

OPDIVO
NIVOLUMAB
Solución inyectable
1 Caja, 1 Vial(es), 4 ml, 40 Miligramos
1 Caja, 1 Vial(es), 10 ml, 100 Miligramos
FÓRMULA CUALICUANTITATIVA:
OPDIVO™ (Nivolumab) se suministra en frascos ampolla/viales de un solo uso de 40 mg/4 mL y 100 mg/10 mL.
Cada frasco ampolla/vial de 10 mL contiene: 100 mg de nivolumab y excipientes c.s.
Cada frasco ampolla/vial de 4 mL contiene: 40 mg de nivolumab y excipientes c.s.
Cada mililitro de solución de OPDIVO™ contiene nivolumab 10 mg, manitol (30 mg), ácido pentético (0,008 mg), polisorbato 80 (0,2 mg), cloruro de sodio (2,92 mg), citrato de sodio dihidrato (5,88 mg) y agua para uso inyectable, USP. Puede contener ácido clorhídrico y/o hidróxido de sodio para ajustar el pH a 6.
¿CUÁLES SON LOS INGREDIENTES DE OPDIVO™?
Ingrediente activo: Nivolumab
Ingredientes inactivos: Manitol, ácido pentético, polisorbato 80, cloruro de sodio, citrato de sodio dihidrato y agua para uso inyectable. Puede contener ácido clorhídrico y/o hidróxido de sodio.
Consulte a su médico.
OPDIVO™ es una marca registrada de Bristol-Myers Squibb Company.
Fabricado por:
Bristol-Myers Squibb Holdings Pharma, Ltd. Liability Company
Manatí, Puerto Rico, EEUU
Bajo licencia de: Bristol-Myers Squibb Company - EEUU
BRISTOL-MYERS SQUIBB
¿QUÉ ES OPDIVO™?
OPDIVO™ es un medicamento de venta bajo receta usado para tratar:
• Un tipo de cáncer de piel llamado melanoma que se ha extendido o no puede eliminarse por cirugía (melanoma avanzado). Usted puede recibir OPDIVO™ solo o en combinación con ipilimumab.
• Un tipo de cáncer de pulmón en estadio avanzado (llamado cáncer de pulmón de células no pequeñas) OPDIVO™ puede ser usado cuando su cáncer de pulmón:
– Se ha extendido o ha crecido, y
– Usted ha utilizado quimioterapia que contiene platino, y esta no funcionó o ya no está funcionando.
Si su tumor tiene un gen EGFR o ALK anormal, usted también debe haber intentado una terapia aprobada para tumores con estos genes anormales, y esta no funcionó o ya no está funcionando.
• Cáncer de riñón (carcinoma de células renales):
– OPDIVO™ se puede usar cuando su cáncer se ha extendido o ha crecido luego del tratamiento con otras medicaciones contra el cáncer.
• Un tipo de cáncer sanguíneo que afecta los glóbulos blancos, conocidos como linfocitos (denominado linfoma de Hodgkin clásico) OPDIVO™ puede ser usado si:
– Su cáncer se ha vuelto a manifestar o se ha extendido luego de un tipo de trasplante de células madre que utiliza sus propias células madre (autólogo), y o usted usó el medicamento brentuximab vedotina luego de su trasplante de células madre.
• Cáncer de cabeza y cuello OPDIVO™ se puede utilizar cuando su cáncer de cabeza y cuello:
– Ha vuelto a aparecer o se ha diseminado, y
– Usted ha probado una quimioterapia que contiene platino y no funcionó o ya no está funcionando.
• Cáncer de vejiga (carcinoma urotelial): OPDIVO™ se puede utilizar cuando su cáncer de vejiga:
– Se ha diseminado o ha crecido, y
– Usted ha probado una quimioterapia que contiene platino y no funcionó o ya no está funcionando.
Se desconoce si OPDIVO™ es seguro y efectivo en niños de menos de 18 años de edad.
¿QUÉ DEBO DECIRLE A MI MÉDICO ANTES DE RECIBIR OPDIVO™?
Antes de recibir OPDIVO™, informe a su médico si usted:
• Tiene problemas del sistema inmunológico, tales como enfermedad de Crohn, colitis ulcerosa o lupus
• Ha tenido un trasplante de órgano
• Tiene problemas pulmonares o respiratorios
• Tiene problemas hepáticos
• Tiene alguna otra afección médica
• Está embarazada o planea quedar embarazada. OPDIVO™ puede dañar a su bebé por nacer.
– Las mujeres que puedan quedar embarazadas deben usar un método efectivo de control natal durante el tratamiento y durante al menos 5 meses luego de la última dosis de OPDIVO™. Consulte a su médico sobre los métodos anticonceptivos que puede usar durante este período.
– Informe a su médico inmediatamente si queda embarazada durante el tratamiento con OPDIVO™.
• Está amamantando o planea amamantar. Se desconoce si OPDIVO™ pasa a la leche materna. No amamante durante su tratamiento con OPDIVO™.
Informe a su médico sobre todos los medicamentos que toma, incluidos los medicamentos de venta bajo receta y de venta libre, vitaminas, y suplementos a base de hierbas.
Conozca los medicamentos que toma. Lleve una lista de ellos para mostrarle a su médico y farmacéutico cuando obtenga un nuevo fármaco.
¿CÓMO RECIBIRÉ OPDIVO™?:
• Su médico le administrará OPDIVO™ a través de una vía intravenosa (IV) durante 60 minutos.
• OPDIVO™ se administra generalmente cada 2 semanas.
• Cuando se usa en combinación con ipilimumab, OPDIVO™ generalmente se administra cada 3 semanas, por un total de 4 dosis. Ipilimumab se administrará el mismo día. Luego de esto, OPDIVO™ se administrará como agente único cada 2 semanas.
• Su médico decidirá el tratamiento adecuado para usted.
• Su médico le realizará análisis de sangre para detectar efectos secundarios.
• Si usted falta a alguna cita, llame a su médico lo antes posible para reprogramarla.
PRESENTACIÓN/ALMACENAMIENTO Y MANIPULACIÓN:
OPDIVO™ (Nivolumab) se presenta de la siguiente forma:
Forma de presentación:
Caja con 1 Vial/Frasco ampolla de vidrio Tipo I con Solución inyectable por 4 mL (40 mg/4 mL).
Caja con 1 Vial/Frasco ampolla de vidrio Tipo I con Solución inyectable por 10 mL (100 mg/10 mL).
Conservar OPDIVO™ en condiciones de refrigeración a 2 °C-8 °C. Proteger OPDIVO™ de la luz conservándolo en su envase original hasta el momento de usarlo.
No congelar ni agitar.
¿CUÁLES SON LOS POSIBLES EFECTOS SECUNDARIOS DE OPDIVO™?
OPDIVO™ puede causar efectos adversos serios que incluyen:
• Véase “¿Cuál es la información más importante que debo saber sobre OPDIVO™?”
• Reacciones graves a la perfusión intravenosa. Infórmele inmediatamente a su medico o enfermera si usted tiene estos síntomas durante la administración de OPDIVO™:
– Escalofríos o temblores
– Picazón o erupción
– Enrojecimiento o dificultad para respirar
– Mareos o fiebre
– Sensación de desmayo
• Complicaciones de trasplante de células madres que utiliza un donante de células madres (trasplante alogénico) luego del tratamiento con OPDIVO™. Estas complicaciones pueden ser severas e incluso producir la muerte. Su médico deberá monitorear si usted presenta signos de estas complicaciones si usted ha tenido un trasplante alogénico de células madres.
Los efectos secundarios más comunes de OPDIVO™ cuando se utiliza como agente único incluyen:
• Sensación de cansancio
• Erupción
• Dolor en músculos, huesos y articulaciones
• Picazón de piel
• Diarrea
• Náuseas
• Tos
• Disnea (sensación de falta de aire)
• Constipación
• Disminución del apetito
• Dolor de espalda
• Infección de vías respiratorias superiores
• Fiebre
• Debilidad
Éstos no son todos los posibles efectos secundarios de OPDIVO™. Para más información, consulte con su médico o farmacéutico. Comuníquese con su médico para obtener asesoramiento sobre efectos secundarios.
Información general sobre el uso seguro y efectivo de OPDIVO™:
Los medicamentos a veces se recetan con fines distintos de los enumerados en un folleto de Información para el Paciente. Si usted desea recibir más información sobre OPDIVO™, consulte con su médico. Puede pedirle a su médico la información sobre OPDIVO™ que se destina a los profesionales médicos.
FORMA FARMACÉUTICA Y CONCENTRACIÓN:
Solución Inyectable de 10 mg/mL.
ACCIÓN TERAPÉUTICA:
Anticuerpo monoclonal humano que bloquea la interacción entre PD-1 y sus ligandos, PD-L1 y PD-L2. Inmunoglobulina IgG4 kappa. Código ATC: L01XC17.
Nota: Se han omitido secciones, subsecciones y/o tablas numeradas de la información completa sobre prescripción por no ser aplicables.
INDICACIONES Y USO:
Melanoma irresecable o metastásico:
• OPDIVO™ como agente único está indicado para el tratamiento de pacientes con melanoma irresecable o metastásico sin mutación BRAF V600 (wildtype) [véase Estudios clínicos (Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado)].
• OPDIVO™ como agente único está indicado para el tratamiento de pacientes con melanoma irresecable o metastásico, positivo para la mutación BRAF V600 [véase Estudios clínicos (Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado)].*
• OPDIVO™, en combinación con ipilimumab, está indicado para el tratamiento de pacientes con melanoma irrese-cable o metastásico [véase Estudios clínicos (Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado)].*
* Esta indicación ha sido aprobada bajo “Aprobación acelerada de la FDA”, en base a la sobrevida libre de progresión. La continuidad de la aprobación para esta indicación - puede ser condicionada a la verificación y descripción del beneficio clínico en los ensayos clínicos confirmatorios.
Cáncer de pulmón metastásico de células no pequeñas (NSCLC): OPDIVO™ está indicado para el tratamiento de pacientes con cáncer de pulmón metastásico de células no pequeñas (NSCLC, por sus siglas en inglés) que muestra progresión durante o después de la quimioterapia basada en platino. Previo a recibir OPDIVO™, los pacientes con mutaciones tumorales genómicas de EGFR o ALK deben haber presentado progresión de la enfermedad con una terapia aprobada para estas mutaciones [véase Estudios clínicos (Cáncer de pulmón metastásico de células no pequeñas (NSCLC))].
Carcinoma de células renales (RCC): OPDIVO™ está indicado para el tratamiento de pacientes con carcinoma avanzado de células renales (RCC, por sus siglas en inglés) que han recibido terapia anti-angiogénica previa [véase Estudios clínicos (Carcinoma de células renales)].
Linfoma de Hodgkin clásico (cHL); OPDIVO™ está indicado para el tratamiento de pacientes con linfoma de Hodgkin clásico (cHL, por sus siglas en inglés) que ha presentado recidiva o progresión luego del trasplante autólogo de células madre hematopoyéticas (HSCT, por sus siglas en inglés) y tratamiento con brentuximab vedotina post-trasplante [véase
Estudios clínicos (Linfoma de Hodgkin clásico)].**
** Esta indicación ha sido aprobada bajo “Aprobación acelerada de la FDA” en base a la tasa de respuesta global. La continuidad de la aprobación para esta indicación - puede depender de la verificación y descripción del beneficio clínico en los ensayos confirmatorios [véase Estudios clínicos (Linfoma de Hodgkin clásico)].
Carcinoma de células escamosas de cabeza y cuello (SCCHN): OPDIVO™ está indicado para el tratamiento de pacientes con carcinoma de células escamosas de cabeza y cuello (SCCHN, por sus siglas en inglés) recurrente o metastásico luego de una terapia basada en platino [véase Estudios clínicos (. Carcinoma metastásico o recurrente de células escamosas de cabeza y cuello (SCCHN))].
Carcinoma urotelial: OPDIVO™ está indicado para el tratamiento de pacientes con carcinoma urotelial localmente avanzado o metastásico que:
• Tienen progresión de la enfermedad durante o después de una quimioterapia que contiene platino,
• Tienen progresión de la enfermedad dentro de los 12 meses del tratamiento neoadyuvante o adyuvante con una quimioterapia que contiene platino.***
*** Esta indicación está aprobada bajo aprobación acelerada en función de la tasa de respuesta tumoral y la duración de la respuesta. La aprobación continuada para esta indicación podrá depender de la verificación y la descripción de un be-neficio clínico en ensayos confirmatorios. [véase Estudios clínicos (Linfoma de Hodgkin clásico)]
INFORMACIÓN PARA EL PACIENTE:
OPDIVO™
(Nivolumab)
Solución inyectable para perfusión intravenosa 10 mg/mL
Lea esta información para el paciente antes de iniciar su tratamiento con OPDIVO™ y antes de cada perfusión intravenosa, ya que puede haber nueva información. Si su médico le prescribe OPDIVO™ en combinación con ipilimumab, lea también el Información para el Paciente que se entrega con ipilimumab. Esta Información para el Paciente no reemplaza la conversación con su médico acerca de su condición médica o su tratamiento.
INFORMACIÓN FARMACÉUTICA:
Lista de excipientes:
• Citrato de sodio dihidrato
• Cloruro de sodio
• Manitol
• Ácido pentético
• Polisorbato 80
• Agua para inyección
• Para ajuste a pH 6: ácido clorhídrico y/o hidróxido de sodio c.s.
Incompatibilidades: Ninguna conocida
Vida útil: 24 meses.
No utilizar el producto luego de la fecha de expira impresa en el envase.
POSOLOGÍA/DOSIS Y ADMINISTRACIÓN:
Dosis recomendada para melanoma: La dosis recomendada de OPDIVO™ como agente único, es de 240 mg administrada en forma de perfusión intravenosa durante 60 minutos cada 2 semanas, hasta la aparición de progresión de la enfermedad o de una toxicidad inaceptable. La dosis recomendada de OPDIVO™ es de 1 mg/kg administrada en forma de perfusión intravenosa durante 60 minutos, seguida por ipilimumab el mismo día, cada 3 semanas por 4 dosis [véase Estudios clínicos (Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado)]. La dosis subsiguiente recomendada de OPDIVO™, como agente único, es de 240 mg administrada en forma de perfusión intravenosa durante 60 minutos cada 2 semanas, hasta la aparición de progresión de la enfermedad o de una toxicidad inaceptable. Leer la Información Completa sobre Prescripción para ipilimumab antes de iniciar tratamiento.
Dosis recomendada para NSCLC: La dosis recomendada de OPDIVO™ es de 240 mg administrada por perfusión intravenosa durante 60 minutos cada 2 semanas, hasta la aparición de progresión de la enfermedad o de una toxicidad inaceptable.
Dosis recomendada para RCC: La dosis recomendada de OPDIVO™ es de 240 mg administrada en forma de perfusión intravenosa durante 60 minutos cada 2 semanas, hasta la aparición de progresión de la enfermedad o de una toxicidad inaceptable.
Dosis recomendada para cHL: La dosis recomendada de OPDIVO™ es de 3 mg/kg administrada en forma de perfusión intravenosa durante 60 minutos cada 2 semanas, hasta la aparición de progresión de la enfermedad o de una toxicidad inaceptable.
Dosis recomendada para SCCHN: La dosis recomendada de OPDIVO™ es de 3 mg/kg administrada en forma de perfusión intravenosa durante 60 minutos cada 2 semanas, hasta la aparición de progresión de la enfermedad o de una toxicidad inaceptable.
Dosis recomendada para carcinoma urotelial: La dosis recomendada de OPDIVO™ es de 240 mg administrada en forma de infusión intravenosa durante 60 minutos cada 2 semanas, hasta la aparición de progresión de la enfermedad o de una toxicidad inaceptable.
Modificaciones de la dosis recomendada: Las recomendaciones para modificar la dosis de OPDIVO™ se proporcionan en la Tabla 1. Cuando OPDIVO™ se administra en combinación con ipilimumab, si se suspende OPDIVO™, también se debe suspender ipilimumab.
No hay modificaciones de dosis recomendadas para hipotiroidismo o hipertiroidismo.
Interrumpir o disminuir la velocidad de la perfusión intravenosa en pacientes con reacciones leves o moderadas a la administración. Discontinuar OPDIVO™ en pacientes con reacciones severas o potencialmente mortales a la perfusión intravenosa.
Tabla 1: Modificaciones de dosis recomendadas para OPDIVO™
|
Reacción adversa |
Severidad* |
Modificación de la dosis |
|
Colitis |
Diarrea o colitis de Grado 2 |
Suspender la dosisa |
|
Diarrea o colitis de Grado 3 |
Suspender la dosisa cuando se administra como agente único |
|
|
Discontinuar permanentemente cuando se administra con ipilimumab |
||
|
Diarrea o colitis de Grado 4 |
Discontinuar permanentemente |
|
|
Neumonitis |
Neumonitis de Grado 2 |
Suspender la dosisa |
|
Neumonitis de Grado 3 o 4 |
Discontinuar permanentemente |
|
|
Hepatitis |
Aspartato aminotransferasa (AST) o alanina aminotransferasa (ALT) más de 3 y hasta 5 veces el límite superior del rango normal, o bilirrubina total más de 1,5 y hasta 3 veces el límite superior del rango normal |
Suspender la dosisa |
|
AST o ALT más de 5 veces el límite superior del rango normal o bilirrubina total más de 3 veces el límite superior del rango normal |
Discontinuar permanentemente |
|
|
Hipofisitis |
Hipofisitis de Grado 2 o 3 |
Suspender la dosisa |
|
Hipofisitis de Grado 4 |
Discontinuar permanentemente |
|
|
Insuficiencia adrenal |
Insuficiencia adrenal de Grado 2 |
Suspender la dosisa |
|
Insuficiencia adrenal de Grado 3 o 4 |
Discontinuar permanentemente |
|
|
Diabetes mellitus tipo 1 |
Hiperglucemia de Grado 3 |
Suspender la dosisa |
|
Hiperglucemia de Grado 4 |
Discontinuar permanentemente |
|
|
Nefritis y disfunción renal |
Creatinina sérica más de 1,5 y hasta 6 veces el límite superior del rango normal |
Suspender la dosisa |
|
Creatinina sérica más de 6 veces el límite superior del rango normal |
Discontinuar permanentemente |
|
|
Dérmicos |
Erupción de Grado 3 o sospecha de síndrome de Stevens-Johnson (SJS) o necrólisis epidérmica tóxica (TEN) |
Suspender la dosisa |
|
Erupción de Grado 4 o confirmación de SJS o TEN |
Discontinuar permanentemente |
|
|
Encefalitis |
Signos o síntomas neurológicos moderados o severos de reciente aparición |
Suspender la dosisa |
|
Encefalitis mediada por la respuesta inmune |
Discontinuar permanentemente |
|
|
Otras |
Otras reacciones adversas de Grado 3 Primera ocurrencia Recurrencia de las mismas reacciones adversas de Grado 3 |
Suspender la dosisa Discontinuar permanentemente |
|
Reacción adversa potencialmente mortal o de Grado 4 |
Discontinuar permanentemente |
|
|
Requisito de 10 mg por día o más de prednisona o equivalente durante más de 12 semanas |
Discontinuar permanentemente |
|
|
Reacciones adversas persistentes de Grado 2 o 3 que duran 12 semanas o más |
Discontinuar permanentemente |
|
|
* La toxicidad se calificó según los Criterios de Terminología Común para Eventos Adversos del Instituto Nacional del Cáncer, versión 4.0 (NCI CTCAE v4). a Reiniciar el tratamiento cuando la reacción adversa vuelva a Grado 0 o 1. |
||
Preparación y administración: Inspeccionar visualmente la solución del producto farmacológico en busca de partículas y decoloración antes de su administración. OPDIVO™ es una solución de transparente a opalescente, entre incolora y de color amarillo pálido. Descartar el vial si la solución se presenta turbia, decolorada, o contiene material particulado extraño distinto de algunas partículas proteináceas translúcidas a blancas. No agitar el vial.
Preparación:
• Retirar el volumen requerido de OPDIVO™ y transferirlo a una bolsa para perfusión intravenosa.
• Diluir OPDIVO™ con cloruro de sodio al 0,9% para uso inyectable USP, o con dextrosa al 5% para uso inyectable USP, para preparar una solución con una concentración final de 1 mg/mL a 10 mg/mL.
• Mezclar la solución diluida invirtiendo el envase suavemente. No agitar.
• Descartar los viales parcialmente usados o los viales vacíos de OPDIVO.
Almacenamiento de la solución diluida: El producto no contiene conservantes.
Luego de su preparación, conservar la solución diluida de OPDIVO en alguna de las siguientes condiciones:
• A temperatura ambiente durante no más de 4 horas desde el momento de la preparación. Esto incluye el almace-namiento a temperatura ambiente de la solución diluida en el recipiente IV y el tiempo para la administración por perfusión intravenosa, o
• En condiciones de refrigeración a 2°C - 8°C durante no más de 24 horas desde el momento de la preparación.
No congelar.
Administración: Administrar la solución diluida durante 60 minutos a través de una vía intravenosa que contenga un filtro en línea estéril, no pirogénica, de baja unión a proteínas (tamaño de poro de 0,2 micrómetros a 1,2 micrómetros). No coadministrar otros fármacos a través de la misma vía intravenosa. Enjuagar la vía intravenosa al finalizar la administración. Cuando se administra en combinación con ipilimumab, infundir OPDIVO™ primero, seguido por ipilimumab el mismo día. Usar bolsas y filtros separados para cada producto.
CONTRAINDICACIONES:
OPDIVO™™ está contraindicado en pacientes con hipersensibilidad previamente demostrada a nivolumab o a cualquier componente del producto.
REACCIONES ADVERSAS:
Las siguientes reacciones adversas se analizan en mayor detalle en otras secciones del prospecto.
• Neumonitis mediada por la respuesta inmune [véase Advertencias y Precauciones (Neumonitis mediada por la respuesta inmune)]
• Colitis mediada por la respuesta inmune [véase Advertencias y Precauciones (Colitis mediada por la respuesta inmune)]
• Hepatitis mediada por la respuesta inmune [véase Advertencias y Precauciones (Hepatitis mediada por la respuesta inmune)]
• Endocrinopatías mediadas por la respuesta inmune [véase Advertencias y Precauciones (Endocrinopatías mediadas por la respuesta inmune)]
• Nefritis y disfunción renal mediadas por la respuesta inmune [véase Advertencias y Precauciones (Nefritis y disfunción renal mediadas por la respuesta inmune)]
• Reacciones adversas dérmicas mediadas por la respuesta inmune [véase Advertencias y Precauciones (Reacciones adversas dérmicas mediadas por la respuesta inmune)]
• Encefalitis mediada por la respuesta inmune [véase Advertencias y Precauciones (Encefalitis mediada por la respuesta inmune)]
• Otras reacciones adversas mediadas por la respuesta inmune [véase Advertencias y Precauciones (Otras reacciones adversas mediadas por la respuesta inmune)]
• Reacciones a la perfusión intravenosa [véase Advertencias y Precauciones (Reacciones a la perfusión intravenosa)]
• Complicaciones del HSCT alogénico tras OPDIVO™ [véase Advertencias y Precauciones (Complicaciones del HSCT alogénico tras OPDIVO)]
Experiencia en estudios clínicos: Debido a que los ensayos clínicos se llevan a cabo en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas de los ensayos clínicos de otro fármaco, y pueden no reflejar las tasas observadas en la práctica.
Los datos en la sección de Advertencias y Precauciones reflejan la exposición a OPDIVO™, como agente único, para reacciones adversas clínicamente significativas en 1994 pacientes enrolados en los Ensayos 1 a 8, o en un ensayo de rama única en NSCLC (n=117) en donde se administró OPDIVO™ como agente único [véase Advertencias y Precauciones (Neumonitis mediada por la respuesta inmune, Otras reacciones adversas mediadas por la respuesta inmune)]. Además, las reacciones adversas clínicamente significativas de OPDIVO™ administrado con ipilimumab fueron evaluadas en 407 pacientes con melanoma enrolados en el Ensayo 6 (n=313) o en un estudio randomizado de Fase 2 (n=94), en donde se administró OPDIVO™ con ipilimumab, complementadas por reportes de reacciones adversas media-das por la respuesta inmune en ensayos clínicos en curso [véase Advertencias y Precauciones (Neumonitis mediada por la respuesta inmune, Otras reacciones adversas mediadas por la respuesta inmune]. Los datos descritos a continuación reflejan la exposición a OPDIVO™ como agente único en los Ensayos 1, 4 y 6, y a OPDIVO™ con ipilimumab en el Ensayo 6, los cuales son ensayos randomizados, con control activo, realizados en pacientes con melanoma irresecable o metastásico. También se describen a continuación los datos de OPDIVO™ como agente único de los Ensayos 2 y 3, que son ensayos randomizados realizados en pacientes con NSCLC metastásico, y del Ensayo 5, que es un ensayo randomizado en pacientes con RCC avanzado, de los Ensayos 7 y 8, que son ensayos abiertos, de múltiples cohortes, en pacientes con cHL, del Ensayo 9, un ensayo randomizado en pacientes con SCCHN recurrente o metastásico, y el Ensayo 10, que es un ensayo de rama única en pacientes con carcinoma urotelial.
Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado. La seguridad de OPDIVO™ como agente único fue evaluada en el Ensayo 1, un estudio randomizado, abierto, en el cual 370 pacientes con melanoma irresecable o metastásico recibieron OPDIVO™ 3 mg/kg cada 2 semanas (n=268) o quimioterapia a elección del investigador (n=102), ya sea dacarbazina 1000 mg/m2 cada 3 semanas o la combinación de carboplatino AUC 6 cada 3 semanas más paclitaxel 175 mg/m2 cada 3 semanas [véase Estudios clínicos (Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado)]. La mediana de la duración de la exposición fue de 5,3 meses (rango: 1 día a 13,8+ meses) en pacientes tratados con OPDIVO™ y fue de 2 meses (rango: 1 día a 9,6+ meses) en pacientes tratados con quimioterapia. En este ensayo en curso, el 24% de los pacientes recibieron OPDIVO™ durante más de 6 meses, y el 3% de los pacientes recibieron OPDIVO™ durante más de 1 año. En el Ensayo 1, los pacientes tenían progresión documentada de la enfermedad luego del tratamiento con ipilimumab y, si eran positivos para la mutación BRAF V600, un inhibidor de BRAF. El ensayo excluyó pacientes con enfermedad autoinmune, reacciones adversas previas de Grado 4 relacionadas con ipilimumab (excepto por endocrinopatías) o reacciones adversas de Grado 3 relacionadas con ipilimumab que no se habían resuelto o que estaban inadecuadamente controladas dentro de las 12 semanas de iniciado el evento, pacientes con una afección que requería tratamiento sistémico crónico con corticosteroides (>10 mg diarios de equivalente de prednisona) u otras medicaciones inmunosupresoras, resultado positivo en la prueba de he-patitis B o C, y antecedentes de VIH. Las características de la población del ensayo en el grupo de OPDIVO™ y el grupo de quimioterapia eran similares: 66% de hombres, mediana de edad 59,5 años, 98% de raza blanca, estado funcional según el Eastern Cooperative Oncology Group (ECOG) en condición basal 0 (59%) o 1 (41%), 74% con enfermedad en estadio M1c, 73% con melanoma cutáneo, 11% con melanoma mucosal, 73% recibió dos terapias previas o más para la enferme-dad avanzada o metastásica, y 18% tenía metástasis cerebral. Había más pacientes en el grupo de OPDIVO™ con nivel elevado de LDH en condición basal (51% vs. 38%). OPDIVO™ fue discontinuado por reacciones adversas en el 9% de los pacientes. El 26% de los pacientes que recibieron OPDIVO™ tuvieron una demora del fármaco debido a una reacción adversa. Se produjeron reacciones adversas serias en el 41% de los pacientes que recibieron OPDIVO™. Se registraron reacciones adversas de Grado 3 y 4 en el 42% de los pacientes que recibieron OPDIVO™. Las reacciones adversas de Grado 3 y 4 más frecuentes reportadas en 2% a menos del 5% de los pacientes que recibieron OPDIVO™ fueron dolor abdominal, hiponatremia, aumento de aspartato aminotransferasa y aumento de lipasa.
La Tabla 2 resume las reacciones adversas que ocurrieron en al menos el 10% de los pacientes tratados con OPDIVO™ en el Ensayo 1. La reacción adversa más común (reportada en al menos el 20% de los pacientes) fue erupción.
Tabla 2: Reacciones adversas que se ocurrieron en ≥10% de los pacientes tratados con OPDIVO™ y con una mayor in-cidencia que en la rama de quimioterapia (diferencia entre ramas ≥5% [todos los grados] o ≥2% [Grados 3-4]) (Ensayo 1)
|
Reacción adversa |
OPDIVO (n=268) |
Quimioterapia (n=102) |
||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Porcentaje (%) de pacientes |
||||
|
Trastornos de la piel y el tejido subcutáneo |
||||
|
Erupcióna |
21 |
0,4 |
7 |
0 |
|
Prurito |
19 |
0 |
3,9 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos |
17 |
0 |
6 |
0 |
|
Infecciones |
||||
|
Infección del tracto respiratorio superiorb |
11 |
0 |
2,0 |
0 |
|
Trastornos generales y afecciones en el sitio de administración |
||||
|
Edema periférico |
10 |
0 |
5 |
0 |
La toxicidad se calificó según NCI CTCAE v4.
a Erupción es un término compuesto que incluye erupción máculopapular, erupción eritematosa, erupción prurítica, erupción folicular, erupción macular, erupción papular, erupción pustular, erupción vesicular y dermatitis acneiforme.
b Infección del tracto respiratorio superior es un término compuesto que incluye rinitis, faringitis y nasofaringitis.
Otras reacciones adversas clínicamente importantes ocurridas en menos del 10% de los pacientes tratados con OPDIVO en el Ensayo 1 fueron las siguientes:
Trastornos cardíacos: arritmia ventricular
Trastornos oculares: iridociclitis
Trastornos generales y afecciones en el sitio de administración: reacciones relacionadas con la perfusión intravenosa
Investigaciones: aumento de amilasa, aumento de lipasa
Trastornos del sistema nervioso: mareos, neuropatía periférica y sensorial
Trastornos de la piel y el tejido subcutáneo: dermatitis exfoliativa, eritema multiforme, vitiligo, psoriasis
Tabla 3: Anormalidades de laboratorio de empeoramiento respecto del nivel basal que ocurrieron en ≥10% de los pacientes tratados con OPDIVO™ y con una mayor incidencia que en la rama de quimio-terapia (diferencia entre ramas ≥5% [todos los grados] o ≥2% [Grados 3-4]) (Ensayo 1)
|
Anormalidad de laboratorio |
Porcentaje de pacientes con un empeoramiento en los análisis de laboratorio desde la condición basala |
|||
|
OPDIVO™ |
Quimioterapia |
|||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Aumento de AST |
28 |
2,4 |
12 |
1,0 |
|
Aumento de fosfatasa alcalina |
22 |
2,4 |
13 |
1,1 |
|
Hiponatremia |
25 |
5 |
18 |
1,1 |
|
Aumento de ALT |
16 |
1,6 |
5 |
0 |
|
Hiperpotasemia |
15 |
2,0 |
6 |
0 |
|
a La incidencia de cada análisis se basa en el número de pacientes que tenían disponible una medición de laboratorio basal y al menos una medi-ción durante el estudio: grupo de OPDIVO™ (rango: 252 a 256 pacientes) y grupo de quimioterapia (rango: 94 a 96 pacientes). |
||||
Melanoma metastásico no tratado previamente: Ensayo 4. La seguridad de OPDIVO™ también fue evaluada en el Ensayo 4, un ensayo randomizado, doble ciego, con control activo, en el cual 411 pacientes con melanoma irresecable o metas-tásico sin mutación BRAF V600 (wild-type), no tratados previamente, recibieron OPDIVO™ 3 mg/kg cada 2 semanas (n=206) o dacarbazina 1000 mg/m2 cada 3 semanas (n=205) [véase Estudios Clínicos (14.1)]. La mediana de la duración de la exposición fue de 6,5 meses (rango: 1 día a 16,6 meses) en pacientes tratados con OPDIVO™. En este ensayo, el 47% de los pacientes recibieron OPDIVO™ durante más de 6 meses, y el 12% de los pacientes recibieron OPDIVO™ durante más de 1 año. El ensayo excluyó a pacientes con enfermedad autoinmune y a pacientes que requerían tratamiento sistémico crónico con corticosteroides (>10 mg diarios de equivalente de prednisona) u otras medicaciones inmunosupresoras. Las características de la población de ensayo en el grupo de OPDIVO™ y en el de dacarbazina fueron: 59% de pacientes de sexo masculino, mediana de edad de 65 años, 99,5% de raza blanca, 61% con enfermedad en estadio M1c, 74% con melanoma cutáneo, 11% con melanoma mucosal, 4% con metástasis cerebral, y 37% con nivel elevado de LDH en condición basal. Hubo más pacientes en el grupo de OPDIVO™ con un estado funcional ECOG 0 (71% versus 59%). Las reac-ciones adversas condujeron a la discontinuación permanente de OPDIVO™ en el 7% de los pacientes y a la interrupción de la dosis en el 26% de los pacientes; ningún tipo único de reacción adversa representó la mayoría de las discontinuaciones de OPDIVO™. Se produjeron reacciones adversas serias en el 36% de los pacientes que recibieron OPDIVO™. Se produjeron reacciones adversas de Grado 3 y 4 en el 41% de los pacientes que recibieron OPDIVO™. Las reacciones adversas de Grado 3 y 4 más frecuentes reportadas en al menos 2% de los pacientes que recibieron OPDIVO™ fueron aumento de gammaglutamiltransferasa (3,9%) y diarrea (3,4%). La Tabla 4 sintetiza reacciones adversas seleccionadas que ocurrieron en al menos 10% de los pacientes tratados con OPDIVO™. Las reacciones adversas más comunes (reportadas en al menos 20% de los pacientes y con mayor incidencia que en la rama de dacarbazina) fueron fatiga, dolor musculoesquelético, erupción y prurito.
Tabla 4. Reacciones adversas que ocurrieron en ≥10% de los pacientes tratados con OPDIVO™ y con mayor incidencia que en la rama de dacarbazina (diferencia entre ramas ≥5% [todos los grados] o ≥2% [Grados 3-4]) (Ensayo 4)
|
Reacción adversa |
OPDIVO (n=206) |
Dacarbazina (n=205) |
||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Porcentaje (%) de pacientes |
||||
|
Trastornos generales y afecciones en el sitio de administración |
||||
|
Fatiga |
49 |
1,9 |
39 |
3,4 |
|
Edemaa |
12 |
1,5 |
4,9 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Dolor musculoesqueléticob |
32 |
2,9 |
25 |
2,4 |
|
Trastornos de la piel y el tejido subcutáneo |
||||
|
Erupciónc |
28 |
1,5 |
12 |
0 |
|
Prurito |
23 |
0,5 |
12 |
0 |
|
Eritema |
10 |
0 |
2,9 |
0 |
|
Vitíligo |
11 |
0 |
0,5 |
0 |
|
Infecciones |
||||
|
Infección del tracto respiratorio superiord |
17 |
0 |
6 |
0 |
|
La toxicidad se calificó según NCI CTCAE v4. a Incluye edema periorbital, edema de rostro, edema generalizado, edema gravitacional, edema localizado, edema periférico, edema pulmonar y linfedema. b Incluye dolor de espalda, dolor óseo, dolor de pecho musculoesquelético, malestar musculoesquelético, mialgia, dolor de cuello, dolor de extremidades, dolor mandibular y dolor espinal. c Incluye erupción máculopapular, erupción eritematosa, erupción prurítica, erupción folicular, erupción macular, erupción papular, erupción pustular, erupción vesicular, dermatitis, dermatitis alérgica, dermatitis exfoliativa, dermatitis acneiforme, erupción medicamentosa y reacción dérmica. d Incluye rinitis, rinitis viral, faringitis y nasofaringitis. |
||||
Otras reacciones adversas clínicamente importantes observadas en menos del 10% de los pacientes tratados con OPDIVO™ en el Ensayo 4 fueron las siguientes:
Trastornos del sistema nervioso: neuropatía periférica
Tabla 5. Anormalidades de laboratorio de empeoramiento respecto del nivel basal que ocurrieron en ≥10% de los pacientes tratados con OPDIVO™ y con mayor incidencia que en la rama de dacarbazina (diferencia entre ramas ≥5% [to-dos los grados] o ≥2% [Grados 3-4]) (Ensayo 4)
|
Anormalidad de laboratorio |
Porcentaje de pacientes con empeoramiento de los análisis de laboratorio desde la condición basala |
|||
|
OPDIVO™ |
Dacarbazina |
|||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Aumento de ALT |
25 |
3,0 |
19 |
0,5 |
|
Aumento de AST |
24 |
3,6 |
19 |
0,5 |
|
Aumento de fosfatasa alcalina |
21 |
2,6 |
14 |
1,6 |
|
Aumento de bilirrubina |
13 |
3,1 |
6 |
0 |
|
a La incidencia de cada análisis se basa en el número de pacientes que tenían disponible una medición de laboratorio basal y al menos una medi-ción de laboratorio durante el estudio: grupo de OPDIVO™ (rango: 194 a 197 pacientes) y grupo de dacarbazina (rango: 186 a 193 pacientes). |
||||
Ensayo 6. La seguridad de OPDIVO™, administrado con ipilimumab o como agente único, fue evaluada en el Ensayo 6 [véase Estudios clínicos (Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado)], un ensayo randomizado (1:1:1), a doble ciego, en el cual 937 pacientes con melanoma irresecable o metastásico no tratados previamente recibieron:
• OPDIVO™ 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas por 4 dosis, seguido por OPDIVO™ 3 mg/kg como agente único cada 2 semanas (rama de OPDIVO™ más ipilimumab; n=313),
• OPDIVO™ 3 mg/kg cada 2 semanas (rama de OPDIVO™; n=313), o
• Ipilimumab 3 mg/kg cada 3 semanas por hasta 4 dosis (rama de ipilimumab; n=311).
La mediana de la duración de la exposición a OPDIVO™ fue de 2,8 meses (rango: 1 día a 18,8 meses) para la rama de OPDIVO™ más ipilimumab, y de 6,6 meses (rango: 1 día a 17,3 meses) para la rama de OPDIVO™. En la rama de OPDIVO™ más ipilimumab, el 39% estuvo expuesto a OPDIVO™ durante ≥6 meses y el 24% estuvo expuesto durante >1 año. En la rama de OPDIVO™, el 53% estuvo expuesto durante ≥6 meses y el 32% durante >1 año. El Ensayo 6 excluyó a pacientes con enfermedad autoinmune, una afección que requiriera tratamiento sistémico con corticosteroides (más de 10 mg diarios de equivalentes de prednisona) u otra medicación inmunosupresora dentro de los 14 días del inicio de la terapia del estudio, resultado positivo en la prueba de hepatitis B o C, o antecedentes de VIH. Las características de la población de ensayo fueron las siguientes: 65% de sexo masculino, mediana de edad 61 años, 97% de raza blanca, estado funcional ECOG en condición basal 0 (73%) o 1 (27%), 93% con enfermedad en Estadio IV AJCC, 58% con enfermedad en estadio M1c; 36% con nivel elevado de LDH en condición basal, 4% con antecedentes de metástasis cerebral, y 22% habían recibido terapia adyuvante. En el Ensayo 6, las reacciones adversas serias (73% versus 37%), las reacciones adversas que condujeron a la discontinuación permanente (43% y 14%) o la demora de la dosis (55% y 28%), y las reacciones adversas de Grado 3 o 4 (72% y 44%) se produjeron todas con mayor frecuencia en pacientes de la rama de OPDIVO™ más ipilimumab que en la rama de OPDIVO™. Las reacciones adversas serias más frecuentes (≥10%) en la rama de OPDIVO™ más ipilimumab y en la rama de OPDIVO™, respectivamente, fueron diarrea (13% y 2,6%), colitis (10% y 1,6%) y pirexia (10% y 0,6%). Las reacciones adversas más frecuentes que condujeron a la discontinuación de ambos fármacos en la rama de OPDIVO™ más ipilimumab y de OPDIVO™ en la rama de OPDIVO™, respectivamente, fueron diarrea (8% y 1,9%), colitis (8% y 0,6%), aumento de ALT (4,8% y 1,3%), aumento de AST (4,5% y 0,6%) y neumonitis (1,9% y 0,3%). Las reac-ciones adversas más comunes (≥20%) en la rama de OPDIVO™ más ipilimumab fueron fatiga, erupción, diarrea, náuseas, pirexia, vómitos y disnea. Las reacciones adversas más comunes (≥20%) en la rama de OPDIVO™ fueron fatiga, erupción, diarrea y náuseas. La Tabla 6 sintetiza la incidencia de reacciones adversas que se produjeron en al menos el 10% de los pacientes de cualquiera de las ramas que contenían OPDIVO™ en el Ensayo 6.
Tabla 6. Reacciones adversas que ocurrieron en ≥10% de los pacientes de la rama de OPDIVO™ más ipilimumab o la rama de OPDIVO™ y con mayor incidencia que en la rama de ipilimumab (diferencia entre ramas de ≥5% [todos los gra-dos] o ≥2% [Grados 3-4]) (Ensayo 6)
|
Reacción adversa |
Porcentaje (%) de pacientes |
|||||
|
OPDIVO™ más ipilimumab (n=313) |
OPDIVO™ (n=313) |
Ipilimumab (n=311) |
||||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
Todos los gra-dos |
Grados 3-4 |
|
|
Trastornos generales y afecciones en el sitio de administración |
||||||
|
Fatigaa |
59 |
6 |
53 |
1,9 |
50 |
3,9 |
|
Pirexia |
37 |
1,6 |
14 |
0 |
17 |
0,6 |
|
Trastornos de la piel y el tejido subcutáneo |
||||||
|
Erupciónb |
53 |
5 |
40 |
1,6 |
42 |
3,9 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
52 |
11 |
31 |
3,8 |
46 |
8 |
|
Náuseas |
40 |
3,5 |
28 |
0,6 |
29 |
1,9 |
|
Vómitos |
28 |
3,5 |
17 |
1,0 |
16 |
1,6 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||
|
Disnea |
20 |
2,2 |
12 |
1,3 |
13 |
0,6 |
|
La toxicidad se calificó según NCI CTCAE v4. a Fatiga es un término compuesto que incluye astenia y fatiga. b Erupción es un término compuesto que incluye erupción pustular, dermatitis, dermatitis acneiforme, dermatitis alérgica, dermatitis atópica, dermatitis bullosa, dermatitis exfoliativa, dermatitis psoriasiforme, erupción medicamentosa, eritema, erupción exfoliativa, erupción eritematosa, erupción generalizada, erupción macular, erupción máculopapular, erupción morbiliforme, erupción papular, erupción papuloescamosa, erupción prurítica y dermatitis seborreica. |
||||||
Otras reacciones adversas importantes desde el punto de vista clínico en menos del 10% de los pacientes tratados con OPDIVO™ con ipilimumab o con OPDIVO™ como agente único en el Ensayo 6 fueron las siguientes:
Trastornos gastrointestinales: estomatitis, perforación intestinal
Trastornos de la piel y el tejido subcutáneo: vitiligo
Trastornos musculoesqueléticos y del tejido conectivo: miopatía, síndrome de Sjogren, espondiloartropatía
Trastornos del sistema nervioso: neuritis, parálisis del nervio peroneo
Tabla 7. Anormalidades de laboratorio de empeoramiento respecto del nivel basal que ocurrieron en ≥20% de los pacientes tratados con OPDIVO™ con ipilimumab o con OPDIVO™ como agente único y con una mayor incidencia que en la rama de ipilimumab (diferencia entre ramas ≥5% [todos los grados] o ≥2% [Grados 3-4]) (Ensayo 6)
|
Anormalidad de laboratorio |
Porcentaje (%) de pacientesa |
|||||
|
OPDIVO™ más ipilimumab |
OPDIVO™ |
Ipilimumab |
||||
|
Cualquier grado |
Grado 3-4 |
Cualquier grado |
Grado 3-4 |
Cualquier grado |
Grado 3-4 |
|
|
Química |
||||||
|
Aumento de ALT |
53 |
15 |
23 |
3,0 |
28 |
2,7 |
|
Aumento de AST |
47 |
13 |
27 |
3,7 |
27 |
1,7 |
|
Hiponatremia |
42 |
9 |
20 |
3,3 |
25 |
7 |
|
Aumento de lipasa |
41 |
20 |
29 |
9 |
23 |
7 |
|
Aumento de fosfatasa alcalina |
40 |
6 |
24 |
2,0 |
22 |
2,0 |
|
Hipocalcemia |
29 |
1,1 |
13 |
0,7 |
21 |
0,7 |
|
Aumento de amilasa |
25 |
9,1 |
15 |
1,9 |
14 |
1,6 |
|
Aumento de creatinina |
23 |
2,7 |
16 |
0,3 |
16 |
1,3 |
|
Hematología |
||||||
|
Anemia |
50 |
2,7 |
39 |
2,6 |
40 |
6 |
|
Linfopenia |
35 |
4,8 |
39 |
4,3 |
27 |
3,4 |
|
Química |
||||||
|
Aumento de ALT |
53 |
15 |
23 |
3,0 |
28 |
2,7 |
|
Aumento de AST |
47 |
13 |
27 |
3,7 |
27 |
1,7 |
|
a La incidencia de cada análisis se basa en el número de pacientes que tenían disponible una medición de laboratorio basal y al menos una medición durante el estudio: OPDIVO™ más ipilimumab (rango: 241 a 297); OPDIVO™ (rango: 260 a 306); ipilimumab (rango: 253 a 304). |
||||||
Cáncer de pulmón de células no pequeñas metastásico: La seguridad de OPDIVO™ en el NSCLC metastásico fue evaluada en el Ensayo 2, un ensayo multicéntrico, abierto, randomizado, realizado en pacientes con NSCLC escamoso metastásico y progresión de la enfermedad durante o después de un régimen de quimioterapia dual basado en platino previo y en el Ensayo 3, un ensayo randomizado, de diseño abierto, multicéntrico, realizado en pacientes con NSCLC no escamoso metastásico y progresión durante o después de un régimen de quimioterapia dual previo basado en platino [véase Estudios Clínicos (14.2)]. Los pacientes recibieron 3 mg/kg de OPDIVO™ administrado por vía intravenosa durante 60 minutos cada 2 semanas o docetaxel administrado por vía intravenosa a razón de 75 mg/m2 cada 3 semanas. La mediana de la duración de la terapia en pacientes tratados con OPDIVO™ en el Ensayo 2 fue de 3,3 meses (rango: 1 día a 21,7+ meses) y en el Ensayo 3 fue de 2,6 meses (rango: 0 a 24,0+ meses). En el Ensayo 2, el 36% de los pacientes recibieron OPDIVO™ durante al menos 6 meses y el 18% de los pacientes recibieron OPDIVO™ durante al menos 1 año, y en el Ensayo 3, el 30% de los pacientes recibieron OPDIVO™ durante más de 6 meses, y el 20% de los pacientes recibieron OPDIVO™ durante más de 1 año. El Ensayo 2 y el Ensayo 3 excluyeron a pacientes con enfermedad autoinmune activa, afecciones médicas que requirieran inmunosupresión sistémica o enfermedad pulmonar intersticial sintomática. En ambos ensayos, la me-diana de la edad de los pacientes tratados con OPDIVO™ fue de 61 años (rango: 37 a 85); el 38% tenían ≥65 años de edad, el 61% eran de sexo masculino, y el 91% eran de raza blanca. El 10% de los pacientes tenían metástasis cerebral, y su estado funcional ECOG era de 0 (26%) o 1 (74%). OPDIVO™ fue discontinuado en el 11% de los pacientes, y fue demorado en el 28% de los pacientes por una reacción adversa. Se produjeron reacciones adversas serias en el 46% de los pacientes que recibieron OPDIVO™. Las reacciones adversas serias más frecuentes reportadas en al menos 2% de los pacientes que recibieron OPDIVO™ fueron neumonía, embolia pulmonar, disnea, pireaxia, derrame pleural, neumonitis y falla respiratoria. En el Ensayo 3, en la rama de OPDIVO™, siete muertes se debieron a infección, incluido un caso de neumonía por Pneumocystis jirovecii, cuatro muertes se debieron a embolia pulmonar, y una muerte se debió a encefalitis límbica. En ambos ensayos, las reacciones adversas más comunes (reportadas en al menos 20% de los pacientes) fue-ron fatiga, dolor musculoesquelético, tos, disnea y disminución del apetito. La Tabla 8 sintetiza las reacciones adversas seleccionadas que se produjeron con mayor frecuencia en al menos el 10% de los pacientes tratados con OPDIVO™.
Tabla 8. Reacciones adversas que ocurrieron en ≥10% de los pacientes tratados con OPDIVO™ y con mayor incidencia que con docetaxel (diferencia entre ramas de ≥5% [todos los grados] o ≥2% [Grados 3-4]) (Ensayos 2 y 3)
|
Reacción adversa |
OPDIVO™ (n=418) |
Docetaxel (n=397) |
||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Porcentaje (%) de pacientes |
||||
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos |
31 |
0,7 |
24 |
0 |
|
Trastornos del metabolismo y la nutrición |
||||
|
Disminución del apetito |
28 |
1,4 |
23 |
1,5 |
|
Trastornos de la piel y el tejido subcutáneo |
||||
|
Prurito |
10 |
0,2 |
2,0 |
0 |
|
La toxicidad se calificó según NCI CTCAE v4. |
||||
Otras reacciones adversas clínicamente importantes observadas en pacientes tratados con OPDIVO™ y que se produjeron con una incidencia similar en pacientes tratados con docetaxel y que no se enumeran en otra parte de la sección 6 incluyen: fatiga/astenia (48% de Grado 1-4, 5% de Grado 3-4), dolor musculoesquelético (33%), derrame pleural (4,5%), embolia pulmonar (3,3%).
Tabla 9. Anormalidades de laboratorio de empeoramiento respecto del nivel basal que ocurrieron en ≥10% de los pacientes tratados con OPDIVO™ para todos los grados de NCI CTCAE y con una mayor incidencia que con docetaxel (diferencia entre ramas ≥5% [todos los grados] o ≥2% [Grados 3-4]) (Ensayos 2 y 3)
|
Anormalidad de laboratorio |
Porcentaje de pacientes con un empeoramiento en los análisis de labora-torio desde la condición basala |
|||
|
OPDIVO™ |
Docetaxel |
|||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Química |
||||
|
Hipopotasemia |
35 |
7 |
34 |
4,9 |
|
Aumento de AST |
27 |
1,9 |
13 |
0,8 |
|
Aumento de fosfatasa alcalina |
26 |
0,7 |
18 |
0,8 |
|
Aumento de ALT |
22 |
1,7 |
17 |
0,5 |
|
Aumento de creatinina |
18 |
0 |
12 |
0,5 |
|
Aumento de TSHb |
14 |
N/A |
6 |
N/A |
|
a La incidencia de cada análisis se basa en el número de pacientes que tenían disponible una medición de laboratorio basal y al menos una medición durante el estudio: grupo de OPDIVO™ (rango: 405 a 417 pacientes) y grupo de docetaxel (rango: 372 a 390 pacientes); TSH: grupo de OPDIVO™ (n=314) y grupo de docetaxel (n=297). b No calificado según NCI CTCAE v4. |
||||
Carcinoma de células renales. La seguridad de OPDIVO™ fue evaluada en el Ensayo 5, un ensayo randomizado, de diseño abierto, en el cual 803 pacientes con RCC avanzado que habían experimentado progresión de la enfermedad durante o después de al menos un régimen de tratamiento antiangiogénico recibieron OPDIVO™ 3 mg/kg cada 2 semanas (n=406) o everolimus 10 mg/kg diariamente (n=397) [véase Estudios clínicos (Carcinoma de células renales)]. La mediana de la duración del tratamiento fue de 5,5 meses (rango: 1 día a 29,6+ meses) en pacientes tratados con OPDIVO™ y de 3,7 meses (rango: 6 días a 25,7+ meses) en pacientes tratados con everolimus. La terapia del estudio fue discontinuada por reacciones adversas en el 16% de los pacientes tratados con OPDIVO™ y en el 19% de los pacientes tratados con everolimus. El 44% de los pacientes que recibieron OPDIVO™ tuvieron una demora en la administración de la dosis por una reacción adversa. Se produjeron reacciones adversas serias en el 47% de los pacientes que recibieron OPDIVO™. Las reacciones adversas más frecuentes reportadas en al menos el 2% de los pacientes fueron lesión renal aguda, derrame pleural, neumonía, diarrea e hipercalcemia. La tasa de mortalidad durante el tratamiento o dentro de los 30 días posteriores a la última dosis del fármaco del estudio fue del 4,7% en la rama de OPDIVO™ versus 8,6% en la rama de everolimus. Las reacciones adversas más comunes (reportadas en al menos el 20% de los pacientes) fueron afecciones asténicas, tos, náuseas, erupción, disnea, diarrea, constipación, disminución del apetito, dolor de espalda y artralgia. La Tabla 10 sintetiza las reacciones adversas que se produjeron en más del 15% de los pacientes tratados con OPDIVO™.
Tabla 10. Reacciones adversas de Grado 1-4 en >15% de los pacientes que recibieron OPDIVO™ (Ensayo 5)
|
OPDIVO (n=406) |
Everolimus (n=397) |
|||
|
Porcentaje (%) de pacientes |
||||
|
Grados 1-4 |
Grados 3-4 |
Grados 1-4 |
Grados 3-4 |
|
|
Reacción adversa |
98 |
56 |
96 |
62 |
|
Trastornos generales y afecciones en el sitio de administración |
||||
|
Afecciones asténicasa |
56 |
6 |
57 |
7 |
|
Pirexia |
17 |
0,7 |
20 |
0,8 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos/tos productiva |
34 |
0 |
38 |
0,5 |
|
Disnea/disnea de esfuerzo |
27 |
3,0 |
31 |
2,0 |
|
Infección respiratoria superiorb |
18 |
0 |
11 |
0 |
|
Trastornos gastrointestinales |
||||
|
Náuseas |
28 |
0,5 |
29 |
1 |
|
Diarreac |
25 |
2,2 |
32 |
1,8 |
|
Constipación |
23 |
0,5 |
18 |
0,5 |
|
Vómitos |
16 |
0,5 |
16 |
0,5 |
|
Trastornos de la piel y el tejido subcutáneo |
||||
|
Erupciónd |
28 |
1,5 |
36 |
1,0 |
|
Prurito/prurito generalizado |
19 |
0 |
14 |
0 |
|
Trastornos del metabolismo y la nutrición |
||||
|
Disminución del apetito |
23 |
1,2 |
30 |
1,5 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Artralgia |
20 |
1,0 |
14 |
0,5 |
|
Dolor de espalda |
21 |
3,4 |
16 |
2,8 |
|
La toxicidad se calificó según NCI CTCAE v4. a Afecciones asténicas que abarcan los términos preferidos (PT) astenia, disminución de la actividad, fatiga y malestar. b Incluye nasofaringitis, faringitis, rinitis e infección respiratoria superior de origen viral. c Incluye colitis, enterocolitis y gastroenteritis. d Incluye dermatitis, dermatitis acneiforme, erupción eritematosa, erupción generalizada, erupción macular, erupción máculopapular, erupción papular, erupción prurítica, eritema multiforme y eritema. |
||||
Otras reacciones adversas clínicamente importantes en el Ensayo 5 fueron las siguientes:
Trastornos generales y afecciones en el sitio de administración: edema periférico/edema
Trastornos gastrointestinales: dolor/malestar abdominal
Trastornos musculoesqueléticos y del tejido conectivo: dolor de extremidades, dolor musculoesquelético
Trastornos del sistema nerviosos: cefalea/migraña, neuropatía periférica
Investigaciones: descenso de peso
Trastornos dérmicos: Eritrodisestesia palmo-plantar
Las anormalidades de laboratorio más comunes que empeoraron en comparación con la condición basal en ≥30% de los pacientes incluyen aumento de creatinina, linfopenia, anemia, aumento de AST, aumento de fosfatasa alcalina, hiponatremia, nivel elevado de triglicéridos e hiperpotasemia. La Tabla 11 sintetiza las anormalidades de laboratorio que se produjeron en más del 15% de los pacientes tratados con OPDIVO™.
Tabla 11. Valores de laboratorio de Grado 1-4 de empeoramiento respecto del nivel basal en >15% de los pacientes tratados con OPDIVO™ (Ensayo 5)
|
Anormalidad de laboratorio |
Porcentaje de pacientes con empeoramiento de los valores de laboratorio desde la condición basala |
|||
|
OPDIVO™ |
Everolimus |
|||
|
Grados 1-4 |
Grados 3-4 |
Grados 1-4 |
Grados 3-4 |
|
|
Hematología |
||||
|
Linfopenia |
42 |
6 |
53 |
11 |
|
Anemia |
39 |
8 |
69 |
16 |
|
Química |
||||
|
Aumento de creatinina |
42 |
2,0 |
45 |
1,6 |
|
Aumento de AST |
33 |
2,8 |
39 |
1,6 |
|
Aumento de fosfatasa alcalina |
32 |
2,3 |
32 |
0,8 |
|
Hiponatremia |
32 |
7 |
26 |
6 |
|
Hiperpotasemia |
30 |
4,0 |
20 |
2,1 |
|
Hipocalcemia |
23 |
0,9 |
26 |
1,3 |
|
Aumento de ALT |
22 |
3,2 |
31 |
0,8 |
|
Hipercalcemia |
19 |
3,2 |
6 |
0,3 |
|
Lípidos |
||||
|
Aumento de triglicéridos |
32 |
1,5 |
67 |
11 |
|
Aumento de colesterol |
21 |
0,3 |
55 |
1,4 |
|
a La incidencia de cada análisis se basa en el número de pacientes que tenían disponible una medición de laboratorio basal y al menos una medición de laboratorio durante el estudio: grupo de OPDIVO™ (rango: 259 a 401 pacientes) y grupo de everolimus (rango: 257 a 376 pacientes). |
||||
Asimismo, entre los pacientes con TSH menor al ULN en condición basal, una mayor proporción de pacientes experi-mentaron una elevación de TSH emergente del tratamiento superior al ULN en el grupo de OPDIVO™ en comparación con el grupo de everolimus (26% y 14%, respectivamente).
Linfoma de Hodgkin clásico. La seguridad de OPDIVO™ 3 mg/kg cada 2 semanas fue evaluada en 263 pacientes adultos con cHL (240 pacientes en el Ensayo 7 y 23 pacientes en el Ensayo 8). El tratamiento pudo continuar hasta la progresión de la enfermedad, el máximo beneficio clínico o una toxicidad inaceptable. La mediana de la edad fue de 34 años (rango: 18 a 72), el 98% de los pacientes habían recibido HSCT autólogo, ninguno había recibido HSCT alogénico, y el 74% había recibido brentuximab vedotina. La mediana del número de regímenes sistémicos previos fue 4 (rango: 1 a 15). Los pacientes recibieron una mediana de 10 dosis (ciclos) de OPDIVO™ (rango: 1 a 48), con una mediana de la duración de la terapia de 4,8 meses (rango: 0,3 a 24 meses). OPDIVO™ fue discontinuado debido a reacciones adversas en el 4,2% de los pacientes. El 23% de los pacientes tuvieron una demora de la dosis por una reacción adversa. Se produjeron reacciones adversas serias en el 21% de los pacientes. Las reacciones adversas serias más frecuentes reportadas en al menos el 1% de los pacientes fueron reacciones relacionadas con la perfusión, neumonía, derrame pleural, pirexia, erupción y neumonitis. Diez pacientes murieron por causas no relacionadas a la progresión de la enfermedad, incluidos 6 que murieron a raíz de complicaciones del HSCT alogénico. Las reacciones adversas más comunes (reportadas en al menos el 20%) entre todos los pacientes (población de seguridad), fueron fatiga, infección del tracto respiratorio superior, pirexia, diarrea y tos. En el subconjunto de pacientes de la población de eficacia, las reacciones adversas más comunes también incluyeron erupción, dolor musculoesquelético, prurito, náuseas, artralgia y neuropatía periférica. Se produjeron reacciones adversas serias en el 27% de estos pacientes. La Tabla 12 sintetiza las reacciones adversas que se produje-ron en al menos el 10% de los pacientes de la población de seguridad (n=263) y la población de eficacia (n=95). Hay una mayor incidencia de reacciones adversas en el subconjunto de pacientes evaluados en eficacia; estos pacientes recibieron una mediana de 17 dosis de OPDIVO™ y una mediana de 5 regímenes sistémicos previos [véase Estudios clínicos (Linfoma de Hodgkin clásico)].
Tabla 12. Reacciones adversas no hematológicas que ocurrieron en ≥10% de los pacientes con cHL (Ensayos 7 y 8)
|
OPDIVO™ cHL Población de seguridad (n=263) |
OPDIVO™ cHL Población de eficacia (n=95) |
|||
|
Porcentaje (%) de pacientes |
||||
|
Reacción adversaa |
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
Trastornos generales y afecciones en el sitio de administración |
||||
|
Fatigab |
32 |
1,1 |
43 |
1,1 |
|
Pirexia |
24 |
0,8 |
35 |
1,1 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
23 |
0,8 |
30 |
1,1 |
|
Náuseas |
17 |
0 |
23 |
0 |
|
Vómitos |
15 |
0,8 |
16 |
1,1 |
|
Dolor abdominalc |
11 |
0,8 |
13 |
2,1 |
|
Constipación |
9 |
0,4 |
14 |
0 |
|
Infecciones |
||||
|
Infección del tracto respiratorio superiord |
28 |
0,4 |
48 |
1,1 |
|
Neumonía/bronconeumoníae |
9 |
3,0 |
19 |
5,3 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos/tos productiva |
22 |
0 |
35 |
0 |
|
Disnea/disnea de esfuerzo |
10 |
0,8 |
16 |
2,1 |
|
Trastornos de la piel y el tejido subcutáneo |
||||
|
Erupciónf |
19 |
1,5 |
31 |
3,2 |
|
Prurito |
17 |
0 |
25 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Dolor musculoesqueléticog |
19 |
1,1 |
27 |
1,1 |
|
Artralgia |
11 |
0 |
21 |
0 |
|
Trastornos endocrinos |
||||
|
Hipotiroidismo/tiroiditis |
12 |
0 |
17 |
0 |
|
Hiperglucemia/Aumento de glucosa en sangre |
9 |
0,4 |
14 |
1,1 |
|
Trastornos del sistema nervioso |
||||
|
Cefalea |
12 |
0,4 |
12 |
1,1 |
|
Neuropatía periféricah |
11 |
0,4 |
21 |
0 |
|
Lesiones, intoxicación y complicaciones de los procedimientos |
||||
|
Reacción relacionada con la perfusión |
12 |
0,4 |
18 |
0 |
|
La toxicidad se calificó según NCI CTCAE v4. a Incluye eventos que ocurrieron hasta 30 días después de la última dosis de nivolumab, independientemente de la causalidad. Luego de una reacción adversa mediada por la respuesta inmune, se incluyeron las reacciones que le siguieron a la reinstauración de nivolumab si se produjeron hasta 30 días después de completado el régimen inicial de nivolumab. b Incluye astenia. c Incluye malestar abdominal y dolor abdominal superior. d Incluye nasofaringitis, faringitis, rinitis y sinusitis. e Incluye neumonía bacteriana, neumonía micoplásmica y neumonía por Pneumocystis jirovecii. f Incluye dermatitis, dermatitis acneiforme, dermatitis exfoliativa, y erupción descrita como macular, papular, máculopapular, prurítica, exfoliativa o acneiforme. g Incluye dolor de espalda, dolor óseo, dolor de pecho musculoesquelético, malestar musculoesquelético, mialgia, dolor de cuello y dolor de extremidades. h Incluye hiperestesia, hipoestesia, parestesia, disestesia, neuropatía motriz periférica, neuropatía sensorial periférica y polineuropatía. |
||||
Información adicional sobre reacciones adversas clínicamente importantes:
Neumonitis mediada por la respuesta inmune: En los Ensayos 7 y 8, se produjo neumonitis, incluida enfermedad pulmonar intersticial, en el 4,9% (13/263) de los pacientes que recibieron OPDIVO™. Se produjo neumonitis mediada por la respuesta inmune en el 3,4% (9/263) de los pacientes que recibieron OPDIVO™ (un caso de Grado 3 y ocho casos de Grado 2). La mediana del tiempo hasta su presentación fue de 2,2 meses (rango: 1 día a 10,1 meses). Los nueve pacientes recibieron corticosteroides sistémicos, y se observó la resolución en siete de ellos. Un paciente discontinuó permanentemente OPDIVO™ debido a neumonitis de Grado 2. Se produjo un retraso de la dosis en tres pacientes. Cinco pacientes reiniciaron OPDIVO™, de los cuales ninguno presentó recurrencia de la neumonitis. Neuropatía periférica: En los Ensayos 7 y 8, se observó neuropatía periférica en el 11% (30/263) de todos los pacientes que recibieron OPDIVO™. Veintidós pacientes (8%) tuvieron neuropatía periférica de nuevo inicio, y cuatro pacientes tuvieron empeoramiento desde la condición basal. Cuatro pacientes más con neuropatía periférica de base (sacar “en condición basal”) (tres de Grado 1 y uno de Grado 2) no empeoraron. Todos los eventos fueron de Grado 1 o 2, excepto por 1 evento de Grado 3 (0,4%). Complicaciones del HSCT alogénico tras OPDIVO™: [véase Advertencias y Precauciones (Complicaciones del HSCT alogénico tras OPDIVO™)].
Tabla 13. Anormalidades de laboratorio de empeoramiento respecto del nivel basal que ocurrieron en ≥10% de los pacientes con cHL tratados con OPDIVO™ (Ensayos 7 y 8)
|
Anormalidad de laboratorio |
OPDIVO™ cHL Población de seguridada |
OPDIVO™ cHL Población de eficaciab |
||
|
Porcentaje (%) de pacientesc |
||||
|
Todos los grados |
Grados 3-4 |
Todos los grados |
Grados 3-4 |
|
|
Hematología |
||||
|
Neutropenia |
29 |
3,6 |
37 |
6 |
|
Trombocitopenia |
28 |
2,4 |
33 |
3,2 |
|
Linfopenia |
24 |
8 |
32 |
7 |
|
Anemia |
22 |
2,8 |
27 |
2,1 |
|
Química |
||||
|
Aumento de ALT |
24 |
2,0 |
25 |
2,1 |
|
Aumento de AST |
23 |
2,4 |
32 |
3,2 |
|
Amento de fosfatasa alcalina |
17 |
1,6 |
21 |
2,1 |
|
Aumento de lipasa |
16 |
6,5 |
28 |
12 |
|
Hiponatremia |
14 |
0,8 |
15 |
1,1 |
|
Hipopotasemia |
11 |
1,6 |
14 |
3,2 |
|
Hipocalcemia |
11 |
0,4 |
14 |
1,1 |
|
Hipomagnesemia |
10 |
0,4 |
15 |
1,3 |
|
Aumento de creatinina |
10 |
0 |
15 |
0 |
|
Aumento de bilirrubina |
9 |
0,8 |
10 |
0 |
|
a El número de pacientes evaluables para la población de seguridad oscila de 226 a 253. b El número de pacientes evaluables para la población de eficacia oscila de 80 a 85. c Incluye eventos que ocurrieron hasta 30 días después de la última dosis de nivolumab. Luego de una reacción adversa mediada por la respuesta inmune, se incluyeron las reacciones que le siguieron a la reinstauración de nivolumab si se produjeron dentro de los 30 días de completado el régimen inicial de nivolumab. |
||||
Carcinoma de células escamosas de cabeza y cuello recurrente o metastásico: La seguridad de OPDIVO™ fue evaluada en el Ensayo 9, un ensayo randomizado, con control activo, abierto, multicéntrico, en pacientes con SCCHN recurrente o metastásico y progresión durante o dentro de los 6 meses después de haber recibido una terapia previa basada en platino [véase Estudios clínicos (Carcinoma metastásico o recurrente de células escamosas de cabeza y cuello (SCCHN).)]. Los pacientes recibieron 3 mg/kg de OPDIVO™ (n=236) administrado por vía intravenosa (IV) durante 60 minutos cada 2 semanas, o un agente a elección del investigador:
• Cetuximab (n=13), dosis de carga IV de 400 mg/m2 seguida por 250 mg/m2 semanalmente, o
• Metotrexato (n=46) de 40 a 60 mg/m2 semanalmente por vía IV, o
• Docetaxel (n=52) de 30 a 40 mg/m2 semanalmente por vía IV.
La mediana de la duración de la exposición a nivolumab fue de 1,9 meses (rango: 1 día a 16,1+ meses) en pacientes tratados con OPDIVO™. En este ensayo, el 18% de los pacientes recibieron OPDIVO™ durante más de 6 meses y el 2,5% de los pacientes recibieron OPDIVO™ durante más de 1 año. El Ensayo 9 excluyó a pacientes con enfermedad autoinmune activa, afecciones médicas que requirieran inmunosupresión sistémica, o carcinoma recurrente o metastásico de nasofaringe, carcinoma de células escamosas de histología primaria desconocida, de glándulas salivales o de histologías no escamosas (por ejemplo, melanoma de mucosa). La mediana de la edad de todos los pacientes randomizados fue de 60 años (rango: 28 a 83); el 28% de los pacientes del grupo de OPDIVO™ tenían ≤65 años de edad, y el 37% del grupo comparador tenían ≥65 años de edad; el 83% eran de sexo masculino, y el 83% blancos, el 12% asiáticos y el 4% negros. El estado funcional ECOG en condición basal fue 0 (20%) o 1 (78%); el 45% de los pacientes recibieron una sola línea de terapia sistémica previa, mientras que el 55% restante de los pacientes recibieron dos o más líneas de terapia previas, y el 90% recibió radioterapia previa. OPDIVO™ fue discontinuado en el 14% de los pacientes y fue demorado en el 24% de los pacientes por una reacción adversa. Se produjeron reacciones adversas serias en el 49% de los pacientes que recibieron OPDIVO™. Las reacciones adversas serias más frecuentes reportadas en al menos el 2% de los pacientes que recibieron OPDIVO™ fueron neumonía, disnea, insuficiencia respiratoria, infección de las vías respiratorias y septicemia. Las reacciones adversas y anormalidades de laboratorio ocurridas en pacientes con SCCHN generalmente fueron similares a las ocurridas en pacientes con melanoma y NSCLC. Las reacciones adversas más comunes ocurridas en >10% de los pacientes tratados con OPDIVO™ y con mayor incidencia que con el agente a elección del investigador fueron tos y disnea. Las anormalidades de laboratorio más comunes ocurridas en ≥10% de los pacientes tratados con OPDIVO™ y con mayor incidencia que con el agente a elección del investigador fueron aumento de fosfatasa alcalina, aumento de amilasa, hipercalcemia, hiperpotasemia y aumento de TSH.
Carcinoma urotelial: La seguridad de OPDIVO™ fue evaluada en el Ensayo 10, un estudio de rama única en el cual 270 pacientes con carcinoma urotelial localmente avanzado o metastásico que tuvieron progresión de la enfermedad durante o después de una quimioterapia con contenido de platino o que tuvieron progresión de la enfermedad dentro de los 12 meses del tratamiento neoadyuvante o adyuvante con una quimioterapia que contenía platino recibieron OPDIVO™ 3 mg/kg cada 2 semanas hasta la progresión de la enfermedad o una toxicidad inaceptable. La mediana de la duración del tratamiento fue de 3,3 meses (rango: 0 a 13,4+). El 46% de los pacientes tuvieron una demora en la administración del fármaco por una reacción adversa. Catorce pacientes (5,2%) murieron por causas distintas de progresión de la enfermedad. Esto incluye a 4 pacientes (1,5%) que murieron por neumonitis o insuficiencia cardiovascular que se atribuyó al tratamiento con OPDIVO™. OPDIVO™ fue discontinuado por reacciones adversas en el 17% de los pacientes. Se produjeron reacciones adversas serias en el 54% de los pacientes. Las reacciones adversas serias más frecuentes reportadas en al menos el 2% de los pacientes fueron infección del tracto urinario, septicemia, diarrea, obstrucción del intestino delgado y deterioro del estado físico general. Veinticinco pacientes (9%) recibieron una dosis oral de prednisona equivalente a ≥40 mg diarios por una reacción adversa mediada por la respuesta inmune [véase Advertencias y Precauciones]. Las reacciones adversas más comunes (reportadas en al menos el 20% de los pacientes) fueron fatiga, dolor musculoesquelético, náuseas y disminución del apetito. La Tabla 14 sintetiza las reacciones adversas que se produjeron en más del 10% de los pacientes.
Tabla 14. Reacciones adversas ocurridas en ≥10% de los pacientes (Ensayo 10)
|
OPDIVO™ Carcinoma Urotelial |
||
|
Porcentaje (%) de pacientes |
||
|
Todos los grados |
Grados 3-4 |
|
|
Reacción adversa |
99 |
51 |
|
Trastornos generales y afecciones en el sitio de administración |
||
|
Astenia/fatiga/malestar |
46 |
7 |
|
Pirexia/fiebre asociada con el tumor |
17 |
0,4 |
|
Edema/edema periférico/ inflamación periférica |
13 |
0,4 |
|
Infecciones e infestaciones |
||
|
Tracto urinario Infección/eschericia/ infección fúngica del tracto urinario |
17 |
7 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Tos/tos productiva |
18 |
0 |
|
Disnea/disnea por esfuerzo |
14 |
3,3 |
|
Trastornos gastrointestinales |
||
|
Náuseas |
22 |
0,7 |
|
Diarrea |
17 |
2,6 |
|
Constipación |
16 |
0,4 |
|
Dolor abdominala |
13 |
1,5 |
|
Vómitos |
12 |
1,9 |
|
Trastornos de la piel y el tejido subcutáneo |
||
|
Erupción cutáneab |
16 |
1,5 |
|
Prurito |
12 |
0 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||
|
Dolor musculoesqueléticoc |
30 |
2,6 |
|
Artralgia |
10 |
0,7 |
|
Trastornos del metabolismo y la nutrición |
||
|
Disminución del apetito |
22 |
2,2 |
|
Trastornos endocrinos |
||
|
Trastornos tiroideosd |
15 |
0 |
|
La toxicidad se calificó según los criterios NCI CTCAE v4. a Incluye malestar abdominal, dolor abdominal inferior y superior. b Incluye dermatitis, dermatitis acneiforme, dermatitis bulosa y erupción cutánea descrita como generalizada, macular, máculopapular o prurítica. c Incluye dolor de espalda, dolor óseo, dolor de pecho musculoesquelético, malestar musculoequelético, mialgia, dolor de cuello, dolor de extremidades y dolor espinal. d Incluye tiroiditis autoinmune, disminución de TSH en sangre, aumento deTSH en sangre, hipertiroidismo, hipotiroidismo, tiroiditis, disminución de tiroxina, aumento de tiroxina libre, aumento de tiroxina, aumento de tri-iodotironina libre, aumento de tri-iodotironina. |
||
Tabla 15. Empeoramiento de anormalidades de laboratorio desde la condición basal ocurridas en ≥10% de los pacientes (Ensayo 10)
|
OPDIVO™ Carcinoma Uroteliala |
||
|
Porcentaje (%) de pacientes |
||
|
Todos los grados |
Grados 3-4 |
|
|
Hematología |
||
|
Linfopenia |
42 |
9 |
|
Anemia |
40 |
7 |
|
Trombocitopenia |
15 |
2,4 |
|
Leucopenia |
11 |
0 |
|
Química |
||
|
Hiperglucemia |
42 |
2,4 |
|
Hiponatremia |
41 |
11 |
|
Aumento de creatinina |
39 |
2,0 |
|
Aumento de fosfatasa alcalina |
33 |
5,5 |
|
Hipocalcemia |
26 |
0,8 |
|
Aumento de AST |
24 |
3,5 |
|
Hiperpotasemia |
19 |
1,2 |
|
Aumento de ALT |
18 |
1,2 |
|
Hipomagnesemia |
16 |
0 |
|
Aumento de lipasa |
20 |
7 |
|
Aumento de amilasa |
18 |
4,4 |
|
a La incidencia de cada análisis se basa en el número de pacientes que tuvieron disponible una medición de laboratorio basal y al menos una durante el estudio: rango: 84 a 256 pacientes. |
||
Inmunogenicidad: Al igual que con todas las proteínas terapéuticas, existe la posibilidad de inmunogenicidad. De 2022 pacientes que fueron tratados con OPDIVO™ 3 mg/kg cada 2 semanas y que fueron evaluables en cuanto a la presencia de anticuerpos antinivolumab, 231 pacientes (11,4%) dieron positivo para anticuerpos antinivolumab emergentes del tratamiento mediante un ensayo de electroquimioluminiscencia (ECL), y quince pacientes (0,7%) tuvieron anticuerpos neutralizantes contra nivolumab. No hubo evidencia de una alteración en el perfil de farmacocinética ni un aumento de la incidencia de reacciones a la perfusión con desarrollo de anticuerpos antinivolumab. De 394 pacientes que fueron tratados con OPDIVO™ con ipilimumab y evaluables en cuanto a la presencia de anticuerpos antinivolumab, 149 pacientes (37,8%) dieron positivo en cuanto a anticuerpos antinivolumab emergentes del tratamiento mediante un ensayo de ECL, y 18 pacientes (4,6%) tuvieron anticuerpos neutralizantes contra nivolumab. De los 391 pacientes evaluables en cuanto a la presencia de anticuerpos antiipilimumab, 33 pacientes (8,4%) dieron positivo para anticuerpos antiipilimumab emergentes del tratamiento mediante un ensayo ECL, y un paciente (0,3%) tuvo anticuerpos neutralizantes contra ipilimumab. No hubo evidencia de una mayor incidencia de reacciones a la perfusión con desarrollo de anticuerpos antinivolumab. La detección de la formación de anticuerpos es altamente dependiente de la sensibilidad y especificidad del ensayo. Además, la incidencia observada de positividad de anticuerpos (incluidos anticuerpos neutralizantes) en un ensayo puede ser influenciada por varios factores, que incluyen la metodología del ensayo, la manipulación de muestras, el cronograma de muestreo, las medicaciones concomitantes y la enfermedad subyacente. Por estos motivos, la comparación de la incidencia de anticuerpos contra OPDIVO™ con la incidencia de anticuerpos contra otros productos puede ser engañosa.
EFECTOS SOBRE LA CAPACIDAD DE CONDUCIR Y USAR MAQUINARIAS:
No indica.
INTERACCIONES MEDICAMENTOSAS:
No se han realizado estudios farmacocinéticos formales de interacciones medicamentosas con OPDIVO™.
ESTUDIOS CLÍNICOS:
Melanoma irresecable o metastásico. Melanoma metastásico previamente tratado.:El Ensayo 1 fue un estudio multicéntrico, abierto, que randomizó (2:1) pacientes con melanoma irresecable o metastásico para recibir OPDIVO™ administrado por vía intravenosa a razón de 3 mg/kg cada 2 semanas o una quimioterapia a elección del investigador, ya sea el agente único dacarbazina 1000 mg/m2 cada 3 semanas o la combinación de carboplatino AUC 6 cada 3 semanas más paclitaxel 175 mg/m2 cada 3 semanas. Los pacientes debían tener progresión de la enfermedad durante o después del tratamiento con ipilimumab y, si eran positivos para la mutación BRAF V600, un inhibidor de BRAF. El ensayo excluyó a los pacientes con enfermedad autoinmune, afecciones médicas que requerían inmunosupresión sistémica, melanoma ocular, metástasis cerebral activa, o antecedentes de reacciones adversas de Grado 4 relacionadas con ipilimumab (excepto por endocrinopatías) o reacciones adversas de Grado 3 relacionadas con ipilimumab que no se habían resuelto o que estaban inadecuadamente controladas dentro de las 12 semanas de iniciado el evento. Se llevaron a cabo evaluaciones tumorales 9 semanas después de la randomización, luego cada 6 semanas durante el primer año, y cada 12 semanas de allí en más.
La eficacia se evaluó en un análisis preliminar planeado de rama única, no comparativo, de los primeros 120 pacientes que recibieron OPDIVO™ en el Ensayo 1 y en quienes la duración mínima del seguimiento fue de 6 meses. Las medidas principales de los resultados de eficacia en esta población fueron la tasa de respuesta objetiva (ORR, por sus siglas en inglés) confirmada, según se mide por revisión central independiente bajo ciego usando los Criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST 1.1, por sus siglas en inglés) y la duración de la respuesta.
Entre los 120 pacientes tratados con OPDIVO™, la mediana de la edad fue de 58 años (rango: 25 a 88), el 65% de los pacientes eran de sexo masculino, el 98% eran de raza blanca, y el estado funcional (PS) según el ECOG era 0 (58%) o 1 (42%). Las características de la enfermedad eran enfermedad M1c (76%), positividad para mutación BRAF V600 (22%), nivel elevado de LDH (56%), antecedentes de metástasis cerebral (18%), y dos o más terapias sistémicas previas para la enfermedad metastásica (68%). La ORR fue del 32% (intervalo de confianza [IC] del 95%: 23, 41), consistente en 4 res-puestas completas y 34 respuestas parciales en pacientes tratados con OPDIVO™. De 38 pacientes con respuesta, 33 pacientes (87%) tenían respuestas en curso con duraciones que oscilaban entre 2,6+ y 10+ meses, que incluyeron a 13 pacientes con respuestas en curso de 6 meses o más. Hubo respuestas objetivas en pacientes con y sin melanoma positivo para la mutación BRAF V600.
Melanoma metastásico no tratado previamente: Ensayo 4. El Ensayo 4 fue un ensayo multicéntrico, doble ciego, rando-mizado (1:1), realizado en pacientes con melanoma irresecable o metastásico sin mutación BRAF V600 (wild-type). Los pacientes fueron randomizados para recibir OPDIVO™ 3 mg/kg por perfusión intravenosa cada 2 semanas o dacarbazina 1000 mg/m2 por perfusión intravenosa cada 3 semanas hasta la progresión de la enfermedad o la aparición de una toxicidad inaceptable. La randomización se estratificó por estado PD-L1 (mayor o igual al 5% de tinción en membrana de células tumorales por inmunohistoquímica, versus menor al 5% o resultado indeterminado) y estadio M (M0/M1a/M1b versus M1c). Los criterios de elegibilidad clave incluyeron melanoma cutáneo, mucosal o acral irresecable o metastásico histológicamente confirmado; ausencia de terapia previa para la enfermedad metastásica; finalización de la terapia adyuvante o neoadyuvante previa al menos 6 semanas antes de la randomización; estado funcional ECOG 0 o 1; ausencia de enfermedad autoinmune; y ausencia de metástasis cerebrales o leptomeníngeas activas. El ensayo excluyó a pacientes con melanoma ocular. Las evaluaciones tumorales se realizaron 9 semanas después de la randomización, luego cada 6 semanas durante el primer año y después cada 12 semanas de allí en adelante. El principal criterio de valoración de la eficacia fue la sobrevida global (OS, por sus siglas en inglés). Las mediciones adicionales de resultados incluyeron la sobrevida libre de progresión (PFS, por sus siglas en inglés) y la tasa de respuesta objetiva (ORR, por sus siglas en inglés) evaluadas por el investigador según los criterios RECIST v1.1. Un total de 418 pacientes fueron randomizados a OPDIVO™ (n=210) o dacarbazina (n=208). La mediana de la edad fue de 65 años (rango: 18 a 87), el 59% eran hombres, y el 99,5% eran de raza blanca. Las características de la enfermedad fueron enfermedad en estadio M1c (61%), melanoma cutáneo (74%), melanoma mucosal (11%), nivel elevado de LDH (37%), PD-L1 mayor o igual al 5% de expresión en membrana de células tumorales (35%) y antecedentes de metástasis cerebral (4%). Más pacientes en la rama de OPDIVO™ tuvieron un estado funcional ECOG de 0 (71% versus 58%). El Ensayo 4 demostró una mejora estadísticamente significativa en la OS para la rama de OPDIVO™ en comparación con la rama de dacarbazina en un análisis preliminar basado en el 47% de los eventos planeados totales para OS. La Tabla 16 y la Figura 1 sintetizan los resultados de eficacia.
Tabla 16. Resultados de eficacia - Ensayo 4
|
OPDIVO™ (n=210) |
Dacarbazina (n=208) |
|
|
Sobrevida global |
||
|
Muertes (%) |
50 (24) |
96 (46) |
|
Mediana, meses (IC 95%) |
No alcanzada |
10,8 (9,3; 12,1) |
|
Relación de riesgo (IC 95%)a |
0,42 (0,30; 0,60) |
|
|
Valor pb,c |
<0,0001 |
|
|
Sobrevida libre de progresión |
||
|
Eventos (%) |
108 (51) |
163 (78) |
|
Mediana, meses (IC 95%) |
5,1 (3,5; 10,8) |
2,2 (2,1; 2,4) |
|
Relación de riesgo (IC 95%)a |
0,43 (0,34; 0,56) |
|
|
Valor pb,c |
<0,0001 |
|
|
Tasa de respuesta objetiva |
34% |
9% |
|
(IC 95%) |
(28; 41) |
(5; 13) |
|
Tasa de respuesta completa |
4% |
1% |
|
Tasa de respuesta parcial |
30% |
8% |
|
a Basado en un modelo de riesgos proporcionales estratificado. b Basado en una prueba de rango logarítmico estratificada. c El valor p se compara con el valor alfa asignado de 0,0021 para este análisis preliminar. |
||
Figura 1: Curvas de Kaplan-Meier de la sobrevida global - Ensayo 4
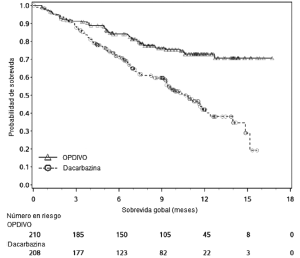
Al momento del análisis, el 88% (63/72) de los pacientes tratados con OPDIVO™ tenían respuestas en curso, incluidos 43 pacientes con respuestas en curso de 6 meses o más.
Ensayo 6. El Ensayo 6 fue un estudio multicéntrico, a doble ciego, que randomizó (1:1:1) pacientes con melanoma irrese-cable o metastásico, no tratado previamente a una de las siguientes ramas: OPDIVO™ más ipilimumab, OPDIVO™ o ipilimumab. Los pacientes debían haber completado el tratamiento adyuvante o neoadyuvante al menos 6 semanas antes de la randomización, no haber sido tratados antes con un anticuerpo anti-CTLA-4, y no presentar evidencia de metástasis cerebral activa, enfermedad autoinmune o afecciones médicas que requirieran inmunosupresión sistémica.
Los pacientes fueron randomizados para recibir:
• OPDIVO™ 1 mg/kg con ipilimumab 3 mg/kg cada 3 semanas por 4 dosis, seguido por OPDIVO™ 3 mg/kg como agente único cada 2 semanas (rama de OPDIVO™ más ipilimumab),
• OPDIVO™ 3 mg/kg cada 2 semanas (rama de OPDIVO™), o
• Ipilimumab 3 mg/kg cada 3 semanas por 4 dosis, seguido por placebo cada 2 semanas (rama de ipilimumab).
La randomización fue estratificada por expresión de PD-L1 (≥5% vs. <5% de expresión en membrana celular tumoral) según se determina mediante un ensayo clínico, estado de mutación BRAF V600, y estadio M según el sistema de estadi-ficación del Comité Conjunto Estadounidense sobre Cáncer (AJCC) (M0, M1a, M1b vs. M1c). Las evaluaciones tumorales se realizaron 12 semanas después de la randomización, luego cada 6 semanas durante el primer año, y cada 12 semanas de allí en adelante. Las mediciones de resultados de eficacia principales fueron la PFS evaluada por el investigador según los criterios RECIST v1.1 y la OS. Las mediciones de resultados de eficacia adicionales fueron la ORR confirmada y la duración de la respuesta. Un total de 945 pacientes fueron randomizados: 314 pacientes a la rama de OPDIVO™ más ipilimumab, 316 a la rama de OPDIVO™ y 315 a la rama de ipilimumab. Las características de la población del ensayo fueron las siguientes: mediana de edad 61 años (rango: 18 a 90); 65% de sexo masculino; 97% de raza blanca; estado funcional ECOG de 0 (73%) o 1 (27%). Las características de la enfermedad fueron: enfermedad en Estadio IV AJCC (93%); enfermedad M1c (58%); nivel elevado de LDH (36%); antecedentes de metástasis cerebral (4%); melanoma positivo para mutación BRAF V600 (32%); PD-L1 ≥5% de expresión en membrana celular tumoral según se determina por ensayo clínico (46%); y terapia adyuvante previa (22%). El Ensayo 6 demostró mejoras estadísticamente significativas en la PFS para los pacientes randomizados a cualquiera de las ramas que contenían OPDIVO™ en comparación con la rama de ipilimumab. Los resultados de eficacia se presentan en la Tabla 17 y la Figura 2.
Tabla 17. Resultados de eficacia en el Ensayo 6
|
OPDIVO™ más ipili-mumab (n=314) |
OPDIVO™ (n=316) |
Ipilimumab (n=315) |
|
|
Sobrevida libre de progresión |
|||
|
Progresión de la enfermedad o muerte |
151 |
174 |
234 |
|
Mediana en meses (IC 95%) |
11,5 (8,9; 16,7) |
6,9 (4,3; 9,5) |
2,9 (2,8; 3,4) |
|
Relación de riesgoa (vs. ipilimumab) |
0,42 |
0,57 |
|
|
(IC 95%) |
(0,34; 0,51) |
(0,47; 0,69) |
|
|
Valor pb,c |
<0,0001 |
<0,0001 |
|
|
Tasa de respuesta objetiva confirmada |
50% |
40% |
14% |
|
(IC 95%) |
(44; 55) |
(34; 46) |
(10; 18) |
|
Valor pd |
<0,0001 |
<0,0001 |
|
|
Respuesta completa |
8,9% |
8,5% |
1,9% |
|
Respuesta parcial |
41% |
31% |
12% |
|
Duración de la respuesta |
|||
|
Proporción ≥6 meses de duración |
76% |
74% |
63% |
|
Rango (meses) |
1,2+ a 15,8+ |
1,3+ a 14,6+ |
1,0+ a 13,8+ |
|
a Basado en un modelo de riesgos proporcionales estratificado. b Basado en una prueba de rango logarítmico estratificada. c El valor p se compara con 0,005 del valor alfa asignado para las comparaciones de PFS final entre tratamientos. d Basado en la prueba de Cochran-Mantel-Haenszel estratificada. |
|||
Figura 2: Sobrevida libre de progresión: Melanoma irresecable o metastásico - Ensayo 6
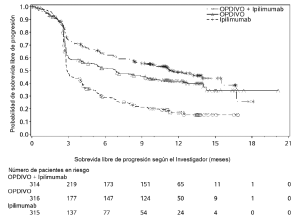
Las Figuras 3 y 4 presentan análisis exploratorios de eficacia por subgrupos de la PFS sobre la base de niveles de ex-presión definidos de PD-L1 determinados en muestras tumorales de archivo usando el ensayo PD-L1 IHC 28-8 pharmDx. Las muestras tumorales estuvieron disponibles para la evaluación retrospectiva en el 97% de la población del estudio; el estado de expresión de PD-L1 se determinó para el 89% de la población del estudio, mientras que en el 6% de los pacientes, la melanina impidió la evaluación del estado de expresión de PD-L1. El estado de expresión de PD-L1 fue des-conocido para el 5% de la población del estudio debido al retiro del consentimiento o a muestras faltantes.
Figura 3: Sobrevida libre de progresión por expresión de PD-L1 (<1%) - Ensayo 6
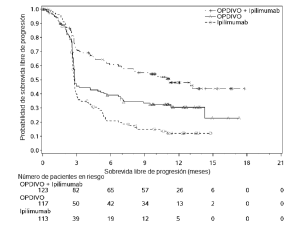
Figura 4: Sobrevida libre de progresión por expresión de PD-L1 (≥1%) - Ensayo 6
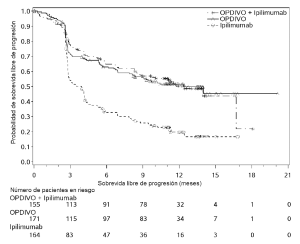
Los datos presentados en la figura a continuación sintetizan los resultados de análisis exploratorios que comparan las dos ramas que contienen OPDIVO™ en subgrupos definidos por expresión tumoral de PD-L1.
Figura 5: Gráfico de bosque: PFS basada en la expresión de PD-L1 que compara ramas que contienen OPDIVO™ - Ensayo 6
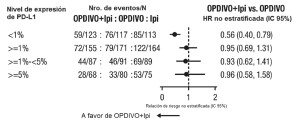
Cáncer de pulmón metastásico de células no pequeñas (NSCLC): Tratamiento de segunda línea del NSCLC escamoso metastásico. El Ensayo 2 fue un estudio randomizado (1:1), abierto, que enroló a 272 pacientes con NSCLC escamoso metastásico que habían experimentado progresión de la enfermedad durante o después de un régimen de quimioterapia dual previo basado en platino. Los pacientes recibieron OPDIVO™ (n=135) administrado por vía intravenosa a razón de 3 mg/kg cada 2 semanas o docetaxel (n=137) administrado por vía intravenosa a razón de 75 mg/m2 cada 3 semanas. La randomización se estratificó por tratamiento previo con paclitaxel versus otro tratamiento previo, y por región (EE.UU./Canadá versus Europa versus Resto del Mundo). Este estudio incluyó pacientes independientemente de su estado PD-L1. El ensayo excluyó pacientes con enfermedad autoinmune, afecciones médicas que requirieran inmunosupresión sistémica, enfermedad pulmonar intersticial sintomática o metástasis cerebral no tratada. Los pacientes con metástasis cerebral tratada eran elegibles si retornaban al estado neurológico basal al menos 2 semanas antes del enrolamiento, y no estaban recibiendo corticosteroides, o se encontraban con una dosis estable o en disminución de <10 mg de equivalentes de prednisona por día. Las primeras evaluaciones tumorales se llevaron a cabo 9 semanas después de la randomización y continuaron cada 6 semanas de allí en adelante. La medición del resultado de eficacia principal fue la sobrevida global (OS). Las mediciones adicionales de los resultados de eficacia fueron la ORR y la PFS evaluadas por el investigador. En el Ensayo 2, la mediana de la edad fue de 63 años (rango: 39 a 85), con un 44% de ≥65 años de edad y un 11% de ≥75 años de edad. La mayoría de los pacientes era de raza blanca (93%) y sexo masculino (76%); la mayoría de los pacientes fueron enrolados en Europa (57%), y los restantes en EE.UU./Canadá (32%) y el resto del mundo (11%). El estado funcional ECOG en condición basal fue 0 (24%) o 1 (76%), y el 92% de los pacientes eran exfumadores/ fumadores actuales. Las características basales de la enfermedad de la población según reportaron los investigadores fueron Estadio IIIb (19%), Estadio IV (80%) y metástasis cerebrales (6%). Todos los pacientes recibieron terapia previa con un régimen dual basado en platino, y el 99% de los pacientes tenían tumores con histología de células escamosas. El ensayo demostró una mejora estadísticamente significativa en la OS para los pacientes randomizados a OPDIVO™ en comparación con docetaxel en el análisis preliminar preespecificado cuando se observaron 199 eventos (el 86% de la cantidad planeada de eventos para el análisis final) (Tabla 18 y Figura 6).
Tabla 18. Resultados de eficacia en el Ensayo 2
|
OPDIVO™ (n=135) |
Docetaxel (n=137) |
|
|
Sobrevida global |
||
|
Muertes (%) |
86 (64%) |
113 (82%) |
|
Mediana (meses) (IC 95%) |
9,2 (7,3; 13,3) |
6,0 (5,1; 7,3) |
|
Relación de riesgo (IC 95%)a |
0,59 (0,44; 0,79) |
|
|
Valor pb,c |
0,0002 |
|
|
Tasa de respuesta objetiva |
27 (20%) |
12 (9%) |
|
(IC 95%) |
(14; 28) |
(5; 15) |
|
Valor pd |
0,0083 |
|
|
Respuesta completa |
1 (0,7%) |
0 |
|
Mediana de la duración de la respuesta, meses (IC 95%) |
NR (9,8; NR) |
8,4 (3,6; 10,8) |
|
Sobrevida libre de progresión |
||
|
Progresión de la enfermedad o muerte (%) |
105 (78%) |
122 (89%) |
|
Mediana (meses) |
3,5 |
2,8 |
|
Relación de riesgo (IC 95%)a |
0,62 (0,47; 0,81) |
|
|
Valor pb |
0,0004 |
|
|
a Basado en un modelo de riesgos proporcionales estratificado. b Basado en una prueba de rango logarítmico estratificada. c El valor p se compara con 0,0315 del valor alfa asignado para este análisis preliminar. d Basado en la prueba de Cochran-Mantel-Haenszel estratificada. |
||
Figura 6: Sobrevida global - Ensayo 2
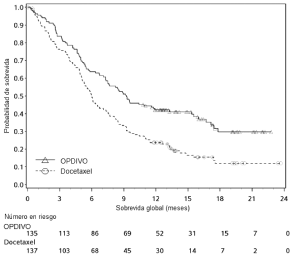
Las muestras tumorales de archivo fueron evaluadas retrospectivamente en cuanto a la expresión de PD-L1. En toda la población del estudio, el 17% (47/272) de los pacientes tuvieron resultados no cuantificables. Entre los 225 pacientes con resultados cuantificables, el 47% (106/225) tuvieron NSCLC de células escamosas PD-L1 negativo, definido como <1% de células tumorales que expresaban PD-L1, y el 53% (119/225) tuvieron NSCLC de células escamosas PD-L1 positivo, definido como ≥1% de células tumorales que expresaban PD-L1. En los análisis exploratorios preespecificados por subgrupos, las relaciones de riesgo para sobrevida fueron 0,58 (IC 95%: 0,37; 0,92) en el subgrupo PD-L1 negativo y 0,69 (IC 95%: 0,45; 1,05) en el subgrupo de NSCLC PD-L1 positivo.
Tratamiento de segunda línea del NSCLC no escamoso metastásico: El Ensayo 3 fue un estudio randomizado (1:1), de diseño abierto, de 582 pacientes con NSCLC no escamoso metastásico, que habían experimentado progresión de la enfermedad durante o después de un régimen de quimioterapia dual previo basado en platino. Se permitió una terapia dirigida previa apropiada en pacientes con mutación EGFR sensibilizante conocida o translocación de ALK. Los pacientes recibieron OPDIVO™ (n=292) administrado por vía intravenosa a razón de 3 mg/kg cada 2 semanas o docetaxel (n=290) administrado por vía intravenosa a razón de 75 mg/m2 cada 3 semanas. La randomización fue estratificada por terapia de mantenimiento previa (sí versus no) y número de terapias previas (1 versus 2). El ensayo excluyó a pacientes con enfermedad autoinmune, afecciones médicas que requirieran inmunosupresión sistémica, enfermedad pulmonar intersticial sintomática o metástasis cerebral no tratada. Los pacientes con metástasis cerebral tratada fueron elegibles si eran neurológicamente estables. Las primeras evaluaciones tumorales se llevaron a cabo 9 semanas después de la randomización y continuaron cada 6 semanas de allí en adelante. La principal medición del resultado de eficacia fue la sobrevida global (OS). Las mediciones adicionales del resultado de eficacia fueron la tasa de respuesta objetiva (ORR) y la sobrevida libre de progresión (PFS) evaluadas por el investigador. Asimismo, se llevaron a cabo análisis preespecificados en subgrupos definidos por la expresión de PD-L1. En el Ensayo 3, la mediana de la edad fue de 62 años (rango: 21 a 85), con el 42% de los pacientes ≥65 años y el 7% de los pacientes ≥75 años. La mayoría de los pacientes eran de raza blanca (92%) y sexo masculino (55%); la mayoría de los pacientes fueron enrolados en Europa (46%), seguida por EE.UU./Canadá (37%) y el resto del mundo (17%). El estado funcional ECOG basal fue 0 (31%) o 1 (69%), el 79% eran ex-fumadores o fumadores actuales, el 3,6% tenía NSCLC con redisposición de ALK, el 14% tenía NSCLC con mutación EGFR, y el 12% tenía metástasis cerebral previamente tratada. La terapia previa incluyó un régimen dual basado en platino (100%), y el 40% recibió terapia de mantenimiento como parte del régimen de primera línea. Los subtipos histológicos incluyeron adenocarcinoma (93%), de células grandes (2,4%) y broncoalveolar (0,9%). El Ensayo 3 demostró una mejora estadísticamente significativa en la OS para los pacientes randomizados a OPDIVO™ en comparación con docetaxel en el análisis preliminar preespecificado, cuando se observaron 413 eventos (93% del número planeado de eventos para el análisis final) (Tabla 19 y Figura 7).
Tabla 19. Resultados de eficacia en el Ensayo 3
|
OPDIVO™ (n=292) |
Docetaxel (n=290) |
|
|
Sobrevida global |
||
|
Muertes (%) |
190 (65%) |
223 (77%) |
|
Mediana (meses) (IC 95%) |
12,2 (9,7; 15,0) |
9,4 (8,0; 10,7) |
|
Relación de riesgo (IC 95%)a |
0,73 (0,60; 0,89) |
|
|
Valor pb,c |
0,0015 |
|
|
Tasa de respuesta objetiva |
56 (19%) |
36 (12%) |
|
(IC 95%) |
(15; 24) |
(9; 17) |
|
Valor pd |
0,02 |
|
|
Respuesta completa |
4 (1,4%) |
1 (0,3%) |
|
Mediana de la duración de la respuesta (meses) (IC 95%) |
17 (8,4; NR) |
6 (4,4; 7,0) |
|
Sobrevida libre de progresión |
||
|
Progresión de la enfermedad o muerte (%) |
234 (80%) |
245 (84%) |
|
Mediana (meses) |
2,3 |
4,2 |
|
Relación de riesgo (IC 95%)a |
0,92 (0,77; 1,11) |
|
|
Valor pb |
0,39 |
|
|
a Basado en un modelo de riesgos proporcionales estratificado. b Basado en una prueba de rango logarítmico estratificada. c El valor p se compara con 0,0408 del valor alfa asignado para este análisis preliminar. d Basado en la prueba de Cochran-Mantel-Haenszel estratificada. |
||
Figura 7. Sobrevida global - Ensayo 3
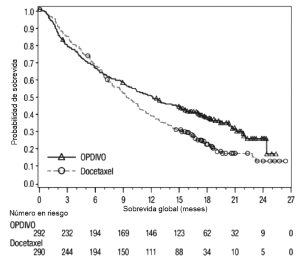
Las muestras tumorales de archivo fueron evaluadas en cuanto a la expresión de PD-L1 tras completar el ensayo. En toda la población de estudio, el 22% (127/582) de los pacientes tuvieron resultados no cuantificables. De los 455 pacientes restantes, la proporción de pacientes en subgrupos determinados retrospectivamente sobre la base del análisis de PD-L1 usando el ensayo PD-L1 IHC 28-8 pharmDx fueron: 46% (209/455) PD-L1 negativos, definidos como <1% de células tumorales que expresan PD-L1, y 54% (246/455) con expresión de PD-L1, definidos como ≥1% de células tumorales que expresan PD-L1. Entre los 246 pacientes con tumores que expresan PD-L1, el 26% (65/246) tenía ≥1%, pero <5% de células tumorales con tinción positiva, el 7% (16/246) tenía ≥5% pero <10% de células tumorales con tinción positiva, y el 67% (165/246) tenía un porcentaje mayor o igual al 10% de células tumorales con tinción positiva. La Figura 8 resume los resultados de análisis preespecificados de la sobrevida en subgrupos determinados por el porcentaje de células tumorales que expresan PD-L1. La Figura 9 resume los resultados de análisis preespecificados de la sobrevida libre de progresión en subgrupos determinados por el porcentaje de células tumorales que expresan PD-L1.
Figura 8. Diagrama de bosque: SG (OS, por sus siglas en inglés) basada en la expresión de PD-L1 - Ensayo 3
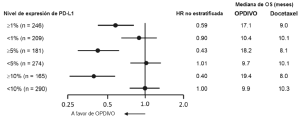
Figura 9. Diagrama de bosque: PFS basada en la expresión de PD-L1 - Ensayo 3
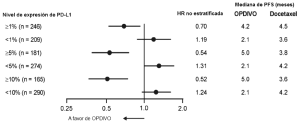
Carcinoma de células renales: El Ensayo 5 fue un estudio randomizado (1:1), de diseño abierto, en pacientes con RCC avanzado que experimentaron progresión de la enfermedad durante o después de uno o dos regímenes de terapia anti-angiogénica previa. Los pacientes debían tener un puntaje de rendimiento de Karnofsky (KPS) ≥70%, y fueron incluidos independientemente de su estado PD-L1. El Ensayo 5 excluyó a pacientes con cualquier antecedente o cuadro concurrente de metástasis cerebral, tratamiento previo con un inhibidor de mTOR, enfermedad autoinmune activa o afección médica que requiriera inmunosupresión sistémica. Los pacientes fueron estratificados por región, grupo de riesgo según el Memorial Sloan Kettering Cancer Center (MSKCC) y número de terapias anti-angiogénicas previas. Los pacientes fueron randomizados a OPDIVO™ (n=410) administrado por vía intravenosa a razón de 3 mg/kg cada 2 semanas o everolimus (n=411) administrado por vía oral a razón de 10 mg por día. La mediana de la edad fue de 62 años (rango: 18 a 88), con un 40% ≥65 años de edad y un 9% ≥75 años de edad. La mayoría de los pacientes eran de sexo masculino (75%) y raza blanca (88%), y el 34% y 66% de los pacientes tenían un KPS basal del 70% al 80% y del 90% al 100%, respectivamente. La mayoría de los pacientes (77%) fueron tratados con una terapia anti-angiogénica previa. La distribución de pacientes por grupos de riesgo MSKCC fue 34% favorable, 47% intermedio y 19% malo. Las primeras evaluaciones tumorales se llevaron a cabo 8 semanas después de la randomización y continuaron cada 8 semanas de allí en adelante durante el primer año y luego cada 12 semanas hasta la progresión o la discontinuación del tratamiento, lo que sucediera más tarde. El principal criterio de valoración de la eficacia fue la sobrevida global (OS). El ensayo demostró una mejora estadísticamente significativa en la OS para los pacientes randomizados a OPDIVO™ en comparación con everolimus en el análisis preliminar preespecificado cuando se observaron 398 eventos (el 70% del número planeado de eventos para el análisis final) (Tabla 20 y Figura 10). El beneficio de la OS se observó independientemente del nivel de expresión de PD-L1. Otros criterios de valoración incluyen las tasas de respuesta objetiva confirmada, que también se presentan en la Tabla 20.
Tabla 20. Resultados de eficacia - Ensayo 5
|
OPDIVO™ (n=410) |
Everolimus (n=411) |
|
|
Sobrevida global |
||
|
Muertes (%) |
183 (45) |
215 (52) |
|
Mediana de sobrevida en meses (IC 95%) |
25,0 (21,7; NE) |
19,6 (17,6; 23,1) |
|
Relación de riesgo (IC 95%)a |
0,73 (0,60; 0,89) |
|
|
Valor pb,c |
0,0018 |
|
|
Tasa de respuesta objetiva confirmada (IC 95%) |
21,5% (17,6; 25,8) |
3,9% (2,2; 6,2) |
|
Mediana de la duración de la respuesta en meses (IC 95%) |
23,0 (12,0; NE) |
13,7 (8,3; 21,9) |
|
Mediana del tiempo hasta el inicio de la respuesta confirmada en meses (mín., máx.) |
3,0 (1,4; 13,0) |
3,7 (1,5; 11,2) |
|
a Basado en un modelo de riesgos proporcionales estratificado. b Basado en una prueba de rango logarítmico estratificada. c El valor p se compara con 0,148 del valor alfa asignado para este análisis preliminar. |
||
Figura 10. Sobrevida global - Ensayo 5
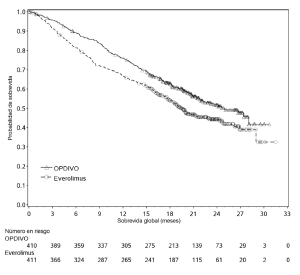
Linfoma de Hodgkin clásico: Dos estudios evaluaron la eficacia de OPDIVO™ como agente único en pacientes con cHL luego del fracaso del HSCT autólogo y de tratamiento con brentuximab vedotina post-trasplante. El Ensayo 7 fue un estudio de rama única, diseño abierto, multicéntrico y múltiples cohortes realizado en cHL. El Ensayo 8 fue un estudio de diseño abierto, multicéntrico, de escalación de dosis, que incluyó cHL. Ambos estudios incluyeron pacientes independientemente de su estado PD-L1 tumoral, y excluyeron pacientes con un estado funcional ECOG de 2 o más, enfermedad autoinmune, enfermedad pulmonar intersticial sintomática, transaminasas hepáticas en un nivel de más de 3 veces el límite superior del rango normal (ULN), clearance de creatinina inferior a 40 mL/min, HSCT alogénico previo, o irradiación de tórax dentro de las 24 semanas previas. Asimismo, ambos estudios requirieron una capacidad de difusión ajustada de los pulmones para monóxido de carbono (DLCO) de más del 60% en pacientes con toxicidad pulmonar previa. Los pacientes recibieron 3 mg/kg de OPDIVO™ administrado por vía intravenosa durante 60 minutos cada 2 semanas hasta la progresión de la enfermedad, el máximo beneficio clínico o una toxicidad inaceptable. Un ciclo consistió en una dosis. No se permitió la reducción de dosis. La eficacia fue evaluada por la tasa de respuesta objetiva (ORR), según la determinación de un comité independiente de revisión radiográfica (IRRC). Las mediciones de resultados adicionales incluyeron la duración de la respuesta. La eficacia fue evaluada en 95 pacientes en los Ensayos 7 y 8 combinados, quienes habían recibido brentuximab vedotina tras el fracaso del HSCT autólogo. La mediana de la edad fue de 37 años (rango: 18 a 72). La mayoría de los pacientes eran de sexo masculino (64%) y raza blanca (87%). Los pacientes habían recibido una mediana de 5 regímenes sistémicos previos (rango: 3 a 15). Los resultados se muestran en la Tabla 21. Los pacientes recibieron una mediana de 17 dosis de OPDIVO™ (rango: 3 a 48), con una mediana de la duración de la terapia de 8,3 meses (rango: 1,9 a 24 meses).
Tabla 21. Eficacia en cHL luego del HSCT autólogo y brentuximab vedotina
|
Ensayo 7 y Ensayo 8 (n=95) |
|
|
Tasa de respuesta objetiva, n (%)a (IC 95%) |
62 (65%) (55; 75) |
|
Tasa de remisión completa (IC 95%) |
7 (7%) (3; 15) |
|
Tasa de remisión parcial (IC 95%) |
55 (58%) (47; 68) |
|
Mediana de la duración de la respuesta (meses) (IC 95%) Rango |
8,7 (6,8; NE) 0,0+; 23,1+ |
|
Mediana del tiempo hasta la respuesta (meses) Rango |
2,1 0,7; 5,7 |
|
a Según los criterios revisados de 2007 del Grupo de Trabajo Internacional. |
|
Carcinoma metastásico o recurrente de células escamosas de cabeza y cuello (SCCHN): El Ensayo 9 fue un estudio randomizado (2:1), con control activo, abierto, que enroló pacientes con SCCHN metastásico o recurrente que habían experimentado progresión de la enfermedad durante o dentro de los 6 meses después de recibir una terapia previa basada en platino administrada en el entorno adyuvante, neoadyuvante, primario (irresecable localmente avanzado) o metastásico. El ensayo excluyó a pacientes con enfermedad autoinmune, afecciones médicas que requirieran inmunosupresión, carcinoma recurrente o metastásico de nasofaringe, carcinoma de células escamosas de histología primaria desconocida, de glándulas salivales o de histologías no escamosas (por ejemplo, melanoma de mucosa), o con metástasis cerebral no tratada. Los pacientes con metástasis cerebral tratada eran elegibles si presentaban una condición neurológica estable. Los pacientes fueron randomizados para recibir OPDIVO™ administrado por vía intravenosa (IV) en una dosis de 3 mg/kg cada 2 semanas, o un agente a elección del investigador:
• Una dosis de carga IV de cetuximab 400 mg/m2, seguida por 250 mg/m2 semanalmente,
• Metotrexato de 40 a 60 mg/m2 por vía IV semanalmente, o
• Docetaxel de 30 a 40 mg/m2 por vía IV semanalmente.
La randomización se estratificó por tratamiento previo con cetuximab (sí/no). Las primeras evaluaciones tumorales se llevaron a cabo 9 semanas después de la randomización y continuaron cada 6 semanas de allí en adelante. La principal medición de resultados de eficacia fue la OS. Las mediciones adicionales de los resultados de eficacia fueron la PFS y la ORR. En el Ensayo 9, un total de 361 pacientes fueron randomizados: 240 pacientes para recibir OPDIVO™ y 121 pacientes para recibir el agente a elección del investigador (el 45% recibieron docetaxel, el 43% recibieron metotrexato y el 12% recibieron cetuximab). La mediana de la edad fue de 60 años (rango: 28 a 83), con un 31% ≥65 años de edad; 83% eran blancos, 12% eran asiáticos y 4% eran negros; y 83% de sexo masculino. El estado funcional ECOG en condición basal fue 0 (20%) o 1 (78%); el 76% eran exfumadores o fumadores actuales, el 90% tenían enfermedad en Estadio IV; el 45% de los pacientes recibieron sólo una línea de terapia sistémica previa, mientras que el 55% restante recibió dos o más líneas de terapia sistémica previa; el 25% tenía tumores HPV p16 positivos, el 24% tenía tumores HPV p16 negativos, y el 51% tenía estado desconocido. El ensayo demostró una mejoría estadísticamente significativa en la OS para pacientes randomizados a OPDIVO™ en comparación con el agente a elección del investigador en un análisis preliminar especificado previamente (78% del número previsto de eventos para el análisis final). Los resultados se sobrevida se muestran en la Tabla 22 y la Figura 11. No hubo diferencias estadísticamente significativas entre ambas ramas para la PFS (HR=0,89; IC 95%: 0,70; 1,13) o la ORR (13,3% [IC 95%: 9,3; 18,3] versus 5,8% [IC 95%: 2,4; 11,6] para nivolumab y el agente a elección del investigador, respectivamente).
Tabla 22. Sobrevida global en el Ensayo 9
|
OPDIVO™ (n=240) |
Agente a elección del investi-gador (n=121) |
|
|
Sobrevida global |
||
|
Muertes (%) |
133 (55%) |
85 (70%) |
|
Mediana (meses) (IC 95%) |
7,5 (5,5; 9,1) |
5,1 (4,0; 6,0) |
|
Relación de riesgo (IC 95%)a |
0,70 (0,53; 0,92) |
|
|
Valor pb,c |
0,0101 |
|
|
a Basado en un modelo de riesgos proporcionales estratificado. b Basado en una prueba de rango logarítmico estratificada. c El valor p se compara con 0,0227 del alfa asignado para este análisis preliminar. |
||
Figura 11. Sobrevida global - Ensayo 9
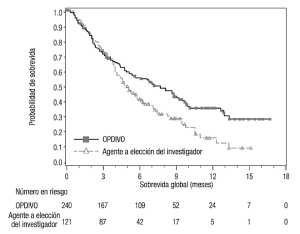
Las muestras tumorales de archivo fueron evaluadas retrospectivamente en cuanto a la expresión de PD-L1 usando el ensayo PD-L1 IHC 28-8 pharmDx. En toda la población de estudio, el 28% (101/361) de los pacientes tuvieron resultados no cuantificables. Entre los 260 pacientes con resultados cuantificables, el 43% (111/260) tuvieron SCCHN PD-L1 negativo, definido como <1% de células tumorales que expresaban PD-L1, y el 57% (149/260) tuvieron SCCHN PD-L1 positivo, definido como ≥1% de células tumorales que expresaban PD-L1. En análisis exploratorios preespecificados por subgrupos, la relación de riesgo para la sobrevida fue de 0,89 (IC 95%: 0,54; 1,45), con medianas de sobrevida de 5,7 y 5,8 meses para las ramas de nivolumab y quimioterapia, respectivamente, en el subgrupo PD-L1 negativo. La HR para la sobrevida fue de 0,55 (IC 95%: 0,36; 0,83), con medianas de sobrevida de 8,7 y 4,6 meses para las ramas de nivolumab y quimioterapia, respectivamente, en el subgrupo de SCCHN PD-L1 positivo.
Carcinoma urotelial: En el Ensayo 10, 270 pacientes con carcinoma urotelial localmente avanzado o metastásico que tuvieron progresión de la enfermedad durante o después de una quimioterapia con contenido de platino o que tuvieron progresión de la enfermedad dentro de los 12 meses del tratamiento con un régimen de quimioterapia neoadyuvante o adyuvante que contenía platino fueron tratados con OPDIVO™. Se excluyó a pacientes con metástasis cerebral o leptomeníngea activa, enfermedad autoinmune activa, afecciones médicas que requirieran inmunosupresión sistémica y estado funcional ECOG >1. Los pacientes recibieron una infusión intravenosa de 3 mg/kg de OPDIVO™ cada 2 semanas hasta la aparición de una toxicidad inaceptable o progresión radiográfica o clínica. Se llevaron a cabo evaluaciones de la respuesta tumoral cada 8 semanas durante las primeras 48 semanas y luego cada 12 semanas de allí en adelante. Las principales mediciones de resultados de eficacia incluyeron la tasa de respuesta objetiva (ORR) confirmada según evaluó un comité independiente de revisión radiográfica (IRRC) usando los Criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST v1.1) y la duración de la respuesta (DOR). La mediana de la edad fue de 66 años (rango de 38 a 90), el 78% eran varones, y el 86% de los pacientes eran de raza blanca. El 27% tenía carcinoma urotelial distinto de vejiga, y el 84% tenía metástasis visceral. El 34% de los pacientes tenían progresión de la enfermedad luego de una terapia neoadyuvante o adyuvante previa que contenía platino. El 29% de los pacientes habían recibido ≥2 regímenes sistémicos previos en el entorno metastásico. El 36% de los pacientes recibieron cisplatino previo solamente, el 23% recibieron carboplatino previo solamente, y el 7% fueron tratados con cisplatino y carboplatino en el entorno metastásico. El 46% de los pacientes tenían un estado funcional ECOG de 1. El 18% de los pacientes tenían un nivel de hemoglobina <10 g/dl, y el 28% de los pacientes tenían metástasis hepática en condición basal. Los pacientes fueron incluidos independientemente de su estado de PD-L1. Las muestras tumorales fueron evaluadas prospectivamente usando el en-sayo PD-L1 IHC 28-8 pharmDx en un laboratorio central, y los resultados se usaron para definir subgrupos para los análisis preespecificados. De los 270 pacients, el 46% se definieron como con expresión de PD-L1 ≥1% (definida como ≥1% de células tumorales que expresan PD-L1). El restante 54% de los pacientes fueron clasificados como con expresión de PD-L1 <1% (definida como <1% de células tumorales que expresan PD-L1). La ORR confirmada en todos los pacientes y los dos subgrupos de PD-L1 se sintetizan en la Tabla 23. La mediana del tiempo hasta la respuesta fue de 1,9 meses (rango: 1,6-7,2). En 77 pacientes que recibieron terapia sistémica previa solamente en el entorno neoadyuvante o adyuvante, la ORR fue del 23,4% (IC del 95%: 14,5%; 34,4%).
Tabla 23. Resultados de eficacia en el Ensayo 10
|
Todos los pacientes |
PD-L1 <1% |
PD-L1 ≥1% |
|
|
N=270 |
N=146 |
N=124 |
|
|
Tasa de respuesta objetiva confirmada, n (%) (IC del 95%) |
53 (19,6%) (15,1; 24,9) |
22 (15,1%) (9,7; 21,9) |
31 (25,0%) (17,7; 33,6) |
|
Tasa de respuesta completa |
7 (2,6%) |
1 (0,7%) |
6 (4,8%) |
|
Tasa de respuesta parcial |
46 (17,0%) |
21 (14,4%) |
25 (20,2%) |
|
Mediana de la duración de la respuestaa (meses) (rango) |
10,3 (1,9+; 12,0+) |
7,6 (3,7; 12,0+) |
NE (1,9+; 12,0+) |
|
a Estimado a partir de la curva de Kaplan-Meier. |
|||
TOXICOLOGÍA NO CLÍNICA:
Carcinogénesis, mutagénesis, disfunción de la fertilidad: No se han llevado a cabo estudios para evaluar el potencial de carcinogenicidad o genotoxicidad de nivolumab. No se han realizado estudios de fertilidad con nivolumab. En estudios de toxicología con dosis repetidas de 1 mes y 3 meses realizados en monos, no hubo efectos notables en los órganos reproductivos masculinos y femeninos; sin embargo, la mayoría de los animales de estos estudios no había alcanzado la madurez sexual.
Toxicología y/o farmacología en animales: En modelos de animales, la inhibición de la señalización de PD-1 aumentó la severidad de algunas infecciones e intensificó las respuestas inflamatorias. Los ratones PD-1 knockout infectados con M. tuberculosis exhiben una sobrevida marcadamente menor en comparación con los controles de tipo salvaje, que se correlacionó con un aumento de la proliferación bacteriana y las respuestas inflamatorias en estos animales. Los ratones PD-1 knockout también han demostrado una menor sobrevida luego de la infección con el virus de coriomeningitis linfocítica.
ADVERTENCIAS Y PRECAUCIONES:
Neumonitis mediada por la respuesta inmune: OPDIVO™™ puede causar neumonitis mediada por la respuesta inmune, que se define por requerir el uso de corticosteroides sin una etiología alternativa clara. Se han informado casos mortales. Monitorear a los pacientes en busca de signos y síntomas de neumonitis a través de imágenes radiográficas. Administrar corticosteroides en una dosis de 1 a 2 mg/kg/día de equivalentes de prednisona para los casos de neumonitis moderada (Grado 2) o más severa (Grados 3-4), seguido por la reducción gradual de los corticosteroides. Discontinuar OPDIVO™™ en forma permanente en caso de neumonitis severa (Grado 3) o potencialmente mortal (Grado 4), y suspender OPDIVO™™ hasta la resolución en caso de neu-monitis moderada (Grado 2) [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)].
OPDIVO™ como agente único. En pacientes que recibieron OPDIVO™ como agente único, se produjo neumonitis mediada por la respuesta inmune en el 3,1% (61/1994) de los pacientes. La mediana del tiempo hasta el inicio de la neumonitis mediada por la respuesta inmune fue de 3,5 meses (rango: 1 día a 22,3 meses). La neumonitis mediada por la respuesta inmune condujo a la discontinuación permanente de OPDIVO™ en el 1,1%, y a la suspensión de OPDIVO™ en el 1,3% de los pacientes. Aproximadamente el 89% de los pacientes con neumonitis recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 26 días (rango: 1 día a 6 meses). Se produjo la resolución completa de los síntomas luego de la disminución gradual de los corticosteroides en el 67% de los pacientes. Aproximadamente el 8% de los pacientes tuvieron recurrencia de la neumonitis tras la reiniciación de OPDIVO™. OPDIVO™ con ipilimumab. En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo neumonitis mediada por la respuesta inmune en el 6% (25/407) de los pacientes. La mediana del tiempo hasta el inicio de la neumonitis mediada por la respuesta inmune fue de 1,6 meses (rango: 24 días a 10,1 meses). La neumonitis mediada por la respuesta inmune condujo a la discontinuación permanente o a la suspensión de OPDIVO™ con ipilimumab en el 2,2% y 3,7% de los pacientes, respectivamente. Aproximadamente el 84% de los pacientes con neumonitis recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 30 días (rango: 5 días a 11,8 meses). Se produjo la resolución completa en el 68% de los pacientes. Aproximadamente el 13% de los pacientes tuvieron recurrencia de la neumonitis tras la reiniciación de OPDIVO™ con ipilimumab.
Colitis mediada por la respuesta inmune: OPDIVO™ puede causar colitis mediada por la respuesta inmune, definida por la necesidad de usar corticosteroides sin una etiología alternativa clara. Monitorear a los pacientes en busca de signos y síntomas de colitis. Administrar corticosteroides en una dosis de 1 a 2 mg/kg/día de equivalentes de prednisona, seguido por la reducción gradual de los corticosteroides, en caso de colitis severa (Grado 3) o potencialmente mortal (Grado 4). Administrar corticosteroides en una dosis de 0,5 a 1 mg/kg/día de equivalentes de prednisona, seguido por la disminución gradual de los corticosteroides en caso de colitis moderada (Grado 2) de más de 5 días de duración; si se produce un empeoramiento o no se registra mejoría a pesar de haber iniciado los corticosteroides, aumentar la dosis a 1 a 2 mg/ kg/día de equivalentes de prednisona. Suspender OPDIVO™ por colitis moderada o severa (Grado 2 o 3). Discontinuar permanentemente OPDIVO™ en caso de colitis potencialmente mortal (Grado 4) o colitis recurrente tras reiniciar OPDIVO™ [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. Cuando se administra en combinación con ipilimumab, suspender OPDIVO™ e ipilimumab por colitis moderada (Grado 2). Discontinuar permanentemente OPDIVO™ e ipilimumab en caso de colitis severa o potencialmente mortal (Gra-do 3 o 4), o por colitis recurrente [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. OPDIVO™ como agente único. En pacientes que recibieron OPDIVO™ como agente único, se produjo colitis mediada por la respuesta inmune en el 2,9% (58/1994) de los pacientes; la mediana del tiempo hasta el inicio fue de 5,3 meses (rango: 2 días a 20,9 meses). La colitis mediada por la respuesta inmune condujo a la discontinuación permanente de OPDIVO™ en el 0,7% y a la suspensión de OPDIVO™ en el 1% de los pacientes. Aproximadamente el 91% de los pacientes con colitis recibieron altas dosis de corticosteroi-des (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 23 días (rango: 1 día a 9,3 meses). Cuatro pacientes requirieron la adición de infliximab a los corticosteroides en altas dosis. Se produjo la resolución completa en el 74% de los pacientes. Aproximadamente el 16% de los pacientes tuvieron recurrencia de colitis tras la reiniciación de OPDIVO™. OPDIVO™ con ipilimumab. En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo colitis mediada por la respuesta inmune en el 26% (107/407) de los pacientes, incluidos tres casos mortales. La mediana del tiempo hasta el inicio de la colitis mediada por la respuesta inmune fue de 1,6 meses (rango: 3 días a 15,2 meses). La colitis mediada por la respuesta inmune condujo a la discontinuación permanente o a la suspensión de OPDIVO™ con ipilimumab en el 16% y 7% de los pacientes, respectivamente. Aproximadamente el 96% de los pacientes con colitis recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 1,1 meses (rango: 1 día a 12 meses). Aproximadamente el 23% de los pacientes requirieron la adición de infliximab a los corticosteroides en altas dosis. Se produjo la resolución completa en el 75% de los pacientes. Aproximadamente el 28% de los pacientes tuvieron recurrencia de la colitis tras la reiniciación de OPDIVO™ con ipilimumab.
Hepatitis mediada por la respuesta inmune: OPDIVO™ puede causar hepatitis mediada por la respuesta inmune, definida por la necesidad de usar corticosteroides sin una etiología alternativa clara. Monitorear a los pacientes por anormalidades en las pruebas hepáticas antes del trata-miento y periódicamente durante el tratamiento. Administrar corticosteroides en una dosis de 1 a 2 mg/kg/día de equiva-lentes de prednisona, seguido por la disminución gradual de los corticosteroides, en caso de elevación de transaminasas severa (Grado 3) o potencialmente mortal (Grado 4), con o sin elevación concomitante de la bilirrubina total. Admi-nistrar corticosteroides en una dosis de 0,5 a 1 mg/kg/día de equivalentes de prednisona en caso de elevación de tran-saminasas moderada (Grado 2). Suspender OPDIVO™ en caso de hepatitis moderada mediada por la respuesta inmune (Grado 2) y discontinuar permanentemente OPDIVO™ en casos severos (Grado 3) o con riesgo de muerte (Grado 4) [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. OPDIVO™ como agente único. En pacientes que recibieron OPDIVO™ como agente único, se produjo hepatitis mediada por la respuesta inmune en el 1,8% (35/1994) de los pacientes; la mediana del tiempo hasta el inicio fue de 3,3 meses (rango: 6 días a 9 meses). La hepatitis mediada por la respuesta inmune condujo a la discontinuación permanente de OPDIVO™ en el 0,7% y a la suspensión de OPDIVO™ en el 1% de los pacientes. Todos los pacientes con hepatitis recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona) durante una mediana de 23 días (rango: 1 día a 2 meses). Dos pacientes requirieron la adición de ácido micofenólico a los corticosteroides en altas dosis. Se produjo la resolución completa en el 74% de los pacientes. Aproximadamente el 29% de los pacientes tuvieron recurrencia de la hepatitis tras la reiniciación de OPDIVO™. OPDIVO™ con ipilimumab. En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo hepatitis mediada por la respuesta inmune en el 13% (51/407) de los pacientes; la mediana del tiempo hasta el inicio fue de 2,1 meses (rango: 15 días a 11 meses). La hepatitis mediada por la respuesta inmune condujo a la discontinuación permanente o a la suspensión de OPDIVO™ con ipilimumab en el 6% y el 5% de los pacientes, respectivamente. Aproximadamente el 92% de los pacientes con hepatitis recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 1,1 meses (rango: 1 día a 13,2 meses). Se produjo la resolución completa en el 75% de los pacientes. Aproximada-mente el 11% de los pacientes tuvieron recurrencia de la hepatitis tras la reiniciación de OPDIVO™ con ipilimumab.
Endocrinopatías mediadas por la respuesta inmune:
Hipofisitis: OPDIVO™ puede causar hipofisitis mediada por la respuesta inmune. Monitorear a los pacientes en busca de signos y síntomas de hipofisitis. Administrar terapia de reemplazo hormonal, según esté clínicamente indicado, y corticosteroides en una dosis de 1 mg/kg/día de equivalentes de prednisona, seguido por la disminución gradual de los corticosteroides, en caso de hipofisitis moderada (Grado 2) o mayor. Suspender OPDIVO™ por hipofisitis moderada (Grado 2) o severa (Grado 3). Discontinuar permanentemente OPDIVO™ por hipofisitis potencialmente mortal (Grado 4) [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. En pacientes que recibieron OPDIVO™ como agente único, se produjo hipofisitis en el 0,6% (12/1994) de los pacientes; la mediana del tiempo hasta el inicio fue de 4,9 meses (rango: 1,4 a 11 meses). La hipofisitis condujo a la discontinuación permanente de OPDIVO™ en el 0,1% y a la suspensión de OPDIVO™ en el 0,2% de los pacientes. Aproximadamente el 67% de los pacientes con hipofisitis recibieron terapia de reemplazo hormonal, y el 33% recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 14 días (rango: 5 a 26 días). En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo hipofisitis en el 9% (36/407) de los pacientes; la mediana del tiempo hasta el inicio fue de 2,7 meses (rango: 27 días a 5,5 meses). La hipofisitis condujo a la discontinuación permanente o a la suspensión de OPDIVO™ con ipilimumab en el 1,0% y el 3,9% de los pacientes, respectivamente. Aproximadamente el 75% de los pacientes con hipofisitis recibieron terapia de reemplazo hormonal, y el 56% recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 19 días (rango: 1 día a 2,0 meses).
Insuficiencia adrenal: OPDIVO™ puede causar insuficiencia adrenal mediada por la respuesta inmune. Monitorear a los pacientes en busca de signos y síntomas de insuficiencia adrenal. Administrar corticosteroides en una dosis de 1 a 2 mg/kg/día de equivalentes de prednisona, seguido por la disminución gradual de los corticosteroides, en caso de insuficiencia adrenal severa (Grado 3) o potencialmente mortal (Grado 4). Suspender OPDIVO™ en caso de insuficiencia adrenal moderada (Grado 2), y discontinuar permanentemente OPDIVO™ en caso de insuficiencia adrenal severa (Grado 3) o potencialmente mortal (Grado 4) [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. En pacientes que recibieron OPDIVO™ como agente único, se produjo insuficiencia adrenal en el 1% (20/1994) de los pacientes, y la mediana del tiempo hasta el inicio fue de 4,3 meses (rango: 15 días a 21 meses). La insuficiencia adrenal condujo a la discontinuación permanente de OPDIVO™ en el 0,1% y a la suspensión de OPDIVO™ en el 0,5% de los pacientes. Aproximadamente el 85% de los pacientes con insuficiencia adrenal recibieron terapia de reemplazo hormonal, y el 25% recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 11 días (rango: 1 día a 1 mes). En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo insuficiencia adrenal en el 5% (21/407) de los pacientes, y la mediana del tiempo hasta el inicio fue de 3,0 meses (rango: 21 días a 9,4 meses). La insuficiencia adrenal condujo a la discontinuación permanente o a la suspensión de OPDIVO™ con ipilimumab en el 0,5% y el 1,7% de los pacientes, respectivamente. Aproximadamente el 57% de los pacientes con insuficiencia adrenal recibieron terapia de reemplazo hormonal, y el 33% recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 9 días (rango: 1 día a 2,7 meses).
Hipotiroidismo e hipertiroidismo: OPDIVO™ puede causar trastornos tiroideos autoinmunes. Monitorear la función tiroidea antes y periódicamente durante el tratamiento con OPDIVO™. Administrar terapia de reemplazo hormonal en caso de hipotiroidismo. Iniciar tratamiento médico para el control del hipertiroidismo. No hay ajustes de dosis recomendados de OPDIVO™ para hipotiroidismo o hipertiroidismo. En pacientes que recibieron OPDIVO™ como agente único, se produjo hipotiroidismo o tiroiditis que condujo a hipotiroidismo en el 9% (171/1994) de los pacientes; la mediana del tiempo hasta el inicio fue de 2,9 meses (rango: 1 día a 16,6 meses). Aproximadamente el 79% de los pacientes con hipotiroidismo recibieron levotiroxina, y el 4% también requirieron corticosteroides. Se produjo la resolución en el 35% de los pacientes. Se produjo hipertiroidismo en el 2,7% (54/1994) de los pacientes que recibieron OPDIVO™ como agente único; la mediana del tiempo hasta el inicio fue de 1,5 meses (rango: 1 día a 14,2 meses). Aproximadamente el 26% de los pacientes con hipertiroidismo recibieron metimazol, el 9% recibieron carbimazol, el 4% recibieron propiltiouracilo, y el 9% recibieron corticosteroides. Se produjo la resolución en el 76% de los pacientes. En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo hipotiroidismo o tiroiditis que condujo a hipotiroidismo en el 22% (89/407) de los pacientes; la mediana del tiempo hasta el inicio fue de 2,1 meses (rango: 1 día a 10,1 meses). Aproximadamente el 73% de los pacientes con hipotiroidismo o tiroiditis recibieron levotiroxina. Se produjo la resolución en el 45% de los pacientes. Se produjo hipertiroidismo en el 8% (34/407) de los pacientes que recibieron OPDIVO™ con ipilimumab: la mediana del tiempo hasta el inicio fue de 23 días (rango: 3 días a 3,7 meses). Aproximadamente el 29% de los pacientes con hipertiroidismo recibieron metimazol, y el 24% recibieron carbimazol. Se produjo la resolución en el 94% de los pacientes.
Diabetes mellitus tipo 1: OPDIVO™ puede causar diabetes mellitus tipo 1. Monitorear a los pacientes para detectar la aparición de hiperglucemia. Suspender OPDIVO™ en caso de hiperglucemia severa (Grado 3) hasta alcanzar el control metabólico. Discontinuar OPDIVO™ en forma permanente en caso de hiperglucemia con riesgo de muerte (Grado 4) [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. En pacientes que recibieron OPDIVO™ como agente único, se produjo diabetes en el 0,9% (17/1994) de los pacientes, incluidos dos casos de cetoacidosis diabética. La mediana del tiempo hasta el inicio fue de 4,4 meses (rango: 15 días a 22 meses). En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo diabetes en el 1,5% (6/407) de los pacientes; la mediana del tiempo hasta el inicio fue de 2,5 meses (rango: 1,3 a 4,4 meses). OPDIVO™ con ipilimumab se suspendió en un paciente y se discontinuó permanentemente en un segundo paciente que desarrolló diabetes.
Nefritis y disfunción renal mediadas por la respuesta inmune: OPDIVO™ puede causar nefritis mediada por la respuesta inmune, definida como disfunción renal o aumento de creatinina ≥Grado 2, requisito de corticosteroides y ausencia de una etiología alternativa clara. Monitorear a los pacientes para detectar una elevación de la creatinina sérica antes y periódicamente durante el tratamiento. Administrar corticosteroides en una dosis de 1 a 2 mg/kg/día de equivalentes de prednisona, seguido por la disminución gradual de los corticosteroides en caso de aumento de creatinina sérica potencialmente mortal (Grado 4). Administrar corticosteroides en una dosis de 0,5 a 1 mg/kg/día de equivalentes de prednisona en caso de aumento de creatinina sérica moderado (Grado 2) o severo (Grado 3); si el cuadro empeora o no se produce una mejoría, aumentar la dosis de corticosteroides a 1 a 2 mg/kg/día de equivalentes de prednisona. Suspender OPDIVO™ en caso de aumento de creatinina sérica moderado (Gra-do 2) o severo (Grado 3). Discontinuar permanentemente OPDIVO™ en caso de aumento de creatinina sérica potencial-mente mortal (Grado 4) [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada) y Reacciones adversas (Experiencia en estudios clínicos)]. OPDIVO™ como agente único. En pacientes que recibieron OPDIVO™ como agente único, se produjo nefritis y disfunción renal mediadas por la respuesta inmune en el 1,2% (23/1994) de los pacientes; la mediana del tiempo hasta el inicio fue de 4,6 meses (rango: 23 días a 12,3 meses). La nefritis y disfunción renal mediadas por la respuesta inmune condujeron a la discontinuación permanente de OPDIVO™ en el 0,3% y a la suspensión de OPDIVO™ en el 0,8% de los pacientes. Todos los pacientes recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 21 días (rango: 1 día a 15,4 meses). Se produjo la resolución completa en el 48% de los pacientes. Ningún paciente presentó recurrencia de nefritis o disfunción renal tras la reiniciación de OPDIVO™. OPDIVO™ con ipilimumab. En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo nefritis y disfunción renal mediadas por la respuesta inmune en el 2,2% (9/407) de los pacientes; la mediana del tiempo hasta el inicio fue de 2,7 meses (rango: 9 días a 7,9 meses). La nefritis y disfunción renal mediadas por la respuesta inmune condujeron a la discontinuación permanente o a la sus-pensión de OPDIVO™ con ipilimumab en el 0,7% y el 0,5% de los pacientes, respectivamente. Aproximadamente el 67% de los pacientes recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) duran-te una mediana de 13,5 días (rango: 1 día a 1,1 meses). Se produjo la resolución completa en todos los pacientes. Dos pacientes reiniciaron OPDIVO™ con ipilimumab, sin recurrencia de la nefritis o disfunción renal.
Reacciones adversas dérmicas mediadas por la respuesta inmune: OPDIVO™ puede causar erupción mediada por la respuesta inmune, incluido síndrome de Stevens-Johnson (SJS) y necrólisis epidérmica tóxica (TEN), algunos casos con desenlace mortal. En caso de signos o síntomas de SJS o TEN, suspender OPDIVO™ y remitir al paciente para recibir atención especializada para su evaluación y tratamiento. Si se confirma SJS o TEN, discontinuar OPDIVO™ en forma permanente. En caso de erupción mediada por la respuesta inmune, administrar corticosteroides en una dosis de 1 a 2 mg/kg/día de equivalentes de prednisona, seguido por la disminución gradual de los corticosteroides, en caso de erupción severa (Grado 3) o potencialmente mortal (Grado 4). Suspender OPDIVO™ en caso de erupción severa (Grado 3) y discontinuar permanentemente OPDIVO™ en caso de erupción potencialmente mortal (Grado 4) [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. OPDIVO™ como agente único. En pacientes que recibieron OPDIVO™ como agente único, se produjo erupción mediada por la respuesta inmune en el 9% (171/1994) de los pacientes; la mediana del tiempo hasta el inicio fue de 2,8 meses (rango: <1 día a 25,8 meses). La erupción mediada por la respuesta inmune condujo a la discontinuación permanente de OPDIVO™ en el 0,3% y a la suspensión de OPDIVO™ en el 0,8% de los pacientes. Aproximadamente el 16% de los pacientes con erupción recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 12 días (rango: 1 día a 8,9 meses), y el 85% recibieron corticosteroides tópicos. Se produjo la resolución completa en el 48% de los pacientes. Se produjo la recurrencia de la erupción en el 1,4% de los pacientes que reiniciaron OPDIVO™ tras la resolución de la erupción. OPDIVO™ con ipilimumab. En pacientes que recibieron OPDIVO™ con ipilimumab, se produjo erupción mediada por la respuesta inmune en el 22,6% (92/407) de los pacientes; la mediana del tiempo hasta el inicio fue de 18 días (rango: 1 día a 9,7 meses). La erupción mediada por la respuesta inmune condujo a la discontinuación permanente o a la suspensión de OPDIVO™ con ipilimumab en el 0,5% y el 3,9% de los pacientes, respectivamente. Aproximadamente el 17% de los pacientes con erupción recibieron altas dosis de corticosteroides (al menos 40 mg de equivalentes de prednisona por día) durante una mediana de 14 días (rango: 2 días a 4,7 meses). Se produjo la resolución completa en el 47% de los pacientes. Aproximadamente el 6% de los pacientes que reiniciaron OPDIVO™ e ipilimumab tras la resolución presentaron recurrencia de la erupción.
Encefalitis mediada por la respuesta inmune: OPDIVO™ puede causar encefalitis mediada por la respuesta inmune, sin una etiología alternativa clara. La evaluación de pacientes con síntomas neurológicos puede incluir, entre otras cosas, consulta con un neurólogo, estudio por resonancia magnética del cerebro y punción lumbar. Suspender OPDIVO™ en pacientes con signos o síntomas neurológicos de inicio reciente moderados a severos, y evaluar para descartar causas infecciosas u otras causas de deterioro neurológico moderado a severo. Si se descartan otras etiologías, administrar corticosteroides en una dosis de 1 a 2 mg/kg/día de equivalentes de prednisona para pacientes con encefalitis mediada por la respuesta inmune, seguido por la disminución gradual de los corticosteroides. Discontinuar permanentemente OPDIVO™ por encefalitis mediada por la respuesta inmune [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. OPDIVO™ como agente único. En los pacientes que recibieron OPDIVO™ como agente único, se produjo encefalitis en el 0,2% (3/1994). Se produjo encefalitis límbica fatal en un paciente luego de 7,2 meses de exposición a pesar de la discontinuación de OPDIVO™ y la administración de corticosteroides. En los otros dos pacientes, se produjo encefalitis post-HSCT alogénico [véase Advertencias y Precauciones (Complicaciones del HSCT alogénico tras OPDIVO™)]. OPDIVO™ con ipilimumab. Se produjo encefalitis en un paciente que recibió OPDIVO™ con ipilimumab (0,2%) luego de 1,7 meses de exposición.
Otras reacciones adversas mediadas por la respuesta inmune: OPDIVO™ puede causar otras reacciones adversas mediadas por la respuesta inmune clínicamente significativas. Pueden ocurrir reacciones adversas mediadas por la respuesta inmune luego de la discontinuación de la terapia con OPDIVO™. Para cualquier presunta reacción adversa mediada por la respuesta inmune, excluir otras causas. En función de la severidad de la reacción adversa, discontinuar permanentemente o suspender OPDIVO™, administrar corticosteroides en altas dosis y, si corresponde, iniciar terapia de reemplazo hormonal. Tras la mejoría hasta alcanzar el Grado 1 o menor, disminuir los corticosteroides gradualmente y continuar con dicha disminución durante al menos 1 mes. Considerar reiniciar OPDIVO™ luego de completar la disminución gradual de los corticosteroides, según la severidad del evento [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. En los ensayos clínicos de OPDIVO™ administrado como agente único o en combinación con ipilimumab, se produjeron las siguientes reacciones adversas mediadas por la respuesta inmune, clínica-mente significativas en menos del 1,0% de los pacientes que recibieron OPDIVO™: uveítis, iritis, pancreatitis, parálisis facial y del nervio abducens, desmielinización, polimialgia reumática, neuropatía autoinmune, síndrome de Guillain-Barré, hipopituitarismo, síndrome de respuesta inflamatoria sistémica, gastritis, duodenitis, sarcoidosis, linfadenitis necrotizante histiocítica (linfadenitis de Kikuchi), miositis, miocarditis, rabdomiólisis, disfunción motriz, vasculitis y síndrome miasténico.
Reacciones a la perfusión intravenosa: OPDIVO™ puede causar reacciones severas a la perfusión intravenosa, que se han reportado en menos del 1,0% de los pacientes en los ensayos clínicos. Discontinuar OPDIVO™ en pacientes con reacciones severas o potencialmente mortales a la perfusión intravenosa. Interrumpir o demorar la velocidad de perfusión intravenosa en pacientes con reacciones leves o moderadas, a la administración [véase Posología/Dosis y Administración (Modificaciones de la dosis recomendada)]. OPDIVO™ como agente único. En los pacientes que recibieron OPDIVO™ como agente único, se produjeron reacciones relacionadas con la perfusión en el 6,4% (127/1994) de los pacientes. OPDIVO™ con ipilimumab. En los pacientes que recibieron OPDIVO™ con ipilimumab, se produjeron reacciones relacionadas con la perfusión en el 2,5% (10/407) de los pacientes.
Complicaciones del HSCT alogénico tras OPDIVO™: Se produjeron complicaciones, incluidos eventos fatales, en pacientes que recibieron HSCT alogénico tras recibir OPDIVO™. Se evaluaron los resultados en 17 pacientes de los Ensayos 8 y 9 que fueron sometidos a HSCT alogénico tras discontinuar OPDIVO™ (15 con condicionamiento de intensidad reducida y 2 con acondicionamiento mieloablativo). La mediana de la edad al momento del HSCT fue de 33 (rango: 18 a 56), y se había administrado una mediana de 9 dosis de OPDIVO™ (rango: 4 a 16). Seis de 17 pacientes (35%) murieron a raíz de complicaciones del HSCT alogénico después de recibir OPDIVO™. Cinco muertes se produjeron en el contexto de enfermedad de injerto versus huésped (GVHD) severa o refractaria. Se reportó GVHD aguda de Grado 3 o mayor en 5/17 pacientes (29%). Se reportó GVHD hiperaguda, definida como GVHD ocurrida dentro de los 14 días luego de la perfusión de células madre, en 2 pacientes (20%). Se reportó síndrome febril que requirió esteroides, sin una causa infecciosa identificada, en seis pacientes (35%) dentro de las primeras 6 semanas post-trasplante, con cinco pacientes que respondieron a los esteroides. Se reportaron dos casos de encefalitis: un caso de encefalitis linfocítica de Grado 3 sin una causa infecciosa identificada, que se produjo y se resolvió con esteroides, y un caso de encefalitis presuntamente de origen viral de Grado 3 que se resolvió con trata-miento antiviral. Se produjo enfermedad veno-oclusiva (VOD) hepática en un paciente, quien recibió HSCT alogénico con condicionamiento de intensidad reducida, y murió por GVHD y falla multi-orgánica. Otros casos de VOD hepática tras el HSCT alogénico con condicionamiento de intensidad reducida también se han reportado en pacientes con linfoma que recibieron un anticuerpo bloqueador del receptor de PD-1 antes del trasplante. También se han reportado casos de GVHD hiperaguda fatal. Estas complicaciones podrían ocurrir, a pesar de la terapia interviniente, entre el bloqueo de PD-1 y el HSCT alogénico. Seguir a los pacientes de cerca para obtener evidencia temprana de complicaciones relacionadas con el trasplante, tales como GVHD hiperaguda, GVHD aguda severa (Grado 3 a 4), síndrome febril que requiere esteroides, VOD hepática, y otras reacciones adversas mediadas por la respuesta inmune, e intervenir pronta-mente.
Toxicidad embriofetal: Sobre la base de su mecanismo de acción y los datos de estudios en animales, OPDIVO™ puede causar daño fetal cuando se administra a una mujer embarazada. En los estudios de reproducción en animales, la administración de nivolumab a monos cynomolgus desde el comienzo de la organogénesis hasta el parto dio como resultado un aumento de los abor-tos y las muertes prematuras de la cría. Advertir a las mujeres embarazadas sobre el potencial riesgo para el feto. Aconsejar a las mujeres en edad fértil que usen un método anticonceptivo efectivo durante el tratamiento con un régimen que contiene OPDIVO™ y durante al menos 5 meses después de la última dosis de OPDIVO™ [véase Uso en Poblaciones específicas (Embarazo. Resumen del riesgo, Hombres y mujeres en edad fértil)].
USO EN POBLACIONES ESPECÍFICAS:
Embarazo. Resumen del riesgo: Sobre la base de su mecanismo de acción y los datos de estudios realizados en animales, OPDIVO™ puede causar daño fetal cuando es administrado a una mujer embarazada [véase Farmacología clínica (Mecanismo de acción)]. En los estudios de reproducción animal, la administración de nivolumab a monos cynomolgus desde el inicio de la organogénesis hasta el parto dio como resultado un aumento de los abortos y las muertes prematuras de la cría [véase Datos]. Se sabe que la IgG4 humana atraviesa la barrera placentaria, y el nivolumab es una inmunoglobulina G4 (IgG4); por lo tanto, nivolumab tiene el potencial de ser transmitido de la madre al feto en desarrollo. Los efectos de OPDIVO™ probablemente sean mayores durante el segundo y el tercer trimestre del embarazo. No se dispone de datos en humanos que informen sobre el riesgo asociado con el fármaco. Advertir a las mujeres en edad fértil sobre el riesgo potencial para el feto. Se desconoce el riesgo de referencia de defectos graves del nacimiento y aborto espontáneo para la población indicada; sin embargo, el riesgo de referencia en la población general de EE.UU. de defectos graves del nacimiento es del 2% a 4% y de aborto espontáneo es del 15% a 20% de los embarazos clínicamente reconocidos. Datos. Datos en animales. Una función central de la vía PD-1/PD-L1 es preservar el embarazo, manteniendo la tolerancia inmune materna al feto. Se ha demostrado en modelos murinos de embarazo que el bloqueo de la señalización de PD-L1 altera la tolerancia al feto y aumenta los casos de pérdida del feto. Los efectos de nivolumab sobre el desarrollo prenatal y postnatal fueron evaluados en monos que recibieron nivolumab dos veces por semana desde el inicio de la organogénesis hasta el parto, a niveles de exposición entre 9 y 42 veces mayores que aquellos observados con la dosis clínica de 3 mg/kg de nivolumab (sobre la base del AUC). La administración de nivolumab dio como resultado un aumento no relacionado con la dosis de los abortos espontáneos y un aumento de las muertes neonatales. Sobre la base de su mecanismo de acción, la exposición fetal a nivolumab puede aumentar el riesgo de desarrollar trastornos mediados por la respuesta inmune o de alterar la respuesta inmune normal, y se han informado trastornos mediados por la respuesta inmune en ratones PD-1 knockout. En las crías sobrevivientes de monos cynomolgus tratados con nivolumab (18 de 32, en comparación con 11 de 16 crías expuestas al vehículo), no hubo malformaciones evidentes ni efectos sobre los parámetros de neuroconducta, inmunológicos o de patología clínica durante el período postnatal de 6 meses.
Mujeres en período de lactancia:
Resumen del riesgo. Se desconoce si OPDIVO™ está presente en la leche humana. Dado que muchos fármacos, incluidos los anticuerpos, se excretan en la leche humana y debido al potencial de reacciones adversas serias en los lactantes a raíz de OPDIVO™, se debe advertir a las mujeres que discontinúen la lactancia durante el tratamiento con OPDIVO™.
Hombres y mujeres en edad fértil:
Anticoncepción: En función de su mecanismo de acción, OPDIVO™ puede causar daño fetal cuando es administrado a una mujer embarazada [véase Uso en poblaciones específicas (Embarazo. Resumen del riesgo)]. Indicar a las mujeres en edad fértil que deben usar un método anticonceptivo efectivo durante el tratamiento con OPDIVO™ y durante al menos 5 meses luego de la última dosis de OPDIVO™.
Uso pediátrico: No se ha establecido la seguridad y la efectividad de OPDIVO™ en pacientes pediátricos.
Uso geriátrico: De los 1359 pacientes randomizados para recibir OPDIVO™ como agente único en los Ensayos 2 a 6, el 39% tenía 65 años de edad o más, y el 9% tenía 75 años o más. No se reportaron diferencias generales en la seguridad ni la efectividad entre pacientes geriátricos y pacientes más jóvenes. En el Ensayo 10 (carcinoma urotelial), el 55% de los pacientes tenía 65 años de edad o más, y el 14% tenía 75 años o más. No se reportaron diferencias generales en la seguridad ni la efectividad entre pacientes geriátricos y pacientes más jóvenes. Los Ensayos 1, 7, 8 y 9 no incluyeron suficiente cantidad de pacientes de 65 años de edad o más para determinar si responden de manera diferente a los pacientes más jóvenes. De los 314 pacientes randomizados para recibir OPDIVO™ administrado con ipilimumab en el Ensayo 6, el 41% tenía 65 años de edad o más, y el 11% tenía 75 años de edad o más. No se reportaron diferencias generales en la seguridad ni la efectividad entre pacientes geriátricos y pacientes más jóvenes.
Insuficiencia renal: Sobre la base de un análisis de farmacocinética poblacional, no se recomienda ningún ajuste de dosis en pacientes con deterioro renal [véase Farmacología clínica (Farmacocinética)].
Insuficiencia hepática: Sobre la base de un análisis de farmacocinética poblacional, no se recomienda ningún ajuste de dosis en pacientes con deterioro hepático leve. OPDIVO™ no ha sido estudiado en pacientes con deterioro hepático moderado o severo [véase Farmacología clínica (Farmacocinética)].
SOBREDOSIS:
No se cuenta con información sobre la sobredosis de OPDIVO™.
DESCRIPCIÓN:
Nivolumab es un anticuerpo monoclonal humano que bloquea la interacción entre PD-1 y sus ligandos, PD-L1 y PD-L2. Nivolumab es una inmunoglobulina IgG4 kappa, cuya masa molecular se calcula en 146 kDa. OPDIVO™ es un líquido esteril, libre de conservantes, no pirogénico, de transparente a opalescente, entre incoloro y color amarillo pálido, que puede contener partículas livianas (pocas). OPDIVO™ inyectable para perfusión intravenosa se presenta en viales para dosis única. Cada mililitro de solución de OPDIVO™ contiene nivolumab 10 mg, manitol (30 mg), ácido pentético (0,008 mg), polisorbato 80 (0,2 mg), cloruro de sodio (2,92 mg), citrato de sodio dihidrato (5,88 mg) y agua para uso inyectable, USP. Puede contener ácido clorhídrico y/o hidróxido de sodio para ajustar el pH a 6.
FARMACOLOGÍA CLÍNICA:
Mecanismo de acción: La unión de los ligandos de PD-1, PD-L1 y PD-L2, al receptor de PD-1 hallado en las células T inhibe la proliferación de células T y la producción de citoquinas. En algunos tumores se produce la sobrerregulación de los ligandos de PD-1, y la señalización a través de esta vía puede contribuir a la inhibición de la vigilancia inmunológica activa de células T de los tumores. Nivolumab es un anticuerpo monoclonal humano de inmunoglobulina G4 (IgG4) que se une al receptor de PD-1 y bloquea su interacción con PD-L1 y PD-L2, liberando la inhibición mediada por la vía de PD-1 de la respuesta inmune, incluida la respuesta inmune antitumoral. En modelos de tumor en ratones singénicos, el bloqueo de la actividad de PD-1 dio como resultado una disminución del crecimiento tumoral. La inhibición mediada por la combinación de nivolumab (anti-PD-1) e ipilimumab (anti-CTLA-4) da por resultado una mejor función de las células T, que es mayor que los efectos de cualquiera de los anticuerpos solo, y causa mejores respuestas antitumorales en el melanoma metastásico. En modelos de tumor singénico murino, el bloqueo dual de PD-1 y CTLA-4 dio como resultado una mayor actividad antitumoral.
Farmacodinámica: Sobre la base de las relaciones de dosis/exposición en cuanto a eficacia y seguridad, no existen diferencias clínicamente significativas en la seguridad y la eficacia entre una dosis de nivolumab de 240 mg y una de 3 mg/kg cada 2 semanas en pacientes con melanoma, NSCLC, RCC y carcinoma urotelial.
Farmacocinética: La farmacocinética (PK) de nivolumab fue evaluada usando un enfoque de PK poblacional tanto para OPDIVO™ como agente único como para OPDIVO™ combinado con ipilimumab.
OPDIVO™ como agente único: La PK de nivolumab como agente único fue estudiada en pacientes dentro de un rango de dosis de 0,1 a 20 mg/kg administra-dos en forma de dosis única o de dosis múltiples de OPDIVO™ cada 2 o 3 semanas. El aclaramiento (CL) de nivolumab disminuye con el tiempo, con una reducción máxima media (% de coeficiente de variación [CV%]) desde los valores basales de aproximadamente 24,5% (47,6%), lo que da como resultado un aclaramiento en estado estacionario (CLss) por media geométrica (CV%) de 8,2 mL/h, 53,9%); la disminución del CLss no se considera clínicamente relevante. El volumen de distribución por media geométrica en estado estacionario (Vss) (CV%) es de 6,8 L (27,3%), y la vida media de eliminación por media geométrica (t1/2) es de 25 días (77,5%). Las concentraciones de nivolumab en estado estacionario se alcanzaron a las 12 semanas aproximadamente cuando el fármaco se administró a razón de 3 mg/kg cada 2 semanas, y la acumulación sistémica fue de aproximadamente 3,7 veces. La exposición a nivolumab aumentó de manera proporcional a la dosis dentro del rango de dosis de 0,1 a 10 mg/kg administrados cada 2 semanas.
OPDIVO™ con ipilimumab: El CL por media geométrica (CV%), el Vss y la vida media terminal de nivolumab fueron 10,0 mL/h (50,3%), 7,92 L (30,1%) y 24,8 días (94,3%), respectivamente. Cuando se administró en combinación, el CL de nivolumab aumentó un 24%, mientras que no hubo efecto sobre el aclaramiento de ipilimumab. Cuando se administró en combinación, el aclaramiento de nivolumab aumentó un 42% en presencia de anticuerpos antinivolumab. No hubo efecto de los anticuerpos antiipilimumab sobre el aclaramiento de ipilimumab.
Poblaciones específicas: El análisis de PK poblacional sugirió que los siguientes factores no tenían un efecto clínicamente importante sobre el aclaramiento de nivolumab: edad (29 a 87 años), peso (35 a 160 kg), género, raza, LDH basal, expresión de PD-L1, tipo de tumor sólido, tamaño del tumor, deterioro renal y deterioro hepático leve.
Deterioro renal: El efecto del deterioro renal sobre el aclaramiento de nivolumab fue evaluado por un análisis de PK poblacional en pacientes con deterioro renal leve (eGFR 60 a 89 mL/min/1,73 m2; n=313), moderado (eGFR 30 a 59 mL/min/1,73 m2; n=140) o severo (eGFR 15 a 29 mL/min/1,73 m2; n=3). No se observaron diferencias clínicamente importantes en el aclaramiento de nivolumab entre pacientes con deterioro renal y pacientes con función renal normal [véase Uso en poblaciones específicas (Insuficiencia renal)].
Deterioro hepático: El efecto del deterioro hepático sobre el aclaramiento de nivolumab fue evaluado por análisis de PK poblacional en pacientes con deterioro hepático leve (bilirrubina total [TB] menor o igual al límite superior del rango normal [ULN] y AST mayor al ULN o TB menor a 1 a 1,5 veces el ULN y cualquier valor de AST; n=92). No se hallaron diferencias clínicamente importantes en el aclaramiento de nivolumab entre pacientes con deterioros hepáticos leves y pacientes con función hepática normal. Nivolumab no ha sido estudiado en pacientes con deterioro hepático moderado (TB mayor a 1,5 a 3 veces el ULN y cualquier valor de AST) o severo (TB mayor a 3 veces el ULN y cualquier valor de AST) [véase Uso en poblaciones específicas (Insuficiencia hepática.)].
INFORMACIÓN DE ASESORAMIENTO PARA EL PACIENTE:
Informar a los pacientes sobre el riesgo de reacciones adversas mediadas por la respuesta inmune que pueden requerir tratamiento con corticosteroides y la suspensión o discontinuación de OPDIVO™, las cuales incluyen:
• Neumonitis: Indicar a los pacientes que deben comunicarse de inmediato con su médico por cualquier cuadro nuevo o empeoramiento de tos, dolor de pecho o falta de aliento [véase Advertencias y precauciones (Neumonitis mediada por la respuesta inmune)].
• Colitis: Indicar a los pacientes que deben comunicarse de inmediato con su médico en caso de diarrea o do-lor abdominal severo [véase Advertencias y precauciones (Colitis mediada por la respuesta inmune)].
• Hepatitis: Indicar a los pacientes que deben comunicarse de inmediato con su médico en caso de ictericia, náuseas o vómitos severos, dolor en el cuadrante derecho del abdomen, letargo, o formación fácil de hematomas o hemorragias [véase Advertencias y precauciones (Hepatitis mediada por la respuesta inmune)].
• Endocrinopatías: Indicar a los pacientes que deben comunicarse de inmediato con su médico en caso de signos o síntomas de hipofisitis, insuficiencia adrenal, hipotiroidismo, hipertiroidismo, y diabetes mellitus [véase Advertencias y precauciones (Endocrinopatías mediadas por la respuesta inmune)].
• Nefritis y disfunción renal: Indicar a los pacientes que deben comunicarse de inmediato con su médico en caso de signos o síntomas de nefritis, que incluyen disminución de la producción de orina, sangre en la orina, hinchazón de tobillos, pérdida del apetito, y cualquier otro síntoma de disfunción renal [véase Advertencias y precauciones (Nefritis y disfunción renal mediadas por la respuesta inmune)].
• Reacciones adversas dérmicas: Indicar a los pacientes que deben comunicarse de inmediato con su médico en caso de erupción [véase Advertencias y precauciones (Reacciones adversas dérmicas mediadas por la respuesta inmune)].
• Encefalitis: Indicar a los pacientes que deben comunicarse de inmediato con su médico en caso de signos o síntomas neurológicos de encefalitis [véase Advertencias y precauciones (Reacciones adversas dérmicas mediadas por la respuesta inmune)].
• Reacciones a la perfusión intravenosa: Advertir a los pacientes sobre el potencial riesgo de reacciones a la perfusión intravenosa [véase Advertencias y precauciones (Reacciones a la perfusión intravenosa)].
• Complicaciones del HSCT alogénico tras OPDIVO™: Advertir a los pacientes sobre el potencial riesgo de complicaciones post-trasplante [véase Advertencias y precauciones (Complicaciones del HSCT alogénico tras OPDIVO)].
• Mujeres en edad fértil: Advertir a las mujeres en edad fértil sobre el potencial riesgo para el feto y que deben informar a su médico en caso de embarazo o sospecha de embarazo [véase Advertencias y precauciones (Toxicidad embriofetal), Uso en poblaciones específicas (Embarazo. Resumen del riesgo.)]. Indicar a las mujeres en edad fértil que deben usar un método anticonceptivo efectivo durante el tratamiento con OPDIVO™ y durante al menos 5 meses luego de la última dosis de OPDIVO™ [véase Uso en poblaciones específicas (Hombres y mujeres en edad fértil)].
• Lactancia: Advertir a las mujeres que no deben amamantar mientras toman OPDIVO™ [véase Uso en poblaciones específicas (Mujeres en período de lactancia)].
CONDICIONES DE ALMACENAMIENTO:
Conservar el vial en condiciones de refrigeración a 2 °C - 8 °C.
Proteger de la luz conservándolo en su envase original hasta el momento de usarlo.
Luego de su preparación conservar la solución en alguna de las siguientes condiciones:
• A temperatura ambiente durante no más de 4 horas desde el momento de la preparación. Esto incluye el almacenamiento a temperatura ambiente de la solución diluida en el recipiente IV y el tiempo para la administración por perfusión intravenosa, o
- En condiciones de refrigeración a 2 °C - 8 °C durante no más de 24 horas desde el momento de la preparación.
No congelar. No agitar.
Información especial para el desecho: No almacenar la solución no utilizada para su reutilización. La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa del establecimiento de salud.
¿CUÁL ES LA INFORMACIÓN MÁS IMPORTANTE QUE DEBO SABER SOBRE OPDIVO™?:
OPDIVO™ es un medicamento que puede tratar su melanoma, cáncer de pulmón, cáncer de riñón, cáncer sanguíneo, cáncer de cabeza y cuello, o cáncer de vejiga al trabajar junto con su sistema inmunológico. OPDIVO™ puede hacer que su sistema inmunológico ataque órganos y tejidos normales en muchas áreas del cuerpo, y puede afectar la manera en que éstos funcionan. Estos problemas a veces pueden volverse serios o poner en riesgo la vida, y pueden resultar mortales. Estos problemas pueden ocurrir en cualquier momento durante el tratamiento, o incluso después de finalizado el tratamiento. Algunos de estos problemas pueden ocurrir con más frecuencia cuando OPDIVO™ se usa en combinación con ipilimumab.
Llame a su médico o visítelo de inmediato si desarrolla cualquier síntoma de los siguientes problemas, o si estos síntomas empeoran:
Problemas pulmonares (neumonitis): Los síntomas de la neumonitis pueden incluir:
• Nuevo cuadro o empeoramiento de tos
• Dolor de pecho
• Disnea (sensación de falta de aire)
Problemas intestinales (colitis), que pueden llevar a desgarros o perforaciones en los intestinos: Los signos y síntomas de colitis pueden incluir:
• Diarrea (heces blandas) o más movimientos intestinales que los habituales
• Sangre en las heces o con aspecto alquitranado, heces oscuras y pegajosas
• Dolor o molestia severa en el área del estómago (abdomen)
Problemas hepáticos (hepatitis): Los signos y síntomas de hepatitis pueden incluir:
• Color amarillento de la piel o la parte blanca de los ojos
• Náuseas o vómitos severos
• Dolor en el lado derecho del área del estómago (abdomen)
• Aturdimiento
• Orina oscura (color té)
• Sangrado o formación de hematomas más fácilmente de lo habitual
• Sensación de menor apetito del habitual
Problemas de las glándulas hormonales (en especial, tiroides, pituitaria, glándulas suprarrenales y páncreas): Los signos y síntomas de que sus glándulas hormonales no están funcionando bien pueden incluir los siguientes:
• Dolores de cabeza que no desaparecen o dolores de cabeza inusuales
• Cansancio extremo
• Aumento de peso o pérdida de peso
• Mareos o desmayos
• Cambios en el estado de ánimo o la conducta, tales como menor impulso sexual, irritabilidad u olvidos
• Pérdida del cabello
• Sensación de frío
• Constipación
• Voz más grave
• Sed excesiva o abundante cantidad de orina
Problemas renales, incluidas nefritis y falla renal: Los signos de problemas renales pueden incluir:
• Menor producción de orina
• Sangre en la orina
• Hinchazón de tobillos
• Pérdida del apetito
Problemas de piel: Los signos de estos problemas pueden incluir:
• Erupción
• Picazón
• Aparición de ampollas en la piel
• Úlceras en la boca u otras membranas mucosas
Inflamación del cerebro (encefalitis): Los signos y síntomas de encefalitis pueden incluir:
• Dolor de cabeza
• Fiebre
• Cansancio o debilidad
• Confusión
• Problemas de la memoria
• Somnolencia
• Ilusiones ópticas o auditivas (alucinaciones)
• Convulsiones
• Rigidez en el cuello
Problemas en otros órganos: Los signos de estos problemas pueden incluir:
• Alteración de la visión
• Dolor muscular o articular severo o persistente
• Debilidad muscular severa
La obtención de tratamiento médico inmediato puede evitar que estos problemas se tornen más serios.
Su médico lo analizará para detectar estos problemas durante el tratamiento con OPDIVO™. Podrá tratarlo con corticoste-roides o terapia de reemplazo hormonal. Si usted presenta efectos secundarios severos, su médico quizá también deba demorar o interrumpir completamente el tratamiento con OPDIVO™.


