
GIOTRIF 20 MG
AFATINIB, LACTOSA
Comprimidos recubiertos
Caja, Blíster de PVC aluminio, 7,14,28 Comprimidos recubiertos, 20 Miligramos
Caja , Blíster de PVC aluminio , 35,42,49,56 Comprimidos recubiertos , 20 Miligramos
COMPOSICIÓN CUALI-CUANTITATIVA:
GIOTRIF 20 mg
Cada COMPRIMIDO RECUBIERTO contiene:
Afatinib 20 mg (como dimaleato),
Excipiente con efectos conocidos: Un comprimido recubierto contiene 118 mg de lactosa (como monohidrato).
GIOTRIF 30 mg
Cada COMPRIMIDO RECUBIERTO contiene:
Afatinib 30 mg (como dimaleato),
Excipiente con efectos conocidos: Un comprimido recubierto contiene 176 mg de lactosa (como monohidrato).
GIOTRIF 40 mg
Cada COMPRIMIDO RECUBIERTO contiene:
Afatinib 40 mg (como dimaleato),
Excipiente con efectos conocidos: Un comprimido recubierto contiene 235 mg de lactosa (como monohidrato).
GIOTRIF 50 mg
Cada COMPRIMIDO RECUBIERTO contiene:
Afatinib 50 mg (como dimaleato),
Excipiente con efectos conocidos: Un comprimido recubierto contiene 294 mg de lactosa (como monohidrato).
Para consultar el listado completo de excipientes Véase sección Listado de excipientes.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Un COMPRIMIDO RECUBIERTO contiene: 20 mg de afatinib (como dimaleato).
Excipiente con efecto conocido: un comprimido recubierto contiene 123,86 mg de lactosa (como monohidrato).
Para consultar la lista completa de excipientes, ver Lista de excipientes.
FORMA FARMACÉUTICA: Comprimido recubierto.
LISTA DE EXCIPIENTES
Núcleo del comprimido:
• Lactosa monohidrato.
• Celulosa microcristalina.
• Sílice coloidal anhidra.
• Crospovidona Tipo A.
• Estearato de magnesio.
Cubierta:
• Hipromelosa 2910.
• Macrogol 400.
• Dióxido de titanio.
• Talco.
• Polisorbato 80.
ADVERTENCIAS Y PRECAUCIONES DE USO ESPECIALES:
Evaluación del estado de mutación del EGFR: Cuando se evalúe el estado de mutación del EGFR de un paciente, es importante elegir una metodología bien validada y robusta para evitar resultados falsos negativos o falsos positivos.
Diarrea: Se ha informado diarrea, lo que incluye cuadros de diarrea grave, durante el tratamiento con GIOTRIF (véase la sección Reacciones adversas). La diarrea puede conducir a una deshidratación con o sin insuficiencia renal, cuadro éste que, en casos raros, ha tenido un desenlace fatal. La diarrea usualmente se presentó dentro de las 2 primeras semanas de tratamiento. La diarrea de Grado 3 se presentó con mayor frecuencia dentro de las primeras 6 semanas de tratamiento.
Es importante el tratamiento proactivo de la diarrea que incluya la hidratación adecuada combinada con medicamentos antidiarreicos, en especial dentro de las primeras 6 semanas de tratamiento, y debe iniciarse ante la aparición de los primeros signos de diarrea. Deben utilizarse medicamentos antidiarreicos (p. ej., loperamida) y, de ser necesario, debe aumentarse gradualmente la dosis hasta llegar a la dosis aprobada recomendada más alta. Los medicamentos antidiarreicos deben estar fácilmente asequibles para los pacientes, de manera que el tratamiento pueda iniciarse ante los primeros signos de diarrea y continuarse hasta que las deposiciones blandas hayan cesado por un lapso de 12 horas. Los cuadros de diarrea grave pueden requerir la interrupción del tratamiento y la reducción de la dosis o bien la discontinuación del tratamiento con GIOTRIF (véase la sección Posología y forma de administración). En el caso de deshidratación, puede ser necesaria la administración de electrolitos y líquidos por vía intravenosa.
Eventos adversos cutáneos: En pacientes tratados con este medicamento se ha informado exantema/acné (véase la sección Reacciones adversas). En general, el exantema se manifiesta como una erupción eritematosa y acneiforme leve o moderada, que puede producirse o empeorar en las zonas expuestas al sol. Se aconseja el uso de ropa que brinde una protección adecuada y el uso de pantalla solar en los pacientes que se expongan al sol. La intervención temprana (tal como el uso de emolientes o antibióticos) de las reacciones dermatológicas puede facilitar la continuación del tratamiento con GIOTRIF. Los pacientes con reacciones cutáneas graves también pueden requerir la interrupción temporaria del tratamiento, una reducción de la dosis (véase la sección Reacciones adversas), intervenciones terapéuticas adicionales o la derivación a un especialista con experiencia en el manejo de estos efectos dermatológicos.
Se han notificado cuadros cutáneos bullosos, vesiculares y exfoliativos, incluidos casos raros sugestivos de síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. Se debe interrumpir o discontinuar el tratamiento con este medicamento si el paciente presenta cuadros bullosos, vesiculares o exfoliativos graves (véase la sección Reacciones adversas).
Sexo femenino, bajo peso corporal e insuficiencia renal subyacente: Se ha observado una mayor exposición a afatinib en pacientes de sexo femenino, en pacientes con bajo peso corporal y en pacientes con insuficiencia renal subyacente (véase la sección Propiedades farmacocinéticas). Esto podría dar como resultado un mayor riesgo de desarrollar reacciones adversas, en particular diarrea, exantema/acné y estomatitis. Se recomienda un monitoreo más estrecho en los pacientes con dichos factores de riesgo.
Enfermedad pulmonar intersticial: Se han informado casos de ILD o reacciones adversas similares a la ILD (como infiltración pulmonar, neumonitis, síndrome de distrés respiratorio agudo, alveolitis alérgica), incluidos casos con desenlace fatal, en pacientes que recibieron GIOTRIF para el tratamiento del NSCLC. Las reacciones adversas similares a la ILD se informaron en el 0,7% de los pacientes tratados con GIOTRIF entre todos los estudios clínicos (incluido el 0,5% de los pacientes con reacciones adversas similares a la ILD de Grado ≥3 según los CTCAE). No se han estudiado pacientes con antecedentes de ILD.
Se debe realizar una evaluación cuidadosa de todos los pacientes que presenten un inicio agudo y/o un empeoramiento inexplicable de los síntomas pulmonares (disnea, tos, fiebre) para excluir la posibilidad de un cuadro de ILD. El tratamiento con este medicamento debe suspenderse hasta tanto estén disponibles los resultados de la investigación de dichos síntomas. En el caso de un diagnóstico de ILD, debe suspenderse en forma definitiva la administración de GIOTRIF y debe iniciarse el tratamiento adecuado según sea necesario (véase la sección Posología y forma de administración).
Insuficiencia hepática grave: Se han informado casos de insuficiencia hepática, incluidos casos con desenlace fatal, durante el tratamiento con este medicamento en menos del 1% de los pacientes. En dichos pacientes, existían factores de confusión tales como hepatopatía preexistente y/o comorbilidades asociadas con la progresión de la enfermedad maligna subyacente. Se recomienda la realización periódica de pruebas de función hepática en los pacientes con patologías hepáticas preexistentes. En los estudios pivote, se observaron elevaciones de Grado 3 en los valores de aminotransferasa alanina (ALT) y de aspartato aminotransferasa (AST) en el 2,4% (LUX-Lung-3) y en el 1,6% (LUX-Lung 8) de los pacientes que tenían valores normales en las pruebas de función hepática del nivel basal y fueron tratados con 40 mg/día. En LUX-Lung 3, las elevaciones de Grado 3 en los valores de ALT/AST fueron aproximadamente 3,5 veces más altos en los pacientes que tenían resultados anómalos en las pruebas de función hepática del nivel basal. En LUX-Lung 8, no hubo elevaciones de Grado 3 en los valores de ALT/AST en los pacientes que tenían resultados anómalos en las pruebas de función hepática del nivel basal (véase la sección Reacciones adversas). Podría ser necesaria una interrupción de la administración de este medicamento en los pacientes que presenten un empeoramiento de su función hepática (véase la sección Posología y forma de administración). Se debe discontinuar el tratamiento con GIOTRIF en los pacientes que desarrollen una insuficiencia hepática grave mientras reciban dicho medicamento.
Queratitis: Los pacientes que presenten síntomas como inflamación ocular aguda o un empeoramiento de inflamación ocular, lagrimeo, sensibilidad a la luz, visión borrosa, dolor ocular y/o enrojecimiento ocular deben ser derivados sin demora a un oftalmólogo. Si se confirma un diagnóstico de queratitis ulcerosa, se debe interrumpir o bien discontinuar el tratamiento. Si se diagnostica queratitis, se deberán sopesar cuidadosamente los riesgos y beneficios de continuar el tratamiento. Este medicamento debe utilizarse con precaución en pacientes con antecedentes de queratitis, queratitis ulcerosa y sequedad ocular grave. El uso de lentes de contacto constituye también un factor de riesgo para la queratitis y la ulceración (véase la sección Reacciones adversas).
Función ventricular izquierda: La disfunción ventricular izquierda se ha asociado con la inhibición del HER2. Sobre la base de los datos de estudios clínicos disponibles, no existen indicios de que este medicamento provoque reacciones adversas sobre la contractilidad cardíaca. Sin embargo, este medicamento no ha sido estudiado en pacientes con anomalías de la fracción de eyección ventricular izquierda (LVEF) o antecedentes de afecciones cardíacas importantes. En los pacientes que tengan factores de riesgo cardíacos y en aquellos con trastornos que puedan afectar la LVEF, debe considerarse un control cardíaco, incluida una evaluación de la LVEF al inicio del tratamiento y durante el mismo. En los pacientes que desarrollen signos/síntomas cardíacos relevantes durante el tratamiento, debe considerarse un control cardíaco que incluya la evaluación de la LVEF.
En los pacientes cuya fracción de eyección sea menor que el límite normal inferior de la institución, debe considerarse la realización de una consulta cardiológica y la interrupción o discontinuación del tratamiento.
Interacciones con la glucoproteína P (P-gp): El tratamiento concomitante con inductores potentes de la P-gp puede reducir la exposición a afatinib (véase la sección Interacción con otros medicamentos y otras formas de interacción).
Lactosa: Este medicamento contiene lactosa. Los pacientes con trastornos hereditarios raros de intolerancia a la galactosa, deficiencia de lactasa Lapp o mala absorción de glucosa-galactosa no deben tomar este medicamento.
INDICACIONES TERAPÉUTICAS:
GIOTRIF administrado como monoterapia está indicado para el tratamiento de pacientes con las siguientes afecciones:
• Carcinoma de pulmón de células no pequeñas (non-small cell lung cancer, NSCLC) localmente avanzado o metastásico con mutación(es) activadora(s) del receptor del factor de crecimiento epidérmico (epidermal growth factor receptor, EGFR) que no han recibido tratamiento previo con un inhibidor de la tirosina quinasa (tyrosine kinase inhibitor, TKI) del EGFR.
• NSCLC localmente avanzado o metastásico de histología escamosa que progrese durante o tras quimioterapia basada en platino (véase la sección Propiedades farmacodinámicas).
DATOS FARMACÉUTICOS:
Listado de excipientes:
• Núcleo del comprimido:
– Lactosa, monohidrato
– Celulosa microcristalina (E460)
– Sílice coloidal anhidro (E551)
– Crospovidona tipo A
– Estearato de magnesio (E470b)
• Recubrimiento:
GIOTRIF 20 mg Comprimidos recubiertos
– Hipromelosa (E464)
– Macrogol 400
– Dióxido de titanio (E171)
– Talco (E553b)
– Polisorbato 80 (E433)
GIOTRIF 30, 40 y 50 mg Comprimidos recubiertos
– Hipromelosa (E464)
– Macrogol 400
– Dióxido de titanio (E171)
– Talco (E553b)
– Polisorbato 80 (E433)
– Hidróxido de aluminio de carmín de índigo (E132)
Vida útil: 36 meses.
“No consumir el producto una vez alcanzada la fecha de vencimiento indicada en los rotulados“.
Precauciones de conservación especiales: El producto debe conservarse en el envase original para protegerlo de la humedad y de la luz.
No almacenar a una temperatura superior a 30 °C.
“Consulte a su médico o farmacéutico, según proceda, para cualquier aclaración sobre la utilización del producto“.
PROPIEDADES FARMACOCINÉTICAS
Absorción: Después de la administración oral de GIOTRIF se observaron Cmax de afatinib aproximadamente de 2 a 5 horas después de la administración. Los valores de Cmax y AUC0-8 aumentaron algo más que proporcionalmente en el intervalo de dosis entre 20 mg y 50 mg de GIOTRIF. La exposición sistémica a afatinib disminuye un 50% (Cmax) y un 39% (AUC0-8) cuando se administra con una comida rica en grasas en comparación con la administración en ayunas. Según datos farmacocinéticos poblacionales obtenidos en estudios clínicos de diversos tipos de tumores, el AUCt,ss disminuyó un promedio de un 26% cuando se ingirieron alimentos en las 3 horas anteriores y en la hora siguiente a la toma de GIOTRIF. Por lo tanto, no se deben ingerir alimentos al menos 3 horas antes y 1 hora después de tomar GIOTRIF (ver Posología y forma de administración e Interacción con otros medicamentos y otras formas de interacción).
Distribución: La unión in vitro de afatinib a proteínas plasmáticas humanas es de aproximadamente el 95%. Afatinib se une a las proteínas tanto de forma no covalente (unión típica de las proteínas) y covalente.
Biotransformación: Las reacciones metabólicas mediadas por enzimas desempeñan un papel insignificante para afatinib in vivo. Los principales metabolitos circulantes de afatinib fueron aductos covalentes con proteínas.
Eliminación: En el ser humano, afatinib se excreta principalmente por las heces. Después de la administración de una solución oral de 15 mg de afatinib, se recuperó el 85,4% de la dosis en las heces y el 4,3% en orina. El 88% de la dosis recuperada correspondió al compuesto original de afatinib. La semivida terminal de afatinib fue de aproximadamente 37 horas. Se alcanzaron concentraciones plasmáticas en estado estacionario de afatinib en un plazo de 8 días después de la administración múltiple de afatinib, dando lugar a una acumulación de 2,77 veces (AUC0-8) y de 2,11 veces (Cmax).
Poblaciones especiales:
• Insuficiencia renal: De una dosis única de afatinib, menos del 5% se excreta por vía renal. No se han realizado estudios específicos de seguridad, farmacocinética y eficacia de GIOTRIF en pacientes con insuficiencia renal. Según datos farmacocinéticos poblacionales de estudios clínicos en diversos tipos de tumores no es necesario ajustar las dosis en pacientes con insuficiencia renal leve o moderada (ver más abajo “Análisis farmacocinético poblacional en poblaciones especiales” y Posología y forma de administración).
• Insuficiencia hepática: Afatinib se elimina principalmente por vía biliar y fecal. La exposición en sujetos con insuficiencia hepática leve (grado A de Child Pugh) o moderada (grado B de Child Pugh) fue similar a la de voluntarios sanos después de una dosis única de 50 mg de GIOTRIF. Esto concuerda con los datos farmacocinéticos poblacionales de estudios clínicos en diversos tipos de tumores (ver más abajo “Análisis farmacocinético poblacional en poblaciones especiales”). Aparentemente no es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada (ver Posología y forma de administración). No se ha estudiado la farmacocinética de afatinib en sujetos con insuficiencia hepática grave (grado C de Child Pugh) (ver Advertencias y precauciones especiales de empleo).
• Análisis farmacocinético poblacional en poblaciones especiales: Se llevó a cabo un análisis farmacocinético poblacional en 927 pacientes con cáncer (764 con CPNM) tratados con GIOTRIF en monoterapia. No se consideró necesario ajustar la dosis inicial para ninguna de las covariables siguientes estudiadas.
— Edad: No se observó un impacto significativo de la edad (intervalo: 28 años-87 años) en la farmacocinética de afatinib.
— Peso corporal: La exposición plasmática (AUCt,ss) aumentó un 26% en un paciente de 42 kg (percentil 2,5) y disminuyó un 22% en un paciente de 95 kg (percentil 97,5) respecto a un paciente que pesa 62 kg (peso corporal mediano en la población total de pacientes).
— Sexo: La exposición plasmática fue un 15% mayor (AUCt,ss, corregido para el peso corporal) en las pacientes mujeres respecto a los pacientes varones.
— Raza: La raza careció de efecto sobre la farmacocinética de afatinib según un análisis farmacocinético poblacional que incluyó a pacientes asiáticos, de raza blanca y raza negra. Los datos sobre grupos raciales negros fueron escasos.
— Insuficiencia renal: La exposición a afatinib aumentó moderadamente con valores decrecientes de aclaramiento de la creatinina (CrCL, calculado según la fórmula de Cockcroft Gault), es decir, para un paciente con un CrCL de 60 ml/min o 30 ml/min, la exposición (AUCt,ss) a afatinib aumentó un 13% y un 42%, respectivamente y disminuyó un 6% y un 20% para un paciente con un CrCL de 90 ml/min o 120 ml/min, respectivamente, en comparación con un paciente con un CrCL de 79 ml/min (CrCL mediano de los pacientes en la población total de pacientes analizada).
— Insuficiencia hepática: Los pacientes con una insuficiencia hepática leve y moderada, diagnosticada mediante pruebas de función hepática anormales, no se relacionaron con cambios significativos en la exposición a afatinib. Se dispone de pocos datos en relación con la insuficiencia hepática moderada y grave.
— Otras características/factores intrínsecos de los pacientes: Otras características/factores intrínsecos de los pacientes con un impacto significativo en la exposición a afatinib fueron: estado funcional ECOG, niveles de lactato deshidrogenasa, niveles de fosfatasa alcalina y proteínas totales. No se consideró que los tamaños de los efectos individuales de estas covariables fueran clínicamente significativos. El tabaquismo, el consumo de alcohol (pocos datos) o la presencia de metástasis hepáticas no tuvieron un impacto significativo en la farmacocinética de afatinib.
Información adicional sobre las interacciones con otros medicamentos:
• Interacciones con sistemas de transporte por captación: Los datos in vitro obtenidos indicaron una escasa probabilidad de interacciones de afatinib con otros medicamentos debido a la inhibición de los transportadores OATB1B1, OATP1B3, OATP2B1, OAT1, OAT3, OCT1, OCT2 y OCT3.
• Interacciones con las enzimas del citocromo P450 (CYP): En humanos se halló que las reacciones metabólicas catalizadas por enzimas juegan un papel insignificante en el metabolismo de afatinib. Aproximadamente el 2% de la dosis de afatinib fue metabolizada por la FMO3 y la N-desmetilación dependiente de CYP3A4 fue demasiado baja como para ser detectada a nivel cuantitativo. Afatinib no es un inhibidor ni un inductor de las enzimas del CYP. Por lo tanto, es improbable que este medicamento interaccione con otros medicamentos que modulan o se metabolizan por las enzimas del CYP.
• Efecto de la inhibición de la UDP-glucuronosiltransferasa 1A1 (UGT1A1) sobre afatinib: Los datos in vitro indicaron que es poco probable que se produzcan interacciones de otros medicamentos con afatinib debido a la inhibición de la UGT1A1.
PROPIEDADES FARMACODINÁMICAS
Grupo farmacoterapéutico: agentes antineoplásicos, inhibidores de la proteína quinasa, código ATC: L01XE13.
Mecanismo de acción: Afatinib es un inhibidor potente, selectivo e irreversible de receptores de la familia ErbB. Afatinib se une covalentemente y bloquea de forma irreversible las vías de señalización de todos los homodímeros y heterodímeros formados por los miembros de la familia ErbB, EGFR (ErbB1), HER2 (ErbB2), ErbB3 y ErbB4.
Efectos farmacodinámicos: Las vías de señalización aberrantes del ErbB desencadenadas por mutaciones del receptor, y/o por amplificación, y/o por sobreexpresión de ligandos del receptor contribuyen a la aparición de un fenotipo maligno. La mutación en el EGFR define un subtipo molecular distinto de cáncer de pulmón.
En modelos preclínicos de enfermedad con desregulación de las vías del ErbB, afatinib como agente único bloquea eficazmente las vías de señalización del receptor ErbB lo que inhibe el crecimiento tumoral e induce la regresión del tumor. Los tumores de CPNM con mutaciones comunes activadoras del EGFR (Del 19, L858R) y varias mutaciones menos comunes del EGFR en el exón 18 (G719X) y en el exón 21(L861Q) son especialmente sensibles al tratamiento con afatinib en escenarios preclínicos y clínicos.
Afatinib conserva una actividad antitumoral significativa en líneas celulares de CPNM in vitro y/o en modelos tumorales in vivo (xenoinjertos o modelos transgénicos) activados por isoformas mutantes del EGFR con resistencia conocida a los inhibidores reversibles del EGFR, erlotinib y gefitinib, como la T790M o T854A. A nivel clínico, también se ha mostrado actividad en tumores que albergan la mutación T790M en el exón 20. Se ha observado actividad limitada preclínica y/o clínica en tumores de CPNM con mutaciones de inserción en el exón 20.
Eficacia clínica y seguridad:
• LUX-Lung 3: En el contexto de tratamiento de primera línea, se evaluó la eficacia y la seguridad de GIOTRIF en pacientes con CPNM localmente avanzado o metastásico (estadio IIIB o IV) con mutaciones del EGFR en un estudio global, aleatorizado, multicéntrico, abierto. Se seleccionó a los pacientes en función de la presencia de 29 mutaciones diferentes del EGFR con un método basado en reacciones en cadena de la polimerasa (PCR) (TheraScreen : EGFR29 Mutation Kit, Qiagen Manchester Ltd). Se aleatorizó a los pacientes (2:1) para recibir GIOTRIF 40 mg una vez al día o hasta 6 ciclos de pemetrexed/cisplatino.
De los pacientes aleatorizados, el 65% fueron mujeres con una edad mediana de 61 años, el estado funcional ECOG basal fue 0 (39%) o 1 (61%), el 26% eran caucásicos y el 72% eran asiáticos.
La variable principal fue la supervivencia libre de progresión (SLP) según una revisión independiente.
En el momento de llevar a cabo el análisis principal, un total de 45 (20%) pacientes tratados con GIOTRIF y 3 (3%) pacientes tratados con quimioterapia habían sobrevivido libres de progresión, por lo que sus datos se excluyeron de la figura 1 y de la Tabla 4.
Figura 1: Curva de Kaplan-Meier de la SLP según una revisión independiente por grupo de tratamiento en el estudio LUX-Lung 3 (población total)

|
Tabla 4: Resultados de eficacia de GIOTRIF vs. pemetrexed/cisplatino en el estudio LUX-Lung 3 en base al análisis principal (revisión independiente) |
||||
|
GIOTRIF |
Pemetrexed/ Cisplatino (N=115) |
Razón de riesgos (HR)/ Razón de posibilidades (OR) (IC del 95%) |
Valor de p |
|
|
SLP, población de estudio total |
||||
|
Meses (mediana) |
11,1 |
6,9 |
HR 0,58 (0,43-0,78) |
0,0004 |
|
Tasa de SLP al cabo de 1 año |
46,5% |
22,0% |
- |
- |
|
Tasa de respuesta objetiva (RC+RP)1 |
56,1% |
22,6% |
OR 4,66 (2,77-7,83) |
<0,0001 |
|
Supervivencia Global (SG) en meses (mediana)2 |
28,1% |
28,2% |
HR 0,91 (0,66-1,25) |
0,55 |
|
1. RC=respuesta completa; RP=respuesta parcial 2. Análisis de SG actualizado en enero 2013 |
||||
En el subgrupo predefinido de mutaciones comunes (Del 19, L858R) para GIOTRIF (N= 204) y quimioterapia (N= 104), la SLP mediana fue 13,6 meses vs. 6,9 meses (HR 0,47; 95% IC 0,34-0,65; p< 0,0001) y la SG mediana fue 30,3 meses vs. 26,2 meses (HR 0,82; 95% IC 0,59-1,14; p= 0,2244).
El aumento de la SLP se acompañó de una mejoría de los síntomas relacionados con la enfermedad y se retrasó el tiempo hasta el deterioro (ver Tabla 5). Las puntuaciones medias a lo largo del tiempo para la calidad de vida en general, el estado de salud global y las funciones físicas, de rol y cognitivas fueron significativamente mejores para GIOTRIF.
|
Tabla 5: Resultados de síntomas para GIOTRIF vs. quimioterapia en el estudio LUX-Lung 3 (EORTC QLQ-C30 & QLQ-LC13) |
|||
|
Tos |
Disnea |
Dolor |
|
|
% de pacientes que mejorarona |
67% vs. 60%; p=0,2444 |
64% vs. 50%; p=0,0103 |
59% vs. 48%; p=0,0513 |
|
Retraso del tiempo hasta el deterioro (meses)a |
NAb vs. 8,0 HR 0.60; p=0,0072 |
10,3 vs. 2,9 HR 0,68; p=0,0145 |
4,2 vs. 3,1 HR 0,83; p=0,1913 |
|
a. Valores presentados para GIOTRIF vs. quimioterapia. b. NA= no alcanzado. |
|||
• LUX-Lung 2: El estudio LUX-Lung 2 fue un ensayo de fase II con un único grupo en el que participaron 129 pacientes con adenocarcinoma de pulmón en estadio IIIB o IV con mutaciones del EGFR no tratados previamente con EGFR TKI. Se asignó a los pacientes a un tratamiento de primera línea (N=61) o a un tratamiento de segunda línea (N=68) (es decir, después del fracaso de un régimen quimioterápico previo). En 61 pacientes sometidos a tratamiento de primera línea, la TRO confirmada fue del 65,6% y la TCE fue del 86,9% de acuerdo con la revisión independiente. La SLP mediana fue de 12,0 meses según la revisión independiente. La eficacia alcanzó niveles similares en el grupo de pacientes sometidos a quimioterapia previa (N=68; TRO 57,4%; SLP mediana de 8 meses según la revisión independiente). La SG mediana actualizada para primera y segunda línea fue 31,7 meses y 23,6 meses, respectivamente.
Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con este medicamento en los diferentes grupos de la población pediátrica en las indicaciones del CPNM (ver Posología y forma de administración para consultar la información sobre el uso en la población pediátrica).
PROPIEDADES FARMACOLÓGICAS:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: agentes antineoplásicos, inhibidores de la proteína quinasa, código ATC: L01XE13.
Mecanismo de acción: Afatinib es un bloqueador irreversible potente y selectivo de la familia ErbB. Afatinib se une mediante enlace covalente y bloquea en forma irreversible la señalización de todos los homo- y heterodímeros formados por los siguientes integrantes de la familia ErbB: EGFR (ErbB1), HER 2 (ErbB2), ErbB3 y ErbB4.
Efectos farmacodinámicos: Las vías de señalización aberrantes del ErbB desencadenadas por mutaciones del receptor, y/o por amplificación, y/o por sobreexpresión de ligandos del receptor contribuyen a la aparición de un fenotipo maligno. La mutación en el EGFR define un subtipo molecular distinto de cáncer de pulmón.
En modelos preclínicos de enfermedad con desregulación de las vías del ErbB, afatinib como agente único bloquea eficazmente las vías de señalización del receptor ErbB lo que inhibe el crecimiento tumoral e induce la regresión del tumor. Los tumores de CPNM con mutaciones comunes activadoras del EGFR (Del 19, L858R) y varias mutaciones menos comunes del EGFR en el exón 18 (G719X) y en el exón 21(L861Q) son especialmente sensibles al tratamiento con afatinib en escenarios preclínicos y clínicos. Se ha observado actividad limitada preclínica y/o clínica en tumores de CPNM con mutaciones de inserción en el exón 20.
La adquisición de una mutación T790M secundaria es el principal mecanismo de resistencia adquirida a afatinib y la dosis génica del alelo que contiene la mutación T790M se correlaciona con el grado de resistencia in vitro. La mutación T790M se encuentra de forma en aproximadamente en el 50% de los tumores de los pacientes tras la progresión de la enfermedad con afatinib, para los que los TKI del EGFR dirigidos a la mutación T790M se pueden considerar como una opción de línea de tratamiento posterior. Desde la preclínica, se han sugerido otros posibles mecanismos de resistencia a afatinib y, desde la clínica, se ha observado la amplificación del gen MET.
Eficacia y seguridad clínicas: GIOTRIF en pacientes con cáncer de pulmón de células no pequeñas con mutaciones del EGFR
LUX Lung 3: En el contexto del tratamiento de primera línea, la eficacia y la seguridad de GIOTRIF en pacientes con NSCLC localmente avanzado o metastásico (estadío IIIB o IV) positivo para mutación del EGFR se evaluó en un estudio internacional, aleatorizado, multicéntrico, de diseño abierto. Se seleccionó a los pacientes en función de la presencia de 29 mutaciones del EGFR diferentes utilizando un método de reacción en cadena de polimerasa (polymerase chain reaction, PCR) (TheraScreen®: EGFR29 Mutation Kit, Qiagen Manchester Ltd.). Los pacientes fueron aleatorizados (en una proporción de 2:1) a recibir 40 mg de GIOTRIF una vez al día o bien un total de hasta 6 ciclos de pemetrexed/cisplatino. Entre los pacientes aleatorizados, el 65% eran de sexo femenino, la mediana de la edad era 61 años, el puntaje de estado funcional ECOG basal era 0 (39%) o 1 (61%), el 26% eran caucásicos y el 72% eran de raza asiática. El 89% de los pacientes presentó mutaciones comunes del EGFR (Del 19 o L858R).
El criterio de valoración primario fue la sobrevida libre de progresión (progression free survival, PFS) determinada mediante una revisión independiente; los criterios de valoración secundarios incluyeron la sobrevida general (overall survival, OS) y la tasa de respuesta objetiva (objective response rate, ORR). Al momento de la realización del análisis, el 14 de noviembre de 2013, 176 pacientes (76,5%) del grupo tratado con afatinib y 70 pacientes (60,9%) del grupo tratado con quimioterapia presentaron un episodio, es decir, progresión de la enfermedad determinada por una revisión central independiente o muerte, que contribuyó al análisis de la PFS. Los resultados de eficacia se incluyen en la figura 1 y en las tablas 5 y 6.
LUX-Lung 6: Se evaluó la eficacia y la seguridad de GIOTRIF en pacientes asiáticos con adenocarcinoma de pulmón localmente avanzado o metastásico en estadío IIIB/IV, positivo para mutación del EGFR, en un estudio aleatorizado, multicéntrico, de diseño abierto. De manera similar a lo que ocurrió en el estudio LUX-Lung 3, se seleccionaron pacientes con NSCLC no tratados con anterioridad en función de la presencia de mutaciones del EGFR utilizando TheraScreen®: EGFR29 Mutation Kit (Qiagen Manchester Ltd). Entre los pacientes aleatorizados, el 65% eran mujeres, la mediana de edad era de 58 años y todos los pacientes eran de raza asiática. Los pacientes con mutaciones comunes del EGFR representaron el 89% de la población de estudio.
El criterio de valoración primario fue la PFS evaluada por una revisión central independiente; los criterios de valoración secundarios incluyeron la OS y la ORR.
Los dos estudios demostraron una mejora significativa en la FPS de los pacientes con mutación del EGFR tratados con GIOTRIF, en comparación con los tratados con quimioterapia. Los resultados de eficacia se resumen en la figura 1 (LUX-Lung 3) y en las tablas 5 y 6 (LUX-Lung 3 y 6). La tabla 6 muestra los resultados de los subgrupos de pacientes con dos mutaciones comunes del EGFR: Del 19 y L858R.
Figura 1. Curva de Kaplan-Meier para la PFS determinada por revisión independiente, por grupo de tratamiento, en el estudio LUX-Lung 3 (Población total)
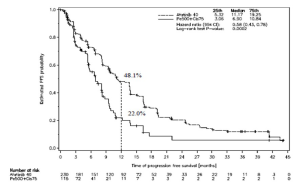
Referencias de la Figura 1:
Estimated PFS probability: Probabilidad de PFS estimada
Time of progression-free survival (months): Tiempo de sobrevida libre de progresión (meses)
25th: Percentil 25
Median: Mediana
75th: Percentil 75
Hazard ratio (95% CI): Razón de riesgos instantáneos (IC 95%)
Log-rank test P-value: Valor p de prueba de rango logarítmico
Number at risk: Número de pacientes en riesgo
Tabla 5. Resultados de eficacia de GIOTRIF frente a pemetrexed/cisplatino (LUX Lung 3) y gemcitabina/cisplatino (LUX-Lung 6) (revisión independiente)
|
LUX-Lung 3 |
LUX-Lung 6 |
|||
|
GIOTRIF (N=230) |
Pemetrexed/cisplatino (N=115) |
GIOTRIF (N=242) |
Gemcitabina/cisplatino (N=122) |
|
|
Sobrevida libre de progresión Meses (mediana) |
11,2 |
6,9 |
11,0 |
5,6 |
|
Razón de riesgos instantáneos (HR) (IC del 95%) |
0,58 (0,43-0,78) |
0,28 (0,20-0,39) |
||
|
Valor p1 |
0,0002 |
<0,0001 |
||
|
Tasa de PFS al cabo de 1 año |
48,1% |
22,0% |
46,7% |
2,1% |
|
Tasa de respuesta objetiva (CR+PR)2 |
56,5% |
22,6% |
67,8% |
23,0% |
|
Cociente de probabilidades (odds ratio, OR) (IC del 95%) |
4,80 (2,89-8,08) |
7,57 (4,52-12,68) |
||
|
Valor p1 |
<0,0001 |
<0,0001 |
||
|
Sobrevida general (OS) Meses (mediana) |
28,2 |
28,2 |
23,1 |
23,5 |
|
Razón de riesgos instantáneos (HR) (IC del 95%) |
0,88 (0,66-1,17) |
0,93 (0,72-1,22) |
||
|
Valor p1 |
0,3850 |
0,6137 |
||
|
1 Valor p para la PFS/OS basado en una prueba de rangos logarítmicos estratificada; valor p para la tasa de respuesta objetiva basada en la regresión logística. 2 CR=respuesta completa (complete response); PR=respuesta parcial (partial response). |
||||
Tabla 6. Resultados de eficacia para la PFS y la OS de GIOTRIF frente a pemetrexed/cisplatino (LUX-Lung 3) y gemcitabina/cisplatino (LUX-Lung 6) en los subgrupos de mutación del EGFR predefinidos Del 19 y L858R (revisión independiente)
|
LUX-Lung 3 |
LUX-Lung 6 |
|||
|
Del 19 |
GIOTRIF (N=112) |
Pemetrexed/Cisplatino (N=57) |
GIOTRIF (N=124) |
Gemcitabina/Cisplatino (N=62) |
|
Sobrevida libre de progresión Meses (mediana) |
13,8 |
5,6 |
13,1 |
5,6 |
|
Razón de riesgos instantáneos (HR) (IC del 95%) |
0,26 (0,17-0,42) |
0,20 (0,13-0,33) |
||
|
Valor p1 |
<0,0001 |
<0,0001 |
||
|
Sobrevida general (OS) Meses (mediana) |
33,3 |
21,1 |
31,4 |
18,4 |
|
Razón de riesgos instantáneos (HR) (IC del 95%) |
0,54 (0,36-0,79) |
0,64 (0,44-0,94) |
||
|
Valor p1 |
0,0015 |
0,0229 |
||
|
L858R |
GIOTRIF (N=91) |
Pemetrexed/Cisplatino (N=47) |
GIOTRIF (N=92) |
Gemcitabina/Cisplatino (N=46) |
|
Sobrevida libre de progresión Meses (mediana) |
10,8 |
8,1 |
9,6 |
5,6 |
|
Razón de riesgos instantáneos (HR) (IC del 95%) |
0,75 (0,48-1,19) |
0,31 (0,19-0,52) |
||
|
Valor p1 |
0,2191 |
<0,0001 |
||
|
Sobrevida general (OS) Meses (mediana) |
27,6 |
40,3 |
19,6 |
24,3 |
|
Razón de riesgos instantáneos (HR) (IC del 95%) |
1,30 (0,80-2,11) |
1,22 (0,81-1,83) |
||
|
Valor p1 |
0,2919 |
0,3432 |
||
|
1 Valor p para la PFS/OS basado en una prueba de rangos logarítmicos estratificada. |
||||
En el subgrupo predefinido de mutaciones comunes (Del 19 y L858R combinadas) para GIOTRIF y para la quimioterapia, la mediana de la PFS fue 13,6 meses frente a 6,9 meses (HR 0,48; IC del 95% 0,35 - 0,66; p< 0,0001; N=307) en el estudio LUX-Lung 3, y de 11,0 meses frente a 5,6 meses (HR 0,24; IC del 95% 0,17 - 0,35; p<0,0001; N=324) en el estudio LUX-Lung 6 respectivamente.
El beneficio en términos de la PFS estuvo acompañado por una mejoría en los síntomas relacionados con la patología y un mayor tiempo hasta el deterioro (véase la Tabla 7). Los puntajes medios a través del tiempo para la calidad de vida general, el estado de salud general y la funcionalidad física, de rol, cognitiva, social y emocional fueron significativamente mejores para GIOTRIF.
Tabla 7. Resultados en términos de síntomas obtenidos para GIOTRIF frente a la quimioterapia en los estudios LUX Lung 3 y LUX-Lung 6 (EORTC QLQ C30 & QLQ LC13)
|
LUX-Lung 3 |
|||
|
Tos |
Disnea |
Dolor |
|
|
% de pacientes con mejoríaa |
67% vs. 60%; p=0,2133 |
65% vs. 50%; p=0,0078 |
60% vs. 48%; p=0,0427 |
|
Retraso de la mediana de tiempo hasta el deterioro (meses)a,b |
27,0 vs. 8,0 HR 0,60; p=0,0062 |
10,4 vs. 2,9 HR 0,68; p=0,0129 |
4,2 vs. 3,1 HR 0,83; p=0,1882 |
|
LUX-Lung 6 |
|||
|
Tos |
Disnea |
Dolor |
|
|
% de pacientes con mejoríaa |
76% vs. 55%; p=0,0003 |
71% vs. 48%; p< 0,0001 |
65% vs. 47%; p=0,0017 |
|
Retraso de la mediana de tiempo hasta el deterioro (meses)a,b |
31,1 vs. 10,3 HR 0,46; p=0,0001 |
7,7 vs. 1,7 HR 0,53; p< 0,0001 |
6,9 vs. 3,4 HR 0,70; p=0,0220 |
|
a Valores presentados para GIOTRIF vs. quimioterapia, valor p basado en la regresión logística. b Valor p para el tiempo hasta el deterioro basado en una prueba de rangos logarítmicos estratificada. |
|||
LUX Lung 2: El estudio LUX Lung 2 fue un estudio de Fase II de rama única realizado en 129 pacientes con adenocarcinoma de pulmón de estadío IIIB o IV con mutaciones del EGFR sin tratamiento previo con un TKI del EGFR. Los pacientes fueron enrolados en un régimen de primera línea (N = 61) o de segunda línea (N = 68) (es decir, después de haber tenido un fracaso terapéutico frente a 1 régimen de quimioterapia previo). En 61 pacientes tratados con el régimen de primera línea, la ORR confirmada fue del 65,6% y la DCR (disease control rate; tasa de control de la enfermedad) fue del 86,9% de acuerdo con la revisión independiente. La mediana de PFS fue 12,0 meses sobre la base de la revisión independiente. La eficacia fue similarmente alta en el grupo de pacientes que había recibido quimioterapia previa (N = 68; ORR 57,4%; mediana de PFS de 8 meses según la revisión independiente). La mediana actualizada de la OS para el régimen de primera línea y para el régimen de segunda línea fue 31,7 meses y 23,6 meses, respectivamente.
LUX-Lung 7: El estudio LUX-Lung 7 es un ensayo internacional, aleatorizado, de diseño abierto, de Fase IIb, en el cual se investigó la eficacia y la seguridad de GIOTRIF en pacientes con adenocarcinoma pulmonar localmente avanzado o metastásico (estadío IIIB o IV) con mutaciones del EGFR en un régimen de primera línea. Los pacientes fueron seleccionados en función de la presencia de mutaciones activadoras del EGFR (Del 19 y/o L858R) usando el método TheraScreen® EGFR RGQ PCR Kit, Qiagen Manchester Ltd.). Los pacientes (N = 319) fueron aleatorizados (1:1) a recibir 40 mg de GIOTRIF® por vía oral una vez al día (N = 160) o 250 mg de gefitinib por vía oral una vez al día (N = 159). La aleatorización se estratificó en función del estado de mutación del EGFR (Del 19; L858R) y de la presencia de metástasis cerebral (sí; no).
Entre los pacientes aleatorizados, el 62% era de sexo femenino, la edad media fue de 63 años, el 16% de los pacientes tenía metástasis cerebral, el estado funcional ECOG inicial fue 0 (31%) o 1 (69%), el 57% eran asiáticos y el 43% eran no-asiáticos. Los pacientes tenían una muestra tumoral con una mutación del EGFR categorizada como deleción del exón 19 (58%) o sustitución del exón 21 L858R (42%).
Los criterios de valoración co-primarios son PFS determinada por la revisión independiente, tiempo hasta el fracaso del tratamiento (TTS) y OS. Los criterios de valoración secundarios incluyen ORR y DCR. El riesgo de progresión se redujo significativamente en el caso de afatinib versus gefitinib (véase Tabla 8) y la ORR fue 70% para afatinib y 56% para gefitinib. El análisis primario de la OS se llevará a cabo luego de que hayan ocurrido la cantidad de eventos requeridos según protocolo.
Tabla 8. Resultados de eficacia de GIOTRIF vs. gefitinib (Estudio LUX-Lung 7) obtenidos sobre la base del análisis primario a agosto de 2015
|
GIOTRIF (N = 160) |
Gefitinib (N = 159) |
Razón de riesgos instantáneos/Cociente de probabilidades (IC del 95%) Valor p2 |
|
|
Mediana de PFS (meses), Población general del estudio Tasa de PFS a 18 meses Tasa de PFS a 24 meses |
11,0 27% 18% |
10,9 15% 8% |
HR 0,73 (0,57-0,95) 0,0165 |
|
Tiempo hasta fracaso del tratamiento (meses) Tasa de TTF a 18 meses Tasa de TTF a 24 meses |
13,7 35% 25% |
11,5 27% 13% |
HR 0,73 (0,58-0,92) 0,0073 |
|
Mediana de OS (meses)1, Población general del estudio |
27,9% |
25% |
HR 0,87 (0,65, 1,15) 0,33 |
|
1 Análisis de OS en desarrollo a agosto de 2015 2 Valor p para la PFS/TTF/OS basado en la prueba de rangos logarítmicos estratificada |
|||
La razón de riesgos instantáneos de la PFS para pacientes con mutaciones DEL 19 y mutaciones L858R fue 0,76 (IC del 95% [0,55, 1,06]; p = 0,1071), y 0,71 (IC del 95% [0,47, 1,06]; p = 0,0856) respectivamente para afatinib vs. gefitinib.
GIOTRIF en pacientes con NSCLC de histología escamosa: La eficacia y la seguridad de GIOTRIF como tratamiento de segunda línea para pacientes con NSCLC avanzado de histología escamosa se investigó en LUX-Lung 8, un estudio de fase III internacional, aleatorizado y abierto. Los pacientes que recibieron al menos 4 ciclos de terapia basada en platino durante el tratamiento de primera línea posteriormente se aleatorizaron 1:1 para recibir 40 mg diarios de GIOTRIF o 150 mg de erlotinib hasta la progresión. La aleatorización se estratificó por raza (asiático oriental frente a no asiático oriental). El criterio de valoración principal fue la PFS; la OS fue el criterio de valoración secundario clave. Otros criterios de valoración secundarios incluyeron la ORR, la DCR, el cambio en el tamaño del tumor y la HRQoL (Health Related Quality of Life; calidad de vida relacionada con la salud).
Entre los 795 pacientes aleatorizados, la mayoría fueron hombres (84%), blancos (73%), fumadores o exfumadores (95%) con estado funcional ECOG basal de 1 (67%) y de 0 (33%).
GIOTRIF en segunda línea mejoró de forma significativa la PFS y la OS de los pacientes con NSCLC de histología escamosa en comparación con erlotinib. Los resultados de eficacia en el momento del análisis principal de la OS que incluía todos los pacientes aleatorizados se resumen en la Figura 2 y en la Tabla 9.
Tabla 9. Resultados de eficacia para GIOTRIF comparado con erlotinib en LUX-Lung 8, sobre la base del análisis primario de la OS, incluidos todos los pacientes aleatorizados.
|
GIOTRIF (N = 398) |
Erlotinib (N = 397) |
Razón de riesgos instantáneos/Cociente de probabilidades (IC del 95%) |
Valor p2 |
|
|
PFS Meses (mediana) |
2,63 |
1,94 |
HR 0,81 (0,69; 0,96) |
0,0103 |
|
OS Meses (mediana) Vivos a los 12 meses Vivos a los 18 meses |
7,92 36,4% 22,0% |
6,77 28,2% 14,4% |
HR 0,81 (0,69; 0,95) |
0,0077 |
|
Tasa de respuesta objetiva (CR+PR)1 |
5,5% |
2,8% |
OR 2,06 (0,98; 4,32) |
0,0551 |
|
Duración de la respuesta Meses (mediana) |
7,29 |
3,71 |
||
|
1 CR=respuesta completa; PR=respuesta parcial. 2 Valor p para la PFS/OS basado en la prueba de rango logarítmico estratificada; valor p para la tasa de respuesta objetiva y tasa de control de la enfermedad basado en la regresión logística. |
||||
La razón de riesgos instantáneos de la sobrevida general en pacientes <65 años de edad fue de 0,68 (IC del 95% 0,55, 0,85) y en pacientes de 65 años de edad o mayores fue de 0,95 (IC del 95% 0,76, 1,19).
Figura 2. Curva de Kaplan-Meier para la OS por grupo de tratamiento en LUX-Lung 8
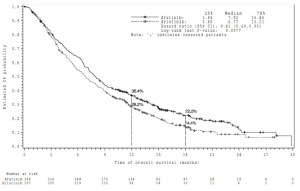
Referencias de la Figura 2:
Estimated OS probability: Probabilidad de OS estimada
Time of overall survival (months): Tiempo de sobrevida general (meses)
Median: Mediana
Hazard ratio (95% CI): Razón de riesgos instantáneos (IC 95%)
Log-rank test P-value: Valor p de prueba de rango logarítmico
Number at risk: Número de pacientes en riesgo
Note: ‘+’ indicates censored patients: Nota: ‘+’ indica pacientes censurados
El beneficio de la PFS estuvo acompañado por la mejoría en los síntomas relacionados con la enfermedad y el retraso del tiempo hasta el deterioro (véase la Tabla 10).
Tabla 10. Resultados de los síntomas para GIOTRIF comparado con erlotinib en el estudio LUX-Lung 8 (QLQ-C30 y QLQ-LC13 de la EORTC)
|
Tos |
Disnea |
Dolor |
|
|
% de pacientes con mejoríaa,c |
43% frente a 35%; p = 0,0294 |
51% frente a 44%; p = 0,0605 |
40% frente a 39%; p = 0,7752 |
|
Retraso del tiempo hasta el deterioro (meses)b,c |
4,5 frente a 3,7 HR 0,89; p = 0,2562 |
2,6 frente a 1,9 HR 0,79; p = 0,0078 |
2,5 frente a 2,4 HR 0,99; p = 0,8690 |
|
a Valores presentados para GIOTRIF comparado con erlotinib, valor p basado en la regresión logística. b Valor p para el tiempo hasta el deterioro basado en la prueba de rango logarítmico estratificada. |
|||
No se ha establecido la eficacia en los tumores negativos para EGFR.
Población pediátrica: La Agencia Europea de Medicamentos (European Medicines Agency, EMA) ha dejado sin efecto la obligación de presentar los resultados de los estudios realizados con este medicamento en todos los subconjuntos de la población pediátrica para las indicaciones de NSCLC (véase la sección Posología y Forma de Administración para obtener información sobre el uso pediátrico).
Propiedades farmacocinéticas:
Absorción: Tras la administración oral de GIOTRIF, las concentraciones máximas (Cmax) de afatinib se observaron aproximadamente 2 a 5 horas después de la administración de la dosis. Los valores de Cmax y AUC0-8 registraron un aumento ligeramente más que proporcional dentro el rango de dosis de GIOTRIF de 20 mg a 50 mg. La exposición sistémica a afatinib se redujo a razón de un 50% (Cmax) y un 39% (AUC0-8) cuando este medicamento se administró junto con una comida de alto contenido graso en comparación con la administración en ayunas. Sobre la base de los datos de farmacocinética poblacional obtenidos a partir de estudios clínicos en diversos tipos de tumores, se observó una disminución promedio del 26% en el AUCt,ss cuando se consumieron alimentos dentro del lapso de 3 horas antes o 1 hora después de la toma de GIOTRIF. Por lo tanto, no se deben consumir alimentos como mínimo 3 horas antes y 1 hora después de haber tomado GIOTRIF (véanse las secciones Posología y forma de administración e Interacción con otros medicamentos y otras formas de interacción).
Distribución: La unión in vitro de afatinib a las proteínas plasmáticas humanas es de aproximadamente un 95%. Afatinib se une a las proteínas mediante enlaces no covalentes (unión a las proteínas tradicional) y covalentes.
Biotransformación: Las reacciones metabólicas catalizadas por enzimas tienen un papel insignificante para afatinib in vivo. Los aductos covalentes a proteínas fueron los principales metabolitos circulantes de afatinib.
Eliminación: En los seres humanos, la excreción de afatinib se produce principalmente a través de las heces. Tras la administración de una solución oral de 15 mg de afatinib, el 85,4% de la dosis se recuperó en las heces y el 4,3% en la orina. El compuesto original afatinib representó el 88% de la dosis recuperada. Afatinib se elimina con una vida media efectiva de aproximadamente 37 horas. Por lo tanto, las concentraciones plasmáticas de afatinib en estado de equilibrio dinámico se alcanzaron dentro de los 8 días de la administración de dosis múltiples de afatinib, lo cual condujo a una acumulación de 2,77 veces (AUC0-8) y 2,11 veces (Cmax). En los pacientes tratados con afatinib durante más de 6 meses, se calculó una vida media terminal de 344 h.
Poblaciones especiales:
Insuficiencia renal: Menos del 5% de una dosis única de afatinib se excreta a través de los riñones.
Se comparó la exposición a afatinib de sujetos con insuficiencia renal y la de voluntarios sanos tras la administración de una dosis única de 40 mg de GIOTRIF®. La exposición de los sujetos con insuficiencia renal moderada (índice de filtración glomerular estimado [eGFR] de 30 a 59 ml/min según la fórmula MDRD) fue 101% (Cmax) y 122% (AUC0-tz) en comparación con los voluntarios sanos. La exposición de los sujetos con insuficiencia renal grave (eGFR de 15 a 29 ml/min de acuerdo con la fórmula MDRD) fue 122% (Cmax) y 150% (AUC0-tz) en comparación con los voluntarios sanos. Según este estudio y el análisis farmacocinético poblacional de los datos obtenidos de ensayos clínicos efectuados en diversos tipos de tumor se concluye que no es necesario ajustar la dosis inicial de los pacientes con insuficiencia renal leve (eGFR 60-89 ml/min), moderada (eGFR 30-59 ml/min) o grave (eGFR 15-29 ml/min), pero que se debe realizar un monitoreo de los pacientes que sufren insuficiencia grave (véase “Análisis farmacocinético poblacional en poblaciones especiales” a continuación y “Posología y administración”). No se ha estudiado GIOTRIF® en pacientes con un eGFR < 15 ml/min o sometidos a diálisis.
Insuficiencia hepática: Afatinib es eliminado principalmente por excreción biliar/fecal. Los sujetos con insuficiencia hepática leve (Child Pugh A) y moderada (Child Pugh B) tuvieron una exposición similar en comparación con los voluntarios sanos después de una dosis única de 50 mg de GIOTRIF. Esto coincide con los datos de farmacocinética poblacional obtenidos de estudios clínicos en diversos tipos de tumores (véase la sección “Análisis de farmacocinética poblacional en poblaciones especiales”, más adelante). No parece ser necesario ningún ajuste de dosis inicial en los pacientes con insuficiencia hepática leve o moderada (véase la sección Posología y forma de administración). No se ha estudiado la farmacocinética de afatinib en sujetos con disfunción hepática grave (Child Pugh C) (véase la sección Advertencias y precauciones de uso especiales).
Análisis farmacocinético poblacional en poblaciones especiales: Se realizó un análisis de farmacocinética poblacional en 927 pacientes con cáncer (764 con NSCLC) que recibieron monoterapia de GIOTRIF. No se consideró necesario realizar ningún ajuste de dosis inicial para ninguna de las siguientes covariables estudiadas.
Edad: No se pudo observar ninguna repercusión significativa de la edad (rango: 28 años a 87 años) sobre la base de la farmacocinética de afatinib.
Peso corporal: La exposición plasmática (AUCt,ss) se incrementó a razón de un 26% para un paciente de 42 kg (percentil 2,5) y se redujo a razón de un 22% para un paciente de 95 kg (percentil 97,5) en relación con un paciente con un peso corporal de 62 kg (mediana del peso corporal de los pacientes en la población total).
Sexo: Las mujeres tuvieron una exposición plasmática (AUCt,ss, con corrección para peso corporal) un 15% más alta que los hombres.
Raza: La raza no tuvo efecto alguno sobre la farmacocinética de afatinib, sobre la base de un análisis de farmacocinética poblacional que abarcó pacientes de raza asiática, raza blanca y raza negra. Los datos sobre los grupos raciales de raza negra fueron limitados.
Insuficiencia renal: La exposición a afatinib se incrementó moderadamente conforme menor fue la depuración de creatinina (creatinine clearance, CrCL; calculada mediante la fórmula de Cockcroft Gault), es decir, para un paciente con una CrCL de 60 ml/min o de 30 ml/min, la exposición (AUCt,ss) a afatinib aumentó un 13% y un 42%, respectivamente, y disminuyó un 6% y un 20% para un paciente con una CrCL de 90 ml/min o de 120 ml/min, respectivamente, en comparación con un paciente con una CrCL de 79 ml/min (mediana de CrCL de los pacientes en la población de pacientes total analizada).
Insuficiencia hepática: La insuficiencia hepática leve y moderada, según lo identificado a través de resultados anómalos en las pruebas de función hepática, no se correlacionó con ningún cambio significativo en la exposición de afatinib. Hubo datos limitados en relación con la insuficiencia hepática moderada y grave.
Otros factores intrínsecos o características de los pacientes: Otros factores intrínsecos o características de los pacientes que tuvieron una repercusión significativa sobre la exposición a afatinib fueron: estado funcional ECOG, niveles de lactato deshidrogenasa, niveles de fosfatasa alcalina y proteína total. Las magnitudes del efecto individual de cada una estas covariables no se consideraron clínicamente relevantes. Los antecedentes de tabaquismo, el consumo de alcohol (datos limitados) o la presencia de metástasis hepáticas no tuvieron ninguna repercusión significativa en la farmacocinética de afatinib.
Otra información sobre interacciones medicamentosas:
Interacciones con los sistemas de transporte y absorción de fármacos: Los datos in vitro indicaron que se considera improbable que se produzcan interacciones medicamentosas con afatinib como consecuencia de la inhibición de los transportadores OATB1B1, OATP1B3, OATP2B1, OAT1, OAT3, OCT1, OCT2 y OCT3.
Interacciones con las enzimas del citocromo P450 (CYP): En los seres humanos, se ha determinado que las reacciones metabólicas catalizadas por enzimas desempeñan un papel insignificante en el metabolismo de afatinib. Aproximadamente el 2% de la dosis de afatinib fue metabolizada por FMO3 y la N-desmetilación dependiente del CYP3A4 fue demasiado baja para ser detectada en forma cuantitativa. Afatinib no es un inhibidor ni un inductor de las enzimas del CYP. Por ende, es improbable que este medicamento interactúe con otros medicamentos que modulan, o bien son metabolizados por, las enzimas CYP.
Efecto de la inhibición de la UDP-glucuronosiltransferasa 1A1 (UGT1A1) sobre afatinib: Sobre la base de datos in vitro, se considera improbable que se produzcan interacciones medicamentosas con afatinib como consecuencia de la inhibición de UGT1A1.
CONTRAINDICACIONES:
Hipersensibilidad al afatinib o a cualquiera de los excipientes.
FERTILIDAD, EMBARAZO Y LACTANCIA:
Mujeres con capacidad para procrear:
Como medida de precaución, se debe advertir a las mujeres con capacidad reproductiva que deben evitar quedar embarazadas mientras reciben tratamiento con GIOTRIF. Se deben utilizar métodos anticonceptivos adecuados durante el tratamiento y durante por lo menos 1 mes después de la última dosis.
Embarazo:
Desde el punto de vista mecanístico, todos los medicamentos que tienen como blanco de acción el EGFR tienen el potencial de causar daños al feto.
Los estudios en animales realizados con afatinib no indicaron efectos nocivos directos ni indirectos en lo que respecta a la toxicidad para la reproducción (véase la sección Datos de seguridad preclínicos). Los estudios en animales no han revelado ningún signo de teratogenia hasta los niveles de dosis maternoletales, inclusive. Los cambios adversos estuvieron restringidos a los niveles de dosis tóxicos. No obstante, las exposiciones sistémicas logradas en los animales se ubicaron dentro de un rango similar o inferior a los niveles observados en los pacientes (véase la sección Datos de seguridad preclínicos).
Existe escasa o ninguna información disponible sobre el uso de este medicamento en las mujeres embarazadas. Se desconoce el riesgo de este medicamento para los seres humanos. Si este medicamento se usa durante el embarazo o si la paciente queda embarazada durante el tratamiento con GIOTRIF o después del mismo, se debe informar a la paciente del potencial riesgo que ello implica para el feto.
Lactancia: Los datos de farmacocinética disponibles obtenidos en animales han indicado que afatinib se excreta en la leche (véase la sección Datos de seguridad preclínicos). Según ello, es probable que afatinib se excrete en la leche humana. No se puede descartar un riesgo para el lactante. Se debe informar a las madres que no deben amamantar mientras estén recibiendo este medicamento.
Fertilidad: No se han realizado estudios de fertilidad con afatinib en los seres humanos. Los datos de toxicología no clínicos disponibles han indicado efectos sobre los órganos reproductores con la administración de dosis elevadas. Por lo tanto, no se puede descartar la posibilidad de que este medicamento tenga efectos adversos sobre la fertilidad en los seres humanos.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR VEHÍCULOS Y OPERAR MAQUINARIA:
GIOTRIF ejerce un efecto mínimo sobre la capacidad para conducir vehículos y operar maquinaria. Durante el tratamiento, se han informado reacciones adversas oculares (conjuntivitis, sequedad ocular, queratitis) en algunos pacientes (véase la sección Reacciones adversas) que podrían afectar la capacidad de los pacientes para conducir vehículos u operar maquinaria.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: La influencia de GIOTRIF sobre la capacidad para conducir y utilizar máquinas es pequeña. Durante el tratamiento se han notificado reacciones adversas oculares en algunos pacientes (conjuntivitis, ojo seco, queratitis) (ver Reacciones adversas) lo que puede afectar la capacidad de los pacientes para conducir y utilizar máquinas.
REACCIONES ADVERSAS:
Resumen del perfil de seguridad: Los tipos de reacciones adversas (adverse reactions, ADR) estuvieron en general asociados con el modo de acción inhibitorio del EGFR que posee afatinib. En la Tabla 2 se presenta un resumen de todas las ADR. Las ADR más frecuentes fueron diarrea y eventos adversos cutáneos (véase la sección Advertencias y precauciones de uso especiales), así como también estomatitis y paroniquia (véase también la Tabla 3 y la Tabla 4). En general, la reducción de la dosis (véase la sección Posología y forma de administración) condujo a una menor frecuencia de reacciones adversas comunes.
En los pacientes tratados con un régimen de una toma diaria de 40 mg de GIOTRIF, las reducciones de la dosis a raíz de ADR se produjeron en el 57% de los pacientes en el estudio LUX-Lung 3 y en el 25% de los pacientes en el estudio LUX-Lung 8. Las discontinuaciones debido a ADR de diarrea y exantema/acné se produjeron en el 1,3% y 0% de los pacientes en el estudio LUX-Lung 3 y en el 3,8% y 2,0% de los pacientes en el estudio LUX-Lung 8, respectivamente.
Las reacciones adversas similares a la ILD se informaron en el 0,7% de los pacientes tratados con afatinib. Se han informado lesiones cutáneas bullosas, vesiculares y exfoliativas, incluidos casos raros sugestivos de síndrome de Stevens Johnson y necrólisis epidérmica tóxica, si bien en dichos casos había otras posibles etiologías (véase la sección Advertencias y precauciones de uso especiales).
Listado tabulado de reacciones adversas: En la Tabla 2 se resumen las frecuencias de ADR combinadas de todos los estudios en NSCLC y de la experiencia posterior a la comercialización con dosis diarias de GIOTRIF de 40 mg o de 50 mg como monoterapia. Se utilizaron los siguientes términos para clasificar a las ADR en función de su frecuencia: muy frecuentes (≥1/10); frecuentes (≥1/100 a <1/10); poco frecuentes (≥1/1.000 a <1/100); raras (≥1/10.000 a <1/1.000); muy raras (<1/10.000). Dentro de cada categoría de frecuencia, las reacciones adversas se presentan en orden decreciente de seriedad.
Tabla 2: Resumen de ADR por categoría de frecuencia
|
Sistema orgánico |
Muy frecuentes (≥ 1/10) |
Frecuentes (≥ 1/100 a < 1/10) |
Poco frecuentes (≥ 1/1.000 a < 1/100) |
Raros (≥ 1/10.000 a < 1/1.000) |
|
Infecciones e infestaciones |
Paroniquia1 |
Cistitis |
||
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito |
Deshidratación Hipopotasemia |
||
|
Trastornos del sistema nervioso |
Disgeusia |
|||
|
Trastornos oculares |
Conjuntivitis Sequedad ocular |
Queratitis |
||
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis |
Rinorrea |
Enfermedad pulmonar intersticial |
|
|
Trastornos gastrointestinales |
Diarrea Estomatitis2 Nauseas Vómitos |
Dispepsia Queilitis |
Pancreatitis |
|
|
Trastornos hepatobiliares |
Elevación de la aminotransferasa alanina Elevación de la aspartato aminotransferasa |
|||
|
Trastornos de la piel y del tejido subcutáneo |
Exantema3 Dermatitis acneiforme4 Prurito5 Piel seca6 |
Alteraciones en las uñas Síndrome de eritrodisestesia palmoplantar |
Síndrome de Stevens-Johnson7 Necrólisis epidérmica tóxica7 |
|
|
Trastornos musculoesquéleticos y del tejido conjuntivo |
Espasmos musculares |
|||
|
Trastornos renales y urinarios |
Insuficiencia renal/ Falla renal |
|||
|
Trastornos generales y afecciones del sitio de administración |
Pirexia |
|||
|
Exploraciones complementarias |
Descenso de peso |
|||
|
1 Incluye paroniquia, infección de uña, infección de lecho ungueal. 2 Incluye estomatitis, estomatitis aftosa, inflamación de mucosa, ulceración de boca, erosión de mucosa bucal, erosión de mucosa, ulceración de mucosa. 3 Incluye un grupo de términos preferentes de exantema. 4 Incluye acné, acné pustuloso, dermatitis acneiforme. 5 Incluye prurito, prurito generalizado. 6 Incluye piel seca, piel descamada. 7 Sobre la base de la experiencia posterior a la comercialización. |
||||
Descripción de las reacciones adversas seleccionadas: Las ADR que fueron muy frecuentes en los pacientes tratados con GIOTRIF y se produjeron en al menos un 10% de los pacientes en el estudio LUX Lung 3 se resumen por Grado de los Criterios Toxicológicos Comunes del Instituto Nacional del Cáncer (National Cancer Institute-Common Toxicity Criteria, NCI CTC) en la Tabla 3.
Tabla 3. ADR muy frecuentes en el estudio LUX Lung 3
|
GIOTRIF (40 mg/día) N = 229 |
Pemetrexed/ Cisplatino N = 111 |
|||||
|
Grado de NCI-CTC |
Cualquier grado |
3 |
4 |
Cualquier grado |
3 |
4 |
|
Término preferente del MedDRA |
% |
% |
% |
% |
% |
% |
|
Infecciones e infestaciones |
||||||
|
Paroniquia1 |
57,6 |
11,4 |
0 |
0 |
0 |
0 |
|
Trastornos del metabolismo y de la nutrición |
||||||
|
Disminución del apetito |
20,5 |
3,1 |
0 |
53,2 |
2,7 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||
|
Epistaxis |
13,1 |
0 |
0 |
0,9 |
0,9 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea Estomatitis2 Queilitis |
95,2 69,9 12,2 |
14,4 8,3 0 |
0 0,4 0 |
15,3 13,5 0,9 |
0 0,9 0 |
0 0 0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||||
|
Exantema3 Dermatitis acneiforme4 Piel seca5 Prurito6 |
70,3 34,9
19,2 |
14 2,6 0,4 0,4 |
0 0 0 0 |
6,3 0 1,8 0,9 |
0 0 0 0 |
0 0 0 0 |
|
Exploraciones complementarias |
||||||
|
Descenso de peso |
10,5 |
0 |
0 |
9,0 |
0 |
0 |
|
1 Incluye paroniquia, infección de uña, infección de lecho ungueal. 2 Incluye estomatitis, estomatitis aftosa, inflamación de mucosa, ulceración de boca, erosión de mucosa bucal, erosión de mucosa, ulceración de mucosa. 3 Incluye un grupo de términos preferentes de exantema. 4 Incluye acné, acné pustuloso, dermatitis acneiforme. 5 Incluye piel seca, piel descamada. 6 Incluye prurito, prurito generalizado. |
||||||
Valores anómalos de función hepática: Se observaron valores anómalos en las pruebas de función hepática (lo que incluye valores elevados de ALT y AST) en los pacientes tratados con GIOTRIF 40 mg. Estas elevaciones fueron mayormente transitorias y no condujeron a la discontinuación del tratamiento. Las elevaciones de los niveles de ALT de Grado 2 (>2,5 a 5,0 veces el límite normal superior (upper limit of normal, ULN)) se produjeron en <8% de los pacientes tratados con este medicamento. Las elevaciones de Grado 3 (>5,0 a 20,0 veces el ULN) se produjeron en <4% de los pacientes tratados con GIOTRIF (véase la sección Advertencias y precauciones de uso especiales).
Informe de sospechas de reacciones adversas: Es importante el informe de los casos de sospechas de reacciones adversas tras la autorización de este medicamento. Ello permite continuar monitoreando el balance de riesgo/beneficio del medicamento. Los profesionales de la salud deben informar todas las sospechas de reacciones adversas a través del Sistema Nacional de Farmacovigilancia.
Descripción de las reacciones adversas seleccionadas: Las RAM que fueron muy frecuentes en los pacientes tratados con GIOTRIF y se produjeron en al menos un
10% de los pacientes en el estudio LUX Lung 8 se resumen por Grado de los Criterios Toxicológicos Comunes del Instituto Nacional del Cáncer en la Tabla 4.
Tabla 4. RAM muy frecuentes en el estudio LUX-Lung 8*
|
GIOTRIF (40 mg/día) N=392 |
Erlotinib N=395 |
|||||
|
Grado de NCI-CTC |
Cualquier grado |
3 |
4 |
Cualquier grado |
3 |
4 |
|
Término preferente del MedDRA |
% |
% |
% |
% |
% |
% |
|
Infecciones e infestaciones |
||||||
|
Paroniquia1 |
11,0 |
0,5 |
0 |
5,1 |
0,3 |
0 |
|
Trastornos del metabolismo y de la nutrición |
||||||
|
Disminución del apetito |
24,7 |
3,1 |
0 |
26,1 |
2,0 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
74,7 |
9,9 |
0,8 |
41,3 |
3,0 |
0,3 |
|
Estomatitis2 |
30,1 |
4,1 |
0 |
10,6 |
0,5 |
0 |
|
Náuseas |
20,7 |
1,5 |
0 |
16,2 |
1,0 |
0,3 |
|
Trastornos de la piel y del tejido subcutáneo |
||||||
|
Exantema3 |
60,7 |
5,4 |
0 |
56,7 |
8,1 |
0 |
|
Dermatitis acneiforme4 |
14,0 |
1,3 |
0 |
18,0 |
2,5 |
0 |
|
* Notificación de la frecuencia de pacientes con EA de cualquier causa. 1 Incluye paroniquia, infección de uña, infección de lecho ungueal. 2 Incluye estomatitis, estomatitis aftosa, inflamación de mucosa, ulceración de boca, erosión de mucosa bucal, erosión de mucosa, ulceración de mucosa. 3 Incluye un grupo de términos preferentes de exantema. 4 Incluye acné, acné pustuloso, dermatitis acneiforme. |
||||||
Valores anómalos de función hepática: Se observaron valores anómalos en las pruebas de función hepática (lo que incluye valores elevados de ALT y AST) en los pacientes tratados con GIOTRIF 40 mg. Estas elevaciones fueron mayormente transitorias y no condujeron a la discontinuación del tratamiento. Las elevaciones de los niveles de ALT de Grado 2 se produjeron en el 1% de los pacientes tratados con este medicamento. Las elevaciones de Grado 3 se produjeron en el 0,8% de los pacientes tratados con GIOTRIF (véase la sección Advertencias y precauciones de uso especiales).
“Por favor comunicarse con su médico o farmacéutico en caso se presente cualquier reacción adversa que no esté descrita en este inserto”.
INCOMPATIBILIDADES: No procede.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN:
Interacciones con los sistemas de transporte de fármacos: Efectos de los inhibidores de la P gp y de la proteína de resistencia del cáncer de mama sobre el afatinib.
Estudios in vitro han demostrado que afatinib es un sustrato de la P gp y de la BCRP. Cuando el ritonavir (200 mg dos veces al día durante 3 días), un inhibidor potente de la P gp y la BCRP, se administró 1 hora antes de una dosis única de 20 mg de GIOTRIF, la exposición a afatinib se incrementó a razón de un 48% (área bajo la curva (AUC0-8)) y un 39% (concentración plasmática máxima (Cmax)). En cambio, cuando ritonavir se administró en forma simultánea o 6 horas después de la administración de 40 mg de GIOTRIF, la biodisponibilidad relativa de afatinib fue del 119% (AUC0-8) y del 104% (Cmax), y del 111% (AUC0-8) y del 105% (Cmax), respectivamente. Por lo tanto, se recomienda que los inhibidores potentes de la P-gp (incluidos, entre otros, ritonavir, ciclosporina A, ketoconazol, itraconazol, eritromicina, verapamilo, quinidina, tacrolimus, nelfinavir, saquinavir y amiodarona) se administren aplicando un régimen alternado, preferentemente con una separación de 6 horas o de 12 horas respecto de la dosis de GIOTRIF (véase la sección Posología y forma de administración).
Efectos de los inductores de la P-gp sobre el afatinib: El pretratamiento con rifampicina (600 mg una vez al día durante 7 días), un inductor potente de la P-gp, redujo la exposición plasmática a afatinib un 34% (AUC0-8) y un 22% (Cmax) tras la administración de una dosis única de 40 mg de GIOTRIF. Los inductores potentes de P-gp (incluidos, entre otros, rifampicina, carbamazepina, fenitoína, fenobarbital o hierba de San Juan (Hypericum perforatum)) pueden reducir la exposición a afatinib (véase la sección Advertencias y precauciones de uso especiales).
Efectos de afatinib sobre los sustratos de la P-gp: Sobre la base de los datos in vitro, se ha determinado que afatinib es un inhibidor moderado de la P-gp. Sin embargo, sobre la base de los datos clínicos, se considera improbable que el tratamiento con GIOTRIF se traduzca en cambios en las concentraciones plasmáticas de otros sustratos de la P gp.
Interacciones con la BCRP: Los estudios in vitro indicaron que afatinib es un sustrato y un inhibidor del transportador BCRP. Afatinib puede incrementar la biodisponibilidad de los sustratos de la BCRP administrados por vía oral (lo que incluye, entre otros, rosuvastatina y sulfasalazina).
Efecto de los alimentos sobre afatinib: La coadministración de una comida de alto contenido graso con GIOTRIF dio como resultado una disminución significativa de la exposición a afatinib de aproximadamente un 50% en lo que respecta a la Cmax y de un 39% en lo que respecta al AUC0-8. Este medicamento debe administrarse lejos de las comidas (véanse las secciones Posología y forma de administración y Propiedades farmacocinéticas).
DATOS DE SEGURIDAD PRECLÍNICOS:
La administración oral de dosis únicas a ratones y a ratas reveló un bajo potencial de toxicidad aguda de afatinib. En estudios de administración de dosis orales repetidas de hasta 26 semanas de duración en ratas, o de 52 semanas en cerdos miniatura, los principales efectos se identificaron en la piel (cambios dérmicos, atrofia epitelial y foliculitis en ratas), en el aparato gastrointestinal (diarrea, erosiones estomacales, atrofia epitelial en ratas y cerdos miniatura) y en los riñones (necrosis papilar en ratas). Según del hallazgo, dichos cambios se produjeron con niveles de exposición ubicados por encima, por debajo o dentro del rango de valores clínicamente relevantes. Además, en diversos órganos se observó atrofia del epitelio mediada farmacodinámicamente en ambas especies.
Toxicidad para la reproducción: Debido a su mecanismo de acción, todos los medicamentos cuyo blanco de acción es el EGFR, entre ellos GIOTRIF, tienen el potencial de causar daño fetal. Los estudios de desarrollo embriofetal realizados con afatinib no revelaron ningún indicio de teratogenia. Los respectivos valores de exposición sistémica total (AUC) de los animales fueron ligeramente más altos (2,2 veces en las ratas) o más bajos (0,3 veces en los conejos) que los niveles observados en los pacientes.
Afatinib radiomarcado administrado por vía oral a ratas en el Día 11 del período de lactancia se excretó en la leche de las madres.
Un estudio de fertilidad efectuado en ratas macho y hembra con diversos niveles de dosis hasta la máxima dosis tolerada no reveló ninguna repercusión significativa sobre la fertilidad. La exposición sistémica total (AUC0-24) observada en las ratas macho y hembra se ubicó dentro, o por debajo, del rango de valores observados en los pacientes (1,3 veces y 0,51 veces, respectivamente).
Un estudio realizado en ratas con diversos niveles de dosis hasta la máxima dosis tolerada no reveló ninguna repercusión significativa en el desarrollo pre- o posnatal. La exposición sistémica total más alta (AUC0-24) observada en las ratas hembra fue inferior a la observada en los pacientes (0,23 veces).
Fototoxicidad: Una prueba de 3T3 in vitro indicó que afatinib podría tener un potencial de fototoxicidad.
Carcinogenia: No se han realizado estudios de carcinogenia con GIOTRIF.
DATOS PRECLÍNICOS SOBRE SEGURIDAD: La administración oral de dosis únicas a ratones y ratas puso de manifiesto una toxicidad potencial aguda baja para afatinib. En estudios a dosis repetidas por vía oral durante un período de hasta 26 semanas en ratas o 52 semanas en cerdos enanos los efectos principales se identificaron en la piel (cambios dérmicos, atrofia epitelial y foliculitis en ratas), el tracto gastrointestinal (diarrea, erosiones en el estómago, atrofia epitelial en ratas y cerdos enanos) y los riñones (necrosis papilar en ratas). Según los hallazgos, estos cambios se produjeron a niveles de exposición bajos, en el intervalo normal o por encima de los niveles clínicamente relevantes. Además, en ambas especies se observó una atrofia epitelial en diversos órganos mediada por efectos farmacodinámicos.
Toxicidad reproductiva: Como consecuencia de su mecanismo de acción, todos los medicamentos dirigidos al EGFR, entre ellos GIOTRIF, pueden causar daño fetal. Los estudios de desarrollo embriofetal realizados con afatinib no mostraron efectos teratógenos. La exposición sistémica total (AUC) respectiva fue ligeramente superior (2,2 veces en las ratas) o inferior (0,3 veces en los conejos) en comparación con los niveles en pacientes.
Afatinib radiomarcado administrado por vía oral a ratas el día 11 del período de lactancia pasó a la leche materna.
Un estudio de fertilidad realizado en ratas machos y hembras con administración del medicamento hasta la dosis máxima tolerada no mostró un impacto significativo en la fertilidad. La exposición sistémica total (AUC0-24) en ratas machos y hembras se situó en el intervalo normal o por debajo de la observada en pacientes (1,3 veces y 0,51 veces, respectivamente).
Un estudio realizado en ratas hasta las dosis máximas toleradas no puso de manifiesto un impacto significativo en el desarrollo prenatal y postnatal. La exposición sistémica total (AUC0-24) máxima en ratas hembras fue inferior a la observada en pacientes (0,23 veces).
Fototoxicidad: Una prueba 3T3 in vitro concluyó que afatinib puede tener potencial fototóxico.
Carcinogenicidad: No se han llevado a cabo estudios de carcinogenicidad con GIOTRIF.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO
Evaluación del estado de mutación del EGFR: Cuando se evalúa el estado de mutación del EGFR de un paciente, es importante elegir una metodología adecuadamente validada y robusta para evitar la obtención de falsos negativos o falsos positivos.
Diarrea: Se han notificado casos de diarrea, incluido diarreas graves, durante el tratamiento con GIOTRIF (ver Reacciones adversas). La diarrea puede dar lugar a deshidratación con o sin insuficiencia renal, lo que en casos raros ha llevado a la muerte. La diarrea apareció habitualmente durante las primeras 2 semanas de tratamiento. La diarrea de grado 3 se produjo sobre todo durante las primeras 6 semanas de tratamiento.
Es importante tomar medidas terapéuticas proactivas contra la diarrea tan pronto como se presenten los primeros síntomas, incluyendo una hidratación adecuada combinada con fármacos antidiarreicos, especialmente durante las primeras 6 semanas del tratamiento. Se deben administrar medicamentos antidiarreicos (p. ej. loperamida) y aumentar progresivamente su dosis hasta la dosis máxima aprobada recomendada si es necesario. Los medicamentos antidiarreicos deben ser fácilmente accesibles para los pacientes para iniciar el tratamiento en cuanto aparezcan los primeros síntomas de diarrea y mantenerlo hasta el cese de la diarrea durante 12 horas. Es posible que los pacientes con diarrea grave tengan que interrumpir y disminuir la dosis o suspender el tratamiento con GIOTRIF (ver Posología y forma de administración). Los pacientes que presenten deshidratación pueden necesitar una reposición hidroelectrolítica por vía intravenosa.
Acontecimientos adversos cutáneos: Se han notificado casos de exantema/acné en pacientes tratados con este medicamento (ver Reacciones adversas). En general, el exantema se manifiesta como un exantema eritematoso y acneiforme leve o moderado que puede aparecer o empeorar en áreas expuestas al sol. Para pacientes que se expongan al sol, se recomienda llevar ropa para protegerse y el uso de protectores solares. La intervención precoz (por ejemplo con emolientes, antibióticos) de las reacciones dermatológicas puede facilitar la continuación del tratamiento con GIOTRIF. Los pacientes con reacciones cutáneas graves pueden requerir la interrupción temporal del tratamiento, reducción de la dosis (ver Posología y forma de administración), intervenciones terapéuticas adicionales y la derivación a un dermatólogo experto en el tratamiento de estos efectos cutáneos.
Se han notificado alteraciones cutáneas ampollosas, vesiculares y exfoliativas, incluyendo casos raros sugestivos de síndrome de Stevens-Johnson. Se debe interrumpir o suspender el tratamiento si el paciente desarrolla alteraciones dermatológicas ampollosas, vesiculares o exfoliativas graves (ver Reacciones adversas).
Sexo femenino, peso corporal bajo e insuficiencia renal subyacente: Se ha observado una mayor exposición a afatinib en pacientes mujeres, pacientes con bajo peso corporal y pacientes con una insuficiencia renal subyacente (ver Propiedades farmacocinéticas). Esto puede resultar en un riesgo mayor de desarrollar reacciones adversas, particularmente diarrea, exantema/acné y estomatitis. Se recomienda un seguimiento más estrecho en pacientes con estos factores de riesgo.
Enfermedad Pulmonar Intersticial (EPI): Se han notificado casos de EPI o reacciones adversas parecidas a la EPI (como infiltración pulmonar, neumonitis, síndrome del distrés respiratorio agudo, alveolitis alérgica), incluyendo fallecimientos, en pacientes que fueron tratados con GIOTRIF para el CPNM. Se notificaron reacciones adversas parecidas a la EPI en un 0,7% de más de 3.800 pacientes tratados. Se notificaron reacciones adversas parecidas a la EPI de grado CTCAE ≥3 en el 0,5% de los pacientes. No se han estudiado pacientes con antecedentes de EPI.
Para descartar una posible EPI se debe evaluar cuidadosamente a todos los pacientes con un inicio agudo y/o un empeoramiento inexplicable de síntomas respiratorios (disnea, tos, fiebre). Se debe interrumpir el tratamiento con este medicamento a la espera de los resultados de la evaluación de estos síntomas. En caso de diagnosticarse una EPI, se suspenderá definitivamente el tratamiento con GIOTRIF y se iniciará un tratamiento adecuado según sea necesario (ver Posología y forma de administración).
Insuficiencia hepática grave: Se han notificado casos de fallo hepático, incluyendo fallecimientos, durante el tratamiento con este medicamento en menos del 1% de los pacientes. En estos pacientes, los factores de confusión incluyeron hepatopatías preexistentes y/o comorbilidades asociadas a la progresión del proceso oncológico subyacente. Se recomienda realizar pruebas de función hepática periódicas en pacientes con hepatopatía preexistente. Se han observado elevaciones de grado 3 de la alanina aminotransferasa (ALT) y de la aspartato aminotransferasa (AST) en el 2,4% de los pacientes con pruebas hepáticas normales a nivel basal tratados con 40 mg/día, y fueron unas 3,5 veces mayores en pacientes con pruebas hepáticas anormales a nivel basal (ver Reacciones adversas). Puede hacerse necesaria la interrupción del tratamiento en pacientes que muestran un empeoramiento de la función hepática (ver Posología y forma de administración). Se suspenderá definitivamente el tratamiento en pacientes que desarrollen una insuficiencia hepática grave durante el tratamiento con GIOTRIF.
Queratitis: La aparición de síntomas como inflamación aguda o empeoramiento de la inflamación ocular, lagrimeo, fotosensiblidad, visión borrosa, dolor ocular y/u ojos enrojecidos debe conllevar la derivación inmediata del paciente a un oftalmólogo. Si el diagnóstico de queratitis ulcerativa se confirma, el tratamiento debe ser interrumpido o suspendido. Si se diagnostica queratitis, se deben evaluar cuidadosamente los beneficios y riesgos de continuar con el tratamiento. Este medicamento debe ser utilizado con precaución en pacientes con antecedentes de queratitis, queratitis ulcerativa o sequedad ocular grave. El uso de lentes de contacto también es un factor de riesgo para queratitis y ulceración (ver Reacciones adversas).
Función ventricular izquierda: Se ha relacionado la disfunción ventricular izquierda con una inhibición del HER2. De acuerdo con los datos de los estudios clínicos disponibles, no existen indicios de que este medicamento cause reacciones adversas sobre la contractilidad cardiaca. Sin embargo, este medicamento no ha sido estudiado en pacientes con una fracción de eyección ventricular izquierda alterada (FEVI) o en pacientes con antecedentes de cardiopatía significativa. En pacientes con factores de riesgo cardiacos y pacientes con enfermedades que pueden afectar la FEVI, se debe considerar la monitorización de la función cardiaca, incluyendo una evaluación de la FEVI en el momento basal y durante el tratamiento. En los pacientes que presentan signos y síntomas cardiacos relevantes durante el tratamiento, se debe considerar una monitorización cardiaca que incluya la evaluación de la FEVI.
En pacientes con una fracción de eyección inferior al límite considerado normal para la institución, se debe solicitar una consulta a cardiología y considerar la posibilidad de interrumpir o suspender el tratamiento.
Interacciones con la glicoproteína P (gp-P): El tratamiento concomitante con inductores potentes de la gp-P puede disminuir la exposición a afatinib (ver Interacción con otros medicamentos y otras formas de interacción).
Lactosa: Este medicamento contiene lactosa. Los pacientes con formas raras de intolerancia hereditaria a la galactosa, de insuficiencia de lactasa de Lapp o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
PRECAUCIONES ESPECIALES DE ELIMINACIÓN: La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Venta bajo receta médica.
Importado por:
BOEHRINGER INGELHEIM PERÚ S.A.C
Av. Canaval y Moreyra 480, Piso 20
San Isidro - Telf.: (51-1) 212-4132
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
El tratamiento con GIOTRIF debe ser iniciado y supervisado por un médico con experiencia en el uso de tratamientos oncológicos.
Debe establecerse el estado del paciente en términos de mutaciones del EGFR antes de iniciarse el tratamiento con GIOTRIF (véase la sección Advertencias y precauciones de uso especiales).
Posología: La dosis recomendada es 40 mg una vez al día.
Este medicamento debe administrarse lejos de las comidas. No se deben consumir alimentos como mínimo 3 horas antes y 1 hora después de haber tomado este medicamento (véanse las secciones Interacción con otros medicamentos y otras formas de interacción y Propiedades farmacocinéticas).
El tratamiento con GIOTRIF debe continuarse hasta la progresión de la enfermedad o hasta que el paciente ya no lo tolere (véase la Tabla 1 a continuación).
Incremento gradual de la dosis: Se puede considerar un incremento gradual de la dosis hasta un máximo de 50 mg/día en los pacientes que toleren una dosis inicial de 40 mg/día (es decir, en ausencia de diarrea, exantema, estomatitis y otras reacciones adversas de Grado > 1 según los CTCAE) durante el primer ciclo de tratamiento (21 días para el NSCLC positivo para mutación del EGFR y 28 días para el NSCLC de histología escamosa). La dosis no debe ser incrementada en los pacientes con una reducción previa de la dosis. La dosis diaria máxima es 50 mg.
Ajuste de dosis por reacciones adversas: Las reacciones adversas sintomáticas (p. ej., diarrea grave o persistente o reacciones adversas cutáneas) pueden manejarse con éxito mediante la interrupción del tratamiento y la aplicación de reducciones de dosis o bien mediante la discontinuación del tratamiento con GIOTRIF según se indica en la Tabla 1 (véanse las secciones Advertencias y precauciones de uso especiales y Reacciones adversas).
Tabla 1. Información sobre ajustes de dosis por reacciones adversas
|
Grado CTCAEa de las reacciones adversas |
Posología recomendada |
|
|
Grado 1 o Grado 2 |
No interrumpirb |
Ningún ajuste de dosis |
|
Grado 2 (prolongadoc o intolerable) o Grado >3 |
Interrumpir hasta Grado 0/1b |
Reanudar reducciones de dosis a razón de 10 mgd |
|
a Criterios Terminológicos Comunes del Instituto Nacional del Cáncer (National Cancer Institute, NCI) para Eventos Adversos. b En el caso de diarrea, debe iniciarse de inmediato la administración de medicamentos antidiarreicos (p. ej., loperamida), y en el caso de diarrea persistente debe continuarse su administración hasta que cesen las deposiciones flojas. c >48 horas de diarrea y/o >7 días de exantema. d Si un paciente no puede tolerar 20 mg/día, se debe considerar la discontinuación permanente de GIOTRIF. |
||
Si un paciente presenta un cuadro agudo de síntomas respiratorios o un empeoramiento de tales síntomas, se debe considerar la posibilidad de enfermedad pulmonar intersticial (interstitial lung disease, ILD), en cuyo caso debe interrumpirse el tratamiento hasta tanto esté disponible el resultado de la evaluación. En el caso de un diagnóstico de ILD, debe discontinuarse la administración de GIOTRIF y debe iniciarse el tratamiento adecuado según sea necesario (véase la sección Advertencias y precauciones de uso especiales).
Dosis omitidas: Si se omite una dosis, la dosis omitida se debe tomar dentro del mismo día tan pronto como el paciente detecte la omisión. No obstante, si la próxima dosis programada se debe tomar dentro de las 8 horas, entonces la dosis omitida se debe saltear.
Uso de inhibidores de la glucoproteína P (P-gp): Si fuera necesario el tratamiento con inhibidores de la P gp, los mismos deben ser administrados aplicando un régimen alternado, es decir, administrando la dosis del inhibidor de la P gp lo más alejada posible de la dosis de GIOTRIF. El lapso ideal de separación entre la administración del inhibidor de la P-gp y la dosis de GIOTRIF es de 6 horas (en el caso de los inhibidores de la P gp que tienen un régimen de administración de dos tomas diarias) o bien de 12 horas (en el caso de aquellos inhibidores de la P gp que tienen un régimen de una toma diaria) (véase la sección Interacción con otros medicamentos y otras formas de interacción).
Pacientes con insuficiencia renal: Se observó un aumento de la exposición a afatinib en los pacientes con insuficiencia renal moderada o grave (véase la sección Farmacocinética). No es necesario realizar ajustes en la dosis inicial de los pacientes con insuficiencia renal leve, moderada o grave (eGFR 15-29 ml/min). Debe realizarse un monitoreo de los pacientes con insuficiencia renal grave y ajustar la dosis de GIOTRIF® en caso de intolerancia. No se recomienda el tratamiento con GIOTRIF® en pacientes con eGFR < 15 ml/min o sometidos a diálisis. (véase la sección Propiedades farmacocinéticas).
Pacientes con insuficiencia hepática: La exposición a afatinib no cambia significativamente en los pacientes con insuficiencia hepática leve (Child Pugh A) o moderada (Child Pugh B) (véase la sección Propiedades farmacocinéticas). No es necesario realizar ajustes de la dosis inicial en los pacientes con insuficiencia hepática leve o moderada. Este medicamento no ha sido estudiado en pacientes con insuficiencia hepática grave (Child Pugh C). No se recomienda el tratamiento con este medicamento en dicha población (véase la sección Advertencias y precauciones de uso especiales).
Población pediátrica: No existe ningún uso relevante de GIOTRIF en la población pediátrica en la indicación de NSCLC. Por lo tanto, no se recomienda el tratamiento con este medicamento en los niños o adolescentes.
Forma de administración: Este medicamento está destinado a la administración por vía oral. Los comprimidos deben tragarse enteros con agua. Si el paciente no pudiera tragar los comprimidos enteros, estos se pueden disolver en aproximadamente 100 ml de agua sin gas. No se deben usar otros líquidos. El comprimido debe colocarse en el agua, sin aplastarlo, y se debe revolver de tanto en tanto el líquido durante un lapso de hasta 15 minutos hasta que el comprimido se desintegre en partículas muy pequeñas. La dispersión se debe ingerir de inmediato. El vaso se debe enjuagar con aproximadamente 100 ml de agua, que también se debe beber. La dispersión también se puede administrar mediante una sonda gástrica.
SOBREDOSIS:
Síntomas: La dosis más alta de afatinib estudiada en un número limitado de pacientes en estudios clínicos de Fase I fue 160 mg una vez al día durante 3 días y 100 mg una vez al día durante 2 semanas. Las reacciones adversas observadas con estas dosis fueron principalmente de tipo dermatológico (exantema/acné) y gastrointestinal (especialmente diarrea). Los casos de sobredosis en 2 adolescentes sanos, que involucraron la ingesta de 360 mg de afatinib cada uno (como parte de una ingesta farmacológica mixta), estuvieron asociados con eventos adversos de náuseas, vómitos, astenia, mareos, cefalea, dolor abdominal y elevación de la amilasa (<1,5 veces el ULN). Ambos individuos se recuperaron de dichos eventos adversos.
Tratamiento: No existe ningún antídoto específico para la sobredosis de este medicamento. Cuando exista una sospecha de sobredosis, deberá suspenderse la administración de GIOTRIF e iniciarse el cuidado de soporte.
"Ante esta eventualidad concurrir al hospital o comunicarse con los centros de toxicología:
En Uruguay: Ante una eventualidad concurrir al hospital, al Centro de Tóxico y Farmacovigilancia, Hospital de Clínicas – Facultad de Medicina, Tel. 1722
Si estuviera indicado, la eliminación del afatinib no absorbido se puede lograr mediante inducción de vómitos o por lavado gástrico.
PERÍODO DE VALIDEZ: 3 años.
No consumir el producto una vez sobrepasada la fecha de vigencia (expiración), incluida en los rotulados.
FORMA DE PRESENTACIÓN: Cajas de cartón x 7,14, 21, 28, 35, 42, 49 y 56 comprimidos recubiertos en envase blister (PVC/PVDC/ALUMINIO) contenido en sobre de aluminio - Pet (Película de tereftalato de polietileno (PET)/Lámina de aluminio/Película de polietileno de baja densidad (LDPE)).
TIPO Y CONTENIDO DEL ENVASE :
Presentaciones en Uruguay: Caja de cartón x 7, 14, 28 comprimidos recubiertos en envase blíster (PVC/PVDC-ALUMINIO) plateado, conteniendo en sobre de aluminio – PET (Película de tereftalato de polietileno (PET)/Lámina de aluminio/Película de Polietileno de baja densidad (LDPE)
Es posible que no se comercialicen todas las presentaciones.
Precauciones especiales para el descarte
Todo remanente del medicamento o material de descarte debe desecharse de acuerdo con los requerimientos locales.
Manténgase fuera del alcance de los niños
Venta con receta médica
Fabricado por:Boehringer Ingelheim Pharma GmbH & Co. KG, Ingelheim am Rheim, Alemania. Industria Alemana.
Bajo licencia de Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Alemania
V07-EMA
Importado por:
Perú:
BOEHRINGER INGELHEIM PERÚ S.A.C.
RUC 20523163320 Tel. (01) 412-5000
Dir. Tec. Q.F. Jesús Peña.
CONDICIONES DE CONSERVACIÓN Y ALMACENAMIENTO: Conservar en su envase original para proteger de la humedad y la luz.
No almacenar a temperatura superior a 30 °C.


