
PRAXBIND 50 MG/ML
IDARUCIZUMAB
Solución inyectable
1 Caja, 2 Vial(es), 50 ml, 50 mg/ml
COMPOSICIÓN:
Cada mL de SOLUCIÓN INYECTABLE y para perfusión contiene: 50 mg de idarucizumab.
Cada VIAL de 50 mL contiene: 2,5 g de Idarucizumab
Excipientes: (Ácido acético glacial, Polisorbato 20, Acetato de sodio trihidrato, Sorbitol, Agua para inyección) c.s.p.
Idarucizumab se produce mediante la tecnología de ADN recombinante en células ováricas de hámster chino.
Excipientes con efecto conocido:
Cada vial de 50 ml contiene 2 g de sorbitol y 25 mg de sodio (ver sección Advertencias y precauciones especiales de empleo). Para consultar la lista completa de excipientes, ver sección Lista de excipientes.
FORMA FARMACÉUTICA:
Solución inyectable
Solución de transparente a ligeramente opalescente y de incolora a ligeramente amarilla.
INDICACIONES TERAPÉUTICAS:
PRAXBIND® es un agente de reversión específico para dabigatrán y está indicado en pacientes adultos tratados con PRADAXA® (dabigatrán etexilato) cuando se necesita una reversión rápida de sus efectos anticoagulantes:
• Para intervenciones quirúrgicas de urgencia o procedimientos urgentes.
• En el caso de hemorragias potencialmente mortales o no controladas.
DATOS FARMACÉUTICOS:
Lista de excipientes:
Acetato de sodio trihidrato Ácido acético glacial
Sorbitol Polisorbato 20
Agua para Inyección
Incompatibilidades: Este medicamento no debe mezclarse con otros.
Tiempo de vida útil: 3 años.
Tras la apertura del vial, se ha demostrado la estabilidad química y física en uso de idarucizumab durante 6 horas a temperatura ambiente.
Desde un punto de vista microbiológico, a menos que el método de apertura excluya el riesgo de contaminación microbiana, el producto se debe utilizar inmediatamente después de abrirlo. Si no se utiliza de inmediato, los tiempos de almacenamiento en uso y las condiciones previas al uso son responsabilidad del usuario.
“No consumir el producto una vez alcanzada la fecha de vencimiento indicada en los rotulados“.
Precauciones especiales de conservación: Almacenar en refrigerador.
Consérvese a una temperatura entre 2 °C a 8 °C.
No congelar.
Conservar en su envase original para protegerlo de la luz.
Antes de su uso, el vial puede mantenerse a temperatura ambiente (hasta 30 °C) hasta 48 horas, si se conserva en el embalaje original para protegerlo de la luz. La solución no debe exponerse a la luz durante más de 6 horas (en el vial sin abrir y/o en uso).
Para las condiciones de conservación tras la apertura del medicamento, ver sección Naturaleza y contenido del envase.
Naturaleza y contenido del envase: 50 ml de solución en un vial de vidrio (vidrio tipo I), con un tapón de goma butílica, con tapa de Flip – off.
Tamaño del envase de 2 viales.
Precauciones especiales de eliminación y otras manipulaciones: Antes de su administración, los medicamentos parenterales como PRAXBIND® se deben someter a una inspección visual para ver si presentan partículas o decoloración.
La eliminación del producto biotecnológico no utilizado o los restos derivados del mismo se realizarán acorde a normativa vigente.
PRAXBIND® no se debe mezclar con otros medicamentos. Para la administración de PRAXBIND® se puede utilizar una vía intravenosa preexistente. Dicha vía debe aclararse con una solución inyectable de 9 mg/ml de cloruro sódico (0,9%) antes y al final de la perfusión. No se debe administrar ninguna otra perfusión en paralelo a través del mismo acceso intravenoso.
PRAXBIND® es para un solo uso y no contiene conservantes (ver sección Tiempo de vida útil).
No se han observado incompatibilidades entre PRAXBIND® y equipos de perfusión de cloruro de polivinilo, polietileno o poliuretano, ni tampoco con jeringas de polipropileno.
Fecha de revisión de texto de la ficha técnica: 9 de noviembre de 2017.
Manténgase fuera del alcance de los niños
Venta con receta médica
PROPIEDADES FARMACOLÓGICAS:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: todos los demás productos terapéuticos, antídotos, código ATC: V03AB37
Mecanismo de acción:
Idarucizumab es un agente de reversión específico para dabigatrán. Es un fragmento de anticuerpo monoclonal humanizado (Fab) que se une a dabigatrán con una afinidad muy alta, aproximadamente 300 veces más potente que la afinidad de unión de dabigatrán a la trombina. El complejo idarucizumab-dabigatrán se caracteriza por una constante de asociación rápida y una constante de disociación extremadamente lenta, lo que da lugar a un complejo muy estable.
Idarucizumab se une de forma potente y específica a dabigatrán y a sus metabolitos y neutraliza su efecto anticoagulante.
Eficacia clínica y seguridad:
Se llevaron a cabo tres estudios de fase I aleatorizados, doble ciego y controlados con placebo en 283 individuos (224 tratados con idarucizumab) con el fin de evaluar la seguridad, la eficacia, la tolerancia, la farmacocinética y la farmacodinámica de idarucizumab, administrado solo o tras la administración de dabigatrán etexilato. La población investigada estaba formada por individuos sanos e individuos que presentaban unas características de población específicas que abarcaban la edad, el peso corporal, la raza, el sexo y la insuficiencia renal. En estos estudios, las dosis de idarucizumab oscilaron entre 20 mg y 8 g y la duración de las perfusiones osciló entre 5 minutos y 1 hora.
Los valores representativos de los parámetros farmacocinéticos y farmacodinámicos se establecieron basándose en individuos sanos de edades comprendidas entre 45 y 64 años que recibieron 5 g de idarucizumab (ver Propiedades farmacodinámicas y Propiedades farmacocinéticas).
Se llevó a cabo un estudio prospectivo, abierto, no aleatorizado y no controlado (RE-VERSE AD) para investigar el tratamiento de pacientes adultos que presentaron una hemorragia no controlada o potencialmente mortal relacionada con dabigatrán (Grupo A) o que necesitaron una intervención quirúrgica o procedimientos urgentes (Grupo B). El criterio de valoración principal fue el porcentaje máximo de reversión del efecto anticoagulante de dabigatrán en las 4 horas posteriores a la administración de idarucizumab, de acuerdo a la determinación por parte de un laboratorio central del tiempo de trombina diluido (TTd) o el tiempo de coagulación de ecarina (TCE). Un criterio de valoración secundario clave fue la restauración de la hemostasia.
El estudio RE-VERSE AD incluyó datos de 503 pacientes: 301 pacientes con sangrado grave (Grupo A) y 202 pacientes que necesitaron un procedimiento/intervención quirúrgica urgente (Grupo B). Aproximadamente la mitad de los pacientes de cada grupo eran varones. La mediana de la edad fue de 78 años y la mediana del aclaramiento de creatinina fue de 52,6 ml/min. El 61,5% de los pacientes del Grupo A y el 62,4% de los pacientes del Grupo B habían sido tratados con 110 mg de dabigatrán dos veces al día.
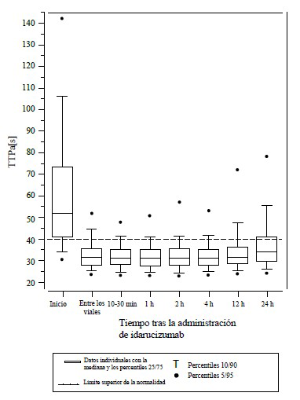
La reversión solo fue evaluable en los pacientes que presentaban tiempos de coagulación prolongados antes del tratamiento con idarucizumab. La mayoría de los pacientes, tanto en el Grupo A como en el B, lograron una reversión completa del efecto anticoagulante de dabigatrán (TTd: 98,7%; TCE: 82,2%; TTPa: 92,5% de los pacientes evaluables, respectivamente) en las primeras 4 horas tras la administración de 5 g de idarucizumab. Los efectos de la reversión fueron evidentes inmediatamente después de la administración.
Figura 1: Reversión de la prolongación del tiempo de coagulación inducida por dabigatrán, determinado mediante el TTd en pacientes del estudio RE-VERSE AD (N = 487)
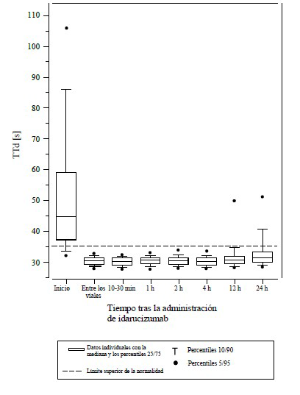
Figura 2: Reversión de la prolongación del tiempo de coagulación inducida por dabigatrán, determinado mediante el TCE en pacientes del estudio RE-VERSE AD (N = 487)
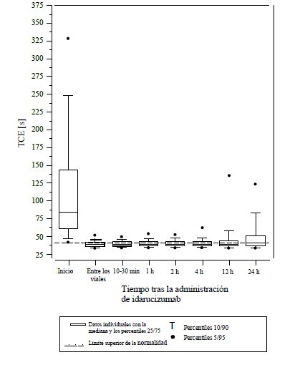
Figura 3: Reversión de la prolongación del tiempo de coagulación inducida por dabigatrán, determinado mediante el TTPa en pacientes del estudio RE-VERSE AD (N = 486)
La restauración de la hemostasia se alcanzó en el 80,3% de los pacientes evaluables que presentaron sangrado grave y se observó una hemostasia normal en el 93,4% de los pacientes que necesitaron un procedimiento urgente.
Del total de 503 pacientes, 101 pacientes murieron. Cada una de las muertes pudo atribuirse a una complicación del evento por el que ha entrado en el ensayo o asociarse a comorbilidades. Se notificaron acontecimientos trombóticos en 34 pacientes (23 de los 34 pacientes no se encontraban en tratamiento antitrombótico en el momento del acontecimiento). En cada uno de estos casos, el acontecimiento trombótico pudo atribuirse al trastorno médico subyacente del paciente. Se notificaron síntomas leves de hipersensibilidad potencial (pirexia, broncoespasmo, hiperventilación, erupción cutánea o prurito). No pudo establecerse una relación causal con idarucizumab.
Efectos farmacodinámicos: Se investigó la farmacodinámica de idarucizumab tras la administración de dabigatrán etexilato en 141 individuos en estudios de fase I, de los cuales se presentan los datos de un subgrupo representativo de 6 individuos sanos de edades comprendidas entre 45 y 64 años que estaban recibiendo una dosis de 5 g en perfusión intravenosa. La mediana de la exposición máxima a dabigatrán en los individuos sanos investigados estaba en el rango de una administración dos veces al día de 150 mg de dabigatrán etexilato en pacientes.
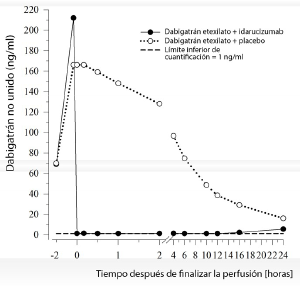
Efecto de idarucizumab sobre la exposición y la actividad anticoagulante de dabigatrán: Inmediatamente después de la administración de idarucizumab, las concentraciones plasmáticas de dabigatrán libre se redujeron en más del 99%, lo que dio lugar a niveles sin actividad anticoagulante.
La mayoría de los pacientes mostró una reversión sostenida de las concentraciones plasmáticas de dabigatrán de hasta 12 horas (≥90%). En un subgrupo de pacientes, se observado la recurrencia de concentraciones plasmáticas de dabigatrán libre y la elevación concomitante de las pruebas de coagulación, posiblemente debido a la redistribución de dabigatrán desde la periferia. Este hecho se produjo entre 1 y 24 horas tras la administración de idarucizumab, principalmente en tiempos ≥12 horas.
Figura 4: Niveles plasmáticos de dabigatrán libre en el grupo representativo de individuos sanos (administración de idarucizumab o placebo a las 0 h)
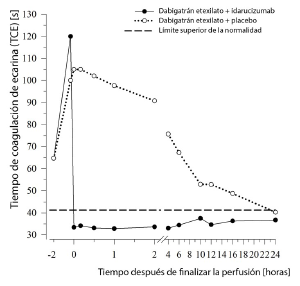
Dabigatrán prolonga el tiempo de coagulación de los marcadores de coagulación, como el tiempo de trombina diluido (TTd), el tiempo de trombina (TT), el tiempo de tromboplastina parcial activado (TTPa) y el tiempo de coagulación de ecarina (TCE), que proporcionan una indicación aproximada de la intensidad de la anticoagulación. Un valor en el rango normal después de la administración de idarucizumab indica que el paciente ya no está anticoagulado. Un valor por encima del rango normal puede reflejar la existencia de dabigatrán residual activo u otros cuadros clínicos, como la presencia de otros fármacos o una coagulopatía por transfusión. Estas pruebas se utilizaron para evaluar el efecto anticoagulante de dabigatrán. Inmediatamente después de la perfusión de idarucizumab, se observó una reversión completa y sostenida de la prolongación del tiempo de coagulación inducida por dabigatrán, que se mantuvo durante todo el periodo de observación de al menos 24 horas.
Figura 5: Reversión de la prolongación del tiempo de coagulación inducida por dabigatrán, determinado mediante el TTd en el grupo representativo de individuos sanos (administración de idarucizumab o placebo a las 0 h)
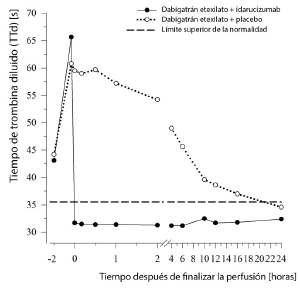
Figura 6: Reversión de la prolongación del tiempo de coagulación inducida por dabigatrán, determinado mediante el TCE en el grupo representativo de individuos sanos (administración de idarucizumab o placebo a las 0 h)
Parámetros de generación de trombina: Dabigatrán ejerce efectos pronunciados sobre los parámetros del potencial endógeno de la trombina (PET). El tratamiento con idarucizumab normalizó ambos, el índice de tiempo de latencia de la trombina y el índice de tiempo hasta el valor máximo a los niveles basales, según se determinó de 0,5 a 12 horas después de finalizar la perfusión de idarucizumab. Idarucizumab solo no ha mostrado ningún efecto procoagulante medido como PET. Eso sugiere que idarucizumab no tiene efecto protrombótico.
Readministración de dabigatrán etexilato: 24 horas después de la perfusión de idarucizumab, la readministración de dabigatrán etexilato dio lugar a la actividad anticoagulante esperada.
Inmunogenicidad: Se evaluaron muestras de suero de 283 individuos en ensayos de fase I (224 voluntarios tratados con idarucizumab) y de 501 pacientes para buscar anticuerpos frente a idarucizumab antes y después del tratamiento. Se detectaron anticuerpos preexistentes con reactividad cruzada frente a idarucizumab en aproximadamente el 12% (33/283) de los individuos de los ensayos de fase I y el 3,8% (19/501) de los pacientes. No se observó ningún impacto sobre la farmacocinética ni sobre el efecto de reversión de idarucizumab, ni tampoco reacciones de hipersensibilidad.
Se observaron títulos bajos de anticuerpos antiidarucizumab posiblemente persistentes relacionados con el tratamiento en el 4% (10/224) de los individuos de los ensayos de fase I y el 1,6% (8/501) de los pacientes, lo que sugiere un bajo potencial inmunógeno de idarucizumab. En un subgrupo de 6 individuos de los ensayos de fase I, idarucizumab se administró por segunda vez, dos meses después de la primera administración. No se detectaron anticuerpos antiidarucizumab en estos individuos antes de la segunda administración. En un individuo, se detectaron anticuerpos antiidarucizumab relacionados con el tratamiento después de la segunda administración. Nueve pacientes recibieron una segunda dosis de idarucizumab. Los 9 pacientes recibieron la segunda dosis en los 6 días siguientes a la primera dosis de idarucizumab. Ninguno de los pacientes que recibió una segunda dosis de idarucizumab mostró un resultado positivo para anticuerpos antiidarucizumab.
Farmacodinámica preclínica: Se realizó un modelo traumático en cerdos utilizando una lesión hepática contusa después de administrar dabigatrán con el fin de alcanzar concentraciones supraterapéuticas de aproximadamente 10 veces los niveles plasmáticos humanos. Idarucizumab revirtió de forma rápida y eficaz la hemorragia potencialmente mortal durante los 15 minutos siguientes a la inyección. Todos los cerdos sobrevivieron a dosis de idarucizumab de aproximadamente 2,5 y 5 g. Sin idarucizumab, la mortalidad en el grupo anticoagulado fue del 100%.
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con PRAXBIND® en uno o más grupos de la población pediátrica en la prevención y el tratamiento de las hemorragias asociadas a dabigatrán (ver sección Posología y forma de administración para consultar la información sobre el uso en la población pediátrica).
Propiedades farmacocinéticas:
La farmacocinética de idarucizumab se investigó en 224 individuos sanos en estudios de fase I, de los cuales se presentan datos de un subgrupo representativo de 6 individuos sanos entre 45 y 64 años de edad que recibieron una dosis de 5 g en perfusión intravenosa.
Distribución: Idarucizumab mostró una cinética de disposición multifase y una distribución extravascular limitada. Tras la perfusión intravenosa de una dosis de 5 g, la media geométrica del volumen de distribución en el estado estacionario (Vss) fue de 8,9 l (coeficiente de variación geométrica (gCV) 24,8%).
Biotransformación: Se han descrito varias vías que pueden contribuir al metabolismo de los anticuerpos. Todas estas vías implican la biodegradación del anticuerpo a moléculas más pequeñas, es decir, pequeños péptidos o aminoácidos que luego se reabsorben y se incorporan a la síntesis general de proteínas.
Eliminación: Idarucizumab se eliminó rápidamente, con un aclaramiento total de 47,0 ml/min (gCV 18,4%), una semivida inicial de 47 minutos (gCV 11,4%) y una semivida terminal de 10,3 horas (gCV 18,9%). Después de la administración intravenosa de 5 g de idarucizumab, se recuperó en la orina el 32,1% (gCV 60,0%) de la dosis, con un periodo de recogida de 6 horas, mientras que en las 18 horas siguientes se recuperó menos del 1%. Se supone que la parte restante de la dosis se elimina mediante el catabolismo de proteínas, principalmente en el riñón.
Se ha observado proteinuria tras el tratamiento con idarucizumab. La proteinuria transitoria es una reacción fisiológica a la llegada de una concentración elevada de proteínas a nivel renal tras la aplicación rápida o a corto plazo de 5 g de idarucizumab por vía intravenosa. La proteinuria transitoria suele alcanzar su máximo aproximadamente 4 horas tras la administración de idarucizumab y se normaliza entre las 12 y 24 horas. En casos individuales, la proteinuria transitoria persistió durante más de 24 horas.
Pacientes con insuficiencia renal: Se ha investigado PRAXBIND® en estudios de fase I en individuos con un aclaramiento de creatinina comprendido entre 44 y 213 ml/min. Los individuos con un aclaramiento de creatinina inferior a 44 ml/min no se han estudiado en la fase I. Dependiendo del grado de insuficiencia renal, el aclaramiento total se redujo en comparación con los individuos sanos, dando lugar a un aumento de la exposición de idarucizumab.
De acuerdo a los datos farmacocinéticos de 347 pacientes con diferentes grados de función renal (aclaramiento medio de creatinina entre 21 y 99 ml/min), se estima que la exposición media a idarucizumab (AUC0 – 24 h) se incrementa en un 38% en los pacientes con insuficiencia renal leve (AcCr 50-<80 ml/min), en un 90% en los pacientes con insuficiencia renal moderada (30-<50 ml/min) y en un 146% en los pacientes con insuficiencia renal grave (0-<30 ml/min). Puesto que dabigatrán también se excreta principalmente a través de los riñones, también se observan incrementos en la exposición a dabigatrán con el empeoramiento de la función renal.
De acuerdo a estos datos y al grado de reversión del efecto anticoagulante de dabigatrán en los pacientes, la insuficiencia renal no tiene impacto en el efecto de reversión de idarucizumab.
Pacientes con insuficiencia hepática: No se ha observado que la insuficiencia hepática, valorada por la lesión hepática determinada por la elevación en las pruebas funcionales hepáticas, afecte a la farmacocinética de idarucizumab.
Idarucizumab se ha estudiado en 58 pacientes con diversos grados de insuficiencia hepática. En comparación con 272 pacientes sin insuficiencia hepática, la mediana del AUC de idarucizumab varió en un -6%, 37% y 10% en pacientes con elevaciones de AST/ALT de 1 a <2 x LSN (N = 34), 2 a <3 x LSN (N = 3) y >3 x LSN (N = 21), respectivamente. De acuerdo con los datos farmacocinéticos de 12 pacientes con enfermedad hepática, el AUC de idarucizumab aumentó en un 10% en comparación con pacientes sin enfermedad hepática.
Personas de edad avanzada/Sexo/Raza: Según los análisis farmacocinéticos poblacionales, ni el sexo, ni la edad ni la raza tienen un efecto clínicamente relevante sobre la farmacocinética de idarucizumab.
CONTRAINDICACIONES:
Ninguna.
FERTILIDAD, EMBARAZO Y LACTANCIA:
Embarazo:
No hay datos relativos al uso de PRAXBIND® en mujeres embarazadas. Dada la naturaleza y el uso clínico previsto del medicamento, no se han realizado estudios de toxicidad para la reproducción ni el desarrollo. PRAXBIND® se puede utilizar durante el embarazo si los beneficios clínicos esperados superan los riesgos potenciales.
Lactancia: Se desconoce si idarucizumab se excreta en la leche materna.
Fertilidad: No hay datos relativos al efecto de PRAXBIND® sobre la fertilidad (ver Datos preclínicos sobre la seguridad ).
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS:
No procede.
REACCIONES ADVERSAS:
Se ha evaluado la seguridad de PRAXBIND® en un ensayo de fase III con 503 pacientes que habían presentado una hemorragia no controlada o necesitado una intervención quirúrgica o procedimiento de urgencia y que estaban en tratamiento con PRADAXA® (dabigatrán etexilato), así como en 224 voluntarios en ensayos de fase I.
No se han identificado reacciones adversas.
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas al Centro Nacional de Farmacovigilancia y Tecnovigilancia.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN:
No se han realizado estudios formales de interacciones con PRAXBIND® y otros medicamentos. De acuerdo a las propiedades farmacocinéticas y a la alta especificidad de la unión a dabigatrán, se considera improbable que se produzcan interacciones clínicamente relevantes con otros medicamentos.
Las investigaciones preclínicas con idarucizumab han demostrado que no se producen interacciones con:
• Expansores de volumen.
• Concentrados de factores de coagulación, tales como concentrados de complejo de protrombina (CCP, por ejemplo, factor 3 y factor 4), CCP activado (CCPa) y factor recombinante VIIa.
• Otros anticoagulantes (por ejemplo, inhibidores de la trombina distintos a dabigatrán, inhibidores del factor Xa incluida la heparina de bajo peso molecular, antagonistas de la vitamina K o heparina). Por tanto, idarucizumab no revertirá los efectos de otros anticoagulantes.
DATOS PRECLÍNICOS SOBRE SEGURIDAD:
Los datos preclínicos no muestran riesgos especiales para los seres humanos según los estudios de toxicidad a dosis repetidas de hasta cuatro semanas en ratas y dos semanas en monos. Los estudios de farmacología de seguridad no han demostrado efectos sobre los sistemas respiratorio, nervioso central o cardiovascular.
No se han realizado estudios para evaluar el potencial mutágeno y carcinógeno de idarucizumab. De acuerdo a su mecanismo de acción y a las características de las proteínas, no se esperan efectos carcinógenos ni genotóxicos.
No se han realizado estudios para evaluar los potenciales efectos de idarucizumab sobre la reproducción. No se han identificado efectos relacionados con el tratamiento en tejidos reproductores de cualquier sexo durante los estudios de toxicidad intravenosa a dosis repetidas de hasta cuatro semanas en ratas y dos semanas en monos. Además, no se observó ninguna unión de idarucizumab a tejidos reproductores humanos en un estudio de reactividad cruzada en tejidos. Por tanto, los resultados preclínicos no sugieren que exista riesgo para la fertilidad o el desarrollo embriofetal.
No se observaron irritaciones locales de los vasos sanguíneos después de la administración intravenosa o paravenosa de idarucizumab. La formulación de idarucizumab no produjo hemólisis de sangre completa humana in vitro.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
Idarucizumab se une de forma específica a dabigatrán y revierte su efecto anticoagulante. No revierte los efectos de otros anticoagulantes (ver Propiedades farmacodinámicas).
El tratamiento con PRAXBIND® se puede utilizar en combinación con otras medidas estándar de soporte, si se considera que estas son médicamente apropiadas.
Hipersensibilidad: El riesgo de utilizar PRAXBIND® en pacientes con hipersensibilidad conocida (por ejemplo, reacción anafilactoide) a idarucizumab o a alguno de los excipientes se debe valorar cuidadosamente frente al beneficio potencial de este tratamiento de urgencia. Si se produce una reacción anafiláctica u otra reacción alérgica grave, la administración de PRAXBIND® se debe suspender de inmediato e iniciar el tratamiento adecuado.
Intolerancia hereditaria a la fructosa: La dosis recomendada de PRAXBIND® contiene 4 g de sorbitol como excipiente. En pacientes con intolerancia hereditaria a la fructosa, la administración parenteral de sorbitol se ha asociado a casos de hipoglucemia, hipofosfatemia, acidosis metabólica, aumento del ácido úrico, insuficiencia hepática aguda con fracaso de la función excretora y sintética y muerte. Por tanto, en pacientes con intolerancia hereditaria a la fructosa, el riesgo de administrar tratamiento con PRAXBIND® se debe valorar frente al beneficio potencial de este tratamiento de urgencia. Si se administra PRAXBIND® en estos pacientes, se requiere intensificar la asistencia médica durante la exposición a PRAXBIND® y dentro de las 24 horas tras la exposición al mismo.
Acontecimientos tromboembólicos: Los pacientes que estén recibiendo tratamiento con dabigatrán presentan enfermedades subyacentes que les predisponen a los acontecimientos tromboembólicos. La reversión del tratamiento con dabigatrán expone a los pacientes al riesgo trombótico derivado de su enfermedad o afección subyacentes. Para reducir este riesgo, se debe considerar la reanudación del tratamiento anticoagulante tan pronto como sea médicamente apropiado (ver Posología y forma de administración).
Análisis de proteínas en orina: PRAXBIND® provoca proteinuria transitoria como reacción fisiológica a la llegada de una concentración elevada de proteínas a nivel renal tras la aplicación rápida (bolo) o a corto plazo de 5 g de idarucizumab por vía intravenosa (ver Propiedades farmacocinéticas). La proteinuria transitoria no es indicativa de daño renal, lo que se debe tener en cuenta en los análisis de orina.
Contenido en sodio: Este medicamento contiene 2,2 mmol (o 50 mg) de sodio por dosis, lo que deberá tenerse en cuenta en el tratamiento de pacientes con dietas pobres en sodio.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
Limitado únicamente a uso hospitalario.
Posología: La dosis recomendada de PRAXBIND® es de 5 g (2 x 2,5 g/50 ml).
En un subconjunto de pacientes, se ha producido la recurrencia de concentraciones plasmáticas de dabigatrán libre y la prolongación concomitante de las pruebas de coagulación hasta 24 horas después de la administración de idarucizumab (ver Propiedades farmacodinámicas ).
Se puede considerar la administración de una segunda dosis de 5 g de PRAXBIND® en las siguientes situaciones:
• Recurrencia de sangrado clínicamente relevante junto con tiempos de coagulación prolongados, o
• Si un posible nuevo sangrado fuese potencialmente mortal y se observa la prolongación de los tiempos de coagulación, o
• Los pacientes necesitan una segunda intervención quirúrgica de urgencia o procedimiento urgente y presentan prolongación de los tiempos de coagulación.
Los principales parámetros de la coagulación son el tiempo de tromboplastina parcial activado (TTPa), el tiempo de trombina diluido (TTd) o el tiempo de coagulación de ecarina (TCE) (ver Propiedades farmacodinámicas).
No se ha investigado una dosis máxima diaria.
Reinicio del tratamiento antitrombótico: El tratamiento con PRADAXA® (dabigatrán etexilato) puede reiniciarse 24 horas después de la administración de PRAXBIND®, siempre y cuando el paciente se encuentre clínicamente estable y se haya alcanzado una hemostasia adecuada.
Tras la administración de PRAXBIND®, es posible iniciar otro tratamiento antitrombótico (por ejemplo, con heparina de bajo peso molecular) en cualquier momento, siempre y cuando el paciente se encuentre clínicamente estable y se haya alcanzado una hemostasia adecuada.
La ausencia de tratamiento antitrombótico expone a los pacientes al riesgo trombótico derivado de su enfermedad o afección subyacentes.
Insuficiencia renal:
No se requiere ajuste de dosis en pacientes con insuficiencia renal. La insuficiencia renal no influyó en el efecto de reversión de idarucizumab (ver Propiedades farmacocinéticas).
Insuficiencia hepática: No se requiere ajuste de dosis en pacientes con insuficiencia hepática (ver Propiedades farmacocinéticas).
Pacientes de edad avanzada: No se requiere ajuste de dosis en pacientes de edad avanzada de 65 años de edad o mayores (ver Propiedades farmacocinéticas).
Población pediátrica: No se ha establecido todavía la seguridad y eficacia de PRAXBIND® en niños menores de 18 años. No se dispone de datos.
Forma de administración: Vía intravenosa.
PRAXBIND® (2 x 2,5 g/50 ml) se administra por vía intravenosa en dos perfusiones consecutivas de entre 5 y 10 minutos cada una o en una inyección rápida (bolo).
Para consultar instrucciones adicionales sobre el uso y la manipulación, ver Lista de excipientes.
SOBREDOSIS:
No existe experiencia clínica con sobredosis de PRAXBIND®.
La dosis única más alta de PRAXBIND® estudiada en individuos sanos fue de 8 g. No se han identificado señales de seguridad en este grupo.
INFORMACIÓN CONTENIDA EN EL INSERTO PARA EL PACIENTE:
PRAXBIND® 2,5 g/50 mL solución inyectable
Idarucizumab
(Uso hospitalario)
Este medicamento está sujeto a seguimiento adicional, lo que agilizará la detección de nueva información sobre su seguridad. Puede contribuir comunicando los efectos adversos que pudiera usted tener. La parte final de la sección Posibles efectos adversos incluye información sobre cómo comunicar estos efectos adversos.
Lea todo el Inserto detenidamente, porque contiene información importante para usted. Tenga en cuenta que este medicamento se utiliza principalmente en situaciones de urgencia en las que su médico ha decidido que lo necesita.
- Conserve este inserto, ya que puede tener que volver a leerlo.
- Si tiene alguna duda, consulte a su médico o enfermero.
- Si experimenta efectos adversos, consulte a su médico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este Inserto. Ver sección Posibles efectos adversos.
Contenido del inserto:
1. Qué es PRAXBIND® y para qué se utiliza
2. Qué necesita saber antes de empezar a recibir PRAXBIND®
3. Cómo usar PRAXBIND®
4. Posibles efectos adversos
5. Conservación de PRAXBIND®
6. Contenido del envase e información adicional
Qué es PRAXBIND® y para qué se utiliza
Qué es PRAXBIND®: PRAXBIND® es un agente de reversión específico para dabigatrán (PRADAXA®), un medicamento que diluye la sangre y bloquea una sustancia del cuerpo que interviene en la formación de coágulos de sangre. PRAXBIND® se utiliza para capturar rápidamente dabigatrán con el fin de anular su efecto.
PRAXBIND® contiene el principio activo idarucizumab.
Para qué se utiliza PRAXBIND®:
PRAXBIND® se utiliza en adultos en situaciones de urgencia cuando su médico decide que es necesario anular rápidamente el efecto de PRADAXA®.
- Para intervenciones quirúrgicas de urgencia o procedimientos urgentes.
- En el caso de hemorragias potencialmente mortales o no controladas.
Qué necesita saber antes de empezar a recibir PRAXBIND®:
Advertencias y precauciones: Informe a su médico o enfermero
- Si es alérgico a idarucizumab o a alguno de los demás componentes incluidos en la sección Contenido del envase e información adicional.
- Si tiene una enfermedad genética llamada intolerancia hereditaria a la fructosa. En este caso, la sustancia sorbitol contenida en este medicamento puede provocar reacciones adversas graves.
Estos profesionales tendrán en cuenta estos factores antes de tratarle con PRAXBIND®.
Este medicamento solo elimina dabigatrán de su cuerpo. No elimina otros medicamentos utilizados para evitar la formación de coágulos de la sangre.
Después de que dabigatrán se haya eliminado de su cuerpo, no está protegido frente a la formación de coágulos de sangre. Su médico seguirá tratándole con medicamentos utilizados para evitar la formación de coágulos de la sangre tan pronto como su situación médica lo permita.
Niños y adolescentes: No existe información sobre el uso de PRAXBIND® en niños.
Otros medicamentos y PRAXBIND®: Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o pudiera tener que tomar cualquier otro medicamento.
Este medicamento se ha desarrollado para unirse solo a dabigatrán. Es improbable que PRAXBIND® interfiera con el efecto de otros medicamentos, o que otros medicamentos interfieran con PRAXBIND®.
Embarazo y lactancia: Si está embarazada o en periodo de lactancia, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico.
No existe información sobre los efectos de este medicamento en mujeres embarazadas o en periodo de lactancia. PRAXBIND® no afecta a ninguna función en el organismo como tal, por lo que puede que su médico decida administrarle este medicamento, si los beneficios esperados superan los riesgos potenciales.
PRAXBIND® contiene sodio: Este medicamento contiene 50 mg de sodio por dosis, lo que debe ser tenido en cuenta en los pacientes con dietas pobres en sodio.
Cómo usar PRAXBIND®:
Este medicamento es únicamente para uso hospitalario. La dosis recomendada es de 5 g (2 viales de 50 ml).
En casos raros, puede seguir teniendo demasiado dabigatrán en su sangre tras una primera dosis de PRAXBIND® y su médico puede decidir administrarle una segunda dosis de 5 g en situaciones específicas.
Su médico o enfermero le administrarán este medicamento mediante inyección o perfusión en una vena.
Después de recibir PRAXBIND®, su médico decidirá si debe continuar con el tratamiento para evitar la formación de coágulos en la sangre. PRADAXA® puede administrarse de nuevo a las 24 horas después de la administración de PRAXBIND®.
Al final de este Inserto se incluyen instrucciones detalladas para el médico o el enfermero sobre cómo administrar PRAXBIND® (ver Instrucciones de manipulación).
Si tiene cualquier otra duda sobre el uso de este medicamento, pregunte a su médico.
Posibles efectos adversos: Al igual que todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Hasta ahora, no se han identificado efectos adversos.
Comunicación de efectos adversos: Si experimenta cualquier tipo de efecto adverso, consulte a su médico o enfermero, incluso si se trata de posibles efectos adversos que no aparecen en este Inserto. También puede comunicarlos directamente a través del Centro Nacional de Farmacovigilancia y Tecnovigilancia. Mediante la comunicación de efectos adversos usted puede contribuir a proporcionar más información sobre la seguridad de este medicamento.
Conservación de PRAXBIND®:
Mantener este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en el vial y en la caja después de “FV”. La fecha de vencimiento es el último día del mes que se indica.
Almacenar en refrigerador (2 °C a 8 °C) .No congelar. Conservar en el embalaje original para protegerlo de la luz. Una vez abierto, PRAXBIND® es para uso inmediato.
“No consumir el producto una vez alcanzada la fecha de vencimiento indicada en los rotulados“.
Contenido del envase e información adicional:
Composición de PRAXBIND®:
Cada VIAL de 50 mL contiene 2,5 g de Idarucizumab
Cada mL de SOLUCIÓN INYECTABLE y para perfusión contiene 50 mg de idarucizumab.
Idarucizumab se produce mediante la tecnología de ADN recombinante en células ováricas de hámster chino.
- El principio activo es idarucizumab.
- Los demás componentes son acetato de sodio trihidrato, ácido acético, sorbitol, polisorbato 20 y agua para preparaciones inyectables.
Aspecto del producto y contenido del envase:
PRAXBIND® solución inyectable y para perfusión es una solución de transparente a ligeramente opalescente y de incolora a ligeramente amarilla que se suministra en un vial de vidrio cerrado con un tapón de goma butílica con tapa de Flip –off.
Cada envase contiene dos viales.
Precauciones especiales de eliminación y otras manipulaciones: Antes de su administración, los medicamentos parenterales como PRAXBIND® se deben someter a una inspección visual para ver si presentan partículas o decoloración.
La eliminación del producto biotecnológico no utilizado o los restos derivados del mismo se realizarán acorde a normativa vigente.
Esta información está destinada únicamente a profesionales del sector sanitario
Instrucciones de manipulación:
PRAXBIND® no se debe mezclar con otros medicamentos. Para la administración de PRAXBIND® se puede utilizar una vía intravenosa preexistente. Dicha vía debe aclararse con una solución inyectable de 9 mg/ml de cloruro sódico (0,9%) antes y al final de la perfusión. No se debe administrar ninguna otra perfusión en paralelo a través del mismo acceso intravenoso.
PRAXBIND® es para un solo uso y no contiene conservantes.
Antes de su uso, el vial puede mantenerse a temperatura ambiente (hasta 30°C) durante hasta 48 horas, si se conserva en el embalaje original para protegerlo de la luz. Tras la apertura del vial, se ha demostrado la estabilidad química y física en uso de idarucizumab durante 6 horas a temperatura ambiente. La solución no debe exponerse a la luz durante más de 6 horas (en el vial sin abrir y/o en uso).
Desde un punto de vista microbiológico, a menos que el método de apertura excluya el riesgo de contaminación microbiana, el medicamento se debe utilizar inmediatamente después de abrirlo. Si no se utiliza de inmediato, los tiempos de almacenamiento en uso y las condiciones previas al uso son responsabilidad del usuario.
No se han observado incompatibilidades entre PRAXBIND® y equipos de perfusión de cloruro de
polivinilo, polietileno o poliuretano, ni tampoco con jeringas de polipropileno.
Manténgase fuera del alcance de los niños
Venta con receta médica
Industria Alemana.
Importado por:
BOEHRINGER INGELHEIM PERÚ S.A.C
RUC: 20523163320 Tel: (01) 412-5000
Dir. Técnico Q.F. Jesús Peña


