esté inhibido si el compuesto antimicrobiano en la sangre alcanza las concentraciones usualmente obtenibles.
Un informe de “Intermedio” indica que el resultado debe considerarse equívoco y, si el microorganismo no es totalmente susceptible a fármacos alternativos y clínicamente viables, la prueba deberá ser repetida. Esta categoría implica una posible aplicabilidad clínica en los lugares del cuerpo donde la droga está fisiológicamente concentrada, o en situaciones en que puede usarse una dosis alta de fármaco. Esta categoría también ofrece una zona de amortiguación que impide que factores técnicos pequeños no controlados provoquen mayores discrepancias de interpretación. Un informe de “Resistente” indica que el patógeno probablemente no se inhibirá si el compuesto antimicrobiano en la sangre alcanza las concentraciones usualmente obtenibles; deberá elegirse otra terapia.
• Control de calidad: Los procedimientos estándar de ensayos de susceptibilidad requieren el uso de microorganismos de control de calidad para controlar los aspectos técnicos de los procedimientos de prueba. El polvo de linezolid estándar deberá proveer el siguiente rango de valores que se indica en el Cuadro 5.
• NOTA:
Cuadro 5. Rangos de control de calidad aceptables para linezolid que se utilizarán en la validación de los resultados de la prueba de susceptibilidad
|
Cepas para control de calidad |
Rangos aceptables para control de calidad |
|
|
Concentración mínima inhibitoria (MIC en µg/mL) |
Difusión en disco (diámetros de zona en mm) |
|
|
Enterococcus faecalis |
1 – 4 |
No aplicable |
|
Staphylococcus aureus |
1 – 4 |
No aplicable |
|
Staphylococcus aureus |
No aplicable |
25 - 32 |
|
Streptococcus pneumoniae |
0.50 - 2 |
25 – 34 |
|
a. Este organismo puede utilizarse para validar los resultados de la prueba de susceptibilidad cuando se evalúa Streptococcus spp. distinto de S. pneumoniae |
||
TOXICOLOGÍA NO CLÍNICA
Carcinogénesis, mutagénesis, deterioro de la fertilidad: No se han realizado estudios de por vida en animales para evaluar el potencial carcinogénico de linezolid. No se ha demostrado potencial mutagénico ni clastogénico en una serie de pruebas que incluyó: ensayos de mutagenicidad (mutación de células CHO y reversión bacteriana Ames), un ensayo in vitro de síntesis de ADN no programada (UDS), un ensayo de aberración cromosómica in vitro en linfocitos humanos, y un ensayo de micronúcleos en ratón in vivo.
Linezolid no afectó la fertilidad o el desempeño reproductivo en ratas hembras adultas, disminuyó la fertilidad y el desempeño reproductivo de manera reversible en ratas machos adultos cuando se administró en dosis ≥50 mg/kg/día, con exposiciones aproximadamente mayores o iguales que el nivel de exposición esperado en humanos (las comparaciones de exposición están basadas en las AUC). Los efectos reversibles en la fertilidad fueron mediados a través de espermatogénesis alterada. Las espermátides afectadas contenían mitocondrias con formación y orientación anormal y no eran viables. Se observó hipertrofia celular epitelial e hiperplasia en el epidídimo, junto con disminución de la fertilidad. No se observaron cambios similares en el epidídimo en perros.
En ratas machos con madurez sexual expuestos al fármaco al igual que las ratas jóvenes se observó leve disminución de la fertilidad después del tratamiento con linezolid en la mayoría de su periodo de desarrollo sexual (50 mg/kg/día desde los días 7 a 36 de edad, y 100 mg/kg/día desde los días 37 a 55 de edad), con exposiciones de hasta 1,7 veces mayores que la media de las AUC observadas en pacientes pediátricos de 3 meses a 11 años. No se observó disminución de la fertilidad con periodos de tratamiento más cortos, que corresponde a la exposición en el útero hasta el periodo neonatal inicial (día de gestación 6 hasta el día postnatal 5), exposición neonatal (días postnatales 5 a 21), o exposición juvenil (días postnatales 22 a 35). Se observaron disminuciones reversibles de la motilidad del esperma y alteraciones en la morfología del esperma en ratas que recibieron tratamiento desde el día postnatal 22 a 35.
Toxicologia animal y/o farmacológica: Los órganos objetivo de la toxicidad de linezolid fueron similares en ratas y perros jóvenes y adultos . Dosis y mielosupresión dependiente del tiempo, evidenciado por hipocelularidad médula ósea/disminución de la hematopoyesis , disminución de la hematopoyesis extramedular en el bazo y el hígado, y la disminución de los niveles de eritrocitos circulantes, leucocitos y plaquetas se han visto en estudios con animales. Depleción linfoide se produjo en el timo, los ganglios linfáticos y el bazo. En general, los hallazgos linfoides se asociaron con anorexia, pérdida de peso, y la supresión del aumento de peso, lo que puede haber contribuido a los efectos observados.
Se observó mínimo a leve degeneración axonal de los nervios ciáticos, no reversibles, en ratas administradas con linezolid a 80 mg/kg/día por vía oral durante 6 meses; también se observó degeneración mínima del nervio ciático en 1 macho a este nivel de dosis en un 3-mes necropsia provisional. Se llevó a cabo la evaluación morfológica de los tejidos sensibles de perfusión-fijos para investigar evidencia de degeneración del nervio óptico. Mínima a moderada degeneración del nervio óptico fue evidente en 2 ratas macho después de 6 meses de dosificación , pero la relación directa a las drogas fue equívoca debido a la naturaleza aguda de la hallazgo y su distribución asimétrica. La degeneración del nervio observado microscópicamente era comparable a la degeneración espontánea del nervio óptico unilateral reportado en ratas de edad avanzada y puede ser una exacerbación de un cambio común.
Estos efectos se observaron a niveles de exposición que son comparables a los observados en algunos sujetos humanos. Los efectos hematopoyéticos y linfoides fueron reversibles, aunque en algunos estudios, la reversión fue incompleta dentro de la duración del período de recuperación
ADVERTENCIAS Y PRECAUCIONES
Mielosupresión: Se ha informado mielosupresión en pacientes que recibieron linezolid (incluidas anemia, leucopenia, pancitopenia y trombocitopenia). En los casos en que se conoce la evolución, tras la descontinuación de linezolid, los parámetros hematológicos afectados aumentaron hasta llegar a los niveles de pre-tratamiento. Se debe monitorear semanalmente con un hemograma completo a los pacientes que reciben linezolid, particularmente a aquéllos que reciben linezolid durante más de dos semanas, aquéllos con mielosupresión pre-existente, aquéllos que reciben fármacos concomitantes que producen depresión de la médula ósea, o aquéllos con infecciones crónicas que han recibido terapia antibiótica previa o concomitante. Se debe considerar la discontinuación de la terapia con linezolid en pacientes que desarrollen o empeoren una condición de mielosupresión.
Neuropatía periférica y óptica: Se ha informado de neuropatía periférica y óptica en pacientes tratados con ZYVOX®, principalmente en aquellos pacientes que recibieron tratamiento por períodos más largos que la duración máxima recomendada de 28 días. En los casos de neuropatía óptica que evolucionaron hacia la pérdida de la visión, los pacientes recibieron tratamiento durante períodos más prolongados que la duración máxima recomendada. Se ha informado de visión borrosa en algunos pacientes tratados con ZYVOX® durante menos de 28 días. Neuroparias óptica y perfiferica han sido reportados en niños.
Se recomienda la evaluación oftalmológica inmediata si aparecen síntomas de deterioro de la visión, tales como alteraciones en la agudeza visual, alteraciones en la visión de los colores, visión borrosa o defectos del campo visual. La función visual debe supervisarse en todos los pacientes que reciben ZYVOX® durante períodos prolongados (≥3 meses) y en todos los pacientes que informen nuevos síntomas visuales independientemente de la duración de la terapia con ZYVOX®. Si se informa de neuropatía periférica u óptica, deberá evaluarse la continuidad del uso de ZYVOX® en estos pacientes frente a los riesgos potenciales
Síndrome de serotonina: Se han presentado informes espontáneos de síndrome serotoninérgico, incluidos casos fatales, asociado con la coadministración de ZYVOX® y agentes serotoninérgicos, incluidos antidepresivos tales como los inhibidores selectivos de la recaptación de serotonina (SSRI) (ver Precauciones: Interacciones medicamentosas).
A menos que sea adecuado clínicamente y los pacientes sean observados cuidadosamente para detectar signos y/o síntomas de síndrome de la serotonina o reacciones similares al síndrome (RSM-similar) neuroléptico maligno, linezolid no se debe administrar a pacientes con síndrome de Down y/o pacientes que toman cualquiera de los siguientes medicamentos carcinoide : inhibidores de la recaptación de serotonina, antidepresivos tricíclicos, los agonistas del receptor de la serotonina 5 HT1 (triptanos), meperidina, bupropión, o buspirona.
En algunos casos, un paciente que ya recibe un antidepresivo serotonérgico o buspirona puede requerir tratamiento urgente con linezolid. Si no están disponibles alternativas al linezolid y los beneficios potenciales del linezolid superan los riesgos del síndrome serotoninérgico o las reacciones del tipo síndrome neuroléptico maligno, debe detenerse con prontitud el antidepresivo serotonérgico y administrarse linezolid. Debe monitorearse al paciente por dos semanas (cinco semanas si se tomó fluoxetina) o hasta 24 horas después de la última dosis de linezolid, lo primero que ocurra. Los síntomas del síndrome serotonérgico o de las reacciones del tipo síndrome neuroléptico maligno incluyen hipertermia, rigidez, mioclonía, inestabilidad autonómica y cambios en el estado mental que incluyen agitación extrema que progresa hasta el delirio y coma. También debe monitorearse al paciente en busca de síntomas de descontinuación del antidepresivo. (vea el inserto del empaque del (de los) agente(s) especificado(s) para conocer la descripción de los síntomas de descontinuación asociados.
Desequilibrio de la mortalidad en un estudio de investigación realizado en pacientes con infecciones circulatorias relacionadas a catéter, incluidas las infecciones en el sitio del catéter: Se observó un desequilibrio de la mortalidad en pacientes tratados con linezolid en relación con vancomicina/dicloxacilina/oxacilina en un estudio abierto con pacientes gravemente enfermos con infecciones intravasculares relacionadas a catéter [78/363 (21,5%) frente a 58/363 (16,0%); razón de probabilidad 1,426, CI de 95% 0,970, 2,098]. Aunque no se estableció la causalidad, este desequilibrio observado se produjo principalmente en pacientes tratados con linezolid en quienes se identificaron patógenos Gram negativos, una combinación de patógenos Gram negativos y Gram positivos, o ningún patógeno identificado al incio, pero no no fue observado en pacientes con infecciones por Gram positivos solamente.
Linezolid no está aprobado y no debe ser usado para el tratamiento de pacientes con infecciones circulatorias relacionadas a catéter o infecciones en el sitio del catéter.
Linezolid no tiene actividad clínica contra los patógenos Gram negativos y no está indicada para el tratamiento de infecciones por Gram negativos. Es vital que se inicie una terapia específica contra Gram negativos si hay documentación o sospecha de un patógeno Gram negativo concomitante (ver Indicaciones y uso).
Clostridium difficile asociado a diarrea: Se ha informado de diarrea asociada con Clostridium difficile (CDAD) con el uso de casi todos los agentes antibacterianos, incluido ZYVOX®, cuya severidad puede variar desde diarrea leve hasta colitis fatal. El tratamiento con agentes antibacterianos altera la flora normal del colon, lo que produce el crecimiento excesivo de C difficile.
C. difficile produce toxinas A y B que contribuyen al desarrollo de CDAD. Las cepas que producen hipertoxinas de C. difficile causan aumento de la morbilidad y de la mortalidad, ya que estas infecciones pueden ser refractarias a la terapia antimicrobiana y pueden requerir colectomía. Debe considerarse la posibilidad de CDAD en todos los pacientes que presenten diarrea después del uso de antibióticos.
Es necesaria una historia médica cuidadosa, ya que se ha reportado que CDAD se presenta más de dos meses después de la administración de agentes antibacterianos.
Si hay sospecha o confirmación de CDAD, puede ser necesario suspender el tratamiento antibiótico en curso no dirigido a C. difficile. Se deberá instituir un manejo adecuado de líquidos y electrolitos, complementos de proteínas, tratamiento antibiótico contra C. difficile y evaluación quirúrgica, según este indicado clínicamente.
Potenciales interacciones serotoninérgicas: A menos que los pacientes sean cuidadosamente observados para detectar signos y/o síntomas del síndrome serotoninérgico o reacciones del tipo síndrome neuroléptico maligno, no debe administrarse linezolid a pacientes con síndrome carcinoide y/o pacientes que estén tomando cualquiera de los siguientes medicamentos: inhibidores de la recaptación de serotonina, antidepresivos tricíclicos, agonistas del receptor 5-HT1 para serotonina (triptanos), meperidina, bupropión o buspirona (ver Precauciones: General e Interacciones medicamentosas).
Potenciales interacciones que produce elevación de la presión arterial: A menos que los pacientes sean monitoreados por posibles aumentos en la presión arterial, linezolid no se debe administrar a pacientes con hipertensión no controlada, feocromocitoma, hipertiroidismo y/o pacientes que toman cualquiera de los siguientes tipos de medicamentos: actuación directa o indirecta de los agentes simpaticomiméticos (por ejemplo, pseudoefedrina), agentes vasopresores (por ejemplo, epinefrina, norepinefrina), agentes dopaminérgicos (por ejemplo, dopamina, dobutamina).
Acidosis láctica: Se ha informado de acidosis láctica con el uso de ZYVOX®. En los casos informados, los pacientes experimentaron repetidos episodios de náuseas y vómitos. Los pacientes que desarrollen náuseas o vómitos recurrentes, acidosis sin explicación o un bajo nivel de bicarbonato mientras reciben ZYVOX® deben recibir inmediata evaluación médica.
Convulsiones: Se ha informado la ocurrencia de convulsiones en pacientes tratados con linezolid. En algunos de estos casos, se informó antecedentes de convulsiones o factores de riesgo para convulsiones.
Hipoglucemia: Casos post comercialización de hipoglucemia sintomática se han informado en pacientes con diabetes mellitus que reciben insulina o agentes hipoglucémicos orales cuando se tratan con linezolid, un inhibidor de la MAO reversible, no selectivo. Algunos inhibidores de la MAO se han asociado con episodios hipoglucémicos en pacientes diabéticos que reciben insulina o agentes hipoglucémicos. Aunque no se ha establecido una relación causal entre linezolid y la hipoglucemia, los pacientes diabéticos deben tener precaución en caso de potenciales reacciones hipoglucémicas cuando se les administra linezolid.
Si ocurre hipoglucemia, puede requerirse una descontinuación en la dosis de insulina o del agente hipoglucémico oral, de la insulina o de linezolid.
Desarrollo de bacterias resistente a medicamentos: La prescripción de ZYVOX® en ausencia de una infección bacteriana comprobada o fuertemente sospechada, o una indicación profiláctica es improbable que aporte beneficios al paciente, y aumenta el riesgo de desarrollar bacterias resistentes a fármacos.
DOSIFICACIÓN Y ADMINISTRACIÓN: En el siguiente cuadro se describe la dosificación recomendada para las formulaciones de ZYVOX® para el tratamiento de infecciones.
Pautas para la dosificación de ZYVOX®
|
Infección* |
Dosificación y vía de administración |
Duración recomendada del tratamiento (días consecutivos) |
|
|
Pacientes pediátricos† (desde el nacimiento hasta los 11 años) |
Adultos y adolescentes (12 años y mayores) |
||
|
Infecciones complicadas de la piel y de la estructura cutánea |
10 mg/kg IV o por vía oral‡ cada 8h |
600 mg IV o por vía oral‡ c 12h |
10 a 14 |
|
Neumonía adquirida en la comunidad, incluida bacteriemia concurrente |
|||
|
Neumonía nosocomial |
|||
|
Infecciones por Enterococcus faecium resistente a la vancomicina, incluida bacteriemia concurrente |
10 mg/kg IV o por vía oral‡ cada 8h |
600 mg IV o por vía oral‡ cada 12h |
14 a 28 |
|
Infecciones no complicadas de la piel y de la estructura cutánea |
<5 años: 10 mg/kg oral‡ cada 8h 5-11 años: 10 mg/kg oral‡ cada 12h |
Adultos: 400 mg por vía oral‡ cada 12h Adolescentes: 600 mg por vía oral‡ cada 12h |
10 a 14 |
|
* Debido a los patógenos designados (ver Indicaciones y uso) † Neonatos <7 días: La mayoría de los neonatos pretérmino <7 días de edad (edad gestacional < 34 semanas) tienen valores menores de depuración sistémica de linezolid y valores mayores de AUC que muchos neonatos nacidos a término y bebés mayores. Estos neonatos deben iniciar con un régimen de dosificación de 10 mg/kg q12h. Se debe considerar el uso de un régimen de 10 mg/kg q8h en neonatos con una respuesta clínica subóptima. Todos los pacientes neonatos deben recibir 10 mg/kg q8h a los 7 días de vida (ver Farmacología clínica: Poblaciones especiales, Pediátricos). ‡ Dosificación oral usando tabletas de ZYVOX®. |
|||
No se requiere ningún ajuste de dosis al cambiar de la administración intravenosa a la oral.
Administración intravenosa (I.V.)
ZYVOX® Solución Inyectable (I.V.) se provee en bolsas plásticas, de una única dosis, para infusión listas para usar. Las bolsas deben ser inspeccionadas visualmente para verificar la ausencia de partículas antes de ser utilizadas. Se aconseja la observación de posibles filtraciones o goteo de las bolsas, luego de ser apretadas por unos minutos. Si esto ocurriera, descartar las bolsas ya que han perdido la esterilidad.
La solución inyectable debe ser administrada por infusión intravenosa en un período de 30 a 120 minutos. No usar conexiones en serie con las bolsas de infusión. No agregar aditivos en la solución. Si ZYVOX® debe administrarse en forma concomitante con otras drogas, cada droga debe ser administrada separadamente de acuerdo con el dosaje y vías recomendadas para cada producto. Particularmente, puede haber incompatibilidades físicas, cuando ZYVOX® Solución inyectable (I.V.) se combina con las siguientes drogas durante la administración simulada en Y: anfotericina B, clorhidrato de clorpromazina, diazepam, isotionato de pentamidina, lactobionato de eritromicina, fenitoína sódica y trimetoprima-sulfametoxazol. Adicionalmente, existe incompatibilidad química en la combinación de ZYVOX® Solución inyectable (I.V.) con ceftriaxona sódica.
Si el mismo sistema de infusión se utiliza para administrar secuencialmente distintas drogas, éste debe ser enjuagado antes y después de la infusión de ZYVOX® Solución inyectable (I.V.), con una solución de infusión compatible con ZYVOX® Solución inyectable (I.V.) y con otra(s) droga(s) administrada(s) por esa vía común.
• Soluciones intravenosas compatibles:
– Solución de Dextrosa Inyectable al 5% USP.
– Solución Inyectable de Cloruro de Sodio al 0,9% USP.
– Solución de Ringer Lactato USP.
USO EN POBLACIONES ESPECíFICAS
Embarazo
• Efectos teratogénicos, Embarazo Categoría C: Linezolid no fue teratogénica en ratones, ratas o conejos con niveles de exposición de 6,5 veces (en ratones), equivalente a (en ratas), o 0,06 veces (en conejos) que el nivel de exposición esperado en humanos, con base en las AUCs. Sin embargo, se observó toxicidad embrionaria y fetal (ver Efectos no teratogénicos).
No existen estudios apropiados y bien controlados en mujeres embarazadas. Debe usarse ZYVOX® en el embarazo sólo si el beneficio potencial justifica el riesgo potencial para el feto.
• Efectos no teratogénicos: En ratones se observaron toxicidades embrionarias y fetales sólo con dosis que causaron toxicidad materna (signos clínicos y disminución de la ganancia de peso corporal). Una dosis de 450 mg/kg/día (6,5 veces el nivel de exposición estimado en seres humanos con base en las AUC) se correlacionó con el aumento de muertes embrionarias post-implantación, incluida pérdida de toda la camada, disminución de los pesos corporales fetales y aumento de la incidencia de fusión del cartílago costal.
En ratas, se observó toxicidad fetal leve con 15 y 50 mg/kg/día (niveles de exposición hasta 0,22 veces aproximadamente a la exposición humana equivalente estimada, respectivamente, con base en las AUC). Los efectos consistieron en disminución de los pesos corporales fetales y disminución de la osificación de esternebras, un hallazgo que se observa con frecuencia en asociación con disminución de los pesos corporales fetales. Se observó ligera toxicidad materna, en forma de disminución de la ganancia de peso corporal, con 50 mg/kg/día.
En conejos, se produjo disminución del peso corporal fetal sólo en presencia de toxicidad materna (signos clínicos, disminución de la ganancia de peso corporal y consumo de alimentos) cuando se administraron dosis de 15 mg/kg/día (0,06 veces la exposición humana estimada con base en las AUC).
Cuando se trató ratas hembras con 50 mg/kg/día (aproximadamente equivalente a la exposición estimada en seres humanos con base en las AUC) de linezolid durante el embarazo y la lactancia, se observó disminución de la supervivencia de las crías en los días postnatales 1 a 4. Las crías machos y hembras que pudieron madurar hasta alcanzar la edad reproductiva, una vez apareadas, mostraron aumento de pérdida antes de la implantación.
Madres que dan de lactar: Linezolid y sus metabolitos son excretados a través de la leche de ratas que dan de lactar. Las concentraciones en la leche fueron similares a aquéllas obtenidas en el plasma materno. Se desconoce si linezolid se excreta a través de la leche humana. Dado que muchos medicamentos son excretados a través de la leche humana, se debe tener cuidado se administre ZYVOX® a mujeres en período de lactancia.
Uso pediátrico: La seguridad y eficacia de ZYVOX® para el tratamiento de pacientes pediátricos con las siguientes infecciones están respaldadas por evidencia proveniente de estudios apropiados y bien controlados realizados en adultos, datos farmacocinéticos de pacientes pediátricos y datos adicionales de un estudio controlado con comparador en infecciones por Gram positivos en pacientes pediátricos con edades que oscilaron entre el nacimiento hasta los 11 años (ver Indicaciones y uso):
• Neumonía nosocomial.
• Infecciones complicadas de la piel y de la estructura cutánea.
• Neumonía adquirida en la comunidad (también respaldada por evidencia proveniente de un estudio no controlado realizado en pacientes cuyas edades oscilaban entre 8 meses y 12 años).
• Infecciones por Enterococcus faecium resistente a la vancomicina.
Se ha establecido la seguridad y eficacia de ZYVOX® para el tratamiento de pacientes pediátricos con la siguiente infección, en un estudio controlado con comparador realizado en pacientes pediátricos cuyas edades oscilaban entre 5 hasta 17 años .
• Infecciones no complicadas de la piel y de la estructura cutánea causadas por Staphylococcus aureus (sólo cepas susceptibles a la meticilina) o Streptococcus pyogenes.
La información farmacocinética generada en pacientes pediátricos con derivaciones ventriculoperitoneales mostró concentraciones variables de linezolid en el líquido cerebrospinal (CSF) después de dosis únicas y múltiples de linezolid; no se alcanzaron o mantuvieron consistentemente concentraciones terapéuticas en el CSF. Por lo tanto, no se recomienda el uso de linezolid para el tratamiento empírico de pacientes pediátricos con infecciones en el sistema nervioso central.
La farmacocinética de linezolid se ha evaluado en pacientes pediátricos desde el nacimiento hasta los 17 años de edad. En general, el aclaramiento basado en el peso de linezolid disminuye gradualmente a medida que aumenta la edad de los pacientes pediátricos. Sin embargo, en recién nacidos prematuros <7 días de edad (edad gestacional <34 semanas), el aclaramiento de linezolid es a menudo más bajo que en los recién nacidos de gestación completa <7 días de edad. En consecuencia, los recién nacidos prematuros <7 días de edad pueden necesitar un régimen de dosificación de linezolid alternativo de 10 mg/kg cada 12 horas.
En la experiencia clínica limitada, 5 de 6 (83%) pacientes pediátricos con infecciones debidas a patógenos Gram positivos con MICs de 4 µg/mL tratados con ZYVOX® tuvieron restablecimiento clínico. Sin embargo, los pacientes pediátricos mostraron una variabilidad más amplia en la depuración de linezolid y exposición sistémica (AUC) en comparación con los adultos. En pacientes pediátricos con una respuesta clínica subóptima, en particular aquéllos con patógenos con MIC de 4 µg/mL, se debe considerar menor exposición sistémica, lugar y severidad de la infección, y la condición médica subyacente cuando se evalúe la respuesta clínica (ver Farmacología clínica: Poblaciones especiales, Pediátricos y Dosificación y administración).
Uso geriátrico: De los 2046 pacientes tratados con ZYVOX® en pruebas clínicas controladas con comparador en Fase 3, 589 (29%) tenían 65 años o más y 253 (12%) tenían 75 años o más. No se observaron diferencias generales en la seguridad o eficacia entre estos pacientes y pacientes más jóvenes.
SOBREDOSIS: En el caso de sobredosis se recomienda tomar medidas de soporte, con el mantenimiento de la filtración glomerular. La hemodiálisis puede facilitar la eliminación más rápida de linezolid. En un ensayo clínico de Fase 1, aproximadamente 30% de una dosis de linezolid se eliminó durante una sesión de hemodiálisis de 3 horas, comenzando 3 horas después de la administración de la dosis de linezolid. No hay datos de eliminación de linezolid con diálisis peritoneal o hemoperfusión. Los signos clínicos de toxicidad aguda en animales fueron disminución de la actividad y ataxia en ratas, y vómitos y temblores en perros tratados con 3000 mg/kg/día y 2000 mg/kg/día, respectivamente.
FARMACOLOGÍA CLÍNICA
Mecanismo de acción: ZYVOX® es un antibiótico (ver Microbiología).
Farmacodinamia: En un exhaustivo estudio QT cruzado positivo y controlado con placebo, 40 sujetos sanos se les administró una dosis única de 600 mg ZYVOX® a través de una hora de infusión intravenosa, una sola dosis de 1200 mg ZYVOX® a través de una hora de infusión intravenosa, placebo y un dosis oral única de control positivo. No se detectó ningún efecto significativo en el intervalo QTc a una concentración pico en plasma o en cualquier otro momento para las dosis de 600 mg y 1200 mg dosis ZYVOX®.
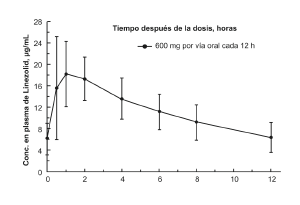
Farmacocinética: La media de los parámetros farmacocinéticos de linezolid en adultos después de dosis únicas y múltiples por vía oral e intravenosa (IV) están resumidos en el Cuadro 1. Las concentraciones en plasma de linezolid en estado estable después de la administración de dosis orales de 600 mg cada 12 horas (q12h) se muestran en la Figura 1.
Cuadro 1. Media (desviación estándar) de los parámetros farmacocinéticos de linezolid en adultos
|
Dosis de linezolid |
Cmax mcg/mL |
Cmin mcg/mL |
Tmax hrs |
AUC* mcg • h/mL |
t1/2 hrs |
CL mL/min |
|
Tableta de 400 mg |
||||||
|
Dosis única |
8,10 (1,83) |
– |
1,52 (1,01) |
55,10 (25,00) |
5,20 (1,50) |
146 (67) |
|
Cada 12 horas |
11,00 (4,37) |
3,08 (2,25) |
1,12 (0,47) |
73,40 (33,50) |
4,69 (1,70) |
110 (49) |
|
Tableta de 600 mg |
||||||
|
Dosis única |
12,70 (3,96) |
– |
1,28 (0,66) |
91,40 (39,30) |
4,26 (1,65) |
127 (48) |
|
Cada 12 horas |
21,20 (5,78) |
6,15 (2,94) |
1,03 (0,62) |
138,00 (42,10) |
5,40 (2,06) |
80 (29) |
|
Inyección 600 mg IV‡ |
||||||
|
Dosis única |
12,90 (1,60) |
– |
0,50 (0,10) |
80,20 (33,30) |
4,40 (2,40) |
138 (39) |
|
Cada 12 horas |
15,10 (2,52) |
3,68 (2,36) |
0,51 (0,03) |
89,70 (31,00) |
4,80 (1,70) |
123 (40) |
|
Suspension 600 mg |
||||||
|
Dosis única |
11,00 (2,76) |
– |
0,97 (0,88) |
80,80 (35,10) |
4,60 (1,71) |
141 (45) |
|
* AUC para dosis única = AUC0-8; para dosis múltiple = AUC0-? † Datos normalizados de dosis de 375 mg ‡ Datos normalizados de dosis de 625 mg, la dosis IV fue administrada como infusión de 0,5-hora. Cmax = concentración máxima en plasma; Cmin= concentración mínima en plasma; Tmax = Tiempo para Cmax; AUC = Área bajo la curva de concentración-tiempo; t1/2 = vida media de eliminación; CL = depuración sistémica |
||||||
Figura 1. Concentraciones en plasma de linezolid en adultos en estado estable después de la dosificación oral cada 12 horas (Media ± Desviación Estándar, n=16)
• Absorción: Linezolid se absorbe rápida y ampliamente después de la administración oral. Las concentraciones máximas en plasma se alcanzan aproximadamente en 1 a 2 horas después de la dosificación y la biodisponibilidad absoluta es de aproximadamente 100%. Por lo tanto, linezolid puede administrarse oralmente o por vía intravenosa sin ajustar la dosis. Linezolid puede administrarse sin tener en cuenta el momento de ingesta de alimentos. El tiempo para alcanzar la concentración máxima sufre un retraso de 1,5 horas a 2,2 horas y la Cmax disminuye aproximadamente 17% cuando se administra linezolid con alimentos ricos en grasas. Sin embargo, la exposición total medida como valores de AUC0-8 es similar en ambas condiciones.
• Distribución: Estudios farmacocinéticos realizados en animales y seres humanos demuestran que linezolid se distribuye rápidamente a tejidos bien perfundidos. Su unión a las proteínas plasmáticas es de aproximadamente 31%, y es independiente de la concentración. El promedio de su volumen de distribución en estado estable fue de 40 a 50 litros en voluntarios adultos saludables.
Las concentraciones de linezolid se han determinado en diversos fluidos a partir de un número limitado de participantes en estudios de Fase I realizados con voluntarios después de dosificaciones múltiples de linezolid. La proporción de la concentración de linezolid en saliva relativa al plasma fue 1,2 a 1 y en sudor en relación al plasma fue de 0,55 a 1.
• Metabolismo: Linezolid se metaboliza principalmente por oxidación del anillo morfolino, que da como resultado dos metabolitos inactivos de ácido carboxílico de anillo abierto: el metabolito de ácido aminoetoxiacético (A) y el metabolito de hidroxietilglicina (B). La formación del metabolito B está mediada por un mecanismo de oxidación químico no enzimático in vitro. Linezolid no es inductora del citocromo P450 (CYP) en ratas y se demostró, a partir de estudios in vitro, que no es metabolizada de modo detectable por el citocromo P450 humano y no inhibe las actividades de las isoformas de CYP humanas clínicamente significativas (1A2, 2C9, 2C19, 2D6, 2E1, 3A4).
• Excreción: La depuración no renal representa aproximadamente 65% de la depuración total de linezolid. En condiciones de estado estable, aproximadamente 30% de la dosis aparece en la orina como linezolid, 40% aparece como metabolito B y 10% como metabolito A. La depuración renal de linezolid es baja (promedio de 40 mL/min) y sugiere reabsorción tubular neta. Virtualmente linezolid no aparece en las heces, aunque aproximadamente 6% de la dosis aparece en las heces como metabolito B y 3% como metabolito A.
Se observó un pequeño grado de no linealidad en la depuración con dosis mayores de linezolid, lo cual al parecer se debe a la menor depuración renal y no renal de linezolid a mayores concentraciones. Sin embargo, la diferencia en la depuración fue pequeña y no se reflejó en la vida media de eliminación aparente.
• Poblaciones especiales:
– Geriátrica: La farmacocinética de linezolid no se afecta significativamente en los pacientes de edad avanzada (65 años o más); por lo tanto, no es necesario ajustar la dosis para pacientes geriátricos.
– Pediátrica: Se investigó la farmacocinética de linezolid después de la administración de una dosis única por vía IV en pacientes pediátricos cuya edad oscilaba entre el nacimiento y los 17 años (incluidos prematuros y neonatos nacidos a término), en adolescentes saludables cuyas edades oscilaban entre 12 y 17 años, y en pacientes pediátricos cuyas edades oscilaban entre 1 semana y 12 años. En el Cuadro 2 se muestra el resumen de los parámetros farmacocinéticos de linezolid para las poblaciones pediátricas estudiadas y participantes adultos saludables después de la administración de dosis únicas IV.
La Cmax y el volumen de distribución (VSS) de linezolid son similares independientemente de la edad en pacientes pediátricos. Sin embargo, la depuración de linezolid varía en función de la edad. Con excepción de los neonatos pre término con menos de 1 semana de vida, la depuración es más rápida en los grupos de menos edad, oscilando entre >1 semana a 11 años, lo que origina una menor exposición sistémica después de dosis única (AUC) y una vida media más corta, en comparación con los adultos. A medida que la edad de los pacientes pediátricos aumenta, la depuración de linezolid disminuye gradualmente; en la adolescencia, la media de los valores de depuración se aproxima a aquéllos observados en la población adulta. Hay una variabilidad interparticipantes más amplia en la depuración de linezolid y la exposición farmacológica sistémica (AUC) en todos los grupos pediátricos en comparación con los adultos. Se han observado valores diarios promedio similares de AUC en pacientes pediátricos desde el nacimiento hasta los 11 años, con una administración cada 8 horas (q8h), en relación con los adolescentes o adultos que recibían el fármaco cada 12 horas (q12h). Por lo tanto, la dosificación para pacientes pediátricos hasta 11 años de edad debe ser de 10 mg/kg q8h. Los pacientes pediátricos de 12 años en adelante deben recibir 600 mg q12h (ver Dosificación y administración).
Cuadro 2. Parámetros farmacocinéticos de linezolid en pacientes pediátricos y adultos después de una infusión intravenosa única de 10 mg/kg o 600 mg de linezolid (Media: (%CV); [Valores Min, Max])
|
Grupo de edad |
Cmax |
VSS |
AUC*, |
t1/2 hr |
CL |
|
Pacientes neonatos Pre-término** A término*** A término*** |
12,7 (30%) 11,5 (24%) 12,9 (28%) |
0,81 (24%) 0,78 (20%) 0,66 (29%) |
108 (47%) 55 (47%) 34 (21%) |
5,6 (46%) 3,0 (55%) 1,5 (17%) |
2,0 (52%) 3,8 (55%) 5,1 (22%) |
|
Pacientes bebés > 28 días a < 3 Meses (N=12 )† |
11,0 (27%) |
0,79 (26%) |
33 (26%) |
1,8 (28%) |
5,4 (32%) |
|
Pacientes pediátricos 3 meses hasta 11 años† (N=59) |
15,1 (30%) |
0,69 (28%) |
58 (54%) |
2,9 (53%) |
3,8 (53%) |
|
Pacientes y participantes adolescentes 12 hasta 17 años‡ (N=36) |
|
0,61 (15%) |
95 (44%) |
4,1 (46%) |
2,1 (53%) |
|
Participantes adultos§ (N= 29) |
12,5 (21%) |
0,65 (16%) |
91 (33%) |
4,9 (35%) |
1,7 (34%) |
|
* AUC = AUC0-8 de dosis única ** En este conjunto de datos se definió “pre-término” como < 34 semanas de edad gestacional. (Nota: sólo un paciente enrolado era pre-término con una edad postnatal entre 1 semana y 28 días) ** En este conjunto de datos, “A término” se definió como una edad gestacional ≥ 34 semanas. † Dosis de 10 mg/kg. ‡ Dosis de 600 mg o 10 mg/kg hasta un máximo de 600 mg. § Dosis normalizada a 600 mg. Cmáx = concentración máxima en plasma; VSS = volumen de distribución; AUC = área bajo la curva de concentracióntiempo; t1/2 = vida media de eliminación aparente, CL = depuración sistémica normalizada para el peso corporal. |
|||||
– Género: La población femenina tiene un volumen de distribución de linezolid ligeramente menor que el de los hombres. Las concentraciones en plasma son mayores en mujeres que en hombres, lo cual se debe en parte a las diferencias en el peso corporal. Después de administrar una dosis de 600 mg, la depuración media oral es aproximadamente 38% menor en mujeres que en hombres; sin embargo, no existen diferencias significativas de género en la media de la constante de la tasa de eliminación aparente o vida media. En consecuencia, no se espera que la exposición al fármaco en mujeres aumente sustancialmente por encima de los niveles que se sabe son bien tolerados. Por lo tanto, no parece ser necesario ajustar la dosis por género.
– Insuficiencia renal: La farmacocinética de la droga original, linezolid, no se modifica en pacientes afectados con cualquier grado de insuficiencia renal; sin embargo, los dos metabolitos principales de linezolid podrían acumularse en pacientes con esa afección, siendo mayor la acumulación al aumentar la severidad de la disfunción renal (ver el Cuadro 3). La importancia clínica de la acumulación de estos dos metabolitos no ha sido determinada en pacientes con insuficiencia renal severa. Debido a que se alcanzan concentraciones en plasma de linezolid similares independientemente de la función renal, se recomienda no hacer ajustes de dosis en pacientes afectados con insuficiencia renal.
No obstante, dada la falta de información sobre la importancia clínica de la acumulación de los metabolitos principales, debe evaluarse el uso de linezolid en pacientes con insuficiencia renal frente a los posibles riesgos derivados de la acumulación de dichos metabolitos. Tanto linezolid como los dos metabolitos se eliminan por diálisis. No existe información disponible sobre el efecto de la diálisis peritoneal en la farmacocinética de linezolid. Aproximadamente 30% de una dosis se eliminó durante una sesión de diálisis de 3 horas, comenzando 3 horas después de la administración de linezolid; por lo tanto, linezolid debe administrarse después de la hemodiálisis.
Cuadro 3. Media (desviación estándar) de las AUC y vidas medias de eliminación de linezolid y metabolitos A y B en pacientes con diversos grados de insuficiencia renal después de la administración de una dosis única oral de 600 mg de linezolid
|
Parámetro |
Participantes saludables CLCR > 80 mL/min |
Insuficiencia renal moderada 30 < CLCR < 80 mL/min |
Insuficiencia renal severa 10 < CLCR < 30 mL/min |
Dependiente de hemodiálisis |
|
|
Fuera de diálisis* |
En diálisis |
||||
|
LINEZOLID |
|||||
|
AUC0-8, µg h/mL |
110 (22) |
128 (53) |
127 (66) |
141 (45) |
83 (23) |
|
t1/2, horas |
6,4 (2,2) |
6,1 (1,7) |
7,1 (3,7) |
8,4 (2,7) |
7,0 (1,8) |
|
Metabolito A |
|||||
|
AUC0-48, µg h/mL |
7,6 (1,9) |
11,7 (4,3) |
56,5 (30,6) |
185 (124) |
68,8 (23,9) |
|
t1/2, horas |
6,3 (2,1) |
6,6 (2,3) |
9,0 (4,6) |
NA |
NA |
|
METABOLITO B1 |
|||||
|
AUC0-48 , µg h/mL |
30,5 (6,2) |
51,1 (38,5) |
203 (92) |
467 (102) |
239 (44) |
|
t1/2, horas |
6,6 (2,7) |
9,9 (7,4) |
11,0 (3,9) |
NA |
NA |
|
1. Metabolito B es el matabolito mayor de linezolid. NA = no aplicable |
|||||
Cuadro 4 . Promedio (desviación estándar) AUCs y vida media de eliminación de linezolid y metabolitos A y B en sujetos con enfermedad renal en etapa terminal (ESRD) después de la administración de 600 mg de linezolid cada 12 horas por 14,5 días
|
Parámetro |
ESRD Sujetos1 |
|
LINEZOLID |
|
|
AUC0-12, mcg h/mL (después de la dosis) |
181 (52,3) |
|
t1/2, h (después de la dosis) |
8,3 (2,4) |
|
METABOLITO A |
|
|
AUC0-12, mcg h/mL (después de la dosis) |
153 (40,6) |
|
t1/2, h (después de la dosis) |
15,9 (8,5) |
|
METABOLITO B2 |
|
|
AUC0-12, mcg h/mL (después de la dosis) |
356 (99,7) |
|
t1/2, h (después de la dosis) |
34,8 (23,1) |
|
2. Metabolito B es el principal metabolito de linezolid. |
|
– Insuficiencia hepática: La farmacocinética de linezolid no está alterada en pacientes (n=7) afectados con insuficiencia hepática leve a moderada (Clase A o B de Child-Pugh). Con base en la información disponible, no es necesario ajustar la dosis en pacientes con insuficiencia hepática leve a moderada. No se han evaluado la farmacocinética de linezolid en pacientes con insuficiencia hepática severa.
Interacciones medicamentosas
• Fármacos metabolizados por el Citocromo P450: Linezolid no es un inductor del citocromo P450 (CYP) en ratas. No es metabolizada de modo detectable por el citocromo humano P450 y no inhibe las actividades de las isoformas de CYP humanas clínicamente significativas (1A2, 2C9, 2C19, 2D6, 2E1, 3A4). Por lo tanto, no se esperan interacciones medicamentosas inducidas por el CYP450 con linezolid. La administración concurrente de linezolid no altera sustancialmente las características farmacocinéticas de (S)-warfarina que es ampliamente metabolizada por CYP2C9. Los fármacos como la warfarina y la fenitoína, que son sustratos de CYP2C9, pueden administrarse con linezolid sin cambios en el régimen de dosis.
• Antibióticos:
– Aztreonam: La farmacocinética de linezolid o aztreonam no se modifica cuando se administran conjuntamente.
– Gentamicina: La farmacocinética de linezolid o gentamicina no se modifican cuando se administran conjuntamente.
• Antioxidantes: El potencial de interacciones farmacológicas con linezolid y los antioxidantes vitamina C y la vitamina E se ha estudiado en voluntarios sanos. A los sujetos se les administraron una dosis oral de 600 mg de linezolid en el Día 1, y otra dosis de 600 mg de linezolid en el Día 8. En los días 2-9, los sujetos se les dio vitamina C (1000 mg/día) o la vitamina E (800 UI/día). El AUC0-8 de linezolid se incrementó un 2,3% cuando se administra conjuntamente con la vitamina C y el 10,9% cuando se administra conjuntamente con la vitamina E. No se recomienda ningún ajuste de la dosis de linezolid durante la administración concomitante con vitamina C o vitamina E.
• Inductores potentes del CYP 3A4
– Rifampicina: Se evaluó el efecto de rifampicina en la farmacocinética de linezolid en un estudio de 16 hombres adultos sanos. Los voluntarios se les administró vía oral linezolid 600 mg dos veces al día durante 5 dosis con y sin rifampicina 600 mg una vez al día durante 8 días. La coadministración de rifampicina con linezolid resultó en una disminución del 21% en la Cmax linezolid [IC del 90%, 15% - 27%] y una disminución del 32% en linezolid AUC0-12 [IC del 90%, 27% - 37%]. La importancia clínica de esta interacción es desconocida. El mecanismo de esta interacción no se entiende completamente y puede estar relacionado con la inducción de enzimas hepáticas. Otros inductores potentes de las enzimas hepáticas (por ejemplo, carbamazepina, fenitoína, fenobarbital) podrían causar una disminución similar o más pequeño en la exposición de linezolid.
• Inhibición de la Monoamino Oxidasa: Linezolid es un inhibidor no selectivo y reversible de la monoamino oxidasa, por lo que tiene una potencial interacción con agentes serotoninérgicos y adrenérgicos.
• Agentes adrenérgicos: Se ha observado una respuesta presora significativa en participantes adultos normales que recibieron dosis de linezolid y tiramina superiores a 100 mg. Por lo tanto, los pacientes tratados con linezolid deben evitar consumir grandes cantidades de alimentos o bebidas con alto contenido de tiramina (ver Precauciones: Información para pacientes).
Se observa un aumento reversible de la respuesta presora del clorhidrato de pseudoefedrina (PSE) o del clorhidrato de fenilpropanolamina (PPA) cuando se administra linezolid a participantes normotensos saludables (ver Precauciones: Interacciones medicamentosas. No se ha efectuado un estudio similar con pacientes hipertensos. Los estudios de interacción realizados en participantes normotensos evaluaron los efectos en la presión arterial y en la frecuencia cardiaca del placebo, PPA o PSE solas, linezolid sola, y la combinación de linezolid en estado estable (600 mg q12h durante 3 días) con dos dosis de PPA (25 mg) o PSE (60 mg) administradas cada 4 horas. La frecuencia cardiaca no fue afectada por ninguno de los tratamientos. La presión arterial aumentó con ambos tratamientos combinados. Se observaron niveles máximos de presión arterial 2 a 3 horas después de la segunda dosis de PPA o PSE, y volvieron al valor basal 2 a 3 horas después de alcanzar el valor máximo. Los resultados del estudio con PPA, donde se muestra la media (y el rango) de la presión arterial sistólica máxima en mm Hg son los siguientes: placebo = 121 (103 a 158); linezolid sola = 120 (107 a 135); PPA sola = 125 (106 a 139); PPA con linezolid = 147 (129 a 176). Los resultados del estudio con PSE fueron similares a los resultados del estudio con PPA. La media del aumento máximo de la presión arterial sistólica sobre la basal fue 32 mm Hg (rango: 20-52 mm Hg) y 38 mm Hg (rango: 18-79 mm Hg) durante la coadministración de linezolid con pseudoefedrina o fenilpropanolamina, respectivamente.
• Agentes serotoninérgicos: Se estudió la interacción medicamentosa potencial con dextrometorfano en voluntarios saludables. Se administró dextrometorfano a los pacientes (dos dosis de 20 mg administradas cada 4 horas) con o sin linezolid. No se han observado efectos de síndrome de la serotonina (confusión, delirio, inquietud, temblores, rubor, diaforesis, hiperpirexia) en participantes normales que recibían linezolid y dextrometorfano.
FORMAS DE PRESENTACIÓN
Solución inyectable: Bolsa de Polietileno de baja densidad x 300 mL contenido en una bolsa de aluminio
Tabletas: Cajas x 10, 20 y 30 tabletas en blíster: Caja x 10, 20 y 30 tabletas en frasco.
CONDICIONES DE ALMACENAMIENTO: Consérvese a temperatura menor de 30 °C.
Tabletas:
Fabricado por:
Pfizer Pharmaceuticals LLC- Puerto Rico
Solución inyectable:
Fabricado por:
Fresenius Kabi Norge AS- Noruega
Importado por:
PFIZER S.A.
Av. Javier Prado este 6230, 2do Piso
La Molina, Lima Perú
Teléfono: 615-2100, Fax: 615-2106
LLD basado en el USPI (3May 2013) V3.
VIDA ÚTIL: 36 meses.


