

SINRESOR
ZOLENDRONATO
Polvo liofilizado
1 Caja, 1 Vial de polvo, 4 Miligramos
COMPOSICIÓN
Cada frasco ampolla contiene:
Ácido zoledrónico 4 mg
(Como ácido zoledrónico monohidrato 4.264 mg)
Excipientes c.s.p.
Cada ampolla de solvente contiene:
Agua para inyectable 5 ml
INDICACIONES
• Prevención de eventos relacionados con el esqueleto (fracturas patológicas, compresión medular, radiación o cirugía ósea, o hipercalcemia inducida por tumor) en pacientes con neoplasias avanzadas con afectación ósea.
• Tratamiento de la hipercalcemia inducida por tumor (HIT).
ACCIÓN FARMACOLÓGICA
El ácido zoledrónico pertenece a la clase de los bisfosfonatos y actúa principalmente en el hueso. Es un inhibidor de la resorción ósea osteoclástica.
La acción ósea selectiva de los bisfosfonatos se basa en su gran afinidad por el hueso mineralizado, pero el mecanismo molecular preciso que da lugar a la inhibición de la actividad osteoclástica aún no está claro. En estudios de larga duración en animales, el ácido zoledrónico inhibe la resorción ósea sin perjudicar la formación, mineralización ni las propiedades mecánicas del hueso.
Además de ser un muy potente inhibidor de la resorción ósea, el ácido zoledrónico también posee varias propiedades antitumorales que pueden contribuir a su eficacia general en el tratamiento de la metástasis ósea. Se han demostrado las siguientes propiedades en ensayos preclínicos:
• In vivo: Inhibición de la resorción ósea osteoclástica, lo que altera el microentorno de la médula ósea haciéndolo menos favorable al crecimiento de la célula tumoral, actividad antiangiogénica y actividad analgésica.
• In vitro: Inhibición de la proliferación osteoblástica, actividad citostática directa y pro- apoptótica sobre las células tumorales, efecto citostático sinérgico con otros fármacos anticancerígenos, actividad antiadhesiva/invasiva.
Resultados de los ensayos clínicos en la prevención de eventos relacionados con el esqueleto en pacientes con neoplasias avanzadas con afectación ósea:
El primer estudio aleatorizado, doble ciego, controlado con placebo comparó ácido zoledrónico con placebo para la prevención de eventos relacionados con el esqueleto (ERE) en pacientes con cáncer de próstata. Ácido zoledrónico 4 mg disminuyó significativamente la proporción de pacientes que experimentaron al menos un evento relacionado con el esqueleto (ERE), retrasó la mediana de tiempo hasta el primer ERE en más de 5 meses y redujo la incidencia anual de eventos por paciente – tasa de morbididad esquelética. El análisis de eventos múltiples mostró una reducción del riesgo del 36% en el desarrollo de ERE en el grupo de ácido zoledrónico en comparación con placebo. Los pacientes que recibieron ácido zoledrónico registraron un menor incremento del dolor que los que recibieron placebo, alcanzando diferencias significativas en los meses 3, 9, 21 y 24. Un menor número de pacientes tratados con SINRESOR sufrió fracturas patológicas. Los efectos del tratamiento fueron menos pronunciados en pacientes con lesiones blásticas. Los resultados de eficacia se muestran en la Tabla 2.
En un segundo estudio, que incluía tumores sólidos diferentes del cáncer de mama y de próstata, ácido zoledrónico 4 mg redujo significativamente la proporción de pacientes con un ERE, retrasó la mediana de tiempo hasta el primer ERE en más de 2 meses y redujo la tasa de morbididad esquelética. El análisis de eventos múltiples mostró una reducción del riesgo del 30,7% en el desarrollo de ERE en el grupo de ácido zoledrónico en comparación con placebo. Los resultados de eficacia se muestran en la Tabla 3.
En un tercer ensayo de fase III aleatorizado, doble ciego, se comparó ácido zoledrónico 4 mg con pamidronato 90 mg cada 3 o 4 semanas en pacientes con mieloma múltiple o cáncer de mama con al menos una lesión ósea. Los resultados demostraron que ácido zoledrónico 4 mg mostraba una eficacia comparable a pamidronato 90 mg en la prevención de ERE. El análisis de eventos múltiples reveló una reducción significativa del riesgo del 16% en pacientes tratados con ácido zoledrónico en comparación con los pacientes que recibieron pamidronato. Los resultados de eficacia se muestran en la Tabla 4.
Se estudió también SINRESOR en un ensayo controlado con placebo, randomizado y doble ciego en 228 pacientes con metástasis óseas documentadas a partir de cáncer de mama, para evaluar el efecto de ácido zoledrónico sobre la tasa de eventos relacionados con el esqueleto (ERE), calculada como el número total de eventos ERE (excluyendo hipercalcemia y ajustado para fractura previa), dividido por el periodo total de riesgo. Los pacientes recibieron 4 mg de SINRESOR o placebo cada cuatro semanas durante un año. Los pacientes se distribuyeron a partes iguales entre los grupos tratados con ácido zoledrónico y con placebo.
Tabla 2: Resultados de eficacia (pacientes con cáncer de próstata que recibían terapia hormonal)
|
Algún ERE (+HIT) |
Fracturas* |
Radioterapia en hueso |
||||
|
SINRESOR 4 mg |
Placebo |
SINRESOR 4 mg |
Placebo |
SINRESOR 4 mg |
Placebo |
|
|
N |
214 |
208 |
214 |
208 |
214 |
208 |
|
Proporción de pacientes con ERE (%) |
38 |
49 |
17 |
25 |
26 |
33 |
|
Valor p |
0,028 |
0,052 |
0,119 |
|||
|
Mediana de tiempo hasta ERE (días) |
488 |
321 |
NA |
NA |
NA |
640 |
|
Valor p |
0,009 |
0,020 |
0,055 |
|||
|
Tasa de morbididad esquelética |
0,77 |
1,47 |
0,20 |
0,45 |
0,42 |
0,89 |
|
Valor p |
0,005 |
0,023 |
0,060 |
|||
|
Reducción del riesgo de sufrir eventos múltiples** (%) |
36 |
- |
NAp |
NAp |
NAp |
Nap |
|
Valor p |
0,002 |
NAp |
NAp |
|||
|
* Incluye fracturas vertebrales y no vertebrales ** Tiene en cuenta todos los eventos esqueléticos, el número total así como el tiempo hasta cada evento durante el ensayo NA No Alcanzado NAp No aplicable |
||||||
Tabla 3: Resultados de eficacia (tumores sólidos distintos de cáncer de mama o próstata)
|
Algún ERE (+HIT) |
Fracturas* |
Radioterapia en hueso |
||||
|
SINRESOR 4 mg |
Placebo |
SINRESOR 4 mg |
Placebo |
SINRESOR 4 mg |
Placebo |
|
|
N |
257 |
250 |
257 |
250 |
257 |
250 |
|
Proporción de pacientes con ERE (%) |
39 |
48 |
16 |
22 |
29 |
34 |
|
Valor p |
0,039 |
0,064 |
0,173 |
|||
|
Mediana de tiempo hasta ERE (días) |
236 |
155 |
NA |
NA |
424 |
307 |
|
Valor p |
0,009 |
0,020 |
0,079 |
|||
|
Tasa de morbididad esquelética |
1,74 |
2,71 |
0,39 |
0,63 |
1,24 |
1,89 |
|
Valor p |
0,012 |
0,066 |
0,099 |
|||
|
Reducción del riesgo de sufrir eventos múltiples** (%) |
30,7 |
- |
NAp |
NAp |
NAp |
NAp |
|
Valor p |
0,003 |
NAp |
NAp |
|||
|
* Incluye fracturas vertebrales y no vertebrales ** Tiene en cuenta todos los eventos esqueléticos, el número total así como el tiempo hasta cada evento durante el ensayo NA No Alcanzado NAp No aplicable |
||||||
Tabla 4: Resultados de eficacia (pacientes con cáncer de mama o mieloma múltiple)
|
Algún ERE (+HIT) |
Fracturas* |
Radioterapia en hueso |
||||
|
SINRESOR 4 mg |
Pam 90 mg |
SINRESOR 4 mg |
Pam 90 mg |
SINRESOR 4 mg |
Pam 90 mg |
|
|
N |
561 |
555 |
561 |
555 |
561 |
555 |
|
Proporción de pacientes con ERE (%) |
48 |
52 |
37 |
39 |
19 |
24 |
|
Valor p |
0,198 |
0,653 |
0,037 |
|||
|
Mediana de tiempo hasta ERE (días) |
376 |
356 |
NA |
714 |
NA |
NA |
|
Valor p |
0,151 |
0,672 |
0,026 |
|||
|
Tasa de morbididad esquelética |
1,04 |
1,39 |
0,53 |
0,60 |
0,47 |
0,71 |
|
Valor p |
0,084 |
0,614 |
0,015 |
|||
|
Reducción del riesgo de sufrir eventos múltiples** (%) |
16 |
- |
NAp |
NAp |
NAp |
NAp |
|
Valor p |
0,030 |
NAp |
NAp |
|||
|
*Incluye fracturas vertebrales y no vertebrales **Tiene en cuenta todos los eventos esqueléticos, el número total así como el tiempo hasta cada evento durante el ensayo NA No Alcanzado / NAp No aplicable |
||||||
La tasa de ERE (eventos/persona año) fue 0,628 para ácido zoledrónico y 1,096 para placebo. La proporción de pacientes con al menos un ERE (excluyendo hipercalcemia) fue de 29,8% en el grupo tratado con SINRESOR frente a 49,6% en el grupo con placebo (p=0,003). La mediana de tiempo hasta el inicio del primer ERE no se alcanzó en el brazo de tratamiento con ácido zoledrónico al final del ensayo y fue significativamente prolongada comparado con placebo (p=0,007). En un análisis de evento múltiple ácido zoledrónico redujo el riesgo de EREs en un 41% (RR=0,59, p=0,019) comparado con placebo.
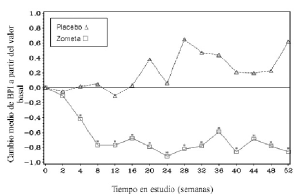
En el grupo tratado con SINRESOR, se observó una mejoría estadísticamente significativa en la puntuación de medida del dolor (utilizando el “Brief Pain Inventory”, BPI) a las 4 semanas y en cada punto de tiempo posterior durante el estudio, en comparación a placebo (Figura 1). La puntuación en la escala de dolor obtenida con SINRESOR fue consistentemente inferior a la basal y la reducción de dolor se acompañó de una tendencia decreciente en las puntuaciones obtenidas en la escala de uso de analgésicos.
Figura 1. Cambios medios en la puntuación de BPI respecto al valor basal. Las diferencias estadísticamente significativas están marcadas (*p<0,05) para las comparaciones entre tratamientos (ácido zoledrónico frente a placebo).
Resultados de los ensayos clínicos en el tratamiento de la HIT: Los ensayos clínicos en hipercalcemia inducida por tumor (HIT) demostraron que el ácido zoledrónico se caracteriza por disminuir el calcio sérico y la excreción urinaria de calcio. En los estudios de Fase I de búsqueda de dosis en pacientes con hipercalcemia inducida por tumor (HIT) de leve a moderada, las dosis efectivas ensayadas estuvieron en el rango de aproximadamente 1,2–2,5 mg.
Para valorar los efectos de ácido zoledrónico frente a 90 mg de pamidronato, se combinaron los resultados de dos ensayos multicéntricos principales en pacientes con HIT en un análisis previamente planificado. Hubo una normalización más rápida de las concentraciones corregidas de calcio sérico en el día 4 para 8 mg de ácido zoledrónico, y en el día 7 para 4 mg y 8 mg de SINRESOR. Se observaron las proporciones de respuesta siguientes:
Tabla 5: Proporción de individuos con respuesta completa por día en estudios combinados de HIT
|
Día 4 |
Día 7 |
Día 10 |
|
|
SINRESOR 4 mg (N=86) |
45,3% (p=0,104) |
82,6% (p=0,005)* |
88,4% (p=0,002)* |
|
SINRESOR 8 mg (N=90) |
55,6% (p=0,021)* |
83,3% (p=0,010)* |
86,7% (p=0,015)* |
|
Pamidronato 90 mg (N=99) |
33,3% |
63,6% |
69,7% |
|
*valores de p comparados con pamidronato. |
|||
La mediana de tiempo hasta la normocalcemia fue de 4 días. La mediana de tiempo hasta la recaída (re-elevación de los valores de calcio sérico corregidos respecto a la albúmina ≥2,9 mmol/l) fue de 30 a 40 días para los pacientes tratados con SINRESOR frente a 17 días para los tratados con 90 mg de pamidronato (valores de p: 0,001 para 4 mg y 0,007 para 8 mg). No hubo diferencias estadísticamente significativas entre las dos dosis de ácido zoledrónico.
En los ensayos clínicos a 69 pacientes que recayeron o fueron refractarios al tratamiento inicial (ácido zoledrónico 4 mg, 8 mg o pamidronato 90 mg) se les repitió el tratamiento con ácido zoledrónico 8 mg. La tasa de respuesta en estos pacientes fue de aproximadamente el 52%. Dado que a estos pacientes se les repitió el tratamiento solo con la dosis de 8 mg, no se dispone de datos que permitan la comparación con la dosis de 4 mg.
En los ensayos clínicos realizados en pacientes con hipercalcemia inducida por tumor (HIT), el perfil de seguridad global de los tres grupos de tratamiento (4 mg y 8 mg de ácido zoledrónico y 90 mg de pamidronato) fue similar en cuanto a tipo y gravedad.
Población pediátrica:
• Resultados del ensayo clínico en el tratamiento de osteogénesis imperfecta grave en pacientes pediátricos de 1 a 17 años de edad: Se compararon los efectos del ácido zoledrónico intravenoso en el tratamiento de pacientes pediátricos (de 1 a 17 años) con osteogénesis imperfecta grave (tipos I, III y IV) con los efectos de pamidronato intravenoso, en un ensayo abierto, internacional, multicéntrico, aleatorizado con 74 y 76 pacientes en cada grupo de tratamiento, respectivamente. El periodo de tratamiento del estudio fue de 12 meses precedidos por un periodo de screening de 4 a 9 semanas durante el cual se tomaron suplementos de vitamina D y calcio elemental durante al menos 2 semanas. En el programa clínico los pacientes de 1 a < 3 años recibieron 0,025 mg/kg de ácido zoledrónico (hasta una dosis única máxima de 0,35 mg) cada 3 meses y los pacientes de 3 a 17 años recibieron 0,05 mg/kg de ácido zoledrónico (hasta una dosis única máxima de 0,83 mg) cada 3 meses. Se llevó a cabo un ensayo de extensión para examinar la seguridad general y renal a largo plazo de la administración de ácido zoledrónico una vez al año o dos veces al año durante el periodo de tratamiento de la extensión de 12 meses en niños que habían completado un año de tratamiento con ácido zoledrónico o pamidronato en el estudio principal.
La variable principal del estudio fue el porcentaje de cambio en la densidad mineral ósea (DMO) de la columna lumbar desde el inicio hasta después de 12 meses de tratamiento. Los efectos del tratamiento sobre la DMO estimados fueron similares, pero el diseño del ensayo no fue suficientemente robusto para establecer la no inferioridad de eficacia para SINRESOR. En particular, no se observó una evidencia clara de eficacia sobre la incidencia de fracturas o de dolor. Se notificaron efectos adversos de fracturas de los huesos largos en las extremidades inferiores en aproximadamente un 24% (fémur) y 14% (tibia) de los pacientes con osteogénesis imperfecta grave tratados con ácido zoledrónico frente a un 12% y 5% de pacientes tratados con pamidronato, independientemente del tipo de enfermedad y de la causalidad pero la incidencia global de fracturas fue comparable para los pacientes tratados con ácido zoledrónico y con pamidronato: 43%(32/74) frente a 41% (31/76). La interpretación del riesgo de fractura se confunde con el hecho que las fracturas son acontecimientos frecuentes en pacientes con osteogénesis imperfecta grave, como parte del proceso de la enfermedad
El tipo de reacciones adversas observadas en esta población fue similar a las observadas anteriormente en adultos con procesos malignos avanzados que afectan al hueso. Las reacciones adversas, agrupadas por frecuencia, se presentan en la Tabla 6. Se utiliza la siguiente clasificación convencional: muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1.000 a <1/100), raras (≥1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
En pacientes pediátricos con osteogénesis imperfecta grave, el ácido zoledrónico parece que está asociado con riesgos más pronunciados de reacción de fase aguda, hipocalcemia y taquicardia no explicada, comparado con pamidronato, pero esta diferencia disminuyó tras las perfusiones posteriores.
La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con ácido zoledrónico en los diferentes grupos de la población pediátrica en el tratamiento de la hipercalcemia inducida por tumor y la prevención de eventos relacionados con el esqueleto en pacientes con neoplasias avanzadas con afectación ósea.
Tabla 6: Reacciones adversas observadas en pacientes pediátricos con osteogénesis imperfecta1
|
Trastornos del sistema nervioso |
|
|
Frecuentes: |
Cefalea |
|
Trastornos cardiacos |
|
|
Frecuentes: |
Taquicardia |
|
Trastornos respiratorios, torácicos y mediastínicos |
|
|
Frecuentes: |
Nasofaringitis |
|
Trastornos gastrointestinales |
|
|
Muy frecuentes: |
Vómitos, náuseas |
|
Frecuentes: |
Dolor abdominal |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|
|
Frecuentes: |
Dolor en las extremidades, artralgia, dolor musculoesquelético |
|
Trastornos generales y alteraciones en el lugar de administración |
|
|
Muy frecuentes: |
Pirexia, fatiga |
|
Frecuentes: |
Reacción de fase aguda, dolor |
|
Exploraciones complementarias |
|
|
Muy frecuentes: |
Hipocalcemia |
|
Frecuentes: |
Hipofosfataemia |
|
1 Las reacciones adversas que aparecieron con frecuencias <5% se evaluaron médicamente y se demostró que estos casos eran consistentes con el perfil de seguridad bien establecido de SINRESOR. |
|
Incompatibilidades: Para evitar incompatibilidades potenciales, la solución reconstituida de SINRESOR se diluye con una solución de cloruro sódico al 0,9% p/V o una solución de glucosa al 5% p/V.
La solución de SINRESOR reconstituida no debe mezclarse con otras soluciones para perfusión que contengan calcio u otros cationes divalentes, como la solución de Ringer lactato, debiendo administrarse como una solución intravenosa única en una vía de perfusión distinta.
Los estudios realizados con frascos de vidrio y con diferentes tipos de bolsas para perfusión y líneas de perfusión de cloruro de polivinilo, polietileno y polipropileno (previamente llenadas con cloruro sódico al 0,9% p/V o solución de glucosa al 5% p/V) no revelaron incompatibilidad con SINRESOR.
Periodo de validez: 3 años.
La solución reconstituida es química y físicamente estable durante 24 horas a 2 °C - 8 °C.
Precauciones especial de conservación: No requiere condiciones especiales de conservación.
Después de la reconstitución y dilución asépticas, conviene utilizar el producto reconstituido y diluido inmediatamente. Si éste no se usa enseguida, el tiempo y las condiciones de conservación antes de su utilización son responsabilidad del manipulador. El tiempo transcurrido entre la reconstitución, dilución y conservación en refrigeración a 2 °C – 8 °C y el final de la administración no debe exceder de 24 horas.
Precauciones especiales de eliminación y otras manipulaciones: Antes de la administración, se deberán diluir 5,0 ml de concentrado de un vial o el volumen de concentrado requerido con 100 ml de una solución para perfusión exenta de calcio (solución de cloruro sódico al 0,9% p/V o solución de glucosa al 5% p/V). La solución, si se ha refrigerado, deberá alcanzar la temperatura ambiente antes de la administración.
Fecha de revisión del texto: Abril 2011
LABORATORIOS BAGÓ DEL PERÚ S.A.
Av. Jorge Chávez Nº 154 Int. 401, Miraflores
Lima 18 – Perú
CONTRAINDICACIONES
Hipersensibilidad al principio activo, a otros bisfosfonatos, o a alguno de los excipientes de la formulación de SINRESOR.
Lactancia.
REACCIONES ADVERSAS
La frecuencia de las reacciones adversas para SINRESOR 4 mg está basada principalmente en la recogida de datos de tratamiento crónico. Las reacciones adversas con SINRESOR son similares a las observadas con otros bisfosfonatos y puede esperarse que tengan lugar en aproximadamente un tercio de los pacientes. La administración intravenosa se ha asociado comúnmente con un síndrome similar a la gripe en aproximadamente el 9% de los pacientes, incluyendo dolor óseo (9,1%), fiebre (7,2%), fatiga (4,1%) y escalofríos (2,9%). Ocasionalmente se han descrito casos de artralgia y mialgia en aproximadamente el 3%. No se dispone de información sobre la reversibilidad de estas reacciones adversas.
Con frecuencia, la reducción de la excreción renal de calcio se acompaña de un descenso de las concentraciones séricas de fosfato (en aproximadamente el 20% de los pacientes), el cual es asintomático y no requiere tratamiento. El calcio sérico puede descender hasta concentraciones hipocalcémicas asintomáticas en aproximadamente el 3% de los pacientes.
Se han descrito reacciones gastrointestinales, como náuseas (5,8%) y vómitos (2,6%), después de la perfusión intravenosa de SINRESOR. Ocasionalmente también se han observado reacciones locales en el punto de perfusión, como enrojecimiento o tumefacción y/o dolor en menos del 1% de los pacientes.
En el 1,5% de los pacientes tratados con SINRESOR 4 mg se ha descrito anorexia. Se han observado pocos casos de erupción o prurito (inferior al 1%).
Al igual que con otros bisfosfonatos, se han descrito casos de conjuntivitis en aproximadamente el 1% de los pacientes.
Ha habido algunos casos de disfunción renal, (2,3%) a pesar de que la etiología parece ser en muchos casos multifactorial.
En base a un análisis conjunto de estudios controlados con placebo, se describió anemia grave (Hb < 8,0 g/dl) en el 5,2% de los pacientes que recibieron SINRESOR, frente al 4,2% de los pacientes con placebo.
Las siguientes reacciones adversas (Tabla 1) se han recopilado de los ensayos clínicos principalmente tras el tratamiento crónico con ácido zoledrónico:
Tabla 1
Las reacciones adversas están ordenadas de mayor a menor frecuencia, utilizando la siguiente estimación: Muy frecuentes (≥1/10), frecuentes (≥1/100 a <1/10), poco frecuentes (≥1/1.000 a <1/100), raras (≥1/10.000 a <1/1.000), muy raras (<1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
|
Trastornos de la sangre y del sistema linfático |
|
|
Frecuente: |
Anemia |
|
Poco frecuente: |
Trombocitopenia, leucopenia |
|
Rara: |
Pancitopenia |
|
Trastornos del sistema nervioso |
|
|
Frecuente: |
Cefalea |
|
Poco frecuente: |
Vértigo, parestesia, alteraciones del gusto, hipoestesia, hiperestesia, temblar |
|
Trastornos psiquiátricos |
|
|
Poco frecuente: |
Ansiedad, alteraciones de sueño |
|
Rara: |
Confusión |
|
Trastornos oculares |
|
|
Frecuente: |
Conjuntivitis |
|
Poco frecuente: |
Visión borrosa |
|
Muy rara: |
Uveitis, episcleritis |
|
Trastornos gastrointestinales |
|
|
Frecuente: |
Náuseas, vómitos, anorexia |
|
Poco frecuente: |
Poco frecuente: Diarrea, estreñimiento, dolor abdominal, dispepsia, estomatitis, sequedad de boca |
|
Trastornos respiratorios, torácicos y mediastínicos |
|
|
Poco frecuente: |
Disnea, tos |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Poco frecuente: |
Poco frecuente prurito, erupción (incluyendo erupción eritematosa y macular), aumento de la sudoración |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|
|
Frecuente: |
Frecuente: Dolor óseo, mialgia, artralgia, dolor generalizado |
|
Poco frecuente: |
Calambres musculares |
|
Trastornos cardiacos |
|
|
Poco frecuente: |
Hipertensión, hipotensión |
|
Rara: |
Bradicardia |
|
Trastornos renales y urinarios |
|
|
Frecuente: |
Insuficiencia renal |
|
Poco frecuente: |
Fallo renal agudo, hematuria, proteinuria |
|
Trastornos del sistema inmunológico |
|
|
Poco frecuente: |
Reacción de hipersensibilidad |
|
Rara: |
Edema angioneurótico |
|
Trastornos generales y alteraciones en el lugar de administración |
|
|
Frecuente: |
Fiebre, síndrome similar a la gripe (incluyendo fatiga, escalofríos, malestar y sofocos) |
|
Poco frecuente: |
Poco frecuente: Astenia, edema periférico, reacciones en el lugar de la inyección (incluyendo dolor, irritación, tumefacción, induración), dolor torácico, aumento de peso |
|
Exploraciones complementarias |
|
|
Muy frecuente: |
Hipofosfatemia Aumento de la creatinina y urea sanguíneas, hipocalcemia |
|
Poco frecuente: |
Hipomagnesemia, hipopotasemia |
|
Rara: |
Hiperpotasemia, hipernatremia |
En un ensayo clínico controlado, doble ciego, aleatorizado y de 3 años de duración que evaluó la eficacia y seguridad de 5 mg de ácido zoledrónico administrados una vez al año frente a placebo en el tratamiento de la osteoporosis (OP) postmenopáusica, la incidencia global de fibrilación auricular en pacientes que recibieron tratamiento con 5 mg de ácido zoledrónico y con placebo fue de un 2,5% (96 de 3.862) y 1,9% (75 de 3.852), respectivamente. La proporción de reacciones adversas graves de fibrilación auricular fue de 1,3% (51 de 3.862) y 0,6% (22 de 3.852) en pacientes que recibieron 5 mg de ácido zoledrónico y placebo, respectivamente. La diferencia observada en este ensayo no se ha descrito en otros ensayos con ácido zoledrónico, incluyendo los ensayos con ácido zoledrónico 4 mg, administrado cada 3-4 semanas en pacientes oncológicos. Se desconoce el mecanismo causante del aumento de la incidencia de fibrilación auricular en este ensayo clínico en particular.
Experiencia post-comercialización: Se han notificado las siguientes reacciones adversas durante el uso post-autorización de ácido zoledrónico. Se han descrito casos de osteonecrosis (especialmente de las mandíbulas) predominantemente en pacientes con cáncer tratados con bisfosfonatos, incluyendo ácido zoledrónico. Muchos de estos pacientes presentaron signos de infección local incluyendo osteomielitis, y la mayoría de los informes hacen referencia a pacientes con cáncer tras una extracción dentaria u otras cirugías dentales. La osteonecrosis de la mandíbula tiene múltiples factores de riesgo documentados, incluyendo cáncer diagnosticado, tratamientos concomitantes (p.ej. quimioterapia, radioterapia, corticosteroides) y situaciones comórbidas (p.ej. anemia, coagulopatías, infección, afección oral preexistente. Aunque no se ha determinado la causalidad, es prudente evitar la cirugía dental ya que la recuperación puede ser larga.
En casos muy raros, se han notificado las siguientes reacciones adversas: síncope o colapso circulatorio secundarios a hipotensión, sobretodo en pacientes con factores de riesgo subyacentes, fibrilación auricular, somnolencia, broncoconstricción, reacción/shock anafiláctico, urticaria, escleritis e inflamación orbital. Debido a que estas notificaciones provienen de una población de un tamaño incierto y están sujetas a factores de confusión, es difícil evaluar la causalidad y estimar las tasas de incidencia de los acontecimientos.
ADVERTENCIAS
Efectos sobre la capacidad para conducir y utilizar máquinas: No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas.
PRECAUCIONES
General: Los pacientes deben ser evaluados antes de la administración de SINRESOR para asegurar que están adecuadamente hidratados.
Debe evitarse la sobre hidratación en pacientes con riesgo de insuficiencia cardíaca.
Los parámetros metabólicos habituales relacionados con la hipercalcemia, como las concentraciones séricas de calcio, fosfato y magnesio, deben ser cuidadosamente vigilados después de iniciar la terapia con SINRESOR. Puede ser necesario un tratamiento adicional a corto plazo si se produce hipocalcemia, hipofosfatemia o hipomagnesemia. Los pacientes con hipercalcemia no tratada, presentan generalmente algún grado de alteración de la función renal, por lo tanto, deberá considerarse la monitorización cuidadosa de la función renal.
SINRESOR contiene el mismo principio activo que Aclasta (ácido zoledrónico). Los pacientes que están siendo tratados con SINRESOR no deberán recibir tratamiento con Aclasta de forma concomitante.
No se ha establecido la seguridad ni la eficacia de SINRESOR en pacientes pediátricos.
Insuficiencia renal: Deberá evaluarse apropiadamente a los pacientes con HIT y evidencia de deterioro de la función renal, teniendo en consideración si el beneficio potencial del tratamiento con SINRESOR supera el posible riesgo.
La decisión de tratar a pacientes con metástasis óseas para la prevención de eventos relacionados con el esqueleto deberá tener en consideración que el inicio del efecto del tratamiento es de 2–3 meses.
Al igual que sucede con otros bisfosfonatos, SINRESOR se ha asociado con descripciones de disfunción renal. Los factores que pueden aumentar el riesgo de deterioro de la función renal incluyen deshidratación, insuficiencia renal preexistente, ciclos múltiples de SINRESOR y otros bisfosfonatos y también el uso de otros fármacos nefrotóxicos. A pesar de que el riesgo se reduce con una dosis de SINRESOR 4 mg administrada durante 15 minutos, puede presentarse todavía deterioro de la función renal. Se han notificado casos de deterioro de la función renal con progresión a insuficiencia renal y diálisis después de la administración de la dosis inicial o de una dosis única de SINRESOR. En algunos pacientes con administración crónica de SINRESOR a las dosis recomendadas para prevención de eventos relacionados con el esqueleto también se presentan aumentos de creatinina sérica, aunque con menor frecuencia.
Antes de cada dosis de SINRESOR deberán valorarse los niveles de creatinina sérica de los pacientes. Al inicio del tratamiento de pacientes con metástasis ósea con insuficiencia renal de leve a moderada, se recomiendan dosis más bajas de SINRESOR. En pacientes que muestren evidencia de deterioro renal durante el tratamiento, deberá interrumpirse la administración de SINRESOR. Solamente deberá reanudarse el tratamiento con SINRESOR cuando la creatinina sérica vuelva a hallarse dentro de un 10% del valor basal.
En vista del impacto potencial de los bisfosfonatos incluyendo SINRESOR sobre la función renal, la ausencia de datos clínicos de seguridad en pacientes con insuficiencia renal grave (definida en los ensayos clínicos como creatinina sérica ≥ 400 µmol/l o ≥ 4,5 mg/dl para pacientes con HIT y ≥ 265 µmol/l o ≥ 3,0 mg/dl para pacientes con cáncer y metástasis óseas, respectivamente) a nivel basal y los limitados datos de farmacocinética en pacientes con insuficiencia renal grave a nivel basal (aclaramiento de creatinina < 30 ml/min), no se recomienda el uso de SINRESOR en pacientes con insuficiencia renal grave.
Insuficiencia hepática: Dado que sólo se dispone de datos clínicos limitados en pacientes con insuficiencia hepática grave, no pueden darse recomendaciones específicas para esta población de pacientes.
Osteonecrosis de mandíbula: Se ha observado osteonecrosis de mandíbula predominantemente en pacientes con cáncer, tratados con bisfosfonatos, incluyendo SINRESOR. Muchos de estos pacientes también recibían quimioterapia y corticosteroides. La mayoría de los casos descritos se han asociado con procesos dentales, tales como una extracción dental. Muchos mostraron signos de infección local incluyendo osteomielitis.
En aquellos pacientes con factores de riesgo concomitante (p.ej. cáncer, quimioterapia, corticosteroides, una higiene oral pobre), deberá considerarse un examen dental con una apropiada odontología preventiva, antes de iniciar el tratamiento con bisfosfonatos.
Durante el tratamiento, si es posible, estos pacientes deben evitar procesos dentales invasivos. La cirugía dental puede agravar la situación en pacientes que desarrollen osteonecrosis de mandíbula durante la terapia con bisfosfonatos. No hay datos disponibles que indiquen si la interrupción del tratamiento con bisfosfonatos reduce el riesgo de osteonecrosis de mandíbula en pacientes que precisen procesos dentales. La valoración clínica del médico, debe orientar sobre cómo proceder con cada paciente según la valoración individual de la relación beneficio-riesgo.
Dolor musculoesquelético: En la experiencia post-comercialización, se han notificado casos de dolor óseo, articular y muscular grave y ocasionalmente incapacitante, en pacientes que toman bisfosfonatos. Sin embargo, estos informes han sido infrecuentes. Esta categoría de fármacos incluye SINRESOR (ácido zoledrónico). El tiempo hasta la aparición de los síntomas varió desde un día hasta varios meses tras el inicio del tratamiento. La mayor parte de los pacientes mejoró al suspender el tratamiento. Un subgrupo presentó recurrencia de los síntomas al administrar otra vez el mismo fármaco u otro bisfosfonato.
Embarazo y lactancia:
• Embarazo: No existen datos suficientes sobre la utilización de ácido zoledrónico en mujeres embarazadas. Estudios de reproducción en animales con ácido zoledrónico han mostrado toxicidad reproductiva. Se desconoce el riesgo en seres humanos. SINRESOR no debe utilizarse durante el embarazo.
• Lactancia: Se desconoce si el ácido zoledrónico se excreta en la leche materna. SINRESOR está contraindicado en mujeres en periodo de lactancia.
Interacción con otros medicamentos y otras formas de interacción: En ensayos clínicos, ácido zoledrónico se ha administrado simultáneamente con agentes anticancerosos, diuréticos, antibióticos y analgésicos utilizados comúnmente sin que ocurrieran interacciones clínicamente evidentes. In vitro, el ácido zoledrónico no se une considerablemente a proteínas plasmáticas y no inhibe las enzimas humanas del citocromo P450, aunque no se han realizado estudios clínicos estrictos de interacciones. Se recomienda precaución cuando se administran bisfosfonatos con aminoglucósidos, dado que ambos agentes pueden ejercer un efecto aditivo, dando como resultado una menor concentración de calcio sérico durante periodos más largos de los necesarios. Se recomienda precaución cuando se utilice SINRESOR junto con otros fármacos potencialmente nefrotóxicos. También debe prestarse atención a la posibilidad de que se desarrolle hipomagnesemia durante el tratamiento.
En los pacientes con mieloma múltiple, el riesgo de disfunción renal puede verse aumentado cuando se utilicen bisfosfonatos por vía intravenosa en combinación con talidomida.
DOSIS Y VÍAS DE ADMINISTRACIÓN
La solución de SINRESOR reconstituida no debe mezclarse con otras soluciones para perfusión que contengan calcio u otros cationes divalentes, como la solución de Ringer lactato, debiendo administrarse como una solución intravenosa única en una vía de perfusión distinta.
Prevención de eventos relacionados con el esqueleto en pacientes con neoplasias avanzadas con afectación ósea:
• Adultos y personas de edad avanzada: La dosis recomendada en la prevención de eventos relacionados con el esqueleto en pacientes con neoplasias avanzadas con afectación ósea es de 4 mg de SINRESOR polvo liofilizado para inyectable, reconstituido y posteriormente diluido (diluido con 100 ml de cloruro sódico al 0,9% p/V estéril o solución de glucosa al 5% p/V), administrados como perfusión intravenosa durante 15 minutos como mínimo cada 3 ó 4 semanas.
Deberá administrarse a los pacientes diariamente un suplemento oral de calcio de 500 mg y 400 UI de vitamina D.
Tratamiento de la HIT:
• Adultos y personas de edad avanzada: La dosis recomendada en hipercalcemia (concentración corregida de calcio sérico respecto a la albúmina ≥ 12,0 mg/dl ó 3,0 mmol/l) es de 4 mg de SINRESOR polvo liofilizado para inyectable, reconstituido y posteriormente diluido (diluido con 100 ml de cloruro sódico al 0,9% p/V estéril o solución de glucosa al 5% p/V), administrados como perfusión intravenosa única durante 15 minutos como mínimo. Los pacientes deben mantenerse bien hidratados antes y después de la administración de SINRESOR.
Insuficiencia renal:
• HIT: En los pacientes con HIT que también sufran insuficiencia renal grave el tratamiento con SINRESOR deberá considerarse solamente tras la evaluación de los riesgos y los beneficios del tratamiento. En los ensayos clínicos, se excluyeron a los pacientes con creatinina sérica > 400 µmol/l ó > 4,5 mg/dl. No se requiere un ajuste de la dosis en los pacientes con HIT con una creatinina sérica < 400 µmol/l ó < 4,5 mg/dl.
• Prevención de eventos relacionados con el esqueleto en pacientes con neoplasias avanzadas con afectación ósea: Cuando se inicia el tratamiento con SINRESOR en pacientes con mieloma múltiple o con lesiones metastásicas óseas de tumores sólidos, se deberá determinar la creatinina sérica y el aclaramiento de creatinina (CLcr). El CLcr se calcula a partir de la creatinina sérica utilizando la fórmula de Cockcroft- Gault. No se recomienda SINRESOR en los pacientes que presenten insuficiencia renal grave, definida para esta población como CLcr < 30 ml/min, antes del inicio del tratamiento. En los ensayos clínicos con ácido zoledrónico, se excluyeron los pacientes con creatinina sérica > 265 µmol/l ó > 3,0 mg/dl.
En pacientes con metástasis óseas que presentaban insuficiencia renal de leve a moderada, definida para esta población como CLcr 30–60 ml/min, antes del inicio de tratamiento se recomienda la siguiente dosis de SINRESOR:
|
Aclaramiento de creatinina basal (ml/min) |
Dosis recomendada de SINRESOR* |
|
> 60 |
4,0 mg |
|
50–60 |
3,5 mg* |
|
40–49 |
3,3 mg* |
|
30–39 |
3,0 mg* |
|
* Las dosis se han calculado asumiendo un AUC objetivo de 0,66 (mg• hr/l) (CLcr = 75 ml/min). Se espera que en los pacientes con insuficiencia renal las dosis reducidas alcancen la misma AUC que la observada en los pacientes con aclaramiento de creatinina de 75 ml/min. |
|
Una vez iniciado el tratamiento deberá medirse la creatinina sérica antes de cada dosis de SINRESOR y el tratamiento deberá interrumpirse si se ha deteriorado la función renal. En los ensayos clínicos, el deterioro renal se definió como se indica a continuación:
• Para pacientes con creatinina sérica basal normal (< 1,4 mg/dl ó < 124 µmol/l), un aumento de 0,5 mg/dl ó 44 µmol/l;
• Para pacientes con creatinina basal anormal (> 1,4 mg/dl ó > 124 µmol/l), un aumento de 1,0 mg/dl ó 88 µmol/l.
En los ensayos clínicos, el tratamiento con ácido zoledrónico se reanudó únicamente cuando el nivel de creatinina volvió a hallarse dentro de un 10% del valor basal. El tratamiento con SINRESOR deberá reanudarse a la misma dosis que tenía antes de la interrupción del tratamiento.
Instrucciones para preparar dosis reducidas de SINRESOR: Retirar un volumen apropiado de la solución reconstituida (4 mg/5 ml) según sea necesario:
• 4,4 ml para una dosis de 3,5 mg
• 4,1 ml para una dosis de 3,3 mg
• 3,8 ml para una dosis de 3,0 mg
Para información sobre la reconstitución y dilución de SINRESOR, ver Precauciones especiales de eliminación y otras manipulaciones. La cantidad de solución reconstituida retirada deberá diluirse en 100 ml de solución estéril de cloruro sódico al 0,9% p/V o en solución de glucosa al 5% p/V. La dosis deberá administrarse como perfusión intravenosa única durante 15 minutos como mínimo.
Se ha estudiado el uso de ácido zoledrónico en pacientes pediátricos en 2 ensayos clínicos en el tratamiento de la osteogénesis imperfecta grave. No deberá utilizarse SINRESOR en la población pediátrica porque no se ha establecido la seguridad ni la eficacia en niños.
TRATAMIENTO EN CASOS DE SOBREDOSIS
La experiencia clínica sobre la sobredosis con SINRESOR es limitada. Los pacientes que han recibido dosis superiores a las recomendadas deben someterse a una monitorización estrecha, dado que se han observado alteración de la función renal (incluyendo insuficiencia renal) y valores anómalos de los electrolitos séricos (incluyendo calcio, fósforo y magnesio). Si se produce una hipocalcemia, debe administrarse perfusiones de gluconato cálcico, según criterio clínico.


