

EPOETINA ALFA 2000 UI/ML
EPOETINA ALFA
Solución inyectable
COMPOSICIÓN
Cada mL contiene:
|
Epoetina alfa (equivalente a 16.8 mcg de epoetina alfa) |
2000 UI |
|
Excipientes: Albúmina, citrato de sodio, ácido cítrico, cloruro de sodio, agua para inyección c.s.p. |
1 mL. |
FORMA FARMACÉUTICA
Solución inyectable.
INDICACIONES TERAPÉUTICAS
Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica (IRC) en pacientes adultos y pediátricos:
• Tratamiento de la anemia asociada a insuficiencia renal crónica en pacientes pediátricos y adultos en hemodiálisis y pacientes adultos en diálisis peritoneal. (ver Advertencias y precauciones especiales de empleo).
• Tratamiento de la anemia grave de origen renal acompañada de síntomas clínicos en pacientes adultos con insuficiencia renal que aún no se someten a diálisis (ver Advertencias y precauciones especiales de empleo).
Tratamiento de la anemia y reducción de las necesidades de transfusión en pacientes adultos que reciben quimioterapia para tumores sólidos, linfoma maligno o mieloma múltiple, con riesgo de transfusión, según la evaluación del estado general del paciente (p. ej.: estado cardiovascular, anemia previa al principio de la quimioterapia).
Epoetina alfa puede emplearse para aumentar el rendimiento de sangre autóloga de aquellos pacientes en programa de predonación. Su uso en esta indicación debe sopesarse contra el riesgo notificado de acontecimientos tromboembólicos. El tratamiento sólo debe administrarse a los pacientes con anemia moderada (hemoglobina (Hb) 10 a 13 g/dL (6,2 a 8,1 mmol/L), sin deficiencia de hierro), si no se dispone de procedimientos para ahorrar sangre o si éstos son insuficientes cuando la intervención quirúrgica electiva mayor programada, requiere un volumen grande de sangre (cuatro o más unidades de sangre en las mujeres o cinco o más en los hombres).
Epoetina alfa puede emplearse para reducir la necesidad de llevar a cabo alotransfusiones de sangre previas a una cirugía ortopédica electiva mayor en pacientes adultos sin deficiencia de hierro, que tienen mayor riesgo de complicaciones debidas a la transfusión. El uso deberá restringirse a los pacientes con anemia moderada (p. ej.: hemoglobina (Hb) de 10 a 13 g/dL o 6,2 a 8,1 mmol/L) que no tienen un programa de predonación autóloga disponible y con una pérdida esperada de sangre moderada de 900 a 1800 mL.
En el entorno periquirúrgico se deben aplicar siempre las buenas prácticas de tratamiento sanguíneo.
ACCIÓN FARMACOLÓGICA
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Antianémico.
Código ATC: B03XA01
Mecanismo de acción: La eritropoyetina es una glucoproteína que estimula la formación de eritrocitos a partir de precursores del compartimento de células madre, actuando como factor estimulante de la mitosis y hormona de diferenciación.
El peso molecular aparente de la eritropoyetina es de 32.000 a 40.000 daltons. La fracción proteica de la molécula contribuye a aproximadamente el 58 % y consiste en 165 aminoácidos. Las cuatro cadenas de carbohidratos están unidas mediante enlaces N-glucosídicos y un enlace O-glucosídico a la proteína. La epoyetina alfa obtenida por tecnología génica, se glucosila y es idéntica en su composición de aminoácidos y carbohidratos a la eritropoyetina humana endógena que se ha aislado de la orina de pacientes anémicos.
Epoetina alfa tiene la pureza más alta posible, conforme al estado actual de la ciencia. En particular, no se detectan residuos de la línea celular usada para la producción a las concentraciones del principio activo que se utilizan en los seres humanos.
Efectos farmacodinámicos: Se ha demostrado la eficacia biológica de la epoyetina alfa en diversos modelos animales in vivo (ratas normales y anémicas, ratones policitémicos). El número de eritrocitos, los valores de Hb y los recuentos de reticulocitos aumentan después de la administración de epoyetina alfa, al igual que la tasa de incorporación de 59Fe.
Se ha observado un aumento de la incorporación de 3H-timidina en células eritroides nucleadas de bazo in vitro (cultivo de células de bazo de ratón), después de la incubación con epoyetina alfa.
Eficacia clínica y seguridad: Podría demostrarse, con ayuda de cultivos de células de la médula ósea humana, que la epoyetina alfa estimula la eritropoyesis específicamente y no afecta a la leucopoyesis. No se pudieron detectar efectos citotóxicos de la epoyetina alfa sobre las células de la médula ósea.
Propiedades farmacocinéticas:
• Absorción: La biodisponibilidad de la epoyetina alfa inyectada por vía subcutánea es mucho más baja que cuando se administra por vía intravenosa: aproximadamente el 20 %.
• Eliminación:
–Vía intravenosa: La determinación de epoyetina alfa después de la administración de varias dosis por vía intravenosa reveló una semivida de aproximadamente cuatro horas en voluntarios normales y una semivida algo más prolongada en pacientes con insuficiencia renal de aproximadamente cinco horas. En niños se ha descrito una semivida de aproximadamente seis horas.
–Vía subcutánea: Después de la inyección subcutánea, las concentraciones de epoyetina alfa en suero son mucho más bajas que las concentraciones alcanzadas después de la inyección intravenosa; las concentraciones aumentan lentamente y alcanzan un valor máximo entre 12 y 18 horas después de la administración de la dosis. El valor máximo es siempre inferior al valor máximo alcanzado por la vía intravenosa (aproximadamente 1/20 del valor).
–No hay acumulación: las concentraciones permanecen iguales, sean determinadas 24 horas después de la primera inyección o 24 horas después de la última inyección.
La semivida es difícil de evaluar con la vía subcutánea y se calcula que es de aproximadamente 24 horas.
Datos preclínicos sobre seguridad: En algunos estudios toxicológicos preclínicos en perros y ratas, pero no en monos, el tratamiento con epoyetina alfa se asoció a fibrosis subclínica de la médula ósea (la fibrosis de la médula ósea es una complicación conocida de la insuficiencia renal crónica en los seres humanos y puede estar relacionada con hiperparatiroidismo secundario o factores desconocidos. La incidencia de fibrosis de la médula ósea no aumentó en un ensayo en pacientes en hemodiálisis que recibieron tratamiento con epoyetina alfa durante tres años, en comparación con un grupo control de pacientes sometidos a diálisis que no habían recibido tratamiento con epoyetina alfa).
En estudios en animales, se ha demostrado que la epoyetina alfa reduce el peso corporal fetal, retrasa la osificación y aumenta la mortalidad fetal cuando se administra a dosis semanales de aproximadamente 20 veces la dosis semanal recomendada para el ser humano. Estos cambios se interpretaron como secundarios a una disminución del aumento de peso corporal materno.
La epoyetina alfa no mostró ningún cambio en las pruebas de mutagenicidad en cultivos de células bacterianas y de mamíferos, y en la prueba de micronúcleos in vivo en ratones.
No se han realizado estudios de carcinogenia a largo plazo. Hay resultados contradictorios en la bibliografía acerca de si las eritropoyetinas pueden desempeñar una función importante como proliferadores tumorales. Estos resultados publicados se basan en observaciones in vitro de muestras de tumores humanos, pero su significación es indeterminada en la situación clínica.
CONTRAINDICACIONES
• Hipersensibilidad al principio activo o a alguno de los excipientes.
• Los pacientes que presentan aplasia eritrocítica pura (AEP) después del tratamiento con cualquier eritropoyetina no deberán recibir Epoetina alfa ni ninguna otra eritropoyetina (ver Aplasia eritrocítica pura).
• Hipertensión no controlada.
• Pacientes quirúrgicos que, por cualquier razón, no pueden recibir profilaxis antitrombótica adecuada.
• En los pacientes tratados con suplementos de epoyetina alfa deben respetarse todas las contraindicaciones asociadas con los programas de predonación de sangre autóloga.
El empleo de epoyetina alfa en los pacientes programados para una intervención quirúrgica ortopédica electiva mayor que no participan en un programa de predonación de sangre autóloga, está contraindicado en los pacientes con enfermedad coronaria, arterial periférica, carotídea o vascular cerebral grave, incluidos los pacientes con infarto de miocardio o accidente vascular cerebral reciente.
FERTILIDAD, EMBARAZO Y LACTANCIA
Embarazo: No hay datos o éstos son limitados relativos al uso de epoyetina alfa en mujeres embarazadas. Los estudios realizados en animales han mostrado toxicidad para la reproducción (ver Datos preclínicos sobre seguridad).
En consecuencia:
• En las pacientes con insuficiencia renal crónica, Epoetina alfa debe utilizarse en el embarazo sólo si los beneficios potenciales superan a los riesgos potenciales para el feto.
• En las pacientes quirúrgicas embarazadas, que participen en un programa de predonación autóloga, no se recomienda el uso de epoyetina alfa.
Lactancia: Se desconoce si la epoyetina alfa se excreta en la leche materna.
La epoyetina alfa debe usarse con precaución en las mujeres en periodo de lactancia. Se debe decidir si es necesario continuar/interrumpir la lactancia o continuar/interrumpir el tratamiento con epoyetina alfa tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoyetina alfa para la madre.
En las pacientes quirúrgicas en periodo de lactancia, que participen en un programa de predonación autóloga, no se recomienda el uso de epoyetina alfa.
Fertilidad: No se dispone de datos sobre fertilidad.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MAQUINAS
La influencia de epoetina alfa sobre la capacidad para conducir y utilizar máquinas es nula o insignificante.
REACCIONES ADVERSAS
Resumen del perfil de seguridad: En los pacientes con cáncer y en los pacientes con insuficiencia renal crónica, la reacción adversa más frecuente durante el tratamiento con epoyetina alfa es un aumento dosis-dependiente de la presión arterial o un agravamiento de una hipertensión existente. Debe realizarse un control de la presión arterial, sobre todo al comienzo del tratamiento (ver Advertencias y precauciones especiales de empleo). Otras reacciones adversas frecuentes, observadas en los ensayos clínicos con epoyetina alfa, son la trombosis venosa profunda, embolia pulmonar, convulsiones, diarrea, náuseas, cefalea, enfermedad de tipo gripal, pirexia, erupción cutánea y vómitos. La aparición de un cuadro pseudogripal, consistente en cefalea, artralgia, mialgia y pirexia, puede tener lugar especialmente al comienzo del tratamiento. Las frecuencias pueden variar dependiendo de la indicación (ver el siguiente cuadro).
En estudios con ampliación del intervalo de dosificación en pacientes adultos con insuficiencia renal que aún no se someten a diálisis, se ha informado congestión de las vías respiratorias, que consta de episodios de congestión de las vías respiratorias altas, congestión nasal y nasofaringitis.
Las reacciones adversas graves al fármaco incluyen trombosis y embolias, venosas y arteriales (incluso algunas con resultados mortales), como trombosis venosa profunda, embolia pulmonar, trombosis arterial (incluso infarto de miocardio e isquemia miocárdica), trombosis de la retina y trombosis de la derivación (incluso del equipo de diálisis). Además, en los ensayos clínicos de la epoyetina alfa se han notificado accidentes cerebrovasculares (incluso infarto cerebral y hemorragia cerebral) y ataques isquémicos transitorios.
Se han notificado aneurismas.
Se han notificado reacciones de hipersensibilidad, incluso casos de erupción cutánea, urticaria, reacción anafiláctica y edema angioneurótico.
Durante el tratamiento con epoyetina alfa en pacientes con presión arterial previamente normal o baja se han producido crisis de hipertensión con encefalopatía y convulsiones, que han precisado la atención inmediata de un médico y cuidados médicos intensivos. Debe prestarse una atención especial a las cefaleas súbitas y lacerantes de tipo migrañoso, como posible señal de advertencia.
En casos raros (en < 1/10.000 casos por paciente año) se ha notificado aplasia eritrocítica pura mediada por anticuerpos, después de meses a años de tratamiento con epoyetina alfa (ver sección Advertencias y precauciones especiales de empleo).
Se evaluó el perfil global de seguridad de la epoyetina alfa en 142 pacientes con insuficiencia renal crónica y en 765 pacientes con cáncer que participaron en ensayos clínicos de registro, controlados con placebo y doble ciego. Las reacciones adversas al fármaco notificadas en ≥ 0,2% de los pacientes tratados con epoyetina alfa durante estos ensayos y durante ensayos clínicos adicionales así como durante la experiencia poscomercialización se incluyen a continuación según la clasificación por órganos y sistemas, y frecuencia.
Lista tabulada de reacciones adversas: Las frecuencias se definen como: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000); muy raras (< 1/10.000), frecuencia no conocida (no puede estimarse a partir de los datos disponibles).
Las reacciones adversas se enumeran en orden decreciente de gravedad dentro de cada intervalo de frecuencia.
|
Clase de sistema orgánico |
Frecuencia |
Reacciones adversas |
|
Trastornos de la sangre y del sistema linfático |
Poco frecuentes |
Trombocitopenia (pacientes con cáncer) |
|
Frecuencia no conocida |
Aplasia eritrocítica pura mediada por anticuerpos contra la eritropoyetina1 Trombocitopenia (pacientes con insuficiencia renal crónica) |
|
|
Trastornos del sistema inmunológico |
Frecuencia no conocida |
Reacción anafiláctica Hipersensibilidad |
|
Trastornos del sistema nervioso |
Muy frecuentes |
Cefaleas (pacientes con cáncer) |
|
Frecuentes |
Convulsiones (pacientes con insuficiencia renal crónica) Cefaleas (pacientes con insuficiencia renal crónica) Apoplejía |
|
|
Poco frecuentes |
Hemorragia cerebral2 Convulsiones (pacientes con cáncer) |
|
|
Frecuencia no conocida |
Accidente cerebrovascular2 Encefalopatía hipertensiva Accidentes isquémicos transitorios |
|
|
Trastornos oculares |
Frecuencia no conocida |
Trombosis retiniana |
|
Trastornos cardíacos |
Frecuencia no conocida |
Infarto de miocardio |
|
Trastornos vasculares |
Frecuentes |
Trombosis venosa profunda2 (pacientes con cáncer) Hipertensión |
|
Frecuencia no conocida |
Trombosis venosa profunda2 (pacientes con insuficiencia renal crónica) Trombosis arterial Crisis hipertensivas |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Frecuentes |
Embolismo pulmonar2 (pacientes con cáncer) |
|
Frecuencia no conocida |
Embolismo pulmonar2 (pacientes con insuficiencia renal crónica) |
|
|
Trastornos gastrointestinales |
Muy frecuentes |
Náuseas |
|
Frecuentes |
Diarrea (pacientes con cáncer) Vómitos |
|
|
Poco frecuentes |
Diarrea (pacientes con insuficiencia renal crónica) |
|
|
Trastornos de la piel y del tejido subcutáneo |
Frecuentes |
Erupción cutánea |
|
Frecuencia no conocida |
Edema angioneurótico Urticaria |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Muy frecuentes |
Artralgia (pacientes con insuficiencia renal crónica) |
|
Frecuentes |
Artralgia (pacientes con cáncer) |
|
|
Poco frecuentes |
Mialgia (pacientes con cáncer) |
|
|
Frecuencia desconocida |
Mialgia (pacientes con insuficiencia renal crónica) |
|
|
Trastornos congénitos, familiares y genéticos |
Frecuencia no conocida |
Porfiria |
|
Trastornos generales y alteraciones en el lugar de administración |
Muy frecuentes |
Pirexia (pacientes con cáncer) Enfermedad de tipo gripal (pacientes con insuficiencia renal crónica) |
|
Frecuentes |
Enfermedad de tipo gripal (pacientes con cáncer) |
|
|
Frecuencia no conocida |
Sustancia ineficaz Edema periférico Pirexia (pacientes con insuficiencia renal crónica) Reacción en el lugar de inyección |
|
|
Exploraciones complementarias |
Frecuencia no conocida |
Anticuerpo anti-eritropoyetina positivo1 |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
Frecuentes |
Trombosis de la derivación arteriovenosa incluído el equipo de diálisis (pacientes con insuficiencia renal crónica) |
|
1 La frecuencia no puede estimarse a partir de los ensayos clínicos 2 Incluyendo casos que fueron mortales |
||
Descripción de reacciones adversas seleccionadas:
• Pacientes con insuficiencia renal crónica: En los pacientes con insuficiencia renal crónica los valores de hemoglobina superiores a 12 g/dL (7,5 mmol/L) pueden asociarse con un aumento del riesgo de acontecimientos cardiovasculares, incluso la muerte (ver Advertencias y precauciones especiales de empleo)
Se han producido trombosis de la derivación arteriovenosa en pacientes en hemodiálisis, especialmente en los que tienen una tendencia a la hipotensión o en aquellos cuyas fístulas presentan complicaciones (p. ej., estenosis, aneurismas, etc.) (ver Advertencias y precauciones especiales de empleo)
• Pacientes con cáncer: En los pacientes con cáncer que reciben estimulantes de la eritropoyesis, incluida la epoyetina alfa, se ha notificado un aumento de la incidencia de acontecimientos tromboembólicos (ver Advertencias y precauciones especiales de empleo).
• Pacientes quirúrgicos: En los pacientes programados para una intervención quirúrgica ortopédica electiva mayor, con una hemoglobina inicial de entre 10 y 13 g/dL (6,2 a 8,1 mmol/L), la incidencia de episodios trombóticos y vasculares (la mayoría de los cuales fueron TVP), en la población total de pacientes de los ensayos clínicos, pareció ser similar en los diferentes grupos de dosificación de epoyetina alfa y en el grupo que recibió placebo, aunque la experiencia clínica es limitada.
Además, en los pacientes con una hemoglobina inicial de > 13 g/dL (8,1 mmol/L), no puede excluirse la posibilidad de que el tratamiento con epoyetina alfa pueda asociarse a un aumento del riesgo de episodios trombóticos o vasculares postoperatorios.
• Notificación de sospecha de reacciones adversas: Es importante notificar sospechas de reacciones adversas tras su aprobación. Ello permite una supervisión continuada del riesgo/beneficio del medicamento. Es por ello que en caso se presente alguna reacción adversa que no se encuentre descrita en el inserto, ésta deberá ser comunicada a su médico tratante o farmacéutico.
INCOMPATIBILIDADES
Este producto no debe mezclarse con otros medicamentos, disolventes o diluyentes.
INTERACCIONES CON OTROS MEDICAMENTOS
No hay pruebas de que el tratamiento con epoyetina alfa altere el metabolismo de otros medicamentos.
Sin embargo, puesto que la ciclosporina se fija a los eritrocitos, existe la posibilidad de interacción entre sustancias. Si la epoyetina alfa se administra concomitantemente con ciclosporina, debe vigilarse las concentraciones sanguíneas de ciclosporina y la dosis de ciclosporina debe ajustarse a medida que el hematocrito aumente.
No hay pruebas de una interacción entre la epoyetina alfa y el factor estimulante de colonias de granulocitos (G-CSF) o el factor estimulante de colonias de granulocitos macrófagos (GM-CSF) en lo que respecta a la diferenciación hematológica o la proliferación de muestras de biopsia de tumor in vitro.
Población pediátrica: Los estudios de interacciones se han realizado sólo en adultos.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO
Advertencias:
• Los Agentes estimulantes de la eritropoyesis sólo deben utilizarse en el tratamiento de anemia causada por quimioterapia, y se debe proceder a su discontinuación cuando ésta termine.
• Los ESA aumentan el riesgo de muerte y la incidencia de eventos cardiovasculares serios cuando se administran para alcanzar valores de hemoglobina (Hb) mayores de 12 g/dL
• Estudios clínicos dieron a conocer que los AEE estimularon el crecimiento del tumor y redujeron la supervivencia en casos de cáncer avanzado de mama, cabeza y cuello, sistema linfático y cáncer pulmonar de células no pequeñas, al administrarse dosis para alcanzar una concentración de hemoglobina de 12 gramos por decilitro (g/dL) o más.
• En pacientes con insuficiencia renal crónica, los AEE se deben utilizar para mantener la concentración de hemoglobina entre 10 g/dL y 12 g/dL. Mantener concentraciones mayores aumenta el riesgo de muerte y otras afecciones graves.
• En pacientes que recibieron AEE en el preoperatorio para reducir la necesidad de transfusiones alogénicas, se incrementó la posibilidad de trombosis venosa profunda. Estos pacientes no habían recibido anticoagulación profiláctica.
Precauciones:
• Usar la dosis más baja de AEE para aumentar gradualmente la concentración de hemoglobina, lo suficiente como para evitar la transfusión de sangre.
• El uso de medicamentos similares a la epoetina alfa, previo a una cirugía mayor, aumenta el riesgo de formación de coágulos.
• Cuando se administran los Agentes Estimulantes de la Eritropoyesis a pacientes oncológicos:
• Se recomienda un especial cuidado en pacientes oncológicos que reciben AEE.
• Recomendación general: No utilizar eritropoyetina en las primeras etapas del desarrollo del tumor en pacientes con esperanza de cura sometidos a quimioterapia tras la extirpación quirúrgica del tumor.
• Con cáncer avanzado de cabeza y cuello que reciben radioterapia, para alcanzar Hb mayores a 12 g/dL acorta el tiempo de progresión de la enfermedad, al favorecer la aparición de complicaciones que llevan al desenlace.
• En pacientes con cáncer de mama metastásico que reciben quimioterapia, la administración de ESAs para alcanzar niveles de Hb > a 12 g/dL, puede acortar la sobrevida global e incrementar en número de muertes.
• En pacientes oncológicos que no reciben quimioterapia o radioterapia aumenta la mortalidad cuando se administra para superar valores de Hb > g/dL, además de no estar indicado en esta población de pacientes.
• Se aconseja a los profesionales de la salud no utilizar Agentes Estimuladores de la Eritropoyesis en determinados tipos de cáncer de mama, de cabeza y cuello.
Generales: En todos los pacientes que reciben epoyetina alfa, debe monitorizarse y controlarse estrechamente la presión arterial según sea necesario. La epoyetina alfa deberá emplearse con precaución en presencia de hipertensión no tratada, tratada insuficientemente o deficientemente controlable. Puede ser necesario añadir o aumentar el tratamiento antihipertensivo. Si la presión arterial no puede controlarse, el tratamiento con epoyetina alfa deberá suspenderse.
La epoyetina alfa también debe emplearse con precaución en presencia de epilepsia e insuficiencia hepática crónica.
En los pacientes con insuficiencia renal crónica y cáncer que reciben tratamiento con epoyetina alfa, los niveles de hemoglobina deben ser medidos regularmente hasta que se alcance un nivel estable y, posteriormente, de manera periódica.
En todos los pacientes, los niveles de hemoglobina deben ser cuidadosamente controlados debido al posible aumento del riesgo de episodios tromboembólicos que pueden en algunos casos conducir a la muerte, cuando se trata a pacientes que presentan un nivel de hemoglobina por encima del valor establecido para la indicación.
Durante el tratamiento con epoyetina alfa, puede haber un aumento moderado dosis-dependiente del recuento de plaquetas, dentro de los límites normales. El recuento vuelve a su nivel anterior durante el transcurso del tratamiento. Además, se ha notificado trombocitemia por encima de los límites normales. Se recomienda monitorizar el recuento de plaquetas con regularidad durante las ocho primeras semanas de tratamiento.
Todas las demás causas de anemia (deficiencia de hierro, hemólisis, pérdida de sangre, deficiencia de vitamina B12 o folato) deben tenerse en cuenta y tratarse antes de iniciar el tratamiento con epoyetina alfa. En la mayoría de los casos, los valores de ferritina en el suero descienden simultáneamente con el volumen de células empaquetadas. A fin de asegurar una respuesta óptima a la epoyetina alfa, deben asegurarse unos depósitos suficientes de hierro:
• Suplementos de hierro, p. ej.: Se recomiendan de 200 a 300 mg Fe2+/día por vía oral (de 100 a 200 mg Fe2+/día en los pacientes pediátricos) para los pacientes con insuficiencia renal crónica cuyos valores de ferritina en suero sean inferiores a 100 ng/mL.
• Se recomiendan suplementos de hierro por vía oral de 200 a 300 mg Fe2+/día para todos los pacientes con cáncer cuya saturación de transferrina sea inferior al 20 %.
Todos estos factores relativos a la anemia también deberán tenerse en cuenta cuidadosamente al decidir si se va a aumentar la dosis de epoyetina alfa en los pacientes con cáncer.
Muy raramente, se ha observado la aparición o la exacerbación de una porfiria en pacientes tratados con epoyetina alfa. La epoyetina alfa debe usarse con precaución en los pacientes con porfiria.
A fin de mejorar la trazabilidad de los estimulantes de la eritropoyesis, el nombre del fármaco administrado debe anotarse (o declararse) claramente en el historial médico del paciente.
El cambio de un estimulante de la eritropoyesis a otro en un paciente debe realizarse siempre bajo una supervisión apropiada.
Aplasia eritrocítica pura (AEP): La AEP mediada por anticuerpos se ha descrito después de meses o años de tratamiento con eritropoyetina por vía subcutánea, principalmente en pacientes con insuficiencia renal crónica.
También se han notificado casos en pacientes con hepatitis C tratada con interferón y ribavirina, cuando se usan epoyetinas simultáneamente. No se ha aprobado Epoetina alfa para el tratamiento de la anemia asociada a hepatitis C.
En los pacientes que presentan una falta súbita de eficacia, caracterizada por un descenso de la hemoglobina (1 a 2 g/dL o 0,62 a 1,25 mmol/L al mes) con un aumento de la necesidad de transfusiones, deberá realizarse un recuento de reticulocitos y deberán investigarse las causas típicas de ausencia de respuesta (deficiencia de p. ej.: hierro, folato o vitamina B12, intoxicación por aluminio, infección o inflamación, pérdida de sangre y hemólisis).
Una disminución paradójica de la hemoglobina y la aparición de anemia grave asociada a recuentos bajos de reticulocitos darán lugar a la suspensión del tratamiento con Epoetina alfa y a la realización de análisis de anticuerpos anti-eritropoyetina. También debe plantearse una exploración de la médula ósea para el diagnóstico de una AEP.
No debe iniciarse una terapia con otro estimulante de la eritropoyesis debido al riesgo de reacción cruzada.
Tratamiento de la anemia sintomática en los pacientes adultos y pediátricos con insuficiencia renal crónica: Los datos de inmunogenia correspondientes a la administración por la vía subcutánea de Epoetina alfa en pacientes con riesgo de AEP inducida por anticuerpos, es decir, los pacientes con anemia renal, no son suficientes. Por lo tanto, en los pacientes con anemia renal, el producto debe administrarse por vía intravenosa.
En los pacientes con insuficiencia renal crónica, la tasa de aumento de la hemoglobina deberá ser de aproximadamente 1 g/dL (0,62 mmol/L) al mes y no deberá ser superior a 2 g/dL (1,25 mmol/L) por mes, a fin de reducir al mínimo los riesgos de un aumento de la hipertensión.
Concentración de hemoglobina: En los pacientes con insuficiencia renal crónica, la concentración de mantenimiento de hemoglobina no debe exceder el límite superior del objetivo de la concentración de hemoglobina tal como se recomienda en Dosis y Administración. En los ensayos clínicos, se observó un aumento del riesgo de muerte, de acontecimientos cardiovasculares o cerebrovasculares graves, incluyendo apoplejía, cuando se administraron ESA para lograr un objetivo de hemoglobina superior a 12 g/dL (7,5 mmol/L).
Los ensayos clínicos controlados no han demostrado ventajas clínicas significativas atribuibles a la administración de epoyetinas cuando la concentración de hemoglobina se aumenta más allá del valor necesario para controlar los síntomas de la anemia y para evitar la transfusión de sangre.
Es posible que algunos pacientes con intervalos de dosificación más ampliados (superiores a una vez a la semana) de epoyetina alfa no mantengan unas concentraciones adecuadas de hemoglobina (ver Propiedades farmacodinámicas) y pueden necesitar un aumento de la dosis de epoetina alfa. Se debe vigilar con regularidad la concentración de hemoglobina.
En los pacientes en hemodiálisis se han producido trombosis arteriovenosas, especialmente en los que tienen una tendencia a la hipotensión o en aquellos cuyas fístulas arteriovenosas presentan complicaciones (por ejemplo, estenosis, aneurismas, etc.). En estos pacientes se recomienda un examen temprano de la derivación y profilaxis de la trombosis, mediante la administración de ácido acetilsalicílico, por ejemplo.
En casos aislados se ha observado hiperpotasemia, aunque no se ha establecido su causalidad. En los pacientes con insuficiencia renal crónica deben monitorizarse los electrolitos en el suero. Si se detecta un aumento de la concentración sérica de potasio, además del adecuado tratamiento de la hiperpotasemia, deberá interrumpirse la administración de epoyetina alfa hasta que la concentración sérica de potasio se haya corregido.
Durante el transcurso del tratamiento con epoyetina alfa se requiere con frecuencia un aumento de la dosis de heparina, a consecuencia del aumento del volumen de células empaquetadas. Es posible la oclusión del sistema de diálisis si la heparinización no es óptima.
Teniendo en cuenta la información disponible hasta la fecha, la corrección de la anemia con epoyetina alfa en pacientes adultos con insuficiencia renal que aún no se someten a diálisis no acelera la velocidad de progresión de la insuficiencia renal.
Tratamiento de los pacientes con anemia inducida por quimioterapia: Las eritropoyetinas son factores de crecimiento que estimulan principalmente la producción de los glóbulos rojos. Los receptores de la eritropoyetina pueden expresarse en la superficie de una variedad de células tumorales. Al igual que con todos los factores de crecimiento, existe la preocupación de que las epoyetinas puedan estimular el crecimiento de cualquier tipo de tumores. En varios ensayos controlados, no se ha comprobado que las epoyetinas mejoren la supervivencia global ni que disminuyan el riesgo de progresión del tumor en los pacientes con anemia asociada al cáncer.
En ensayos clínicos controlados, el uso de epoyetina alfa y de otros estimuladores de la eritropoyesis ha demostrado:
• Disminución del control locorregional en los pacientes con cáncer avanzado de la cabeza y el cuello, que reciben radioterapia, cuando se administra para lograr un objetivo de hemoglobina superior a 14 g/dL (8,7 mmol/L).
• Un acortamiento de la supervivencia global y un aumento de las muertes atribuidas a la progresión de la enfermedad a los cuatro meses en los pacientes con cáncer de mama que reciben quimioterapia cuando se administra para lograr un objetivo de hemoglobina de 12 a 14 g/L (7,5 a 8,7 mmol/L).
• Un aumento del riesgo de fallecimiento cuando se administra para lograr un objetivo de hemoglobina de 12 g/dL (7,5 mmol/L) en los pacientes con enfermedad maligna activa que no reciben ni quimioterapia ni radioterapia. Los estimuladores de la eritropoyesis no están indicados en esta población de pacientes.
Teniendo en cuenta lo anterior, en algunas situaciones clínicas, la transfusión de sangre debe ser el tratamiento de elección para la anemia en pacientes con cáncer. La decisión de administrar eritropoyetinas recombinantes deberá basarse en la evaluación de la relación beneficio/riesgo, en la que se tendrá en consideración a cada paciente individualmente, así como el contexto clínico específico. Los factores que deben considerarse en esta evaluación son el tipo de tumor y su estadio, el grado de anemia, la esperanza de vida, el entorno en el que el paciente está siendo tratado y la preferencia del paciente (ver Propiedades farmacodinámicas).
En los pacientes con cáncer que reciben quimioterapia, las dos o tres semanas que transcurren entre la administración de epoyetina alfa y la aparición de los hematíes inducidos por eritropoyetina deben tenerse en cuenta al evaluar si el tratamiento con epoyetina alfa es adecuado (paciente con riesgo de recibir una transfusión).
Como se ha observado un aumento de la incidencia de episodios vasculares trombóticos (EVT) en los pacientes con cáncer que reciben estimulantes de la eritropoyesis (ver Reacciones adversas), debe valorarse este riesgo frente al beneficio que se derivará del tratamiento (con epoyetina alfa), especialmente en los pacientes con cáncer y con un riesgo aumentado de episodios vasculares trombóticos, como la obesidad y en los pacientes con antecedentes de EVT (p. ej.: trombosis venosa profunda o la embolia pulmonar). Se realizó un estudio investigacional (estudio BEST) en mujeres con cáncer de mama metastásico para determinar si el tratamiento con epoyetina alfa durante un periodo más amplio que el necesario para la corrección de la anemia mejoraba los resultados del tratamiento.
En ese estudio, la incidencia de acontecimientos tromboembólicos mortales fue más alta en las pacientes que recibieron epoyetina alfa que en las que recibieron placebo (ver Propiedades farmacodinámicas).
Pacientes quirúrgicos en un programa de predonación autóloga: Deben respetarse todas las advertencias y precauciones especiales asociadas a los programas de predonación autóloga, especialmente el reemplazo sistemático de volumen.
Pacientes programados para una intervención quirúrgica ortopédica electiva mayor: En los pacientes programados para una intervención quirúrgica ortopédica electiva mayor, debe establecerse la causa de la anemia y debe tratarse, si es posible, antes del inicio del tratamiento con epoyetina alfa. Los episodios trombóticos pueden ser un riesgo en esta población y deberá valorarse frente al beneficio que se obtendrá del tratamiento.
Los pacientes programados para una intervención quirúrgica ortopédica electiva mayor deben recibir profilaxis antitrombótica suficiente, ya que los episodios trombóticos y vasculares pueden producirse en pacientes quirúrgicos, especialmente los que padecen una enfermedad cardiovascular de base. Asimismo debe tenerse especial precaución en aquellos pacientes con predisposición para la aparición de trombosis venosa profunda (TVP). Además, en los pacientes con niveles de hemoglobina inicial de > 13 g/dL (> 8,1 mmol/L), no puede excluirse la posibilidad de que el tratamiento con epoyetina alfa esté asociado con un aumento del riesgo de episodios trombóticos o vasculares postoperatorios. Por lo tanto, no debe emplearse en los pacientes con una hemoglobina inicial > 13 g/dL (> 8,1 mmol/L).
PRECAUCIONES ESPECIALES DE ELIMINACIÓN Y OTRAS MANIPULACIONES
No administrar por perfusión intravenosa o conjuntamente con otras soluciones medicamentosas.
El producto no debe ser utilizado, y debe ser desechado si:
• El precinto está roto,
• El líquido tiene color o se ven partículas flotando en él,
• Sabe o cree que puede haber sido congelado accidentalmente, o
• Se ha producido una avería en el refrigerador.
DROGUERÍA LABORATORIOS AMERICANOS S.A.
Calle Felipe Santiago Salaverry 419 Urb. Industrial El Pino, San Luis.
Telf.: 626-8600 Fax: 326-4793, Lima - Perú
http://www.labot.com.pe
DOSIS Y ADMINISTRACIÓN
El tratamiento con epoetina alfa se tiene que iniciar bajo la supervisión de médicos con experiencia, en el tratamiento de las indicaciones anteriores.
Posología:
Tratamiento de la anemia sintomática en los pacientes adultos y pediátricos con insuficiencia renal crónica: En los pacientes con insuficiencia renal crónica, el medicamento debe administrarse por vía intravenosa (ver Advertencias y precauciones especiales de empleo).
Los síntomas y las secuelas de la anemia pueden variar con la edad, el sexo y las enfermedades concomitantes (co-morbilidad); es necesario que el médico realice una evaluación individualizada de la evolución clínica y del estado de cada paciente.
Epoetina alfa debe administrarse a fin de aumentar la hemoglobina a una concentración no superior a 12 g/dL (7,5 mmol/L).
Deberá evitarse un aumento de la hemoglobina superior a 2 g/dL (1,25 mmol/L) durante un período de cuatro semanas. Si esto ocurre, deberán hacerse los ajustes adecuados de la dosis que sean necesarios.
Debido a la variabilidad intrapaciente, de forma ocasional, se pueden observar en algunos pacientes valores individuales de hemoglobina superiores e inferiores a los valores deseados. La variabilidad de la hemoglobina deberá tratarse mediante el manejo de la dosis, teniendo en cuenta un objetivo de intervalo de la hemoglobina de 10 g/dL (6,2 mmol/L) a 12 g/dL (7,5 mmol/L). En los pacientes pediátricos, el objetivo recomendado del intervalo de la hemoglobina es de 9,5 a 11 g/dL (5,9 a 6,8 mmol/L).
Deberá evitarse una concentración prolongada de hemoglobina superior a 12 g/dL (7,5 mmol/L). Si la hemoglobina aumenta más de 2 g/dL (1,25 mmol/L) al mes o si la hemoglobina prolongada es superior a 12 g/dL (7,5 mmol/L), se debe reducir un 25% la dosis de epoyetina alfa. Si la hemoglobina es superior a 13 g/dL (8,1 mmol/L), se debe suspender el tratamiento hasta que descienda a menos de 12 g/dL (7,5 mmol/L) y, luego, se debe reiniciar el tratamiento con epoyetina alfa a una dosis un 25% inferior al nivel anterior.
Se debe vigilar estrechamente a los pacientes con el fin de asegurar que se emplea la dosis mínima aprobada de epoyetina alfa para proporcionar un control adecuado de los síntomas de la anemia. Deberá evaluarse el nivel de hierro, antes y durante el tratamiento y si es necesario, debe administrarse tratamiento y suplementos de hierro. Además, antes de iniciar el tratamiento con epoyetina alfa, deberán excluirse otras causas de anemia, como deficiencia de vitamina B12 o folato. La ausencia de respuesta al tratamiento con epoyetina alfa puede tener las siguientes causas: deficiencia de hierro, folato o vitamina B12, intoxicación por aluminio, infecciones intercurrentes, episodios inflamatorios o traumáticos, pérdida de hemoglobina, hemólisis y fibrosis de la médula ósea con cualquier origen.
Pacientes adultos en hemodiálisis: El tratamiento se divide en dos fases:
• Fase de corrección: 50 UI/kg: tres veces por semana, por vía intravenosa. En caso de que sea necesario un ajuste de la dosis debe hacerse en pasos de al menos cuatro semanas. En cada paso, el aumento o la reducción de la dosis deberá ser de 25 UI/kg, tres veces por semana.
• Fase de mantenimiento: Ajustar la dosificación a fin de mantener los valores de hemoglobina al nivel deseado: Hb entre 10 y 12 g/dL (de 6,2 a 7,5 mmol/L).
La dosis semanal total recomendada es de entre 75 y 300 UI/kg por vía intravenosa.
Los datos clínicos disponibles sugieren que aquellos pacientes cuya hemoglobina inicial es muy baja (< 6 g/dL o < 3,75 mmol/L) pueden precisar dosis de mantenimiento más altas que aquellos cuya anemia inicial es menos grave (Hb > 8 g/dL o > 5 mmol/L).
Pacientes pediátricos en hemodiálisis: El tratamiento se divide en dos fases:
• Fase de corrección: 50 UI/kg: tres veces por semana, por vía intravenosa. En caso de que sea necesario un ajuste de la dosis debe hacerse en pasos de 25 UI/kg tres veces por semana, a intervalos de al menos cuatro semanas, hasta alcanzar el objetivo deseado.
• Fase de mantenimiento: Ajustar la dosificación a fin de mantener los valores de hemoglobina al nivel deseado: Hb entre 9,5 y 11 g/dL (de 5,9 a 6,8 mmol/L).
Por lo general, los niños con un peso inferior a 30 kg requieren dosis de mantenimiento más altas que los niños con un peso superior a 30 kg y que los adultos.
Se observaron las siguientes dosis de mantenimiento en ensayos clínicos después de seis meses de tratamiento:
|
Dosis (UI/kg administradas 3 veces por semana) |
||
|
Peso (kg) |
Media |
Dosis de mantenimiento usual |
|
<10 |
100 |
75-150 |
|
10-30 |
75 |
60-150 |
|
>30 |
33 |
30-100 |
Los datos clínicos disponibles sugieren que aquellos pacientes pediátricos cuya hemoglobina inicial es muy baja (< 6,8 g/dL o < 4,25 mmol/L) pueden precisar dosis de mantenimiento más altas que aquellos cuya hemoglobina inicial es más elevada (> 6,8 g/dL o > 4,25 mmol/L).
Pacientes adultos en diálisis peritoneal: El tratamiento se divide en dos fases:
• Fase de corrección: Dosis inicial, de 50 UI/kg: dos veces por semana, por vía intravenosa.
• Fase de mantenimiento: Ajustar la dosificación a fin de mantener los valores de hemoglobina al nivel deseado: Hb entre 10 y 12 g/dL (de 6,2 a 7,5 mmol/L). Dosis de mantenimiento entre 25 y 50 UI/kg, dos veces por semana, en dos inyecciones iguales.
Pacientes adultos con insuficiencia renal que aún no se someten a diálisis: El tratamiento se divide en dos fases:
• Fase de corrección: Dosis inicial de 50 UI/kg tres veces por semana, por vía intravenosa, seguida, si es necesario, de un aumento de la dosificación con incrementos de 25 UI/kg (tres veces por semana), hasta alcanzar el objetivo deseado (esto deberá hacerse en pasos de al menos cuatro semanas).
• Fase de mantenimiento: Deberá hacerse un ajuste adecuado de la dosis, a fin de mantener los valores de hemoglobina al nivel deseado: Hb entre 10 y 12 g/dL (de 6,2 a 7,5 mmol/L). La ampliación de los intervalos de dosis puede exigir un aumento de la dosis.
La dosificación máxima no deberá ser superior a 150 UI/kg tres veces por semana.
Pacientes con anemia inducida por la quimioterapia: La epoyetina alfa debe administrarse a los pacientes con anemia por vía subcutánea (por ejemplo, concentración de hemoglobina ≤ 10 g/dL (6,2 mmol/L)). Los síntomas y las secuelas de la anemia pueden variar con la edad, el sexo y la carga total de la enfermedad, por lo que es necesario que el médico evalúe la evolución y el estado clínico del paciente.
Debido a la variabilidad intrapaciente, pueden observarse valores individuales y ocasionales de la hemoglobina superiores e inferiores al valor deseado para un paciente. La variabilidad de la hemoglobina debe abordarse por medio de la gestión de las dosis, teniendo en cuenta un objetivo de valores de la hemoglobina comprendido en un intervalo de 10 g/dL (6,2 mmol/L) a 12 g/dL (7,5 mmol/L).
Deberá evitarse un valor sostenido de la hemoglobina superior a 12 g/dL (7,5 mmol/L); a continuación, se describe una guía para realizar un ajuste correcto de las dosis cuando los valores de hemoglobina son superiores a 12 g/dL (7,5 mmol/L).
El tratamiento con epoyetina alfa deberá continuarse hasta un mes después del fin de la quimioterapia.
La dosis inicial es 150 UI/kg, administrada por vía subcutánea tres veces por semana. Como alternativa, la epoyetina alfa puede administrarse a una dosis inicial de 450 UI/kg, por vía subcutánea, una vez a la semana.
• Si la hemoglobina ha aumentado por lo menos en 1 g/dL (0,62 mmol/L) o si el recuento de reticulocitos ha aumentado ≥ 40.000 células/microlitro (µL) por encima del valor inicial después de cuatro semanas de tratamiento, la dosis deberá mantenerse a 150 UI/kg, tres veces por semana ó 450 UI/kg una vez a la semana.
• Si el aumento de la hemoglobina es < 1 g/dL (< 0,62 mmol/L) y el recuento de reticulocitos ha aumentado < 40.000 células/µL por encima del valor inicial, la dosis debe aumentarse a 300 UI/kg tres veces por semana. Si, después de otras cuatro semanas de tratamiento, a 300 UI/kg tres veces por semana, la hemoglobina ha aumentado ≥ 1 g/dL (≥ 0,62 mmol/L) o si el recuento de reticulocitos ha aumentado ≥ 40.000 células/µL, la dosis deberá mantenerse a 300 UI/kg tres veces por semana. Sin embargo, si la hemoglobina ha aumentado < 1 g/dL (< 0,62 mmol/L) y el recuento de reticulocitos ha aumentado < 40.000 células/µL por encima del valor inicial, la respuesta al tratamiento con epoyetina alfa es improbable y el tratamiento deberá suspenderse.
La pauta de dosificación recomendada se describe en el siguiente diagrama:
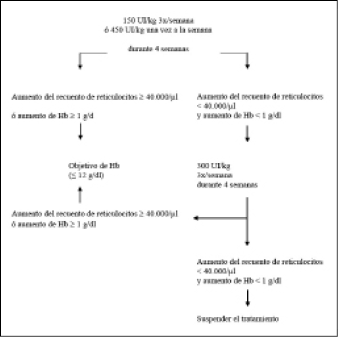
Se debe vigilar estrechamente a los pacientes con el fin de asegurar de que se emplea la dosis mínima aprobada de epoyetina alfa para proporcionar un control adecuado de los síntomas de la anemia.
Ajuste de la posología para mantener una concentración de hemoglobina entre 10 g/dL y 12 g/dL (de 6,2 a 7,5 mmol/L):
Si la hemoglobina aumenta más de 2 g/dL (1,25 mmol/L) al mes o si la hemoglobina es superior a 12 g/dL (7,5 mmol/L), reduzca la dosis de epoyetina alfa en alrededor de un 25 a un 50%. Si la hemoglobina es superior a 13 g/dL (8,1 mmol/L), interrumpa el tratamiento hasta que descienda a 12 g/dL (7,5 mmol/L) y después, reinicie el tratamiento con epoyetina alfa a una dosis un 25% inferior, a la dosis anterior.
Pacientes quirúrgicos adultos en un programa de predonación autóloga: Epoetina alfa debe administrarse por vía intravenosa.
En el momento de la donación de sangre, epoetina alfa debe administrarse después de finalizar la donación de sangre.
Los pacientes con anemia leve (hematocrito del 33 al 39%) y que precisan un depósito previo de ≥ 4 unidades de sangre, deben recibir tratamiento con epoetina alfa a una dosis de 600 UI/kg de peso corporal, dos veces por semana, durante tres semanas antes de la intervención. Utilizando esta pauta, fue posible retirar ≥ 4 unidades de sangre del 81% de los pacientes tratados con epoyetina alfa frente al 37% de los tratados con placebo. La terapia con epoyetina alfa redujo el riesgo de exposición a sangre homóloga en un 50% en comparación con los pacientes que no recibieron epoyetina alfa.
Todos los pacientes que reciben tratamiento con epoetina alfa deben recibir una cantidad suficiente de suplementos de hierro (p. ej., 200 mg de hierro elemental por vía oral al día) durante todo el transcurso del tratamiento.
Los suplementos de hierro deben iniciarse lo antes posible, incluso varias semanas antes de iniciar el depósito previo autólogo, a fin de alcanzar unos depósitos de hierro altos antes de comenzar el tratamiento con epoetina alfa.
Pacientes adultos programados para cirugía ortopédica electiva: Deberá usarse la vía de administración subcutánea.
La pauta de dosificación recomendada es de 600 UI/kg de epoyetina alfa, administrada semanalmente, durante tres semanas (días - 21, - 14 y - 7) antes de la intervención y en el día de la intervención (día 0). En los casos en los que hay una necesidad médica de acortar el tiempo previo a la intervención quirúrgica a menos de tres semanas, deberán administrarse 300 UI/kg de epoyetina alfa durante diez días consecutivos antes de la intervención quirúrgica, en el día de la intervención quirúrgica y durante cuatro días inmediatamente después. Si se realizan evaluaciones hematológicas durante el periodo preoperatorio y la concentración de hemoglobina es igual o superior a 15 g/dL (9,38 mmol/L), la administración de epoyetina alfa deberá suspenderse y no deberán administrarse más dosificaciones adicionales.
Debe asegurarse que, al inicio del tratamiento, los pacientes no tengan deficiencia de hierro.
Todos los pacientes que reciben tratamiento con epoyetina alfa deberán recibir suplementos suficientes de hierro (p. ej.: suplementos de hierro oral de 200 mg Fe2+ diarios) durante todo el periodo de tratamiento con epoyetina alfa. Si es posible, la administración de suplementos de hierro deberá iniciarse antes del tratamiento con epoyetina alfa, a fin de conseguir unos depósitos de hierro suficientes.
Forma de administración: Al igual que con cualquier otro producto inyectable, compruebe que no haya partículas en la solución ni cambios en su coloración.
Debe administrarse la cantidad necesaria.
No lo administre por perfusión intravenosa ni lo mezcle con otros medicamentos (para más información, consulte la Precauciones especiales de eliminación y otras manipulaciones).
• Inyección intravenosa: Durante al menos de uno a cinco minutos, dependiendo de la dosis total. En los pacientes en hemodiálisis, puede administrarse una inyección en bolo durante la sesión de diálisis, a través de un punto de entrada venoso adecuado en la vía de diálisis. O bien, la inyección puede administrarse al final de la sesión de diálisis, por medio del tubo de la aguja de la fístula, seguido de 10 mL de solución salina isotónica para purgar el tubo y asegurar una inyección satisfactoria del producto en la circulación.
Es preferible una inyección más lenta en los pacientes que reaccionan al tratamiento con síntomas “de tipo gripal”.
• Inyección subcutánea: En general, no deberá sobrepasarse un volumen máximo de 1 mL en un lugar de inyección. En caso de volúmenes más grandes, debe elegirse más de un lugar para la inyección. Las inyecciones se administran en las extremidades o en la pared abdominal anterior.
En aquellas situaciones en las que el médico determine que el paciente o su cuidador pueden administrar epoetina alfa por vía subcutánea de forma eficaz y segura, deben proporcionarse las instrucciones pertinentes a su adecuada dosis y administración.
En caso de olvido u omisión de dosis: consultar con su médico tratante
VÍA DE ADMINISTRACIÓN: Intravenosa - Subcutánea.
SOBREDOSIS
El margen terapéutico de la epoyetina alfa es muy amplio. La sobredosis de epoyetina alfa puede producir efectos que son derivados de los efectos farmacológicos de la hormona. Puede realizarse una flebotomía si se produce una concentración excesivamente alta de hemoglobina o un hematocrito excesivamente alto. Pueden proporcionarse las medidas adicionales de apoyo que sean necesarias.
PRECAUCIONES ESPECIALES DE CONSERVACIÓN
Almacenar entre 2 ° a 8 °C. Evitar la congelación. Mantener el producto en su envase original para protegerlo de la luz.
No agitar el vial, dado que puede desnaturalizar la proteína y convertirlo en biológicamente inactivo. Cualquier porción no usada de la solución deberá ser descartada.
Manténgase fuera del alcance de los niños.
Si tiene cualquier duda o no está seguro de algo, pregunte a su médico o farmacéutico.
Comunicar a su médico o farmacéutico cualquier reacción adversa no descrita en este inserto.
No utilizar después de la fecha de vencimiento indicada en el envase.


