CRESTOR
ROSUVASTATINA
Comprimidos recubiertos
1 Caja, 28 Comprimidos recubiertos, 5 Miligramos
1 Caja, 30 Comprimidos recubiertos, 10 Miligramos
1 Caja, 30 Comprimidos recubiertos, 20 Miligramos
1 Caja, 30 Comprimidos recubiertos, 40 Miligramos
1 Caja, 30 Comprimidos recubiertos, 10 Miligramos
COMPOSICIÓN
Cada COMPRIMIDO contiene: 5 mg, 10 mg, 20 mg o 40 mg de rosuvastatina (en forma de sal de calcio).
FORMA FARMACÉUTICA
Comprimidos recubiertos.
Comprimidos redondos de color amarillo (5 mg); comprimidos redondos de color rosado (10 mg y 20 mg); comprimidos ovalados de color rosado (40 mg).
USO EN GRUPOS ESPECÍFICOS DE PACIENTES
Embarazo: Categoría X.
Efectos teratógenos: CRESTOR está contraindicado en mujeres embarazadas o que podrían embarazarse. Las concentraciones séricas de colesterol y triglicéridos aumentan durante el embarazo normal, y los productos derivados del colesterol desempeñan un papel central en el desarrollo fetal. La aterosclerosis es un proceso crónico y la suspensión del tratamiento con hipolipemiantes durante el embarazo debería tener pocos efectos en los resultados a largo plazo del tratamiento de la hiperlipidemia primaria (ver Contraindicaciones).
No se han realizado estudios adecuados y bien controlados sobre CRESTOR en mujeres embarazadas. Se han notificado raramente casos de anomalías congénitas después de la exposición intrauterina a inhibidores de la HMG-CoA reductasa. En un análisis de alrededor de 100 embarazos objeto de una vigilancia prospectiva en mujeres tratadas con otros inhibidores de la HMG-CoA reductasa, las incidencias de anomalías congénitas, abortos espontáneos y mortalidad fetal/mortinatalidad no superaron la incidencia prevista en la población general. No obstante, este estudio sólo permitió descartar un aumento de 3 a 4 veces del riesgo de anomalías congénitas frente a la incidencia de referencia. En el 89% de estos casos, el tratamiento comenzó antes del embarazo y se suspendió durante el primer trimestre, en cuanto se confirmó el embarazo.
La rosuvastatina atraviesa la barrera placentaria en ratas y conejos. En ratas, CRESTOR no tuvo efectos teratógenos con exposiciones sistémicas equivalentes a una dosis terapéutica de 40 mg al día en el ser humano. Con 10 a 12 veces la dosis de 40 mg al día en el ser humano, se observó una disminución de la supervivencia de las crías, del peso fetal de las hembras y retraso de la osificación. En conejos, la viabilidad de las crías disminuyó y la mortalidad materna aumentó con dosis equivalentes a la de 40 mg al día en el ser humano (ver Toxicología preclínica).
CRESTOR puede causar daños al feto si se administra a una mujer embarazada. Si la paciente se embaraza durante el tratamiento con CRESTOR, deben tenerse en cuenta los riesgos potenciales para el feto y el hecho de que no se conocen beneficios clínicos de continuar el tratamiento durante el embarazo.
Lactancia: No se sabe si la rosuvastatina se excreta en la leche materna, pero se ha demostrado que una pequeña cantidad de otro medicamento de esta clase pasa a la leche materna. En ratas, las concentraciones de rosuvastatina en la leche materna son tres veces mayores que las concentraciones plasmáticas; sin embargo, las concentraciones del medicamento en la leche de animales quizás no reflejen exactamente las concentraciones en la leche materna humana. Ya que otro medicamento de esta clase se secreta en la leche materna y que los inhibidores de la HMG-CoA reductasa pueden causar reacciones adversas graves en los lactantes, debe aconsejarse a las mujeres que necesitan un tratamiento con CRESTOR que no amamanten a sus bebés (ver Contraindicaciones).
Uso pediátrico: La seguridad y la eficacia de CRESTOR en pacientes de 10 a 17 años con hipercolesterolemia familiar heterocigota se evaluaron en un estudio clínico controlado de 12 semanas seguido por un periodo de exposición de 40 semanas sin enmascaramiento. Los pacientes tratados con 5 mg, 10 mg y 20 mg al día de CRESTOR mostraron un perfil de reacciones adversas generalmente similar al de los pacientes del grupo placebo (ver Reacciones adversas). Aunque no todas las reacciones adversas identificadas en la población adulta se han observado en los estudios clínicos realizados en niños y adolescentes, deben tenerse en mente las mismas advertencias y precauciones de uso que en los adultos. En pacientes pediátricos de 10 a 17 años, no se observó ningún efecto detectable de CRESTOR en el crecimiento, el peso, el IMC (índice de masa corporal) o la maduración sexual (ver Estudios clínicos). Se debe aconsejar a las adolescentes que usen un método anticonceptivo adecuado durante el tratamiento con CRESTOR (ver Uso en grupos específicos de pacientes). CRESTOR no ha sido objeto de estudios clínicos controlados en pacientes prepúberes o menores de 10 años. Tampoco se han investigado dosis de CRESTOR superiores a 20 mg en la población pediátrica.
En niños y adolescentes con hipercolesterolemia familiar homocigota, la experiencia se limita a 8 pacientes (de 8 años o más).
En un estudio de farmacocinética, 18 pacientes (9 niños y 9 niñas) de 10 a 17 años con HF heterocigota recibieron dosis orales únicas y múltiples de CRESTOR. Los valores de la Cmáx. y el ABC de la rosuvastatina fueron similares a los observados en adultos tratados con las mismas dosis.
Uso geriátrico: De los 10 275 pacientes que participaron en los estudios clínicos de CRESTOR, 3159 (31%) tenían más de 65 años y 698 (6,8%) más de 75 años. No se observaron diferencias de seguridad y eficacia entre estos sujetos y los sujetos más jóvenes, y según la experiencia clínica, no se han identificado diferencias de respuesta entre los ancianos y pacientes más jóvenes, aunque no puede descartarse una mayor sensibilidad de algunas personas de edad avanzada.
El riesgo de miopatía es mayor en los pacientes de edad avanzada, por lo que CRESTOR debe recetarse con precaución en estos pacientes (ver Advertencias y precauciones especiales de uso y Farmacología clínica).
Insuficiencia renal: La insuficiencia renal leve a moderada (depuración de creatinina ≥30 ml/min/1,73 m2) no influye en la exposición a la rosuvastatina; sin embargo, esta última aumenta en un grado clínicamente significativo en los pacientes con insuficiencia renal grave no hemodializados. La dosis de CRESTOR debe ajustarse en los pacientes con insuficiencia renal grave (DEPCr <30 ml/min/1,73 m2) que no requieren hemodiálisis (ver Posología y forma de administración, Advertencias y precauciones especiales de uso y Farmacología clínica).
Insuficiencia hepática: CRESTOR está contraindicado en los pacientes con una enfermedad hepática activa, lo cual puede incluir elevaciones persistentes e inexplicables de las transaminasas hepáticas. Se sabe que la hepatopatía alcohólica crónica aumenta la exposición a la rosuvastatina, por lo que se requiere precaución al utilizar CRESTOR en estos pacientes (ver Contraindicaciones, Advertencias y precauciones especiales de uso y Farmacología clínica).
Pacientes de origen asiático: Los estudios farmacocinéticos demostraron que la mediana de la exposición a la rosuvastatina es aproximadamente 2 veces mayor en sujetos de origen asiático que en sujetos de control de raza blanca. La dosis de CRESTOR debe ajustarse en los pacientes de origen asiático (ver Posología y forma de administración y Farmacología clínica).
TOXICOLOGÍA PRECLÍNICA
Carcinogenia, mutagenia y alteración de la fecundidad: En un estudio de carcinogenia de 104 semanas en ratas que recibieron dosis de 2, 20, 60 u 80 mg/kg/día mediante una sonda esofágica, la incidencia de pólipos del estroma uterino aumentó significativamente en las hembras tratadas con 80 mg/kg/día (exposición sistémica 20 veces mayor que la exposición humana tras la administración de 40 mg/día basándose en la comparación de las ABC). No se observó una mayor incidencia de pólipos con las dosis inferiores.
En un estudio de carcinogenia de 107 semanas en ratones tratados con 10, 60 o 200 mg/kg/día mediante una sonda esofágica, se observó un aumento de la incidencia de adenoma/carcinoma hepatocelular con la dosis de 200 mg/kg/día (exposición sistémica 20 veces mayor que la exposición humana tras la administración de 40 mg/día basándose en la comparación de las ABC). No se observó una mayor incidencia de tumores hepatocelulares con las dosis inferiores.
La rosuvastatina no mostró efectos mutagénicos o clastogénicos, con o sin activación metabólica, en la prueba de Ames con Salmonella typhimurium y Escherichia coli, en el ensayo con células de linfoma de ratón y en el ensayo de aberraciones cromosómicas en células pulmonares de hámster chino. La rosuvastatina produjo resultados negativos en el ensayo de micronúcleos de ratón in vivo.
En estudios de fecundidad en ratas tratadas con dosis orales de 5, 15 y 50 mg/kg/día mediante una sonda esofágica, los machos recibieron el tratamiento durante 9 semanas antes y durante el apareamiento mientras que las hembras lo recibieron 2 semanas antes y durante el apareamiento hasta el día 7 de la gestación. No se observaron efectos adversos en la fecundidad con la dosis de 50 mg/kg/día (exposición sistémica de hasta 10 veces la exposición humana tras dosis de 40 mg/día basándose en la comparación de las ABC). En los testículos de perros tratados con 30 mg/kg/día de rosuvastatina durante un mes, se observaron células espermatídicas gigantes. También se observaron células espermatídicas gigantes en monos después de 6 meses de tratamiento con 30 mg/kg/día, además de vacuolación del epitelio de los túbulos seminíferos. Las exposiciones fueron 20 veces mayores en perros y 10 veces mayores en monos que la exposición humana tras la administración de 40 mg/día, basándose en una comparación de las dosis por unidad de superficie corporal. Se han observado hallazgos similares con otros medicamentos de esta clase.
Toxicología o farmacología en animales:
• Desarrollo embrionario y fetal: En ratas, la rosuvastatina atraviesa la barrera placentaria y se recupera en el tejido fetal y el líquido amniótico con concentraciones del 3% y 20%, respectivamente, de la concentración registrada en el plasma materno tras una dosis única de 25 mg/kg administrada mediante una sonda esofágica el día 16 de la gestación. Se observó una mayor distribución en el tejido fetal (25% de la concentración plasmática materna) en conejas después de una dosis oral única de 1 mg/kg administrada mediante una sonda esofágica el día 18 de la gestación.
La administración de dosis orales de 5, 15 y 50 mg/kg/día de rosuvastatina mediante una sonda esofágica a ratas hembras antes del apareamiento y hasta el día 7 poscoito, produjo una disminución de la supervivencia de los fetos (crías hembras) y un retraso de la osificación con la dosis alta (exposición sistémica 10 veces mayor que la exposición humana tras la administración de 40 mg al día, basándose en la comparación de las ABC).
La administración a ratas gestantes de dosis orales de 2, 10 y 50 mg/kg/día mediante una sonda esofágica desde el día 7 de la gestación hasta el día 21 de la lactancia (destete), produjo una disminución del peso de las crías en los grupos tratados con 50 mg/kg/día (exposición sistémica ≥12 veces que la exposición humana tras la administración de 40 mg al día, basándose en la comparación de las dosis por unidad de superficie corporal).
La administración a conejas gestantes de dosis orales de 0,3, 1 y 3 mg/kg/día mediante una sonda esofágica desde el día 6 de la gestación hasta el día 18 de la lactancia (destete), produjo una disminución de la viabilidad fetal y mortalidad materna (exposición equivalente a la exposición humana tras la administración de 40 mg/día, basándose en la comparación de las dosis por unidad de superficie corporal).
La rosuvastatina no tuvo efectos teratógenos en ratas tratadas con dosis ≤25 mg/kg/día ni en conejos tratados con dosis ≤3 mg/kg/día (exposiciones sistémicas equivalentes a la exposición humana tras la administración de 40 mg al día, basándose en la comparación de las ABC o de las dosis por unidad de superficie corporal, respectivamente).
• Toxicidad en el sistema nervioso central: En perros tratados con otros miembros de esta clase de medicamentos se han observado lesiones vasculares del SNC caracterizadas por hemorragias perivasculares, edema e infiltración de células mononucleares en el espacio perivascular. Un medicamento de esta clase con una estructura química similar produjo degeneración del nervio óptico en función de la dosis en perros (degeneración walleriana de las fibras retinogeniculadas), con una dosis que dio lugar a concentraciones plasmáticas del medicamento, aproximadamente 30 veces mayores que la concentración media en seres humanos tratados con la máxima dosis recomendada. Se observaron edema, hemorragia y necrosis parcial del intersticio del plexo coroideo en una perra sacrificada en estado moribundo el día 24 que recibió 90 mg/kg/día mediante una sonda esofágica (exposición sistémica 100 veces mayor que la exposición humana tras una dosis de 40 mg al día, basándose en la comparación de las ABC). Se observó opacidad de la córnea en perros tratados durante 52 semanas con 6 mg/kg/día mediante una sonda esofágica (exposición sistémica 20 veces mayor que la exposición humana tras la administración de 40 mg al día, basándose en la comparación de las ABC). Se observaron cataratas en perros tratados durante 12 semanas con 30 mg/kg/día mediante una sonda esofágica (exposición sistémica 60 veces mayor que la exposición humana tras la administración de 40 mg al día, basándose en la comparación de las ABC). Se observaron displasia retiniana y pérdida de la retina en perros tratados durante 4 semanas con 90 mg/kg/día mediante una sonda esofágica (exposición sistémica 100 veces mayor que la exposición humana tras la administración de 40 mg al día, basándose en la comparación de las ABC). La administración de dosis ≤30 mg/kg/día (exposición sistémica ≤60 veces que la exposición humana tras la administración de 40 mg al día, basándose en la comparación de las ABC) no produjo hallazgos retinianos después de un periodo de tratamiento de hasta un año.
INDICACIONES TERAPÉUTICAS
Hiperlipidemia y dislipidemia mixta: CRESTOR está indicado como coadyuvante de la dieta para reducir las concentraciones elevadas de C-total, C-LDL, ApoB, C-no HDL y triglicéridos, y para aumentar las concentraciones de C-HDL en adultos con hiperlipidemia primaria o dislipidemia mixta. La dieta pobre en grasas saturadas y colesterol debe complementarse con medicamentos que modifican el perfil de lípidos cuando ha sido inadecuada la respuesta a la dieta y a intervenciones no farmacológicas por sí solas.
Pacientes pediátricos de 10 a 17 años con hipercolesterolemia familiar heterocigota: CRESTOR está indicado como coadyuvante de la dieta para reducir las concentraciones de C-total, C-LDL y ApoB en adolescentes de 10 a 17 años con hipercolesterolemia familiar heterocigota, de ambos sexos, al menos un año después de la menarquia en el caso de las mujeres, siempre que se observen las siguientes condiciones después de un intento adecuado de intervención dietética: C-LDL > 190 mg/dl, o bien, C-LDL > 160 mg/dl junto con antecedentes familiares de enfermedad cardiovascular prematura o dos o más factores de riesgo cardiovasculares.
Disbetalipoproteinemia primaria (hiperlipoproteinemia de tipo III): CRESTOR está indicado como coadyuvante de la dieta para el tratamiento de la disbetalipoproteinemia primaria (hiperlipoproteinemia de tipo III).
Hipercolesterolemia familiar homocigota: CRESTOR está indicado como coadyuvante de otros tratamientos hipolipemiantes (p. ej., aféresis de LDL), o sólo cuando no están disponibles tales tratamientos, para reducir las concentraciones de C-LDL, C-total y ApoB en adultos con hipercolesterolemia familiar homocigota.
Retraso de la progresión de la aterosclerosis: CRESTOR está indicado como coadyuvante de la dieta, para retrasar la progresión de la aterosclerosis en adultos en el marco de una estrategia terapéutica destinada a reducir el C-total y el C-LDL hasta las concentraciones deseadas.
Prevención primaria de la enfermedad cardiovascular: En los individuos que no presentan manifestaciones clínicas de cardiopatía coronaria, pero que tienen un riesgo elevado de enfermedad cardiovascular debido a su edad (≥50 años en los varones o ≥60 años en las mujeres), una concentración de hsCRP ≥2 mg/l y la presencia de al menos un factor de riesgo cardiovascular adicional (hipertensión, baja concentración de C-HDL, tabaquismo o antecedentes familiares de cardiopatía coronaria prematura),
CRESTOR está indicado para:
• Reducir el riesgo de accidente vascular cerebral.
• Reducir el riesgo de infarto de miocardio.
• Reducir el riesgo de procedimientos de revascularizacion arterial.
Limitaciones de uso: CRESTOR no se ha investigado en las dislipidemias de tipos I y V según la clasificación de Fredrickson.
INFORMACIÓN PARA EL PACIENTE
Efectos osteomusculares: Se debe advertir a los pacientes que señalen inmediatamente a su médico cualquier dolor, sensibilidad, o debilidad musculares inexplicables, especialmente si se acompañan de malestar general o fiebre.
Coadministración de antiácidos: Al combinar CRESTOR con un antiácido que contiene una asociación de hidróxido de aluminio e hidróxido de magnesio, el antiácido debe tomarse al menos 2 horas después de CRESTOR.
Embarazo: Si una paciente se embaraza durante el tratamiento, deben tenerse en cuenta los riesgos potenciales para el feto y el hecho de que no se conocen beneficios clínicos de continuar el tratamiento durante el embarazo.
Enzimas hepáticas: Se recomienda efectuar pruebas de la función hepática antes de empezar el tratamiento y 12 semanas después de empezarlo o de aumentar la dosis, y luego periódicamente (p. ej., cada semestre).
Plazo de caducidad: Véase la fecha de caducidad en la caja de cartón externa.
Precauciones de conservación especiales: No conservar a más de 30 °C.
Tamaño del envase: Véase el tamaño del envase en la caja de cartón externa.
AstraZeneca UK Limited, Macclesfield, Cheshire, Reino Unido.
Fabricado por:
IPR Pharmaceuticals Inc. Puerto Rico.
CRESTOR es una marca
registrada del grupo AstraZeneca
CONTRAINDICACIONES
CRESTOR está contraindicado en las siguientes situaciones:
• Hipersensibilidad conocida a uno de los componentes de este producto. Se han observado reacciones de hipersensibilidad con CRESTOR que han incluido exantema, prurito, urticaria y edema angioneurótico (ver Reacciones adversas).
• Enfermedad hepática activa, que puede incluir elevaciones persistentes e inexplicables de las transaminasas hepáticas (ver Advertencias y precauciones especiales de uso).
• Embarazo o posibilidad de embarazo. Dado que los inhibidores de la HMG-CoA reductasa disminuyen la síntesis del colesterol y posiblemente de otras sustancias derivadas del colesterol que ejercen una actividad biológica, CRESTOR puede causar lesiones fetales si se administra a mujeres embarazadas. Además, el tratamiento no parece tener efectos beneficiosos durante el embarazo y no se ha establecido su inocuidad en mujeres embarazadas. Si una paciente se embaraza durante el tratamiento, deben tenerse en cuenta los riesgos potenciales para el feto y el hecho de que no se conocen beneficios clínicos de continuar el tratamiento durante el embarazo (ver Uso en grupos específicos de pacientes y Toxicología preclínica).
• Lactancia. Dado que otros medicamentos de esta clase se secretan en la leche materna y que los inhibidores de la HMG-CoA reductasa pueden provocar reacciones adversas graves en los lactantes, se debe aconsejar a las mujeres que necesitan un tratamiento con CRESTOR que no amamanten a sus bebés (ver Uso en grupos específicos de pacientes).
REACCIONES ADVERSAS
Las siguientes reacciones adversas son objeto de comentarios detallados en otras secciones de la información de prescripción:
• Rabdomiólisis con mioglobinuria e insuficiencia renal aguda y miopatía (incluyendo miositis) (ver Advertencias y precauciones especiales de uso).
• Anomalías de las enzimas hepáticas (ver Advertencias y precauciones especiales de uso).
En la base de datos de los estudios clínicos controlados de CRESTOR (estudios controlados con placebo o con un fármaco de referencia) que incluyeron 5394 pacientes que recibieron el tratamiento durante un período medio de 15 semanas, el 1,4% de los pacientes suspendieron el tratamiento debido a reacciones adversas. A continuación figuran las reacciones adversas más frecuentes que condujeron a la suspensión del tratamiento:
• Mialgia.
• Dolor abdominal.
• Náuseas.
Las reacciones adversas más frecuentes (incidencia ≥2%) que figuran en la base de datos de los estudios clínicos controlados de CRESTOR que incluyeron 5394 pacientes, fueron:
• Cefalea.
• Mialgia.
• Dolor abdominal.
• Astenia.
• Náuseas.
Experiencia acumulada durante los estudios clínicos: Ya que los estudios clínicos se realizan en condiciones muy variables, la incidencia de las reacciones adversas observadas en los estudios clínicos de un medicamento no pueden compararse directamente con las incidencias registradas en estudios clínicos de otro medicamento, y pueden diferir de las incidencias observadas en la práctica clínica.
La Tabla 1 presenta las reacciones adversas que se notificaron en ≥2% de los pacientes, con una incidencia superior a la que se observó con el placebo en los estudios clínicos controlados con placebo. Estos estudios tuvieron una duración de hasta 12 semanas.
Tabla 1. Reacciones adversas* notificadas en ≥2% de los pacientes tratados con CRESTOR, con una incidencia superior a la observada con el placebo en los estudios controlados con placebo (% de pacientes)
|
Reacciones adversas |
CRESTOR |
CRESTOR |
CRESTOR |
CRESTOR |
Total CRESTOR |
Placebo |
|
Cefalea |
5,5 |
4,9 |
3,1 |
8,5 |
5,5 |
5,0 |
|
Náuseas |
3,8 |
3,5 |
6,3 |
0 |
3,4 |
3,1 |
|
Mialgia |
3,1 |
2,1 |
6,3 |
1,9 |
2,8 |
1,3 |
|
Astenia |
2,4 |
3,2 |
4,7 |
0,9 |
2,7 |
2,6 |
|
Estreñimiento |
2,1 |
2,1 |
4,7 |
2,8 |
2,4 |
2,4 |
|
* Reacciones adversas clasificadas por término preferido COSTART. |
||||||
Las otras reacciones adversas notificadas en los estudios clínicos consistieron en dolor abdominal, mareo, hipersensibilidad (que puede incluir exantema, prurito, urticaria y edema angioneurótico) y pancreatitis. También se han notificado las siguientes anomalías de laboratorio: proteinuria (determinada por resultados positivos de la prueba con tira reactiva) y hematuria microscópica (ver Advertencias y precauciones de uso), elevación de creatinfosfocinasa, transaminasas, glucosa, glutamil-transpeptidasa, fosfatasa alcalina y bilirrubina, así como anomalías de la función tiroidea.
En el estudio METEOR en el que participaron 981 pacientes tratados con 40 mg de rosuvastatina (n = 700) o un placebo (n = 281), y cuya duración media fue de 1,7 años, el 5,6% de los sujetos tratados con CRESTOR frente al 2,8% de los que recibieron un placebo suspendieron el tratamiento debido a reacciones adversas. Las reacciones adversas más frecuentes que condujeron a la suspensión del tratamiento consistieron en mialgia, elevación de las enzimas hepáticas, cefalea y náuseas (ver Estudios clínicos).
La Tabla 2 muestra las reacciones adversas notificadas en ≥2% de los pacientes, con una incidencia superior a la observada con el placebo.
Tabla 2. Reacciones adversas* notificadas en ≥2% de los pacientes tratados con CRESTOR, con una incidencia superior a la observada con el placebo en el estudio METEOR (% de pacientes)
|
Reacciones adversas |
CRESTOR 40 mg |
Placebo |
|
Mialgia |
12,7 |
12,1 |
|
Artralgia |
10,1 |
7,1 |
|
Cefalea |
6,4 |
5,3 |
|
Mareo |
4,0 |
2,8 |
|
Elevación de CPK |
2,6 |
0,7 |
|
Dolor abdominal |
2,4 |
1,8 |
|
ALT >3 x LSN† |
2,2 |
0,7 |
|
* Reacciones adversas clasificadas por término preferido del MedDRA. † Frecuencia registrada como un valor anormal de laboratorio. |
||
En el estudio JUPITER, 17802 participantes recibieron 20 mg de rosuvastatina (n = 8901) o un placebo (n = 8901) durante un período de 2 años en promedio. La proporción de pacientes que abandonaron el tratamiento del estudio debido a una reacción adversa, independientemente de su relación causal con el tratamiento, fue mayor en el grupo de la rosuvastatina (6,6%) que en el grupo placebo (6,2%). La mialgia fue la reacción adversa más frecuente que provocó la suspensión del tratamiento.
En el estudio JUPITER, la frecuencia de diabetes fue significativamente mayor en el grupo de la rosuvastatina (2,8%) que en el grupo placebo (2,3%). La concentración media de HbA1c aumento significativamente (un 0,1%) en el grupo de la rosuvastatina frente al grupo placebo. El número de pacientes con HbA1c >6,5% al final del estudio fue significativamente mayor en el grupo de la rosuvastatina que en el grupo placebo (ver Advertencias y precauciones especiales de uso y Estudios clínicos).
La Tabla 3 muestra la lista de reacciones adversas notificadas en ≥2% de los pacientes, con una incidencia superior a la observada con el placebo.
Tabla 3. Reacciones adversas* notificadas en ≥2% de los pacientes tratados con CRESTOR, con una incidencia superior a la observada con el placebo en el estudio JUPITER
(% de pacientes)
|
Reacciones adversas |
CRESTOR 20 mg |
Placebo |
|
Mialgia |
7,6 |
6,6 |
|
Artralgia |
3,8 |
3,2 |
|
Estreñimiento |
3,3 |
3,0 |
|
Náuseas |
2,4 |
2,3 |
|
* Reacciones adversas que ocurrieron durante el tratamiento, clasificadas por término preferido del diccionario MedDRA. |
||
Pacientes pediátricos de 10 a 17 años: En un estudio controlado de 12 semanas que se llevó a cabo en adolescentes (varones y mujeres posmenárquicas), el perfil de seguridad y tolerabilidad de CRESTOR (de 5 a 20 mg al día) fue generalmente similar al del placebo (ver Estudios clínicos y Uso en grupos específicos de pacientes: Uso pediátrico).
Sin embargo, la frecuencia de elevaciones de las concentraciones séricas de creatinfosfocinasa (CK) >10 x LSN fue mayor en el grupo de la rosuvastatina que en el grupo placebo. Cuatro de 130 adolescentes (3%) tratados con la rosuvastatina (2 con 10 mg y 2 con 20 mg) presentaron elevaciones de la CK > 10 x LSN, frente a ninguno de los 46 adolescentes que recibieron el placebo.
Experiencia adquirida durante la farmacovigilancia: Desde su aprobación, se han identificado las siguientes reacciones con CRESTOR: artralgia, hepatitis, ictericia y pérdida de memoria. Dado que estas reacciones se notifican espontáneamente y que se desconoce el tamaño exacto de la población, no siempre es posible estimar de manera fiable su frecuencia o establecer una relación de causa y efecto con la exposición al medicamento.
INTERACCIONES FARMACOLÓGICAS
Ciclosporina: La ciclosporina aumentó de manera significativa la exposición a la rosuvastatina. No se recomienda la asociación de la ciclosporina con dosis de CRESTOR de 10 a 40 mg (ver Posología y forma de administración, Advertencias y precauciones especiales de uso y Farmacología clínica).
Gemfibrozilo: El gemfibrozilo aumentó de manera significativa la exposición a la rosuvastatina. Por lo tanto, debe evitarse la asociación de CRESTOR con el gemfibrozilo. Si aún así se utiliza, la dosis de CRESTOR debe limitarse a 10 mg una vez al día (ver Posología y forma de administración y Farmacología clínica).
Inhibidores de la proteasa: La coadministración de la rosuvastatina con ciertos inhibidores de la proteasa combinados con el ritonavir puede tener diferentes efectos en la exposición a la rosuvastatina. Las asociaciones de inhibidores de la proteasa lopinavir/ritonavir y atazanavir/ritonavir aumentan hasta 3 veces la exposición a la rosuvastatina (ABC) (ver Tabla 3 Farmacología clínica). En caso de coadministración con estas asociaciones, la dosis de CRESTOR debe limitarse a 10 mg. Las asociaciones de tipranavir/ritonavir o fosamprenavir/ritonavir producen poca o ninguna variación de la exposición a la rosuvastatina. Debe tenerse precaución al coadministrar la rosuvastatina con inhibidores de la proteasa combinados con el ritonavir (ver Posología y forma de administración, Advertencias y precauciones especiales de uso y Farmacología clínica).
Anticoagulantes cumarínicos: CRESTOR aumentó de manera significativa el INR en pacientes tratados con anticoagulantes cumarínicos. Por lo tanto, debe tenerse precaución al coadministrar anticoagulantes cumarínicos con CRESTOR. En los pacientes que toman anticoagulantes cumarínicos y CRESTOR en forma concomitante, debe determinarse el INR antes de empezar el tratamiento con CRESTOR y con una frecuencia suficiente al principio del mismo para asegurarse de que no se produzca una modificación importante del INR (ver Posología y forma de administración y Farmacología clínica).
Niacina: La coadministración de CRESTOR y niacina puede elevar el riesgo de efectos osteomusculares; en esta situación clínica debe considerarse una reducción de la dosis de CRESTOR (ver Advertencias y precauciones especiales de uso).
Fenofibrato: La coadministracion de CRESTOR y fenofibrato no produjo aumentos de importancia clínica del ABC de la rosuvastatina o del fenofibrato. Conviene sopesar cuidadosamente los beneficios de las modificaciones adicionales de las concentraciones de lípidos que se consiguen asociando CRESTOR con fibratos, frente a los riesgos potenciales que plantea esta asociación farmacológica (ver Posología y forma de administración y Farmacología clínica).
ESTUDIOS CLÍNICOS
Hiperlipidemia y dislipidemia mixta: CRESTOR reduce las concentraciones de C-total, C-LDL, ApoB, C-no HDL y TG, y aumenta las de C-HDL en pacientes adultos con hiperlipidemia y dislipidemia mixta.
• Estudio de búsqueda de la dosis terapéutica: En un estudio multicéntrico de búsqueda de la dosis, con un diseño doble ciego, controlado con placebo, que se llevó a cabo en pacientes con hiperlipidemia, el tratamiento con una sola dosis diaria de CRESTOR durante 6 semanas redujo significativamente las concentraciones de C-total, C-LDL, C-no HDL y ApoB con todas las dosis examinadas (Tabla 6).
Tabla 6. Relación dosis-respuesta en pacientes con hiperlipidemia (media ajustada del porcentaje de variación entre los valores iniciales y los de la semana 6)
|
Dosis |
N |
C-total |
C-LDL |
C-no HDL |
ApoB |
TG |
C-HDL |
|
Placebo |
13 |
-5 |
-7 |
-7 |
-3 |
-3 |
3 |
|
5 mg de CRESTOR |
17 |
-33 |
-45 |
-44 |
-38 |
-35 |
13 |
|
10 mg de CRESTOR |
17 |
-36 |
-52 |
-48 |
-42 |
-10 |
14 |
|
20 mg de CRESTOR |
17 |
-40 |
-55 |
-51 |
-46 |
-23 |
8 |
|
40 mg de CRESTOR |
18 |
-46 |
-63 |
-60 |
-54 |
-28 |
10 |
• Estudio controlado con un fármaco de referencia: CRESTOR se comparó con tres inhibidores de la HMG-CoA reductasa (atorvastatina, simvastatina y pravastatina) en un estudio multicéntrico de búsqueda de la dosis con un diseño abierto en el que participaron 2240 pacientes con hiperlipidemia o dislipidemia mixta. Después de la aleatorización, los pacientes recibieron durante 6 semanas una sola dosis diaria de CRESTOR, atorvastatina, simvastatina o pravastatina (Figura 1 y Tabla 7).
Figura 1. Porcentaje de variación del C-LDL por dosis de CRESTOR, atorvastatina, simvastatina y pravastatina después de 6 semanas en pacientes con hiperlipidemia o dislipidemia mixta
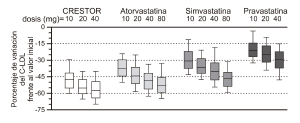
Los cuadros representan los percentiles 25, 50 y 75 y los bigotes los percentiles 10 y 90.
Media inicial de C-LDL = 189 mg/dL.
Tabla 7. Porcentaje de variación del C-LDL entre el valor inicial y la semana 6 (medias de mínimos cuadrados*) por grupo de tratamiento (muestras de 156-167 pacientes por grupo)
|
Dosis diaria del tratamiento |
||||
|
Tratamiento |
10 mg |
20 mg |
40 mg |
80 mg |
|
CRESTOR |
-46† |
-52‡ |
-55§ |
--- |
|
Atorvastatina |
-37 |
-43 |
-48 |
-51 |
|
Simvastatina |
-28 |
-35 |
-39 |
-46 |
|
Pravastatina |
-20 |
-24 |
-30 |
--- |
|
* Los errores estándares correspondientes son de aproximadamente 1,00. † El tratamiento con 10 mg de CRESTOR redujo las concentraciones de C-LDL significativamente más que 10 mg de atorvastatina, 10 mg, 20 mg y 40 mg de pravastatina, y 10 mg, 20 mg y 40 mg de simvastatina (p< 0,002). ‡ El tratamiento con 20 mg de CRESTOR redujo las concentraciones de C-LDL significativamente más que 20 mg y 40 mg de atorvastatina, 20 mg y 40 mg de pravastatina, y 20 mg, 40 mg y 80 mg de simvastatina (p<0,002). § El tratamiento con 40 mg de CRESTOR redujo las concentraciones de C-LDL significativamente más que 40 mg de atorvastatina, 40 mg de pravastatina, y 40 mg y 80 mg de simvastatina (p<0,002). |
||||
Hipercolesterolemia familiar heterocigota:
• Estudio controlado con un fármaco de referencia: En un estudio realizado en pacientes con HF heterocigota (media inicial de las concentraciones de LDL: 291), los pacientes se distribuyeron al azar en dos grupos: 20 mg de CRESTOR o 20 mg de atorvastatina. La dosis se incrementó cada 6 semanas. Todas las dosis produjeron reducciones significativas del C-LDL con respecto a los valores iniciales en ambos grupos de tratamiento (Tabla 8).
Tabla 8. Media del porcentaje de variación de la concentración de C-LDL con respecto al valor inicial
|
CRESTOR (n=435) |
Atorvastatina (n=187) |
||
|
Semana 6 |
20 mg |
-47% (-49%, -46%) |
-38% (-40%, -36%) |
|
Semana 12 |
40 mg |
-55% (-57%, -54%) |
-47% (-49%, -45%) |
|
Semana 18 |
80 mg |
NA |
-52% (-54%, -50%) |
|
* Media MC = Media de mínimos cuadrados ajustada en función del valor inicial del C-LDL. |
|||
Disbetalipoproteinemia primaria (hiperlipoproteinemia de tipo III): En un estudio multicéntrico con un diseño aleatorizado, doble ciego y cruzado, 32 pacientes (27 con mutación ?2/?2 y 4 con mutación de apo E [Arg145Cys]) con disbetalipoproteinemia (hiperlipoproteinemia de tipo III) ingresaron en un período de introducción de 6 semanas durante el cual siguieron la dieta de modificación terapéutica del modo de vida del NCEP. Al cabo de este período de introducción con intervención dietética, los pacientes fueron distribuidos al azar entre dos secuencias de tratamiento en complemento de la dieta durante un período de 6 semanas cada uno: 10 mg de rosuvastatina seguidos de 20 mg de rosuvastatina, o 20 mg de rosuvastatina seguidos de 10 mg de rosuvastatina. CRESTOR redujo el C-no HDL (variable principal) y las concentraciones de lipoproteínas circulantes residuales. Los resultados se presentan en la tabla a continuación.
Tabla 9. Efectos modificadores de lípidos de la rosuvastatina (10 mg y 20 mg) en la disbetalipoproteinemia primaria (hiperlipoproteinemia de tipo III) después de seis semanas en función de la mediana del porcentaje de variación (IC del 95%) con respecto al valor inicial (N = 32).
|
Mediana inicial (mg/dl) |
Mediana del porcentaje de variación respecto al valor inicial (IC del 95%) |
Mediana del porcentaje de variación respecto al valor inicial (IC del 95%) |
|
|
C-total |
342,5 |
-43,3 |
-47,6 |
|
Triglicéridos |
503,5 |
-40,1 |
-43,0 |
|
C-no HDL |
294,5 |
-48,2 |
-56,4 |
|
C-VLDL + C-IDL |
209,5 |
-46,8 |
-56,2 |
|
C-LDL |
112,5 |
-54,4 |
-57,3 |
|
C-HDL |
35,5 |
10,2 |
11,2 |
|
C-LPR |
82,0 |
-56,4 |
-64,9 |
|
Apo E |
16,0 |
-42,9 |
-42,5 |
Hipercolesterolemia familiar homocigota:
• Estudio de ajuste de la dosis: En un estudio con un diseño abierto y con ajuste forzoso de la dosis, se evaluó la respuesta de pacientes con HF homocigota (n = 40, 8-63 años) al tratamiento con dosis de CRESTOR de 20 a 40 mg ajustadas cada 6 semanas. En la población total, la reducción media del C-LDL fue del 22% con respecto al valor inicial. Alrededor de un tercio de los pacientes obtuvieron beneficios con el aumento de la dosis de 20 mg a 40 mg, pues consiguieron una reducción adicional del C-LDL de más del 6%. En los 27 pacientes que consiguieron una reducción del 15% o más del C-LDL, la reducción media fue del 30% (mediana del 28%). De los 13 pacientes que consiguieron una reducción del C-LDL <15%, 3 no presentaron ningún cambio o un aumento del C-LDL. Se observaron reducciones del C-LDL del 15% o más en 3 de 5 pacientes con receptores negativos.
Pacientes pediátricos con hipercolesterolemia familiar heterocigota: En un estudio multicéntrico con un diseño aleatorizado, doble ciego, controlado con placebo de 12 semanas de duración, 176 niños y adolescentes (97 varones y 79 mujeres) con hipercolesterolemia familiar heterocigota recibieron 5, 10 o 20 mg de rosuvastatina al día o un placebo. La edad de los pacientes oscilaba entre 10 y 17 años (mediana de 14 años), alrededor del 30% tenía entre 10 y 13 años y aproximadamente el 17%, 18%, 40% y 25% correspondían a los estadios II, III, IV y V, respectivamente, según la clasificación de Tanner. Las mujeres debían haber tenido su primera menstruación al menos 1 año antes. La media del C-LDL inicial fue de 233 mg/dl (mínimo 129, máximo 399). Tras la fase de 12 semanas con un diseño doble ciego, tuvo lugar una fase de ajuste de la dosis sin enmascaramiento de 40 semanas en la que todos los pacientes (n = 173) recibieron 5 mg, 10 mg o 20 mg diarios de rosuvastatina.
La rosuvastatina redujo significativamente las concentraciones de C-LDL (variable principal), colesterol total y ApoB con cada dosis frente al placebo. Los resultados figuran en la Tabla 10 a continuación.
Tabla 10. Efectos modificadores de lípidos de la rosuvastatina en pacientes pediátricos de 10 a 17 años con hipercolesterolemia familiar heterocigota (media de mínimos cuadrados del porcentaje de variación entre el valor inicial y la semana 12)
|
Dosis (mg) |
N |
C-LDL |
C-HDL |
C-total |
TGa |
ApoB |
|
Placebo |
46 |
-1% |
+7% |
0% |
-7% |
-2% |
|
5 |
42 |
-38% |
+4%b |
-30% |
-13%b |
-32% |
|
10 |
44 |
-45% |
+11%b |
-34% |
-15%b |
-38% |
|
20 |
44 |
-50% |
+9%b |
-39% |
-16%b |
-41% |
|
a. Mediana del porcentaje de variación. b. La diferencia con respecto al placebo no fue estadísticamente significativa. |
||||||
Al final del período de tratamiento de 12 semanas con un diseño doble ciego, el porcentaje de pacientes que alcanzaron el objetivo de C-LDL < 110 mg/dl (2,8 mmol/l) fue del 0% con el placebo, del 12% con 5 mg de rosuvastatina, del 41% con 10 mg de rosuvastatina y del 41% con 20 mg de rosuvastatina. Para la fase de 40 semanas sin enmascaramiento, el tratamiento del 71% de los pacientes se ajustó hasta la dosis máxima de 20 mg y el 41% de los pacientes alcanzaron el objetivo de C-LDL < 110 mg/dl.
No se ha demostrado la eficacia a largo plazo del tratamiento con la rosuvastatina, iniciado durante la niñez para reducir la morbimortalidad a la edad adulta.
Retraso de la progresión de la aterosclerosis: En el estudio METEOR (medición de los efectos del tratamiento con 40 mg de rosuvastatina en el espesor de las túnicas íntima y media), el efecto del tratamiento con CRESTOR en la aterosclerosis de la carótida se evaluó por ecografía en modo B en pacientes con concentraciones elevadas de C-LDL, con un bajo riesgo de cardiopatía coronaria sintomática (riesgo de Framingham <10% en diez años) y con aterosclerosis subclínica confirmada por la medición del espesor de las túnicas íntima y media de la arteria carótida (ETIMAC). En este estudio clínico controlado con placebo, con un diseño doble ciego, 984 pacientes fueron aleatorizados (y 876 analizados) en una proporción de 5:2 entre el tratamiento con 40 mg de CRESTOR o un placebo una vez al día. Las ecografías de las paredes de la carótida se emplearon para determinar la variación anualizada por paciente del ETIMAC de 12 segmentos entre los valores iniciales y los valores medidos después de dos años. La diferencia estimada entre las velocidades de variación del ETIMAC máximo anualizado de los 12 segmentos de la arteria carótida entre el grupo tratado con CRESTOR y el grupo placebo fue de –0,00145 mm/año (IC del 95%: –0,0196, –0,0093; p<0,0001).
La velocidad de variación anualizada en el grupo placebo fue de +0,0131 mm/año (p<0,0001), mientras que en el grupo tratado con CRESTOR fue de -0,0014 mm/año (p=0,32).
Si consideramos los pacientes individuales del grupo tratado con CRESTOR, el 52,1% no mostró ninguna progresión de la enfermedad (velocidad de variación anualizada negativa), frente al 37,7% de los pacientes del grupo placebo.
Prevención primaria de la enfermedad cardiovascular: El estudio JUPITER (justificación del uso de estatinas para la prevención primaria: un estudio de intervención para evaluar la rosuvastatina) evaluó el efecto de CRESTOR (rosuvastatina cálcica) en la incidencia de acontecimientos cardiovasculares graves en 17 802 pacientes (varones≥50 años y mujeres ≥60 años) sin manifestaciones clínicas de enfermedad cardiovascular, con concentraciones de C-LDL < 130 mg/dl (3,3 mmol/l) y concentraciones de hsCRP ≥2 mg/d. Según los criterios de riesgo de Framingham, la población del estudio tenía un riesgo inicial de cardiopatía coronaria estimado del 11,6% a 10 años e incluía una proporción importante de pacientes con factores de riesgo adicionales, tales como hipertensión (58%), bajas concentraciones de C-HDL (23%), tabaquismo (16%) o antecedentes familiares de cardiopatía coronaria prematura (12%). Las medianas iniciales de las concentraciones de C-LDL y de hsCRP de los participantes en el estudio eran de 108 mg/dl y 4,3 mg/l, respectivamente. Los participantes del estudio fueron distribuidos al azar entre el placebo (n = 8901) y el tratamiento con 20 mg de rosuvastatina una vez al día (n = 8901); el período de observación duró 2 años en promedio. El Consejo de Supervisión de los datos de seguridad decidió suspender prematuramente el estudio JUPITER tras haberse cumplido los criterios predefinidos de suspensión al confirmarse la eficacia del tratamiento con la rosuvastatina.
Como criterio de valoración principal se utilizó una variable compuesta del tiempo transcurrido hasta la primera ocurrencia de uno de los siguientes acontecimientos cardiovasculares graves: muerte de origen cardiovascular, infarto de miocardio no mortal, accidente vascular cerebral no mortal, hospitalización debida a angina de pecho inestable o procedimiento de revascularización arterial.
La rosuvastatina redujo significativamente el riesgo de acontecimientos cardiovasculares graves (252 acontecimientos en el grupo placebo frente a 142 en el grupo de la rosuvastatina), con una reducción estadísticamente significativa del 44% del riesgo relativo (p<0,001) y una reducción del riesgo absoluto del 1,2% (ver Figura 2). La reducción del riesgo correspondiente a la variable principal fue uniforme en todos los subgrupos predefinidos basados en los siguientes criterios: edad, sexo, raza, tabaquismo, antecedentes familiares de cardiopatía coronaria prematura, índice de masa corporal y concentraciones de C-LDL, C-HDL y hsCRP.
Figura 2. Tiempo hasta la primera ocurrencia de un acontecimiento cardiovascular grave en el estudio JUPITER
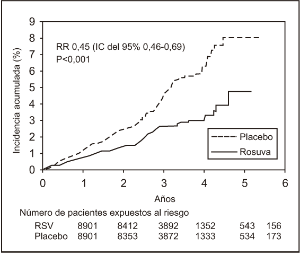
Los componentes individuales de la variable principal se presentan en la Figura 3. La rosuvastatina redujo significativamente el riesgo de infarto de miocardio no mortal, de accidente vascular cerebral no mortal y de procedimientos de revascularización arterial. No hubo diferencias significativas entre los grupos de la rosuvastatina y del placebo en la mortalidad de origen cardiovascular ni en la hospitalización debida a angina de pecho inestable.
La rosuvastatina redujo significativamente el riesgo de infarto de miocardio (6 acontecimientos mortales y 62 no mortales en el grupo placebo frente a 9 acontecimientos mortales y 22 no mortales en el grupo tratado con la rosuvastatina), así como el riesgo de accidente vascular cerebral (6 acontecimientos mortales y 58 no mortales en el grupo placebo frente a 3 acontecimientos mortales y 30 no mortales en el grupo tratado con la rosuvastatina).
En un análisis de subgrupos post hoc de los sujetos del estudio JUPITER (n = 1405, rosuvastatina 725, placebo 680) con concentraciones de hsCRP ≥2 mg/l y ningún otro factor de riesgo tradicional aparte de la edad (tabaquismo, PA ≥140/90 o tratamiento antihipertensivo, bajas concentraciones de C-HDL), tras un ajuste para tomar en cuenta las concentraciones elevadas de C-HDL, no se observó un beneficio significativo del tratamiento con la rosuvastatina.
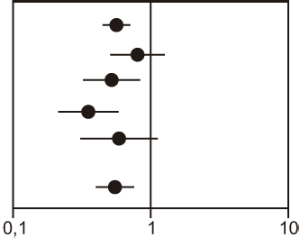
Figura 3. Acontecimientos cardiovasculares graves por grupo de tratamiento en el estudio JUPITER
|
Variable |
Número de acontecimientos |
RR (IC del 95%) |
Valor de p |
|||
|
Rosuva 20 mg |
Placebo 20 mg |
Razón de riesgos |
(–– 95% ––) |
|||
|
Variable principal |
142 (7,6) |
252 (13,6) |
0,56 (0,46, 0,69) |
<0,001 |
|
|
|
Muerte de origen cardiovascular** |
35 (1,9) |
44 (2,4) |
0,80 (0,51, 1,24) |
0,315 |
||
|
Accidente vascular cerebral no mortal |
30 (1,6) |
58 (3,1) |
0,52 (0,33, 0,80) |
0,003 |
||
|
IM no mortal |
22 (1,2) |
62 (3,3) |
0,35 (0,22, 0,58) |
<0,001 |
||
|
Hospitalización debida a angina inestable |
16 (0,9) |
27 (1,5) |
0,59 (0,32, 1,10) |
0,093 |
||
|
Revascularización arterial |
71 (3,8) |
131 (7,1) |
0,54 (0,41, 0,72) |
<0,001 |
||
|
* Incidencia de acontecimientos por 1000 años-paciente. ** La muerte de origen cardiovascular incluyó: IM mortal, accidente vascular cerebral mortal, muerte súbita y otras causas de muerte cardiovascular. |
||||||
Después de 1 año, la rosuvastatina aumentó las concentraciones séricas de C-HDL y redujo las de C-LDL, hsCRP, colesterol total y triglicéridos (todas las variables: p<0,001 frente al placebo).
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE USO
Efectos osteomusculares: Se han notificado casos de miopatía y rabdomiólisis con insuficiencia renal aguda secundaria a una mioglobinuria con los inhibidores de la HMG-CoA reductasa, incluyendo CRESTOR. Estos riesgos pueden aparecer con cualquier dosis, pero son mayores con la dosis máxima (40 mg).
CRESTOR debe recetarse con precaución en pacientes con factores de predisposición a la miopatía (p. ej., edad ≥65 años, hipotiroidismo mal tratado, insuficiencia renal).
El riesgo de miopatía durante el tratamiento con CRESTOR puede aumentar si se coadministran otros tratamientos hipolipemiantes (fibratos o niacina), gemfibrozilo, ciclosporina, o las asociaciones lopinavir/ritonavir o atazanavir/ritonavir (ver Posología y forma de administración e Interacciones farmacológicas).
El tratamiento con CRESTOR debe suspenderse si se producen elevaciones pronunciadas de las concentraciones de creatincinasa o si se diagnostica o se sospecha de una miopatía. El tratamiento con CRESTOR también debe interrumpirse temporalmente en cualquier paciente que presente un estado agudo y grave que indique una miopatía o que predisponga a una insuficiencia renal secundaria a una rabdomiólisis (p. ej., sepsis, hipotensión, deshidratación, cirugía importante, traumatismo, trastornos metabólicos, endocrinos y electrolíticos graves o convulsiones no controladas). Se debe advertir a todos los pacientes que señalen inmediatamente cualquier dolor, sensibilidad o debilidad muscular inexplicable, especialmente si van acompañados de malestar general o fiebre.
Anomalías y control de las enzimas hepáticas: Se recomienda efectuar determinaciones de las enzimas hepáticas antes de empezar el tratamiento y 12 semanas después de empezarlo o de aumentar la dosis, y luego periódicamente (p. ej., cada semestre).
Se han notificado aumentos de las concentraciones séricas de transaminasas [AST (TGO) o ALT (TGP)] con los inhibidores de la HMG-CoA reductasa, entre ellos CRESTOR. En la mayoría de los casos, las elevaciones fueron transitorias y se resolvieron o mejoraron sin interrumpir el tratamiento o después de una breve interrupción del mismo. Se registraron dos casos de ictericia en los que no pudo determinarse la relación con CRESTOR y que se resolvieron después de interrumpir el tratamiento. En estos estudios no se observaron casos de insuficiencia hepática o de hepatopatía irreversible.
En un análisis combinado de los estudios controlados con placebo, se produjeron aumentos de las concentraciones séricas de transaminasas a más de 3 veces el límite superior normal en el 1,1% de los pacientes tratados con CRESTOR frente al 0,5% de los pacientes que recibieron un placebo.
Los pacientes que presentan elevaciones de las transaminasas deben vigilarse de cerca hasta la resolución de las anomalías. Si persiste una elevación de la ALT o AST >3 x LSN, se recomienda reducir la dosis o interrumpir el tratamiento con CRESTOR.
CRESTOR debe emplearse con precaución en los pacientes que consumen cantidades importantes de alcohol o que tienen antecedentes hepáticos crónicos (ver Farmacología clínica). La presencia de una enfermedad hepática activa, que puede incluir elevaciones persistentes e inexplicables de las transaminasas, constituye una contraindicación al uso de CRESTOR (ver Contraindicaciones).
Tratamiento concomitante con anticoagulantes cumarínicos: Debe tenerse precaución al coadministrar anticoagulantes con CRESTOR debido a la potenciación de los efectos de prolongación del tiempo de protrombina/INR de los anticoagulantes cumarínicos. En los pacientes que toman anticoagulantes cumarínicos y CRESTOR en forma concomitante, debe determinarse el INR antes de empezar el tratamiento con CRESTOR y con una frecuencia suficiente al principio del mismo para asegurarse de que no se produzca una modificación importante del INR (ver Interacciones farmacológicas).
Proteinuria y hematuria: Durante el programa de estudios clínicos de CRESTOR se observaron proteinuria (resultados positivos en la prueba con tira reactiva) y hematuria (examen microscópico) en pacientes tratados con CRESTOR. Estos hallazgos fueron más frecuentes en los pacientes tratados con 40 mg que en los que recibieron dosis más bajas de CRESTOR o inhibidores de la HMG-CoA de comparación, aunque el efecto generalmente fue transitorio y no se acompañó de un deterioro de la función renal. Aunque se desconoce la importancia clínica de este hallazgo, debe considerarse una reducción de la dosis en los pacientes tratados con 40 mg de CRESTOR cuyos análisis de orina regulares muestren proteinuria y/o hematuria persistentes e inexplicables.
Efectos endocrinos: Se han notificado elevaciones de la HbA1c y de la glucemia en ayunas con inhibidores de la HMG-CoA reductasa, lo cual incluye a CRESTOR (ver Reacciones adversas).
Si bien los estudios clínicos demostraron que, por sí solo, CRESTOR no reduce la concentración plasmática basal de cortisol y no altera las reservas suprarrenales, debe tenerse precaución al coadministrar CRESTOR con medicamentos capaces de reducir las concentraciones o la actividad de las hormonas esteroides endógenas, tales como el ketoconazol, la espironolactona y la cimetidina.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
Información general sobre la posología: La dosis de CRESTOR varía de 5 a 40 mg una vez al día por vía oral. La dosis inicial usual es de 10 a 20 mg.
CRESTOR puede administrarse en una sola dosis a cualquier hora del día, con o sin alimentos.
Al empezar el tratamiento con CRESTOR o al reemplazar otro inhibidor de la HMG-CoA reductasa por CRESTOR, debe utilizarse primero la dosis inicial adecuada de CRESTOR y sólo entonces ajustarla en función de la respuesta del paciente y del objetivo individualizado del tratamiento.
Tras iniciar el tratamiento con CRESTOR o modificar la dosis, las concentraciones de lípidos deben determinarse en un plazo de 2 a 4 semanas, ajustando la dosis en consecuencia.
La dosis de 40 mg de CRESTOR se reserva exclusivamente a los pacientes que no han alcanzado su objetivo de C-LDL con la dosis de 20 mg (ver Advertencias y precauciones especiales de uso).
Hipercolesterolemia familiar heterocigota en pacientes pediátricos (de 10 a 17 años): La dosis usual de CRESTOR es de 5 a 20 mg al día y la dosis máxima recomendada es de 20 mg al día (no se han investigado dosis superiores a 20 mg en esta población de pacientes).
Las dosis deben ajustarse individualmente en función del objetivo terapéutico recomendado (ver Farmacología clínica e Indicaciones terapéuticas). Los ajustes de la dosis deben efectuarse en intervalos de 4 semanas o más.
Hipercolesterolemia familiar homocigota: La dosis inicial recomendada de CRESTOR es de 20 mg una vez al día. La evaluación de la respuesta al tratamiento debe basarse en las concentraciones de C-LDL determinadas antes de la aféresis.
Posología en pacientes de origen asiático: En los pacientes de origen asiático debe considerarse una dosis inicial de CRESTOR de 5 mg una vez al día (ver Uso en grupos específicos de pacientes y Farmacología clínica).
Coadministración con la ciclosporina o con las asociaciones lopinavir/ritonavir o atazanavir/ritonavir: En los pacientes que toman ciclosporina, la dosis de CRESTOR debe limitarse a 5 mg una vez al día (ver Advertencias y precauciones especiales de uso e Interacciones farmacológicas). En los pacientes que toman una asociación de lopinavir y ritonavir o de atazanavir y ritonavir, la dosis de CRESTOR debe limitarse a 10 mg una vez al día (ver Advertencias y precauciones especiales de uso e Interacciones farmacológicas).
Tratamiento hipolipemiante concomitante: La asociación de CRESTOR con la niacina o el fenofibrato puede aumentar el riesgo de efectos osteomusculares; en esta situacion clínica, debe considerarse una reducción de la dosis de CRESTOR (ver Advertencias y precauciones especiales de uso e Interacciones farmacológicas).
Debe evitarse la asociación de CRESTOR con el gemfibrozilo porque el uso concomitante de estos dos medicamentos aumenta la exposición a CRESTOR; al asociar CRESTOR con el gemfibrozilo, la dosis de CRESTOR debe limitarse a 10 mg una vez al día (ver Advertencias y precauciones especiales de uso e Interacciones farmacológicas).
Posología en pacientes con insuficiencia renal grave: En los pacientes con insuficiencia renal grave (DEPCr < 30 ml/min/1,73m2) no hemodializados, la dosis de CRESTOR debe ser de 5 mg una vez al día inicialmente, sin superar 10 mg una vez al día (ver Uso en grupos específicos de pacientes y Farmacología clínica).
SOBREDOSIS
No existe un tratamiento específico en caso de sobredosis. El paciente debe recibir un tratamiento sintomático y las medidas de apoyo que convengan. La hemodiálisis no aumenta de manera significativa la depuración de la rosuvastatina.
DESCRIPCIÓN
CRESTOR (rosuvastatina cálcica) es un hipolipemiante sintético para administración oral.
La denominación química de la rosuvastatina cálcica es: sal cálcica del ácido bis[(E)-7-[4-(4-fluorofenil)-6-isopropil-2-[metil(metilsulfonil)amino] pirimidin-5-il](3R,5S)-3,5-dihidroxihept-6-enoico] y su fórmula estructural es la siguiente:
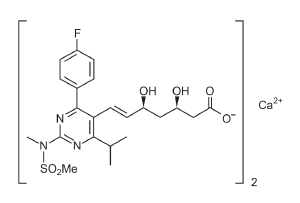
La fórmula empírica de la rosuvastatina cálcica es C22H27FN3O6S)2Ca y su masa molecular es de 1001,14. La rosuvastatina cálcica es un polvo blanco amorfo, bastante soluble en agua y metanol y poco soluble en etanol. Es un compuesto hidrofílico con un coeficiente de partición (octanol/agua) de 0,13 en condiciones de pH de 7,0.
Componentes inactivos: Cada comprimido contiene celulosa microcristalina NF, lactosa monohidratada NF, fosfato de calcio tribásico NF, crospovidona NF, estearato de magnesio NF, hipromelosa NF, triacetina NF, dióxido de titanio USP, óxido de hierro amarillo y óxido de hierro rojo NF.
FARMACOLOGÍA CLÍNICA
Mecanismo de acción: La rosuvastatina es un inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima que convierte la 3-hidroxi-3-metilglutaril coenzima A en mevalonato, un precursor del colesterol, y que limita la síntesis del mismo. Los estudios in vivo en animales y los estudios in vitro en cultivos de células animales y humanas han demostrado que la rosuvastatina se absorbe principalmente en el hígado y que ejerce un efecto selectivo en este órgano efector de la reducción del colesterol. En los estudios in vivo e in vitro, la rosuvastatina modifica el perfil de lípidos mediante dos mecanismos. Primero, aumenta el número de receptores hepáticos de LDL en la superficie celular para promover la captación y el catabolismo de las LDL y, en segundo lugar, inhibe la síntesis hepática de VLDL reduciendo la cantidad total de partículas de VLDL y LDL.
Farmacocinética:
• Absorción: En los estudios de farmacología clínica en el ser humano, las concentraciones plasmáticas máximas de rosuvastatina se alcanzaron de 3 a 5 horas después de la administración oral. La concentración máxima (Cmáx.) y el área bajo la curva de las concentraciones plasmáticas en función del tiempo (ABC) aumentaron de manera aproximadamente proporcional a la dosis de CRESTOR. La biodisponibilidad absoluta de la rosuvastatina es de aproximadamente un 20%.
La administracion de CRESTOR con alimentos no tuvo ningún efecto en el ABC de la rosuvastatina.
El ABC de la rosuvastatina no varía al administrar la rosuvastatina por la mañana o la noche.
• Distribución: El volumen medio de distribución de la rosuvastatina en el estado de equilibrio es de aproximadamente 134 litros. La rosuvastatina se une en un 88% a las proteínas plasmáticas, sobre todo a la albúmina. Esta unión es reversible e independiente de las concentraciones plasmáticas.
• Metabolismo: El metabolismo de la rosuvastatina es limitado; aproximadamente un 10% de una dosis radiactiva se recupera en forma de metabolitos. El principal metabolito es la N-desmetil-rosuvastatina cuya formación es mediada principalmente por la enzima 2C9 del citocromo P450. Los estudios in vitro han demostrado que la N-desmetilrosuvastatina tiene aproximadamente entre una sexta parte y la mitad de la actividad inhibidora de la HMG-CoA reductasa del compuesto original. En total, el compuesto original representa más del 90% de la actividad inhibidora de la HMG-CoA reductasa en el plasma.
• Excreción: Después de la administración oral, la rosuvastatina y sus metabolitos se excretan principalmente en las heces (90%). La vida media de eliminación (t1/2) de la rosuvastatina es de aproximadamente 19 horas.
Después de una dosis intravenosa, alrededor del 28% del medicamento se elimina del organismo por la vía renal y el 72% por la vía hepática.
• Raza: Un análisis farmacocinético poblacional no reveló diferencias farmacocinéticas de importancia clínica entre los grupos de pacientes de raza blanca, hispanoamericanos y de raza negra o afrocaribeña. Sin embargo, los estudios farmacocinéticos, incluido uno realizado en los Estados Unidos, revelaron una elevación de aproximadamente 2 veces de la mediana de la exposición (ABC y Cmáx.) en sujetos de origen asiático respecto al grupo de control de raza blanca.
• Sexo: No se observaron diferencias entre las concentraciones plasmáticas de rosuvastatina de varones y mujeres.
• Pacientes de edad avanzada: No se observaron diferencias entre las concentraciones plasmáticas de rosuvastatina de la población de edad avanzada (edad ≥65 años) y del resto de la población.
• Insuficiencia renal: La insuficiencia renal leve a moderada (depuración de creatinina ≥30 ml/min/1,73 m2) no tuvo ninguna influencia en las concentraciones plasmáticas de rosuvastatina. Sin embargo, las concentraciones plasmáticas de rosuvastatina aumentaron en un grado clínicamente significativo (aproximadamente 3 veces) en los pacientes con insuficiencia renal grave (DEPCr < 30 ml/min/1,73 m2) no hemodializados con respecto a voluntarios sanos (DEPCr > 80 ml/min/ 1,73 m2).
• Hemodiálisis: En pacientes sometidos a hemodiálisis crónica, las concentraciones plasmáticas de rosuvastatina en el estado de equilibrio fueron aproximadamente 50% mayores que las registradas en voluntarios sanos con una función renal normal.
• Insuficiencia hepática: En pacientes con una hepatopatía alcohólica crónica, las concentraciones plasmáticas de rosuvastatina aumentaron moderadamente.
En pacientes de la clase A de Child-Pugh, la Cmáx. el ABC aumentaron un 60% y un 5%, respectivamente, con respecto a los pacientes con una función hepática normal. En los pacientes de la clase B de Child-Pugh, la Cmáx. y el ABC aumentaron un 100% y un 21%, respectivamente, con respecto a pacientes con una función hepática normal.
Interacciones farmacológicas:
• Enzima 3A4 del citocromo P450: La depuración de la rosuvastatina no depende en un grado clínicamente significativo del metabolismo mediado por la enzima 3A4.
Tabla 4. Efecto de medicamentos coadministrados en la exposición sistémica a la rosuvastatina
|
Medicamento coadministrado y posología |
Rosuvastatina |
||
|
Dosis (mg)* |
Variación del ABC |
Variación de la Cmáx. |
|
|
Ciclosporina, dosis estable de 75 mg–200 mg dos veces al día |
10 mg al día durante 10 días |
↑ x 7† |
↑ x 11† |
|
Gemfibrozilo, 600 mg dos veces al día durante 7 días |
80 mg |
↑ x 1,9† |
↑ x 2,2† |
|
Asociación de lopinavir/ritonavir, 400 mg/100 mg dos veces al día durante 10 días |
20 mg al día durante 7 días |
↑ x 2† |
↑ x 5† |
|
Asociación de atazanavir/ritonavir, 300 mg/100 mg una vez al día durante 7 días |
10 mg |
↑ x 3† |
↑ x 7† |
|
Asociación de tipranavir/ritonavir, 500 mg/200 mg dos veces al día durante 11 días |
10 mg |
↑ 26% |
↑ x 2 |
|
Asociación de fosamprenavir/ritonavir, 700 mg/100 mg dos veces al día durante 7 días |
10 mg |
↑ 8% |
↑ 45% |
|
Fenofibrato, 67 mg tres veces al día durante 7 días |
10 mg |
↑ 7% |
↑ 21% |
|
Antiácido a base de una asociación de hidróxido de aluminio e hidróxido de magnesio Administración simultánea Administración con un intervalo de 2 horas |
40 mg 40 mg |
↓ 54%† ↓ 22% |
↓ 50%† ↓ 16% |
|
Eritromicina, 500 mg cuatro veces al día durante 7 días |
80 mg |
↓ 20% |
↓ 31% |
|
Ketoconazol, 200 mg dos veces al día durante 7 días |
80 mg |
↑ 2% |
↓ 5% |
|
Itraconazol, 200 mg al día durante 5 días |
10 mg |
↑ 39% |
↑ 36% |
|
Fluconazol, 200 mg al día durante 11 días |
80 mg |
↑ 14% |
↑ 9% |
|
* Dosis únicas (a menos que se especifique lo contrario). ** Razón media (con/sin la coadministración del medicamento y ninguna variación = x 1) o porcentaje de variación (con/sin la coadministración del medicamento y ninguna variación = 0%); los simbolos ↑ y ↓ indican un aumento o una disminución de la exposición, respectivamente. † Variación de importancia clínica (ver Posología y forma de administración, Advertencias y precauciones especiales de uso e Interacciones farmacológicas). |
|||
Tabla 5. Efecto de la coadministración de la rosuvastatina en la exposición sistémica a otros medicamentos
|
Posología de la rosuvastatina |
Medicamento coadministrado |
||
|
Denominación y dosis |
Variación del ABC |
Variación de la Cmáx. |
|
|
40 mg al día durante 10 días |
Warfarina*, dosis única de 25 mg |
R-Warfarina ↑ 4% |
R-Warfarina ↓ 1% |
|
40 mg al día durante 12 días |
Digoxina, dosis única de 0,5 mg |
↑ 4% |
↑ 4% |
|
40 mg al día durante 28 días |
Anticonceptivo oral (0,035 mg de etinilestradiol y 0,180, 0,215 y 0,50 mg de norgestrel) al día durante 21 días |
EE ↑ 26% |
EE ↑ 25% |
|
EE = etinilestradiol, NG = norgestrel * Efectos farmacodinámicos de importancia clínica (ver Advertencias y precauciones especiales de uso). |
|||