JARDIANCE 10 MG
EMPAGLIFLOZINA
Comprimidos recubiertos
Blíster de PVC aluminio , Comprimidos recubiertos , 10 Miligramos
1 3 Blíster, 10 Comprimidos recubiertos, 10 Miligramos
1 3 Blíster, 10 Comprimidos recubiertos, 10 Miligramos
Blíster de PVC aluminio , Comprimidos recubiertos , 10 Miligramos
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada COMPRIMIDO contiene: 10 mg de empagliflozina.
Para consultar la lista completa de excipientes, ver Lista de excipientes.
LISTA DE EXCIPIENTES
• Lactosa monohidratada.
• Celulosa microcristalina.
• Hidroxipropilcelulosa.
• Croscarmelosa sódica.
• Sílice coloidal anhidro.
• Estearato de magnesio.
• Opadry amarillo.
INDICACIONES TERAPÉUTICAS: JARDIANCE está indicado en el tratamiento de la diabetes mellitus tipo 2 para mejorar el control glucémico en adultos en:
• Monoterapia: Cuando la dieta y el ejercicio por sí solos no proporcionen un control glucémico adecuado en pacientes en los que el uso de metformina se considera inapropiado debido a una intolerancia.
• Tratamiento adicional en combinación: En combinación con otros medicamentos hipoglucemiantes, incluida la insulina, cuando estos, junto con la dieta y el ejercicio, no proporcionen un control glucémico adecuado.
INDICACIONES Y USO:
JARDIANCE® está indicado:
• Como complemento de la dieta y el ejercicio para mejorar el control glucémico en adultos con diabetes mellitus tipo 2.
• Para reducir el riesgo de muerte cardiovascular en pacientes adultos con diabetes mellitus tipo 2 y enfermedad cardiovascular establecida.
Limitaciones del uso: No se recomienda el uso de JARDIANCE® en pacientes con diabetes tipo 1 y tampoco para el tratamiento de la cetoacidosis diabética.
DATOS FARMACÉUTICOS:
Incompatibilidades: No procede.
Período de validez: 3 años
“No consumir el producto una vez alcanzada la fecha de vencimiento indicada en los rotulados”.
Precauciones especiales de conservación: No requiere condiciones especiales de conservación.
“Consulte a su médico o farmacéutico, según proceda, para cualquier aclaración sobre la utilización del producto”.
Precauciones especiales de almacenamiento:
Manténgase a una temperatura no superior a 30 °C.
Precauciones especiales de eliminación: La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Manténgase fuera del alcance de los niños
Venta con receta médica.
PROPIEDADES FARMACOCINÉTICAS
Absorción: La farmacocinética de empagliflozina se ha caracterizado extensivamente en voluntarios sanos y en pacientes con diabetes tipo 2. Tras la administración oral, empagliflozina se absorbió rápidamente, alcanzándose concentraciones plasmáticas máximas a una mediana de tmax de 1,5 horas después de la dosis. Después, las concentraciones plasmáticas disminuyeron de forma bifásica con una fase de distribución rápida y una fase terminal relativamente lenta. La AUC plasmática media en estado estacionario y la Cmax fueron de 1870 nmol.h y 259 nmol/l con empagliflozina 10 mg y de 4740 nmol.h y 687 nmol/l con empagliflozina 25 mg una vez al día. La exposición sistémica de empagliflozina aumentó de forma proporcional a la dosis. Los parámetros farmacocinéticos de dosis única y de estado estacionario de empagliflozina fueron similares, lo que sugiere una farmacocinética lineal respecto al tiempo. No hubo diferencias clínicamente relevantes en la farmacocinética de empagliflozina entre los voluntarios sanos y los pacientes con diabetes tipo 2.
La administración de empagliflozina 25 mg después de la ingesta de una comida rica en grasas y alta en calorías dio lugar a una exposición ligeramente inferior; la AUC disminuyó en aproximadamente el 16% y la Cmax disminuyó en aproximadamente un 37% en comparación con las condiciones de ayunas. El efecto observado de los alimentos sobre la farmacocinética de la empagliflozina no se consideró clínicamente relevante, por lo que la empagliflozina puede administrarse con o sin alimentos.
Distribución: En base al análisis farmacocinético poblacional, se calculó que el volumen de distribución aparente en estado estacionario era de 73,8 litros. Después de la administración de una solución oral de [14C]-empagliflozina a voluntarios sanos, la distribución de los glóbulos rojos fue de aproximadamente un 37% y la unión a proteínas plasmáticas, del 86%.
Biotransformación: No se detectaron metabolitos importantes de empagliflozina en el plasma humano y los metabolitos más abundantes fueron tres conjugados glucurónidos (2-, 3- y 6-O glucurónido). La exposición sistémica de cada metabolito fue inferior al 10% del material total relacionado con el fármaco. Los estudios in vitro sugirieron que la principal vía metabólica de empagliflozina en humanos es la glucuronidación por las uridina 5"-difosfo-glucuronosiltransferasas UGT2B7, UGT1A3, UGT1A8 y UGT1A9.
Eliminación: En base al análisis farmacocinético poblacional, se calculó que la semivida de eliminación terminal aparente de la empagliflozina era de 12,4 horas y que el aclaramiento oral aparente era de 10,6 l/hora.
Las variabilidades interindividual y residual para el aclaramiento oral de empagliflozina fueron del 39,1% y del 35,8%, respectivamente. Con una pauta posológica de una vez al día, las concentraciones plasmáticas de empagliflozina en estado estacionario se alcanzaron en la quinta dosis.
Acorde con la semivida, en el estado estacionario se observó una acumulación de hasta el 22% de acumulación con respecto al AUC plasmática. Tras la administración de una solución oral de [14C]-empagliflozina a voluntarios sanos, aproximadamente el 96% de la radioactividad relacionada con el fármaco se eliminó por las heces (41%) o la orina (54%). La mayor parte de la radioactividad relacionada con el fármaco que se recuperó en las heces fue el fármaco original sin cambios y aproximadamente la mitad de la radioactividad relacionada con el fármaco excretado por la orina fue el fármaco original sin cambios.
Poblaciones especiales:
• Insuficiencia renal: En pacientes con insuficiencia renal leve, moderada o grave (TFGe <30 - <90 ml/min/1,73 m2) y pacientes con fallo renal/enfermedad renal terminal (ERT), el AUC de empagliflozina aumentó en aproximadamente el 18%, 20%, 66% y 48% respectivamente en comparación con los sujetos con una función renal normal. Los niveles plasmáticos máximos de empagliflozina fueron similares en los sujetos con insuficiencia renal moderada y fallo renal/ERT en comparación con los pacientes con una función renal normal. Los niveles plasmáticos máximos de empagliflozina fueron aproximadamente un 20% más altos en los sujetos con insuficiencia renal leve y grave en comparación con los sujetos con una función renal normal. El análisis farmacocinético poblacional demostró que el aclaramiento oral aparente de empagliflozina disminuía con un descenso en el TFGe, dando lugar a un aumento en la exposición al fármaco.
• Insuficiencia hepática: En sujetos con insuficiencia hepática leve, moderada y grave según la clasificación Child-Pugh, el AUC de la empagliflozina aumentó en aproximadamente el 23%, 47%, y 75% y la Cmax aumentó en el 4%, 23% y 48% respectivamente en comparación con los sujetos con función hepática normal.
• Índice de masa corporal: El índice de masa corporal no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de empagliflozina según un análisis farmacocinético poblacional. En este análisis, se estimó que el AUC era un 5,82%, 10,4% y 17,3% inferior en sujetos con un IMC de 30, 35 y 45 kg/m2 respectivamente, en comparación con sujetos con un índice de masa corporal de 25 kg/m2.
• Sexo: El sexo no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de empagliflozina según un análisis farmacocinético poblacional.
• Raza: En el análisis farmacocinético poblacional, se estimó que el AUC era un 13,5% más alta en asiáticos con un índice de masa corporal de 25 kg/m2 en comparación con los no asiáticos con un índice de masa corporal de 25 kg/m2.
• Pacientes de edad avanzada: La edad no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de empagliflozina según un análisis farmacocinético poblacional.
• Pacientes pediátricos: No se han realizado ensayos que caractericen la farmacocinética de empagliflozina en pacientes pediátricos.
PROPIEDADES FARMACODINÁMICAS
Grupo farmacoterapéutico: Fármacos usados en la diabetes, otros fármacos hipoglucemiantes, excl. insulinas, código ATC: A10BX12
Mecanismo de acción: Empagliflozina es un inhibidor competitivo reversible y selectivo altamente potente (IC50 de 1,3 nmol) del cotransportador de sodio-glucosa tipo 2 (SGLT2). Empagliflozina no inhibe otros transportadores de glucosa importantes para el transporte de glucosa a los tejidos periféricos y es 5000 veces más selectivo para el SGLT2 que para el SGLT1, el transportador más importante responsable de la absorción de glucosa en el intestino. El SGLT2 se encuentra altamente expresado en el riñón, mientras que la expresión en otros tejidos es inexistente o muy baja. Es responsable, como transportador predominante, de la reabsorción de glucosa tras la filtración glomerular para devolverla a la circulación. En los pacientes con diabetes tipo 2 e hiperglucemia, se filtra y reabsorbe una mayor cantidad de glucosa.
La empagliflozina mejora el control glucémico en pacientes con diabetes tipo 2 al reducir la reabsorción renal de glucosa. La cantidad de glucosa eliminada por el riñón mediante este mecanismo glucurético depende de la concentración de glucosa en sangre y de la TFG. La inhibición del SLGT2 en pacientes con diabetes tipo 2 e hiperglucemia conduce a un exceso de excreción de glucosa por la orina.
En pacientes con diabetes tipo 2, la excreción de glucosa por la orina aumentó inmediatamente después de la primera dosis de empagliflozina y se mantuvo continua durante el intervalo de administración de 24 horas. El aumento en la eliminación de glucosa por la orina se mantuvo al final del periodo de tratamiento de 4 semanas, con un promedio de aproximadamente 78 g/día. El aumento en la eliminación de glucosa por la orina dio lugar a una reducción inmediata de los niveles de glucosa plasmática en pacientes con diabetes tipo 2.
La empagliflozina mejora los niveles de glucosa en plasma, tanto en ayunas como posprandiales. El mecanismo de acción de la empagliflozina es independiente de la función de las células beta y de la vía de la insulina, y esto contribuye a un bajo riesgo de hipoglucemia. Se observó una mejora de los marcadores indirectos de la función de las células beta, incluido el Modelo Homeostático ß para la evaluación de la resistencia a la insulina (HOMA-ß). Además, la excreción de glucosa por la orina desencadena una pérdida de calorías, que se asocia a una pérdida de grasa corporal y a una reducción de peso corporal. La glucosuria observada con empagliflozina se ve acompañada por una leve diuresis, que puede contribuir a la reducción sostenida y moderada de la presión arterial.
Eficacia clínica y seguridad: En 10 ensayos clínicos doble ciego, controlados con placebo y con activo, se trataron un total de 11 250 pacientes con diabetes tipo 2, de los cuales 6015 recibieron empagliflozina (empagliflozina 10 mg: 3021 pacientes; empagliflozina 25 mg: 3994 pacientes). Cuatro estudios tuvieron una duración del tratamiento de 24 semanas; las extensiones de estos y otros estudios incluyeron a pacientes expuestos a empagliflozina hasta un máximo de 102 semanas.
El tratamiento con empagliflozina como monoterapia y en combinación con metformina, pioglitazona, una sulfonilurea, inhibidores de la DPP-4 e insulina proporcionó mejoras clínicamente significativas en HbA1c, glucosa plasmática en ayunas (GPA), peso corporal y presión arterial sistólica y diastólica.
Con la administración de empagliflozina 25 mg, una mayor proporción de pacientes logró el objetivo de alcanzar una HbA1c inferior al 7% y hubo menos pacientes que necesitaron un rescate glucémico en comparación con empagliflozina 10 mg y placebo. Se asoció un nivel basal más alto de HbA1c con una mayor reducción de la HbA1c.
Monoterapia: Se evaluó la eficacia y la seguridad de empagliflozina en monoterapia en un ensayo doble ciego controlado con placebo y con activo de 24 semanas de duración, con pacientes que no habían recibidoningún tratamiento anterior. El tratamiento con empagliflozina dio lugar a una reducción estadísticamente significativa (p<0,0001) de HbA1c en comparación con el placebo (Tabla 2), así como a una disminución clínicamente significativa de la GPA.
En un análisis pre-especificado de pacientes (N = 201) con un nivel basal de la HbA1c ≥8,5%, el tratamiento provocó una reducción de la HbA1c respecto a los valores basales del -1,44% para empagliflozina 10 mg, del -1,43% para empagliflozina 25 mg y del -1,04% para sitagliptina, así como un aumento del 0,01% para el placebo.
En la extensión doble ciego controlada con placebo de este ensayo, las reducciones en la HbA1c, el peso corporal y la presión arterial se mantuvieron hasta la semana 52.
|
Tabla 2: Resultados de eficacia de un ensayo controlado con placebo de 24 semanas de duración de empagliflozina como monoterapiaa |
||||
|
Placebo |
JARDIANCE |
Sitagliptina |
||
|
10 mg |
25 mg |
100 mg |
||
|
N |
228 |
224 |
224 |
223 |
|
HbA1c (%) |
||||
|
Valor basal (media) |
7,91 |
7,87 |
7,86 |
7,85 |
|
Cambio respecto al valor basal1 |
0,08 |
-0,66 |
-0,78 |
-0,66 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-0,74* |
-0,85* |
-0,73 |
|
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de la HbA1c ≥7%2 |
12,0 |
35,3 |
43,6 |
37,5 |
|
N |
228 |
224 |
224 |
223 |
|
Peso corporal (kg) |
||||
|
Valor basal (media) |
78,23 |
78,35 |
77,80 |
79,31 |
|
Cambio respecto al valor basal1 |
-0,33 |
-2,26 |
-2,48 |
0,18 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
1,93* |
-2,15* |
0,52 |
|
|
N |
228 |
224 |
224 |
223 |
|
PAS (mm Hg)4 |
||||
|
Valor basal (media) |
130,4 |
133,0 |
129,9 |
132,5 |
|
Cambio respecto al valor basal1 |
-0,3 |
-2,9 |
-3,7 |
0,5 |
|
Diferencia respecto al placebo1 (IC del 95%) |
-2,6* |
-3,4* |
0,8 |
|
|
a. Grupo completo de análisis (FAS) utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico. 1. Media ajustada respecto al valor basal. 2. No evaluado en cuanto a significación estadística debido al resultado del procedimiento de prueba confirmatorio secuencial. 3. IC del 95%. 4. LOCF, valores censurados estadísticamente después del rescate antihipertensivo. *valor p<0,0001 |
||||
Tratamiento de combinación:
• Empagliflozina como tratamiento en combinación con metformina, sulfonilurea, pioglitazona: La empagliflozina como tratamiento de adición a metformina, metformina y una sulfonilurea o pioglitazona con o sin metformina dio lugar a reducciones estadísticamente significativas (p<0,0001) en la HbA1c y en el peso corporal en comparación con el placebo (Tabla 3). Además, también dio lugar a una reducción clínicamente significativa en la GPA y la presión arterial sistólica y diastólica en comparación con el placebo.
En la extensión doble ciego controlada con placebo de estos estudios, la reducción en la HbA1c, el peso corporal y la presión arterial se mantuvo hasta la semana 52.
|
Tabla 3: Resultados de eficacia de los ensayos controlados con placebo de 24 semanasa |
|||
|
Placebo |
JARDIANCE |
||
|
10 mg |
25 mg |
||
|
Tratamiento de adición a metformina |
|||
|
N |
207 |
217 |
213 |
|
HbA1c (%) |
|||
|
Valor basal (media) |
7,90 |
7,94 |
7,86 |
|
Cambio respecto al valor basal1 |
-0,13 |
-0,70 |
-0,77 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-0,57* |
-0,64* |
|
|
N |
184 |
199 |
191 |
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de la HbA1c ≥7%2 |
12,5 |
37,7 |
38,7 |
|
N |
207 |
217 |
213 |
|
Peso corporal (kg) |
|||
|
Valor basal (media) |
79,73 |
81,59 |
82,21 |
|
Cambio respecto al valor basal1 |
-0,45 |
-2,08 |
-2,46 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-1,63* |
-2,01* |
|
|
N |
207 |
217 |
213 |
|
PAS (mm Hg)2 |
|||
|
Valor basal (media) |
128,6 |
129,6 |
130,0 |
|
Cambio respecto al valor basal1 |
-0,4 |
-4,5 |
-5,2 |
|
Diferencia respecto al placebo1 (IC del 95%) |
-4,1* (-6,2, -2,1) |
-4,8* |
|
|
Tratamiento de adición a metformina y una sulfonilurea |
|||
|
N |
225 |
225 |
216 |
|
HbA1c (%) |
|||
|
Valor basal (media) |
8,15 |
8,07 |
8,10 |
|
Cambio respecto al valor basal1 |
-0,17 |
-0,82 |
-0,77 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-0,64* |
-0,59* |
|
|
N |
216 |
209 |
202 |
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de la HbA1c ≥7%2 |
9,3 |
26,3 |
32,2 |
|
N |
225 |
225 |
216 |
|
Peso corporal (kg) |
|||
|
Valor basal (media) |
76,23 |
77,08 |
77,50 |
|
Cambio respecto al valor basal1 |
-0,39 |
-2,16 |
-2,39 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-1,76* (-2,25, -1,28) |
-1,99* |
|
|
N |
225 |
225 |
216 |
|
PAS (mm Hg)2 |
|||
|
Valor basal (media) |
128,8 |
128,7 |
129,3 |
|
Cambio respecto al valor basal1 |
-1,4 |
-4,1 |
-3,5 |
|
Diferencia respecto al placebo1 (IC del 95%) |
-2,7 (-4,6, -0,8) |
-2,1 |
|
|
Tratamiento con pioglitazona +/- metformina |
|||
|
N |
165 |
165 |
168 |
|
HbA1c (%) |
|||
|
Cambio respecto al valor basal |
-0,11 |
-0,59 |
-0,72 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-0,48* (-0,69, -0,27) |
-0,61* |
|
|
N |
155 |
151 |
160 |
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de la HbA1c ≥7%2 |
7,7 |
24 |
30 |
|
N |
165 |
165 |
168 |
|
Peso corporal (kg) |
|||
|
Valor basal (media) |
78,1 |
77,97 |
78,93 |
|
Cambio respecto al valor basal1 |
0,34 |
-1,62 |
-1,47 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-1,95* (-2,64, -1,27) |
-1,81* |
|
|
N |
165 |
165 |
168 |
|
Valor basal (media) |
125,7 |
126,5 |
126 |
|
Cambio respecto al valor basal1 |
0,7 |
-3,1 |
-4,0 |
|
Diferencia respecto al placebo1 (IC del 95%) |
-3,9 (-6,23, -1,50) |
-4,7 |
|
|
a. Grupo completo de análisis (FAS) utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico. 1. Media ajustada respecto al valor basal. 2. No evaluado en cuanto a significación estadística debido al resultado del procedimiento de prueba confirmatorio secuencial. 3. LOCF, valores censurados estadísticamente después del rescate antihipertensivo. * valor p<0,0001 |
|||
• Datos de empagliflozina de 24 meses, como tratamiento de adición a metformina en comparación con glimepirida: En un ensayo que comparó la eficacia y la seguridad de empagliflozina 25 mg frente a glimepirida (hasta 4 mg al día) en pacientes con un control glucémico inadecuado con metformina sola, el tratamiento diario con empagliflozina dio lugar a una mayor reducción de la HbA1c (Tabla 4) y a una reducción clínicamente significativa de la GPA en comparación con la glimepirida. La administración diaria de empagliflozina provocó una reducción estadísticamente significativa del peso corporal y de la presión arterial sistólica y diastólica, y una proporción inferior de pacientes estadísticamente significativa con episodios hipoglucémicos en comparación con la glimepirida (1,6% para empagliflozina, 20,4% para glimepirida, p<0,0001).
|
Tabla 4: Resultados de eficacia en la semana 104 de un ensayo controlado con activo que comparó empagliflozina con glimepirida en adición a metforminaa |
||
|
Empagliflozina 25 mg |
Glimepiridab |
|
|
N |
765 |
780 |
|
HbA1c (%) |
||
|
Valor basal (media) |
7,92 |
7,92 |
|
Cambio respecto al valor basal1 |
-0,66 |
-0,55 |
|
Diferencia respecto a la glimepirida1 (IC del 97,5%) |
-0,11* (-0,20, -0,01) |
|
|
N |
690 |
715 |
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de la HbA1c ≥7%2 |
33,6 |
30,9 |
|
N |
765 |
780 |
|
Peso corporal (kg) |
||
|
Valor basal (media) |
82,52 |
83,03 |
|
Cambio respecto al valor basal1 |
-3,12 |
1,34 |
|
Diferencia respecto a la glimepirida1 (IC del 97,5%) |
-4,46** (-4,87, -4,05) |
|
|
N |
765 |
780 |
|
PAS (mm Hg)2 |
||
|
Valor basal (media) |
133,4 |
133,5 |
|
Cambio respecto al valor basal1 |
-3,1 |
2,5 |
|
Diferencia respecto a la glimepirida1 (IC del 97,5%) |
-5,6** (-7,0,-4,2) |
|
|
a. Grupo completo de análisis (FAS) utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico. b. Hasta 4 mg de glimepirida. 1. Media ajustada respecto al valor basal. 2. LOCF, valores censurados estadísticamente después del rescate antihipertensivo. * valor p <0,0001 para no inferioridad, y valor p = 0,0153 para superioridad. ** valor p<0,0001 |
||
Tratamiento de adición a insulina:
• Empagliflozina añadida a dosis diarias múltiples de insulina: Se evaluó la eficacia y la seguridad de la empagliflozina como tratamiento de adición a dosis diarias múltiples de insulina con o sin metformina concomitante en un ensayo doble ciego controlado con placebo de 52 semanas de duración. Durante las 18 primeras semanas y las 12 últimas semanas, la dosis de insulina se mantuvo estable, pero se ajustó entre las semanas 19 y 40 para alcanzar niveles de glucosa preprandial <100 mg/dl [5,5 mmol/l] y niveles de glucosa posprandial <140 mg/dl [7,8 mmol/l].
En la semana 18, empagliflozina presentó una mejora estadísticamente significativa de la HbA1c en comparación con el placebo (Tabla 5).
En la semana 52, el tratamiento con empagliflozina provocó una reducción estadísticamente significativa de la HbA1c y un ahorro de insulina en comparación con el placebo, así como una reducción en la GPA y el peso corporal.
|
Tabla 5: Resultados de eficacia en las semanas 18 y 52 de un ensayo controlado con placebo de empagliflozina como tratamientode adición a dosis múltiples diarias de insulina con o sin metformina |
|||
|
Placebo |
JARDIANCE |
||
|
10 mg |
25 mg |
||
|
N |
188 |
186 |
189 |
|
HbA1c (%) en la semana 18 |
|||
|
Valor basal (media) |
8,33 |
8,39 |
8,29 |
|
Cambio respecto al valor basal1 |
-0,50 |
-0,94 |
-1,02 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
0,44* |
-0,52* |
|
|
N |
115 |
119 |
118 |
|
HbA1c (%) en la semana 522 |
|||
|
Valor basal (media) |
8,25 |
8,40 |
8,37 |
|
Cambio respecto al valor basal1 |
-0,81 |
-1,18 |
-1,27 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-0,38*** (-0,62, -0,13) |
-0,46* |
|
|
N |
113 |
118 |
118 |
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de la HbA1c ≥7% en la semana 52 |
26,5 |
39,8 |
45,8 |
|
N |
115 |
118 |
117 |
|
Dosis de insulina (UI/día) en la semana 522 |
|||
|
Valor basal (media) |
89,94 |
88,57 |
90,38 |
|
Cambio respecto al valor basal1 |
10,16 |
1,33 |
-1,06 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-8,83# |
-11,22** |
|
|
N |
115 |
119 |
118 |
|
Peso corporal (kg) en la semana 522 |
|||
|
Valor basal (media) |
96,34 |
96,47 |
95,37 |
|
Cambio respecto al valor basal1 |
0,44 |
-1,95 |
-2,04 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-2,39* |
-2,48* |
|
|
1. Media ajustada respecto al valor basal. 2. Semana 19-40: pauta de tratamiento hasta alcanzar el objetivo con ajuste de la dosis de insulina para conseguir niveles objetivo de glucosa predefinidos (preprandial <100 mg/dl (5,5 mmol/l), posprandial <140 mg/dl (7,8 mmol/l). * valor p<0,0001 ** valor p = 0,0003 *** valor p = 0,0005 # valor p = 0,0040 |
|||
• Empagliflozina como tratamiento de adición a insulina basal: Se evaluó la eficacia y la seguridad de empagliflozina como tratamiento de adición a insulina basal con o sin metformina y/o una sulfonilurea en un ensayo doble ciego controlado con placebo de 78 semanas de duración. Durante las 18 primeras semanas, la dosis de insulina se mantuvo estable, pero se ajustó para lograr una GPA <110 mg/dl en las 60 semanas siguientes.
En la semana 18, empagliflozina presentó una mejora estadísticamente significativa en la HbA1c (Tabla 6).
En la semana 78, empagliflozina provocó una disminución estadísticamente significativa de la HbA1c y del ahorro de insulina en comparación con placebo. Además, empagliflozina dio lugar a una reducción de la GPA, el peso corporal y la presión arterial.
|
Tabla 6: Resultados de eficacia en las semanas 18 y 78 de un ensayo controlado con placebo de empagliflozina como tratamiento de adición a insulina basal con o sin metformina o una sulfonilureaa |
|||
|
Placebo |
Empagliflozina |
||
|
10 mg |
25 mg |
||
|
N |
125 |
132 |
117 |
|
HbA1c (%) en la semana 18 |
|||
|
Valor basal (media) |
8,10 |
8,26 |
8,34 |
|
Cambio respecto al valor basal1 |
-0,01 |
-0,57 |
-0,71 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-0,56* (-0,78, -0,33) |
-0,70* |
|
|
N |
112 |
127 |
110 |
|
HbA1c (%) en la semana 78 |
|||
|
Valor basal (media) |
8,09 |
8,27 |
8,29 |
|
Cambio respecto al valor basal1 |
-0,02 |
-0,48 |
-0,64 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-0,46* |
-0,62* (-0,90, -0,34) |
|
|
N |
112 |
127 |
110 |
|
Dosis de insulina basal (UI/día) en la semana 78 |
|||
|
Valor basal (media) |
47,84 |
45,13 |
48,43 |
|
Cambio respecto al valor basal1 |
5,45 |
-1,21 |
-0,47 |
|
Diferencia respecto al placebo1 (IC del 97,5%) |
-6,66** |
-5,92** |
|
|
a. Grupo completo de análisis (FAS): pacientes que completaron el estudio utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico. 1. Media ajustada respecto al valor basal. * valor p<0,0001 ** valor p <0,025 |
|||
Pacientes con insuficiencia renal, datos controlados con placebo de 52 semanas: Se evaluó la eficacia y la seguridad de empagliflozina como tratamiento de adición a un tratamiento antidiabético en pacientes con insuficiencia renal en un estudio doble ciego controlado con placebo de 52 semanas de duración. En la semana 24, el tratamiento con empagliflozina provocó una reducción estadísticamente significativa de la HbA1c (Tabla 7) y una mejora clínicamente significativa en la GPA en comparación con placebo. La mejora en la HbA1c, el peso corporal y la presión arterial se mantuvo durante 52 semanas.
|
Tabla 7: Resultados en la semana 24 de un estudio controlado con placebo de empagliflozina en pacientes con diabetes tipo 2 e insuficiencia renala |
|||||
|
Placebo |
Empagliflozina 10 mg |
Empagliflozina 25 mg |
Placebo |
Empagliflozina 25 mg |
|
|
TFGe ≥60 a <90 ml/min/1,73 m2 |
TFGe ≥45 a <60 ml/min/1,73 m2 |
||||
|
N |
95 |
98 |
97 |
89 |
91 |
|
HbA1c (%) |
|||||
|
Valor basal (media) |
8,09 |
8,02 |
7,96 |
8,08 |
8,12 |
|
Cambio respecto al valor basal1 |
0,06 |
-0,46 |
- 0,63 |
-0,08 |
-0,54 |
|
Diferencia respecto al placebo1 (IC del 95%) |
-0,52* |
-0,68* |
-0,46 |
||
|
N |
89 |
94 |
91 |
84 |
86 |
|
Pacientes (%) que logran un valor de HbA1c <7% con un valor basal de la HbA1c ≥7%2 |
6,7 |
17,0 |
24,2 |
10,7 |
15,1 |
|
N |
95 |
98 |
97 |
89 |
91 |
|
Peso corporal (kg)2 |
|||||
|
Valor basal (media) |
86,00 |
92,05 |
88,06 |
83,20 |
84,90 |
|
Cambio respecto al valor basal1 |
-0,33 |
-1,76 |
-2,33 |
-0,11 |
-1,39 |
|
Diferencia respecto al placebo1 (IC del 95%) |
-1,43 |
-2,00 |
-1,28 |
||
|
N |
95 |
98 |
97 |
89 |
91 |
|
PAS (mm Hg)2 |
|||||
|
Valor basal (media) |
134,69 |
137,37 |
133,68 |
137,29 |
135,04 |
|
Cambio respecto al valor basal1 |
0,65 |
-2,92 |
-4,47 |
0,37 |
-5,69 |
|
Diferencia respecto al placebo1 (IC del 95%) |
-3,57 |
-5,12 |
-6,07 |
||
|
a. Grupo completo de análisis (FAS) utilizando la última observación considerada (LOCF) antes del tratamiento de rescate glucémico. 1. Media ajustada respecto al valor basal. 2. No evaluado en cuanto a significación estadística debido al resultado del procedimiento de prueba confirmatorio secuencial. * p<0,0001 |
|||||
Seguridad cardiovascular: En un metanálisis prospectivo pre-especificado de episodios cardiovasculares adjudicados de manera independiente procedentes de 12 estudios clínicos de fase 2 y 3 en 10 036 pacientes con diabetes tipo 2, empagliflozina no aumentó el riesgo cardiovascular.
Glucosa plasmática en ayunas: En cuatro ensayos controlados con placebo, el tratamiento con empagliflozina en monoterapia o como tratamiento de adición a metformina, pioglitazona o metformina más una sulfonilurea provocó cambios medios respecto al valor basal de GPA de -20,5 mg/dl [-1,14 mmol/l] para empagliflozina 10 mg y de -23,2 mg/dl [-1,29 mmol/l] para empagliflozina 25 mg en comparación con el placebo (7,4 mg/dl [0,41 mmol/l]). Este efecto se observó después de 24 semanas y se mantuvo durante 76 semanas.
Glucosa posprandial a las 2 horas: El tratamiento con empagliflozina como adición a metformina o metformina y una sulfonilurea provocó una reducción clínicamente significativa de la glucosa posprandial a las 2 horas (prueba de tolerancia a la glucosa) a las 24 semanas (tratamiento de adición a metformina: placebo +5,9 mg/dl, empagliflozina 10 mg: -46,0 mg/dl, empagliflozina 25 mg: -44,6 mg/dl, tratamiento de adición a metformina y una sulfonilurea: placebo -2,3 mg/dl, empagliflozina 10 mg: -35,7 mg/dl, empagliflozina 25 mg: -36,6 mg/dl).
Pacientes con un valor basal alto de la HbA1c >10%: En un análisis conjunto pre-especificado de tres estudios de fase 3, el tratamiento abierto con empagliflozina 25 mg en pacientes con hiperglucemia grave (N=257, valor basal medio de HbA1c 11,26%) provocó una reducción clínicamente significativa en la HbA1c respecto al valor basal del 3,27%; en estos estudios no se incluyó ningún brazo de placebo ni de empagliflozina 10 mg.
Peso corporal: En un análisis conjunto pre-especificado de 4 estudios controlados con placebo, el tratamiento con empagliflozina provocó una reducción del peso corporal (-0,24 kg para placebo, -2,04 kg para empagliflozina 10 mg y -2,26 kg para empagliflozina 25 mg) en la semana 24, que se mantuvo hasta la semana 52 (-0,16 kg para placebo, -1,96 kg para empagliflozina 10 mg y -2,25 kg para empagliflozina 25 mg).
Presión arterial: La eficacia y la seguridad de empagliflozina se evaluaron en un ensayo doble ciego controlado con placebo de 12 semanas de duración, en pacientes con diabetes tipo 2 y presión arterial alta que seguían diferentes tratamientos antidiabéticos y hasta 2 tratamientos antihipertensivos. El tratamiento con empagliflozina una vez al día provocó una mejora estadísticamente significativa en la HbA1c, y en la presión arterial sistólica y diastólica media de 24 horas, determinada mediante controles ambulatorios de la presión arterial (Tabla 8). El tratamiento con empagliflozina provocó reducciones en la PAS y PAD en posición sentada.
|
Tabla 8: Resultados de eficacia en la semana 12 de un ensayo controlado con placebo de empagliflozina en pacientes con diabetes tipo 2 y presión arterial no controladaa |
|||
|
Placebo |
JARDIANCE |
||
|
10 mg |
25 mg |
||
|
N |
271 |
276 |
276 |
|
HbA1c (%) en la semana 121 |
|||
|
Valor basal (media) |
7,90 |
7,87 |
7,92 |
|
Cambio respecto al valor basal2 |
0,03 |
-0,59 |
-0,62 |
|
Diferencia respecto al placebo2 (IC del 95%) |
0,62* |
-0,65* |
|
|
PAS a las 24 horas en la semana 123 |
|||
|
Valor basal (media) |
131,72 |
131,34 |
131,18 |
|
Cambio respecto al valor basal4 |
0,48 |
-2,95 |
-3,68 |
|
Diferencia respecto al placebo4 (IC del 95%) |
-3,44* (-4,78, -2,09) |
-4,16* (-5,50, -2,83) |
|
|
PAD a las 24 horas en la semana 123 |
|||
|
Valor basal (media) |
75,16 |
75,13 |
74,64 |
|
Cambio respecto al valor basal5 |
0,32 |
-1,04 |
-1,40 |
|
Diferencia respecto al placebo5 (IC del 95%) |
-1,36** |
-1,72* (-2,51, -0,93) |
|
|
a. Grupo completo de análisis (FAS). 1. LOCF, valores censurados estadísticamente después del tratamiento de rescate antidiabético. 2. Media ajustada respecto a la HbA1c basal, la TFGe basal, la región geográfica y el número de medicamentos antihipertensivos. 3. LOCF, valores censurados estadísticamente después del tratamiento de rescate antidiabético o de cambiar el tratamiento de rescate antihipertensivo. 4. Media ajustada respecto a la PAS basal la HbA1c basal, la TFGe basal, la región geográfica y el número de medicamentos antihipertensivos. 5. Media ajustada respecto a la PAD basal, la HbA1c basal, la TFGe basal, la región geográfica y el número de medicamentos antihipertensivos. * valor p<0,0001 ** valor p <0,001 |
|||
En un análisis conjunto pre-especificado de 4 estudios controlados con placebo, el tratamiento con empagliflozina provocó una reducción de la presión arterial sistólica (empagliflozina 10 mg: -3,9 mm Hg; empagliflozina 25 mg: -4,3 mm Hg) en comparación con placebo (-0,5 mm Hg) y de la presión arterial diastólica (empagliflozina 10 mg: -1,8 mm Hg; empagliflozina 25 mg: -2,0 mm Hg) en comparación con placebo (-0,5 mm Hg) en la semana 24 y esta reducción se mantuvo hasta la semana 52.
Población pediátrica: La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con JARDIANCE en uno o más subgrupos de población pediátrica con diabetes mellitus tipo 2.
CONTRAINDICACIONES:
• Antecedentes de reacción de hipersensibilidad seria a la empagliflozina o a cualquiera de los excipientes presentes en JARDIANCE® (ver Advertencias y precauciones).
• Deterioro renal severo, enfermedad renal terminal o diálisis (ver Uso en poblaciones específicas).
FERTILIDAD, EMBARAZO Y LACTANCIA
Embarazo: No hay datos sobre el uso de empagliflozina en mujeres embarazadas. Los estudios realizados en animales muestran que la empagliflozina atraviesa la placenta durante la última fase de la gestación en un grado muy limitado, pero no indican efectos perjudiciales directos ni indirectos en lo que respecta al desarrollo embrionario temprano. No obstante, los estudios realizados en animales han mostrado efectos adversos en el desarrollo posnatal. Como medida de precaución, es preferible evitar el uso de JARDIANCE durante las primeras etapas del embarazo. El uso de JARDIANCE no está recomendado durante el segundo y el tercer trimestre del embarazo.
Lactancia: No se dispone de datos en humanos sobre la excreción de la empagliflozina en la leche materna. Los datos toxicológicos disponibles en animales han mostrado que la empagliflozina se excreta en la leche.
No se puede excluir el riesgo para los recién nacidos o los lactantes. JARDIANCE no debe utilizarse durante la lactancia.
Fertilidad: No se han realizado estudios sobre el efecto de JARDIANCE en la fertilidad humana. Los estudios realizados en animales no sugieren efectos perjudiciales directos o indirectos sobre la fertilidad.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: La influencia de JARDIANCE sobre la capacidad para conducir y utilizar máquinas es pequeña. Se debe advertir a los pacientes que tomen las debidas precauciones para evitar una hipoglucemia mientras conducen y utilizan máquinas, sobre todo cuando JARDIANCE se use en combinación con una sulfonilurea y/o con insulina.
REACCIONES ADVERSAS:
Las siguientes reacciones adversas importantes se describen a continuación y en otras secciones del prospecto:
• Hipotensión (ver Advertencias y precauciones)
• Cetoacidosis (ver Advertencias y precauciones)
• Lesión renal aguda y deterioro de la función renal (ver Advertencias y precauciones)
• Urosepticemia y pielonefritis (ver Advertencias y precauciones)
• Hipoglucemia con el uso concomitante con insulina y secretagogos de insulina (ver Advertencias y precauciones)
• Infecciones micóticas genitales (ver Advertencias y precauciones)
• Reacciones de hipersensibilidad (ver Advertencias y precauciones)
• Niveles elevados de colesterol de lipoproteína de baja densidad (LDL-C) (ver Advertencias y precauciones)
“Por favor comunicarse con su médico o farmacéutico en caso se presente cualquier reacción adversa que no esté descrita en este inserto”.
Experiencia de estudios clínicos: Dado que los estudios clínicos se llevan a cabo en condiciones sumamente variables, las incidencias de reacciones adversas observadas en los estudios clínicos de un fármaco no pueden compararse en forma directa con las incidencias observadas en los estudios clínicos de otro fármaco, y pueden no reflejar las tasas de incidencia observadas en la práctica.
Datos combinados de estudios con control de placebo en los que se evaluó JARDIANCE® 10 y 25 mg: Los datos que se brindan en la Tabla 1 se obtuvieron a partir de un cúmulo de datos combinados de cuatro estudios controlados con placebo de 24 semanas de duración y datos de 18 semanas obtenidos a partir de un estudio con control de placebo de administración junto con insulina. JARDIANCE® se utilizó como monoterapia en un estudio y como tratamiento complementario en cuatro estudios (ver Estudios clínicos).
Estos datos reflejan la exposición de 1976 pacientes a JARDIANCE®, con una duración media de exposición de aproximadamente 23 semanas. Los pacientes recibieron placebo (N = 995), JARDIANCE® 10 mg (N = 999) o JARDIANCE® 25 mg (N = 977) una vez al día. La media de la edad de la población fue 56 años, y el 3% de los pacientes eran mayores de 75 años de edad. Más de la mitad (55%) de la población era de sexo masculino; el 46% era de raza blanca, el 50% era de raza asiática y el 3% era de raza negra o afroamericana. En el nivel basal del estudio, el 57% de la población tenía diabetes que databa de más de 5 años y un valor medio de hemoglobina A1c (HbA1c) de 8%. Las complicaciones microvasculares establecidas de la diabetes presentes en el nivel basal del estudio incluyeron nefropatía diabética (7%), retinopatía (8%) o neuropatía (16%). La función renal en el nivel basal fue normal o ligeramente deteriorada en el 91% de los pacientes y moderadamente deteriorada en el 9% de los pacientes (media de eGFR 86,8 mL/min/1,73 m2).
En la Tabla 1 se presentan las reacciones adversas frecuentes (excluyendo la hipoglucemia) asociadas con el uso de JARDIANCE®. Dichas reacciones adversas no estaban presentes al inicio del estudio, ocurrieron más comúnmente con JARDIANCE® que con el placebo y se produjeron en una proporción mayor o igual al 2% de los pacientes tratados con JARDIANCE® 10 mg o JARDIANCE® 25 mg.
Tabla 1. Reacciones adversas informadas en ≥ 2% de los pacientes tratados con JARDIANCE® y en un porcentaje mayor que con el placebo en el combinado de estudios clínicos controlados con placebo de JARDIANCE® como monoterapia o terapia combinada
|
Cantidad (%) de pacientes |
|||
|
Placebo N = 995 |
JARDIANCE® 10 mg N = 999 |
JARDIANCE® 25 mg N = 977 |
|
|
Infección de las vías urinariasa |
7,6% |
9,3% |
7,6% |
|
Infecciones micóticas genitales femeninasb |
1,5% |
5,4% |
6,4% |
|
Infección de las vías respiratorias superiores |
3,8% |
3,1% |
4,0% |
|
Aumento de la micciónc |
1,0% |
3,4% |
3,2% |
|
Dislipidemia |
3,4% |
3,9% |
2,9% |
|
Artralgia |
2,2% |
2,4% |
2,3% |
|
Infecciones micóticas genitales masculinasd |
0,4% |
3,1% |
1,6% |
|
Náuseas |
1,4% |
2,3% |
1,1% |
|
a Agrupación predefinida de eventos adversos, incluyendo, sin carácter taxativo, infección de las vías urinarias, bacteriuria asintomática y cistitis. b Las infecciones micóticas genitales femeninas incluyen las siguientes reacciones adversas: infección micótica vulvovaginal, infección vaginal, vulvitis, candidiasis vulvovaginal, infección genital, candidiasis genital, infección genital por hongos, infección urogenital, vulvovaginitis, cervicitis, infección urogenital por hongos y vaginitis bacteriana. Porcentajes calculados con la cantidad de sujetos de sexo femenino de cada grupo como denominador: placebo (N = 481), JARDIANCE® 10 mg (N = 443), JARDIANCE® 25 mg (N = 420). c Agrupación predefinida de eventos adversos, incluyendo, sin carácter taxativo, poliuria, polaquiuria y nicturia. d Las infecciones micóticas genitales masculinas incluyen las siguientes reacciones adversas: balanopostitis, balanitis, infecciones genitales por hongos, infecciones urogenitales, balanitis por cándidas, absceso escrotal, infección peneana. Porcentajes calculados con la cantidad de sujetos de sexo masculino de cada grupo como denominador: placebo (N = 514), JARDIANCE® 10 mg (N = 556), JARDIANCE® 25 mg (N = 557). |
|||
Se informó sed (incluyendo polidipsia) en el 0%, 1,7% y 1,5% de los pacientes para el placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente.
Depleción del volumen: JARDIANCE® provoca diuresis osmótica, lo cual puede conducir a contracción del volumen intravascular y reacciones adversas relacionadas con depleción del volumen. En el grupo combinado de cinco estudios clínicos controlados con placebo, las reacciones adversas relacionadas con depleción de volumen (p. ej., presión arterial (ambulatoria) disminuida, presión arterial sistólica disminuida, deshidratación, hipotensión, hipovolemia, hipotensión ortostática y síncope) fueron informadas por el 0,3%, 0,5% y 0,3% de los pacientes tratados con placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente. JARDIANCE® puede incrementar el riesgo de hipotensión en pacientes con riesgo de contracción del volumen (ver Advertencias y precauciones y Uso en poblaciones específicas).
Aumento de la micción: En el grupo combinado de cinco estudios clínicos controlados con placebo, las reacciones adversas de aumento de la micción (por ejemplo, poliuria, polaquiuria y nicturia) ocurrieron con mayor frecuencia con JARDIANCE® que con el placebo (ver la Tabla 1). Específicamente, la nicturia fue informada por el 0,4%, 0,3% y 0,8% de los pacientes tratados con placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente.
Deterioro agudo de función renal: El tratamiento con JARDIANCE® se asoció con aumentos de los niveles de creatinina sérica y reducciones de la eGFR (ver la Tabla 2). Los pacientes con deterioro renal moderado en el nivel basal tuvieron medias de cambio de mayores (ver Advertencias y precauciones y Uso en poblaciones específicas).
En un estudio de desenlaces cardiovasculares a largo plazo, se observó una reversión del deterioro agudo de la función renal tras la interrupción del tratamiento, lo cual sugiere que los cambios hemodinámicos agudos desempeñan un papel en los cambios de la función renal observados con empagliflozina.
Tabla 2. Cambios respecto del nivel basal en la creatinina sérica y la eGFRa en el grupo combinado de cuatro estudios con control de placebo de 24 semanas de duración y un estudio de deterioro renal
|
Grupo combinado de estudios con control de placebo de 24 semanas de duración |
||||
|
Placebo |
JARDIANCE® 10 mg |
JARDIANCE® 25 mg |
||
|
Media del nivel basal |
N |
825 |
830 |
822 |
|
Creatinina (mg/dL) |
0,84 |
0,85 |
0,85 |
|
|
eGFR (mL/min/1,73 m2) |
87,3 |
87,1 |
87,8 |
|
|
Cambio de la Semana 12 |
N |
771 |
797 |
783 |
|
Creatinina (mg/dL) |
0,00 |
0,02 |
0,01 |
|
|
eGFR (mL/min/1,73 m2) |
-0,3 |
-1,3 |
-1,4 |
|
|
Cambio de la Semana 24 |
N |
708 |
769 |
754 |
|
Creatinina (mg/dL) |
0,00 |
0,01 |
0,01 |
|
|
eGFR (mL/min/1,73 m2) |
-0,3 |
-0,6 |
-1,4 |
|
|
Deterioro renal moderadob |
||||
|
Placebo |
JARDIANCE® 25 mg |
|||
|
Media del nivel basal |
N |
187 |
-- |
187 |
|
Creatinina (mg/dL) |
1,49 |
-- |
1,46 |
|
|
eGFR (mL/min/1,73 m2) |
44,3 |
-- |
45,4 |
|
|
Cambio de la Semana 12 |
N |
176 |
-- |
179 |
|
Creatinina (mg/dL) |
0,01 |
-- |
0,12 |
|
|
eGFR (mL/min/1,73 m2) |
0,1 |
-- |
-3,8 |
|
|
Cambio de la Semana 24 |
N |
170 |
-- |
171 |
|
Creatinina (mg/dL) |
0,01 |
-- |
0,10 |
|
|
eGFR (mL/min/1,73 m2) |
0,2 |
-- |
-3,2 |
|
|
Cambio de la Semana 52 |
N |
164 |
-- |
162 |
|
Creatinina (mg/dL) |
0,02 |
-- |
0,11 |
|
|
eGFR (mL/min/1,73 m2) |
-0,3 |
-- |
-2,8 |
|
|
Cambio postratamientoc |
N |
98 |
-- |
103 |
|
Creatinina (mg/dL) |
0,03 |
-- |
0,02 |
|
|
eGFR (mL/min/1,73 m2) |
0,16 |
-- |
1,48 |
|
|
a Casos observados durante el tratamiento. b Subconjunto de pacientes del estudio de deterioro renal con eGFR de 30 a menos de 60 mL/min/1,73 m2. c Aproximadamente 3 semanas después del fin del tratamiento. |
||||
Hipoglucemia: La incidencia de hipoglucemia por estudio se presenta en la Tabla 3. La incidencia de hipoglucemia se incrementó cuando JARDIANCE® se administró con insulina o con una sulfonilurea (ver Advertencias y precauciones).
Tabla 3. Incidencia de eventos de hipoglucemia en generala y severosb en los estudios clínicos controlados con placeboc
|
Monoterapia (24 semanas) |
Placebo (n = 229) |
JARDIANCE® 10 mg (n = 224) |
JARDIANCE® 25 mg (n = 223) |
|
Total general (%) |
0,4 % |
0,4 % |
0,4 % |
|
Severos (%) |
0% |
0% |
0% |
|
En combinación con Metformina (24 semanas) |
Placebo + Metformina (n = 206) |
JARDIANCE® 10 mg + Metformina (n = 217) |
JARDIANCE® 25 mg + Metformina (n = 214) |
|
Total general (%) |
0,5 % |
1,8 % |
1,4 % |
|
Severos (%) |
0% |
0% |
0% |
|
En combinación con Metformina + Sulfonilurea (24 semanas) |
Placebo (n = 225) |
JARDIANCE® 10 mg + Metformina + Sulfonilurea (n = 224) |
JARDIANCE® 25 mg + Metformina + Sulfonilurea (n = 217) |
|
Total general (%) |
8,4 % |
16,1 % |
11,5 % |
|
Severos (%) |
0% |
0% |
0% |
|
En combinación con Pioglitazona +/- Metformina (24 semanas) |
Placebo (n = 165) |
JARDIANCE® 10 mg + Pioglitazona +/- Metformina (n = 165) |
JARDIANCE® 25 mg + Pioglitazona +/- Metformina (n = 168) |
|
Total general (%) |
1,8 % |
1,2 % |
2,4 % |
|
Severos (%) |
0% |
0% |
0% |
|
En combinación con Insulina Basal +/- Metformina (18 semanasd) |
Placebo (n = 170) |
JARDIANCE® 10 mg (n = 169) |
JARDIANCE® 25 mg (n = 155) |
|
Total general (%) |
20,6 % |
19,5 % |
28,4 % |
|
Severos (%) |
0% |
0% |
1,3 % |
|
En combinación con Insulina en IDM +/- Metformina (18 semanasd) |
Placebo (n = 188) |
JARDIANCE® 10 mg (n = 186) |
JARDIANCE® 25 mg (n = 189) |
|
Total general (%) |
37,2 % |
39,8 % |
41,3 % |
|
Severos (%) |
0,5 % |
0,5 % |
0,5 % |
|
a Eventos de hipoglucemia en general: glucosa plasmática o capilar menor o igual a 70 mg/dL. b Eventos de hipoglucemia severos: requieren asistencia independientemente del nivel de glucosa en sangre. c Conjunto tratado (pacientes que habían recibido al menos una dosis del fármaco en estudio). d La dosis de insulina no podía ajustarse durante el período de tratamiento inicial de 18 semanas. |
|||
Infecciones micóticas genitales: En el grupo combinado de cinco estudios clínicos controlados con placebo, la incidencia de infecciones micóticas genitales (p. ej., infección micótica vaginal, infección vaginal, infección genital por hongos, candidiasis vulvovaginal y vulvitis) estuvo incrementada en los pacientes tratados con JARDIANCE® en comparación con aquellos tratados con placebo; tales eventos se produjeron en el 0,9%, 4,1% y 3,7% de los pacientes aleatorizados a placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente. La discontinuación del estudio debido a infección genital se produjo en el 0% de los pacientes tratados con placebo y en el 0,2% de los pacientes tratados con JARDIANCE® 10 o 25 mg.
Las infecciones micóticas genitales se produjeron con mayor frecuencia en las pacientes de sexo femenino que en los pacientes de sexo masculino (ver Tabla 1).
La fimosis se produjo con mayor frecuencia en los pacientes de sexo masculino tratados con JARDIANCE®
10 mg (menos del 0,1%) y JARDIANCE® 25 mg (0,1%) que en aquellos tratados con placebo (0%).
Infecciones de las vías urinarias: En el grupo combinado de cinco estudios clínicos controlados con placebo, la incidencia de infecciones urinarias (p. ej., infección de las vías urinarias, bacteriuria asintomática y cistitis) estuvo incrementada en los pacientes tratados con JARDIANCE® en comparación con aquellos tratados con el placebo (ver Tabla 1). Los pacientes con antecedentes de infecciones de las vías urinarias crónicas o recurrentes fueron más propensos a tener una infección de las vías urinarias. La tasa de interrupción del tratamiento debido a infecciones de las vías urinarias fue 0,1%, 0,2% y 0,1% para el placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente.
Las infecciones de las vías urinarias se produjeron con mayor frecuencia en las pacientes de sexo femenino. La incidencia de infecciones de las vías urinarias en las pacientes de sexo femenino aleatorizadas a placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg fue del 16,6%, 18,4% y 17,0%, respectivamente. La incidencia de infecciones de las vías urinarias en los pacientes de sexo masculino aleatorizados a placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg fue del 3,2%, 3,6% y 4,1%, respectivamente (ver Advertencias y precauciones y Uso en poblaciones específicas).
Pruebas de laboratorio:
Elevación de los niveles de colesterol de lipoproteína de baja densidad (LDL-C): Se observaron incrementos relacionados con la dosis en los niveles de colesterol de lipoproteína de baja densidad (LDL-C) en los pacientes tratados con JARDIANCE®. El LDL-C se incrementó a razón de un 2,3%, 4,6% y 6,5% en los pacientes tratados con placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente (ver Advertencias y precauciones). El rango de medias de los niveles de LDL-C del nivel basal fue de entre 90,3 y 90,6 mg/dL en los diferentes grupos de tratamiento.
Elevación del hematocrito: En un grupo combinado de cuatro estudios controlados con placebo, la mediana del hematocrito se redujo un 1,3% en los pacientes tratados con placebo y se incrementó a razón de un 2,8% en los pacientes tratados con JARDIANCE® 10 mg y un 2,8% en los pacientes tratados con JARDIANCE® 25 mg. Al final del tratamiento, el 0,6%, el 2,7% y el 3,5% de los pacientes con valores de hematocrito inicialmente dentro del rango de referencia tenían valores por encima del límite superior del rango de referencia con placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente.
Experiencia post-comercialización: Se han identificado reacciones adversas adicionales durante el uso post-aprobación de JARDIANCE®. En vista de que estas reacciones han sido informadas en forma voluntaria a partir de una población cuyo tamaño es incierto, en general no es posible estimar de manera confiable su frecuencia o establecer una relación causal entre las mismas y la exposición al fármaco.
• Cetoacidosis (ver Advertencias y precauciones)
• Urosepticemia y pielonefritis (ver Advertencias y precauciones)
• Angioedema (ver Advertencias y precauciones)
• Reacciones cutáneas (p. ej., exantema, urticaria)
INCOMPATIBILIDADES: No procede.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN
Interacciones farmacodinámicas:
• Diuréticos: Empagliflozina puede aumentar el efecto diurético de las tiazidas y de los diuréticos del asa y puede aumentar el riesgo de deshidratación e hipotensión.
• Insulina y secretagogos de insulina: La insulina y los secretagogos de insulina, como las sulfonilureas, pueden aumentar el riesgo de hipoglucemia. Por lo tanto, puede necesitarse una dosis más baja de insulina o de un secretagogo de insulina para disminuir el riesgo de hipoglucemia cuando éstos se usan en combinación con empagliflozina.
Interacciones farmacocinéticas:
• Efectos de otros medicamentos sobre la empagliflozina: Los datos in vitro sugieren que la principal vía metabólica de la empagliflozina en humanos es la glucuronidación por las uridina 5"-difosfoglucuronosiltransferasas UGT1A3, UGT1A8, UGT1A9 y UGT2B7. La empagliflozina es un sustrato de los transportadores de captación humanos OAT3, OATP1B1, y OATP1B3, pero no de OAT1 y OCT2. La empagliflozina es un sustrato de la glicoproteína-P (gp-P) y la proteína de resistencia al cáncer de mama (BCRP).
La administración conjunta de empagliflozina con probenecid, un inhibidor de las enzimas UGT y del OAT3, dio lugar a un aumento del 26% en las concentraciones plasmáticas máximas (Cmax) de empagliflozina y a un aumento del 53% en el área bajo la curva concentración-tiempo (AUC). Estos cambios no se consideraron clínicamente significativos.
No se ha estudiado el efecto de la inducción de la UGT sobre la empagliflozina. La medicación concomitante con inductores de las enzimas UGT debe evitarse debido al riesgo potencial de que disminuya la eficacia.
Un estudio de interacción con gemfibrozil, un inhibidor in vitro de los transportadores OAT3 y OATP1B1/1B3, mostró que la Cmax de empagliflozina aumentaba en un 15% y el AUC aumentaba en un 59% después de la administración conjunta. Estos cambios no se consideraron clínicamente significativos.
La inhibición de los transportadores OATP1B1/1B3 mediante la administración conjunta de rifampicina dio lugar a un aumento del 75% en la Cmax y un aumento del 35% en el AUC de la empagliflozina. Estos cambios no se consideraron clínicamente significativos.
La exposición a empagliflozina fue similar con y sin la administración conjunta de verapamilo, un inhibidor de la gp-P, lo que indica que la inhibición de la gp-P no tiene un efecto clínicamente relevante sobre la empagliflozina.
Los estudios de interacción realizados en voluntarios sanos sugieren que la farmacocinética de la empagliflozina no se vio influida por la administración conjunta de metformina, glimepirida, pioglitazona, sitagliptina, linagliptina, warfarina, verapamilo, ramipril, simvastatina, torasemida e hidroclortiazida.
• Efectos de la empagliflozina sobre otros medicamentos: En base a los estudios in vitro, la empagliflozina no inhibe, inactiva ni induce las isoformas del CYP450. La empagliflozina no inhibe la UGT1A1. Por lo tanto, se considera improbable que se produzcan interacciones farmacológicas que impliquen a las principales isoformas del CYP450 o a la UGT1A1 con empagliflozina y a los sustratos de estas enzimas administrados de forma conjunta. No se ha estudiado el potencial de la empagliflozina para inhibir la UGT2B7.
La empagliflozina no inhibe la gp-P a dosis terapéuticas. En base a los estudios in vitro, se considera improbable que la empagliflozina provoque interacciones con fármacos que sean sustratos de la gp-P.
Un estudio de interacción con gemfibrozil, un inhibidor in vitro de los transportadores OAT3 y OATP1B1/1B3, mostró que la Cmax de empagliflozina aumentaba en un 15% y el AUC aumentaba en un 59% después de la administración conjunta. Estos cambios no se consideraron clínicamente significativos.
La inhibición de los transportadores OATP1B1/1B3 mediante la administración conjunta de rifampicina dio lugar a un aumento del 75% en la Cmax y un aumento del 35% en el AUC de la empagliflozina. Estos cambios no se consideraron clínicamente significativos.
La exposición a empagliflozina fue similar con y sin la administración conjunta de verapamilo, un inhibidor de la gp-P, lo que indica que la inhibición de la gp-P no tiene un efecto clínicamente relevante sobre la empagliflozina.
Los estudios de interacción realizados en voluntarios sanos sugieren que la farmacocinética de la empagliflozina no se vio influida por la administración conjunta de metformina, glimepirida, pioglitazona, sitagliptina, linagliptina, warfarina, verapamilo, ramipril, simvastatina, torasemida e hidroclortiazida.
• Efectos de la empagliflozina sobre otros medicamentos: En base a los estudios in vitro, la empagliflozina no inhibe, inactiva ni induce las isoformas del CYP450. La empagliflozina no inhibe la UGT1A1. Por lo tanto, se considera improbable que se produzcan interacciones farmacológicas que impliquen a las principales isoformas del CYP450 o a la UGT1A1 con empagliflozina y a los sustratos de estas enzimas administrados de forma conjunta. No se ha estudiado el potencial de la empagliflozina para inhibir la UGT2B7.
La empagliflozina no inhibe la gp-P a dosis terapéuticas. En base a los estudios in vitro, se considera improbable que la empagliflozina provoque interacciones con fármacos que sean sustratos de la gp-P. La administración conjunta de digoxina, un sustrato de la gp-P, con empagliflozina dio lugar a un aumento del 6% en el AUC y un aumento del 14% en la Cmax de la digoxina. Estos cambios no se consideraron clínicamente significativos.
La empagliflozina no inhibe in vitro a los transportadores de captación humanos, tales como OAT3, OATP1B1 y OATP1B3 a concentraciones plasmáticas clínicamente relevantes y, como tales, las interacciones farmacológicas con sustratos de estos transportadores de captación se consideran improbables.
Los estudios de interacción realizados en voluntarios sanos sugieren que la empagliflozina no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de la metformina, la glimepirida, la pioglitazona, la sitagliptina, la linagliptina, la simvastatina, la warfarina, el ramipiril, la digoxina, los diuréticos y los anticonceptivos orales.
INTERACCIONES MEDICAMENTOSAS
Evaluación in vitro de las interacciones medicamentosas: La empagliflozina no inhibe, inactiva ni induce las isoformas del CYP450. Los datos in vitro sugieren que la principal vía metabólica de la empagliflozina en los seres humanos es su glucuronidación a través de las uridina 5"-difosfo-glucuronosiltransferasas UGT1A3, UGT1A8, UGT1A9 y UGT2B7. La empagliflozina no inhibe la UGT1A1, la UGT1A3, la UGT1A8, la UGT1A9 ni la UGT2B7. Por lo tanto, no se anticipa ningún efecto de la empagliflozina administrada en forma concomitante con fármacos que son sustratos de las principales isoformas del CYP450 o las UGT1A1, UGT1A3, UGT1A8, UGT1A9 y UGT2B7. El efecto de la inducción de la UGT (p. ej., inducción por rifampicina o cualquier otro inductor enzimático de la UGT) en la exposición a la empagliflozina no ha sido evaluado.
La empagliflozina es un sustrato de la glucoproteína P (P-gp) y de la proteína de resistencia al cáncer de mama (BCRP), pero no inhibe estos transportadores de eflujo en las dosis terapéuticas. Sobre la base de los estudios in vitro, se considera improbable que la empagliflozina tenga alguna interacción con los fármacos que son sustratos de la P-gp. La empagliflozina es un sustrato de los transportadores de entrada humanos OAT3, OATP1B1 y OATP1B3, pero no así, en cambio, de OAT1 y OCT2. La empagliflozina no inhibe ninguno de estos transportadores de entrada humanos en las concentraciones plasmáticas clínicamente relevantes y, por lo tanto, no se anticipa ningún efecto de la empagliflozina sobre los fármacos que son sustratos de estos transportadores de entrada en el caso de su administración concomitante.
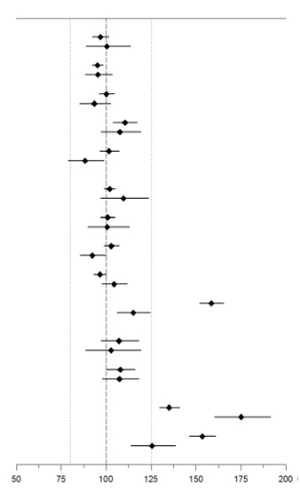
Evaluación in vivo de las interacciones medicamentosas: No se recomienda ningún ajuste de la dosis de JARDIANCE® cuando este medicamento se coadministra con productos medicinales comúnmente prescriptos, sobre la base de los resultados de los estudios de farmacocinética descritos. La farmacocinética de la empagliflozina fue similar con y sin la coadministración de metformina, glimepirida, pioglitazona, sitagliptina, linagliptina, warfarina, verapamilo, ramipril y simvastatina en voluntarios sanos, y con y sin coadministración de hidroclorotiazida y torasemida en pacientes con diabetes tipo 2 (ver Figura 1). Los incrementos observados en la exposición total (AUC) de la empagliflozina tras la coadministración con gemfibrozil, rifampicina o probenecid no son clínicamente relevantes. En los sujetos con función renal normal, la coadministración de empagliflozina con probenecid produjo una disminución del 30% en la fracción de empagliflozina excretada en la orina sin ningún efecto sobre la excreción de glucosa urinaria de 24 horas. Se desconoce la relevancia de esta observación para los pacientes con deterioro renal.
Figura 1. Efecto de diferentes medicamentos sobre la farmacocinética de la empagliflozina, presentado como intervalo de confianza del 90% de los cocientes de medias geométricas de AUC y Cmax (las líneas de referencia indican el 100% (80% - 125%))
|
Cociente de medias geométricas (intervalo de confianza del 90%) |
||
|
Fármacos antidiabéticos |
||
|
Metformina, 1000 mg, dos veces al díaa |
AUC Cmax |
|
|
Glimepirida, 1 mg, dosis únicaa |
AUC Cmax |
|
|
Pioglitazona, 45 mg, una vez al díaa |
AUC Cmax |
|
|
Sitagliptina, 100 mg, una vez al díaa |
AUC Cmax |
|
|
Linagliptina, 5 mg, una vez al díaa |
AUC Cmax |
|
|
Otros |
||
|
Simvastatina, 40 mg, dosis únicab |
AUC Cmax |
|
|
Warfarina, 25 mg, dosis únicac |
AUC Cmax |
|
|
Verapamilo, 120 mg, dosis únicab |
AUC Cmax |
|
|
Ramipril, 5 mg, una vez al díac |
AUC Cmax |
|
|
Gemfibrozil, 600 mg, dos veces al díab |
AUC Cmax |
|
|
Hidroclorotiazida, 25 mg, una vez al díac |
AUC Cmax |
|
|
Torsemida, 5 mg, una vez al díac |
AUC Cmax |
|
|
Rifampicina, 600 mg, dosis únicad |
AUC Cmax |
|
|
Probenecid, 500 mg, dos veces al díad |
AUC Cmax |
|
|
Cambio respecto de empagliflozina sola |
||
|
a empagliflozina, 50 mg, una vez al día; b empagliflozina, 25 mg, dosis única; c empagliflozina, 25 mg, una vez al día; d empagliflozina, 10 mg, dosis única |
||
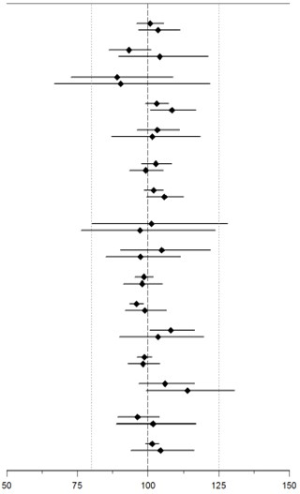
La empagliflozina no tuvo ningún efecto clínicamente relevante en la farmacocinética de la metformina, la glimepirida, la pioglitazona, la sitagliptina, la linagliptina, la warfarina, la digoxina, el ramipril, la simvastatina, la hidroclorotiazida, la torasemida ni los anticonceptivos orales cuando se coadministró en voluntarios sanos (ver Figura 2).
Figura 2. Efecto de la empagliflozina sobre la farmacocinética de diversos medicamentos, presentado como intervalo de confianza del 90% de los cocientes de medias geométricas de AUC y Cmax (las líneas de referencia indican el 100% (80% - 125%))
|
Cociente de medias geométricas (intervalo de confianza del 90%) |
||
|
Fármacos antidiabéticos |
||
|
Metformina, 1000 mg, dos veces al díaa |
AUC Cmax |
|
|
Glimepirida, 1 mg, dosis únicaa |
AUC Cmax |
|
|
Pioglitazona, 45 mg, una vez al díaa |
AUC Cmax |
|
|
Sitagliptina, 100 mg, una vez al díaa |
AUC Cmax |
|
|
Linagliptina, 5 mg, una vez al díaa |
AUC Cmax |
|
|
Anticonceptivos orales |
||
|
Etinilestriadol, 30 mcg, una vez al díab,f |
AUC Cmax |
|
|
Levonorgestrel, 150 mgc, una vez al díab,f |
AUC Cmax |
|
|
Otros |
||
|
Simvastatina, 40 mg, dosis únicab |
AUC Cmax |
|
|
Simvastatina ácidad |
AUC Cmax |
|
|
R-Warfarina. 25 mg, dosis únicab,e |
AUC Cmax |
|
|
S-Warfarina. 25 mg, dosis únicab,e |
AUC Cmax |
|
|
Ramipril, 5 mg, una vez al díab |
AUC Cmax |
|
|
Ramiprilatog |
AUC Cmax |
|
|
Digoxina, 0,5 mg, dosis únicab |
AUC Cmax |
|
|
Hidroclorotiazida, 25 mg, una vez al díab |
AUC Cmax |
|
|
Probenecid, 500 mg, dos veces al díad |
AUC Cmax |
|
|
Torsemida, 5 mg, una vez al díab |
AUC Cmax |
|
|
a empagliflozina, 50 mg, una vez al día; b empagliflozina, 25 mg, una vez al día; c empagliflozina, 25 mg, dosis única; d administrada como simvastatina; e administrada como mezcla racémica de warfarina; f administrado como Microgynon®; g administrado como ramipril |
||
INTERACCIONES MEDICAMENTOSAS:
Diuréticos: La coadministración de empagliflozina con diuréticos dio lugar a un incremento en el volumen urinario y la frecuencia de las micciones, lo cual podría incrementar el potencial de depleción del volumen (ver Advertencias y precauciones).
Insulina o secretagogos de insulina: La coadministración de empagliflozina con insulina o secretagogos de insulina aumenta el riesgo de hipoglucemia (ver Advertencias y precauciones).
Prueba de glucosa en orina positiva: No se recomienda monitorear el control glucémico con pruebas de glucosa en orina en los pacientes que están tomando inhibidores del SGLT2, ya que los inhibidores del SGLT2 aumentan la excreción de glucosa urinaria y conducirán a resultados positivos en las pruebas de glucosa en orina. Deben utilizarse métodos alternativos para monitorear el control glucémico.
Interferencia con la prueba de 1, 5-anhidroglucitol (1,5-AG)
No se recomienda monitorear el control glucémico con la prueba de 1,5-AG, puesto que las mediciones de 1,5-AG no son confiables para evaluar el control glucémico en los pacientes que están tomando inhibidores del SGLT2. Deben utilizarse métodos alternativos para monitorear el control glucémico.
ESTUDIOS CLÍNICOS:
Control glucémico: JARDIANCE® se ha estudiado como monoterapia y en combinación con metformina, sulfonilurea, pioglitazona, linagliptina e insulina. JARDIANCE® también ha sido estudiado en pacientes con diabetes tipo 2 con deterioro renal leve o moderado.
En los pacientes con diabetes tipo 2, el tratamiento con JARDIANCE® redujo los niveles de hemoglobina A1c (HbA1c), en comparación con el placebo. La reducción de los niveles de HbA1c logrados por JARDIANCE® en comparación con el placebo se observó en todos los subgrupos, incluyendo los definidos por sexo, raza, región geográfica, IMC del nivel basal y duración de la enfermedad.
Monoterapia: Un total de 986 pacientes con diabetes tipo 2 participaron en un estudio doble ciego, controlado con placebo, destinado a evaluar la eficacia y seguridad de la monoterapia de JARDIANCE®.
Pacientes con diabetes tipo 2 inadecuadamente controlada que no habían recibido ningún tratamiento para tal afección fueron ingresados en un período de preinclusión de placebo de diseño abierto de 2 semanas de duración. Al final de dicho período de preinclusión, los pacientes que continuaban con un control inadecuado y tenían un valor de HbA1c de entre 7 y 10% fueron aleatorizados a placebo, JARDIANCE® 10 mg, JARDIANCE® 25 mg o un comparador de referencia.
En la Semana 24, el tratamiento con JARDIANCE® 10 mg o 25 mg a diario proporcionó reducciones estadísticamente significativas en los valores de HbA1c (valor p < 0,0001), glucosa plasmática en ayunas (GPA) y peso corporal en comparación con el placebo (ver Tabla 4 y Figura 3).
Tabla 4. Resultados de la Semana 24 obtenidos a partir de un estudio controlado con placebo de monoterapia de JARDIANCE®
|
JARDIANCE® 10 mg N = 224 |
JARDIANCE® 25 mg N = 224 |
Placebo N = 228 |
|
|
HbA1c (%)a |
|||
|
Nivel basal (media) |
7,9 |
7,9 |
7,9 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,7 |
-0,8 |
0,1 |
|
Diferencia respecto del placebo (media ajustada) (CI 97,5 %) |
-0,7b (-0,9; -0,6) |
-0,9b (-1,0; -0,7) |
-- |
|
Pacientes (n (%)) que lograron un valor de HbA1c < 7 % |
72 (35 %) |
88 (44 %) |
25 (12 %) |
|
GPA (mg/dL)c |
|||
|
Nivel basal (media) |
153 |
153 |
155 |
|
Cambio respecto del nivel basal (media ajustada) |
-19 |
-25 |
12 |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-31 (-37; -26) |
-36 (-42; -31) |
-- |
|
Peso corporal |
|||
|
Nivel basal (media) en kg |
78 |
78 |
78 |
|
Cambio porcentual respecto del nivel basal (media ajustada) |
-2,8 |
-3,2 |
-0,4 |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-2,5b (-3,1; -1,9) |
-2,8b (-3,4; -2,2) |
-- |
|
Nivel basal (media) en kg |
78 |
78 |
78 |
|
Cambio porcentual respecto del nivel basal (media ajustada) |
-2,8 |
-3,2 |
-0,4 |
|
Diferencia respecto del placebo (media ajustada) (CI 95%) |
-2,5b (-3,1; -1,9) |
-2,8b (-3,4; -2,2) |
|
|
a Población por intención de tratamiento modificada (mITT). Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 24. En la Semana 24, se imputó un 9,4%, 9,4% y 30,7% para los pacientes aleatorizados a JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo, respectivamente. b Valor p derivado de ANCOVA < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento, función renal y región. Peso corporal y GPA: se utilizó el mismo modelo que para la HbA1c, pero además se incluyó el valor de peso corporal del nivel basal/GPA del nivel basal, respectivamente). c GPA (mg/dL); para JARDIANCE® 10 mg, n = 223, para JARDIANCE® 25 mg, n = 223 y para el placebo, n = 226. |
|||
Figura 3. Media ajustada del cambio en la HbA1c en cada punto temporal (pacientes que completaron) y en la Semana 24 (Población mITT) - LOCF
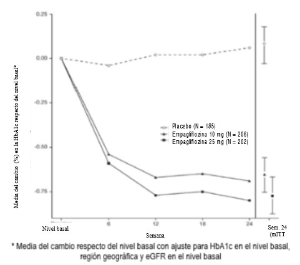
En la Semana 24, se observó una reducción estadísticamente significativa de la presión arterial sistólica en comparación con el placebo, cuya magnitud fue de -2,6 mmHg (valor p con ajuste para placebo = 0,0231) en los pacientes aleatorizados a 10 mg de JARDIANCE® y de -3,4 mmHg (valor p con corrección para placebo = 0,0028) en los pacientes aleatorizados a 25 mg de JARDIANCE®.
Terapia combinada complementaria con metformina: Un total de 637 pacientes con diabetes tipo 2 participaron en un estudio doble ciego, controlado con placebo, destinado a evaluar la eficacia y seguridad de JARDIANCE® en combinación con metformina.
Pacientes con diabetes tipo 2 inadecuadamente controlada en tratamiento con al menos 1500 mg de metformina por día fueron ingresados en un período de preinclusión de placebo de diseño abierto de 2 semanas de duración. Al final de dicho período de preinclusión, los pacientes que continuaban con un control inadecuado y tenían una valor de HbA1c de entre 7 y 10% fueron aleatorizados a placebo, JARDIANCE® 10 mg o JARDIANCE® 25 mg.
En la Semana 24, el tratamiento con JARDIANCE® 10 mg o 25 mg a diario proporcionó reducciones estadísticamente significativas en los valores de HbA1c (valor p < 0,0001), GPA y peso corporal en comparación con el placebo (ver Tabla 5).
Tabla 5. Resultados de la Semana 24 obtenidos a partir de un estudio controlado con placebo de JARDIANCE® utilizado en combinación con metformina
|
JARDIANCE® 10 mg + Metformina N = 217 |
JARDIANCE® 25 mg + Metformina N = 213 |
Placebo + Metformina N = 207 |
|
|
HbA1c (%)a |
|||
|
Nivel basal (media) |
7,9 |
7,9 |
7,9 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,7 |
-0,8 |
-0,1 |
|
Diferencia respecto de placebo + metformina (media ajustada) (CI 95 %) |
-0,6b (-0,7; -0,4) |
-0,6b (-0,8; -0,5) |
-- |
|
Pacientes (n (%)) que lograron un valor de HbA1c < 7 % |
75 (38 %) |
74 (39 %) |
23 (13 %) |
|
GPA (mg/dL)c |
|||
|
Nivel basal (media) |
155 |
149 |
156 |
|
Cambio respecto del nivel basal (media ajustada) |
-20 |
-22 |
6 |
|
Diferencia respecto de placebo + metformina (media ajustada) |
-26 |
-29 |
-- |
|
Peso corporal |
|||
|
Media del nivel basal en kg |
82 |
82 |
80 |
|
Cambio porcentual respecto del nivel basal (media ajustada) |
-2,5 |
-2,9 |
-0,5 |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-2,0b (-2,6; -1,4) |
-2,5b (-3,1; -1,9) |
-- |
|
a Población por intención de tratamiento modificada (mITT). Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 24. En la Semana 24, se imputó un 9,7%, 14,1% y 24,6% para los pacientes aleatorizados a JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo, respectivamente. b Valor p derivado de ANCOVA < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento, función renal y región. Peso corporal y GPA: se utilizó el mismo modelo que para la HbA1c, pero además se incluyó el valor de peso corporal del nivel basal/GPA del nivel basal, respectivamente). c GPA (mg/dL); para JARDIANCE® 10 mg, n = 216, para JARDIANCE® 25 mg, n = 213 y para el placebo, n = 207. |
|||
En la Semana 24, se observó una reducción estadísticamente significativa de la presión arterial sistólica en comparación con el placebo, cuya magnitud fue de -4,1 mmHg (valor p con corrección para placebo < 0,0001) para JARDIANCE® 10 mg y de -4,8 mmHg (valor p con corrección para placebo < 0,0001) para JARDIANCE® 25 mg.
Terapia combinada inicial con metformina: Un total de 1364 pacientes con diabetes tipo 2 participaron en un estudio doble ciego, aleatorizado, con control activo, destinado a evaluar la eficacia y seguridad de JARDIANCE® en combinación con metformina como terapia inicial en comparación con los correspondientes componentes en forma individual.
Pacientes con diabetes tipo 2 inadecuadamente controlada que no habían recibido ningún tratamiento para tal afección fueron ingresados en un período de preinclusión de placebo de diseño abierto de 2 semanas de duración. Al final de dicho período de preinclusión, los pacientes que continuaban con un control inadecuado y tenían un valor de HbA1c de entre 7 y 10,5% fueron aleatorizados a uno de los 8 grupos de tratamiento activo. JARDIANCE® 10 mg o 25 mg; metformina 1000 mg o 2000 mg; JARDIANCE® 10 mg en combinación con 1000 mg o 2000 mg de metformina; o JARDIANCE® 25 mg en combinación con 1000 mg o 2000 mg de metformina.
En la Semana 24, la terapia inicial de JARDIANCE® en combinación con metformina proporcionó reducciones estadísticamente significativas en la HbA1c (valor p < 0,01) en comparación con los componentes individuales (ver Tabla 6).
Tabla 6. Parámetros de glucemia a las 24 semanas en un estudio comparativo de JARDIANCE® y metformina frente a los componentes individuales como terapia inicial
|
JARDIANCE 10 mg + Metformina 1000 mga N = 161 |
JARDIANCE 10 mg + Metformina 2000 mga N = 167 |
JARDIANCE 25 mg + Metformina 1000 mga N = 165 |
JARDIANCE 25 mg + Metformina 2000 mga N = 169 |
JARDIANCE 10 mg N = 169 |
JARDIANCE 25 mg N = 163 |
Metformina 1000 mga N = 167 |
Metformina 2000 mga N = 162 |
|
|
HbA1c (%) |
||||||||
|
Nivel basal (media) |
8,7 |
8,7 |
8,8 |
8,7 |
8,6 |
8,9 |
8,7 |
8,6 |
|
Cambio respecto del nivel basal (media ajustada) (CI 95 %) |
-2,0 |
-2,1 |
-1,9 |
-2,1 |
-1,4 |
-1,4 |
-1,2 |
-1,8 |
|
Comparación vs. JARDIANCE (media ajustada) (CI 95 %) |
-0,6b (-0,9; -0,4) |
-0,7b (-1,0; -0,5) |
-0,6c (-0,8; -0,3) |
-0,7c (-1,0; -0,5) |
-- |
-- |
-- |
-- |
|
Comparación vs. metformina (media ajustada) |
-0,8b (-1,0; -0,6) |
-0,3b (-0,6; -0,1) |
-0,8c (-1,0; -0,5) |
-0,3c (-0,6; -0,1) |
-- |
-- |
-- |
-- |
|
a Dosis diaria total de metformina, dividida en partes iguales y administrada en dos tomas diarias. b Valor p ≤ 0,0062 (población por intención de tratamiento modificada (casos observados) El modelo MMRM incluyó tratamiento, función renal, región, visita, interacción entre visita y tratamiento y HbA1c del nivel basal). c Valor p ≤ 0,0056 (población por intención de tratamiento modificada (casos observados) El modelo MMRM incluyó tratamiento, función renal, región, visita, interacción entre visita y tratamiento y HbA1c del nivel basal). |
||||||||
Terapia combinada complementaria con metformina y sulfonilurea: Un total de 666 pacientes con diabetes tipo 2 participaron en un estudio doble ciego, controlado con placebo, destinado a evaluar la eficacia y seguridad de JARDIANCE® en combinación con metformina más una sulfonilurea.
Pacientes con diabetes tipo 2 inadecuadamente controlada en tratamiento con al menos 1500 mg de metformina por día y una sulfonilurea fueron ingresados en un período de preinclusión de placebo de diseño abierto de 2 semanas de duración. Al final de dicho período de preinclusión, los pacientes que continuaban con un control inadecuado y tenían un valor de HbA1c de entre 7% y 10% fueron aleatorizados a placebo, JARDIANCE® 10 mg o JARDIANCE® 25 mg.
El tratamiento con JARDIANCE® 10 mg o 25 mg a diario proporcionó reducciones estadísticamente significativas en los valores de HbA1c (valor p < 0,0001), GPA y peso corporal en comparación con el placebo (ver Tabla 7).
Tabla 7. Resultados de la Semana 24 obtenidos a partir de un estudio controlado con placebo de JARDIANCE® en combinación con metformina y sulfonilurea
|
JARDIANCE 10 mg + Metformina + SU N = 225 |
JARDIANCE 25 mg + Metformina + SU N = 216 |
Placebo + Metformina + SU N = 225 |
|
|
HbA1c (%)a |
|||
|
Nivel basal (media) |
8,1 |
8,1 |
8,2 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,8 |
-0,8 |
-0,2 |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-0,6b (-0,8; -0,5) |
-0,6b (-0,7; -0,4) |
-- |
|
Pacientes (n (%)) que lograron un valor de HbA1c < 7 % |
55 (26 %) |
65 (32 %) |
20 (9 %) |
|
GPA (mg/dL)c |
|||
|
Nivel basal (media) |
151 |
156 |
152 |
|
Cambio respecto del nivel basal (media ajustada) |
-23 |
-23 |
6 |
|
Diferencia respecto del placebo (media ajustada) |
-29 |
-29 |
-- |
|
Peso corporal |
|||
|
Media del nivel basal en kg |
77 |
78 |
76 |
|
Cambio porcentual respecto del nivel basal (media ajustada) |
-2,9 |
-3,2 |
-0,5 |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-2,4b (-3,0; -1,8) |
-2,7b (-3,3; -2,1) |
-- |
|
a Población por intención de tratamiento modificada (mITT). Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 24. En la Semana 24, se imputó un 17,8%, 16,7% y 25,3% para los pacientes aleatorizados a JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo, respectivamente. b Valor p derivado de ANCOVA < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento, función renal y región. Peso corporal y GPA: se utilizó el mismo modelo que para la HbA1c, pero además se incluyó el valor de peso corporal del nivel basal/GPA del nivel basal, respectivamente). c GPA (mg/dL); para JARDIANCE® 10 mg, n = 225, para JARDIANCE® 25 mg, n = 215 y para el placebo, n = 224. |
|||
En combinación con linagliptina como complemento de la terapia de metformina: Un total de 686 pacientes con diabetes tipo 2 participaron en un estudio doble ciego, con control activo, destinado a evaluar la eficacia y seguridad de JARDIANCE® 10 mg o 25 mg en combinación con linagliptina 5 mg en comparación con los componentes individuales.
Pacientes con diabetes tipo 2 inadecuadamente controlada en tratamiento con al menos 1500 mg de metformina por día fueron ingresados en un período de preinclusión de placebo de cegamiento simple de 2 semanas de duración. Al final de dicho período de preinclusión, los pacientes que continuaban con un control inadecuado y tenían un valor de HbA1c de entre 7 y 10,5% fueron aleatorizados en una proporción de 1:1:1:1:1 a uno de los 5 grupos de tratamiento activo de JARDIANCE® 10 mg o 25 mg, linagliptina 5 mg, o linagliptina 5 mg en combinación con 10 mg o 25 mg de JARDIANCE® como un comprimido de combinación de dosis fijas.
En la Semana 24, JARDIANCE® 10 mg o 25 mg usado en combinación con linagliptina 5 mg proporcionó una mejoría estadísticamente significativa en los parámetros de HbA1c (valor p < 0,0001) y GPA (valor p < 0,001) en comparación con los componentes individuales en pacientes que habían tenido un control inadecuado con metformina. El tratamiento con JARDIANCE®/linagliptina 25 mg/5 mg o JARDIANCE®/linagliptina 10 mg/5 mg a diario también condujo a una reducción estadísticamente significativa en el peso corporal en comparación con linagliptina 5 mg (valor p < 0,0001). No hubo ninguna diferencia estadísticamente significativa en el peso corporal en comparación con JARDIANCE® administrado solo.
Estudio con control activo frente a glimepirida en combinación con metformina: La eficacia de JARDIANCE® se evaluó en un estudio doble ciego, controlado con glimepirida, en 1545 pacientes con diabetes tipo 2 con control glucémico insuficiente a pesar de la terapia de metformina.
Pacientes con control glucémico inadecuado y un valor de HbA1c de entre 7% y 10% tras un período de preinclusión de 2 semanas fueron aleatorizados a glimepirida o JARDIANCE® 25 mg.
En la Semana 52, el régimen de JARDIANCE® 25 mg y glimepirida condujo a una reducción de los valores de HbA1c y GPA (ver Tabla 8, Figura 4). La diferencia en la magnitud del efecto observado entre JARDIANCE® 25 mg y glimepirida excluyó el margen de no inferioridad preespecificado del 0,3%. La dosis diaria media de glimepirida fue 2,7 mg y la dosis máxima aprobada en los Estados Unidos es 8 mg por día.
Tabla 8. Resultados de la Semana 52 obtenidos a partir de un estudio con control activo en el que se comparó JARDIANCE® frente a la glimepirida como terapia complementaria en pacientes controlados inadecuadamente con metformina
|
JARDIANCE 25 mg + Metformina N = 765 |
Glimepirida + Metformina N = 780 |
|
|
HbA1c (%)a |
||
|
Nivel basal (media) |
7,9 |
7,9 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,7 |
-0,7 |
|
Diferencia respecto de glimepirida (media ajustada) (CI 97,5 %) |
-0,07b (-0,15; 0,01) |
-- |
|
GPA (mg/dL)d |
||
|
Nivel basal (media) |
150 |
150 |
|
Cambio respecto del nivel basal (media ajustada) |
-19 |
-9 |
|
Diferencia respecto de glimepirida (media ajustada) |
-11 |
-- |
|
Peso corporal |
||
|
Media del nivel basal en kg |
82,5 |
83 |
|
Cambio porcentual respecto del nivel basal (media ajustada) |
-3,9 |
2,0 |
|
Diferencia respecto de glimepirida (media ajustada) (CI 95 %) |
-5,9c (-6,3; -5,5) |
-- |
|
a Población por intención de tratamiento modificada (mITT). Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 52. En la Semana 52, los datos se imputaron para el 15,3% y 21,9% de los pacientes aleatorizados a JARDIANCE® 25 mg y glimepirida, respectivamente. b No inferior, valor p derivado del ANCOVA < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento, función renal y región). c Valor p derivado del ANCOVA < 0,0001 (Peso corporal y GPA: se utilizó el mismo modelo que para la HbA1c, pero además se incluyó el valor de peso corporal del nivel basal/GPA del nivel basal, respectivamente). d GPA (mg/dL); para JARDIANCE® 25 mg, n = 764, para placebo, n = 779. |
||
Figura 4. Media ajustada del cambio en la HbA1c en cada punto temporal (pacientes que completaron) y en la Semana 52 (Población mITT) - LOCF
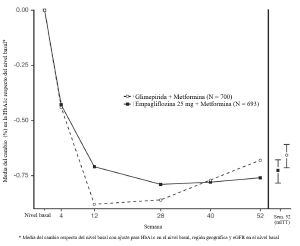
En la Semana 52, la media ajustada del cambio respecto del nivel basal en la presión arterial sistólica fue -3,6 mmHg, en comparación con 2,2 mmHg para glimepirida. Las diferencias entre los grupos de tratamiento para la presión arterial sistólica fueron estadísticamente significativas (valor p < 0,0001).
En la Semana 104, la media ajustada del cambio respecto del nivel basal en la HbA1c fue -0,75% para JARDIANCE® 25 mg y -0,66% para glimepirida. La media ajustada de la diferencia entre los tratamientos fue -0,09% con un intervalo de confianza del 97,5% de (-0,32%; 0,15%), excluyendo el margen de no inferioridad preespecificado del 0,3%. La dosis diaria media de glimepirida fue 2,7 mg y la dosis máxima aprobada en los Estados Unidos es 8 mg por día. El análisis de la Semana 104 incluyó datos con y sin administración concomitante de medicación de rescate para la glucemia, así como también datos obtenidos fuera del tratamiento. Los datos faltantes para los pacientes que no suministraban ninguna información en la visita se imputaron sobre la base de los datos observados fuera del tratamiento. En este análisis de imputación múltiple, se imputó el 13,9% de los datos para JARDIANCE® 25 mg y el 12,9% para glimepirida.
En la Semana 104, el régimen de JARDIANCE® 25 mg a diario condujo a una diferencia estadísticamente significativa en el cambio respecto del nivel basal para el peso corporal en comparación con la glimepirida (-3,1 kg para JARDIANCE® 25 mg vs. +1,3 kg para glimepirida; ANCOVA-LOCF, valor de p < 0,0001).
Terapia de combinación complementaria con pioglitazona con o sin metformina: Un total de 498 pacientes con diabetes tipo 2 participaron en un estudio doble ciego, controlado con placebo, destinado a evaluar la eficacia y seguridad de JARDIANCE® en combinación con pioglitazona, con o sin metformina.
Pacientes con diabetes tipo 2 inadecuadamente controlada en tratamiento con metformina en una dosis de al menos 1500 mg al día y pioglitazona en una dosis de menos de 30 mg por día fueron ingresados en un período de preinclusión de placebo de diseño abierto de 2 semanas de duración. Pacientes con un control glucémico inadecuado y un valor de HbA1c de entre 7% y 10% tras el período de preinclusión fueron aleatorizados a placebo, JARDIANCE® 10 mg o JARDIANCE® 25 mg.
El tratamiento con JARDIANCE® 10 mg o 25 mg a diario proporcionó reducciones estadísticamente significativas en los valores de HbA1c (valor p < 0,0001), GPA y peso corporal en comparación con el placebo (ver Tabla 9).
Tabla 9. Resultados del estudio controlado con placebo para JARDIANCE® en terapia de combinación con pioglitazona
|
JARDIANCE 10 mg + Pioglitazona N = 165 |
JARDIANCE 25 mg + Pioglitazona N = 168 |
Placebo + Pioglitazona N = 165 |
|
|
HbA1c (%)a |
|||
|
Nivel basal (media) |
8,1 |
8,1 |
8,2 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,6 |
-0,7 |
-0,1 |
|
Diferencia respecto de placebo + pioglitazona (media ajustada) (CI 95 %) |
-0,5b (-0,7; -0,3) |
-0,6b (-0,8; -0,4) |
-- |
|
Pacientes (n (%)) que lograron un valor de HbA1c < 7 % |
36 (24 %) |
48 (30 %) |
12 (8 %) |
|
GPA (mg/dL)c |
|||
|
Nivel basal (media) |
152 |
152 |
152 |
|
Cambio respecto del nivel basal (media ajustada) |
-17 |
-22 |
7 |
|
Diferencia respecto de placebo + pioglitazona |
|||
|
(media ajustada) (CI 97,5 %) |
-23b (-31,8; -15,2) |
-28b (-36,7; -20,2) |
-- |
|
Peso corporal |
|||
|
Media del nivel basal en kg |
78 |
79 |
78 |
|
Cambio porcentual respecto del nivel basal (media ajustada) |
-2,0 |
-1,8 |
0,6 |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-2,6b (-3,4; -1,8) |
-2,4b (-3,2; -1,6) |
-- |
|
a Población por intención de tratamiento modificada (mITT). Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 24. En la Semana 24, se imputó un 10,9%, 8,3% y 20,6% para los pacientes aleatorizados a JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo, respectivamente. b Valor p derivado de ANCOVA < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento, función renal y medicación de base. Peso corporal y GPA: se utilizó el mismo modelo que para la HbA1c, pero además se incluyó el valor de peso corporal del nivel basal/GPA del nivel basal, respectivamente). c GPA (mg/dL); para JARDIANCE® 10 mg, n = 163. |
|||
Terapia de combinación complementaria con insulina con o sin metformina y/o sulfonilureas: Un total de 494 pacientes con diabetes tipo 2 inadecuadamente controlada con insulina, o insulina en combinación con medicamentos orales, participaron en un estudio doble ciego, controlado con placebo, destinado a evaluar la eficacia de JARDIANCE® como tratamiento complementario a la insulina durante 78 semanas.
Los pacientes ingresaron en un período de preinclusión de placebo de 2 semanas en tratamiento con insulina basal (p. ej., insulina glargina, insulina detemir o insulina NPH) con o sin terapia de base de metformina y/o sulfonilurea. Luego del período de preinclusión, los pacientes que tenían un control glucémico inadecuado fueron aleatorizados a la incorporación de JARDIANCE® 10 mg, JARDIANCE® 25 mg o placebo. Los pacientes se mantuvieron con una dosis estable de insulina previo al enrolamiento, durante el período de preinclusión y durante las primeras 18 semanas de tratamiento. Durante las 60 semanas restantes, se podía ajustar la dosis de la insulina. La media de la dosis diaria total de insulina en el nivel basal del estudio para JARDIANCE® 10 mg, 25 mg y placebo fue 45 UI, 48 UI y 48 UI, respectivamente.
JARDIANCE® usado en combinación con insulina (con o sin metformina y/o sulfonilurea) proporcionó reducciones estadísticamente significativas en la HbA1c y la GPA en comparación con el placebo al cabo de 18 y 78 semanas de tratamiento (ver Tabla 10). JARDIANCE® 10 mg o 25 mg a diario condujo además a una reducción porcentual del peso corporal estadística y significativamente mayor en comparación con el placebo.
Tabla 10. Resultados de la Semana 18 y 78 obtenidos a partir de un estudio controlado con placebo para JARDIANCE® en combinación con insulina
|
18 semanas (sin ajuste de la insulina) |
78 semanas (dosis de insulina ajustable luego de las 18 semanas) |
|||||
|
JARDIANCE 10 mg + Insulina N = 169 |
JARDIANCE 25 mg + Insulina N = 155 |
Placebo + Insulina N = 170 |
JARDIANCE 10 mg + Insulina N = 169 |
JARDIANCE 25 mg + Insulina N = 155 |
Placebo + Insulina N = 170 |
|
|
HbA1c (%)a |
||||||
|
Nivel basal (media) |
8,3 |
8,3 |
8,2 |
8,3 |
8,3 |
8,2 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,6 |
-0,7 |
0 |
-0,4 |
-0,6 |
0,1 |
|
Diferencia respecto del placebo (media ajustada) |
-0,6b (-0,8; -0,4) |
-0,7b (-0,9; -0,5) |
-- |
-0,5b (-0,7; -0,3) |
-0,7b (-0,9; -0,5) |
-- |
|
Pacientes (%) que lograron un valor de HbA1c < 7 % |
18,0 |
19,5 |
5,5 |
12,0 |
17,5 |
6,7 |
|
GPA (mg/dL) |
||||||
|
Nivel basal (media) |
138 |
146 |
142 |
138 |
146 |
142 |
|
Cambio respecto del nivel basal (media ajustada, SE) |
-17,9 (3,2) |
-19,1 (3,3) |
10,4 (3,1) |
-10,1 (3,2) |
-15,2 (3,4) |
2,8 (3,2) |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-28,2b (-37,0; -19,5) |
-29,5b (-38,4; -20,6) |
-- |
-12,9c (-21,9; 3,9) |
-17,9b (-27,0; -8,8) |
-- |
|
Peso corporal |
||||||
|
Media del nivel basal en kg |
92 |
95 |
90 |
92 |
95 |
90 |
|
Cambio porcentual respecto del nivel basal (media ajustada) |
-1,8 |
-1,4 |
-0,1 |
-2,4 |
-2,4 |
0,7 |
|
Diferencia respecto del placebo (media ajustada) (CI 95 %) |
-1,7d (-3,0; -0,5) |
-1,3e (-2,5; -0,0) |
-- |
-3,0b (-4,4; -1,7) |
-3,0b (-4,4; -1,6) |
-- |
|
a Población por intención de tratamiento modificada (mITT). Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 18 y 78. En la Semana 18, se imputó un 21,3%, 30,3% y 21,8% para los pacientes aleatorizados a JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo, respectivamente. En la Semana 78, se imputó un 32,5%, 38,1%, 42,4% para los pacientes aleatorizados a JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo, respectivamente. b Valor p derivado de ANCOVA < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento y región; GPA: el modelo MMRM incluye GPA del nivel basal, HbA1c del nivel basal, tratamiento, región, visita e interacción de visita por tratamiento. Peso corporal: El modelo MMRM incluye peso corporal del nivel basal, HbA1c del nivel basal, tratamiento, región, visita e interacción de visita por tratamiento. c Valor p = 0,0049. d Valor p = 0,0052. e Valor p = 0,0463. |
||||||
Terapia de combinación complementaria con IDM de insulina con o sin metformina: Un total de 563 pacientes con diabetes tipo 2 inadecuadamente controlada con inyecciones diarias múltiples (IDM) de insulina (dosis diaria total > 60 UI), sola o en combinación con metformina, participaron en un estudio doble ciego, controlado con placebo, destinado a evaluar la eficacia de JARDIANCE® como tratamiento complementario de las IDM de insulina a lo largo de 18 semanas.
Los pacientes ingresaron en un período de preinclusión de placebo de 2 semanas de duración en tratamiento con IDM de insulina con o sin una terapia de base de metformina. Luego del período de preinclusión, los pacientes que tenían un control glucémico inadecuado fueron aleatorizados a la incorporación de JARDIANCE® 10 mg, JARDIANCE® 25 mg o placebo. Los pacientes se mantuvieron con una dosis estable de insulina previo al enrolamiento, durante el período de preinclusión y durante las primeras 18 semanas de tratamiento. La media de la dosis diaria total de insulina en el nivel basal del estudio para JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo fue 88,6 UI, 90,4 UI y 89,9 UI, respectivamente.
JARDIANCE® 10 mg o 25 mg a diario usado en combinación con IDM de insulina (con o sin metformina) proporcionó reducciones estadísticamente significativas en la HbA1c en comparación con el placebo luego de 18 semanas de tratamiento (ver Tabla 11).
Tabla 11. Resultados de la Semana 18 obtenidos a partir de un estudio controlado con placebo para JARDIANCE® en combinación con insulina y con o sin metformina
|
JARDIANCE 10 mg + Insulina +/- Metformina N = 186 |
JARDIANCE 25 mg + Insulina +/- Metformina N = 189 |
Placebo + Insulina +/- Metformina N = 188 |
|
|
HbA1c (%)a |
|||
|
Nivel basal (media) |
8,4 |
8,3 |
8,3 |
|
Cambio respecto del nivel basal (media ajustada) |
-0,9 |
-1,0 |
-0,5 |
|
Diferencia respecto del placebo (media ajustada) (CI 95%) |
-0,4b (-0,6; -0,3) |
-0,5b (-0,7; -0,4) |
-- |
|
a Población por intención de tratamiento modificada (mITT). Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 18. En la Semana 18, se imputó un 23,7%, 22,8% y 23,4% para los pacientes aleatorizados a JARDIANCE® 10 mg, JARDIANCE® 25 mg y placebo, respectivamente. b Valor p derivado de ANCOVA < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento, función renal, región geográfica y medicación de base). |
|||
Durante un período de extensión con tratamiento por un lapso de hasta 52 semanas, se podía ajustar la dosis de la insulina para lograr los niveles objetivo de glucosa definidos. El cambio respecto del nivel basal en la HbA1c se mantuvo de las 18 a las 52 semanas con JARDIANCE® 10 mg y con JARDIANCE® 25 mg. Luego de 52 semanas, JARDIANCE® 10 mg o 25 mg a diario condujo a una reducción porcentual del peso corporal estadísticamente mayor en comparación con el placebo (valor p < 0,0001). La media del cambio en el peso corporal respecto del nivel basal fue -1,95 kg para JARDIANCE® 10 mg y -2,04 kg para JARDIANCE® 25 mg.
Deterioro renal: Un total de 738 pacientes con diabetes tipo 2 y un valor de eGFR en el nivel basal de menos de 90 mL/min/1,73 m2 participaron en un estudio aleatorizado, doble ciego, con control de placebo, de grupos paralelos, destinado a evaluar la eficacia y seguridad de JARDIANCE® en pacientes con diabetes tipo 2 y deterioro renal. La población de estudio estuvo conformada por 290 pacientes con deterioro renal leve (eGFR de 60 a menos de 90 mL/min/1,73 m2), 374 pacientes con deterioro renal moderado (eGFR de 30 a menos de 60 mL/min/1,73 m2) y 74 pacientes con deterioro renal severo (eGFR de menos de 30 mL/min/1,73 m2). Un total de 194 pacientes con deterioro renal moderado tenían un valor de eGFR en el nivel basal de 30 a menos de 45 mL/min/1,73 m2 y 180 pacientes tenían un valor de eGFR en el nivel basal de 45 a menos de 60 mL/min/1,73 m2.
En la Semana 24, JARDIANCE® 25 mg proporcionó una reducción estadísticamente significativa en la HbA1c en comparación con el placebo en los pacientes con deterioro renal leve a moderado (ver Tabla 12). También se observó una reducción estadísticamente significativa en comparación con el placebo con JARDIANCE® 25 mg en los pacientes con deterioro renal leve (-0,7 (CI 95%: -0,9; -0,5)) o moderado (-0,4 (CI 95%: -0,6; -0,3)) y con JARDIANCE® 10 mg en los pacientes con deterioro renal leve (-0,5 (CI 95%: -0,7; -0,3)).
La eficacia hipoglucemiante de JARDIANCE® 25 mg disminuyó conforme menor fue el nivel de función renal dentro del rango de deterioro leve a moderado. Las medias de mínimos cuadrados de los cambios en la HbA1c a las 24 semanas fueron -0,6%, -0,5% y -0,2% para los pacientes con una eGFR en el nivel basal de 60 a menos de 90 mL/min/1,73 m2, de 45 a menos de 60 mL/min/1,73 m2 y de 30 a menos de 45 mL/min/1,73 m2, respectivamente (ver Posología y administración y Uso en poblaciones específicas). Para el placebo, las medias de mínimos cuadrados de los cambios en la HbA1c a las 24 semanas fueron 0,1%, -0,1% y 0,2% para los pacientes con una eGFR en el nivel basal de 60 a menos de 90 mL/min/1,73 m2, de 45 a menos de 60 mL/min/1,73 m2 y de 30 a menos de 45 mL/min/1,73 m2, respectivamente.
Tabla 12. Resultados de la Semana 24 (LOCF) obtenidos a partir de un estudio controlado con placebo para JARDIANCE® en pacientes con diabetes tipo 2 y deterioro renal
|
Deterioro leve y moderadob |
|
|
JARDIANCE® 25 mg |
|
|
HbA1c |
|
|
Cantidad de pacientes |
n = 284 |
|
Comparación vs. placebo (media ajustada) (CI 95%) |
-0,5a (-0,6; -0,4) |
|
a Valor p < 0,0001 (HbA1c: el modelo ANCOVA incluye HbA1c del nivel basal, tratamiento, función renal y medicación de base). b eGFR de 30 a menos de 90 mL/min/1,73 m2 - Población por intención de tratamiento modificada. Se utilizó la última observación obtenida durante el estudio (LOCF) para imputar los datos faltantes en la Semana 24. En la Semana 24, se imputó un 24,6% y 26,2% para los pacientes aleatorizados a JARDIANCE® 25 mg y placebo, respectivamente. |
|
Para los pacientes con deterioro renal severo, los análisis de los cambios en la HbA1c y la GPA no evidenciaron ningún efecto discernible del tratamiento para JARDIANCE® 25 mg en comparación con el placebo (ver Posología y administración y Uso en poblaciones específicas).
Desenlaces cardiovasculares en pacientes con diabetes mellitus tipo 2 y enfermedad cardiovascular ateroesclerótica: El efecto de JARDIANCE® sobre el riesgo cardiovascular en pacientes adultos con diabetes tipo 2 y enfermedad cardiovascular ateroesclerótica estable y establecida se evaluó en el estudio EMPA-REG OUTCOME, un ensayo multicéntrico, multinacional, aleatorizado, doble ciego, de grupos paralelos. Este estudio comparó el riesgo de experimentar un evento adverso cardiovascular importante (MACE) entre JARDIANCE® y placebo cuando éstos se sumaron a, y se utilizaron concomitantemente con, los tratamientos del estándar de atención para la diabetes y la enfermedad cardiovascular ateroesclerótica. Las medicaciones antidiabéticas coadministradas debían mantenerse estables durante las primeras 12 semanas del estudio. A partir de ese momento, las terapias para la diabetes y las enfermedades ateroescleróticas podían ser ajustadas, a criterio de los investigadores, a fin de asegurar que los pacientes fueran tratados de acuerdo con el estándar de atención para tales afecciones.
Un total de 7020 pacientes fueron tratados (JARDIANCE® 10 mg = 2345; JARDIANCE® 25 mg = 2342; placebo = 2333) y sometidos a seguimiento durante una mediana de 3,1 años. Aproximadamente el 72% de la población del estudio era caucásica, el 22% era asiática y el 5% era de raza negra. La media de la edad fue 63 años y aproximadamente el 72% de los pacientes eran de sexo masculino.
Todos los pacientes del estudio tenían diabetes mellitus tipo 2 inadecuadamente controlada en el nivel basal (HbA1c mayor o igual a 7%). La media de la HbA1c en el nivel basal fue de 8,1%, y el 57% de los participantes habían tenido diabetes por más de 10 años. Aproximadamente el 31%, 22% y 20% de los pacientes informaron antecedentes de neuropatía, retinopatía y nefropatía a los investigadores, respectivamente, y la media de eGFR fue 74 mL/min/1,73 m2. En el nivel basal del estudio, los pacientes estaban en tratamiento con una (~ 30%) o más (~ 70%) medicaciones antidiabéticas, incluyendo metformina (74%), insulina (48%) y sulfonilurea (43%).
Todos los pacientes tenían enfermedad cardiovascular ateroesclerótica establecida en el nivel basal, incluyendo uno (82%) o más (18%) de los siguientes factores: antecedentes documentados de arteriopatía coronaria (76%), accidente cerebrovascular (23%) o enfermedad arterial periférica (21%). En el nivel basal, la media de presión arterial sistólica fue 136 mmHg, la media de presión arterial diastólica fue 76 mmHg, la media de LDL fue 86 mg/dL, la media de HDL fue 44 mg/dL y la media del cociente de albúmina-creatinina en orina (UACR) fue 175 mg/g. En el nivel basal, aproximadamente el 81% de los pacientes estaban siendo tratados con inhibidores del sistema de renina-angiotensina, el 65% con beta bloqueantes, el 43% con diuréticos, el 77% con estatinas y el 86% con antiplaquetarios (mayormente aspirina).
El criterio de valoración primario en el estudio EMPA-REG OUTCOME fue el tiempo hasta el primer acaecimiento de un evento adverso cardíaco importante (MACE). Un evento adverso cardíaco importante se definió como el acaecimiento de una muerte cardiovascular o de un infarto de miocardio (IM) no fatal o un accidente cerebrovascular no fatal. El plan de análisis estadístico había preespecificado que las dosis de 10 y 25 mg se combinarían. Se utilizó un modelo de riesgos proporcionales de Cox para evaluar la no inferioridad en función del margen de riesgo preespecificado de 1,3 para el cociente de riesgo de MACE y la superioridad en términos del MACE en el caso de que se demostrara la no inferioridad. El error de tipo 1 se controló a través de pruebas múltiples utilizando una estrategia de pruebas jerárquicas.
JARDIANCE® redujo significativamente el riesgo de primer acaecimiento del criterio de valoración compuesto primario de muerte cardiovascular, infarto de miocardio no fatal o accidente cerebrovascular no fatal (cociente de riesgo (HR): 0,86; CI 95% 0,74; 0,99). El efecto del tratamiento se debió a una reducción significativa en el riesgo de muerte cardiovascular en los sujetos aleatorizados a empagliflozina (HR: 0,62;
CI 95% 0,49; 0,77), sin ningún cambio en el riesgo de infarto de miocardio no fatal o accidente cerebrovascular no fatal (ver Tabla 13 y Figuras 5 y 6). Los resultados para las dosis de empagliflozina de 10 mg y 25 mg fueron concordantes con los resultados obtenidos para los grupos de dosis combinados.
Tabla 13. Efecto del tratamiento para el criterio de valoración compuesto primario y sus componentesa
|
Placebo N = 2333 |
JARDIANCE® N = 4687 |
Cociente de riesgo vs. placebo (CI 95%) |
|
|
Criterio de valoración compuesto de muerte cardiovascular, infarto de miocardio no fatal y accidente cerebrovascular no fatal (tiempo hasta el primer acaecimiento)b |
282 (12,1%) |
490 (10,5%) |
0,86 (0,74; 0,99) |
|
Infarto de miocardio no fatalc |
121 (5,2%) |
213 (4,5%) |
0,87 (0,70; 1,09) |
|
Accidente cerebrovascular no fatalc |
60 (2,6%) |
150 (3,2%) |
1,24 (0,92; 1,67) |
|
Muerte cardiovascularc |
137 (5,9%) |
172 (3,7%) |
0,62 (0,49; 0,77) |
|
a Conjunto tratado (pacientes que habían recibido al menos una dosis del fármaco en estudio). b Valor p para superioridad (bilateral) 0,04. c Número total de eventos. |
|||
Figura 5. Incidencia acumulada estimada de primer MACE
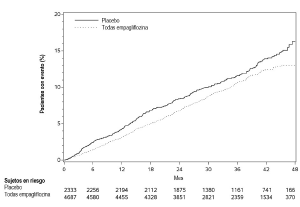
Figura 6. Incidencia acumulada estimada de muerte cardiovascular
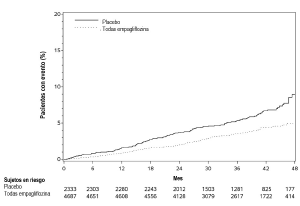
La eficacia de JARDIANCE® en términos de muerte cardiovascular fue en general concordante entre los principales subgrupos demográficos y de enfermedad.
El estado vital se obtuvo para el 99,2% de los sujetos en el ensayo. Se registró un total de 463 muertes durante el estudio EMPA-REG OUTCOME. La mayoría de estas muertes se clasificaron como muertes cardiovasculares. Las muertes no cardiovasculares fueron sólo una pequeña proporción de las muertes y estuvieron equilibradas entre los grupos de tratamiento (2,1% en los pacientes tratados con JARDIANCE® y 2,4% de los pacientes tratados con placebo).
DATOS PRECLÍNICOS SOBRE SEGURIDAD: Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos según los estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas, genotoxicidad, fertilidad y desarrollo embrionario temprano.
En estudios de toxicidad a largo plazo con roedores y perros, se observaron signos de toxicidad a exposiciones iguales o superiores a 10 veces la dosis clínica de empagliflozina. La mayor parte de las toxicidades fueron compatibles con la farmacología secundaria relacionada con pérdida de glucosa por la orina y desequilibrios electrolíticos, incluida la disminución del peso y la grasa corporales, aumento del consumo de alimentos, diarrea, deshidratación, disminución de los niveles de glucosa sérica y aumentos en otros parámetros séricos que reflejan el aumento del metabolismo de las proteínas y la gluconeogénesis, cambios urinarios como la poliuria y la glucosuria y cambios microscópicos, incluida la mineralización en el riñón y en algunos tejidos blandos y vasculares. Las evidencias microscópicas de los efectos de una farmacología exagerada en el riñón que se observaron en algunas especies incluyeron dilatación tubular y mineralización tubular y pélvica a aproximadamente 4 veces la exposición del AUC clínica de la empagliflozina asociada a la dosis de 25 mg.
Empagliflozina no es genotóxica.
En un estudio de carcinogenicidad de 2 años de duración, empagliflozina no aumentó la incidencia de tumores en ratas hembra hasta la dosis máxima de 700 mg/kg/día, lo que corresponde a aproximadamente 72 veces la exposición clínica máxima del AUC a empagliflozina. En las ratas macho, a las dosis más altas se observaron lesiones proliferativas vasculares benignas relacionadas con el tratamiento (hemangiomas) del ganglio linfático mesentérico, pero no a la dosis de 300 mg/kg/día, lo que corresponde a aproximadamente 26 veces la exposición clínica máxima a empagliflozina. Se observaron tumores celulares intersticiales en los testículos con una mayor incidencia en ratas a 300 mg/kg/día o más, pero no a 100 mg/kg/día, lo que corresponde a aproximadamente 18 veces la exposición clínica máxima a empagliflozina. Ambos tumores son frecuentes en ratas, pero es improbable que sean relevantes en los humanos.
Empagliflozina no aumentó la incidencia de tumores en ratones hembra a dosis de hasta 1000 mg/kg/día, lo que corresponde a aproximadamente 62 veces la exposición clínica máxima a empagliflozina. Empagliflozina indujo tumores renales en ratones macho a dosis de 1000 mg/kg/día, pero no a la dosis de 300 mg/kg/día, lo que corresponde a aproximadamente 11 veces la exposición clínica máxima a empagliflozina. El modo de acción de estos tumores depende de la predisposición natural del ratón macho a presentar una patología renal y una vía metabólica que no refleja la de los humanos. Los tumores renales de los ratones macho no se consideran relevantes para los humanos.
A exposiciones suficientemente superiores a la exposición en humanos después de dosis terapéuticas, empagliflozina no tuvo efectos adversos sobre la fertilidad o el desarrollo embrionario temprano. La administración de empagliflozina durante el periodo de organogénesis no fue teratogénica. Solo a dosis tóxicas para la madre, empagliflozina también provocó que los huesos de las extremidades de la rata se doblasen, así como un aumento de la muerte embriofetal en el conejo.
En estudios de la toxicidad prenatal y posnatal en ratas, se observó una reducción en el aumento de peso de la descendencia a exposiciones maternas de aproximadamente 4 veces la exposición clínica máxima a empagliflozina. Dicho efecto no se observó a la exposición sistémica igual a la máxima exposición clínica a la empagliflozina. La relevancia de este hallazgo para los humanos no está clara.
TOXICOLOGÍA NO CLÍNICA:
Carcinogenia, mutagenia y deterioro de la fertilidad:
Carcinogenia: La carcinogenia se evaluó en estudios de 2 años de duración realizados en ratones CD-1 y ratas Wistar. La empagliflozina no incrementó la incidencia de tumores en ratas hembra a las que se les administraron dosis de 100, 300 o 700 mg/kg/día (hasta 72 veces la exposición de la dosis clínica máxima de 25 mg). En las ratas macho, los hemangiomas de los ganglios linfáticos mesentéricos se incrementaron significativamente con el nivel de dosis de 700 mg/kg/día, o aproximadamente 42 veces la exposición de una dosis clínica de 25 mg. La empagliflozina no incrementó la incidencia de tumores en ratones hembra a los que se les administraron dosis de 100, 300 o 1000 mg/kg/día (hasta 62 veces la exposición de una dosis clínica de 25 mg). Se observaron carcinomas y adenomas de los túbulos renales en los ratones machos con el nivel de dosis de 1000 mg/kg/día, que es aproximadamente 45 veces la exposición de la dosis clínica máxima de 25 mg. Estos tumores pueden estar asociados con una vía metabólica predominantemente presente en el riñón del ratón macho.
Mutagenia: La empagliflozina no fue mutagénica ni clastogénica en la prueba de mutagenicidad bacteriana de Ames in vitro con y sin activación metabólica, la prueba de células de linfoma de ratón L5178Y tk+/- in vitro y una prueba de micronúcleo in vivo en ratas.
Deterioro de la fertilidad: La empagliflozina no tuvo ningún efecto sobre el apareamiento, la fertilidad o el desarrollo embrionario temprano en ratas macho o hembra tratadas con niveles de hasta la dosis alta de 700 mg/kg/día (aproximadamente 155 veces la dosis clínica de 25 mg en machos y hembras, respectivamente).
ADVERTENCIAS Y PRECAUCIONES:
Hipotensión:
JARDIANCE® provoca contracción del volumen intravascular. Puede producirse hipotensión sintomática tras el inicio de JARDIANCE® (ver Reacciones adversas, particularmente en pacientes con deterioro renal, pacientes de edad avanzada, pacientes con presión arterial sistólica baja y pacientes tratados con diuréticos. Antes de iniciar JARDIANCE®, debe evaluarse al paciente para determinar si existe contracción del volumen y corregir el estado del volumen en el caso de que estuviera indicado. Debe observarse al paciente en pos de la posible aparición de signos y síntomas de hipotensión tras el inicio de la terapia e incrementarse el control en aquellas situaciones clínicas en las que es dable esperar que se produzca contracción del volumen (ver Uso en poblaciones específicas).
Cetoacidosis:
Se han identificado casos de cetoacidosis, una afección seria y potencialmente fatal que requiere hospitalización de urgencia, en el contexto de la vigilancia post-comercialización en pacientes con diabetes mellitus tipo 1 y tipo 2 que recibían inhibidores del cotransportador de sodio-glucosa tipo 2 (SGLT2), entre ellos JARDIANCE®. Se han informado casos fatales de cetoacidosis en pacientes tratados con JARDIANCE®. JARDIANCE® no está indicado para el tratamiento de pacientes con diabetes mellitus tipo 1 (ver Indicaciones y uso).
Los pacientes tratados con JARDIANCE® que presentan signos y síntomas compatibles con acidosis metabólica severa deben ser evaluados para determinar la posible presencia de cetoacidosis independientemente de los niveles de glucosa en sangre, ya que puede existir cetoacidosis asociada con JARDIANCE® incluso aunque los niveles de glucosa en sangre sean inferiores a 250 mg/dL. En el caso de existir sospecha de cetoacidosis, debe suspenderse la administración de JARDIANCE®, y debe evaluarse al paciente e instituirse tratamiento sin demora. El tratamiento de la cetoacidosis puede requerir reemplazo de carbohidratos, líquidos e insulina.
En muchos de los informes post-comercialización, y en particular en los pacientes con diabetes tipo 1, la presencia de cetoacidosis no fue inmediatamente reconocida y la instauración del tratamiento se demoró debido a que los niveles de glucosa en sangre que presentaba el paciente eran inferiores a los que típicamente se espera para la cetoacidosis diabética (a menudo menos de 250 mg/dL). Los signos y síntomas en la presentación fueron compatibles con deshidratación y acidosis metabólica severa, e incluyeron náuseas, vómitos, dolor abdominal, malestar generalizado y disnea. En algunos casos, pero no todos, se identificaron factores predisponentes para la cetoacidosis tales como reducción de la dosis de insulina, enfermedad febril aguda, reducción de la ingesta calórica debido a enfermedad o cirugía, trastornos pancreáticos sugestivos de deficiencia de insulina (p. ej., diabetes tipo 1, antecedentes de pancreatitis o cirugía pancreática) y consumo excesivo de alcohol.
Antes de iniciar JARDIANCE®, deben considerarse los factores de los antecedentes médicos del paciente que puedan generar una predisposición a la cetoacidosis, entre ellos deficiencia de insulina pancreática por cualquier causa, restricción calórica y consumo excesivo de alcohol. En los pacientes tratados con JARDIANCE®, debe considerarse el monitoreo en pos de cetoacidosis y la suspensión temporaria de la administración de JARDIANCE® en aquellas situaciones clínicas que predisponen al paciente a la cetoacidosis (por ejemplo, ayuno prolongado por enfermedad aguda o cirugía).
Lesión renal aguda y deterioro de la función renal: JARDIANCE® provoca contracción del volumen intravascular (ver Advertencias y precauciones) y puede dar lugar a un deterioro de la función renal (ver Reacciones adversas). Ha habido informes post-comercialización de lesión renal aguda, que en algunos casos requirió hospitalización y diálisis, en pacientes que recibían inhibidores del SGLT2, entre ellos JARDIANCE®; algunos informes involucraron pacientes menores de 65 años de edad.
Antes de iniciar JARDIANCE®, deben considerarse los factores que pueden predisponer a los pacientes a una lesión renal aguda, incluyendo hipovolemia, insuficiencia renal crónica, insuficiencia cardíaca congestiva y medicaciones concomitantes (diuréticos, inhibidores de la ECA, BRAs, AINEs). Debe considerarse la suspensión temporaria de JARDIANCE® en todo contexto de ingesta oral reducida (como enfermedad aguda o ayuno) o pérdida de líquidos (por ejemplo, enfermedad gastrointestinal o exposición excesiva al calor); debe realizarse un monitoreo de los pacientes en pos de signos y síntomas de lesión renal aguda. En el caso de producirse una lesión renal aguda, debe interrumpirse sin demora la administración de JARDIANCE® e instituirse el debido tratamiento.
JARDIANCE® aumenta los niveles de creatinina sérica y reduce la eGFR. Los pacientes con hipovolemia pueden ser más susceptibles a estos cambios. Pueden producirse anomalías de la función renal tras el inicio de JARDIANCE® (ver Reacciones adversas). Debe evaluarse la función renal previo al inicio de JARDIANCE® y realizarse un control periódico en lo sucesivo. Se recomienda un control más frecuente de la función renal en los pacientes con un valor de eGFR por debajo de 60 mL/min/1,73 m2. No se recomienda el uso de JARDIANCE® cuando el valor de eGFR es persistentemente inferior a 45 mL/min/1,73 m2, y está contraindicado en pacientes con un valor de eGFR inferior a 30 mL/min/1,73 m2 (ver Posología y administración, Contraindicaciones y Uso en poblaciones específicas).
Urosepticemia y pielonefritis: Ha habido informes post-comercialización de infecciones serias de las vías urinarias, incluyendo urosepticemia y pielonefritis, que requirieron hospitalización en pacientes que recibían inhibidores del SGLT2, entre ellos JARDIANCE®. El tratamiento con inhibidores del SGLT2 incrementa el riesgo de infecciones de las vías urinarias. Debe evaluarse a los pacientes para detectar la posible presencia de signos y síntomas de infección urinaria e instaurarse tratamiento sin demora, de estar indicado (ver Reacciones adversas).
Hipoglucemia con el uso concomitante con insulina y secretagogos de insulina: Es sabido que la insulina y los secretagogos de insulina causan hipoglucemia. Tal riesgo de hipoglucemia aumenta cuando JARDIANCE® se utiliza en combinación con secretagogos de insulina (por ejemplo, sulfonilurea) o con insulina (ver Reacciones adversas). Por lo tanto, es posible que se requiera una dosis menor del secretagogo de insulina o de la insulina para reducir el riesgo de hipoglucemia cuando se usan en combinación con JARDIANCE®.
Infecciones micóticas genitales: JARDIANCE® incrementa el riesgo de infecciones micóticas genitales (ver Reacciones adversas). Los pacientes con antecedentes de infecciones micóticas genitales crónicas o recurrentes fueron más propensos a desarrollar infecciones micóticas genitales. Debe controlarse a los pacientes e instaurar tratamiento según corresponda.
Reacciones de hipersensibilidad: Ha habido informes post-comercialización de reacciones de hipersensibilidad serias (p. ej., angioedema) en pacientes tratados con JARDIANCE®. En el caso de producirse una reacción de hipersensibilidad, debe discontinuarse la administración de JARDIANCE®; debe instaurarse tratamiento sin demora de conformidad con las pautas del estándar de atención y debe observarse al paciente hasta la resolución de los signos y síntomas. JARDIANCE® está contraindicado en los pacientes con antecedentes de reacción de hipersensibilidad seria a la empagliflozina o a cualquiera de los excipientes presentes en JARDIANCE® (ver Contraindicaciones).
Niveles elevados de colesterol de lipoproteína de baja densidad (LDL-C): Pueden producirse elevaciones de los niveles de LDL-C con JARDIANCE® (ver Reacciones adversas). Debe controlarse a los pacientes e instaurar tratamiento según corresponda.
Amputación de miembros inferiores: Se ha observado un aumento de casos de amputación de miembros inferiores (principalmente en los dedos de los pies) en ensayos clínicos a largo plazo con otro inhibidor SGLT2. Se desconoce si esto constituye un efecto de clase. Como con todos los pacientes diabéticos, es importante aconsejar a los pacientes acerca del cuidado rutinario preventivo del pie.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO
General: JARDIANCE no debe utilizarse en pacientes con diabetes tipo 1 ni para el tratamiento de la cetoacidosis diabética.
Uso en pacientes con insuficiencia renal: El tratamiento con JARDIANCE no debe iniciarse en pacientes con una TFGe inferior a 60 ml/min/1,73 m2 o un CrCl <60 ml/min. En pacientes que toleran la empagliflozina y cuya TFGe se encuentra sistemáticamente por debajo de 60 ml/min/1,73 m2 o con un CrCl < 60 ml/min, la dosis de empagliflozina debe ajustarse o mantenerse en 10 mg una vez al día. El tratamiento con empagliflozina debe interrumpirse cuando la TFGe se encuentre sistemáticamente por debajo de 45 ml/min/1,73 m2 o el CrCl se encuentre sistemáticamente por debajo de 45 ml/min. La empagliflozina no debe utilizarse en pacientes con ERT ni en pacientes sometidos a diálisis, pues no se espera que vaya a ser eficaz en estos pacientes.
Monitorización de la función renal: Debido a su mecanismo de acción, la eficacia de la empagliflozina depende de la función renal. Por lo tanto, se recomienda evaluar la función renal tal como se indica a continuación:
• Antes de iniciar el tratamiento con empagliflozina y periódicamente durante el tratamiento, al menos una vez al año.
• Antes de iniciar el tratamiento con cualquier medicamento concomitante que pueda tener un impacto negativo en la función renal.
Lesión hepática: Se han notificado casos de lesión hepática con el uso de empagliflozina en ensayos clínicos. No se ha establecido una relación causal entre la empagliflozina y la lesión hepática.
Pacientes de edad avanzada: El efecto de la empagliflozina en la eliminación de glucosa por la orina se asocia a la diuresis osmótica, lo que podría afectar al estado de hidratación. Los pacientes de 75 años de edad o mayores pueden presentar un mayor riesgo de hipovolemia. Un mayor número de estos pacientes tuvieron reacciones adversas relacionadas con la hipovolemia en comparación con los tratados con placebo.
La experiencia terapéutica en pacientes de 85 años de edad o mayores es limitada. No se recomienda iniciar el tratamiento con empagliflozina en esta población.
Uso en pacientes con riesgo de hipovolemia: En base al modo de acción de los inhibidores del SGLT-2, la diuresis osmótica que acompaña a la glucosuria terapéutica puede provocar una disminución moderada de la presión arterial. Por lo tanto, se debe tener precaución en los pacientes para los que una caída de la presión arterial inducida por la empagliflozina pudiera suponer un riesgo, tales como pacientes con enfermedad cardiovascular conocida, pacientes en tratamiento antihipertensivo con antecedentes de hipotensión o pacientes de 75 años de edad o mayores.
En caso de enfermedades que puedan conducir a una pérdida de líquidos (por ejemplo, una enfermedad gastrointestinal), se recomienda una estrecha monitorización de la volemia (por ejemplo, exploración física, medición de la presión arterial, pruebas de laboratorio, incluyendo el hematocrito) y de los electrolitos en el caso de pacientes que reciben empagliflozina. Se debe valorar la interrupción temporal del tratamiento con empagliflozina hasta que se corrija la pérdida de líquidos.
Infecciones del tracto urinario: La frecuencia global de infecciones del tracto urinario notificadas como efecto adverso fue similar en los pacientes tratados con empagliflozina 25 mg y en los tratados con placebo, y mayor en los pacientes tratados con empagliflozina 10 mg. Las infecciones complicadas del tracto urinario (por ejemplo, pielonefritis o urosepsis) ocurrieron con una frecuencia similar en los pacientes tratados con empagliflozina en comparación con placebo. Sin embargo, en el caso de pacientes con infecciones complicadas del tracto urinario, debe valorarse la interrupción temporal del tratamiento con empagliflozina.
Insuficiencia cardíaca: La experiencia en la clase I-II de la New York Heart Association (NYHA) es limitada, y no existe experiencia en estudios clínicos con empagliflozina en la clase III-IV de la NYHA.
Análisis de orina: Debido a su mecanismo de acción, los pacientes que están tomando JARDIANCE presentarán un resultado positivo para la glucosa en la orina.
Lactosa: Los comprimidos contienen lactosa. Los pacientes con intolerancia hereditaria a la galactosa, insuficiencia de lactasa de Lapp o problemas de absorción de glucosa-galactosa no deben tomar este medicamento.
PRECAUCIONES ESPECIALES DE ELIMINACIÓN: La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
“Consulte a su médico o farmacéutico, según proceda, para cualquier aclaración sobre la utilización del producto“.
Fabricado por:
Boehringer Ingelheim Pharma GmbH & CO. KG. – Alemania
Importado por:
BOEHRINGER INGELHEIM PERÚ S.A.C
Av. Canaval y Moreyra 480, Piso 20
San Isidro - Telf.: (51-1) 212-4132
POSOLOGÍA Y ADMINISTRACIÓN:
Posología recomendada:
La dosis recomendada de JARDIANCE® es 10 mg una vez al día por la mañana, administrada con o sin alimentos. En los pacientes que toleran JARDIANCE®, la dosis puede aumentarse a 25 mg (ver Estudios clínicos).
En los pacientes con depleción del volumen, se recomienda corregir dicha alteración antes de iniciar JARDIANCE® (ver Advertencias y precauciones, Uso en poblaciones específicas e Información para el asesoramiento del paciente ).
Pacientes con deterioro renal: Se recomienda la evaluación de la función renal previo al inicio de JARDIANCE® y a intervalos periódicos en lo sucesivo.
No debe iniciarse JARDIANCE® en los pacientes con un valor de eGFR inferior a 45 mL/min/1,73 m2.
No es necesario ningún ajuste de dosis en pacientes con un valor de eGFR mayor o igual a 45 mL/min/1,73 m2.
Debe interrumpirse el uso de JARDIANCE® si el valor de eGFR es persistentemente inferior a 45 mL/min/1,73 m2 (ver Advertencias y precauciones y Uso en poblaciones específicas).
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
Posología:
• Monoterapia y tratamiento adicional en combinación: La dosis inicial recomendada es de 10 mg de empagliflozina una vez al día, tanto en monoterapia como en tratamiento adicional en combinación con otros medicamentos hipoglucemiantes, incluida la insulina. En los pacientes que toleran empagliflozina 10 mg una vez al día que tengan una TFGe ≥60 ml/min/1,73 m2 y necesiten un control glucémico más estricto, la dosis puede aumentarse a 25 mg una vez al día. La dosis máxima diaria es de 25 mg.
Cuando la empagliflozina se utiliza en combinación con una sulfonilurea o con insulina, puede considerarse una dosis más baja de la sulfonilurea o de insulina para reducir el riesgo de hipoglucemia.
• Poblaciones especiales:
— Pacientes con insuficiencia renal: Debido a su mecanismo de acción, la eficacia de la empagliflozina depende de la función renal. No se precisa un ajuste de la dosis en pacientes con una tasa de filtración glomerular estimada, TFGe ≥60 ml/min/1,73 m2 o un aclaramiento de creatinina, CrCl ≥60 ml/min.
El tratamiento con empagliflozina no debe iniciarse en pacientes con una TFGe <60 ml/min/1,73 m2 o un CrCl <60 ml/min. En pacientes que toleran la empagliflozina y cuya TFGe desciende sistemáticamente por debajo de 60 ml/min/1,73 m2 o con un CrCl por debajo de 60 ml/min, la dosis de empagliflozina debe ajustarse o mantenerse en 10 mg una vez al día. El tratamiento con empagliflozina debe interrumpirse cuando la TFGe se encuentre sistemáticamente por debajo de 45 ml/min/1,73 m2 o el CrCl se encuentre sistemáticamente por debajo de 45 ml/min.
La empagliflozina no debe utilizarse en pacientes con enfermedad renal terminal (ERT) ni en pacientes sometidos a diálisis, pues no se espera que vaya a ser eficaz en estos pacientes.
— Pacientes con insuficiencia hepática: No se precisa un ajuste de dosis en pacientes con insuficiencia hepática. La exposición a empagliflozina aumenta en pacientes con insuficiencia hepática grave. La experiencia terapéutica en pacientes con insuficiencia hepática grave es limitada y, por lo tanto, no se recomienda su uso en esta población.
— Pacientes de edad avanzada: No se recomienda un ajuste de dosis en función de la edad. En pacientes de 75 años de edad o mayores debe tenerse en cuenta que existe un mayor riesgo de hipovolemia. Dado que la experiencia terapéutica es limitada en pacientes de 85 años de edad o mayores, no se recomienda iniciar el tratamiento con empagliflozina en esta población.
— Población pediátrica: No se ha establecido todavía la seguridad y la eficacia de la empagliflozina en niños y adolescentes.
No hay datos disponibles.
Forma de administración: Los comprimidos pueden tomarse con o sin alimentos y deben tragarse enteros con agua. Si se olvida una dosis, ésta debe tomarse tan pronto como el paciente lo recuerde. No debe tomarse una dosis doble en el mismo día.
USO EN POBLACIONES ESPECÍFICAS:
Embarazo:
Resumen de riesgos: Sobre la base de datos obtenidos en animales que indican efectos adversos renales, no se recomienda el uso de JARDIANCE® durante el segundo y el tercer trimestre del embarazo.
Los limitados datos que existen disponibles en relación con el uso de JARDIANCE® en mujeres embarazadas no son suficientes para determinar el riesgo de anomalías congénitas importantes y aborto espontáneo asociado con el fármaco. Existen riesgos para la madre y el feto asociados con el control deficiente de la diabetes en el embarazo (ver Consideraciones clínicas).
En los estudios en animales, se observaron cambios renales adversos en las ratas cuando empagliflozina se administró durante el período de desarrollo renal correspondiente a la etapa final del segundo trimestre y el tercer trimestre del embarazo de los seres humanos. Dosis equivalentes a aproximadamente 13 veces la dosis clínica máxima provocaron dilataciones de los túbulos renales y de la pelvis renal que fueron reversibles. La empagliflozina no fue teratogénica en ratas y conejos en dosis de hasta 300 mg/kg/día, valor éste que equivale a aproximadamente 48 veces y 128 veces, respectivamente, la dosis clínica máxima de 25 mg, cuando se administró durante la organogénesis (ver Datos).
El riesgo de base estimado para el acaecimiento de anomalías congénitas importantes es 6-10% en las mujeres con diabetes pregestacional con un valor de HbA1c > 7, y se han informado incluso valores de nada menos que 20-25% en las mujeres con niveles de HbA1c > 10. Se desconoce el riesgo de base estimado de aborto espontáneo para la población objetivo de la indicación. En la población general de los Estados Unidos, el riesgo de base estimado de anomalías congénitas importantes y aborto espontáneo en embarazos clínicamente reconocidos es del 2-4% y del 15-20%, respectivamente.
Consideraciones clínicas:
Riesgo materno y/o embriofetal asociado con la enfermedad: La diabetes mal controlada durante el embarazo aumenta el riesgo materno de cetoacidosis diabética, preeclampsia, abortos espontáneos, parto prematuro, mortinatalidad y complicaciones del parto. La diabetes mal controlada aumenta el riesgo fetal de anomalías congénitas importantes, mortinatalidad y morbilidad asociada con macrosomía.
Datos:
Datos obtenidos en animales: La empagliflozina administrada en forma directa a ratas jóvenes desde el día posnatal (DPN) 21 hasta el DPN 90 en dosis de 1, 10, 30 y 100 mg/kg/día causó incrementos del peso de los riñones y dilatación de túbulos renales y pelvis renal en el nivel de dosis de 100 mg/kg/día, que es aproximadamente equivalente a 13 veces la dosis clínica máxima de 25 mg, sobre la base del AUC. Estos hallazgos no se observaron tras un período de recuperación sin administración del fármaco de 13 semanas. Estos desenlaces se produjeron con una exposición al fármaco coincidente con períodos de desarrollo renal de las ratas que corresponden a fines del segundo trimestre y tercer trimestre del desarrollo renal del ser humano.
En los estudios de desarrollo embriofetal en ratas y conejos, la empagliflozina se administró durante intervalos que coincidieron con el período de organogénesis del primer trimestre en el ser humano. Dosis de hasta 300 mg/kg/día, nivel éste equivalente a aproximadamente 48 veces (ratas) y 128 veces (conejos) la dosis clínica máxima de 25 mg (sobre la base del AUC), no produjeron efectos adversos en el desarrollo. En las ratas, con dosis más altas de empagliflozina que causaron toxicidad materna, las malformaciones de los huesos de las extremidades aumentaron en los fetos con el nivel de dosis de 700 mg/kg/día o 154 veces la dosis clínica máxima de 25 mg. La empagliflozina atraviesa la barrera placentaria y se distribuye al tejido fetal en las ratas. En los conejos, dosis más altas de empagliflozina condujeron a toxicidad materna y fetal en el nivel de dosis de 700 mg/kg/día, o el equivalente a 139 veces la dosis clínica máxima de 25 mg.
En estudios de desarrollo pre- y post-natal en ratas preñadas, la empagliflozina se administró desde el día 6 de la gestación hasta el día 20 de la lactancia (destete) en niveles de dosis de hasta 100 mg/kg/día (aproximadamente 16 veces la dosis clínica máxima de 25 mg) sin que se observara toxicidad materna. Se observó un menor peso corporal en las crías con la administración de niveles de dosis iguales o superiores a 30 mg/kg/día (aproximadamente 4 veces la dosis clínica máxima de 25 mg).
Lactancia:
Resumen de riesgos: No existe información respecto de la presencia de JARDIANCE® en la leche humana, los efectos de JARDIANCE® en los lactantes o los efectos sobre la producción de leche. La empagliflozina está presente en la leche de ratas en período de lactancia (ver Datos). Puesto que la maduración del riñón humano se produce in utero y durante los 2 primeros años de vida, que es cuando puede producirse la exposición a través de la lactancia, puede haber riesgo para el riñón humano en desarrollo.
Debido al potencial de que se produzcan reacciones adversas serias en el lactante, lo que incluye el potencial de que la empagliflozina afecte el desarrollo renal posnatal, las mujeres deben ser informadas respecto de que el uso de JARDIANCE® está desaconsejado durante la lactancia.
Datos: La empagliflozina estuvo presente en un nivel bajo en los tejidos fetales de las ratas luego de una dosis oral única administrada a hembras preñadas en el día de gestación 18. En la leche de las ratas, la media del cociente leche-plasma se ubicó en un rango de 0,634 a 5, y fue mayor que 1 dentro del lapso de 2 a 24 horas posdosis. El valor de media máximo de 5 del cociente leche-plasma se produjo a las 8 horas posdosis, lo cual sugiere la acumulación de empagliflozina en la leche. En las ratas jóvenes expuestas en forma directa a empagliflozina, se evidenció un riesgo para el riñón en desarrollo (dilataciones de los túbulos renales y de la pelvis renal) durante la maduración.
Uso pediátrico: La seguridad y efectividad de JARDIANCE® en pacientes pediátricos menores de 18 años de edad no ha sido establecida.
Uso geriátrico: No se recomienda ningún cambio en la posología de JARDIANCE® en función de la edad (ver Posología y administración). En estudios en los que se evaluó la eficacia de empagliflozina en términos de la mejora del control glucémico en pacientes con diabetes tipo 2, un total de 2721 (32%) pacientes tratados con empagliflozina tenían 65 años de edad o más, y 491 (6%) tenían 75 años de edad o más. Se prevé que JARDIANCE® tendrá una eficacia glucémica reducida en los pacientes de edad avanzada con deterioro renal (ver Uso en poblaciones específicas). El riesgo de reacciones adversas relacionadas con depleción del volumen se incrementó en los pacientes de 75 años de edad o más a valores de 2,1%, 2,3% y 4,4% para placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg. El riesgo de infecciones de las vías urinarias se incrementó en los pacientes de 75 años de edad o más a valores del 10,5%, 15,7% y 15,1% en los pacientes aleatorizados a placebo, JARDIANCE® 10 mg y JARDIANCE® 25 mg, respectivamente (ver Advertencias y precauciones y Reacciones adversas).
Deterioro renal: La eficacia y la seguridad de JARDIANCE® se evaluaron en un estudio de pacientes con deterioro renal leve y moderado (ver Estudios clínicos). En este estudio, 195 pacientes expuestos a JARDIANCE® tenían una eGFR de entre 60 y 90 mL/min/1,73 m2, 91 pacientes expuestos a JARDIANCE® tenían una eGFR de entre 45 y 60 mL/min/1,73 m2 y 97 pacientes expuestos a JARDIANCE® tuvieron una eGFR de entre 30 y 45 mL/min/1,73 m2. El beneficio de la reducción de los niveles de glucosa aportado por JARDIANCE® 25 mg se redujo en los pacientes con empeoramiento de la función renal. Los riesgos del deterioro renal (ver Advertencias y precauciones), las reacciones adversas de depleción de volumen y las reacciones adversas relacionadas con infección de las vías urinarias se incrementaron conforme mayor fue el deterioro de la función renal.
En un estudio a gran escala de desenlaces cardiovasculares, hubo 1819 pacientes con eGFR por debajo de 60 mL/min/1,73 m2. Los hallazgos de muerte cardiovascular en este subgrupo fueron concordantes con los hallazgos generales (ver Estudios clínicos).
La eficacia y la seguridad de JARDIANCE® no han sido establecidas en pacientes con deterioro renal severo, con enfermedad renal terminal o en tratamiento de diálisis. No se prevé que JARDIANCE® sea eficaz en estas poblaciones de pacientes (ver Posología y administración, Contraindicaciones y Advertencias y precauciones).
Deterioro hepático: JARDIANCE® puede ser utilizado en pacientes con deterioro hepático (ver Farmacología clínica).
SOBREDOSIS:
En el caso de producirse una sobredosis con JARDIANCE®, póngase en contacto con el Centro de Control de Intoxicaciones. Deben aplicarse las medidas de soporte habituales (p. ej., eliminar del aparato gastrointestinal el material no absorbido, implementar un monitoreo clínico e instituir tratamiento de soporte) según sea necesario en función del estado clínico del paciente. La eliminación de la empagliflozina del organismo mediante hemodiálisis no ha sido estudiada.
En Uruguay: Ante una eventualidad concurrir al hospital, al Centro de Tóxico y Farmacovigilancia, Hospital de Clínicas-Facultad de Medicina, Tel. 1722.
INFORMACIÓN CONTENIDA EN EL INSERTO PARA EL PACIENTE:
JARDIANCE®
Empagliflozina
Comprimido recubierto
¿Cuál es la información más importante que debo conocer sobre JARDIANCE®?
JARDIANCE® puede provocar efectos secundarios serios, entre ellos los siguientes:
Deshidratación. JARDIANCE® puede provocar deshidratación (pérdida de agua y sales del organismo) en algunas personas. La deshidratación puede hacer que usted se sienta mareado, débil, con vahídos o sensación de desmayo inminente, especialmente al ponerse de pie (hipotensión ortostática).
Usted puede tener un mayor riesgo de deshidratación si:
– Tiene presión arterial baja;
– Toma medicamentos para bajar la presión arterial, incluyendo diuréticos (píldoras contra la retención de líquido);
– Está siguiendo una dieta baja en sodio (sal);
– Tiene problemas renales;
– Tiene 65 años de edad o más.
• Infección vaginal por levaduras: Las mujeres que toman JARDIANCE® pueden desarrollar infecciones vaginales por levaduras. Los síntomas de una infección vaginal por levaduras incluyen:
– Mal olor vaginal;
– Flujo vaginal blanco o amarillento (el flujo puede ser grumoso o como queso cottage);
– Prurito vaginal.
• Infección peneana por levaduras (balanitis o balanopostitis): Los hombres que toman JARDIANCE® pueden desarrollar una infección por levaduras en la piel que cubre el pene. Algunos hombres que no están circuncidados pueden presentar una inflamación en el pene que dificulta la retracción de la piel que lo cubre. Otros síntomas de la infección peneana por levaduras son:
– Enrojecimiento, escozor o inflamación del pene;
– Erupción del pene;
– Secreción del pene con olor fétido;
– Dolor en la piel que cubre el pene.
Debe conversar con su médico respecto de lo que debe hacer en el caso de que se presenten síntomas de una infección vaginal o peneana por levaduras. Su médico puede sugerirle el uso de un antimicótico de venta libre. Consulte con su médico sin demora si usa un antimicótico de venta libre y sus síntomas no desaparecen.
¿Qué es JARDIANCE®?
• JARDIANCE® es un medicamento de venta bajo receta que se usa en las siguientes indicaciones:
– Junto con dieta y ejercicio, para reducir el azúcar en sangre en adultos con diabetes tipo 2;
– Para reducir el riesgo de muerte cardiovascular en adultos con diabetes tipo 2 que tienen una enfermedad cardiovascular conocida.
• JARDIANCE® no está indicado para su uso en personas con diabetes tipo 1.
• JARDIANCE® no está indicado para su uso en personas con cetoacidosis diabética (niveles elevados de cuerpos cetónicos en sangre u orina).
Se desconoce si JARDIANCE® es seguro y efectivo en niños menores de 18 años de edad.
¿Quiénes no deben tomar JARDIANCE®?
No tome JARDIANCE® si usted:
Es alérgico a la empagliflozina o a cualquiera de los componentes de JARDIANCE®. Consulte al final de este prospecto el listado completo de los componentes de JARDIANCE®.
Tiene problemas renales severos o está recibiendo diálisis.
¿Qué debo informar a mi médico antes de usar JARDIANCE®?
Antes de tomar JARDIANCE®, usted debe informar a su médico si:
• Tiene problemas renales.
• Tiene problemas hepáticos.
• Tiene antecedentes de infecciones de las vías urinarias o problemas relacionados con la micción.
• Será sometido a una cirugía.
• Está comiendo menos de lo habitual debido a una enfermedad, una cirugía o un cambio en su dieta.
• Tiene o ha tenido problemas con el páncreas, incluyendo pancreatitis o cirugía de páncreas.
• Bebe alcohol muy a menudo, o bebe una gran cantidad de alcohol en un corto plazo (consumo intensivo).
• Tiene alguna otra afección médica.
• Está embarazada o tiene planes de quedar embarazada. JARDIANCE® puede causar daños a su bebé en gestación. Si queda embarazada mientras está tomando JARDIANCE®, infórmelo a su médico tan pronto como sea posible. Converse con su médico acerca de la mejor manera de controlar su nivel de azúcar en sangre durante el embarazo.
• Está en período de lactancia o tiene previsto amamantar. JARDIANCE® puede pasar a la leche materna, y podría causar daño a su bebé. Converse con su médico acerca de la mejor forma de alimentar a su bebé si usted está tomando JARDIANCE®. No amamante mientras esté tomando JARDIANCE®.
Debe informar a su médico acerca de todos los medicamentos que tome, no sólo los medicamentos de venta bajo receta sino también los medicamentos de venta libre, las vitaminas y los suplementos a base de hierbas.
JARDIANCE® puede afectar la acción de otros medicamentos, y viceversa.
Es especialmente importante que informe a su médico si toma:
• Diuréticos (píldoras para la retención de líquidos);
• Insulina u otros medicamentos que puedan reducir los niveles de azúcar en sangre.
Solicite a su médico o farmacéutico un listado de estos medicamentos si no está seguro de si la medicación que usted está tomando está incluida en tal restricción.
Es especialmente importante que informe a su médico si toma: Diuréticos (píldoras para la retención de líquidos); insulina u otros medicamentos que puedan reducir los niveles de azúcar en sangre.
Solicite a su médico o farmacéutico un listado de estos medicamentos si no está seguro de si la medicación que usted está tomando está incluida en tal restricción.
¿Cómo debo tomar JARDIANCE®?
• Debe tomar JARDIANCE® exactamente como se lo haya indicado su médico.
• Debe tomar JARDIANCE® por vía oral 1 vez al día por la mañana, cada día, con o sin alimentos.
• Su médico puede modificar la dosis de ser necesario.
• Si olvida tomar una dosis, tómela tan pronto lo recuerde. Si no detecta la omisión hasta la siguiente dosis programada, debe saltear la dosis que omitió y continuar con su cronograma de tomas habitual. No tome dos dosis de JARDIANCE® juntas. Converse con su médico en el caso de que tenga preguntas respecto de una dosis omitida.
• Su médico puede indicarle que tome JARDIANCE® junto con otros medicamentos para la diabetes. La hipoglucemia (niveles bajos de azúcar en sangre) puede ocurrir con mayor frecuencia cuando JARDIANCE® se toma junto con otros medicamentos para la diabetes. Consulte la sección “¿Cuáles son los posibles efectos secundarios de JARDIANCE®?”.
• Si toma una cantidad JARDIANCE® mayor que la indicada, comuníquese con su médico o concurra al servicio de emergencias del hospital más cercano sin demora.
• Cuando su organismo está sometido a determinados tipos de estrés, como ser fiebre, traumatismo (p. ej., un accidente automovilístico), infección o cirugía, puede suceder que cambie la cantidad de medicación para la diabetes que deba tomar. Debe informar a su médico de inmediato en el caso de verse afectado por cualquiera de las situaciones antes mencionadas, y seguir las instrucciones que éste le brinde.
• Debe controlar sus niveles de azúcar en sangre tal como se lo haya indicado su médico.
• Debe mantener su programa de dieta y ejercicio prescrito mientras esté en tratamiento con JARDIANCE®.
• Debe conversar con su médico acerca de cómo prevenir, reconocer y manejar los niveles de azúcar en sangre bajos (hipoglucemia) o altos (hiperglucemia) y las complicaciones de la diabetes.
• Su médico realizará controles de su diabetes mediante exámenes de sangre periódicos, los cuales incluirán la medición de los niveles de azúcar en sangre y de HbA1c.
Al tomar JARDIANCE®, existe la posibilidad de que tenga azúcar en la orina, lo cual se detectará en un análisis de orina.
¿Cuáles son los posibles efectos secundarios de JARDIANCE®?
JARDIANCE® puede provocar efectos secundarios serios, entre ellos los siguientes: Consulte la sección “¿Cuál es la información más importante que debo conocer sobre JARDIANCE®?”.
Cetoacidosis (aumento de los niveles de cuerpos cetónicos en la sangre o en la orina). Se han producido casos de cetoacidosis durante el tratamiento con JARDIANCE® en personas con diabetes tipo 1 o diabetes tipo 2. La cetoacidosis es una afección seria, que puede requerir atención en el hospital. La cetoacidosis puede conducir a la muerte. La cetoacidosis puede ocurrir con JARDIANCE® incluso si su nivel de azúcar en sangre es inferior a 250 mg/dl. Debe interrumpir la toma de JARDIANCE® y llamar a su médico de inmediato si presenta alguno de los siguientes síntomas:
– Náuseas;
– Cansancio;
– Vómitos;
– Dificultad para respirar.
– Dolor en la zona del estómago (abdominal);
Si presentara alguno de estos síntomas durante el tratamiento con JARDIANCE®, de ser posible, controle sus niveles de cuerpos cetónicos en orina, incluso si su nivel de azúcar en sangre es menos de 250 mg/dl.
Infecciones urinarias serias: Se han producido infecciones urinarias serias con el potencial de requerir hospitalización en personas que estaban tomando JARDIANCE®. Debe informar a su médico en el caso de tener cualquier signo o síntoma de una infección de las vías urinarias, como por ejemplo sensación de ardor al orinar, la necesidad de orinar con frecuencia, una necesidad de orinar imperiosa, dolor en el bajo vientre (pelvis) o presencia de sangre en la orina. En algunos casos, puede presentarse también fiebre, dolor lumbar, náuseas o vómitos.
Niveles bajos de azúcar en sangre (hipoglucemia): Si toma JARDIANCE® junto con otro medicamento que pueda reducir los niveles de azúcar en sangre, como ser una sulfonilurea o insulina, su riesgo de hipoglucemia es mayor. Es posible que durante el tratamiento con JARDIANCE® sea necesario reducir la dosis de la sulfonilurea o la insulina que esté tomando. Los signos y síntomas de la presencia de niveles bajos de azúcar en sangre incluyen:
– Dolor de cabeza;
– Irritabilidad;
– Confusión;
– Mareos;
– Somnolencia;
– Hambre; o temblor o nerviosismo;
– Sudoración.
– Debilidad;
– Ritmo cardíaco acelerado;
Problemas renales: Se ha producido lesión renal súbita en personas que tomaban JARDIANCE®. Consulte a su médico de inmediato si:
– Reduce la cantidad de alimentos o líquido que ingiere, por ejemplo, si está enfermo o no puede comer; o bien
– Comienza a perder los líquidos del organismo, por ejemplo, por tener vómitos o diarrea o por estar al sol demasiado tiempo.
Reacciones alérgicas (hipersensibilidad): Se han producido reacciones alérgicas serias en personas que estaban tomando JARDIANCE®. Los síntomas pueden incluir:
– Hinchazón de la cara, los labios, la garganta y otras zonas de la piel;
– Dificultad para tragar o respirar;
– Zonas de la piel sobre elevadas y con enrojecimiento (ronchas).
Si usted tuviera cualquiera de estos síntomas, debe dejar de tomar JARDIANCE® y debe comunicarse de inmediato con su médico o concurrir al servicio de urgencias del hospital más cercano.
Elevación de los niveles de grasas en sangre (colesterol): Éstos no son todos los posibles efectos secundarios de JARDIANCE®. Para mayor información, consulte a su médico o farmacéutico.
Amputación de miembros inferiores: Se ha observado un aumento de casos de amputación de miembros inferiores (principalmente en los dedos de los pies) en ensayos clínicos a largo plazo con otro inhibidor SGLT2. Se desconoce si esto constituye un efecto de clase. Como con todos los pacientes diabéticos, es importante aconsejar a los pacientes acerca del cuidado rutinario preventivo del pie.
Comuníquese con su médico para recibir asesoramiento médico sobre los efectos secundarios de este medicamento. Usted puede informar los efectos secundarios a DIGEMID llamando (01) 6314300.
Para obtener mayor información sobre JARDIANCE® comuníquese con Boehringer Ingelheim Perú S.A.C y en caso de usted tenga alguna notificación de Reacciones adversas: Usted puede informar al correo pv_local_peru@boehringer-ingelheim.com llamando al (01) 412-5008.
¿Cómo debo conservar JARDIANCE®?
JARDIANCE® debe conservarse a una temperatura no superior a 30 °C.
Información general sobre el uso seguro y eficaz de JARDIANCE®:
En algunas ocasiones los medicamentos se prescriben para indicaciones que no son las que se citan en la Información para el Paciente. JARDIANCE® no debe ser utilizado para el tratamiento de una afección para la cual no esté indicado. No facilite JARDIANCE® a otras personas, aunque presenten los mismos síntomas que usted. Este medicamento puede ser perjudicial para otras personas.
En esta Información para el Paciente se resume la información más importante sobre JARDIANCE®. Si desea obtener información adicional, consulte con su médico. Puede solicitar a su farmacéutico o a su médico información sobre JARDIANCE® dirigida a los profesionales de la salud.
¿Cuáles son los componentes de JARDIANCE®?:
Principio activo: Empagliflozina.
Excipientes: Lactosa monohidratada, Celulosa microcristalina, Hidroxipropilcelulosa, Croscarmelosa sódica, Sílice coloidal anhidro, Estearato de magnesio y Opadry amarillo.
Manténgase fuera del alcance de los niños
Venta con receta médica.
Importado por:
BOEHRINGER INGELHEIM PERÚ S.A.C.
RUC: 20523163320 Tel: (01) 412-5000
Dir. Técnico Q.F. Jesús Peña
DESCRIPCIÓN:
El producto JARDIANCE® comprimidos contiene empagliflozina, un inhibidor del cotransportador de sodio-glucosa tipo 2 (SGLT2) oralmente activo.
El nombre químico de la empagliflozina es D-Glucitol,1,5-anhidro-1-C-(4-cloro-3-((4-(((3S)-tetrahidro-3-furanil)oxi)fenil)metil)fenil)-, (1S).
Su fórmula molecular es C23H27ClO7 y su peso molecular es 450,91. Su fórmula estructural es la siguiente:
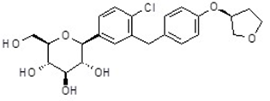
La empagliflozina es un polvo no higroscópico de color entre blanco y amarillento. Es muy levemente soluble en agua, escasamente soluble en metanol, ligeramente soluble en etanol y acetonitrilo; soluble en 50% acetonitrilo/agua; y prácticamente insoluble en tolueno.
Cada COMPRIMIDO RECUBIERTO de JARDIANCE® contiene 10 mg o 25 mg de empagliflozina (base libre) y los siguientes excipientes: lactosa monohidrato, celulosa microcristalina, hidroxipropil celulosa, croscarmelosa sódica, dióxido de silicio coloidal y estearato de magnesio. Asimismo, la película de recubrimiento contiene los siguientes excipientes: hipromelosa, dióxido de titanio, talco, polietilenglicol y óxido férrico amarillo.
FARMACOLOGÍA CLÍNICA:
Mecanismo de acción: El cotransportador de sodio-glucosa tipo 2 (SGLT2) es el principal transportador responsable de la reabsorción de la glucosa a partir del filtrado glomerular y su restitución al torrente sanguíneo. La empagliflozina es un inhibidor del SGLT2. Mediante la inhibición del SGLT2, la empagliflozina reduce la reabsorción renal de la glucosa filtrada y disminuye el umbral renal para la glucosa, e incrementa así la excreción de glucosa urinaria.
Farmacodinamia:
Excreción de glucosa urinaria: En los pacientes con diabetes tipo 2, la excreción de glucosa urinaria se incrementó inmediatamente después de una dosis de JARDIANCE® y se mantuvo al final de un período de tratamiento de 4 semanas, con un valor promedio de aproximadamente 64 gramos por día con la dosis de empagliflozina de 10 mg y de 78 gramos por día con la dosis de 25 mg de JARDIANCE® una vez al día (ver Estudios clínicos).
Volumen urinario: En un estudio de 5 días, la media del aumento del volumen urinario de 24 horas respecto del nivel basal fue 341 mL el Día 1 y 135 mL el Día 5 del régimen de una toma diaria de 25 mg de empagliflozina.
Electrofisiología cardíaca: En un estudio aleatorizado, controlado con placebo, con comparador activo, con cruzamiento, 30 sujetos sanos recibieron una dosis oral única de JARDIANCE® 25 mg, JARDIANCE® 200 mg (8 veces la dosis máxima), moxifloxacina y placebo. No se observó ningún incremento del QTc con empagliflozina 25 mg ni con empagliflozina 200 mg.
Farmacocinética:
Absorción: La farmacocinética de la empagliflozina ha sido caracterizada en voluntarios sanos y en pacientes con diabetes tipo 2, y no se observó ninguna diferencia clínicamente relevante entre estas dos poblaciones. Tras la administración oral, las concentraciones plasmáticas pico de la empagliflozina se alcanzaron a las 1,5 horas posdosis. A partir de ese momento, las concentraciones plasmáticas se redujeron siguiendo un patrón bifásico con una fase de distribución rápida y una fase terminal relativamente lenta. Las medias en estado de equilibrio dinámico del AUC y la Cmax en plasma fueron 1870 nmol·h/L y 259 nmol/L, respectivamente, con el tratamiento de empagliflozina 10 mg una vez al día, y 4740 nmol·h/L y 687 nmol/L, respectivamente, con el tratamiento de empagliflozina 25 mg una vez al día. La exposición sistémica de la empagliflozina se incrementó de manera proporcional a la dosis dentro del rango de dosis terapéutico. Los parámetros farmacocinéticos de las dosis únicas de empagliflozina fueron similares a aquellos del estado de equilibrio dinámico, lo que sugiere una farmacocinética lineal respecto del tiempo.
La administración de 25 mg de empagliflozina tras la ingesta de una comida de alto contenido de grasas y alto contenido calórico condujo a un ligero descenso de la exposición; el AUC se redujo a razón de aproximadamente un 16% y la Cmax se redujo aproximadamente un 37%, en comparación con los valores preprandiales registrados en ayunas. El efecto de los alimentos observado en relación con la farmacocinética de la empagliflozina no se consideró relevante desde el punto de vista clínico, y la empagliflozina puede administrarse con o sin alimentos.
Distribución: El volumen de distribución aparente en estado de equilibrio dinámico se estimó en un valor de 73,8 L sobre la base de un análisis de farmacocinética poblacional. Tras la administración de una solución oral de (14C)-empagliflozina a sujetos sanos, el particionamiento en los glóbulos rojos fue de aproximadamente el 36,8% y el índice de unión a las proteínas plasmáticas fue del 86,2%.
Metabolismo: No se identificó ningún metabolito principal de la empagliflozina en el plasma humano; los metabolitos más abundantes fueron tres conjugados glucurónidos (2-O-, 3-O- y 6-O-glucurónidos). La exposición sistémica de cada metabolito fue de menos del 10% del total del material relacionado con el fármaco. Los estudios in vitro sugirieron que la principal vía metabólica de la empagliflozina en los seres humanos es la glucuronidación a través de las uridino 5"-difosfo-glucuronosiltransferasas UGT2B7, UGT1A3, UGT1A8 y UGT1A9.
Eliminación: La semivida de eliminación terminal aparente de la empagliflozina se estimó en un valor de 12,4 h, y la depuración oral aparente fue de 10,6 L/h sobre la base del análisis de farmacocinética poblacional. Luego de la administración de un régimen de una dosis diaria, en estado de equilibrio dinámico se observó una acumulación de hasta un 22%, en relación con el AUC en plasma, lo cual fue concordante con la semivida de la empagliflozina. Tras la administración de una solución oral de (14C)-empagliflozina a sujetos sanos, aproximadamente el 95,6% de la radioactividad relacionada con el fármaco se eliminó en las heces (41,2%) o en la orina (54,4%). La mayor parte de la radioactividad relacionada con el fármaco recuperada en las heces correspondió a fármaco original inalterado y aproximadamente la mitad de la radioactividad relacionada con el fármaco excretada en la orina fue fármaco original inalterado.
Poblaciones específicas
Deterioro renal: En los pacientes con deterioro renal leve (eGFR: de 60 a menos de 90 mL/min/1,73 m2), moderado (eGFR: de 30 a menos de 60 mL/min/1,73 m2) y severo (eGFR: menos de 30 mL/min/1,73 m2) y en los sujetos con insuficiencia renal/enfermedad renal terminal, el AUC de empagliflozina se incrementó aproximadamente un 18%, 20%, 66% y 48%, respectivamente, en comparación con los sujetos con función renal normal. Los niveles plasmáticos pico de empagliflozina fueron similares en los sujetos con deterioro renal moderado y en los sujetos con insuficiencia renal/enfermedad renal terminal en comparación con los pacientes con función renal normal. Los niveles plasmáticos pico de empagliflozina fueron aproximadamente un 20% más altos en los sujetos con deterioro renal leve y severo en comparación con los sujetos con función renal normal. El análisis de farmacocinética poblacional indicó que la depuración oral aparente de la empagliflozina se redujo, con un descenso en la eGFR que condujo a un incremento en la exposición al fármaco. No obstante ello, la fracción de empagliflozina que se excretó inalterada en la orina, y la excreción de glucosa urinaria, se redujeron conforme menor fue la eGFR.
Deterioro hepático: En los sujetos con deterioro hepático leve, moderado y severo de acuerdo con la clasificación de Child-Pugh, el AUC de la empagliflozina se incrementó aproximadamente un 23%, 47% y 75%, y la Cmax se incrementó aproximadamente un 4%, 23% y 48%, respectivamente, en comparación con los sujetos con función hepática normal.
Efectos de la edad, el índice de masa corporal, el sexo y la raza: Sobre la base del análisis de farmacocinética poblacional, la edad, el índice de masa corporal (IMC), el sexo y la raza (asiática versus principalmente raza blanca) no ejercen ningún efecto clínicamente significativo sobre la farmacocinética de la empagliflozina (ver Uso en poblaciones específicas).
Pacientes pediátricos: No se han realizado estudios para caracterizar la farmacocinética de la empagliflozina en pacientes pediátricos.
CONDICIONES DE ALMACENAMIENTO: Consérvese a una temperatura no superior a 30 °C.
NATURALEZA Y CONTENIDO DEL ENVASE: Comprimidos recubiertos contenidos en blísters de PVC/aluminio.
PRECAUCIONES ESPECIALES DE CONSERVACIÓN: No requiere condiciones especiales de conservación.
No dejar los medicamentos al alcance de los niños
VIDA ÚTIL: 3 años.
“No consumir el producto una vez alcanzada la fecha de vencimiento indicada en los rotulados“.