

ERLHOTIZAM-PD
ERLOTINIB
Comprimidos recubiertos
1 Caja, 30 Comprimidos recubiertos, 150 Miligramos
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA:
Un COMPRIMIDO RECUBIERTO que contiene: 150 mg de erlotinib (como erlotinib clorhidrato).
Para consultar la lista completa de excipientes ver Lista de excipientes.
FORMA FARMACÉUTICA:
Comprimido recubierto
INDICACIONES TERAPÉUTICAS:
Cáncer de Pulmón No Microcítico (CPNM): ERLHOTIZAM-PD® está indicado en el tratamiento de primera línea de pacientes con cáncer de pulmón no microcítico (CPNM) localmente avanzado o metastásico con mutaciones activadoras de EGFR.
ERLHOTIZAM-PD® está también indicado como tratamiento de mantenimiento de pacientes con CPNM localmente avanzado o metastásico con mutaciones activadoras de EGFR y enfermedad estable después de un régimen quimioterápico de primera línea.
ERLHOTIZAM-PD® también está indicado en el tratamiento de pacientes con CPNM localmente avanzado o metastásico tras fallo, al menos, a un tratamiento quimioterápico anterior.
Se deberían considerar los factores asociados con el aumento de la supervivencia cuando se prescriba ERLHOTIZAM-PD®.
No se ha demostrado beneficio en la supervivencia u otros efectos clínicamente relevantes del tratamiento en pacientes con tumores que no expresen el Receptor del Factor de Crecimiento Epidérmico (EGFR) IHQ negativa (ver Propiedades farmacodinámicas).
Cáncer de páncreas: ERLHOTIZAM-PD® en combinación con gemcitabina está indicado en el tratamiento de pacientes con cáncer de páncreas metastásico.
Se deberían considerar los factores asociados con el aumento de la supervivencia cuando se prescriba ERLHOTIZAM-PD® (ver Posología y forma de administración y Propiedades farmacodinámicas).
En pacientes con enfermedad localmente avanzada no se han podido observar ventajas en términos de supervivencia.
DATOS FARMACÉUTICOS:
Lista de excipientes: Lactosa monohidrato, celulosa microcristalina, almidón glicolato de sodio, povidona K-30, lauril estearato de sodio, estearato de magnesio, etanol, opadry II Blanco 85F18422, agua purificada.
Incompatibilidades: No procede.
Período de validez: 36 meses.
Precauciones especiales de conservación: Este medicamento no requiere condiciones especiales de conservación.
Condiciones de almacenamiento: Conservar en envase herméticamente cerrado. Almacenar a temperatura no mayor a 30 °C.
Precauciones especiales de eliminación: Ninguna especial para su eliminación.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Revisado el 24 de mayo del 2017
Fabricado por:
Korea United Pharm Inc.
Importado y distribuido por:
PHARMACEUTICAL DISTOLOZA S.A.
PROPIEDADES FARMACOCINÉTICAS:
Absorción: Tras la administración oral, los niveles plasmáticos máximos de erlotinib se obtienen a las 4 horas, aproximadamente, tras dicha administración. En un ensayo con voluntarios sanos se pudo estimar que la biodisponibilidad absoluta es del 59%. La exposición tras una dosis oral puede verse incrementada por los alimentos.
Distribución: Erlotinib tiene un volumen de distribución aparente medio de 232 l y se distribuye dentro del tejido tumoral en humanos. En un ensayo con 4 pacientes (3 con cáncer de pulmón no microcítico [CPNM] y 1 con cáncer de laringe) que fueron tratados con dosis orales diarias de 150 mg de erlotinib, se determinó los niveles de erlotinib en muestras de tumores obtenidas por extracciones quirúrgicas realizadas el Día 9 del tratamiento, obteniéndose concentraciones medias de erlotinib de 1.185 ng/g de tejido tumoral. Esto correspondía a una media global del 63% (rango 5-161%) de las concentraciones plasmáticas máximas en equilibrio que han sido determinadas. Los metabolitos activos principales estaban presentes en el tumor en concentraciones medias de 160 ng/g de tejido, lo que correspondía a un promedio global del 113% (rango 88-130%) de las concentraciones plasmáticas máximas en equilibrio que han sido determinadas. La unión a las proteínas plasmáticas es aproximadamente de un 95%. Erlotinib se une a la albúmina sérica y a la glicoproteína ácida alfa1 (AAG).
Biotransformación: En humanos, erlotinib se metaboliza en el hígado por los citocromos hepáticos, principalmente por el CYP3A4 y en menor medida por el CYP1A2. También contribuye potencialmente al aclaramiento metabólico de erlotinib el metabolismo extrahepático por el CYP3A4 en el intestino, por el CYP1A1 en pulmones y por el CYP1B1 en tejido tumoral.
Se han identificado tres rutas metabólicas principales: 1) O-demetilación de cualquiera de las cadenas laterales o de ambas, seguida de la oxidación de los ácidos carboxílicos; 2) oxidación del grupo acetileno, seguida de la hidrólisis del ácido aril carboxílico y 3) hidroxilación aromática del grupo fenil-acetileno. Los metabolitos principales de erlotinib, OSI-420 y OSI-413, producidos por la O-demetilación de cualquiera de las cadenas laterales, tuvieron una potencia comparable a erlotinib en ensayos no-clínicos in vitro y en modelos tumorales in vivo. Estos metabolitos están presentes en el plasma a niveles < 10% de erlotinib y muestran una farmacocinética similar a la de erlotinib.
Eliminación: Erlotinib se excreta predominantemente por las heces en forma de metabolitos (>90%) y una pequeña cantidad de una dosis oral se elimina por vía renal (aproximadamente el 9%). Menos del 2% de la dosis administrada oralmente se excreta como sustancia sin alterar. En un análisis farmacocinético poblacional realizado en 591 pacientes que recibieron erlotinib como medicamento único, se determinó un aclaramiento aparente medio de 4,47 l/hora con una mediana de semivida de 36,2 horas. Por lo tanto, el tiempo en alcanzar la concentración plasmática en equilibrio se espera que sea de 7-8 días aproximadamente.
Farmacocinética en poblaciones especiales: En base a los análisis farmacocinéticos poblacionales, no se ha observado relación clínicamente significativa entre el aclaramiento aparente predicho y la edad, peso, sexo y raza del paciente. Los factores de los pacientes que se correlacionaban con la farmacocinética de erlotinib fueron la bilirrubina total sérica, AAG y fumar en la actualidad. Las concentraciones séricas elevadas de bilirrubina total y las concentraciones de AAG se asociaron con una reducción del aclaramiento de erlotinib. No está clara la relevancia clínica de estas diferencias.
Sin embargo, los fumadores tuvieron un índice incrementado de aclaramiento de erlotinib. Esto fue confirmado en un estudio farmacocinético realizado en sujetos sanos que fumaban en el momento de realizar el estudio y en no fumadores, recibiendo ambos grupos una dosis oral única de 150 mg de erlotinib. La media geométrica de la Cmax fue 1056 ng/mL en no fumadores y 689 ng/mL en fumadores con una proporción media de fumadores versus no fumadores del 65,2% (95% CI: 44,3 a 95,9, p = 0,031). La media geométrica del AUC0-inf fue 18726 ng• h/mL en los no fumadores y 6718 ng• h/mL en los fumadores con una proporción media del 35,9% (95% CI: 23,7 a 54,3, p < 0,0001). La media geométrica de la C24h fue 288 ng/mL en no fumadores y 34,8 ng/mL en fumadores con una proporción media del 12,1% (95% CI: 4,82 a 30,2, p = 0,0001).
En un estudio pivotal fase III en fumadores activos con CPNM, se alcanzó una concentración plasmática en estado estacionario de 0,65 µg/mL (n=16), que fue aproximadamente 2 veces menor en aquellos que habían dejado de fumar o en pacientes que nunca habían fumado (1,28 µg/mL n=108). Este efecto se produjo junto a un aumento del 24% en el aclaramiento plasmático aparente de erlotinib. En un estudio fase I de escalada de dosis en pacientes que fueron fumadores activos con CPNM, el análisis de la farmacocinética indicó un aumento proporcional de la exposición a erlotinib en el estado de equilibrio, cuando la dosis de erlotinib se aumento de 150 mg a la dosis máxima tolerada de 300 mg. En este estudio, la concentración plasmática en estado estacionario para una dosis de 300 mg en fumadores activos fue de 1,22 µg/mL (n=17).
En base a los resultados de estudios farmacocinéticos, se debe aconsejar a los fumadores que dejen de fumar mientras estén en tratamiento con erlotinib, ya que, de lo contrario, las concentraciones plasmáticas podrían verse reducidas.
En base a los análisis farmacocinéticos poblacionales, la presencia de un opioide incrementó la exposición alrededor de un 11%.
Se llevó a cabo un segundo análisis farmacocinético poblacional incorporando los datos de erlotinib obtenidos de 204 pacientes con cáncer pancreático que fueron tratados con erlotinib y gemcitabina. Este análisis demostró que las covariantes que afectan al aclaramiento de erlotinib en los pacientes del estudio pancreático eran muy similares a los observados en el análisis farmacocinético previo en monoterapia. No se han identificado nuevos efectos covariantes. La co-administración de gemcitabina no tuvo efecto en el aclaramiento plasmático de erlotinib.
Población pediátrica: No se han realizado estudios específicos en pacientes pediátricos.
Población anciana: No se han realizado estudios específicos en pacientes ancianos.
Insuficiencia hepática: Erlotinib se aclara principalmente en el hígado. En pacientes con tumores sólidos y con insuficiencia hepática moderada (valor Child-Pugh 7- 9) la media geométrica del AUC0-t y la Cmax de erlotinib fue 27000 ng• h/mL y 805 ng/mL, respectivamente, en comparación con 29300 ng• h/mL y 1090 ng/mL en pacientes con buena función hepática incluyendo pacientes con cáncer de hígado primario o metástasis hepáticas. Aunque la Cmax fue más baja de forma estadísticamente significativa en pacientes con insuficiencia hepática moderada, se consideró que esta diferencia no era clínicamente relevante. No hay datos disponibles respecto a la influencia de la disfunción hepática grave en la farmacocinética de erlotinib. En análisis farmacocinéticos poblacionales, el aumento de las concentraciones séricas de la bilirrubina total se asoció con un índice de aclaramiento de erlotinib más lento.
Insuficiencia renal: Erlotinib y sus metabolitos no se excretan significativamente vía renal ya que menos del 9% de una dosis única es excretada en la orina. En análisis farmacocinéticos poblacionales, no se observó una relación significativa entre el aclaramiento de erlotinib y el aclaramiento de creatinina, pero no hay datos disponibles en pacientes con aclaramiento de creatinina < 15 ml/min.
PROPIEDADES FARMACOLÓGICAS:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Agente antineoplásico inhibidor de la proteína quinasa, código ATC: L01XE03
Mecanismo de acción: Erlotinib es un inhibidor del receptor del factor de crecimiento epidérmico/de la tirosina quinasa del receptor del factor de crecimiento epidérmico humano tipo 1 (EGFR, también conocido como HER1). Erlotinib inhibe potentemente la fosforilación intracelular del EGFR. El EGFR se expresa en la superficie de células normales y cancerosas. En modelos no clínicos, la inhibición de la fosfotirosina del EGFR da lugar a que la célula quede en fase de equilibrio y/o conduce a la muerte celular.
Las mutaciones del EGFR pueden conducir a la activación constitutiva de rutas de señalización antiapoptóticas y de proliferación. La potente efectividad de erlotinib para bloquear la señal mediada por el EGFR en estos tumores con mutación positiva del EGFR, se atribuye a la estrecha unión de erlotinib al lugar de unión del ATP en el dominio quinasa mutado del EGFR. Debido al bloqueo de la cascada de señales por debajo del receptor, se detiene la proliferación celular y se induce la muerte celular a través de la ruta intrínseca de apoptosis. Se ha observado regresión tumoral en modelos de ratones con marcada expresión de mutaciones activadoras del EGFR.
Eficacia clínica:
• Tratamiento en primera línea para pacientes con Cáncer de Pulmón No Microcítico (CPNM) con mutaciones activadoras del EGFR (Erlotinib administrado en monoterapia): La eficacia de erlotinib en el tratamiento en primera línea para los pacientes que presentan mutaciones activadoras del EGFR en CPNM, se demostró en un ensayo fase III, aleatorizado, abierto (ML20650, EURTAC). Este estudio se llevó acabo en pacientes caucásicos con CPNM metastásico o localmente avanzado (estadío IIIB y IV) que no habían recibido quimioterapia previa o cualquier terapia antitumoral sistémica para su enfermedad avanzada y que presentaban mutaciones en el dominio de la tirosina kinasa del EGFR (eliminación del exón 19 o mutación del exón 21). Los pacientes fueron aleatorizados 1:1 para ser tratados con erlotinib 150 mg o quimioterapia doble basada en platinos.
La SLP, variable principal del investigador. Los resultados de eficacia se resumen en la Tabla 5.
Figura 1: Curva Kaplan-Meier para datos de SLP evaluada por el investigador en el ensayo ML20650
(EUTARC) (corte abril del 2012)
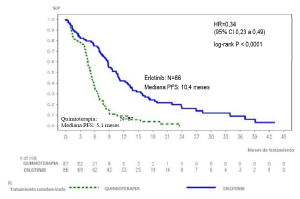
Tabla 5: Resultados de eficacia de Erlotinib vs quimioterapia en el estudio ML20650 (EUTARC)
|
Erlotinib |
Quimioterapia |
Hazard Ratio (95% CI) |
Valor- p |
||
|
Análisis intermedio pre planificado (n=153) Fecha de corte: Agosto 2010 |
n=77 |
n=76 |
|||
|
Variable principal: Supervivencia Libre de Progresión (mediana de SLP en meses)* |
|||||
|
Evaluación del investigador** |
9,4 |
5,2 |
0,42 [0,27-0,64] |
p<0,0001 |
|
|
Revisión independiente ** |
10,4 |
5,4 |
0,47 [0,27-0,78] |
p=0,003 |
|
|
Tasa respuesta objetiva (CR/PR) |
54,5% |
10,5% |
p<0,0001 |
||
|
Supervivencia global (SG) (meses) (35% de eventos) |
22,9 |
18,8 |
0,80 [0,47-1,37] |
p=0,4170 |
|
|
Análisis exploratorio (n=173) Fecha de corte: Enero 2011 |
n=86 |
n=87 |
|||
|
SLP (mediana en meses), Evaluación del investigador |
9,7 |
5,2 |
0,37 [0,27-0,54] |
p<0,0001 |
|
|
Tasa respuesta objetiva (CR/PR) |
58,1% |
14,9% |
p<0,0001 |
||
|
SG (meses) (40% de eventos) |
19,3 |
19,5 |
1,04 [0,65-1,68] |
p=0,8702 |
|
|
Actualización del análisis (n=173) Fecha de corte: Abril 2012 |
n=86 |
n=87 |
|||
|
SLP (mediana en meses) |
10,4 |
5,1 |
0,34 [0,23-0,49] |
p<0,0001 |
|
|
SG*** (meses) (62% de eventos) |
22,9 |
20,8 |
0,93 [0,64-1,36] |
p=0,7149 |
|
|
CR = respuesta completa, PR = respuesta parcial * Se observó una reducción del 58% en el riesgo de progresión de la enfermedad o muerte ** La tasa global de concordancia entre el investigador y la evaluación del Comité Investigador Independiente fue del 70% *** Se observó un elevado número de cruces en el tratamiento. El 82% de los pacientes en el brazo de quimioterapia recibieron en segunda línea un inhibidor de la tirosina quinasa del EGFR (todos menos 2 pacientes recibieron Erlotinib). |
|||||
• Tratamiento de mantenimiento de CPNM tras quimioterapia de primera línea (Erlotinib administrado en monoterapia): Se ha investigado la eficacia y la seguridad de erlotinib como mantenimiento tras una primera línea de quimioterapia, para pacientes con CPNM, en un ensayo (BO18192, SATURN) aleatorizado, doble– ciego, controlado con placebo. Este ensayo fue realizado en 889 pacientes con CPNM localmente avanzado o metastásico que no habían progresado tras 4 ciclos de un doblete de quimioterapia, basada en compuestos de platino. Los pacientes fueron aleatorizados 1:1 para ser tratados con erlotinib 150 mg o placebo por vía oral una vez al día, hasta progresión de la enfermedad. El objetivo primario del ensayo incluía Supervivencia Libre de Progresión (PFS) en todos los pacientes. Las características demográficas basales y de la enfermedad, estuvieron bien equilibradas en los dos brazos de tratamiento. No se incluyeron en el estudio pacientes con ECOG PS>1, con comorbilidad hepática o renal significativa.
En este estudio, la población total mostró un beneficio para la variable principal SLP (HR=0,71 p< 0,0001) y la variable secundaria SG (HR=0,81 p=0,0088). Sin embargo, el mayor beneficio se observó en un análisis exploratorio predefinido en pacientes con mutaciones activadoras de EGFR (n=49) demostrando un beneficio considerable de SLP (HR=0,10, 95% CI 0,04 a 0,25, p<0,0001) y un HR de la supervivencia global de 0,83 (95% CI 0,34 a 2,02). El 67% de los pacientes del subgrupo placebo con mutación EGFR positiva recibieron tratamiento de segunda o sucesivas líneas con EGFR-TKIs.
El estudio BO25460 (IUNO) se realizó en 643 pacientes con CPNM avanzado sin mutaciones activadoras de EGFR (deleción del exón 19 o mutación L858R del exón 21) y que no experimentaron progresión de la enfermedad después de cuatro ciclo de quimioterapia basada en compuestos de platino.
El objetivo del estudio era comparar la supervivencia global del tratamiento de mantenimiento de primera línea con erlotinib versus erlotinib administrado en el momento de progresión de la enfermedad. Este estudio no alcanzó con su variable principal. La SG de erlotinib en el tratamiento de mantenimiento de primera línea no fue superior al tratamiento con erlotinib en segunda línea en pacientes cuyo tumor no albergó una mutación activadora de EGFR (HR=1,02, 95% CI 0,85 a 1,22, p=0,82). La variable secundaria de SLP no mostró ninguna diferencia entre erlotinib y placebo en el tratamiento de mantenimiento (HR=0,94, 95% CI 0,80 a 1,11, p=0,48).
Según los datos del estudio BO25460 (IUNO), el uso de erlotinib no está recomendado en el tratamiento de mantenimiento de primera línea en pacientes sin mutaciones activadoras de EGFR.
• Tratamiento de CPNM tras fallo de al menos un régimen quimioterápico anterior (erlotinib administrado en monoterapia): La eficacia y seguridad de erlotinib en segunda/tercera línea han sido demostradas en un ensayo aleatorizado, doble ciego, controlado con placebo (BR.21) realizado en 731 pacientes con CPNM localmente avanzado o metastático tras el fallo de, al menos, un tratamiento quimioterápico. Los pacientes fueron aleatorizados 2:1 para ser tratados con erlotinib 150 mg o placebo por vía oral una vez al día. Los objetivos del ensayo incluyeron supervivencia global, supervivencia libre de progresión (PFS), índice de respuesta, duración de la respuesta, tiempo hasta el deterioro de los síntomas relacionados con el cáncer de pulmón (tos, disnea y dolor) y seguridad. El objetivo principal fue la supervivencia.
Las características demográficas estuvieron bien equilibradas en los dos grupos de tratamiento. Alrededor de dos tercios de los pacientes fueron hombres y aproximadamente un tercio presentaban un ECOG basal de 2 y un 9% tenían un ECOG basal de 3. El 93% y el 92% de todos los pacientes del grupo erlotinib y del grupo placebo respectivamente, habían sido tratados con una terapia anterior que contenía platino y un 36% y un 37% de todos los pacientes, respectivamente, habían sido tratados anteriormente con taxanos.
El índice de riesgo ajustado por muerte en el grupo tratado con erlotinib en relación con el grupo placebo fue 0,73 (95% CI, de 0,6 a 0,87) (p = 0,001). El porcentaje de pacientes vivos a los 12 meses fue del 31,2% y 21,5% para los grupos tratados con erlotinib y con placebo, respectivamente. La mediana de supervivencia global fue de 6,7 meses (95% CI, de 5,5 a 7,8 meses) en el grupo tratado con erlotinib en comparación con los 4,7 meses observados en el grupo placebo (95% CI, de 4,1 a 6,3 meses).
Se ha investigado el efecto sobre la supervivencia global en distintos subgrupos de pacientes. El efecto de erlotinib sobre la supervivencia global fue similar en pacientes con una ECOG basal de 2 - 3 (HR = 0,77, 95% CI 0,6 - 1,0) o de 0 - 1 (HR = 0,73, 95% CI 0,6 - 0,9), hombres (HR = 0,76, 95% CI 0,6 - 0,9) o mujeres (HR = 0,80, 95% CI 0,6 - 1,1), pacientes menores de 65 años (HR = 0,75, 95% CI 0,6 - 0,9) o mayores (HR = 0,79, 95% CI 0,6 - 1,0), pacientes con un tratamiento anterior (HR = 0,76, 95% CI 0,6 - 1,0) o con más de uno (HR = 0,75, 95% CI 0,6 - 1,0), caucasianos (HR = 0,79, 95% CI 0,6 - 1,0) o asiáticos (HR = 0,61, 95% CI 0,4 - 1,0), pacientes con adenocarcinoma (HR = 0,71, 95% CI 0,6 - 0,9) o carcinoma escamoso (HR = 0,67, 95% CI 0,5 - 0,9), pero no con otras histologías (HR = 1,04, 95% CI 0,7 - 1,5), pacientes con enfermedad en estadio IV diagnosticada (HR = 0,92, 95% CI 0,7 - 1,2) o en estadio < IV (HR = 0,65, 95% CI 0,5 - 0,8). Los pacientes que nunca habían fumado obtuvieron un beneficio mucho mayor con erlotinib (supervivencia HR = 0,42, 95% CI 0,28 - 0,64) en comparación con los fumadores o los ex - fumadores (HR = 0,87, 95% CI 0,71 - 1,05).
La expresión del EGFR se determinó en el 45% de los pacientes, estableciéndose un índice de riesgo de 0,68 (95% CI 0,49 - 0,94) en los pacientes con tumores EGFR - positivos y de 0,93 (95% CI 0,63 - 1,36) en aquellos con tumores EGFR - negativos (definidos mediante ICH empleando el kit EGFR pharmDx y definiendo EGFR - negativo como tinción de menos del 10% de las células tumorales). En el 55% restante, en el que se desconocía la expresión del EGFR, el índice de riesgo fue de 0,77 (95% CI 0,61-0,98).
La mediana de PFS fue de 9,7 semanas en el grupo tratado con erlotinib (95% CI, 8,4 a 12,4 semanas) siendo de 8,0 semanas en el grupo placebo (95% CI, 7,9 a 8,1 semanas).
El índice de respuesta objetiva medido por RECIST fue del 8,9% en el grupo con erlotinib (95% CI, 6,4 a 12,0). Los primeros 330 pacientes que fueron evaluados centralmente (índice de respuesta: 6,2%) y 401 pacientes fueron evaluados por el investigador (índice de respuesta: 11,2%).
La mediana de duración de la respuesta fue de 34,3 semanas, con un rango entre 9,7 y 57,6+ semanas. La proporción de pacientes que presentaron respuesta completa, respuesta parcial o enfermedad estable en el grupo tratado con erlotinib y placebo fue del 44,0% y 27,5% (p = 0,004), respectivamente.
También se observó un beneficio en la supervivencia en pacientes tratados con erlotinib que no alcanzaron una respuesta objetiva tumoral (por RECIST). La evidencia de esta afirmación radica en que el índice de riesgo por muerte fue de 0,82 (95% CI, 0,68 a 0,99) en pacientes cuya mejor respuesta fue enfermedad estable o progresiva.
En el tratamiento con erlotinib se obtuvieron beneficios en los síntomas ya que se prolongó de forma significativa el tiempo hasta el deterioro de los síntomas en forma de tos, disnea y dolor versus placebo.
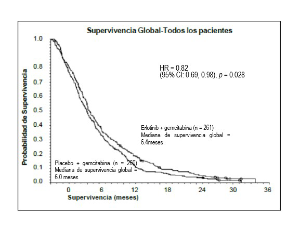
• Cáncer de páncreas (erlotinib administrado en combinación con gemcitabina en el ensayo PA.3): La eficacia y seguridad de erlotinib en combinación con gemcitabina como tratamiento de primera línea fueron evaluadas en un ensayo aleatorizado, doble-ciego, controlado con placebo en pacientes con cáncer de páncreas localmente avanzado, no resecable o metastásico. Los pacientes fueron aleatorizados para ser tratados con erlotinib o placebo una vez al día de forma continua y gemcitabina IV (1000 mg/m2, Ciclo 1 – Días 1, 8, 15, 22, 29, 36 y 43 de un ciclo de 8 semanas; Ciclo 2 y siguientes ciclos – Días 1, 8 y 15 de un ciclo de 4 semanas (ver la Ficha Técnica de gemcitabina para la dosis y posología autorizadas para el cáncer de páncreas). Tanto erlotinib como el placebo fueron administrados por vía oral una vez al día hasta progresión de la enfermedad o toxicidad inaceptable. El objetivo principal del ensayo fue supervivencia global.
Las características demográficas y de enfermedad de los pacientes fueron similares entre los 2 grupos de tratamiento, erlotinib 100 mg y gemcitabina o placebo y gemcitabina, excepto por una ligera mayor proporción de mujeres en el brazo de erlotinib/gemcitabina en comparación con el brazo tratado con placebo/gemcitabina:
|
Situación a nivel basal |
Erlotinib |
Placebo |
|
Mujeres |
51% |
44% |
|
ECOG performance status (PS) = 0 a nivel basal |
31% |
32% |
|
ECOG performance status (PS) = 1 a nivel basal |
51% |
51% |
|
ECOG performance status (PS) = 2 a nivel basal |
17% |
17% |
|
Enfermedad metastásica a nivel basal |
77% |
76% |
La supervivencia se evaluó en la población con intención de ser tratada (“Intend-to-treat population”) en base al seguimiento de los datos de supervivencia. Los resultados se muestran en la tabla siguiente (los datos de los grupos de pacientes con enfermedad metastásica o localmente avanzada se derivan del análisis exploratorio de los subgrupos).
|
Resultado |
Erlotinib (meses) |
Placebo (meses) |
∆ (meses) |
CI del ∆ |
HR |
CI del HR |
Valor de P |
|
Población global |
|||||||
|
Mediana de supervivencia global |
6,4 |
6,0 |
0,41 |
-0,54-1,64 |
0,82 |
0,69-0,98 |
0,028 |
|
Media de supervivencia global |
8,8 |
7,6 |
1,16 |
-0,05-2,34 |
|||
|
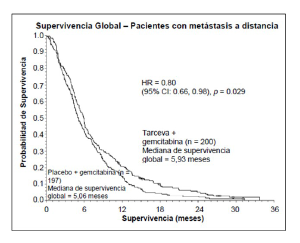
Población con enfermedad metastásica |
|||||||
|
Mediana de supervivencia global |
5,9 |
5,1 |
0,87 |
-0,26-1,56 |
0,80 |
0,66-0,98 |
0,029 |
|
Media de supervivencia global |
8,1 |
6,7 |
1,43 |
0,17-2,66 |
|||
|
Población con enfermedad localmente avanzada |
|||||||
|
Mediana de supervivencia global |
8,5 |
8,2 |
0,36 |
-2,43-2,96 |
0,93 |
0,65-1,35 |
0,713 |
|
Media de supervivencia global |
10,7 |
10,5 |
0,19 |
-2,43-2,69 |
|||
En un análisis de los datos del estudio realizado a posteriori se observó que los pacientes con situación clínica favorable a nivel basal (baja intensidad de dolor, buena calidad de vida y buen PS) pueden obtener más beneficio del tratamiento con erlotinib. El beneficio se relaciona principalmente con la existencia de una puntuación baja de intensidad de dolor.
En un análisis de los datos del estudio realizado a posteriori se observó que los pacientes en tratamiento con erlotinib que desarrollaron rash, tuvieron una supervivencia global más larga comparada con los pacientes que no tuvieron rash (mediana de supervivencia global de 7,2 meses versus 5 meses, HR: 0,61). El 90% de los pacientes desarrollaron rash en los primeros 44 días de tratamiento. La mediana del tiempo de aparición de rash fue de 10 días.
Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con erlotinib en los diferentes grupos de la población pediátrica para las indicaciones de Cáncer de Pulmón No Microcítico y Cáncer Pancreático (ver Posología y forma de administración, para consultar la información sobre el uso en la población pediátrica)
CONTRAINDICACIONES:
Hipersensibilidad a erlotinib o a alguno de los excipientes incluidos en la sección Lista de excipientes.
FERTILIDAD, EMBARAZO Y LACTANCIA:
Embarazo: No hay datos relativos al uso de erlotinib en mujeres embarazadas. Los estudios en animales no han mostrado evidencia de teratogenicidad o parto anormal. Sin embargo, no se puede excluir un efecto adverso en el embarazo, ya que los estudios llevados a cabo en ratas y conejos han mostrado un incremento de la letalidad embrio/fetal (ver Datos preclínicos sobre seguridad). El riesgo potencial en humanos se desconoce.
Mujeres en edad fértil: Se debe aconsejar a las mujeres en edad fértil que eviten quedarse embarazadas mientras estén en tratamiento con erlotinib. Deberán emplearse métodos anticonceptivos adecuados durante el tratamiento y durante al menos las 2 semanas siguientes a su terminación. Sólo se continuará el tratamiento en mujeres embarazadas si el beneficio potencial para la madre supera al riesgo para el feto.
Lactancia: Se desconoce si erlotinib se excreta en la leche humana. Se debe desaconsejar a las madres la lactancia materna mientras sean tratadas con erlotinib por el daño potencial que se pueda causar al niño.
Fertilidad: Los estudios en animales no han mostrado evidencia de alteración de la fertilidad. Sin embargo, no se puede excluir un efecto adverso en la fertilidad, ya que los estudios con animales han mostrado efectos sobre los parámetros reproductivos (ver Datos preclínicos sobre seguridad). No se conoce el riesgo potencial en humanos.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS:
No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas. Sin embargo, no hay asociación entre erlotinib y la alteración de la habilidad mental.
REACCIONES ADVERSAS:
Cáncer de pulmón no microcítico (Erlotinib administrado en monoterapia): En el ensayo aleatorizado doble-ciego (BR.21; erlotinib administrado como segunda línea de tratamiento), las reacciones adversas al medicamento observadas más frecuentemente fueron rash (75%) y diarrea (54%). La mayoría fueron de gravedad grado 1/2 y no necesitaron intervención. Se observaron rash y diarrea de grado 3/4 en un 9% y 6%, respectivamente, de los pacientes tratados con erlotinib y, en ambos casos, supuso el abandono del ensayo en el 1% de los pacientes. Fue necesaria la reducción de la dosis debido a rash y diarrea en un 6% y un 1% de los pacientes, respectivamente. En el ensayo BR.21, la mediana de tiempo que tardó en aparecer el rash fue 8 días y la de la diarrea, 12 días.
Generalmente, la aparición de rash se manifiesta en forma de una erupción eritematosa y papulopustular leve o moderada, que puede producirse o empeorar en las zonas expuestas al sol. Sería aconsejable que los pacientes expuestos al sol usen ropa para protegerse y/o protección solar (por ejemplo que contenga minerales).
En el ensayo pivotal BR.21, las reacciones adversas que se observaron con más frecuencia (≥3%) en los pacientes tratados con erlotinib que en el grupo placebo, y que ocurren en al menos el 10% de los pacientes del grupo tratado con erlotinib, están resumidos en la Tabla 1 en función de los grados según los criterios comunes de toxicidad del Instituto Nacional del Cáncer de Estados Unidos (NCI-CTC).
Se han utilizado los siguientes términos para clasificar los efectos indeseables por frecuencia: muy frecuente (≥1/10); frecuente (≥1/100 a < 1/10); poco frecuente (≥1/1.000 a < 1/100); raros (≥1/10.000 a < 1/1.000); muy raros (< 1/10.000) incluyendo casos aislados.
Dentro de cada grupo de frecuencia, las reacciones adversas son presentadas en orden decreciente de gravedad.
Tabla 1: Reacciones adversas (RA) muy frecuentes observadas en el ensayo BR.21
|
Erlotinib N = 485 |
Placebo N = 242 |
|||||
|
Grado del NCI-CTC |
Cualquier grado |
3 |
4 |
Cualquier grado |
3 |
4 |
|
Término MedDRA |
% |
% |
% |
% |
% |
% |
|
Total de pacientes con cualquier RA |
99 |
40 |
22 |
96 |
36 |
22 |
|
Infecciones e infestaciones |
||||||
|
Infección* |
24 |
4 |
0 |
15 |
2 |
0 |
|
Trastornos del metabolismo y de la nutrición |
||||||
|
Anorexia |
52 |
8 |
1 |
38 |
5 |
< 1 |
|
Trastornos oculares |
||||||
|
Queratoconjuntivitis seca |
2 |
0 |
0 |
3 |
0 |
0 |
|
Conjuntivitis |
12 |
<1 |
0 |
2 |
<1 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||
|
Disnea |
41 |
17 |
11 |
35 |
15 |
11 |
|
Tos |
33 |
4 |
0 |
29 |
2 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea** |
54 |
6 |
< 1 |
18 |
< 1 |
0 |
|
Náuseas |
33 |
3 |
0 |
24 |
2 |
0 |
|
Vómitos |
23 |
2 |
< 1 |
19 |
2 |
0 |
|
Estomatitis |
17 |
< 1 |
0 |
3 |
0 |
0 |
|
Dolor abdominal |
11 |
2 |
< 1 |
7 |
1 |
< 1 |
|
Trastornos de la piel y del tejido subcutáneo |
||||||
|
Rash*** |
75 |
8 |
< 1 |
17 |
0 |
0 |
|
Prurito |
13 |
< 1 |
0 |
5 |
0 |
0 |
|
Sequedad de la piel |
12 |
0 |
0 |
4 |
0 |
0 |
|
Trastornos generales y alteraciones en el lugar de administración |
||||||
|
Fatiga |
52 |
14 |
4 |
45 |
16 |
4 |
|
* Infecciones graves con o sin neutropenia, incluyeron neumonía sepsis y celulitis. ** Puede producir deshidratación, hipopotasemia y fallo renal. *** Rash incluida dermatitis acneiforme. |
||||||
En dos ensayos, doble-ciego, aleatorizados controlados con placebo, Fase III BO18192 (SATURN) y BO25460 (IUNO), erlotinib se administró como tratamiento de mantenimiento tras quimioterapia de primera línea. Estos ensayos se llevaron a cabo en 1.532 pacientes con CPNM avanzado, recurrente o metastásico, tras quimioterapia estándar en primera línea basada en compuestos de platino y no se identificaron nuevas señales de seguridad.
Las reacciones adversas observadas más frecuentemente en pacientes tratados con erlotinib, en los ensayos BO18192 y BO25460 fueron rash y diarrea (ver tabla 2). No se observaron rash o diarrea grado 4 en ninguno de los estudios. El rash y la diarrea supusieron el abandono del tratamiento con erlotinib en el 1% y en <1% de los pacientes respectivamente, en el estudio BO18192, mientras que ningún paciente abandonó el tratamiento por rash o diarrea en el estudio BO25460. Se requirieron modificaciones de dosis (interrupciones o reducciones) por el rash y la diarrea en el 8,3% y 3% de los pacientes, respectivamente, en el estudio BO18192, y 5,6% y 2,8% de los pacientes, respectivamente, en el estudio BO25460.
Tabla 2: Reacciones adversas más frecuentes en los Estudios BO18192 (SATURN) y BO25460 (IUNO)
|
BO18192 (SATURN)* |
BO25460 (IUNO)* |
|||
|
Erlotinib n=433 |
Placebo n=445 |
Erlotinib n=322 |
Placebo n=319 |
|
|
% |
% |
% |
% |
|
|
Rash, todos los grados |
49,2 |
5,8 |
39,4 |
10,0 |
|
Grado 3 |
6,0 |
0 |
5,0 |
1,6 |
|
Diarrea, todos los grados |
20,3 |
4,5 |
24,2 |
4,4 |
|
Grado 3 |
1,8 |
0 |
2,5 |
0,3 |
|
*Análisis de seguridad de la población |
||||
En un estudio fase III, ML20650 abierto, aleatorizado, llevado a cabo en 154 pacientes, la seguridad de erlotinib en primera línea de tratamiento en CPNM con mutaciones activadoras de EGFR fue evaluada en 75 pacientes; no se han observado nuevas señales de seguridad en estos pacientes.
En el estudio ML20650, las RAs más frecuentes observadas en pacientes tratados con erlotinib fueron rash y diarrea (80% y 57% respectivamente, de cualquier grado), la mayoría fueron Grado 1/2 y se manejaron sin necesidad de intervención. Se observaron rash y diarrea de Grado 3 en un 9% y 4% de los pacientes, respectivamente. No se observó rash y diarrea Grado 4. Tanto el rash como la diarrea supusieron el abandono del tratamiento con erlotinib en el 1% de los pacientes. Se necesitaron modificaciones de la dosis (interrupciones o reducciones) por el rash y la diarrea en el 11% y 7% de los pacientes, respectivamente.
Cáncer de páncreas (Erlotinib administrado en combinación con gemcitabina): Las reacciones adversas más frecuentes que se observaron en el ensayo pivotal PA.3 en pacientes con cáncer de páncreas tratados con erlotinib 100 mg y gemcitabina, fueron fatiga, rash y diarrea. En el brazo tratado con erlotinib y gemcitabina, se observó rash Grado 3-4 y diarrea en un 5% de los pacientes, respectivamente. La mediana del tiempo de aparición de rash y diarrea fue de 10 y 15 días, respectivamente. Tanto el rash como la diarrea provocaron la reducción de las dosis en un 2% de los pacientes, y ocasionaron la interrupción del ensayo de hasta el 1% de los pacientes tratados con erlotinib y gemcitabina.
En el ensayo pivotal PA.3, las reacciones adversas que se presentaron con mayor frecuencia (≥3%) en el grupo de pacientes tratados con erlotinib 100 mg y gemcitabina versus el grupo tratado con placebo y gemcitabina y, en al menos el 10% de los pacientes tratados con erlotinib 100 mg y gemcitabina, se resumen en la Tabla 3 siguiendo los criterios de clasificación por grados de toxicidad del Instituto Nacional del Cáncer (NCI-CTC).
Se han utilizado los siguientes términos para clasificar los efectos indeseables por frecuencia: muy frecuente (≥1/10); frecuente (≥1/100 a < 1/10); poco frecuente (≥1/1.000 a < 1/100); raros (≥1/10.000 a < 1/1.000); muy raros (< 1/10.000) incluyendo casos aislados.
Dentro de cada grupo de frecuencia, las reacciones adversas son presentadas en orden decreciente de gravedad.
Tabla 3: Reacciones adversas (RA) muy frecuentes observadas en el ensayo PA.3 (grupo cohorte tratado con 100 mg)
|
Erlotinib N = 259 |
Placebo N = 256 |
|||||
|
Grado según NCI-CTC |
Cualquier grado |
3 |
4 |
Cualquier grado |
3 |
4 |
|
Término MedDRA |
% |
% |
% |
% |
% |
% |
|
Total de pacientes con cualquier RA |
99 |
48 |
22 |
97 |
48 |
16 |
|
Infecciones e infestaciones |
||||||
|
Infección* |
31 |
3 |
< 1 |
24 |
6 |
< 1 |
|
Trastornos del metabolismo y de la nutrición |
||||||
|
Pérdida de peso |
39 |
2 |
0 |
29 |
< 1 |
0 |
|
Trastornos psiquiátricos |
||||||
|
Depresión |
19 |
2 |
0 |
14 |
< 1 |
0 |
|
Trastornos del sistema nervioso |
||||||
|
Neuropatía |
13 |
1 |
< 1 |
10 |
< 1 |
0 |
|
Cefalea |
15 |
<1 |
0 |
10 |
0 |
0 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||
|
Tos |
16 |
0 |
0 |
11 |
0 |
0 |
|
Trastornos gastrointestinales |
||||||
|
Diarrea** |
48 |
5 |
< 1 |
36 |
2 |
0 |
|
Estomatitis |
22 |
< 1 |
0 |
12 |
0 |
0 |
|
Dispepsia |
17 |
< 1 |
0 |
13 |
< 1 |
0 |
|
Flatulencia |
13 |
0 |
0 |
9 |
< 1 |
0 |
|
Trastornos de la piel y tejidos subcutáneos |
||||||
|
Rash*** |
69 |
5 |
0 |
30 |
1 |
0 |
|
Alopecia |
14 |
0 |
0 |
11 |
0 |
0 |
|
Trastornos generales y alteraciones en el lugar de administración |
||||||
|
Fatiga |
73 |
14 |
2 |
70 |
13 |
2 |
|
Fiebre |
36 |
3 |
0 |
30 |
4 |
0 |
|
Escalofríos |
12 |
0 |
0 |
9 |
0 |
0 |
|
* Las infecciones graves, con o sin neutropenia, incluyeron neumonía, sepsis y celulitis. ** Puede producir deshidratación, hipopotasemia y fallo renal. *** Rash incluida dermatitis acneiforme. |
||||||
Otras observaciones: La evaluación de seguridad de erlotinib está basada en los datos obtenidos en más de 1.500 pacientes tratados con, al menos, una dosis de 150 mg de erlotinib en monoterapia y en más de 300 pacientes que recibieron erlotinib 100 o 150 mg en combinación con gemcitabina.
Se han observado las siguientes reacciones adversas en pacientes tratados con erlotinib en monoterapia y en pacientes tratados con erlotinib en combinación con quimioterapia.
Las reacciones adversas observadas muy frecuentemente, en los estudios BR21 y PA 3, se describen en las Tablas 1 y 3 mientras que otras reacciones adversas, incluyendo las observadas en otros estudios, se resumen en la Tabla 4.
Dentro de cada grupo de frecuencia, las reacciones adversas son presentadas en orden decreciente de gravedad.
Tabla 4: Resumen de reacciones adversas por grupo de frecuencia:
|
Sistema Corporal |
Muy Frecuente (≥1/10) |
Frecuente (≥1/100 a < 1/10) |
Poco frecuente (≥1/1.000 a < 1/100) |
Raros (≥1/10.000 a < 1/1.000) |
Muy raros (< 1/10.000) |
|
Transtornos oculares |
-Queratitis -Conjuntivitis1 |
- Cambios en las pestañas2 |
-Perforación de la córnea -Ulceración de la córnea -Uveitis |
||
|
Transtornos respiratorios, torácicos y mediastínicos |
-Epistaxis -Enfermedad pulmonar intersticial grave (EPI)3 |
||||
|
Transtornos gastro intestinales |
-Diarrea7 |
-Hemorragias gastro- intestinales4, 7 |
-Perforación gastro-intestinal7 |
||
|
Transtornos hepatobiliares |
-Anormalidades en el test de función hepática5 |
-Fallo hepático6 |
|||
|
Transtornos de la piel y el tejido subcutáneo |
-Alopecia -Piel seca1 -Paroniquia -Foliculitis -Acné/Dermatitis acneiforme -Grietas en la piel |
-Hirsutismo -Cambios en las cejas -Uñas quebradizas y sueltas -Reacciones cutáneas leves como hiperpigmentación |
-Síndrome eritro-disestesia palmo- plantar |
-Síndrome Stevens- Johnson/necrólis is epidérmica tóxica7 |
|
|
Trastornos renales y urinarios |
-Insuficiencia renal1 |
-Nefritis1 -Proteinuria1 |
|||
|
1 En el ensayo clínico PA.3 2 Incluyendo crecimiento hacia el interior de las pestañas, crecimiento, y engrosamiento excesivo de las pestañas. 3 Incluye fallecimientos, en pacientes que recibieron Erlotinib para el tratamiento de CPNM u otros tumores sólidos avanzados (ver Advertencias y precauciones especiales de empleo). Se ha observado una incidencia alta en pacientes de origen Japonés (ver Advertencias y precauciones especiales de empleo). 4 En ensayos clínicos algunos casos se asociaros a la administración concomitante de warfarina y algunos a la administración concomitante de AINEs (ver Interacción con otros medicamentos y otras formas de interacción) 5 Incluyendo niveles incrementados de alanina aminotransferasa [ALT], aspartamo aminotransferasa [AST] y bilirrubina. Estos fueron muy frecuentes en el ensayo clínico P A 3 y frecuentes en el ensayo clínico BR. 21. Los casos fueron principalmente de gravedad media o moderadaza, de naturaleza transitoria o asociadas a metástasis hepáticas. 6 Incluye fallecimientos. Entre los factores de confusión se ha incluido la existencia previa de enfermedad hepática o la medicación concomitante hepatotóxica (ver Advertencias y precauciones especiales de empleo) 7 Incluye fallecimientos (ver Advertencias y precauciones especiales de empleo) |
|||||
Notificación de sospechas de reacciones adversas: Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. A los profesionales de la salud se les pide que reporten cualquier sospecha de reacciones adversas a través de: farmacovigilancia@digemid.minsa.gob.pe
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN:
Los estudios de interacciones se han realizado sólo en adultos. Erlotinib y otros sustratos del CYP
Erlotinib es un inhibidor potente del citocromo CYP1A1 y un inhibidor moderado del CYP3A4 y
CYP2C8, y es también un inhibidor fuerte de la glucuronidación por UGT1A1 in vitro.
Se desconoce la relevancia fisiológica de la fuerte inhibición del CYP1A1 debido a que la expresión de CYP1A1 es muy limitada en tejidos humanos.
Cuando erlotinib se coadministró con ciprofloxacino, un inhibidor moderado del CYP1A2, la exposición a erlotinib [AUC] aumentó significativamente un 39%, aunque no produjo ningún cambio estadísticamente significativo en Cmáx. Del mismo modo, la exposición al metabolito activo aumentó alrededor de un 60% y un 48% para AUC y Cmáx, respectivamente. No se ha determinado la relevancia clínica de este aumento. Debe tenerse precaución cuando ciprofloxacino o inhibidores potentes del CYP1A2 (por ej. fluvoxamina) se combinen con erlotinib. Si se observan reacciones adversas relacionadas con erlotinib, la dosis de erlotinib puede reducirse.
El tratamiento previo o la co-administración de erlotinib no alteró el aclaramiento de midazolam y eritromicina, sustratos prototípicos del CYP3A4, pero parece que disminuyó la biodisponibilidad oral del midazolam hasta el 24%. En otro ensayo clínico, erlotinib no afectó a la farmacocinética de paclitaxel, sustrato del CYP3A4/2C8, al ser administrado concomitantemente. Por tanto, las interacciones significativas con el aclaramiento de otros sustratos del CYP3A4 son improbables.
La inhibición de la glucuronidación puede provocar interacciones con medicamentos que son sustratos de UGT1A1 y que sólo se eliminan por esta vía. Los pacientes con bajos niveles de expresión de UGT1A1 o alteraciones genéticas de la glucuronidación (por ej. enfermedad de Gilbert) pueden tener un aumento de la concentración de bilirrubina en suero y deben ser tratados con precaución.
En humanos, erlotinib es metabolizado en el hígado por los citocromos hepáticos, principalmente por CYP3A4 y en menor medida por CYP1A2. También contribuye potencialmente al aclaramiento metabólico de erlotinib, el metabolismo extrahepático por CYP3A4 en el intestino, CYP1A1 en pulmones y CYP1B1 en tejido tumoral. Pueden darse interacciones potenciales con sustancias activas que se metabolicen por esas enzimas o sean inhibidores o inductores de dichas enzimas.
Los inhibidores potentes de la actividad del CYP3A4 disminuyen el metabolismo de erlotinib y aumentan la concentración plasmática de erlotinib. En un ensayo clínico, el uso concomitante de erlotinib con ketoconazol (200 mg vía oral dos veces al día durante 5 días), un potente inhibidor del CYP3A4, condujo a un aumento de la exposición a erlotinib (86% del AUC y 69% de la Cmáx). Debe tenerse precaución cuando se combine erlotinib con un inhibidor potente del CYP3A4, como por ej. antifúngicos azoles (como ketoconazol, itraconazol, voriconazol), inhibidores de la proteasa, eritromicina o claritromicina. Si es necesario se deberá reducir la dosis de erlotinib, particularmente si se observa toxicidad.
Los inductores potentes de la actividad del CYP3A4 aumentan el metabolismo de erlotinib y disminuyen significativamente las concentraciones plasmáticas de erlotinib. En un ensayo clínico, el uso concomitante de erlotinib y rifamicina (600 mg vía oral una vez al día durante 7 días), un inductor potente del CYP3A4, produjo una disminución de un 69% de la mediana del AUC de erlotinib. La coadministración de rifampicina con una sola dosis de erlotinib de 450 mg dio lugar a una exposición media de erlotinib (AUC) del 57,5% de la resultante tras una sola dosis de erlotinib de 150 mg sin rifampicina. Por lo tanto, debe evitarse la coadministración de erlotinib con inductores del CYP3A4. Se debe considerar un aumento en la dosis hasta 300 mg en pacientes que requieren tratamiento concomitante de erlotinib con inductores potentes del CYP3A4 tales como rifampicina, siempre que su seguridad sea monitorizada estrechamente (incluyendo la función renal y hepática y los electrolitos séricos), y si ésta se tolera bien durante más de dos semanas, se podría considerar un aumento a 450 mg con una monitorización estrecha de seguridad. También puede darse una exposición reducida con otros inductores como por ej. fenitoína, carbamazepina, barbitúricos o Hipérico (hypericum perforatum, hierba de San Juan). Debe tenerse precaución cuando estas sustancias activas se combinen con erlotinib. Cuando sea posible, se deben considerar tratamientos alternativos evitando los inductores potentes de la actividad del CYP3A4.
Erlotinib y anticoagulantes derivados de la cumarina: En pacientes tratados con Tarcerva se han notificado casos de interacción con anticoagulantes derivados de la cumarina, incluyendo la warfarina, que produjeron aumentos en el Ratio Internacional Normalizado (INR) y hemorragias, que en algunos casos fueron mortales. Los pacientes a los que se les administre anticoagulantes derivados de la cumarina deben ser monitorizados regularmente para detectar cualquier cambio en el tiempo de protombina o en el INR.
Erlotinib y estatinas: La combinación de erlotinib y una estatina puede aumentar el riesgo de miopatía inducida por estatinas, incluyendo rabdomiolisis, que fue observada de forma rara.
Erlotinib y fumadores: Los resultados de un estudio de interacción farmacocinética indicaron que existe una reducción significativa en el AUCinf, Cmax y en la concentración plasmática a las 24 horas de 2,8-, 1,5- y 9-veces, respectivamente, tras la administración de erlotinib en fumadores en comparación con no fumadores (ver Propiedades farmacocinéticas). Por tanto, se debería aconsejar a los pacientes fumadores que dejen de fumar lo antes posible antes de iniciar el tratamiento con erlotinib, puesto que, de lo contrario, la concentración plasmática de erlotinib será más reducida. El efecto clínico de la disminución de dicha exposición no ha sido evaluado de forma determinante pero es probable que sea clínicamente significativo.
Erlotinib e inhibidores de la P-glicoproteína: Erlotinib es sustrato de la P-glicoproteína (transportador de sustancias activas). La administración concomitante de inhibidores de la P-glicoproteína, como p. ej. ciclosporina y verapamilo, puede producir una alteración en la distribución y/o eliminación de erlotinib. No se ha establecido las consecuencias de esta interacción para, p. ej., la toxicidad a nivel del SNC. Deberá tenerse precaución en dichas situaciones.
Erlotinib y medicamentos que alteran el pH: Erlotinib se caracteriza por una disminución de su solubilidad a un pH superior a 5. Los medicamentos que alteran el pH del tracto Gastro-Intestinal (GI) superior pueden alterar la solubilidad de erlotinib y por lo tanto su biodisponibilidad. La coadministración de erlotinib con omeprazol, un inhibidor de la bomba de protones (IBP), disminuyó la exposición [AUC] y la concentración máxima (Cmáx) de erlotinib un 46% y 61%, respectivamente. No hubo cambio alguno del Tmáx o de la vida media. La administración concomitante de erlotinib con 300 mg de ranitidina, un antagonista de los receptores H2, disminuye la exposición de erlotinib [AUC] y las concentraciones máximas [Cmax] un 33% y 54% respectivamente. No es probable que un aumento de la dosis de erlotinib cuando se coadministre con tales agentes compense esta pérdida de exposición. Sin embargo, cuando erlotinib se administró de forma escalonada, 2 horas antes ó 10 horas después de la administración de 150 mg de ranitidina dos veces al día, la exposición de erlotinib [AUC] y las concentraciones máximas [Cmax] disminuyeron sólo un 15% y 17%, respectivamente. No se ha investigado el efecto de antiácidos sobre la absorción de erlotinib pero la absorción puede verse afectada produciendo una disminución de los niveles plasmáticos. En resumen, deberá evitarse la combinación de erlotinib con inhibidores de la bomba de protones. Si se considera necesario el uso de antiácidos durante el tratamiento con erlotinib, deberían tomarse al menos 4 horas antes ó 2 horas después de la dosis diaria de erlotinib. Si se considera necesario el uso de ranitidina, ésta debe administrarse de forma escalonada, es decir, se debe tomar erlotinib al menos 2 horas antes ó 10 horas después de la dosis de ranitidina.
Erlotinib y gemcitabina: En un estudio Fase Ib, no se dieron efectos significativos de gemcitabina sobre la farmacocinética de erlotinib ni tampoco efectos significativos de erlonitib en la farmacocinética de gemcitabina.
Erlotinib y carboplatino/paclitaxel: Erlotinib incrementa las concentraciones de platino. En un ensayo clínico el uso concomitante de erlotinib con carboplatino y paclitaxel dio lugar a un incremento en el AUC0-48 total de platino del 10,6%. Aunque es estadísticamente significativa, la magnitud de esta diferencia no se considera que sea clínicamente relevante. En la práctica clínica puede haber otros factores que produzcan un aumento en la exposición al carboplatino como el trastorno renal. No hubo efectos significativos de carboplatino o placlitaxel en la farmacocinética de erlotinib.
Erlotinib y capecitabina: Capecitabina puede incrementar la concentración de erlotinib. Al administrar erlotinib en combinación con capecitabina, se produjo un aumento estadísticamente significativo en la AUC de erlotinib y un aumento incierto en la Cmax en comparación con los valores observados en otro estudio en el cual se administró erlotinib como único agente. No hubo efectos significativos de erlotinib en la farmacocinética de capecitabina.
Erlotinib e inhibidores del proteasoma: Por su mecanismo de acción, los inhibidores del proteasoma, incluyendo el bortezomib pueden influir en el efecto de los inhibidores del EGFR, incluyendo el erlotinib. Esta influencia se apoya en datos clínicos limitados y en estudios preclínicos que muestran la degradación del EGFR mediante el proteasoma.
DATOS PRECLÍNICOS SOBRE SEGURIDAD:
Los efectos debidos a dosis crónicas observados en al menos una especie animal o estudio incluyeron efectos en la córnea (atrofia, ulceración), piel (degeneración folicular e inflamación, enrojecimiento y alopecia), ovarios (atrofia), hígado (necrosis hepática), riñón (necrosis papilar renal y dilatación tubular), y tracto gastrointestinal (vaciado gástrico retardado y diarrea). Los parámetros de las células rojas sanguíneas disminuyeron y las células blancas sanguíneas, principalmente neutrófilos, aumentaron. Hubo aumentos en ALT, AST y bilirrubina relacionados con el tratamiento. Estos hallazgos se observaron para exposiciones muy por debajo de exposiciones clínicamente relevantes.
En base al mecanismo de acción, erlotinib puede ser potencialmente teratógeno. Los datos obtenidos en pruebas de toxicidad reproductiva realizadas a ratas y conejos a dosis cercanas a la dosis máxima tolerada y/o maternalmente tóxicas mostraron toxicidad reproductiva (embriotoxicidad en ratas, reabsorción del embrión y toxicidad fetal en conejos) y de desarrollo (disminución en el crecimiento de las crías y en la supervivencia en ratas) pero no resultó teratogénico y no dañó la fertilidad. Estos hallazgos fueron observados a exposiciones clínicamente relevantes.
El resultado de estudios de genotoxicidad convencionales con erlotinib fue negativo. Los resultados de carcinogenicidad realizados con erlotinib durante dos años en ratas y ratones expuestos hasta dosis superiores a la exposición terapéutica en humanos (hasta 2 veces y 10 veces más altas, respectivamente, basado en la Cmax y/o AUC) fueron negativos.
En ratas se ha observado una reacción fototóxica media en la piel tras irradiación UV.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
Evaluación del estado mutacional del EGFR: Cuando se evalúa el estado mutacional del EGFR de un paciente, es importante elegir una metodología validada y robusta para evitar la obtención de falsos negativos o falsos positivos.
Fumadores: Se deberá recomendar a los fumadores dejar de fumar ya que las concentraciones plasmáticas de erlotinib se reducen en fumadores en comparación con no fumadores. Es probable que el grado de reducción sea clínicamente significativo (ver Interacción con otros medicamentos y otras formas de interacción).
Enfermedad pulmonar intersticial: Se han observado casos poco frecuentes de reacciones semejantes a enfermedad pulmonar intersticial (EPI), incluyendo fallecimientos, en pacientes que fueron tratados con erlotinib para el cáncer de pulmón no microcítico (CPNM), cáncer de páncreas u otros tumores sólidos avanzados. En el ensayo pivotal BR.21 en CPNM, la incidencia de EPI (0,8%) fue la misma tanto en el grupo al que se le administró placebo como en el tratado con Erlotinib. En el ensayo de cáncer de páncreas en combinación con gemcitabina, la incidencia de reacciones semejantes a EPI fue del 2,5% en el grupo de erlotinib y gemcitabina versus el 0,4% en el grupo tratado con placebo y gemcitabina. La incidencia global en los pacientes tratados con erlotinib de todos los ensayos (incluidos los ensayos no controlados y los ensayos con quimioterapia concurrente) es aproximadamente del 0,6% comparada con el 0,2% en los pacientes con placebo. Los diagnósticos realizados en los pacientes en los que hubo sospecha de que tuvieran reacciones semejantes a EPI incluyeron: neumonitis, neumonitis causada por radiación, neumonitis por hipersensibilidad, neumonía intersticial, enfermedad pulmonar intersticial, bronquiolitis obliterante, fibrosis pulmonar, Síndrome de Distrés Respiratorio Agudo (ARDS), alveolitis e infiltración pulmonar. Los síntomas se presentaron en un intervalo que fue desde unos pocos días tras iniciar la terapia con erlotinib hasta varios meses desde el inicio de ésta. Se dieron frecuentemente factores que pudieron contribuir o confundir el diagnóstico como pueden ser la quimioterapia concomitante o previa, radioterapia previa, enfermedad parenquimal pulmonar preexistente, enfermedad pulmonar metastásica o infecciones pulmonares. Se observó una mayor incidencia de EPI entre los pacientes de origen japonés (aproximadamente el 5% con una tasa de mortalidad del 1,5%).
En pacientes que presenten un comienzo agudo de síntomas pulmonares inexplicables, nuevos y/o progresivos, tales como disnea, tos y fiebre, se debe interrumpir la terapia con erlotinib hasta que se realice una evaluación diagnóstica. Los pacientes tratados con erlotinib y gemcitabina en combinación deberían ser monitorizados cuidadosamente por la posibilidad de desarrollar toxicidad semejante a EPI. Si se diagnostica EPI, se debe suspender el tratamiento con erlotinib e iniciar el tratamiento apropiado necesario (ver Reacciones adversas).
Diarrea, deshidratación, desequilibrio hidroelectrolítico e insuficiencia renal: Se ha observado diarrea (incluyendo casos muy raros con resultado de muerte) en aproximadamente el 50% de los pacientes en tratamiento con erlotinib. La diarrea moderada o grave debe ser tratada con, por ejemplo, loperamida. En algunos casos, puede ser necesaria una reducción de la dosis. En los ensayos clínicos, las dosis fueron reducidas en fracciones de 50 mg. No se han investigado reducciones de dosis en fracciones de 25 mg. En el caso de presentarse diarrea grave o persistente, náuseas, anorexia o vómitos asociados a deshidratación, el tratamiento con erlotinib debe ser interrumpido y deben tomarse las medidas apropiadas para tratar la deshidratación (ver Reacciones adversas).
En raros casos, se ha observado hipopotasemia y fallo renal (incluidos fallecimientos) Algunos casos fueron secundarios a deshidratación grave debida a diarrea, vómitos y/o anorexia, mientras que otros fueron confundidos con la quimioterapia concomitante. La terapia con erlotinib deberá interrumpirse y se deberán adoptar medidas apropiadas para rehidratar por vía intravenosa a los pacientes en muchos casos de diarrea grave o persistente, o en casos que provoquen deshidratación, especialmente en grupos de pacientes con factores de riesgo agravantes (especialmente quimioterapia y otra medicación concomitante, síntomas o enfermedades u otras situaciones que pudieran predisponer, incluyendo la edad avanzada). Además, en los pacientes con riesgo de deshidratación, se deberá monitorizar la función renal y la determinación de electrolitos en suero, incluyendo el potasio.
Hepatitis, fallo hepático: Durante el uso de erlotinib, se han notificado casos raros de fallo hepático (incluidos fallecimientos). Entre los factores de confusión se ha incluido la existencia previa de enfermedad hepática o medicación concomitante hepatotóxica. Por lo tanto, en estos pacientes, se debe considerar la realización de un examen de la función hepática de forma periódica. El tratamiento con erlotinib debe interrumpirse si hay cambios graves en la función hepática (ver Reacciones adversas). No se recomienda el uso de erlotinib en pacientes con disfunción hepática grave.
Perforación gastrointestinal: Se ha observado con poca frecuencia que los pacientes en tratamiento con erlotinib tienen un mayor riesgo de aparición de perforación gastrointestinal (incluyendo algunos casos con resultado de muerte). El riesgo es mayor en los pacientes que reciben de forma concomitante agentes anti- angiogénicos, corticosteroides, AINEs y/o quimioterapia basada en taxanos, o que tengan antecedentes de úlcera péptica o enfermedad diverticular. El tratamiento con erlotinib debe suspenderse permanentemente en aquellos pacientes en los que aparezca perforación gastrointestinal (ver Reacciones adversas).
Trastornos vesiculares y exfoliativos de la piel: Se han notificado casos de alteraciones vesiculares, ampollosas y exfoliativas en la piel, incluyendo casos muy raros indicativos del síndrome de Stevens-Johnson/Necrolisis epidérmica tóxica, que en algunos casos fueron mortales. (ver Reacciones adversas). El tratamiento con erlotinib debe interrumpirse o suspenderse si en el paciente aparecen vesículas, ampollas o exfoliación de carácter grave. Los pacientes con trastornos vesiculares y exfoliativos de la piel, se deben hacer la prueba de infecciones de la piel y ser tratados de acuerdo con las directrices locales.
Trastornos oculares: Los pacientes que presenten signos y síntomas indicadores de queratitis, como una agudización o empeoramiento de: inflamación ocular, lagrimeo, fotosensibilidad ocular, visión borrosa, dolor ocular y/o ojos enrojecidos, deben ser remitidos inmediatamente a un especialista en oftalmología. Si el diagnóstico de queratitis ulcerativa se confirma, el tratamiento con erlotinib debe ser interrumpido o suspendido. Si se diagnostica queratitis, se deben evaluar cuidadosamente los beneficios y riesgos de continuar con el tratamiento. Erlotinib debe ser utilizado con precaución en pacientes con antecedentes de queratitis, queratitis ulcerativa u ojo seco grave. El uso de lentes de contacto también es un factor de riesgo para la queratitis y ulceración. Durante el uso de erlotinib se han notificado casos muy raros de ulceración o perforación de la córnea (ver ver Reacciones adversas).
Interacciones con otros medicamentos: Los inductores potentes del citocromo CYP3A4 pueden reducir la eficacia de erlotinib mientras que los inhibidores potentes del CYP3A4 pueden producir un aumento de la toxicidad. Debe evitarse el tratamiento concomitante con estos tipos de agentes (ver Interacción con otros medicamentos y otras formas de interacción).
Otras formas de interacción: Erlotinib se caracteriza por una disminución de su solubilidad a un pH superior a 5. Los medicamentos que modifican el pH del tracto Gastro–Intestinal (GI) superior, como los inhibidores de la bomba de protones, antagonistas H2 y antiácidos, pueden alterar la solubilidad de erlotinib y, por lo tanto, su biodisponibilidad. No es probable que un aumento de la dosis de erlotinib cuando se co-administre con tales agentes compense la pérdida de exposición. Deberá evitarse la combinación de erlotinib con inhibidores de la bomba de protones. Se desconocen los efectos de la administración concomitante de erlotinib con antagonistas H2 y antiácidos; no obstante, es probable que se reduzca su biodisponibilidad. Por consiguiente, deberá evitarse la administración concomitante de estas combinaciones (ver Interacción con otros medicamentos y otras formas de interacción). Si se considera necesario el uso de antiácidos durante el tratamiento con erlotinib, deberían tomarse al menos 4 horas antes ó 2 horas después de la dosis diaria de erlotinib.
Los comprimidos contienen lactosa y no deben ser administrados a pacientes con intolerancia hereditaria a galactosa, de insuficiencia de lactasa de Lapp o problemas de absorción de glucosa o galactosa.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
El tratamiento con ERLHOTIZAM-PD® debe ser supervisado por un especialista con experiencia en el empleo de terapias anti-cancerosas.
Pacientes con cáncer de pulmón no microcítico: Se debe llevar a cabo el test de la mutación de EGFR antes de iniciar el tratamiento con ERLHOTIZAM-PD® en pacientes con CPNM avanzado o metastásico, que no han sido tratados previamente con quimioterapia.
La dosis diaria recomendada de ERLHOTIZAM-PD® es 150 mg administrada al menos una hora antes o dos después de la ingestión de alimentos.
Pacientes con cáncer de páncreas: La dosis diaria recomendada de ERLHOTIZAM-PD® es 100 mg administrada al menos una hora antes o dos horas después de la ingestión de alimentos, en combinación con gemcitabina (ver la ficha técnica de gemcitabina para la indicación de cáncer de páncreas).
Se debería reevaluar la continuación del tratamiento con ERLHOTIZAM-PD® en pacientes que no desarrollen rash dentro de las primeras 4-8 semanas del tratamiento (ver Propiedades farmacodinámicas).
Cuando sea necesario un ajuste de dosis, ésta se debe reducir en fracciones de 50 mg (ver Advertencias y precauciones especiales de empleo) ERLHOTIZAM-PD®.
El uso concomitante de sustratos y moduladores del citocromo CYP3A4 puede requerir un ajuste de dosis (ver Interacción con otros medicamentos y otras formas de interacción).
Pacientes con insuficiencia hepática: Erlotinib se elimina por metabolismo hepático y excreción biliar. Aunque la exposición a erlotinib fue similar en pacientes con insuficiencia hepática moderada (valor Child-Pugh 7-9) en comparación con la de pacientes con buena función hepática, deberá tenerse precaución cuando se administre ERLHOTIZAM-PD® a pacientes con insuficiencia hepática. Si aparecen reacciones adversas graves, debe considerarse la posibilidad de reducir la dosis o interrumpir la administración de ERLHOTIZAM-PD®. La seguridad y eficacia de erlotinib no ha sido estudiada en pacientes con disfunción hepática grave (AST/SGOT y ALT/SGPT > 5x ULN). No se recomienda usar ERLHOTIZAM-PD® en pacientes con disfunción hepática grave (ver Propiedades farmacocinéticas).
Pacientes con insuficiencia renal: La seguridad y eficacia de erlotinib no ha sido estudiada en pacientes con insuficiencia renal (concentración sérica de creatinina > 1,5 veces el límite superior normal). En base a los datos farmacocinéticos, no parece necesario un ajuste de la dosis en pacientes con insuficiencia renal media o moderada (ver Propiedades farmacocinéticas). No se recomienda el uso de ERLHOTIZAM-PD® en pacientes con insuficiencia renal grave.
Población pediátrica: No se ha establecido la seguridad y eficacia de erlotinib en pacientes menores de 18 años. No está recomendado el uso de ERLHOTIZAM-PD® en pacientes pediátricos.
Fumadores: Se ha demostrado que fumar cigarrillos reduce la exposición a erlotinib en un 50-60%. La dosis máxima tolerada de ERLHOTIZAM-PD® en pacientes fumadores activos con CPNM fue de 300 mg. No se ha establecido la eficacia y seguridad a largo plazo de dosis mayores a las dosis iniciales recomendadas en pacientes que continúan fumando cigarrillos (ver Interacción con otros medicamentos y otras formas de interacción y Propiedades farmacocinéticas). Por lo tanto se debe recomendar a los fumadores activos que dejen de fumar ya que las concentraciones plasmáticas de erlotinib en fumadores se reducen respecto a las que presentan los no fumadores.
SOBREDOSIS:
Síntomas: Se han administrado dosis orales únicas de erlotinib de hasta 1.000 mg de erlotinib en sujetos sanos y hasta 1.600 mg en pacientes con cáncer y éstas se toleraron bien. Las dosis repetidas de 200 mg dos veces al día administradas a sujetos sanos fueron mal toleradas tras sólo unos pocos días de tratamiento. En base a los datos derivados de estos ensayos, por encima de la dosis recomendada podrían darse reacciones adversas graves tales como diarrea, rash y un posible aumento de la actividad de las aminotransferasas hepáticas.
Tratamiento: En caso de que exista sospecha de sobredosis, se debe interrumpir el tratamiento con erlotinib e iniciar un tratamiento sintomático.


