

ECALTA
ANIDULAFUNGINA
Solución para infusión
1 Vial(es), Polvo liofilizado para solución inyectable, 100/30 mg/ml
FORMA FARMACÉUTICA: Polvo liofilizado para solución inyectable.
COMPOSICIÓN CUALI - CUANTITATIVA
Viales conteniendo 100 mg de anidulafungina polvo para solución para infusión y excipientes*.
*Fructuosa, manitol, polisorbato 80, acido tártarico, agua para inyección, hidróxido de sodio, ácido clorhídrico, nitrógeno.
La solución reconstituida contiene 3,33 mg/mL de anidulafungina y la solución diluida contiene 0,77 mg/mL de anidulafungina.
INDICACIONES Y USO
ECALTA está indicado para el uso en adultos en el tratamiento de las siguientes infecciones fúngicas:
• Candidemia y otras formas de infección por Candida (absceso intraabdominal, y peritonitis).
• ECALTA está indicado para el tratamiento de candidemia y las siguientes infecciones de Candida: absceso intra-abdominal y peritonitis (ver Estudios clínicos y Microbiología).
• Candidiasis esofágica: ECALTA está indicado para el tratamiento de candidiasis esofágica (ver Estudios Clínicos, Tabla 7 para índices de recaída mayores fuera de la terapia con ECALTA).
Limitaciones de uso: No se han realizado estudios con ECALTA en el tratamiento de endocarditis, osteomielitis y meningitis causadas por Candida y no se han realizado estudios en cantidades suficientes de pacientes neutropénicos que permitan determinar la eficacia en este grupo.
Los especímenes para cultivo fúngico y otros estudios de laboratorio relevantes (incluyendo histopatología) deben ser obtenidos antes de la terapia para aislar e identificar el organismo (s) causante (s). La terapia puede instituirse antes de conocerse los resultados de los cultivos y otros estudios de laboratorios. Sin embargo, una vez que estos resultados están disponibles, la terapia antifúngica se debe ajustar apropiadamente.
FARMACOCINÉTICA
Características farmacocinéticas generales: La farmacocinética de anidulafungina luego de la administración intravenosa (IV) se ha realizado en sujetos sanos, poblaciones especiales y pacientes. Las exposiciones sistémicas de anidulafungina son proporcionales a la dosis y poseen baja variabilidad interindividual (coeficiente de variación <25%) como se muestra en la Tabla 1. El estado estacionario se alcanza el primer día luego de una dosis de carga (dos veces la dosis diaria de mantenimiento) y el factor de acumulación plasmática estimado en el estado estacionario es aproximadamente 2.
Tabla 1. Parámetros farmacocinéticos promedio en el estado estacionario (%CV) de anidulafungina luego de la administración IV de anidulafungina una vez al día durante 10 días en adultos sanos
|
Parámetro farmacocinéticoa |
Régimen de dosis de |
||
|
70/35c,d |
200/100 (N=10) |
260/130d,e |
|
|
Cmáx., SS [mg/L] |
3,55 (13,2) |
8,6 (16,2) |
10,9 (11,7) |
|
AUCSS [mg• h/L] |
42,3 (14,5) |
111,8 (24,9) |
168,9 (10,8) |
|
CL [L/h] |
0,84 (13,5) |
0,94 (24,0) |
0,78 (11,3) |
|
t1/2 [h] |
43,2 (17,7) |
52,0 (11,7) |
50,3 (9,7) |
|
a. Los parámetros se obtuvieron de diferentes estudios. |
|||
El aclaramiento de anidulafungina es aproximadamente 1 L/h y la anidulafungina tiene una vida media de eliminación terminal de 40-50 horas.
Distribución: La farmacocinética de anidulafungina luego de la administración IV se caracteriza por una vida media de distribución corta (0,5-1 hora) y un volumen de distribución de 30-50 L que es similar al volumen total de los fluidos corporales. La anidulafungina se une extensamente (>99%) a las proteínas plasmáticas humanas.
Metabolismo: No se ha observado metabolismo hepático de anidulafungina. La anidulafungina no es un sustrato, inductor o inhibidor clínicamente relevante de las isoenzimas del citocromo P450 (CYP450). Es poco probable que la anidulafungina tenga efectos clínicamente relevantes en el metabolismo de los medicamentos metabolizados por las isoenzimas del CYP450.
La anidulafungina sufre lenta degradación química a temperatura y pH fisiológicos dando lugar a un péptido de anillo abierto que carece de actividad antifúngica. La vida media de degradación in vitro de anidulafungina bajo condiciones fisiológicas es aproximadamente 24 horas. In vivo, el producto con el anillo abierto es posteriormente convertido a degradantes peptídicos y es eliminado.
Excreción: En un estudio clínico de dosis única, se administró anidulafungina radiomarcada (C14) a pacientes sanos. Aproximadamente 30% de la dosis radioactiva administrada se eliminó en las heces en 9 días, de la cual menos del 10% como fármaco sin modificación. Menos del 1% de la dosis radioactiva administrada se excretó en la orina. Las concentraciones de anidulafungina se ubicaron debajo de los límites inferiores de cuantificación 6 días luego de la dosis. Cantidades no significativas de radioactividad proveniente del fármaco se detectaron en la sangre, orina y heces a las 8 semanas posteriores a la dosis.
Poblaciones específicas:
• Pacientes con infecciones fúngicas: Los análisis farmacocinéticos poblacionales, a partir de cuatro ensayos clínicos incluyendo 107 pacientes varones y 118 mujeres con infecciones fúngicas mostraron que los parámetros farmacocinéticos de anidulafungina no se ven afectados por la edad, raza o presencia de medicaciones concomitantes conocidos por ser sustratos, inhibidores o inductores metabólicos.
La farmacocinética de anidulafungina en pacientes con infecciones fúngicas es similar a la observada en pacientes sanos. Los parámetros farmacocinéticos estimados de anidulafungina utilizando un modelo farmacocinético poblacional luego de la administración IV de una dosis de mantenimiento de 50 mg/día o 100 mg/día (luego de una dosis de carga) se presentan en la Tabla 2.
Tabla 2. Parámetros farmacocinéticos promedio en el estado estacionario (%CV) de anidulafungina luego de la administración IV de anidulafungina en pacientes con infecciones fúngicas estimada utilizando un modelo farmacocinético poblacional
|
Parámetro farmacocinéticoa |
Régimen de dosis de |
|
|
100/50 |
200/100 |
|
|
Cmáx., SS [mg/L] |
4,2 (22,4) |
7,2 (23,3) |
|
Cmin., SS [mg/L] |
1,6 (42.1) |
3,3 (41,8) |
|
AUCSS [mg• h/L] |
55,2 (32,5) |
110,3 (32,5) |
|
CL [L/h] |
1,0 (33,5) |
|
|
t1/2, ß [h]b |
26,5 (28,5) |
|
|
a. Todos los parámetros se estimaron por el modelo poblacional utilizando un modelo bicompartimental con eliminación de primer orden; AUCSS, Cmáx.,SS y Cmin,SS (concentración plasmática mínima en el estado estacionario) se estimaron utilizando parámetros farmacocinéticos individuales y una velocidad de infusión de 1 mg/min para administrar dosis recomendadas de 50 y 100 mg/día. |
||
• Género: No se necesita ajuste de dosis en base al género. Las concentraciones plasmáticas de anidulafungina en varones y mujeres sanos fueron similares. En estudios a pacientes con dosis múltiples, el aclaramiento del fármaco fue ligeramente más rápido (aproximadamente 22%) en varones.
• Geriatría: No se necesita ajuste de dosis en pacientes geriátricos. Los análisis farmacocinéticos poblacionales mostraron que el aclaramiento medio difirió ligeramente entre el grupo de adultos mayores (pacientes ≥65, aclaramiento medio = 1,07 L/h) y el grupo de no adultos mayores (pacientes <65, aclaramiento medio = 1,22 L/h) y el rango del aclaramiento fue similar.
• Raza: No se necesita ajuste de dosis en base a la raza. La farmacocinética de anidulafungina fue similar entre blancos, negros, asiáticos e hispanos.
• Infección por VIH: No se necesita ajuste de dosis si se tiene infección por VIH, independientemente de la terapia antirretroviral concomitante.
• Insuficiencia hepática: No se necesita ajuste de dosis en base a la insuficiencia hepática leve, moderada o severa. La anidulafungina no se metaboliza vía hepática. La farmacocinética de anidulafungina se evaluó en sujetos con insuficiencia hepática, Child-Pugh clase A, B o C. Las concentraciones de anidulafungina no se vieron incrementadas en sujetos con cualquier grado de insuficiencia hepática. Aunque se observó una ligera disminución en el AUC en pacientes con insuficiencia hepática, Child-Pugh clase C, se encontró dentro del rango de estimados poblacionales observados para sujetos sanos.
• Insuficiencia renal: No se necesita ajuste de dosis para pacientes con cualquier grado de insuficiencia renal incluyendo a los que reciben hemodiálisis. La anidulafungina tiene aclaramiento renal no significativo. En un estudio clínico de sujetos con insuficiencia renal leve, moderada, severa o en etapa final (dependiente de diálisis), la farmacocinética de anidulafungina fue similar a la observada en sujetos con función renal normal. La anidulafungina no es dializable y puede ser administrada sin considerar el tiempo de hemodiálisis.
• Pediatría: Se investigó la farmacocinética de anidulafungina luego de dosis diarias en pacientes pediátricos (2 durante 11 años) y adolescentes (12 durante 17 años) inmunocomprometidos con neutropenia. Se alcanzó el estado estacionario el primer día después de la administración de la dosis de carga (dos veces la dosis de mantenimiento), y el Cmáx. y AUCSS se incrementaron de forma proporcional a la dosis. Las concentraciones y exposiciones luego de la administración de dosis de mantenimiento de 0,75 y 1,5 mg/kg/día en esta población fueron similares a las observadas en adultos luego de dosis de mantenimiento de 50 y 100 mg/día, respectivamente (como se muestra en la Tabla 3).
Tabla 3. Parámetros farmacocinéticos en el estado estacionario promedio (%CV) de anidulafungina luego de la administración IV de anidulafungina una vez al día en sujetos pediátricos
|
Parámetro farmacocinéticoa |
Régimen de dosis de |
|||
|
1,5/0,75 |
3,0/1,5 |
|||
|
Grupo etario |
2-11 años |
12-17 años |
2-11 años |
12-17 años |
|
Cmáx., SS [mg/L] |
3,32 (50,0) |
4,35 (22,5) |
7,57 (34,2) |
6,88 (24,3) |
|
AUCSS [mg• h/L] |
41,1 (38,4) |
56,2 (27,8) |
96,1 (39,5) |
102,9 (28,2) |
|
a. Los datos se recolectaron el día 5. b. LD/MD: dosis de carga/dosis de mantenimiento diario. |
||||
FARMACOLOGÍA Y TOXICOLOGÍA ANIMAL: En estudios de 3 meses, se observó toxicidad hepática, incluyendo necrosis hepatocelular en célula única, hipertrofia hepatocelular y pesos hepáticos incrementados en monos y ratas en dosis equivalentes a 5-6 veces la exposición en humanos. Para ambas especies, la hipertrofia hepatocelular aún se observó un mes después del término de la dosificación.
CONTRAINDICACIONES: ECALTA está contraindicado en personas con hipersensibilidad conocida a anidulafungina, cualquier componente de ECALTA, u otra equinocandina.
REACCIONES ADVERSAS
General:
Las reacciones adversas más graves relacionadas con ECALTA son:
• Efectos hepáticos (ver Precauciones)
• Hipersensibilidad (ver Precauciones)
Experiencia de seguridad total con ECALTA: Dado que los ensayos clínicos se realizan bajo condiciones muy diferentes, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas de los ensayos clínicos de otro medicamento y pueden no reflejar las tasas observadas en la práctica.
La seguridad de ECALTA para inyectable se evaluó en 929 personas, incluyendo 257 sujetos sanos y 672 pacientes en estudios clínicos de candidemia, otras formas de infección con Candida y candidiasis esofágica. Un total de 633 pacientes recibieron ECALTA en dosis diarias de ya sea 50 o 100 mg. Un total de 481 pacientes recibió ECALTA durante ≥14 días.
Candidemia/otras infecciones por Candida: En tres estudios (uno comparativo vs. fluconazol, dos no comparativos) se evaluó la eficacia y seguridad de ECALTA (100 mg) en pacientes con candidemia y otras infecciones por Candida.
Los datos mostrados a continuación reflejan la exposición a ECALTA y fluconazol en 127 y 118 pacientes, respectivamente, con candidemia y otras formas de candidiasis invasiva, en un ensayo randomizado comparativo de la seguridad y eficacia de ECALTA y de fluconazol. En los pacientes tratados con ECALTA, el rango de edad estaba comprendido entre 16 y 89 años, la distribución de sexos era de 51% hombres y 49% mujeres, y la distribución de razas era de 72% de blancos, 18% de negros/afroamericanos, 9% de otras razas. Los pacientes fueron randomizados para recibir ECALTA por vía I.V. una vez al día (dosis de carga de 200 mg seguida por una dosis de mantenimiento de 100 mg) o fluconazol por vía I.V. (dosis de carga de 800 mg seguida por una dosis de mantenimiento de 400 mg). El tratamiento tuvo una duración mínima de 14 días y no superior a 42 días.
La cantidad de pacientes con reacciones adversas que ocasionaron la suspensión de la medicación del estudio fue de 11,5% en el brazo de ECALTA y de 21,6% en el plazo de fluconazol. Las reacciones adversas más comunes que ocasionaron la suspensión de la medicación del estudio fueron disfunción orgánica múltiple e infección sistémica con Candida en el brazo de ECALTA.
La Tabla 4 presenta las reacciones adversas reportadas en ≥5% de los sujetos a quienes se administró la terapia con ECALTA o fluconazol en este ensayo.
Tabla 4: Reacciones adversas reportadas en ≥5% de sujetos querecibieron ECALTA o fluconazol para el tratamiento de la candidemia/otras infecciones por Candida*, **
|
ECALTA |
Fluconazol |
|
|
N (%) |
N (%) |
|
|
Sujetos con al menos una reacción adversa |
130 (99) |
122 (98) |
|
Infecciones e infestaciones |
82 (63) |
80 (64) |
|
Bacteriemia |
23 (18) |
23 (18) |
|
Infección del tracto urinario |
19 (15) |
22 (18) |
|
Sepsis |
9 (7) |
11 (9) |
|
Neumonía |
8 (6) |
19 (15) |
|
Trastornos gastrointestinales |
81 (62) |
72 (58) |
|
Náuseas |
32 (24) |
15 (12) |
|
Diarrea |
24 (18) |
23 (18) |
|
Vómitos |
23 (18) |
12 (10) |
|
Estreñimiento |
11 (8) |
14 (11) |
|
Dolor abdominal |
8 (6) |
16 (13) |
|
Trastornos generales y alteraciones en el lugar de administración |
70 (53) |
76 (61) |
|
Pirexia |
23 (18) |
23 (18) |
|
Edema periférico |
14 (11) |
16 (13) |
|
Dolor torácico |
7 (5) |
6 (5) |
|
Trastornos respiratorios, torácicos y mediástinicos |
67 (51) |
55 (44) |
|
Disnea |
15 (12) |
4 (3) |
|
Derrame pleural |
13 (10) |
11 (9) |
|
Tos |
9 (7) |
7 (6) |
|
Dificultad respiratoria |
8 (6) |
2 (2) |
|
Investigaciones |
66 (50) |
46 (37) |
|
Aumento de la fosfatasa alcalina en sangre |
15 (12) |
14 (11) |
|
Leucocitos elevados |
11 (8) |
3 (2) |
|
Enzimas hepáticas elevadas |
7 (5) |
14 (11) |
|
Creatinina elevada en sangre |
7 (5) |
1 (1) |
|
Trastornos metabólicos y de nutrición |
61 (47) |
61 (49) |
|
Hipocaliemia |
33 (25) |
24 (19) |
|
Hipomagnesemia |
15 (12) |
14 (11) |
|
Hipoglucemia |
9 (7) |
10 (8) |
|
Hiperkaliemia |
8 (6) |
14 (11) |
|
Hiperglucemia |
8 (6) |
8 (6) |
|
Deshidratación |
8 (6) |
2 (2) |
|
Trastornos vasculares |
50 (38) |
41 (33) |
|
Hipotensión |
19 (15) |
18 (14) |
|
Hipertensión |
15 (12) |
5 (4) |
|
Trombosis venosa profunda |
13 (10) |
9 (7) |
|
Trastornos psiquiátricos |
48 (37) |
45 (36) |
|
Insomnio |
20 (15) |
12 (10) |
|
Estado confusional |
10 (8) |
10 (8) |
|
Depresión |
8 (6) |
5 (4) |
|
Trastornos del aparato circulatorio y el sistema linfático |
34 (26) |
36 (29) |
|
Anemia |
12 (9) |
20 (16) |
|
Trombocitemia |
8 (6) |
1 (1) |
|
Leucocitosis |
7 (5) |
6 (5) |
|
Trastornos de la piel y del tejido subcutáneo |
30 (23) |
32 (26) |
|
Úlcera de decúbito |
7 (5) |
10 (8) |
|
Trastornos del sistema nervioso |
27 (21) |
31 (25) |
|
Dolor de cabeza |
11 (8) |
10 (8) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
27 (21) |
25 (20) |
|
Dolor de espalda |
7 (5) |
13 (10) |
|
*Un paciente que experimentó múltiples reacciones con una clase de sistema orgánico (SOC) o término preferido fue contado una vez para esa clase, una vez para el término preferido y una vez para los “sujetos con una reacción adversa por lo menos”. ** Este ensayo no fue diseñado para respaldar alegaciones comparativas de ECALTA para las reacciones adversas incluidas en esta tabla. |
||
Candidiasis esofágica: Los datos mostrados a continuación reflejan la exposición a ECALTA y fluconazol en 300 y 301 pacientes, respectivamente, con candidiasis esofágica en un ensayo randomizado que comparó la seguridad y eficacia de ECALTA y de fluconazol oral. En los pacientes tratados con ECALTA, el rango de edad estaba comprendido entre 18 y 68 años, la distribución de sexos era de 42% hombres y 58% mujeres, y la distribución de razas era de 15% de blancos, 49% de negros/afroamericanos, 15% de asiáticos, 0,3% de hispanos, 21% de otras razas. Los pacientes fueron randomizados para recibir ECALTA por vía IV (100 mg el día 1, seguida por 50 mg al día) o fluconazol oral (200 mg el día 1, seguida de 100 mg al día) durante siete días después de la resolución de los síntomas (rango de 14 -21 días).
Veintiocho (9%) pacientes del brazo de ECALTA y 36 (12%) del brazo de fluconazol sufrieron reacciones adversas que causaron la suspensión del medicamento del estudio. Las reacciones adversas más comunes que ocasionaron la suspensión de la medicación del estudio fueron erupción maculopapular para el brazo de ECALTA. Las reacciones adversas más comunes que ocasionaron la suspensión de la medicación del estudio para el brazo de fluconazol fueron erupción y AST elevado.
La Tabla 5 presenta las reacciones adversas reportadas en ≥5% de los sujetos a quienes se administró la terapia con ECALTA.
Tabla 5: Reacciones adversas reportadas en ≥5% de los sujetos a quienes se administró la terapia con ECALTA o Fluconazol para Candidiasis esofágica*, **
|
ECALTA |
Fluconazole |
|
|
N (%) |
N (%) |
|
|
Sujetos con al menos una reacción adversa |
239 (80) |
227 (75) |
|
Infecciones e infestaciones |
115 (38) |
99 (33) |
|
Candidiasis oral |
15 (5) |
10 (3) |
|
Trastornos gastrointestinales |
106 (35) |
113 (38) |
|
Diarrea |
27 (9) |
26 (9) |
|
Vómitos |
27 (7) |
30 (10) |
|
Náuseas |
20 (7) |
23 (8) |
|
Dispepsia |
20 (7) |
21 (7) |
|
Trastornos del aparato circulatorio y el sistema linfático |
55 (18) |
50 (17) |
|
Anemia |
25 (8) |
22 (7) |
|
Trastornos metabólicos y de nutrición |
50 (17) |
46 (15) |
|
Hipocaliemia |
14 (5) |
17 (6) |
|
Trastornos generales y alteraciones en el lugar de administración |
49 (16) |
54 (18) |
|
Pirexia |
27 (9) |
28 (9) |
|
Trastornos del sistema nervioso |
39 (13) |
36 (12) |
|
Dolor de cabeza |
25 (8) |
20 (7) |
|
* Un paciente que experimentó múltiples reacciones con una clase de sistema orgánico (SOC) o término preferido fue contado una vez para esa clase, una vez para el término preferido y una vez para los “sujetos con una reacción adversa por lo menos”. ** Este ensayo no fue diseñado para respaldar alegaciones comparativas de ECALTA para las reacciones adversas incluidas en esta tabla. |
||
Reacciones adversas menos comunes
Las siguientes reacciones adversas seleccionadas se presentaron en <2% de los pacientes:
• Sanguíneo o linfático: Coagulopatía, trombocitopenia.
• Cardíaco: Fibrilación auricular, bloqueo completo de rama derecha, arritmia sinusal, extrasístoles ventriculares.
• Ocular: Dolor ocular, visión borrosa, alteraciones visuales.
• General y sitio de administración: Reacción relacionada con la infusión, edema periférico, escalofríos.
• Hepatobiliar: Pruebas de función hepática anormales, colestasis, necrosis hepática.
• Infecciones: Infecciones clostridiales.
• Investigaciones: Aumento de amilasa, aumento de bilirrubina, aumento de CPK, aumento de creatinina, electrocardiograma QT prolongado, aumento de gamma-glutamil transferasa, aumento de lipasa, disminución de magnesio, aumento del recuento de plaquetas, disminución del recuento de plaquetas, disminución de potasio, tiempo de protrombina prolongado, aumento de urea.
• Sistema nervioso: Convulsiones, vértigo.
• Respiratorio, torácico y mediastinal: Tos.
• Tejido cutáneo y subcutáneo: Edema angioneurótico, eritema, prurito, sudoración incrementada, urticaria.
• Vascular: Enrojecimiento, rubor con sensacion de calor intensa, tromboflebitis superficial.
Experiencia posterior a la comercialización: Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de anidulafungina. Dado que dichas reacciones son reportadas de forma voluntaria por una población de tamaño indeterminado, no siempre es posible estimar fiablemente su frecuencia o establecer una relación causa-efecto con la exposición al fármaco.
Inmune: Shock anafiláctico, reaccion anafiláctica, broncoespasmo (ver Precauciones).
INTERACCIÓN CON MEDICAMENTOS: Los estudios in vitro mostraron que la anidulafungina no es metabolizada por la citocromo P450 humana o hepatocitos humanos aislados, y no inhibe de forma significativa las actividades de las isoformas humanas del CYP (1A2, 2B6, 2C8, 2C9, 2C19, 2D6 y 3A) a concentraciones clínicamente relevantes. No se observó interacciones entre medicamentos clínicamente relevantes con fármacos que podrían ser administrados con anidulafungina.
Ciclosporina (sustrato CYP3A4): En un estudio en el que 12 adultos sanos recibieron una dosis de mantenimiento de 100 mg/día de anidulafungina luego de una dosis de carga de 200 mg (en los días 1 a 8) y en combinación con ciclosporina oral 1,25 mg/kg dos veces al día (en los días 5 a 8), el Cmáx. en el estado estacionario de anidulafungina no se vio significativamente alterado por la ciclosporina; el AUC en el estado estacionario de anidulafungina se incrementó en 22%. Un estudio in vitro aparte mostró que la anidulafungina no tiene efecto sobre el metabolismo de la ciclosporina. No se necesita ajuste de dosis de ninguno de los fármacos cuando sean coadministrados (ver Precauciones: Interacción con medicamentos).
Voriconazol (sustrato e inhibidor CYP2C19, CYP2C9, CYP3A4): En un estudio en el que 17 sujetos sanos recibieron una dosis de mantenimiento de 100 mg/día de anidulafungina luego de una dosis de carga de 200 mg, voriconazol oral 200 mg dos veces al día (luego de dos dosis de carga de 400 mg) y ambas en combinación, el Cmáx. y AUC en el estado estacionario de anidulafungina y voriconazol no se vieron alterados de forma significativa por la coadministración. No se justifica que se realice ajuste de dosis de ninguno de los fármacos cuando sean coadministrados (ver Precauciones: Interacción con medicamentos).
Tacrolimus (sustrato CYP3A4): En un estudio en el que 35 sujetos sanos recibieron una dosis oral única de 5 mg de tacrolimus (en el día 1), dosis de mantenimiento de 100 mg/día de anidulafungina luego de una dosis de carga de 200 mg (en los días 4 a 12) y ambas en combinación (el día 13), el Cmáx. y AUC en el estado estacionario de anidulafungina y tacrolimus no se vieron significativamente alterados por la coadministración. No se justifica el realizar un ajuste de dosis de ninguno de los fármacos cuando sean coadministrados (ver Precauciones: Interacción con medicamentos).
Anfotericina B liposomal inyectable: La farmacocinética de anidulafungina se evaluó en 27 pacientes a los que se les coadministró anfotericina B liposomal. El análisis de farmacocinética poblacional mostró que cuando se compara con los datos de pacientes que no recibieron anfotericina B, la farmacocinética de anidulafungina no se vio alterada de forma significativa por la coadministración con anfotericina B. No se justifica el realizar un ajuste de dosis de anidulafungina (ver Precauciones: Interacción con medicamentos).
Rifampicina (potente inductor del CYP450): La farmacocinética de anidulafungina se evaluó en 27 pacientes a los que se les coadministró anidulafungina y rifampicina. El análisis farmacocinético poblacional mostró que cuando se comparó con los datos de pacientes que no recibieron rifampicina, la farmacocinética de anidulafungina no se vio significativamente alterada por coadministración con rifampicina. No se justifica el realizar un ajuste de dosis de anidulafungina (ver Precauciones: Interacción con medicamentos).
MICROBIOLOGÍA
Mecanismo de acción: La anidulafungina es una equinocandina semisintética con actividad antifúngica. La anidulafungina inhibe la sintasa de glucano, una enzima presente en las células fúngicas pero no en las de los mamíferos. Esto da como resultado la inhibición de la formación del 1,3-ß-D-glucano, un componente esencial de la pared celular fúngica.
Actividad in vitro: La anidulafungina ha demostrado que es activa contra Candida albicans, C. glabrata, C. parapsilosis, y C. tropicalis tanto in vitro como en infecciones clínicas, como se explica en Indicaciones y uso.
Debido al potencial para una susceptibilidad reducida a la anidulafungina, se recomienda que la susceptibilidad sea determinada por un método estandarizado1.
Las concentraciones inhibitorias mínimas (CIM) de anidulafungina se determinaron para aislamientos de Candida spp. obtenidos durante estudios clínicos según el método estándar1. Sin embargo, no se ha establecido la correlación entre la actividad in vitro como se ha determinado por este método y el resultado clínico.
1. Clinical and Laboratory Standards Institute (CLSI). Reference Method for Broth Dilution Antifungal Susceptibility Testing of Yeasts; Approved Standard – Third Edition. CLSI Document M27-A3; Wayne PA. 2009.
Resistencia al fármaco: La resistencia a la equinocandina se debe a mutaciones puntuales en la codificación de los genes (FKS1 y FKS2) para las subunidades del complejo enzimático glucán sintasa. Ha habido informes aislados de Candida con sensibilidad reducida a anidulafungina, lo que sugiere un potencial para el desarrollo de resistencia a la droga. La importancia clínica de esta observación no se comprende totalmente.
PRECAUCIONES
Efectos hepáticos: Se ha observado anormalidades en las pruebas de laboratorio para la función hepática en voluntarios sanos y pacientes tratados con ECALTA. En algunos pacientes con condiciones médicas severas subyacentes que recibieron medicaciones múltiples concomitantes junto con ECALTA, se presentaron anormalidades hepáticas clínicamente significativas. Se ha reportado casos aislados de disfunción hepática significativa, hepatitis, o insuficiencia hepática en pacientes; no se ha establecido una relación causal con ECALTA. Los pacientes que presentaron resultados anormales en las pruebas de función hepática durante la terapia con ECALTA deben ser monitorizados con respecto a la observación de empeoramiento de la función hepática y evaluados para el riesgo/beneficio de continuación de la terapia con ECALTA.
Hipersensibilidad: Se han descrito casos de reacciones anafilácticas, incluido shock, con el uso de ECALTA. Si se produce una reacción de esta clase, debe interrumpirse el tratamiento con ECALTA y administrarse un tratamiento apropiado (ver Reacciones adversas).
Se han descrito reacciones adversas a ECALTA relacionadas con la infusión, posiblemente con histamina como mediador, incluidas erupciones, urticaria, eritema, prurito, broncoespasmo, disnea e hipotensión (ver Reacciones adversas). Para evitar en lo posible estas reacciones, no se debe sobrepasar la tasa de infusión de ECALTA de 1,1 mg/minuto (ver Dosificación y administración).
Interacciones con medicamentos:
• Ciclosporina: La administración de dosis múltiples de anidulafungina y ciclosporina a sujetos sanos no produjo una alteración significativa de la farmacocinética en estado de equilibrio de ninguno de los dos fármacos. No es necesario ajustar la dosis de ciclosporina ni de anidulafungina cuando se administran conjuntamente ambos medicamentos (ver Farmacología clínica: Interacciones con medicamentos).
• Voriconazol: La administración de dosis múltiples de anidulafungina y voriconazol a sujetos sanos no produjo una alteración significativa de la farmacocinética en estado de equilibrio de ninguno de los dos fármacos. No es necesario ajustar la dosis de voriconazol ni de anidulafungina cuando se administran conjuntamente ambos medicamentos (ver Farmacología clínica: Interacciones con medicamentos).
• Tacrolimus: La administración de dosis múltiples de anidulafungina y una dosis única de tacrolimus a sujetos sanos no produjo una alteración significativa de la farmacocinética en estado de equilibrio de ninguno de los dos fármacos. No es necesario ajustar la dosis de tacrolimus ni de anidulafungina cuando se administran conjuntamente ambos medicamentos (ver Farmacología clínica: Interacciones con medicamentos).
• Rifampicina: La administración de dosis múltiples de anidulafungina y rifampicina a sujetos sanos no produjo una alteración significativa de la farmacocinética en estado de equilibrio de ninguno de la anidulafungina. No es necesario ajustar la dosis de anidulafungina cuando se administra conjuntamente con rifampicina (ver Farmacología clínica: Interacciones con medicamentos).
• Anfotericina B liposomal inyectable: La administración de dosis múltiples de anidulafungina y anfotericina B liposomal a sujetos sanos no produjo una alteración significativa de la farmacocinética en estado de equilibrio de ninguno de la anidulafungina. No es necesario ajustar la dosis de anidulafungina cuando se administra conjuntamente con anfotericina B liposomal (ver Farmacología clínica: Interacciones con medicamentos).
Carcinogénesis, mutagénesis, disfunción de la fertilidad: No se ha llevado a cabo estudios de carcinogenicidad animal a largo plazo con anidulafungina.
La anidulafungina no fue genotóxica en los siguientes estudios in vitro: ensayos de mutación reversa bacteriana, un ensayo de aberración cromosómica con células ováricas de hámster chino, y un ensayo de mutación anterógrada de genes con células de linfoma de ratón. La anidulafungina no fue genotóxica en la prueba de micronúcleos in vivo en ratones.
La anidulafungina no produjo efectos adversos sobre la fertilidad en ratas hembra o macho a dosis intravenosas de 20 mg/kg/día (equivalente a 2 veces la dosis de mantenimiento terapéutica propuesta de 100 mg/día en base al área de superficie corporal relativa).
Embarazo: Embarazo - categoría B
Se llevaron a cambio estudios de desarrollo embriofetal con dosis de hasta 20 mg/kg/día en ratas y conejos (equivalente a 2 y 4 veces, respectivamente, la dosis de mantenimiento terapéutica propuesta de 100 mg/día en base al área de superficie corporal relativa). En ratas, la anidulafungina cruzó la barrera placentaria, se detectó en el plasma fetal y se pudo relacionar con una osificación incompleta de algunos huesos.
No existen estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios de reproducción en animales no siempre son predictivos de la respuesta humana, ECALTA debe ser utilizado durante el embarazo solo en caso de necesidad evidente.
Lactancia: La anidulafungina se encontró en la lecha de ratas lactantes. No se sabe si la anidulafungina se excreta en la leche humana. Muchos medicamentos se excretan en la leche humana, por lo que deben tomarse precauciones cuando se administre ECALTA a una mujer en periodo de lactancia.
Uso pediátrico: Aún no se han establecido la seguridad ni la efectividad de ECALTA en pacientes de menos de 16 años (ver Farmacología clínica: Poblaciones específicas/pediátricas).
Uso en geriatría: No es necesario ajustar la dosis para los pacientes geriátricos (ver Farmacología clínica: Poblaciones específicas/pediátricas).
De la cantidad total de sujetos (N = 197) incluidos en los estudios clínicos fundamentales de anidulafungina, un 35% tenían 65 años o más, mientras que un 18% tenían 75 o más años. No se observaron diferencias generales en cuanto a la seguridad o eficacia entre estos sujetos y otros más jóvenes, y otras experiencias clínicas reportadas no identificaron diferencias en las respuestas entre los pacientes ancianos y los más jóvenes, pero no se puede descartar una mayor sensibilidad de algunas personas mayores.
Insuficiencia hepática: No es necesario ajustar la dosis de los pacientes con insuficiencia hepática de cualquier grado. La anidulafungina no se metaboliza por vía hepática. Se examinó la farmacocinética de la anidulafungina en sujetos con insuficiencia hepática Child-Pugh clase A, B o C. Las concentraciones de anidulafungina no aumentaron en los sujetos con insuficiencia hepática de cualquier grado. Aunque se observó una leve disminución del AUC en pacientes con insuficiencia hepática clase C de la escala de Child-Pugh, se mantuvo dentro del rango de las estimaciones para la población de voluntarios sanos (ver Farmacología clínica: Poblaciones específicas/pediátricas).
Insuficiencia renal: No es necesario realizar ajustes de la dosis para los pacientes con algún grado de insuficiencia renal, incluidos los pacientes que deben realizarse hemodiálisis. La anidulafungina tiene un aclaramiento renal despreciable (<1%). En un estudio clínico realizado en pacientes con insuficiencia renal leve, moderada, grave o terminal (dependiente de diálisis), la farmacocinética de la anidulafungina fue similar a la que se observó en los sujetos con función renal normal. La anidulafungina no es dializable y puede ser administrada independientemente del momento de la hemodiálisis (ver Farmacología clínica: Poblaciones específicas/pediátricas).
DOSIS Y ADMINISTRACIÓN
Dosis recomendada:
• Candidemia y otras infecciones por Candida (absceso intraabdominal y peritonitis): La dosis recomendada es una dosis de carga única de 200 mg de ECALTA el día 1, seguida por una dosis diaria posterior de 100 mg. La duración del tratamiento debe basarse en la respuesta clínica del paciente. En general, la terapia antifúngica debe continuar durante al menos 14 días después del último cultivo positivo.
• Candidiasis esofágica: La dosis recomendada es una dosis de carga única de 100 mg de ECALTA el día 1, seguida por una dosis posterior de 50 mg diaria. Los pacientes deben ser tratados durante un mínimo de 14 días y como mínimo 7 días luego de la resolución de los síntomas. La duración del tratamiento debe basarse en la respuesta clínica del paciente. Debido al riesgo de recaída por candidiasis esofágica en pacientes con infecciones por VIH, se puede considerar la terapia antifúngica supresora luego de un curso de tratamiento.
Preparación para la administración
ECALTA para inyectable debe ser reconstituido con agua para inyectable estéril y posteriormente ser diluida solamente con inyección de Dextrosa 5%, USP o inyección de cloruro de sodio 0.9%, USP (salina normal). No se ha establecido la compatibilidad de ECALTA reconstituido con sustancias intravenosas, aditivos o medicamentos diferentes a la inyección de Dextrosa 5%, USP o inyección de cloruro de sodio 0.9%, USP (salina normal).
• Reconstitución 50 mg/vial: Reconstituir de forma aséptica cada vial de 50 mg con 15 mL de agua para inyectable estéril para proporcionar una concentración de 3.33 mg/mL. La solución reconstituida puede almacenarse hasta por 24 horas a temperaturas de hasta 25 °C (77 °F) antes de la dilución en la solución de infusión.
• Reconstitución 100 mg/vial: Reconstituir de forma aséptica cada vial de 100 mg con 30 mL de agua para inyectable estéril para obtener una concentración de 3.33 mg/mL. La solución reconstituida puede almacenarse hasta por 24 horas a 25 °C (77 °F) antes de la dilución en la solución de infusión.
• Dilución e infusión: Transferir de forma aséptica los contenidos del (los) vial (es) reconstituido (s) en una bolsa para infusión IV (o frasco) de tamaño adecuado que contenga ya sea inyección de dextrosa 5%, USP o inyección de cloruro de sodio 0.9%, USP (salina normal). Ver Tabla 6 para las instrucciones de dilución e infusión para cada dosis.
Tabla 6. Requisitos de dilución para la administración de ECALTA
|
Dosis |
Número de viales requeridos |
Volumen requerido del reconstituido total |
Volumen de infusióna |
Volumen de infusión totalb |
Velocidad de infusión |
Duración mínima de la infusión |
|
50 mg |
1–50 mg |
15 mL |
50 mL |
65 mL |
1.4 mL/min u 84 mL/hora |
45 min |
|
100 mg |
2–50 mg o |
30 mL |
100 mL |
130 mL |
1,4 mL/min u 84 mL/hora) |
90 min |
|
200 mg |
4–50 mg o |
60 mL |
200 mL |
260 mL |
1,4 mL/min u 84 mL/hora) |
180 min |
|
a. Ya sea inyección de dextrosa 5%, USP o inyección de cloruro de sodio 0,9%, USP (salina normal). b. La concentración de la solución de infusión es 0,77 mg/mL Precaución: La velocidad de infusión no debe exceder 1.1 mg/minuto (equivalente a 1.4 mL/minuto u 84 ml/hora cuando se reconstituye y diluye por instrucciones). |
||||||
Si la solución de infusión no se utiliza de forma inmediata, debe almacenarse en un refrigerador a 2 °C – 8 °C (36 °F – 46 °F). No congelar. La solución de infusión debe ser administrada dentro de las 24 horas de haber sido preparada.
Los productos parenterales deben ser inspeccionados visualmente con respecto a la materia particulada y decoloración antes de la administración, siempre que la solución y el envase lo permitan. Si se observa materia particulada o decoloración, descartar la solución.
SOBREDOSIS: Durante estudios clínicos, una dosis única de 400 mg de ECALTA fue administrada de forma inadvertida como una dosis de carga. No se reportó eventos adversos clínicos. En un estudio con 10 sujetos sanos a los que se les administró una dosis de carga de 260 mg seguido por 130 mg diarios, ECALTA fue en general bien tolerado; 3 de 10 sujetos experimentaron elevaciones transitorias asintomáticas de transaminasas (≤3 el límite superior normal) (ver Precauciones).
La anidulafungina no es dializable.
La dosis no letal máxima de anidulafungina en ratas fue 50 mg/kg, una dosis que es equivalente a 10 veces la dosis diaria recomendada para candidiasis esofágica (50 mg/día) o equivalente a 5 veces la dosis diaria recomendada para candidemia y otras infecciones por Candida (100 mg/día), en base a la comparacion del área de superficie corporal relativa.
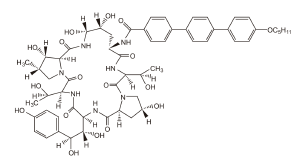
DESCRIPCIÓN: ECALTA para inyectable es un producto liofilizado estéril para infusión intravenosa (IV) que contiene anidulafungina. ECALTA (anidulafungina) es un lipopéptido semisintético sintetizado a partir de un producto de fermentación de Aspergillus nidulans. Anidulafungina es una equinocandina, una clase de fármaco antifúngico que inhibe la síntesis del 1,3-ß-D-glucano, un componente esencial de las paredes celulares fúngicas.
ECALTA (anidulafungina) es la 1-[(4R5R)-4,5-dihidroxi-N2-[[4"-(pentiloxi)[1,1":4",1"- terfenil]-4-il]carbonil]-L-ornitina]equinocandina B. La anidulafungina es un polvo blanco a casi blanco prácticamente insoluble en agua y ligeramente soluble en etanol.
La fórmula empírica de anidulafungina es C58H73N7O17 y el peso de la fórmula es 1140,3.
La fórmula estructural es:
Antes de la administración, ECALTA para inyectable debe ser reconstituido con agua para inyectable estéril y posteriormente diluido con ya sea inyección de dextrosa 5%, USP o inyección de cloruro de sodio 0.9%, USP (salina normal).
NO diluir con otras soluciones ni administrar en infusión concomitantemente con otros medicamentos o electrolitos (ver Dosis y administración).
FARMACOLOGÍA CLÍNICA
Mecanismo de acción: La anidulafungina es un fármaco antifúngico (ver Microbiología).
FORMAS DE PRESENTACIÓN: Caja por un vial de vidrio conteniendo polvo liofilizado para solución inyectable.
ALMACENAMIENTO
Viales no reconstituidos: Los viales de ECALTA no reconstituidos deben almacenarse en refrigeración entre 2 °C– 8 °C (36 °F – 46 °F). No congelar.
Solución reconstituida: La solución reconstituida de ECALTA se puede almacenar en refrigeración entre 2 °C -8 °C(36 °F- 46 °F) hasta por una hora. No congelar.
Se ha demostrado la estabilidad en uso química y física de la solución reconstituida durante 1 hora1 a 5 ºC (41 ºF). .
Solución para infusión: La solución para infusión de ECALTA se puede almacenar en refrigeración entre 2 °C -8 °C(36 °F- 46 °F), pero debe administrarse dentro de 24 horas. No congelar.
Se ha demostrado la estabilidad en uso química y física en uso de la solución para infusión durante 24 horas a 5 ºC (41 °F).
Importado por:
PFIZER S.A.
Av. Javier Prado Este 6230, 2do Piso
La Molina, Lima, Perú
Teléfono: 615-2100, Fax: 615-2106
LLD Basado en USPI (20.07.2012) V2.
VIDA ÚTIL: 36 meses


