
SIFROL
PRAMIPEXOL
Comprimidos
Caja , Envase , 30 Comprimidos , 0,25 Miligramos
Caja , Envase , 30 Comprimidos , 1 Miligramos
Comprimidos , 0,250 Miligramos
Comprimidos , 1 Miligramos
COMPOSICIÓN
Concentración 0,250 mg
Cada COMPRIMIDO contiene:
Pramipexol diclorhidrato monohidrato 0,250 mg
En un excipiente de manitol, almidón de maíz, dióxido de silicio coloidal, povidona y estearato de magnesio, c.s.
Concentración 1 mg
Cada COMPRIMIDO contiene:
Pramipexol diclorhidrato monohidrato 1 mg
En un excipiente de manitol, almidón de maíz, dióxido de silicio coloidal, povidona y estearato de magnesio, c.s.
ACCIÓN TERAPÉUTICA: Antiparkinsoniano.
INDICACIONES Y USO
Enfermedad de Parkinson: SIFROL® comprimidos está indicado para el tratamiento de los signos y síntomas de la enfermedad de Parkinson idiopática.
Síndrome de las Piernas Inquietas: SIFROL® comprimidos está indicado para el tratamiento del Síndrome de las Piernas Inquietas (RLS) primario de grado moderado a severo.
INFORMACIÓN PARA EL PACIENTE
Lea esta Información para el Paciente antes de empezar a tomar SIFROL® y cada vez que resurta su receta. Puede haber nueva información. Esta información no reemplaza el hecho de hablar con su médico sobre su condición médica o su tratamiento.
¿Qué es SIFROL®?
SIFROL® es un medicamento de venta con receta usado para tratar:
• Signos y síntomas de enfermedad de Parkinson (PD).
• Síndrome de las Piernas Inquietas (RLS) moderado a severo.
No se sabe si SIFROL® es seguro y efectivo en niños.
¿Qué debo informar a mi médico antes de tomar SIFROL®?
Antes de tomar SIFROL®, informe a su médico si:
• Siente somnolencia durante el día a raíz de un problema del sueño diferente del Síndrome de las Piernas Inquietas.
• Tiene presión arterial baja, o si se siente mareado o se desmaya, especialmente cuando se levanta después de estar sentado o recostado.
• Tiene problemas para controlar sus músculos (disquinesia).
• Tiene problemas renales.
• Bebe alcohol. El alcohol puede aumentar la probabilidad de que SIFROL® le haga sentir somnolencia o quedarse dormido cuando debe estar despierto.
• Tiene cualquier otra condición médica.
• Está embarazada o planea embarazarse. No se sabe si SIFROL® causará daño a su bebé no nacido.
• Está dando de lactar o planea dar de lactar. No se sabe si SIFROL® pasa a la leche materna. Usted y su médico deben decidir si usted tomará SIFROL® o dará de lactar. No debe hacer ambas cosas.
Informe a su médico sobre todos los medicamentos que toma, incluyendo los medicamentos de venta con receta y sin receta, vitaminas y suplementos herbales.
La combinación de SIFROL® y otros medicamentos puede causar afectación recíproca y puede causar efectos secundarios. SIFROL® puede afectar el modo en que actúan los medicamentos, y otros medicamentos pueden afectar el modo en que actúa SIFROL®.
Informe especialmente a su médico si toma:
• Medicamentos llamados neurolépticos (fenotiazinas, butirofenonas, tioxantenos) o metoclopramida. SIFROL® puede no actuar tan bien si usted toma estos medicamentos.
• Pramipexol de liberación extendida (SIFROL® ER). Pramipexol es el principio activo tanto en SIFROL® como en SIFROL® ER. Si está tomando SIFROL® ER, no debe tomar SIFROL®.
• Cualquier otro medicamento que le haga sentir somnolencia o pueda aumentar los efectos de SIFROL®, como cimetidina (Tagamet).
Pida a su médico una lista de estos medicamentos si no está seguro.
Conozca los medicamentos que toma. Conserve una lista de ellos y muéstrela a su médico y farmacéutico cuando obtenga un nuevo medicamento.
¿Cómo debo tomar SIFROL®?
• Tome SIFROL® exactamente como le dice su médico que lo tome.
• Su médico le dirá qué cantidad de SIFROL® tomar y cuándo tomarlo. No tome una cantidad mayor o menor de SIFROL® de lo que su médico le diga.
• Su médico puede cambiar su dosis si es necesario.
• SIFROL® puede tomarse con o sin alimentos. Tomar SIFROL® comprimidos con alimentos puede reducir sus probabilidades de sentir náuseas.
• Si toma una mayor cantidad de SIFROL® de la que su médico recomienda, llame a su médico o vaya a la sala de emergencia del hospital más cercano de inmediato.
• Si omite una dosis, no duplique su siguiente dosis. No tome la dosis que omitió y tome su siguiente dosis normal.
• Si tiene enfermedad de Parkinson y su médico le dice que deje de tomar SIFROL®, debe suspender SIFROL® de manera lenta como lo indique su médico. Si interrumpe SIFROL® demasiado rápido puede tener síntomas de abstinencia como:
— Fiebre.
— Confusión.
— Rigidez muscular severa.
No deje de tomar SIFROL® sin hablar con su médico.
¿Qué debo evitar mientras tomo SIFROL®?
• No tome alcohol mientras toma SIFROL®. El alcohol puede aumentar su probabilidad de experimentar efectos secundarios serios. Vea “¿Cuáles son los posibles efectos secundarios de SIFROL®?”
• No conduzca, opere maquinaria, o realice otras actividades peligrosas hasta que sepa cómo lo afecta SIFROL®. La somnolencia causada por SIFROL® puede suceder tan tarde como 1 año después de que inicie su tratamiento.
¿Cuáles son los posibles efectos secundarios de SIFROL®?
SIFROL® puede causar efectos secundarios serios, incluyendo:
• Quedarse dormido durante la realización de actividades diarias normales. SIFROL® puede causar que se quede dormido mientras está realizando actividades diarias como conducir, hablar con otras personas o comer.
— Algunas personas que toman el medicamento SIFROL® han tenido accidentes automovilísticos debido a que se quedaron dormidos mientras conducían.
— Algunos pacientes no sintieron somnolencia antes de quedarse dormidos mientras conducían. Usted podría quedarse dormido sin ninguna advertencia.
Informe a su médico de inmediato si se queda dormido mientras está realizado actividades como hablar, comer, conducir, o si siente más sueño de lo normal para usted.
• Presión arterial baja cuando se sienta o para rápidamente. Puede tener:
— Mareos.
— Náuseas.
— Desmayos.
— Sudoración.
Siéntese y póngase de pie lentamente después de haber estado sentado o recostado.
• Impulsos poco usuales. Algunas personas que toman ciertos medicamentos para tratar enfermedad de Parkinson, incluyendo SIFROL®, han reportado problemas, como apostar, comer compulsivamente, comprar compulsivamente y aumento del deseo sexual.
Si usted o los miembros de su familia notan que está desarrollando impulsos o comportamientos poco usuales, hable con su médico.
• Ver visiones, escuchar sonidos o sentir sensaciones que no son reales (alucinaciones). Su probabilidad de tener alucinaciones es mayor si usted es un adulto mayor (mayor de 65 años de edad).
Si tiene alucinaciones, hable con su médico de inmediato.
• Movimientos súbitos no controlados (disquinesia).
Si tiene una nueva disquinesia o su disquinesia existente empeora informe a su médico.
• Cáncer de piel (melanoma). Algunas personas con enfermedad de Parkinson pueden tener una mayor probabilidad de tener melanoma que las personas que no tienen enfermedad de Parkinson. No se sabe si la probabilidad de tener melanoma es mayor debido a los medicamentos para tratar la enfermedad de Parkinson, como SIFROL®, o por la enfermedad de Parkinson. Las personas que toman SIFROL® deben tener exámenes cutáneos regulares para revisar la presencia de melanoma.
Los efectos secundarios más comunes en personas que toman SIFROL® para Síndrome de las Piernas Inquietas son náuseas y dolor de cabeza.
Los efectos secundarios más comunes en personas que toman SIFROL® para enfermedad de Parkinson son:
• Náuseas.
• Mareos.
• Insomnio.
• Estreñimiento.
• Debilidad muscular.
• Sueños anormales.
• Confusión.
• Problemas de memoria (amnesia).
• Orinar de manera más frecuente que lo normal.
Éstos no son todos los efectos secundarios posibles de SIFROL®. Informe a su médico si tiene algún efecto secundario que lo molesta.
Llame a su médico para solicitar consejo médico sobre efectos secundarios.
¿Cómo debo almacenar SIFROL®?
• Conservar en su envase original, y no almacenar a temperatura superior a 30 °C.
• Proteger de la luz.
• Almacenar en lugar seguro fuera del alcance de los niños.
Información General sobre el uso seguro y efectivo de SIFROL®
Los medicamentos se recetan a veces para propósitos diferentes a los listados en un folleto de Información para el Paciente. No use SIFROL® para una condición para la cual no fue recetado. No de SIFROL® a otras personas, incluso si éstas tienen los mismos síntomas que usted tiene. El medicamento puede dañarlos.
Este folleto de Información para el Paciente resume la información más importante sobre SIFROL®. Si desea obtener más información, hable con su médico. Puede solicitar a su farmacéutico o médico la información sobre SIFROL® que está escrita para los profesionales de salud.
¿Cuáles son los ingredientes en SIFROL®?
• Principio activo: Pramipexol diclorhidrato monohidrato
• Ingredientes inactivos: Manitol, almidón de maíz, dióxido de silicio coloidal, povidona y estearato de magnesio.
Bajo licencia de:
Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Alemania.
Elaborado por
Boehringer Ingelheim Pharma GmbH & Co. KG, Alemania. Industria Alemana
Acondicionado por:
Boehringer Ingelheim do Brasil Química e Farmacéutica Ltda. – Brasil.
Importado y distribuido por:
BOEHRINGER INGELHEIM PERÚ S.A.C
Av. Canaval y Moreyra 480, Piso 20
San Isidro - Telf.: (51-1) 212-4132
CONTRAINDICACIONES: Ninguna.
REACCIONES ADVERSAS
Las siguientes reacciones adversas se discuten con más detalle en otras secciones de la etiqueta:
• Quedarse dormido durante la realización de actividades de la vida diaria (ver Advertencias y precauciones: Quedarse dormido durante la realización de actividades de la vida diaria).
• Hipotensión ortostática sintomática (ver Advertencias y precauciones: Hipotensión ortostática sintomática).
• Control de impulsos/Comportamientos compulsivos (ver Advertencias y precauciones: Control de impulsos/Comportamientos compulsivos).
• Alucinaciones (ver Advertencias y precauciones: Alucinaciones).
• Disquinesia (ver Advertencias y precauciones: Disquinesia).
• Insuficiencia renal (ver Advertencias y precauciones: Insuficiencia renal).
• Rabdomiólisis (ver Advertencias y precauciones: Rabdomiólisis).
• Patología retiniana(ver Advertencias y precauciones: Patología retiniana).
• Eventos reportados con la terapia dopaminérgica (ver Advertencias y precauciones: Eventos reportados con la terapia dopaminérgica).
Experiencia en ensayos clínicos: Debido a que los ensayos clínicos se llevan a cabo bajo condiciones muy variables, las tasas de evento adverso observadas en los ensayos clínicos de un fármaco no pueden compararse de manera directa con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
• Enfermedad de Parkinson: Durante el desarrollo previo a la comercialización de pramipexol, pacientes con enfermedad de Parkinson ya sea temprana o avanzada fueron enrolados en ensayos clínicos. Aparte de la severidad y duración de su enfermedad, las dos poblaciones difirieron en su uso de terapia concomitante con levodopa. Los pacientes con enfermedad temprana no recibieron terapia concomitante con levodopa durante el tratamiento con pramipexol; aquéllos con enfermedad de Parkinson avanzada recibieron tratamiento concomitante con levodopa. Debido a que estas dos poblaciones pueden tener riesgos diferenciales para varios eventos adversos, esta sección presentará, en general, datos de eventos adversos para estas dos poblaciones de manera separada.
Debido a que todos los ensayos controlados llevados a cabo durante el desarrollo previo a la comercialización usaron un diseño de titulación, con una confusión resultante con respecto al tiempo y la dosis, fue imposible evaluar adecuadamente los efectos de la dosis en la incidencia de eventos adversos.
• Enfermedad de Parkinson Temprana: En los tres ensayos controlados con placebo, en doble ciego de pacientes con enfermedad de Parkinson temprana, los eventos adversos observados de manera más común (>5%) que fueron numéricamente más frecuentes en el grupo tratado con SIFROL® comprimidos fueron náuseas, mareos, somnolencia, insomnio, estreñimiento, astenia y alucinaciones.
Aproximadamente 12% de 388 pacientes con enfermedad de Parkinson temprana y tratados con SIFROL® comprimidos que participaron en los ensayos controlados con placebo, en doble ciego descontinuaron el tratamiento debido a eventos adversos comparado con 11% de 235 pacientes que recibieron placebo. Los eventos adversos que causaron de manera más común la descontinuación del tratamiento estuvieron relacionados con el sistema nervioso (alucinaciones [3,1% con SIFROL® comprimidos frente a 0,4% con placebo]; mareos [2,1% con SIFROL® comprimidos frente a 1% con placebo]; somnolencia [1,6% con SIFROL® comprimidos frente a 0% con placebo]; síndrome extrapiramidal [1,6% con SIFROL comprimidos frente a 6,4% con placebo]; dolor de cabeza y confusión [1,3% y 1,0%, respectivamente con SIFROL® comprimidos frente a 0% con placebo]); y sistema gastrointestinal (náuseas [2,1% con SIFROL® comprimidos frente a 0,4% con placebo]).
— Incidencia de eventos adversos en estudios clínicos controlados en enfermedad de Parkinson Temprana: El Cuadro 4 lista los eventos adversos surgidos durante el tratamiento que ocurrieron en los estudios controlados con placebo, en doble ciego en enfermedad de Parkinson temprana que fueron reportados por ≥1% de pacientes tratados con SIFROL® comprimidos y que fueron numéricamente más frecuentes que en el grupo placebo. En estos estudios, los pacientes no recibieron levodopa concomitante. La intensidad de los eventos adversos fue usualmente leve o moderada.
El médico que prescribe debe ser consciente de que estas cifras no pueden usarse para pronosticar la incidencia de eventos adversos en el transcurso de la práctica médica usual donde las características de los pacientes y otros factores difieren de los que prevalecieron en los estudios clínicos. Asimismo, las frecuencias citadas no pueden compararse con las cifras obtenidas de otras investigaciones clínicas que involucran diferentes tratamientos, usos e investigadores. Sin embargo, las cifras citadas sí proporcionan al médico que prescribe cierta base para estimar la contribución relativa de factores farmacológicos y no farmacológicos con la tasa de incidencia de eventos adversos en la población estudiada.
|
Cuadro 4: Incidencia de eventos adversos surgidos durante el tratamiento* en ensayos controlados con placebo, en doble ciego en enfermedad de Parkinson temprana (eventos ≥1% de pacientes tratados con SIFROL® comprimidos y numéricamente más frecuentes que en el grupo placebo) |
||
|
Sistema Corporal/Evento Adverso |
SIFROL® |
Placebo |
|
Cuerpo como un todo |
||
|
Astenia |
14 |
12 |
|
Edema general |
5 |
3 |
|
Malestar general |
2 |
1 |
|
Reacción no evaluable |
2 |
1 |
|
Fiebre |
1 |
0 |
|
Sistema digestivo |
||
|
Náuseas |
28 |
18 |
|
Estreñimiento |
14 |
6 |
|
Anorexia |
4 |
2 |
|
Disfagia |
2 |
0 |
|
Sistema Metabólico y Nutricional |
||
|
Edema periférico |
5 |
4 |
|
Disminución del peso |
2 |
0 |
|
Sistema Nervioso |
||
|
Mareos |
25 |
24 |
|
Somnolencia |
22 |
9 |
|
Insomnio |
17 |
12 |
|
Alucinaciones |
9 |
3 |
|
Confusión |
4 |
1 |
|
Amnesia |
4 |
2 |
|
Hipoestesia |
3 |
1 |
|
Distonía |
2 |
1 |
|
Acatisia |
2 |
0 |
|
Pensamiento anormal |
2 |
0 |
|
Disminución de la líbido |
1 |
0 |
|
Mioclono |
1 |
0 |
|
Órganos de los Sentidos |
||
|
Visión anormal |
3 |
0 |
|
Sistema Urogenital |
||
|
Impotencia |
2 |
1 |
|
* Los pacientes pueden haber reportado múltiples experiencias adversas durante el estudio o al momento de la descontinuación; por lo tanto, los pacientes pueden estar incluidos en más de una categoría. |
||
Otros eventos reportados por 1% o más pacientes con enfermedad de Parkinson temprana y tratados con SIFROL® comprimidos pero reportados de manera igual o más frecuente en el grupo placebo fueron infección, lesión accidental, dolor de cabeza, dolor, temblor, dolor de espalda, síncope, hipotensión postural, hipertonía, depresión, dolor abdominal, ansiedad, dispepsia, flatulencia, diarrea, rash, ataxia, sequedad de boca, síndrome extrapiramidal, calambres en las piernas, fasciculación, faringitis, sinusitis, sudoración, rinitis, infección del tracto urinario, vasodilatación, síndrome gripal, aumento de la salivación, enfermedad dental, disnea, aumento de la tos, anormalidades de la marcha, frecuencia urinaria, vómitos, reacción alérgica, hipertensión, prurito, hipoquinesia, aumento de la creatina PK, nerviosismo, sueños anormales, dolor torácico, dolor de cuello, parestesia, taquicardia, vértigo, alteración de la voz, conjuntivitis, parálisis, anormalidades de la acomodación, tinito, diplopía y alteración del gusto.
En un estudio de dosis fija en enfermedad de Parkinson temprana, la frecuencia de ocurrencia de los siguientes eventos aumentó conforme se incrementó la dosis a lo largo del rango de 1,5 mg/día a 6 mg/día: hipotensión postural, náuseas, estreñimiento, somnolencia y amnesia. La frecuencia de estos eventos fue generalmente 2 veces mayor que placebo para dosis de pramipexol mayores de 3 mg/día. La incidencia de somnolencia con pramipexol a una dosis de 1,5 mg/día fue comparable a la reportada para placebo.
• Enfermedad de Parkinson Avanzada: En los cuatro ensayos controlados con placebo, en doble ciego de pacientes con enfermedad de Parkinson avanzada, los eventos adversos observados de manera más común (>5%) que fueron numéricamente más frecuentes en el grupo tratado con SIFROL® comprimidos y levodopa concomitante fueron hipotensión (ortostática) postural, disquinesia, síndrome extrapiramidal, insomnio, mareos, alucinaciones, lesión accidental, sueños anormales, confusión, estreñimiento, astenia, somnolencia, distonía, anormalidad de la marcha, hipertonía, sequedad de boca, amnesia y frecuencia urinaria.
Aproximadamente 12% de 260 pacientes con enfermedad de Parkinson avanzada que recibieron SIFROL® comprimidos y levodopa concomitante en los ensayos controlados con placebo, en doble ciego descontinuaron el tratamiento debido a eventos adversos comparado con 16% de 264 pacientes que recibieron placebo y levodopa concomitante. Los eventos que causaron de manera más común la descontinuación del tratamiento estuvieron relacionados con el sistema nervioso (alucinaciones [2.7% con SIFROL® comprimidos frente a 0,4% con placebo]; disquinesia [1,9% con SIFROL® comprimidos frente a 0.8% con placebo]; síndrome extrapiramidal [1.5% con SIFROL® comprimidos frente a 4.9% con placebo]; mareos [1,2% con SIFROL® comprimidos frente a 1,5% con placebo]; confusión [1,2% con SIFROL® comprimidos frente a 2,3% con placebo]); y sistema cardiovascular (hipotensión [ortostática] postural [2,3% con SIFROL® comprimidos frente a 1,1% con placebo]).
— Incidencia de eventos adversos en estudios clínicos controlados en enfermedad de Parkinson Avanzada: El Cuadro 5 lista los eventos adversos surgidos durante el tratamiento que ocurrieron en los estudios controlados con placebo, en doble ciego en enfermedad de Parkinson avanzada que fueron reportados por ≥1% de pacientes tratados con SIFROL® comprimidos y que fueron numéricamente más frecuentes que en el grupo placebo. En estos estudios, se administró SIFROL® comprimidos o placebo a los pacientes que también estuvieron recibiendo levodopa concomitante. La intensidad de los eventos adversos fue usualmente leve o moderada.
El médico que prescribe debe ser consciente de que estas cifras no pueden usarse para pronosticar la incidencia de eventos adversos en el transcurso de la práctica médica usual donde las características de los pacientes y otros factores difieren de los que prevalecieron en los estudios clínicos. Asimismo, las frecuencias citadas no pueden compararse con las cifras obtenidas de otras investigaciones clínicas que involucran diferentes tratamientos, usos e investigadores. Sin embargo, las cifras citadas sí proporcionan al médico que prescribe cierta base para estimar la contribución relativa de factores farmacológicos y no farmacológicos con la tasa de incidencia de eventos adversos en la población estudiada.
|
Cuadro 5: Incidencia de eventos adversos surgidos durante el tratamiento* en ensayos controlados con placebo, en doble ciego en enfermedad de parkinson avanzada (eventos ≥1% de pacientes tratados con SIFROL® comprimidos y numéricamente más frecuentes que en el grupo placebo) |
||
|
Sistema Corporal/Evento Adverso |
SIFROL®† |
Placebo† |
|
Cuerpo como un Todo |
||
|
Lesión accidental |
17 |
15 |
|
Astenia |
10 |
8 |
|
Edema general |
4 |
3 |
|
Dolor torácico |
3 |
2 |
|
Malestar general |
3 |
2 |
|
Sistema Cardiovascular |
||
|
Hipotensión postural |
53 |
48 |
|
Sistema Digestivo |
||
|
Estreñimiento |
10 |
9 |
|
Sequedad de boca |
7 |
3 |
|
Sistema Metabólico y Nutricional |
||
|
Edema periférico |
2 |
1 |
|
Aumento de creatina PK |
1 |
0 |
|
Sistema Musculoesquelético |
||
|
Artritis |
3 |
1 |
|
Fasciculación |
2 |
0 |
|
Bursitis |
2 |
0 |
|
Miastenia |
1 |
0 |
|
Sistema Nervioso |
||
|
Disquinesia |
47 |
31 |
|
Síndrome extrapiramidal |
28 |
26 |
|
Insomnio |
27 |
22 |
|
Mareos |
26 |
25 |
|
Alucinaciones |
17 |
4 |
|
Sueños anormales |
11 |
10 |
|
Confusión |
10 |
7 |
|
Somnolencia |
9 |
6 |
|
Distonía |
8 |
7 |
|
Anormalidades de la marcha |
7 |
5 |
|
Hipertonía |
7 |
6 |
|
Amnesia |
6 |
4 |
|
Acatisia |
3 |
2 |
|
Pensamiento anormal |
3 |
2 |
|
Reacción paranoide |
2 |
0 |
|
Delirio |
1 |
0 |
|
Alteraciones del sueño |
1 |
0 |
|
Sistema Respiratorio |
||
|
Disnea |
4 |
3 |
|
Rinitis |
3 |
1 |
|
Neumonía |
2 |
0 |
|
Piel y Apéndices |
||
|
Trastornos cutáneos |
2 |
1 |
|
Órganos de los Sentidos |
||
|
Anormalidades de la acomodación |
4 |
2 |
|
Visión anormal |
3 |
1 |
|
Diplopía |
1 |
0 |
|
Sistema Urogenital |
||
|
Frecuencia urinaria |
6 |
3 |
|
Infección del tracto urinario |
4 |
3 |
|
Incontinencia urinaria |
2 |
1 |
|
* Los pacientes pueden haber reportado múltiples experiencias adversas durante el estudio o al momento de la descontinuación; por lo tanto, los pacientes pueden estar incluidos en más de una categoría. † Los pacientes recibieron levodopa concomitante. |
||
Otros eventos reportados por 1% o más pacientes con enfermedad de Parkinson avanzada y tratados con SIFROL comprimidos pero reportados de manera igual o más frecuente en el grupo placebo fueron náuseas, dolor, infección, dolor de cabeza, depresión, temblor, hipoquinesia, anorexia, dolor de espalda, dispepsia, flatulencia, ataxia, síndrome gripal, sinusitis, diarrea, mialgia, dolor abdominal, ansiedad, rash, parestesia, hipertensión, aumento de la salivación, trastorno dental, apatía, hipotensión, sudoración, vasodilatación, vómitos, aumento de la tos, nerviosismo, prurito, hipoestesia, dolor de cuello, síncope, artralgia, disfagia, palpitaciones, faringitis, vértigo, calambres en las piernas, conjuntivitis y problemas de lagrimeo.
• Síndrome de las Piernas Inquietas: SIFROL® comprimidos para el tratamiento de RLS ha sido evaluado para seguridad en 889 pacientes, incluyendo 427 tratados durante más de seis meses y 75 durante más de un año.
La evaluación de seguridad global se centra en los resultados de tres ensayos controlados con placebo, en doble ciego, en los cuales se trató 575 pacientes con RLS con SIFROL® comprimidos hasta por 12 semanas. Los eventos adversos observados de manera más común con SIFROL® comprimidos en el tratamiento de RLS (observados en >5% de pacientes tratados con pramipexol y a una tasa de al menos el doble que la observada en los pacientes tratados con placebo) fueron náuseas y somnolencia. La ocurrencia de náuseas y somnolencia en los ensayos clínicos fue generalmente leve y transitoria.
Aproximadamente 7% de 575 pacientes tratados con SIFROL® comprimidos durante los periodos en doble ciego de tres ensayos controlados con placebo descontinuaron el tratamiento debido a eventos adversos comparado con 5% de 223 pacientes que recibieron placebo. El evento adverso que causó de manera más común la descontinuación del tratamiento fue náuseas (1%).
El Cuadro 6 lista los eventos surgidos durante el tratamiento que ocurrieron en tres estudios controlados con placebo, en doble ciego en pacientes con RLS que fueron reportados por ≥2% de pacientes tratados con SIFROL® comprimidos y que fueron numéricamente más frecuentes que en el grupo placebo.
El médico que prescribe debe ser consciente de que estas cifras no pueden usarse para pronosticar la incidencia de eventos adversos en el transcurso de la práctica médica usual donde las características de los pacientes y otros factores difieren de los que prevalecieron en los estudios clínicos. Asimismo, las frecuencias citadas no pueden compararse con las cifras obtenidas de otras investigaciones clínicas que involucran diferentes tratamientos, usos e investigadores. Sin embargo, las cifras citadas sí proporcionan al médico que prescribe cierta base para estimar la contribución relativa de factores farmacológicos y no farmacológicos con la tasa de incidencia de eventos adversos en la población estudiada.
|
Cuadro 6: Incidencia de eventos adversos surgidos durante el tratamiento* en ensayos controlados con placebo, en doble ciego en síndrome de las piernas inquietas (eventos ≥2% de pacientes tratados con SIFROL® comprimidos y numéricamente más frecuentes que en el grupo placebo) |
||
|
Sistema Corporal/ Evento Adverso |
SIFROL® |
Placebo |
|
Trastornos gastrointestinales |
||
|
Náuseas |
16 |
5 |
|
Estreñimiento |
4 |
1 |
|
Diarrea |
3 |
1 |
|
Sequedad de boca |
3 |
1 |
|
Trastornos generales y cond. del sitio de administración |
||
|
Fatiga |
9 |
7 |
|
Infecciones e infestaciones |
||
|
Gripe |
3 |
1 |
|
Trastornos del sistema nervioso |
||
|
Dolor de cabeza |
16 |
15 |
|
Somnolencia |
6 |
3 |
|
* Los pacientes pueden haber reportado múltiples experiencias adversas durante el estudio o al momento de la descontinuación; por lo tanto, los pacientes pueden estar incluidos en más de una categoría. |
||
Otros eventos reportados por 2% o más de los pacientes con RLS tratados con SIFROL® comprimidos pero reportados de manera igual o más frecuente en el grupo placebo fueron: vómitos, nasofaringitis, dolor de espalda, dolor en las extremidades, mareos e insomnio.
El Cuadro 7 resume datos para eventos adversos que parecieron estar relacionados con la dosis en el estudio de dosis fija de 12 semanas.
|
Cuadro 7: Eventos adversos relacionados con la dosis en un estudio de dosis fija controlado con placebo, en doble ciego, de 12 semanas en síndrome de las piernas inquietas (ocurrieron en ≥5% de todos los pacientes en la fase de tratamiento) |
||||
|
Sistema Corporal/ Evento Adverso |
SIFROL® 0,25 mg |
SIFROL® 0,5 mg |
SIFROL® 0,75 mg |
Placebo |
|
Trastornos gastrointestinales |
||||
|
Náuseas |
11 |
19 |
27 |
5 |
|
Diarrea |
3 |
1 |
7 |
0 |
|
Dispepsia |
3 |
1 |
4 |
7 |
|
Infecciones e Infestaciones |
||||
|
Gripe |
1 |
4 |
7 |
1 |
|
Trastornos generales y cond. del sitio de administración |
||||
|
Fatiga |
3 |
5 |
7 |
5 |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
9 |
9 |
13 |
9 |
|
Sueños anormales |
2 |
1 |
8 |
2 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||
|
Congestión nasal |
0 |
3 |
6 |
1 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Dolor en las extremidades |
3 |
3 |
7 |
1 |
• General:
— Eventos Adversos: Relación con la Edad, Sexo y Raza: Entre los eventos adversos surgidos durante el tratamiento en pacientes tratados con SIFROL® comprimidos, la alucinación pareció mostrar una relación positiva con la edad en pacientes con enfermedad de Parkinson. Aunque no se observó diferencias relacionadas con el sexo en pacientes con enfermedad de Parkinson, las náuseas y la fatiga, ambas generalmente transitorias, fueron reportadas de manera más frecuente por pacientes con RLS de sexo femenino que de sexo masculino. Menos de 4% de pacientes enrolados fueron no caucásicos: por lo tanto, no es posible una evaluación de eventos adversos relacionados con la raza.
• Pruebas de Laboratorio: Durante el desarrollo de SIFROL® comprimidos, no se notó anormalidades sistemáticas en las pruebas de laboratorio de rutina. Por lo tanto, no se ofrece una guía específica con respecto al monitoreo de rutina; el médico conserva la responsabilidad de determinar cómo monitorear mejor el cuidado del paciente.
— Otros Eventos adversos observados durante los ensayos clínicos de Fase 2 y 3: Se ha administrado SIFROL® comprimidos a 1620 pacientes con enfermedad de Parkinson y a 889 pacientes con RLS en ensayos clínicos de Fase 2 y 3. Durante estos ensayos, todos los eventos adversos fueron registrados por los investigadores clínicos usando terminología de su propia elección; los tipos similares de eventos se agruparon en un número más pequeño de categorías estandarizadas usando la terminología del diccionario MedDRA. Estas categorías se usan en la lista a continuación. Los eventos adversos que no están listados más arriba pero que ocurrieron al menos en dos ocasiones (una ocasión si el evento fue serio) en los 2509 sujetos expuestos a SIFROL® comprimidos se listan a continuación. Los eventos reportados a continuación están incluidos independientemente de la determinación de una relación causal con SIFROL comprimidos.
— Trastornos de la sangre y del sistema linfático: Anemia, anemia por deficiencia de hierro, leucocitosis, leucopenia, linfadenitis, linfadenopatía, trombocitemia, trombocitopenia.
— Trastornos cardiacos: Angina pectoris, arritmia supraventricular, fibrilación auricular, bloqueo auriculoventricular de primer grado, bloqueo auriculoventricular de segundo grado, bradicardia, bloqueo de rama, paro cardiaco, insuficiencia cardiaca, insuficiencia cardiaca congestiva, cardiomegalia, oclusión de la arteria coronaria, cianosis, extrasístoles, insuficiencia ventricular izquierda, infarto de miocardio, arritmia nodal, arritmia sinusal, bradicardia sinusal, taquicardia sinusal, extrasístoles supraventriculares, taquicardia supraventricular, taquicardia, fibrilación ventricular, extrasístoles ventriculares, hipertrofia ventricular.
— Trastornos congénitos, familiares y genéticos: Defecto del tabique auricular, malformación congénita del pie, malformación de la columna.
— Trastornos del oído y del laberinto: Sordera, dolor de oído, deterioro de la audición, hipoacusia, enfermedad motora, ataxia vestibular.
— Trastornos endocrinos: Bocio, hipertiroidismo, hipotiroidismo.
— Trastornos oculares: Amaurosis fugaz, blefaritis, blefaroespasmo, catarata, dacrioestenosis adquirida, ojo seco, hemorragia ocular, irritación ocular, dolor ocular, edema de párpado, ptosis de párpado, glaucoma, queratitis, degeneración macular, miopía, fotofobia, desprendimiento de retina, trastorno vascular retiniano, escotoma, visión borrosa, disminución de la agudeza visual, flotadores vítreos.
— Trastornos gastrointestinales: Malestar abdominal, distensión abdominal, estomatitis aftosa, ascitis, queilitis, colitis, colitis ulcerativa, úlcera duodenal, hemorragia por úlcera duodenal, enteritis, eructo, incontinencia fecal, úlcera gástrica, hemorragia por úlcera gástrica, gastritis, hemorragia gastrointestinal, enfermedad por reflujo esofágico, gingivitis, hematemesis, hematoquecia, hemorroides, hernia de hiato, hiperclorhidria, íleo, hernia inguinal, obstrucción intestinal, síndrome de colon irritable, espasmo esofágico, estenosis esofágica, esofagitis, pancreatitis, periodontitis, hemorragia rectal, esofagitis por reflujo, edema de lengua, ulceración de la lengua, odontalgia, hernia umbilical.
— Trastornos generales: Malestar torácico, escalofríos, muerte, síndrome de abstinencia de fármacos, edema facial, sensación de frío, sensación de calor, sensación de euforia, alteración de la marcha, problemas de cicatrización, enfermedad parecida a la gripe, irritabilidad, edema localizado, edema, edema con fóvea, sed.
— Trastornos hepatobiliares: Cólico biliar, colecistitis, colecistitis crónica, colelitiasis.
— Trastornos del sistema inmune: Hipersensibilidad a fármacos.
— Infecciones e infestaciones: Absceso, amigdalitis aguda, apendicitis, bronquiolitis, bronquitis, bronconeumonía, celulitis, cistitis, caries dental, diverticulitis, infección de oído, infección ocular, foliculitis, infección fúngica, forúnculo, gangrena, gastroenteritis, infección gingival, herpes simple, herpes zóster, orzuelo, disquitis intervertebral, laringitis, neumonía lobar, infección ungueal, onicomicosis, candidiasis oral, orquitis, osteomielitis, otitis externa, otitis media, paroniquia, pielonefritis, pioderma, sepsis, infección cutánea, amigdalitis, absceso dental, infección dental, infección del tracto respiratorio alto, uretritis, candidiasis vaginal, infección vaginal, infección viral, infección de la herida.
— Lesión, envenenamiento y complicaciones relacionadas con el procedimiento: Caídas accidentales, toxicidad del fármaco, epicondilitis, accidente de tráfico, eritema solar, rotura de tendón.
— Trastornos metabólicos y nutricionales: Caquexia, disminución del apetito, deshidratación, diabetes mellitus, retención de líquidos, gota, hipercolesterolemia, hiperglucemia, hiperlipidemia, hiperuricemia, hipocalcemia, hipoglucemia, hipocalemia, hiponatremia, hipovitaminosis, aumento del apetito, alcalosis metabólica.
— Trastornos musculoesqueléticos y del tejido conectivo: Dolor óseo, fascitis, dolor en los flancos, trastorno del disco intervertebral, protrusión del disco intervertebral, efusión articular, rigidez articular, inflamación articular, monoartritis, rigidez muscular, espasmos musculares, rigidez musculoesquelética, miopatía, miositis, rigidez de nuca, osteoartritis, osteonecrosis, osteoporosis, polimialgia, artritis reumatoidea, dolor de hombros, osteoartritis espinal, tendinitis, tenosinovitis.
— Neoplasias benignas, malignas y no especificadas: Neoplasia abdominal, adenocarcinoma, adenoma benigno, carcinoma basocelular, cáncer de vejiga, cáncer de mama, neoplasia de mama, leucemia linfocítica crónica, cáncer de colon, cáncer colorrectal, cáncer endometrial, cáncer de vesícula biliar, cáncer gástrico, neoplasia gastrointestinal, hemangioma, neoplasia hepática, neoplasia hepática maligna, cáncer de labio y/o cavidad oral, neoplasia pulmonar maligna, cáncer de pulmón metastático, linfoma, melanoma maligno, nevo melanocítico, metástasis de pulmón, mieloma múltiple, neoplasia oral benigna, neoplasia, neoplasia maligna, neoplasia de próstata, neoplasia de piel, neuroma, cáncer de ovario, cáncer de próstata, adenoma prostático, pseudolinfoma, neoplasia renal, cáncer de piel, papiloma cutáneo, carcinoma de células escamosas, neoplasia tiroidea, leiomioma uterino.
— Trastorno del sistema nervioso: Ageusia, aquinesia, síndrome anticolinérgico, afasia, alteración del equilibrio, edema cerebral, oclusión de al arteria carótida, síndrome del túnel carpiano, embolia de la arteria cerebral, hemorragia cerebral, infarto cerebral, isquemia cerebral, córea, trastorno cognitivo, coma, convulsión, coordinación anormal, demencia, depresión del nivel de consciencia, problemas de atención, mareos posturales, disartria, disgrafía, parálisis facial, convulsión de tipo gran mal, hemiplejía, hiperestesia, hiperquinesia, hiperreflexia, hiporreflexia, hipotonía, letargo, pérdida de consciencia, deterioro de la memoria, migraña, contracciones musculares involuntarias, narcolepsia, neuralgia, neuropatía, nistagmo, parosmia, hiperactividad psicomotora, ciática, sedación, alteración sensorial, alteración del ritmo de las fases del sueño, hablar dormido, estupor, síncope vasovagal, dolor de cabeza tensional.
— Trastornos psiquiátricos: Labilidad emocional, agresión, agitación, bradifrenia, bruxismo, suicidio, delirio, trastorno delirante de tipo persecutorio, desorientación, disociación, dificultad emocional, estado de ánimo eufórico, alucinación auditiva, alucinación visual, insomnio inicial, aumento de la líbido, manía, insomnio medio, estado de ánimo alterado, pesadillas, pensamientos obsesivos, trastorno obsesivo-compulsivo, reacción de pánico, parasomnia, trastorno de la personalidad, trastorno psicótico, inquietud, caminar dormido, pensamiento suicida.
— Trastornos renales y urinarios: Cromaturia, disuria, glicosuria, hematuria, urgencia, nefrolitiasis, vejiga neurogénica, nocturia, oliguria, polaquiuria, proteinuria, estenosis de la arteria renal, cólico renal, quiste renal, insuficiencia renal, deterioro renal, retención urinaria.
— Trastornos del sistema reproductivo y de las mamas: Amenorrea, dolor de mamas, dismenorrea, epididimitis, ginecomastia, síntomas menopáusicos, menorragia, metrorragia, quiste ovárico, priapismo, prostatitis, disfunción sexual, hemorragia uterina, secreción vaginal, hemorragia vaginal.
— Trastornos respiratorios, torácicos y mediastinales: Apnea, aspiración, asma, atragantamiento, enfermedad pulmonar obstructiva crónica, garganta seca, disfonía, disnea de esfuerzo, epistaxis, hemoptisis, hipo, hiperventilación, aumento de la secreción bronquial, laringoespasmo, sequedad nasal, pólipos nasales, trastorno obstructivo de las vías aéreas, dolor faringolaríngeo, pleuresía, neumonía por aspiración, neumotórax, goteo nasal posterior, tos productiva, embolia pulmonar, edema pulmonar, alcalosis respiratoria, dificultad respiratoria, insuficiencia respiratoria, congestión del tracto respiratorio, rinitis alérgica, rinorrea, congestión sinusal, síndrome de apnea del sueño, estornudos, ronquido, taquipnea, silbido.
— Trastornos de la piel y del tejido subcutáneo: Acné, alopecia, sudor frío, quiste dérmico, dermatitis, dermatitis ampollosa, dermatitis de contacto, piel seca, equimosis, eccema, eritema, hiperqueratosis, livedo reticularis, sudores nocturnos, edema periorbitario, petequias, reacción alérgica de fotosensibilidad, psoriasis, púrpura, rash eritematoso, rash maculopapular, rash papular, rosácea, seborrea, dermatitis seborreica, sensación de ardor cutáneo, decoloración cutánea, exfoliación cutánea, hiperpigmentación cutánea, hipertrofia cutánea, irritación cutánea, nódulo cutáneo, olor cutáneo anormal, úlcera cutánea, urticaria.
— Trastornos vasculares: Aneurisma, angiopatía, arteriosclerosis, colapso circulatorio, trombosis venosa profunda, embolia, hematoma, sofoco, crisis hipertensiva, linfedema, palidez, flebitis, fenómeno de Raynaud, shock, tromboflebitis, trombosis, vena varicosa.
Experiencia posterior a la comercialización: Además de los eventos adversos reportados durante los ensayos clínicos, se ha identificado las siguientes reacciones adversas durante el uso de SIFROL® comprimidos posterior a su aprobación, principalmente en pacientes con enfermedad de Parkinson. Debido a que estas reacciones se reportan de manera voluntaria a partir de una población de tamaño incierto, no siempre es posible estimar de manera fiable su frecuencia o establecer una relación causal con la exposición al fármaco. La decisión de incluir estos eventos en el inserto se basan típicamente en uno o más de los siguientes factores: (1) seriedad de la reacción, (2) frecuencia de reporte o (3) fuerza de la relación causal con pramipexol comprimidos. Los tipos similares de reacciones se agruparon en un número más pequeño de categorías estandarizadas usando la terminología de MedDRA: comportamiento anormal, sueños anormales, accidentes (incluyendo caída), pérdida de consciencia, compulsión por comprar, fatiga, alucinaciones (todos los tipos), hipotensión (incluyendo hipotensión postural), secreción inadecuada de la hormona antidiurética (SIADH), consumo mayor de alimentos (incluyendo atracones de comida, comer compulsivamente e hiperfagia), trastornos de la líbido (incluyendo aumento y disminución de la líbido, e hipersexualidad), ludopatía, prurito, síncope, vómitos y aumento de peso.
INTERACCIONES MEDICAMENTOSAS
Ver Dosis y Administración: Enfermedad de Parkinson y Farmacología clínica: Farmacocinética.
Antagonistas de la dopamina: Debido a que pramipexol es un agonista de la dopamina, es posible que los antagonistas de la dopamina, como los neurolépticos (fenotiazinas, butirofenonas, tioxantenos) o metoclopramida, puedan disminuir la eficacia de SIFROL® comprimidos.
Interacciones con fármacos/Pruebas de laboratorio: No hay interacciones conocidas entre pramipexol y las pruebas de laboratorio.
ESTUDIOS CLÍNICOS
Enfermedad de Parkinson: Se evaluó la eficacia de SIFROL® comprimidos en el tratamiento de enfermedad de Parkinson en un programa multinacional de desarrollo del fármaco consistente en siete ensayos controlados, randomizados. Tres se llevaron a cabo en pacientes con enfermedad de Parkinson temprana que no estuvieron recibiendo levodopa concomitante y cuatro se llevaron a cabo en pacientes con enfermedad de Parkinson avanzada que estuvieron recibiendo levodopa concomitante. Entre estos siete estudios, tres estudios proporcionan la evidencia más persuasiva de eficacia de pramipexol en el manejo de pacientes con enfermedad de Parkinson que estuvieron y no estuvieron recibiendo levodopa concomitante. Dos de estos tres ensayos enrolaron pacientes con enfermedad de Parkinson temprana (que no estuvieron recibiendo levodopa) y uno enroló pacientes con enfermedad de Parkinson avanzada que estuvieron recibiendo dosis máximas toleradas de levodopa.
En todos los estudios, la Escala Unificada de Clasificación de Enfermedad de Parkinson (UPDRS), o una o más de sus subpartes, sirvió como la medida de evaluación de resultado primario. La UPDRS es una escala de clasificación de múltiples ítems de cuatro partes para evaluar la mentación (parte I), Actividades de la Vida Diaria (ADL) (parte II), rendimiento motor (parte III) y complicaciones de la terapia (parte IV).
La parte II de la UPDRS contiene 13 preguntas que se relacionan con ADL, las cuales están puntuadas de 0 (normal) a 4 (severidad máxima) para un puntaje máximo (peor) de 52. La Parte III de la UPDRS contiene 27 preguntas (para 14 ítems) y está puntuada como se describe para la parte II. Está diseñada para evaluar la severidad de los hallazgos motores cardinales en pacientes con enfermedad de Parkinson (p. ej., temblor, rigidez, bradiquinesia, inestabilidad postural, etc.), puntuada para diferentes regiones del cuerpo, y tiene un puntaje máximo (peor) de 108.
• Estudios en Pacientes con Enfermedad de Parkinson Temprana: Los pacientes (N=599) en los dos estudios de enfermedad de Parkinson temprana tuvieron una duración media de enfermedad de 2 años, exposición previa limitada o ausente a levodopa (generalmente ninguna en los 6 meses previos), y no estuvieron experimentando el fenómeno “on-off” y la disquinesia característicos de etapas posteriores de la enfermedad.
Uno de los dos estudios de enfermedad de Parkinson temprana (N=335) fue un ensayo paralelo, controlado placebo, en doble ciego consistente en un periodo de aumento de dosis de 7 semanas y un periodo de mantenimiento de 6 meses. Los pacientes pudieron estar bajo tratamiento con selegiline, anticolinérgicos, o ambos, pero no pudieron estar bajo tratamiento con productos de levodopa o amantadina. Se randomizó a los pacientes para recibir SIFROL® comprimidos o placebo. Los pacientes tratados con SIFROL® comprimidos tuvieron una dosis inicial diaria de 0,375 mg y fueron titulados hasta una dosis máxima tolerada, pero no mayor de 4,5 mg/día en tres dosis divididas. Al final del periodo de mantenimiento de 6 meses, la mejora media con respecto a la basal en el puntaje total de la parte II (ADL) de la UPDRS fue 1,9 en el grupo que recibió SIFROL comprimidos y -0,4 en el grupo placebo, una diferencia que fue estadísticamente significativa. La mejora media con respecto a la basal en el puntaje total de la parte III de la UPDRS fue 5,0 en el grupo que recibió SIFROL® comprimidos y -0,8 en el grupo placebo, una diferencia que también fue estadísticamente significativa. Se vio una diferencia estadísticamente significativa entre grupos a favor de SIFROL® comprimidos que comenzó en la semana 2 de la parte II (dosis máxima de 0,75 mg/día) de la UPDRS y en la semana 3 de la parte III (dosis máxima de 1,5 mg/día) de la UPDRS.
El segundo estudio de enfermedad de Parkinson temprana (N=264) fue un ensayo paralelo, controlado con placebo, en doble ciego consistente en un periodo de aumento de la dosis de 6 semanas y un periodo de mantenimiento de 4 semanas. Los pacientes pudieron estar bajo tratamiento con selegiline, anticolinérgicos, amantadina, o cualquier combinación de éstos, pero no pudieron estar bajo tratamiento con productos de levodopa. Se randomizó a los pacientes para recibir 1 de 4 dosis fijas de SIFROL® comprimidos (1,5 mg, 3,0 mg, 4,5 mg o 6,0 mg por día) o placebo. Al final del periodo de mantenimiento de 4 semanas, la mejora media con respecto a la basal en el puntaje total de la parte II de la UPDRS fue 1,8 en los pacientes tratados con SIFROL® comprimidos, independientemente del grupo de dosis asignado, y 0,3 en los pacientes tratados con placebo. La mejora media con respecto a la basal en el puntaje total de la parte III de la UPDRS fue 4,2 en pacientes tratados con SIFROL® comprimidos y 0,6 en los pacientes tratados con placebo. No se demostró una relación dosis-respuesta. Las diferencias entre tratamientos en ambas partes de la UPDRS fueron estadísticamente significativas a favor de SIFROL® comprimidos para todas las dosis.
No se detectó diferencias en la eficacia en base a la edad o sexo. Hubo muy pocos pacientes caucásicos para evaluar el efecto de la raza. Los pacientes que recibieron selegiline o anticolinérgicos tuvieron respuestas similares a los pacientes que no recibieron estos fármacos.
• Estudios en Pacientes con Enfermedad de Parkinson Avanzada: En el estudio de enfermedad de Parkinson avanzada, las evaluaciones primarias fueron la UPDRS y los registros diarios que cuantificaron las cantidades de tiempo “on” y “off”.
Los pacientes en el estudio de enfermedad de Parkinson avanzada (N=360) tuvieron una duración media de enfermedad de 9 años, habían estado expuestos a levodopa durante periodos prolongados de tiempo (media de 8 años), usaron levodopa concomitante durante el ensayo y tuvieron periodos “on-off”.
El estudio de enfermedad de Parkinson avanzada fue un ensayo paralelo, controlado con placebo, en doble ciego consistente en un periodo de aumento de la dosis de 7 semanas y un periodo de mantenimiento de 6 meses. Todos los pacientes fueron tratados con productos de levodopa concomitantes y pudieron estar adicionalmente bajo tratamiento concomitante con selegiline, anticolinérgicos, amantadina, o cualquier combinación. Los pacientes tratados con SIFROL® comprimidos tuvieron una dosis inicial de 0,375 mg/día y fueron titulados hasta una dosis máxima tolerada, pero no mayor de 4,5 mg/día en tres dosis divididas. En momentos seleccionados durante el periodo de mantenimiento de 6 meses, se pidió a los pacientes que registraran la cantidad de tiempo “off”, “on” o “con disquinesia” por día durante varios días consecutivos. Al final del periodo de mantenimiento de 6 meses, la mejora media con respecto a la basal en el puntaje total de la parte II de la UPDRS fue 2,7 en el grupo tratado con SIFROL® comprimidos y 0,5 en el grupo placebo, una diferencia que fue estadísticamente significativa. La mejora media con respecto a la basal en el puntaje total de la parte III de la UPDRS fue 5,6 en el grupo tratado con SIFROL® comprimidos y 2,8 en el grupo placebo, una diferencia que fue estadísticamente significativa. Se vio una diferencia estadísticamente significativa entre grupos a favor de SIFROL® comprimidos en la semana 3 de la parte II de la UPDRS (dosis máxima de 1.5 mg/día) y en la semana 2 de la parte III de la UPDRS (dosis máxima de 0,75 mg/día). Se permitió la reducción de la dosis de levodopa durante este estudio si se desarrolló disquinesia (o alucinaciones); la reducción de la dosis de levodopa ocurrió en 76% de pacientes tratados con SIFROL® comprimidos frente a 54% de pacientes que recibieron placebo. En promedio, la dosis de levodopa se redujo en 27%.
El número medio de horas “off” por día durante la basal fue 6 horas para ambos grupos de tratamiento. Durante todo el ensayo, los pacientes tratados con SIFROL® comprimidos tuvieron una media de 4 horas “off” por día, mientras que los pacientes tratados con placebo continuaron experimentando 6 horas “off” por día.
No se detectó diferencias en la eficacia en base a la edad o sexo. Hubo muy pocos pacientes caucásicos para evaluar el efecto de la raza.
Síndrome de las Piernas Inquietas: Se evaluó la eficacia de SIFROL® comprimidos en el tratamiento de RLS en un programa multinacional de desarrollo del fármaco consistente en 4 ensayos controlados con placebo, en doble ciego, randomizados. Este programa incluyó aproximadamente 1000 pacientes con RLS moderado a severo; se excluyó a los pacientes con RLS secundario a otras condiciones (p. ej., embarazo, insuficiencia renal y anemia). Todos los pacientes recibieron SIFROL® comprimidos (0,125 mg, 0,25 mg, 0,5 mg o 0,75 mg) o placebo una vez al día 2-3 horas antes de acostarse. A lo largo de los 4 estudios, la duración media de RLS fue 4,6 años (rango de 0 a 56 años), la edad media fue aproximadamente 55 años (rango de 18 a 81 años) y aproximadamente 66,6% fueron mujeres.
Los criterios de diagnóstico clave para RLS son: una urgencia de mover las piernas usualmente acompañada o causada por sensaciones incómodas y desagradables en las piernas; los síntomas comienzan o empeoran durante periodos de reposo o inactividad como recostarse o sentarse; los síntomas son parcial o totalmente aliviados por movimientos como caminar o estirarse al menos mientras continúe la actividad; y los síntomas son peores u ocurren solamente en la tarde o noche. La dificultad para quedarse dormido puede estar frecuentemente asociada con síntomas de RLS.
Las dos medidas de resultado usadas para evaluar el efecto de tratamiento fueron la Escala Internacional de Clasificación de RLS (Escala IRLS) y una evaluación de la Mejora de la Impresión Clínica Global (CGI-I). La Escala IRLS contiene 10 ítems diseñados para evaluar la severidad de los síntomas sensoriales y motores, alteración del sueño, somnolencia diurna e impacto en las actividades de la vida diaria y estado de ánimo asociado con RLS. El rango de puntajes es 0 a 40, siendo 0 ausencia de síntomas de RLS y 40 los síntomas más severos. La CGI-I está diseñada para evaluar el progreso clínico (mejora global) en una escala de 7 puntos.
En el Estudio 1, se comparó dosis fijas de SIFROL® comprimidos con placebo en un estudio de 12 semanas de duración. Un total de 344 pacientes fueron randomizados de igual manera a los 4 grupos de tratamiento. Los pacientes tratados con SIFROL® comprimidos (n=254) tuvieron una dosis inicial de 0,125 mg/día y fueron titulados a una de las tres dosis randomizadas (0,25, 0,5, 0,75 mg/día) en las primeras tres semanas del estudio. La mejora media con respecto a la basal en el puntaje total de la Escala IRLS y el porcentaje de pacientes que presentaron respuesta CGI-I para cada uno de los grupos de tratamiento con SIFROL® comprimidos comparado con placebo se resumen en el Cuadro 8. Todos los grupos de tratamiento alcanzaron superioridad estadísticamente significativa comparada con placebo para ambos criterios de valoración. No hubo evidencia clara de una respuesta a la dosis a lo largo de los 3 grupos de dosis randomizados.
|
Cuadro 8: Cambios Medios de la Basal a la Semana 12 en el Puntaje IRLS y CGI-I (Estudio 1) |
|||||
|
SIFROL® 0,25 mg |
SIFROL® 0,5 mg |
SIFROL® 0,75 mg |
SIFROL® Total |
Placebo |
|
|
No. de Pacientes |
88 |
79 |
87 |
254 |
85 |
|
Puntaje IRLS |
-13,1 |
-13,4 |
-14,4 |
-13,6 |
-9,4 |
|
Pacientes que presentaron respuesta CGI-I* |
74,7% |
67,9% |
72,9% |
72,0% |
51,2% |
|
* Pacientes que presentaron respuesta CGI = “mucho mejor” y “sumamente mejor”. |
|||||
El Estudio 2 fue un estudio de retiro randomizado, diseñado para demostrar la eficacia sostenida de pramipexol para tratamiento de RLS después de un periodo de seis meses. Los pacientes con RLS que respondieron a tratamiento con SIFROL® comprimidos en una fase previa de tratamiento de etiqueta abierta de 6 meses (definido como tener una clasificación CGI-I de “sumamente mejor” o “mucho mejor” comparado con la basal y un puntaje IRLS de 15 o menor) fueron randomizados para recibir ya sea tratamiento activo continuo (n=78) o placebo (n=69) durante 12 semanas. El criterio de valoración primario de este estudio fue el tiempo para el fracaso del tratamiento, definido como cualquier empeoramiento en el puntaje CGI-I junto con un puntaje total en la Escala IRLS mayor de 15.
En pacientes que habían respondido al tratamiento de etiqueta abierta de 6 meses con SIFROL® comprimidos, la administración de placebo condujo a una disminución rápida en sus condiciones globales y retorno de sus síntomas RLS. Al final del periodo de observación de 12 semanas, 85% de pacientes tratados con placebo habían experimentado fracaso del tratamiento, comparado con 21% de pacientes tratados con pramipexol ciego, una diferencia que fue alta y estadísticamente significativa. La mayoría de fracasos de tratamiento ocurrieron dentro de los 10 días de randomización. Para los pacientes randomizados, la distribución de las dosis fue: 7 bajo 0,125 mg, 44 bajo 0,25 mg, 47 bajo 0,5 mg y 49 bajo 0,75 mg.
El Estudio 3 fue un estudio de 6 semanas, que comparó una dosis flexible de SIFROL® comprimidos con placebo. En este estudio, 345 pacientes fueron randomizados en una proporción 2:1 a SIFROL® comprimidos o placebo. La mejora media con respecto a la basal en el puntaje total en la Escala IRLS fue -12 para pacientes tratados con SIFROL® y -6 para pacientes tratados con placebo. El porcentaje de pacientes que presentaron respuesta CGI-I fue 63% para pacientes tratados con SIFROL® y 32% para pacientes tratados con placebo. Las diferencias entre grupos fueron estadísticamente significativas para ambas mediciones de resultado. Para los pacientes randomizados a SIFROL® comprimidos, la distribución de las dosis logradas fue: 35 bajo 0,125 mg, 51 bajo 0,25 mg, 65 bajo 0,5 mg y 69 bajo 0,75 mg.
El Estudio 4 fue un estudio de 3 semanas, que comparó 4 dosis fijas de SIFROL® comprimidos, 0,125 mg, 0,25 mg, 0,5 mg y 0,75 mg, con placebo. Aproximadamente 20 pacientes fueron randomizados a cada uno de los 5 grupos de dosis. La mejora media con respecto a la basal en el puntaje total de la Escala IRLS y el porcentaje de pacientes que presentaron respuesta CGI-I para cada uno de los grupos de tratamiento con SIFROL® comprimidos comparado con placebo se resumen en el Cuadro 9. En este estudio, el grupo que recibió la dosis de 0,125 mg no fue estadísticamente diferente de placebo. En promedio, el grupo que recibió la dosis de 0,5 mg se comportó mejor que el grupo que recibió la dosis de 0,25 mg, pero no hubo diferencia entre los grupos que recibieron las dosis de 0,5 mg y 0,75 mg..
|
Cuadro 9: Cambios Medios de la Basal a la Semana 3 en el Puntaje IRLS y CGI-I (Estudio 4) |
||||||
|
SIFROL® 0,125 mg |
SIFROL® 0,25 mg |
SIFROL® 0,5 mg |
SIFROL® 0,75 mg |
SIFROL® Total |
Placebo |
|
|
No. Pacientes |
21 |
22 |
22 |
21 |
86 |
21 |
|
Puntaje IRLS |
-11,7 |
-15,3 |
-17,6 |
-15,2 |
-15,0 |
-6,2 |
|
Pacientes que presentaron respuesta CGI-I* |
61,9% |
68,2% |
86,4% |
85,7% |
75,6% |
42,9% |
|
* Pacientes que presentaron respuesta CGI = “mucho mejor” y “sumamente mejor”. |
||||||
No se detectó diferencias en la eficacia en base a la edad o sexo. Hubo muy pocos pacientes caucásicos para evaluar el efecto de la raza.
Instrucciones de dosificación: Indicar a los pacientes que tomen SIFROL® comprimidos solo como se prescribe. Si se omite una dosis, informar a los pacientes no duplicar la siguiente dosis.
SIFROL® comprimidos puede tomarse con o sin alimentos. Si los pacientes desarrollan náuseas, debe informárseles que tomar SIFROL® comprimidos con alimentos puede reducir la ocurrencia de náuseas.
Pramipexol es el principio activo que está tanto en SIFROL comprimidos como en SIFROL® ER comprimidos de liberación prolongada. Asegurarse de que los pacientes no tomen tanto SIFROL® ER como SIFROL®.
Efectos sedantes: Los pacientes deben ser alertados sobre los potenciales efectos sedantes asociados con SIFROL® comprimidos, incluyendo somnolencia y la posibilidad de quedarse dormido mientras se realiza actividades de la vida diaria. Debido a que la somnolencia es un evento adverso frecuente con consecuencias potencialmente serias, los pacientes no deben conducir ni participar en otras actividades potencialmente peligrosas hasta que hayan ganado suficiente experiencia con SIFROL® comprimidos para medir si afecta o no su rendimiento mental y/o motor de manera adversa. Aconsejar a los pacientes que si experimentan, en cualquier momento durante el tratamiento, un aumento de la somnolencia o nuevos episodios de quedarse dormidos durante la realización de actividades de la vida diaria (p. ej., conversar o comer), no deben conducir o participar en actividades potencialmente peligrosas hasta que se hayan comunicado con su médico. Debido a los posibles efectos aditivos, se recomienda precaución cuando los pacientes estén tomando otros fármacos sedantes o alcohol en combinación con SIFROL® comprimidos y cuando se tome fármacos concomitantes que aumentan los niveles plasmáticos de pramipexol (p. ej., cimetidina) (ver Advertencias y precauciones: Quedarse dormido durante la realización de actividades de la vida diaria)].
Síntomas de control de impulsos incluyendo comportamientos compulsivos: Los pacientes y las personas encargadas de su cuidado deben ser informados sobre la posibilidad de que puedan experimentar impulsos intensos de gastar dinero incontrolablemente, impulsos intensos de apostar, aumento del instinto sexual, darse atracones de comida y/u otros impulsos intensos y la incapacidad de controlar estos impulsos mientras toman SIFROL® (ver Advertencias y precauciones: Control de impulsos/Comportamientos compulsivos).
Alucinaciones: Los pacientes deben ser informados que puede ocurrir alucinaciones y que los paciente adultos mayores están en un mayor riesgo que los pacientes más jóvenes con enfermedad de Parkinson (ver Advertencias y precauciones: Alucinaciones). En ensayos clínicos, los pacientes con RLS tratados con pramipexol rara vez reportaron alucinaciones.
Hipotensión (Ortostática) Postural: Informar a los pacientes que pueden desarrollar hipotensión (ortostática) postural, con o sin síntomas como mareos, náuseas, desmayo o pérdida del conocimiento, y algunas veces, sudoración. La hipotensión puede ocurrir de manera más frecuente durante la terapia inicial. En consecuencia, advertir a los pacientes sobre levantarse rápidamente después de haber estado sentado o recostado, especialmente si han estado haciéndolo durante periodos prolongados y especialmente al inicio del tratamiento con SIFROL® comprimidos (ver Advertencias y precauciones: Hipotensión ortostática sintomática).
Embarazo: Debido a que no se ha establecido completamente el potencial teratogénico de pramipexol en animales de laboratorio, y debido a que la experiencia en humanos es limitada, aconsejar a las mujeres que notifiquen a sus médicos si se embarazan o pretenden embarazarse durante la terapia (ver Uso en poblaciones específicas: Embarazo).
Madres lactantes: Debido a la posibilidad de que pramipexol pueda excretarse en la leche materna, aconsejar a las mujeres que notifiquen a sus médicos si pretenden dar de pecho o están dando pecho a un lactante (ver Uso en poblaciones específicas: Madres lactantes).
TOXICOLOGÍA NO CLÍNICA
Carcinogénesis, mutagénesis y deterioro de la fertilidad: Se ha llevado a cabo estudios de carcinogenicidad de dos años con pramipexol en ratones y ratas. Se administró pramipexol en la dieta a ratones a dosis hasta de 10 mg/kg/día (o aproximadamente 10 veces la dosis máxima recomendada en humanos (MRHD) para enfermedad de Parkinson de 4,5 mg/día sobre una base de mg/m2). Se administró pramipexol en la dieta a ratas a dosis hasta de 8 mg/kg/día. Estas dosis estuvieron asociadas con AUC en plasma hasta de aproximadamente 12 veces la observada en humanos a la MRHD. No ocurrió aumentos significativos en los tumores en ninguna especie.
Pramipexol no fue mutagénico o clastogénico en una serie de ensayos in vitro (mutación inversa en bacterias, mutación génica V79/HGPRT, aberración cromosómica en células CHO) e in vivo (micronúcleo de ratón).
En estudios de fertilidad en la rata, pramipexol a dosis de 2,5 mg/kg/día (5 veces la MRHD sobre una base de mg/m2) prolongó los ciclos estrales e inhibió la implantación. Estos efectos estuvieron asociados con reducciones en los niveles séricos de prolactina, una hormona necesaria para la implantación y mantenimiento de las primeras etapas de la preñez en ratas.
Toxicología y/o Farmacología animal:
• Patología retiniana en ratas: Se observó cambios patológicos (degeneración y pérdida de células fotorreceptoras) en la retina de ratas albinas en el estudio de carcinogenicidad de 2 años con pramipexol. Estos hallazgos fueron observados por primera vez durante la semana 76 y fueron dependientes de la dosis en animales que recibieron 2 ú 8 mg/kg/día (AUC en plasma igual a 2.5 y 12.5 veces el AUC en humanos a la MRHD). En un estudio similar de ratas pigmentadas con 2 años de exposición a pramipexol a 2 ú 8 mg/kg/día, no se observó degeneración retiniana. Los animales que recibieron el fármaco experimentaron un adelgazamiento en la capa nuclear externa de la retina que solo fue ligeramente mayor (por análisis morfométrico) que el visto en ratas de control.
Estudios de investigación demostraron que pramipexol redujo la tasa de derramamiento de disco de las células bastones fotorreceptoras de la retina en ratas albinas, lo cual se asoció con una sensibilidad aumentada a los efectos dañinos de la luz. En un estudio comparativo, ocurrió degeneración y pérdida de células fotorreceptoras en ratas albinas después de 13 semanas de tratamiento con 25 mg/kg/día de pramipexol (54 veces la MRHD sobre una base de mg/m2) y luz constante (100 lux) pero no en ratas pigmentadas expuestas a la misma dosis e intensidades mayores de luz (500 lux). Por lo tanto, se considera que la retina de las ratas albinas es particularmente sensible a los efectos dañinos de pramipexol y la luz. No ocurrieron cambios similares en la retina en un estudio de carcinogenicidad de 2 años en ratones albinos tratados con 0,3, 2 o 10 mg/kg/día (0,3, 2,2 y 11 veces la MRHD sobre una base de mg/m2). La evaluación de las retinas de monos que recibieron 0,1, 0,5 o 2,0 mg/kg/día de pramipexol (0,4, 2,2 y 8,6 veces la MRHD sobre una base de mg/m2) durante 12 meses y cerdos enanos que recibieron 0,3, 1 o 5 mg/kg/día de pramipexol durante 13 semanas tampoco detectó cambios.
No se ha establecido el significado potencial de este efecto en humanos, pero no puede ignorarse debido a que la alteración de un mecanismo que está universalmente presente en los vertebrados (es decir, derramamiento de disco) puede estar involucrado.
• Lesiones proliferativas fibro-óseas en ratones: Ocurrió un aumento en la incidencia de lesiones proliferativas fibro-óseas en el fémur de ratones hembra tratados durante 2 años con 0.3, 2.0 o 10 mg/kg/día (0.3, 2.2 y 11 veces la MRHD sobre una base de mg/m2). No se observó lesiones similares en ratones o ratas macho y monos de cualquier sexo que fueron tratados crónicamente con pramipexol. No se conoce el significado de esta lesión para los humanos.
ADVERTENCIAS Y PRECAUCIONES
Quedarse dormido durante la realización de actividades de la vida diaria: Los pacientes tratados con pramipexol han reportado quedarse dormidos mientras se encontraban realizando actividades de la vida diaria, incluyendo la operación de vehículos motorizados lo cual resultó a veces en accidentes. Aunque muchos de estos pacientes reportaron somnolencia mientras estuvieron bajo tratamiento con pramipexol comprimidos, algunos percibieron que no tuvieron signos de advertencia como somnolencia excesiva, y creyeron estar alerta inmediatamente antes del evento. Algunos de estos eventos se reportaron tan tarde como un año después del inicio del tratamiento.
La somnolencia es una ocurrencia común en pacientes que reciben pramipexol a dosis por encima de 1,5 mg/día (0,5 mg TID) para enfermedad de Parkinson. En ensayos clínicos controlados en pacientes con RLS tratados con SIFROL® comprimidos a dosis de 0,25-0,75 mg una vez al día, la incidencia de somnolencia fue 6% comparado con una incidencia de 3% para pacientes tratados con placebo (ver Reacciones adversas: Experiencia en ensayos clínicos). Muchos expertos clínicos creen que quedarse dormido mientras se realiza actividades de la vida diaria ocurre siempre en un entorno de somnolencia preexistente, aunque los pacientes pueden no proporcionar esta historia. Por esta razón, los médicos que prescriben deben reevaluar de manera continua a los pacientes con respecto a la ocurrencia de somnolencia o sopor, especialmente debido a que algunos de los eventos ocurren bastante después del inicio del tratamiento. Los médicos que prescriben también deben ser conscientes de que los pacientes pueden no reconocer la somnolencia o sopor hasta que se les pregunte directamente sobre la somnolencia o sopor durante la realización de actividades específicas.
Antes de iniciar tratamiento con SIFROL® comprimidos, informar a los pacientes del potencial para desarrollar somnolencia y preguntar específicamente sobre factores que pueden aumentar el riesgo con SIFROL® comprimidos como el uso de fármacos sedantes concomitantes o alcohol, la presencia de alteraciones del sueño y fármacos concomitantes que aumenten los niveles plasmáticos de pramipexol (p. ej., cimetidina) (ver Farmacología clínica: Farmacocinética). Si un paciente desarrolla somnolencia diurna significativa o episodios de quedarse dormido durante la realización de actividades que requieren participación activa (p. ej., conversar, comer, etc.), normalmente debe descontinuarse la administración de SIFROL® comprimidos. Si se toma la decisión de continuar SIFROL® comprimidos, recomendar a los pacientes que no manejen y que eviten otras actividades potencialmente peligrosas. Si bien la reducción de la dosis reduce el grado de somnolencia, no existe información suficiente para establecer que la reducción de la dosis eliminará los episodios de quedarse dormido mientras se realiza actividades de la vida diaria.
Hipotensión ortostática sintomática: Los agonistas de la dopamina, en estudios clínicos y en la experiencia clínica, parecen deteriorar la regulación sistémica de la presión arterial, con hipotensión ortostática resultante, especialmente durante el aumento de la dosis. Además, los pacientes con enfermedad de Parkinson parecer tener una capacidad deteriorada para responder a un reto ortostático. Por estas razones, tanto los pacientes con enfermedad de Parkinson como los pacientes con RLS que son tratados con agonistas dopaminérgicos normalmente requieren un monitoreo cuidadoso con respecto a la presencia de signos y síntomas de hipotensión ortostática, especialmente durante el aumento de la dosis, y deben ser informados sobre este riesgo (ver Información de orientación para el paciente).
Sin embargo, en ensayos clínicos de pramipexol, y pese a los claros efectos ortostáticos en voluntarios normales, la incidencia reportada de hipotensión ortostática clínicamente significativa no fue mayor entre aquéllos asignados a pramipexol comprimidos que entre aquéllos asignados a placebo. Este resultado, especialmente con las dosis más altas usadas en enfermedad de Parkinson, es claramente inesperado a la luz de la experiencia previa con los riesgos de la terapia con agonistas de la dopamina.
Si bien este hallazgo podría reflejar una propiedad única de pramipexol, también podría ser explicado por las condiciones del estudio y la naturaleza de la población enrolada en los ensayos clínicos. Los pacientes fueron titulados de manera muy cuidadosa, y se excluyó a los pacientes con enfermedad cardiovascular activa o hipotensión ortostática significativa en la basal. Además, los ensayos clínicos en pacientes con RLS no incorporaron retos ortostáticos con monitoreo intensivo de la presión arterial realizado en proximidad temporal cercana a la dosificación.
Control de impulsos/Comportamientos compulsivos: Los reportes de caso y los resultados de un estudio transversal sugieren que los pacientes pueden experimentar impulsos intensos de apostar, aumento del instinto sexual, impulsos intensos de gastar dinero incontrolablemente, darse atracones de comida y/u otros impulsos intensos y la incapacidad de controlar estos impulsos mientras toman uno o más de los fármacos, incluyendo SIFROL®, que aumentan el tono dopaminérgico central y que son generalmente usados para el tratamiento de la enfermedad de Parkinson. En algunos casos, aunque no en todos, se reportó que estos impulsos se detuvieron cuando se redujo la dosis o se descontinuó el fármaco. Debido a que los pacientes pueden no reconocer estos comportamientos como anormales es importante que los médicos que prescriben pregunten específicamente a los pacientes o a las personas encargadas de su cuidado sobre el desarrollo, nuevo o aumentado, de impulsos de apostar, instinto sexual, gastar incontrolablemente u otros impulsos mientras están siendo tratados con SIFROL®. Los médicos deben considerar la reducción de la dosis o suspender el fármaco si un paciente desarrolla estos impulsos mientras toma SIFROL® (ver Información de orientación para el paciente).
Alucinaciones: En los tres ensayos controlados con placebo, en doble ciego, en enfermedad de Parkinson temprana, se observó alucinaciones en 9% (35 de 388) de pacientes que recibieron SIFROL comprimidos, comparado con 2,6% (6 de 235) de pacientes que recibieron placebo. En los cuatro ensayos controlados con placebo, en doble ciego, en enfermedad de Parkinson avanzada, donde los pacientes recibieron SIFROL® comprimidos y levodopa concomitante, se observó alucinaciones en 16,5% (43 de 260) de pacientes que recibieron SIFROL® comprimidos comparado con 3,8% (10 de 264) de pacientes que recibieron placebo. Las alucinaciones fueron de severidad suficiente para causar la descontinuación del tratamiento en 3,1% de los pacientes con enfermedad de Parkinson temprana y 2,7% de los pacientes con enfermedad de Parkinson avanzada comparado con aproximadamente 0,4% de pacientes que recibieron placebo en ambas poblaciones.
La edad parece aumentar el riesgo de alucinaciones atribuible a pramipexol. En pacientes con enfermedad de Parkinson temprana, el riesgo de alucinaciones fue 1,9 veces mayor que con placebo en pacientes menores de 65 años y 6,8 veces mayor que con placebo en pacientes mayores de 65 años. En pacientes con enfermedad de Parkinson avanzada, el riesgo de alucinaciones fue 3,5 veces mayor que con placebo en pacientes menores de 65 años y 5,2 veces mayor que con placebo en pacientes mayores de 65 años.
En el programa clínico de RLS, un paciente tratado con pramipexol (de 889) reportó alucinaciones; este paciente descontinuó el tratamiento y los síntomas desaparecieron.
Disquinesia: SIFROL® comprimidos puede potenciar los efectos secundarios dopaminérgicos de levodopa y puede causar o exacerbar la disquinesia preexistente.
Insuficiencia renal: Debido a que pramipexol se elimina a través de los riñones, debe tenerse cuidado cuando se prescriba SIFROL® comprimidos a pacientes con insuficiencia renal (ver Dosis y Administración: Síndrome de las Piernas Inquietas, Uso en poblaciones específicas: Pacientes con Insuficiencia Renal y Farmacología clínica: Farmacocinética).
Rabdomiólisis: Ocurrió un solo caso de rabdomiólisis en un hombre de 49 años de edad con enfermedad de Parkinson avanzada tratado con SIFROL® comprimidos. El paciente fue hospitalizado con CPK elevada (10.631 UI/L). Los síntomas desaparecieron con la descontinuación del fármaco.
Patología retiniana:
• Datos en humanos: Un estudio de seguridad con grupos paralelos, randomizado, de etiqueta abierta, de dos años de deterioro retiniano y visión comparó SIFROL® comprimidos con ropinirol de liberación inmediata. Doscientos treinta y cuatro pacientes con enfermedad de Parkinson (115 bajo pramipexol, dosis media 3,0 mg/día y 119 bajo ropinirol, dosis media 9,5 mg/día) fueron evaluados usando un panel de evaluaciones oftalmológicas clínicas. De 234 pacientes que fueron evaluables, 196 habían sido tratados durante dos años y se consideró que 29 habían desarrollado anormalidades clínicas que se consideraron significativas (19 pacientes en cada brazo de tratamiento habían recibido tratamiento durante menos de dos años). No hubo diferencia estadística en el deterioro retiniano entre los brazos de tratamiento; sin embargo, el estudio solo fue capaz de detectar una diferencia muy grande entre tratamientos. Además, debido a que el estudio no incluyó un grupo de comparación sin tratar (tratado con placebo), no se sabe si los hallazgos reportados en los pacientes tratados con cualquiera de los fármacos son mayores que la tasa de datos históricos en una población que envejece.
• Datos en animales: Se observó cambios patológicos (degeneración y pérdida de células fotorreceptoras) en la retina de ratas albinas en el estudio de carcinogenicidad de 2 años. Si bien no se diagnosticó degeneración retiniana en las ratas pigmentadas tratadas durante 2 años, un adelgazamiento de la capa nuclear externa de la retina fue ligeramente mayor en las ratas que recibieron el fármaco comparado con los controles. La evaluación de las retinas de ratones albinos, monos y cerdos enanos no reveló cambios similares. No se ha establecido el significado potencial de este efecto en humanos, pero no puede pasarse por alto debido a que la alteración de un mecanismo que está universalmente presente en los vertebrados (es decir, derramamiento de disco) puede estar involucrado (ver Toxicología no clínica: Toxicología y/o farmacología animal).
Eventos reportados con la terapia dopaminérgica: Aunque los eventos enumerados a continuación pueden no haber sido reportados en asociación con el uso de pramipexol en su programa de desarrollo, éstos están asociados con el uso de otros fármacos dopaminérgicos. Sin embargo, la incidencia esperada de estos eventos es tan baja que incluso si pramipexol causara estos eventos a tasas similares a las atribuibles a otras terapias dopaminérgicas, sería poco probable que hubiese ocurrido aunque sea un solo caso en una cohorte del tamaño expuesto a pramipexol en los estudios hasta la fecha.
• Hiperpirexia y confusión relacionadas con la abstinencia: Aunque no se han reportado con pramipexol en el programa de desarrollo clínico, se ha reportado un complejo de síntomas que se asemeja al síndrome neuroléptico maligno (caracterizado por temperatura elevada, rigidez muscular, alteración de la conciencia e inestabilidad autónoma), sin otra etiología obvia, en asociación con una reducción rápida de la dosis, abstinencia o cambio en la terapia antiparkinsoniana.
• Complicaciones fibróticas: Se ha reportado casos de fibrosis retroperitoneal, infiltrados pulmonares, efusión pleural, engrosamiento pleural, pericarditis y valvulopatía cardiaca en pacientes tratados con agentes dopaminérgicos derivados del ergot. Si bien estas complicaciones pueden resolverse cuando se descontinúa el fármaco, la resolución completa no siempre ocurre.
Aunque se cree que estos eventos adversos están relacionados con la estructura ergolina de estos compuestos, no se sabe si otros agonistas de la dopamina no derivados del ergot pueden causarlos.
Se ha reportado casos de posibles complicaciones fibróticas, incluyendo fibrosis peritoneal, fibrosis pleural y fibrosis pulmonar en la experiencia posterior a la comercialización con SIFROL® comprimidos. Si bien la evidencia no es suficiente para establecer una relación causal entre SIFROL® comprimidos y estas complicaciones fibróticas, no puede descartarse completamente una contribución de SIFROL® comprimidos.
• Melanoma: Estudios epidemiológicos han demostrado que los pacientes con enfermedad de Parkinson tienen un riesgo mayor (aproximadamente 2 a 6 veces mayor) de desarrollar melanoma que la población general. No es claro si el aumento observado en el riesgo se debió a la enfermedad de Parkinson o a otros factores, como fármacos usados para tratar la enfermedad de Parkinson.
Por las razones mencionadas anteriormente, se recomienda a pacientes y profesionales de la salud monitorear con respecto a la presencia de melanomas de manera frecuente y regular cuando se usa SIFROL® comprimidos para cualquier indicación. Idealmente, debe realizarse exámenes cutáneos periódicos por personas adecuadamente calificadas (p. ej., dermatólogos).
• Rebote y Aumento en RLS: Los reportes en la literatura indican que el tratamiento de RLS con fármacos dopaminérgicos puede resultar en rebote: un empeoramiento de los síntomas luego del cese del tratamiento con mayor intensidad que lo descrito antes de iniciar el tratamiento. En un ensayo clínico, controlado con placebo, de 26 semanas, en pacientes con RLS, un empeoramiento del puntaje de los síntomas (IRLS) más allá de los niveles basales sin tratar fue reportado de manera más frecuente por los pacientes que abandonaron repentinamente SIFROL® (hasta 0,75 mg una vez al día) comparado con el grupo asignado a placebo (10% frente a 2%, respectivamente). El empeoramiento de los síntomas de RLS fue considerado generalmente leve.
El aumento también se ha descrito durante la terapia para RLS. Aumento referido al inicio más temprano de los síntomas en la noche (o incluso en la tarde), aumento en los síntomas y diseminación de síntomas para involucrar otras extremidades. En un ensayo clínico controlado con placebo de 26 semanas en pacientes con RLS, el aumento fue reportado con mayor frecuencia por pacientes tratados con SIFROL® (hasta 0,75 mg una vez al día) comparado con pacientes que recibieron placebo (12% frente a 9%, respectivamente). La incidencia de aumento se incrementó con una duración de exposición cada vez mayor a SIFROL® y a placebo.
La frecuencia y severidad del aumento y/o rebote después de uso a largo plazo de SIFROL® comprimidos así como el manejo apropiado de estos eventos no han sido evaluadas adecuadamente en ensayos clínicos controlados.
ABUSO Y DEPENDENCIA DEL FÁRMACO
Sustancia controlada: Pramipexol no es una sustancia controlada.
Abuso y dependencia: Pramipexol no ha sido sistemáticamente estudiado en animales o humanos con respecto a su potencial para abuso, tolerancia o dependencia física. Sin embargo, en un modelo en la rata bajo autoadministración de cocaína, pramipexol tuvo un efecto escaso o nulo.
DOSIS Y ADMINISTRACIÓN
Consideraciones de dosificación generales: SIFROL® comprimidos se toma por vía oral, con o sin alimentos.
Si ha ocurrido una interrupción significativa en la terapia con SIFROL® comprimidos, el reajuste de la terapia puede estar justificado.
Enfermedad de Parkinson: En todos los estudios clínicos, la dosis se inició a un nivel su terapéutico para evitar efectos adversos intolerables e hipotensión ortostática. SIFROL® comprimidos debe ajustarse gradualmente en todos los pacientes. Debe aumentarse la dosis para lograr un efecto terapéutico máximo, teniendo en cuenta los efectos secundarios principales de disquinesia, alucinaciones, somnolencia y sequedad de boca.
• Dosificación en pacientes con función renal normal:
— Tratamiento inicial: Las dosis deben aumentarse gradualmente a partir de una dosis inicial de 0.375 mg/día administradas en tres dosis divididas y no deben aumentarse de manera más frecuente que cada 5 a 7 días. En el siguiente cuadro se muestra un esquema sugerido de dosificación ascendente que se usó en los estudios clínicos:
|
Cuadro 1: Esquema de Dosificación Ascendente de SIFROL® comprimidos para Enfermedad de Parkinson |
||
|
Semana |
Dosis (mg) |
Dosis Diaria Total (mg) |
|
1 |
0,125 TID |
0,375 |
|
2 |
0,25 TID |
0,75 |
|
3 |
0,5 TID |
1,50 |
|
4 |
0,75 TID |
2,25 |
|
5 |
1 TID |
3,0 |
|
6 |
1,25 TID |
3,75 |
|
7 |
1,5 TID |
4,50 |
— Tratamiento de mantenimiento: SIFROL® comprimidos fue efectivo y bien tolerado a lo largo del rango de dosis de 1,5 a 4,5 mg/día administrado en dosis igualmente divididas tres veces al día con o sin levodopa concomitante (aproximadamente 800 mg/día).
En un estudio de dosis fija en pacientes con enfermedad de Parkinson temprana, no se demostró que dosis de 3 mg, 4,5 mg y 6 mg por día de SIFROL® comprimidos proporcionaron ningún beneficio significativo más allá del logrado a una dosis diaria de 1,5 mg/día. Sin embargo, en el mismo estudio de dosis fija, los siguientes eventos adversos estuvieron relacionados con la dosis: hipotensión postural, náuseas, estreñimiento, somnolencia y amnesia. La frecuencia de estos eventos fue generalmente dos veces mayor que con placebo para dosis de pramipexol mayores de 3 mg/día. La incidencia de somnolencia reportada con pramipexol a una dosis de 1,5 mg/día fue comparable con la de placebo.
Cuando SIFROL® comprimidos se usa en combinación con levodopa, debe considerarse una reducción de la dosis de levodopa. En un estudio controlado en enfermedad de Parkinson avanzada, la dosis de levodopa se redujo en un promedio de 27% con respecto a la basal.
• Dosificación en pacientes con deterioro de la función renal
|
Cuadro 2: Dosificación de SIFROL® comprimidos en Pacientes con Enfermedad de Parkinson con Deterioro de la Función Renal |
||
|
Estado Renal |
Dosis Inicial (mg) |
Dosis Máxima (mg) |
|
Insuficiencia normal a leve (depuración de creatinina >50 mL/min) |
0,125 TID |
1,5 TID |
|
Insuficiencia moderada (depuración de creatinina = 30 a 50 mL/min) |
0,125 BID |
0,75 TID |
|
Insuficiencia severa (depuración de creatinina = 15 a <30 mL/min) |
0,125 QD |
1,5 QD |
|
Insuficiencia muy severa (depuración de creatinina <15 mL/min y pacientes de hemodiálisis) |
No se ha estudiado de manera adecuada el uso de SIFROL® comprimidos en este grupo de pacientes. |
|
• Descontinuación del tratamiento: SIFROL® comprimidos debe disminuirse gradualmente a una tasa de 0,75 mg por día hasta que la dosis diaria se haya reducido a 0,75 mg. De ahí en adelante, la dosis debe reducirse en 0,375 mg por día. Sin embargo, en algunos estudios la descontinuación abrupta no presentó inconveniente.
Síndrome de las Piernas Inquietas: La dosis inicial recomendada de SIFROL® comprimidos es 0,125 mg tomada una vez al día 2-3 horas antes de acostarse. Para pacientes que requieren alivio sintomático adicional, la dosis puede aumentarse cada 4-7 días (Cuadro 3). Aunque la dosis de SIFROL® comprimidos fue aumentada a 0,75 mg en algunos pacientes durante el tratamiento de etiqueta abierta a largo plazo, no existe evidencia de que la dosis de 0,75 mg proporcione beneficio adicional más allá de la dosis de 0,5 mg.
|
Cuadro 3: Esquema de Dosis Ascendente de SIFROL® comprimidos para RLS |
||
|
Paso de Titulación |
Duración |
Dosis (mg) que se tomará una vez al día, 2-3 horas antes de acostarse |
|
1 |
4-7 días |
0,125 |
|
2* |
4-7 días |
0,25 |
|
3* |
4-7 días |
0,5 |
|
* si es necesario |
||
• Pacientes con Insuficiencia Renal: La duración entre pasos de titulación debe aumentar a 14 días en pacientes con RLS con insuficiencia renal severa a moderada (depuración de creatinina 20-60 ml/min) (ver Farmacología clínica: Farmacocinética).
• Descontinuación del tratamiento: En ensayos clínicos de pacientes tratados por RLS con dosis hasta de 0.75 mg una vez al día, SIFROL® comprimidos se descontinuó sin disminución gradual. En un ensayo clínico controlado con placebo de 26 semanas, los pacientes reportaron un empeoramiento de la severidad de los síntomas de RLS comparado con su basal sin tratar cuando el tratamiento con SIFROL® fue retirado abruptamente (ver Advertencias y Precauciones: Eventos reportados con la terapia dopaminérgica).
USO EN POBLACIONES ESPECÍFICAS
Embarazo: Embarazo Categoría C.
No existen estudios adecuados y bien controlados en mujeres embarazadas. SIFROL® comprimidos debe usarse durante el embarazo solo si el beneficio potencial justifica el riesgo potencial para el feto.
Cuando pramipexol se administró a ratas hembra durante la preñez, la implantación se inhibió a una dosis de 2,5 mg/kg/día (5 veces la dosis máxima recomendada en humanos (MRHD) para enfermedad de Parkinson de 4,5 mg/día sobre una base del área de superficie corporal (mg/m2)). La administración de 1.5 mg/kg/día de pramipexol a ratas preñadas durante el periodo de organogénesis (días de gestación 7 a 16) resultó en una incidencia alta de resorción total de embriones. El AUC en plasma en ratas a esta dosis fue 4 veces el AUC en humanos a la MRHD. Se cree que estos hallazgos se deben al efecto reductor de prolactina de pramipexol, debido a que la prolactina es necesaria para la implantación y mantenimiento de las primeras etapas de la preñez en ratas (pero no en conejos o humanos). Debido a la interrupción de la preñez y pérdida embrionaria temprana en estos estudios, no pudo evaluarse adecuadamente el potencial teratogénico de pramipexol. No hubo evidencia de efectos adversos en el desarrollo embriofetal luego de la administración hasta de 10 mg/kg/día a conejas preñadas durante la organogénesis (el AUC en plasma fue 70 veces el AUC en humanos a la MRHD). Se inhibió el crecimiento postnatal en la descendencia de ratas tratadas con 0,5 mg/kg/día (aproximadamente equivalente a la MRHD sobre una base de mg/m2) o más durante la última parte de la preñez y toda la lactancia.
Madres lactantes: Los estudios han demostrado que el tratamiento con pramipexol resultó en una inhibición de la secreción de prolactina en humanos y ratas.
Un estudio radiomarcado, de dosis única mostró que el material relacionado con el fármaco estuvo presente en la leche de rata a concentraciones tres a seis veces mayores que aquéllas en plasma en puntos de evaluación equivalentes. No se sabe si este fármaco se excreta en la leche humana. Debido a que muchos fármacos se excretan en la leche humana y debido al potencial para causar reacciones adversas serias en lactantes de SIFROL®, debe tomarse la decisión si descontinuar la lactancia o descontinuar el fármaco, tomando en cuenta la importancia del fármaco para la madre.
Uso pediátrico: No se ha establecido la seguridad y eficacia de SIFROL® en pacientes pediátricos.
Uso geriátrico: La depuración oral total de pramipexol es aproximadamente 30% menor en personas mayores de 65 años comparada con personas más jóvenes, debido a una disminución en la depuración renal de pramipexol como consecuencia de una reducción relacionada con la edad en la función renal. Esto resultó en un aumento en la vida media de eliminación de aproximadamente 8,5 horas a 12 horas.
En estudios clínicos con pacientes con enfermedad de Parkinson, 38,7% de pacientes fueron mayores de 65 años. No hubo diferencias aparentes en la eficacia o seguridad entre pacientes mayores y más jóvenes, excepto que el riesgo relativo de alucinación asociado con el uso de SIFROL® comprimidos aumentó en el adulto mayor.
En estudios clínicos con pacientes con RLS, 22% de pacientes tuvieron al menos 65 años de edad. No hubo diferencias aparentes en la eficacia o seguridad entre pacientes mayores y más jóvenes.
Pacientes con Insuficiencia Renal: La eliminación de pramipexol es dependiente de la función renal. La depuración de pramipexol es extremadamente baja en pacientes de diálisis, ya que una cantidad mínima de pramipexol es eliminada por diálisis. Debe tenerse cuidado cuando se administre SIFROL® comprimidos a pacientes con enfermedad renal (ver Dosis y administración: Enfermedad de Parkinson, Advertencias y precauciones: Insuficiencia renal y Farmacología clínica: Farmacocinética).
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
0.25 mg: Comprimido, oval, blanca con “BI” en un lado y “P7” en el reverso.
1 mg: Comprimido, redonda, blanca con “BI” en un lado y “P9” en el reverso.
SOBREDOSIS: No existe experiencia clínica con sobredosis significativa. Un paciente tomó 11 mg/día de pramipexol durante 2 días en un ensayo clínico para uso de investigación. La presión arterial permaneció estable aunque la frecuencia del pulso aumentó a entre 100 y 120 latidos/minuto. No se reportó otros eventos adversos relacionados con el aumento de la dosis.
No existe antídoto conocido para sobredosis de un agonista de la dopamina. Si se presenta signos de estimulación del sistema nervioso central, una fenotiazina u otro agente neuroléptico tipo butirofenona puede estar indicado; no se ha evaluado la eficacia de estos fármacos para revertir los efectos de una sobredosis. El manejo de la sobredosis puede requerir medidas de soporte general junto con lavado gástrico, fluidos intravenosos y vigilancia electrocardiográfica.
DESCRIPCIÓN: SIFROL® comprimidos contiene pramipexol, un agonista no ergótico de la dopamina. El nombre químico de pramipexol diclorhidrato es (S)-2-amino-4,5,6,7-tetrahidro-6-(propilamino) benzotiazol diclorhidrato monohidrato. Su fórmula empírica es C10H17N3S • 2HCl • H2O, y su peso molecular es 302,26. La fórmula estructural es:
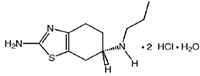
Pramipexol diclorhidrato es una sustancia en polvo de color blanco a blanquecino. La fusión ocurre en el rango de 296 °C a 301 °C, con descomposición. Pramipexol diclorhidrato es más de 20% soluble en agua, aproximadamente 8% en metanol, aproximadamente 0,5% en etanol y prácticamente insoluble en diclorometano.
SIFROL® comprimidos, para administración oral, contiene 0,25 mg y 1 mg de pramipexol diclorhidrato monohidrato. Los ingredientes inactivos consisten en manitol, almidón de maíz, dióxido de silicio coloidal, povidona y estearato de magnesio.
FARMACOLOGÍA CLÍNICA
Mecanismo de acción: Pramipexol es un agonista no ergótico de la dopamina con especificidad relativa alta in vitro y actividad intrínseca completa en la subfamilia D2 de los receptores de dopamina, que se une con mayor afinidad al receptor D3 que a los subtipos de receptores D2 o D4.
• Enfermedad de Parkinson: El mecanismo de acción preciso de pramipexol como tratamiento para enfermedad de Parkinson es desconocido, aunque se cree que está relacionado con su habilidad para estimular los receptores de dopamina en el cuerpo estriado. Esta conclusión está respaldada por estudios electrofisiológicos en animales que han demostrado que pramipexol influye en las tasas de descarga neuronal del cuerpo estriado a través de activación de los receptores de dopamina en el cuerpo estriado y la sustancia negra, el sitio de neuronas que envía proyecciones al cuerpo estriado. Se desconoce la importancia de la unión del receptor D3 en enfermedad de Parkinson.
• Síndrome de las Piernas Inquietas (RLS): El mecanismo de acción preciso de SIFROL® comprimidos como tratamiento para RLS es desconocido. Aunque la patofisiología de RLS es en buena parte desconocida, la evidencia neurofarmacológica sugiere la participación del sistema dopaminérgico primario. Los estudios con Tomografía por Emisión de Positrones (PET) sugieren que una disfunción dopaminérgica presináptica leve en el cuerpo estriado puede estar implicada en la patogénesis de RLS.
Farmacodinámica: Se investigó el efecto de pramipexol en el intervalo QT del ECG en un estudio clínico en 60 voluntarios sanos de sexo masculino y femenino. Todos los participantes iniciaron tratamiento con 0,375 mg de pramipexol comprimidos de liberación extendida administradas una vez al día, y fueron titulados de manera ascendente cada 3 días a 2,25 mg y 4,5 mg diarios. No se observó efecto relacionado con la dosis o exposición en los intervalos QT medios; sin embargo, el estudio no tuvo una evaluación válida de la sensibilidad del ensayo. No se ha evaluado de manera sistemática el efecto de pramipexol en los intervalos QTc a exposiciones mayores logrado ya sea debido a interacciones medicamentosas (p. ej., con cimetidina), insuficiencia renal o a dosis más altas.
Aunque los valores medios se mantuvieron dentro de los rangos de referencia normales durante todo el estudio, la presión arterial sistólica en posición supina (SBP), presión arterial diastólica (DBP) y frecuencia del pulso para los participantes tratados con pramipexol generalmente aumentaron durante la fase de titulación ascendente rápida en 10 mmHg, 7 mmHg y 10 bpm más que para placebo, respectivamente. La SBP, DBP y frecuencias de pulso mayores comparado con placebo se mantuvieron hasta que se redujo gradualmente las dosis de pramipexol; los valores el último día de la reducción gradual fueron generalmente similares a los valores basales. Estos efectos no han sido observados en estudios clínicos con pacientes con enfermedad de Parkinson, que fueron titulados de acuerdo con las recomendaciones de la etiqueta.
Farmacocinética: Pramipexol muestra una farmacocinética lineal a lo largo del rango de dosificación clínica. Su vida media terminal es aproximadamente 8 horas en voluntarios sanos jóvenes y aproximadamente 12 horas en voluntarios adultos mayores. Las concentraciones en estado de equilibrio se logran en el lapso de 2 días de dosificación.
• Absorción: Pramipexol es absorbido de manera rápida, alcanzando concentraciones pico en aproximadamente 2 horas. La biodisponibilidad absoluta de pramipexol es mayor de 90%, indicando que es bien absorbido y que experimenta metabolismo presistémico escaso. Las comidas no afectan el grado de absorción de pramipexol, aunque el tiempo de concentración plasmática máxima (Tmax) aumenta en aproximadamente 1 hora cuando el fármaco se ingiere con una comida.
• Distribución: Pramipexol se distribuye de manera extensa, teniendo un volumen de distribución de aproximadamente 500 L (coeficiente de variación [CV] = 20%). Se une aproximadamente en 15% a las proteínas plasmáticas. Pramipexol se distribuye en las células rojas de la sangre como lo indica una proporción eritrocito/plasma de aproximadamente 2.
• Metabolismo: Pramipexol solo se metaboliza en un grado insignificante (<10%). No se ha identificado ningún metabolito activo específico en plasma u orina humana.
• Eliminación: La excreción urinaria es la vía principal de eliminación de pramipexol, recuperándose 90% de una dosis de pramipexol en la orina, casi todo como fármaco inalterado. La depuración renal de pramipexol es aproximadamente 400 mL/min (CV=25%), aproximadamente tres veces mayor que la tasa de filtración glomerular. Por lo tanto, pramipexol es secretado por los túbulos renales, probablemente por el sistema de transporte de cationes orgánicos.
• Farmacocinética en poblaciones específicas: Debido a que la terapia con SIFROL® comprimidos es iniciada a dosis baja y titulada gradualmente de manera ascendente de acuerdo con la tolerabilidad clínica para obtener el efecto terapéutico óptimo, el ajuste de la dosis inicial en base al sexo, peso, raza o edad no es necesario. Sin embargo, la insuficiencia renal, la cual puede causar una gran disminución en la habilidad para eliminar pramipexol, puede necesitar un ajuste de la dosis (ver Dosis y administración Enfermedad de Parkinson).
— Sexo: La depuración de pramipexol es aproximadamente 30% menor en mujeres que en hombres, pero esta diferencia puede representar diferencias en el peso corporal. No existe diferencia en la vida media entre hombres y mujeres.
— Edad: La depuración de pramipexol disminuye con la edad ya que la vida media y la depuración son aproximadamente 40% más larga y 30% menor, respectivamente, en adultos mayores (mayores de 65 años de edad) comparado con voluntarios sanos jóvenes (menores de 40 años de edad). Es más probable que esta diferencia se deba a la reducción en la función renal con la edad, debido a que la depuración de pramipexol se correlaciona con la función renal, medida por la depuración de creatinina.
— Raza: No se ha identificado diferencias raciales en el metabolismo y eliminación.
— Pacientes con Enfermedad de Parkinson: Una comparación cruzada de datos entre estudios sugiere que la depuración de pramipexol puede estar reducida en aproximadamente 30% en pacientes con enfermedad de Parkinson comparado con voluntarios adultos mayores sanos. La razón para esta diferencia parece ser la reducción de la función renal en pacientes con enfermedad de Parkinson, lo cual puede estar relacionado con su salud general más pobre. La farmacocinética de pramipexol fue comparable entre pacientes con enfermedad de Parkinson temprana y avanzada.
— Pacientes con Síndrome de las Piernas Inquietas: Una comparación cruzada de datos entre estudios sugiere que el perfil farmacocinético de pramipexol administrado una vez al día en pacientes con RLS es similar al perfil farmacocinético de pramipexol en voluntarios sanos.
— Insuficiencia Hepática: No se ha evaluado la influencia de la insuficiencia hepática en la farmacocinética de pramipexol. Debido a que aproximadamente 90% de la dosis recuperada es excretada en la orina como fármaco inalterado, no se esperaría que la insuficiencia hepática tenga un efecto significativo en la eliminación de pramipexol.
— Insuficiencia Renal: La depuración de pramipexol fue aproximadamente 75% menor en pacientes con insuficiencia renal severa (depuración de creatinina aproximadamente 20 mL/min) y aproximadamente 60% menor en pacientes con insuficiencia moderada (depuración de creatinina aproximadamente 40 mL/min) comparado con voluntarios sanos (ver Advertencias y precauciones: Insuficiencia Renal y Dosis y administración: Enfermedad de Parkinson). En pacientes con diversos grados de insuficiencia renal, la depuración de pramipexol se correlaciona bien con la depuración de creatinina. Por lo tanto, la depuración de creatinina puede usarse como un factor de predicción del grado de disminución en la depuración de pramipexol.
• Interacciones medicamentosas:
— Carbidopa/levodopa: Carbidopa/levodopa no influyó en la farmacocinética de pramipexol en voluntarios sanos (N=10). Pramipexol no alteró el grado de absorción (AUC) o la eliminación de carbidopa/levodopa, aunque causó un aumento en la Cmax de levodopa en aproximadamente 40% y una disminución en Tmax de 2,5 a 0,5 horas.
— Selegiline: En voluntarios sanos (N=11), selegiline no influyó en la farmacocinética de pramipexol.
— Amantadina: Los análisis de farmacocinética poblacional sugieren que amantadina puede disminuir ligeramente la depuración oral de pramipexol.
— Cimetidina: Cimetidina, un inhibidor conocido de la secreción tubular renal de bases orgánicas a través del sistema de transporte de cationes, causó un aumento de 50% en el AUC de pramipexol y un aumento de 40% en la vida media (N=12).
— Probenecid: Probenecid, un inhibidor conocido de la secreción tubular renal de ácidos orgánicos a través del transportador aniónico, no influyó notablemente en la farmacocinética de pramipexol (N=12).
— Otros fármacos eliminados a través de secreción renal: El análisis de farmacocinética poblacional sugiere que la coadministración de fármacos que son secretados por el sistema de transporte de cationes (p. ej., cimetidina, ranitidina, diltiazem, triamtereno, verapamil, quinidina y quinina) disminuye la depuración oral de pramipexol en aproximadamente 20%, mientras que es probable que aquéllos secretados por el sistema de transporte de aniones (p. ej., cefalosporinas, penicilinas, indometacina, hidroclorotiazida y clorpropamida) tengan un efecto escaso en la depuración oral de pramipexol. Otros sustratos y/o inhibidores conocidos del transporte de cationes orgánicos (p. ej., cisplatino y procainamida) también pueden reducir la depuración de pramipexol.
— Interacciones con CYP: No se esperaría que los inhibidores de las enzimas del citocromo P450 afecten la eliminación de pramipexol debido a que pramipexol no es considerablemente metabolizado por estas enzimas in vivo o in vitro. Pramipexol no inhibe las enzimas CYP CYP1A2, CYP2C9, CYP2C19, CYP2E1 y CYP3A4. Se observó inhibición de CYP2D6 con un Ki aparente de 30 µm, lo que indica que pramipexol no inhibirá las enzimas CYP a las concentraciones plasmáticas observadas luego de la dosis clínica de 4,5 mg/día (1,5 mg TID).
PRESENTACIONES
SIFROL® 0,25 mg: Envases con 10, 30 y 100 comprimidos.
SIFROL® 1 mg: Envases con 10, 30 y 100 comprimidos.
No dejar los medicamentos al alcance de los niños.
CONDICIONES DE CONSERVACION Y ALMACENAMIENTO: Conservar en su envase original, y no almacenar a temperatura superior a 30 °C.
Proteger de la luz.
Almacenar en lugar seguro fuera del alcance de los niños.
TIEMPO DE VIDA ÚTIL: 3 años.


