

ANTALGINA
METAMIZOL (DIPIRONA)
Solución inyectable
Ampolla , Solución inyectable , 1/2 g/ml
Ampolla , Solución inyectable , 1,5/ 5 g/ml
COMPOSICIÓN:
Cada 2 mL de SOLUCIÓN INYECTABLE contiene: Metamizol sódico 1 g (como metamizol sódico monohidratado)
Excipientes c.s.p. 2 mL
INDICACIONES:
ANTALGINA 1 g/2 mL solución inyectable está indicada en las siguientes condiciones:
• Dolores intensos agudos post-traumáticos o post-operatorios
• Cólicos
• Dolores de origen tumoral
• Otros dolores intensos y agudos o crónicos, cuando otras medidas terapéuticas no están indicadas.
• Fiebre alta que no cede con otras medidas.
La administración parenteral está indicada sólo cuando no cabe la posibilidad de una administración enteral.
ACCIÓN FARMACOLÓGICA:
Propiedades farmacodinámicas: ANTALGINA 1 g/2 mL solución inyectable contiene como único principio activo al metamizol.
Grupo farmacoterapéutico: analgésico, antipirético
Código ATC: N02B B02
El metamizol es un derivado de la pirazolona y posee propiedades analgésicas, antipiréticas y espasmolíticas. No se ha determinado completamente cuál es su mecanismo de acción. Algunos resultados de investigaciones muestran que el metamizol y su metabolito principal (4-N-metil-amino-antipirina) tendrían tanto un mecanismo de acción central como uno periférico.
Propiedades farmacocinéticas: Tras la administración oral, el metamizol es hidrolizado completamente en el metabolito farmacológicamente activo 4-N-metil- amino-antipirina (MAA). La biodisponibilidad de MAA es de aprox. 90% y es un poco más elevada luego de la administración oral que luego de la administración parenteral. La ingesta simultánea de alimentos no tiene ninguna influencia relevante sobre la cinética del metamizol.
La efectividad clínica radica principalmente en el MAA, hasta cierto punto también en el metabolito 4-amino-antipirina (AA). Los valores del AUC para el AA constituyen aprox. el 25% de los valores del AUC para el MAA. Los metabolitos 4-N-acetil-amino- antipirina (AAA) y 4-N-formil-amino-antipirina (FAA) son, aparentemente, farmacológicamente inactivos.
Cabe resaltar que todos los metabolitos poseen una farmacocinética no lineal. Se desconoce la importancia clínica de este fenómeno. La acumulación de los metabolitos no tiene mayor relevancia para el tratamiento a corto plazo.
El metamizol atraviesa la placenta. Los metabolitos del metamizol se excretan en la leche materna.
El enlace proteico es de 58% para el MAA, de 48% para el AA, de 18% para el FAA y de 14% para el AAA.
Luego de la administración intravenosa, la vida media del metamizol en plasma asciende a aproximadamente 14 minutos. Alrededor de 96% de una dosis radiomarcada se detecta en la orina tras una administración intravenosa, y más o menos un 6% en las heces. Después de una dosis única oral, 85% de los metabolitos excretados en la orina fueron identificados. De ellos, 3±1% eran MAA, 6±3% AA, 26±8% AAA y 23±4% FAA. La depuración renal luego de una dosis oral única de 1 g de metamizol ascendió a 5±2 para el MAA, 38±13 para el AA, 61±8 para el AAA y 49±5 mL/min para el FAA. Las correspondientes vidas medias en plasma fueron de 2.7±0.5 horas para el MAA, 3.7±1.3 horas para el AA, 9.5±1.5 horas para el AAA y 11.2±1.5 para el FAA.
Personas de edad avanzada: En los pacientes de edad, el AUC aumenta 2 a 3 veces. Posterior a una dosis única oral, la vida media del MAA y del FAA en pacientes con cirrosis hepática se incrementó en cerca de 3 veces, mientras que la vida media del AA y el AAA no se elevó en la misma proporción. En estos pacientes debería evitarse la administración de dosis altas.
Pacientes con disfunción renal: Los datos disponibles de pacientes con función renal limitada indican una reducción de la velocidad de eliminación para algunos metabolitos (AAA y FAA). Por este motivo, en estos pacientes debería evitarse la administración de dosis altas.
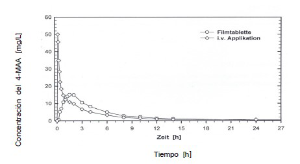
Biodisponibilidad: Una prueba de biodisponibilidad de la solución intramuscular realizada en 1989 en 12 voluntarios arrojó como resultado para el 4-MAA, en comparación con el preparado de referencia (administración intravenosa en 2 minutos):
|
Solución intramuscular (1 g) |
Administración intravenosa (1 g) |
|
|
Concentración máxima en plasma (Cmáx)[mg/L] |
11.4 ± 3.12 |
62.1 ± 15.9 |
|
Momento de la máxima concentración en plasma (tmáx)[h] |
1.67 ± 0.69 |
0.09 ± 0.02 |
|
Área bajo la curva tiempo-concentración (AUC) )[mg h/L] |
64.1 ± 14.8 |
67.8 ± 16.1 |
|
(Valores indicados como valor medio y divergencia estándar) |
||
La biodisponibilidad absoluta de la solución intramuscular, medida en relación con el AUC para las concentraciones del 4-MAA en plasma, asciende a 87%.
La media de los cursos de nivel plasmático en comparación con un preparado de referencia en un diagrama de concentración- tiempo:
Datos preclínicos sobre seguridad: Existen investigaciones sobre la toxicidad subcrónica y crónica en diversas especies animales. Durante 6 meses, se administró por vía oral 100 hasta 900 mg de metamizol por kilogramo de peso corporal a ratas. Con la dosis máxima (900 mg por kilogramo de peso corporal) se observó, al cabo de 13 semanas, una multiplicación de los reticulocitos y de los cuerpos internos de Heinz.
Se usó perros que recibieron durante 6 meses metamizol en dosis de 30 a 600 mg por kilogramo de peso corporal. En función de la dosis, a partir de los 300 mg por kilogramo de peso corporal se observó una anemia hemolítica, así como alteraciones funcionales renales y hepáticas.
Con respecto al metamizol, existen resultados contradictorios de estudios in-vitro e in-vivo en los mismos sistemas de prueba.
Estudios a largo plazo en ratas no arrojaron indicios de un potencial tumorigénico. En dos de tres estudios a largo plazo en ratones, se observaron, a altas dosis, adenomas de células hepáticas en mayor número.
Estudios de embriotoxicidad en ratas y conejos no han arrojado indicios de acciones teratogénicas.
No se observaron efectos letales en embriones de conejos a partir de una dosis diaria no tóxica para la madre de 100 mg por kg de peso corporal. En ratas, se presentaron efectos embrioletales en el rango tóxicas para la madre. Dosis diarias por encima de 100 mg por kilogramo de peso corporal prolongaron la gestación en ratas y tuvieron un efecto negativo sobre el parto con una elevada mortalidad de madres y crías.
Los metabolitos de metamizol se excretan en la leche materna. No existen datos acerca de sus efectos sobre el lactante.
Pruebas de fertilidad mostraron una ligera disminución en la tasa de embarazo de la generación de los padres a una dosis superior a 250 mg por kg de peso corporal y día. La fertilidad de la generación F1 no se vio afectada.
Los metabolitos del metamizol pasan a la leche materna. No existen experiencias sobre sus efectos sobre el lactante.
CONTRAINDICACIONES:
Este medicamento no debe emplearse:
• En caso de hipersensibilidad al metamizol o a otras pirazolonas o pirazolidinas (esto incluye también a pacientes que han reaccionado p.ej. con una agranulocitosis luego de la administración de estas sustancias) o a algún otro componente de metamizol;
• En pacientes con síndrome conocido de asma por analgésicos o intolerancia conocida a los analgésicos, del tipo urticaria- angioedema, es decir, pacientes que reaccionan con broncoespasmo u otras formas de reacción anafilactoide a los salicilatos, al paracetamol o a otros analgésicos no narcóticos, como p.ej. diclofenaco, ibuprofeno, indometacina o naproxeno;
• En caso de trastornos de la función de la médula ósea (p.ej. después de tratamiento citostático) o de enfermedades del sistema hematopoyético;
• En caso de deficiencia de glucosa-6-fosfato deshidrogenasa condicionada genéticamente (riesgo de hemólisis);
• En caso de porfiria hepática aguda intermitente (riesgo de desencadenamiento de ataques de porfiria);
• En el último trimestre del embarazo.
• Lactancia.
• En recién nacidos y en lactantes menores de 3 meses o con un peso corporal por debajo de los 5 kg, debido a que no existe información científica sobre la administración en esta población
• En recién nacidos y en lactantes menores de 3 años o con un peso corporal por debajo de los 5 kg, debido a que no existe información científica sobre la administración
• En lactantes (a partir de los 3 meses) como inyección intravenosa
• En caso de hipotonía existente y condiciones circulatorias inestables
REACCIONES ADVERSAS:
Escala de frecuencias de los efectos adversos:
• Muy frecuentes (≥ 1/10)
• Frecuentes (≥ 1/100, < 1/10)
• Poco frecuentes (≥ 1/1,000, < 1/100) Raras (≥ 1/10,000, < 1/1,000)
• Muy raras (< 1/10,000)
• Frecuencia no conocida (no se puede estimar la frecuencia en base a los datos de los que se dispone)
Trastornos de la sangre y del tejido linfático:
• Raras: Leucopenia.
• Muy raras: Agranulocitosis, trombocitopenia, incluyendo muerte.
• Frecuencia no conocida: anemia aplásica, pancitopenia, incluyendo casos con desenlace fatal.
Estas reacciones también pueden ocurrir incluso cuando anteriormente se ha administrado metamizol sin inconvenientes.
Hay indicios que en ocasiones el riesgo de agranulocitosis puede aumentar cuando metamizol se aplica por más de una semana.
Esta reacción no es dependiente de la dosis y puede ocurrir en cualquier momento durante el tratamiento. Se manifiesta con fiebre alta, escalofríos, dolor de garganta, dificultad para tragar, inflamación de la boca, nariz, garganta, área genital o anal. En los pacientes que recibieron antibióticos, estas características sin embargo pueden ser mínimas. La velocidad de sedimentación globular se acelera en gran medida, los granulocitos se reducen en gran medida o su ausentan totalmente. Generalmente, aunque no siempre, se encuentran valores normales de hemoglobina, eritrocitos y plaquetas.
Para la recuperación del paciente en estos casos, la interrupción inmediata es crucial. Por lo tanto se recomienda altamente interrumpir la administración de metamizol de inmediato, en lugar de esperar los resultados de las pruebas de laboratorio, cuando se observe un deterioro del estado general del paciente, si la fiebre no se resuelve o si ocurren situaciones dolorosas en las mucosas, sobre todo en la boca, nariz y garganta.
En caso que se produzca pancitopenia, el tratamiento debe interrumpirse inmediatamente y se debe monitorear el hemograma hasta que los valores regresen a la normalidad.
Trastornos el sistema inmunológico:
• Raras: Reacciones anafilácticas o anafilactoides*.
• Muy raras: Síndrome asmático inducido por analgésicos. En los pacientes con síndrome asmático inducido por analgésicos, las reacciones de intolerancia se manifiestan normalmente en forma de ataques de asma.
• Frecuencia no conocida: Shock anafiláctico*.
* Estas reacciones pueden ocurrir sobre todo después de la administración parenteral, y pueden ser graves y en algunos casos inclusive fatales. Estas reacciones también pueden ocurrir incluso cuando anteriormente se ha administrado metamizol sin inconvenientes.
Tales reacciones pueden aparecer durante la inyección o inmediatamente después de la ingestión, pero también pueden desarrollarse horas más tarde. Sin embargo, se producen predominantemente durante la primera hora después de la administración. Las reacciones que se manifiestan más típicamente son las reacciones en la piel y en las mucosas (picazón, ardor, enrojecimiento, ronchas, hinchazón), disnea y rara vez molestias gastrointestinales. Tales reacciones leves pueden progresar a más graves como urticaria generalizada, angioedema grave, broncoespasmo grave, arritmias cardiacas, hipotensión, (a veces precedido por el aumento de la presión arterial), shock circulatorio.
Por lo tanto metamizol se debe interrumpir inmediatamente en caso de reacciones cutáneas.
Trastornos vasculares:
• Poco frecuentes: Reacciones de hipotensión durante o después de la aplicación, que puede ir acompañada o no de reacción anafiláctica. Tales reacciones pueden conducir a una disminución severa de la presión arterial. Una inyección intravenosa rápida aumenta el riesgo de hipotensión.
Incluso la hiperpirexia dosis dependiente puede conducir a una caída de la presión arterial sin signos de reacción de hipersensibilidad.
Trastornos de la piel y del tejido subcutáneo:
• Poco frecuentes: Erupciones medicamentosas fijas.
• Raras: Erupciones (p.ej. exantema maculopapular).
• Muy raras: Síndrome de Stevens-Johnson o Necrólisis epidérmica tóxica (Interrumpir tratamiento).
Trastornos renales y urinarios:
• Muy raras: Empeoramiento agudo de la función renal, que muy rara vez puede presentarse con proteinuria, oliguria o anuria o insuficiencia renal aguda, nefritis intersticial aguda.
Trastornos generales y alteraciones en el lugar de administración:
Las inyecciones pueden causar dolor en el sitio de inyección y reacciones locales, muy raramente puede ocurrir flebitis.
En cuanto a la coloración roja en la orina, se ha descrito que puede deberse a las bajas concentraciones de metabolitos de dipirona.
Informar sobre sospechas de reacciones adversas: Los informes de sospechas de reacciones adversas después de la autorización son de gran importancia. Permite el monitoreo continuo de la relación riesgo-beneficio del medicamento. Se invita a los profesionales de la salud a reportar cualquier caso sospechoso de una reacción adversa.
INCOMPATIBILIDADES:
Ante la posibilidad de incompatibilidades, se recomienda no inyectar o infundir la solución inyectable mezclada con otras sustancias terapéuticas (sobre la miscibilidad con soluciones para infusión véase Dosificación, tipo y duración de la administración).
INTERACCIONES MEDICAMENTOSAS:
El metamizol puede causar disminución en los niveles séricos de ciclosporina. Por ello, éstos deben ser vigilados cuando se administra metamizol simultáneamente.
Cuando se administra metamizol y clorpromazina simultáneamente, puede ocurrir una hipotermia severa.
La adición de metamizol a metotrexato puede aumentar la toxicidad hematológica de metotrexato, especialmente en los ancianos. Por lo tanto, se debe evitar esta combinación.
Dipirona puede reducir el efecto del ácido acetilsalicílico sobre la agregación plaquetaria cuando se administran simultáneamente. Por lo tanto metamizol debe utilizarse en pacientes que están tomando dosis bajas de ácido acetilsalicílico para la cardioprotección con precaución.
En relación al grupo de las pirazolonas, se sabe que pueden producir interacciones con anticoagulantes orales, captopril, litio, metotrexato y triamtereno, así como alteraciones en la efectividad de los antihipertensivos y los diuréticos. Aún se desconoce en qué medida también el metamizol puede conllevar a estas interacciones.
Los niveles en sangre de bupropión se pueden bajar por metamizol. Por lo tanto, se debe tener precaución con el uso concomitante de dipirona y el bupropion.
ADVERTENCIAS: Este producto por contener en su fórmula un derivado de la pirazolona puede producir agranulocitosis, debiendo dispensarse por prescripción médica. Las dosis mayores a 1 g incrementan riesgo de hipotensión.
PRECAUCIONES: Durante los tres primeros meses de embarazo y las seis últimas semanas del mismo, así como en lactantes, durante los tres primeros meses de vida o con un peso inferior a 5 Kg no deberá administrarse sin control médico. ANTALGINA® se excreta en la leche materna. Existe riesgo de hipotensión y shock asociados a la infusión endovenosa rápida, por lo tanto la administración deberá ser lenta y no más de 1 mL por minuto.
ADVERTENCIAS Y PRECAUCIONES:
Metamizol contiene el derivado de la pirazolona metamizol y presenta riesgo de shock y agranulocitosis, que son poco frecuentes pero que pueden poner en peligro la vida.
Los pacientes que muestran reacciones anafilactoides a Metamizol también están en especial riesgo de reaccionar de la misma forma a otros analgésicos no narcóticos.
Los pacientes que muestran una reacción anafiláctica u otra mediada inmunológicamente (p.ej. agranulocitosis) a Metamizol, igualmente están en especial riesgo de reaccionar de la misma forma a otras pirazolonas y pirazolidinas.
Agranulocitosis: Cuando aparecen signos de agranulocitosis o trombocitopenia, se debe interrumpir de inmediato la administración de metamizol y se debe controlar el hemograma (incluyendo un hemograma diferencial). No se puede esperar a tener los resultados de los análisis de laboratorio para suspender el tratamiento.
Pancitopenia: En caso que se produzca pancitopenia, se debe interrumpir el tratamiento inmediatamente y debe monitorearse el hemograma hasta regresar a la normalidad. Todos los pacientes deben ser advertidos que deben buscar atención médica inmediatamente si se presentan signos y síntomas durante el tratamiento que puedan sugerir discrasias sanguíneas (p.ej.: malestar general, infección, fiebre, magulladuras, sangrado, palidez).
Reacciones anafilácticas/anafilactoides: Al elegir el método de administración, hay que recordar que la administración parenteral de analgésicos se asocia con un mayor riesgo de reacciones anafilácticas o anafilactoides (véase Precauciones).
El riesgo de posibles reacciones anafilactoides severas a metamizol es significativamente elevado en los pacientes con:
• Síndrome de asma por analgésicos o intolerancia a los analgésicos, del tipo urticaria-angioedema;
• Asma bronquial, particularmente con rinosinusitis existente y pólipos nasales simultáneamente;
• Urticaria crónica;
• Intolerancia a los colorantes (p.ej. tartrazina) o bien a los conservantes (p.ej. benzoatos);
• Intolerancia al alcohol. Estos pacientes reaccionan con síntomas como estornudo, lagrimeo y enrojecimiento facial pronunciado incluso a pequeñas cantidades de bebidas alcohólicas. Una intolerancia al alcohol de este tipo puede ser un indicio de un síndrome de asma por analgésicos no diagnosticado hasta el momento.
Puede producirse shock anafiláctico predominantemente en pacientes sensibles. Por lo tanto se debe tener especial cuidado en pacientes con antecedentes de asma o atopia.
Reacciones cutáneas graves: Se han reportado reacciones cutáneas potencialmente fatales como Síndrome de Stevens-Johnson (SSJ) y Necrólisis epidérmica tóxica (NET) con el uso de metamizol. Si se desarrollan signos y síntomas de SSJ o NET (como erupciones en la piel, a menudo con ampollas, o lesiones de mucosas), el tratamiento con metamizol y luego se puede reintroducir.
Reacciones aisladas de hipotensión: Metamizol puede desencadenar reacciones hipotensoras. Probablemente estas reacciones dependan de la dosis. Hay que contar con ello en la administración parenteral más que en la enteral. El riesgo de tales reacciones es asimismo elevado en:
• Caso de administrarse la inyección intravenosa muy rápidamente.
• Pacientes con p.ej. hipotonía preexistente, con depleción de volumen o deshidratación, inestabilidad circulatoria o deficiencia circulatoria incipiente (como p.ej. en pacientes con infarto de miocardio o politraumatismo);
• Pacientes con fiebre alta.
Por ello, en estos pacientes se requiere una verificación cuidadosa de la indicación y una vigilancia médica estrecha. Pueden ser necesarias medidas preventivas (p.ej. estabilización de la circulación) para reducir el riesgo de reacciones hipotensoras.
Metamizol sólo debe emplearse bajo vigilancia atenta de los parámetros hemodinámicos en los pacientes en los que debe evitarse absolutamente la disminución de la presión arterial, como p.ej. en caso de cardiopatía coronaria grave o estenosis relevante de los vasos sanguíneos que irrigan el cerebro.
Metamizol debería emplearse únicamente luego de una rigurosa valoración riesgo-beneficio y con las correspondientes medidas de precaución en pacientes con trastornos funcionales renales o hepáticos.
Antes de administrar metamizol se debe interrogar específicamente al paciente. En pacientes con riesgo elevado de reacciones anafilactoides, metamizol puede emplearse sólo tras una valoración cuidadosa de los posibles riesgos frente a los beneficios esperados. De administrarse metamizol en estos casos, el médico deberá vigilar estrechamente al paciente y asegurar la disponibilidad de medidas de emergencia.
En la envoltura exterior: Advertencia: Contiene metamizol.
1 mL contiene 1,42 mmol (32,7 mg) de sodio. Esto debe ser considerado en pacientes con dietas pobres en sodio (baja en sodio/bajo de nivel salino)
Embarazo y lactancia:
Embarazo: No existen datos suficientes sobre el empleo de Metamizol en mujeres embarazadas. El metamizol atraviesa la placenta. En estudios experimentales en animales, metamizol no mostró efectos teratogénicos. Dado que no existe experiencia suficiente en el ser humano, no debería administrarse/ingerirse metamizol en el primer trimestre y en el segundo trimestre debería hacerlo únicamente después de que el médico haya realizado una valoración riesgo-beneficio estricta.
Pese a que el metamizol inhibe sólo débilmente la síntesis de las prostaglandinas, no se puede descartar un cierre prematuro del ducto arterioso (Botalli), como tampoco complicaciones perinatales debidas a una reducción de la agregación trombocitaria del neonato y de la madre. Considerando esto, metamizol está contraindicada durante el último trimestre del embarazo.
Lactancia: Los metabolitos del metamizol se excretan en la leche materna. En consecuencia, debe evitarse la lactancia durante la/el ingesta/empleo y hasta por lo menos 48 horas después de la/del última(o) ingesta/empleo de metamizol.
Efectos sobre la capacidad de conducir y utilizar máquinas
Dentro de la gama de dosis recomendada, no se conoce disminución de la capacidad de concentración y reacción. Sin embargo, por una cuestión de seguridad, al menos en el caso de dosis elevadas, se debería considerar la posibilidad de una disminución de esta capacidad y no utilizar máquinas, conducir vehículos o realizar otras actividades peligrosas. Esto es especialmente aplicable cuando se ha consumido alcohol.
DOSIS Y VÍA DE ADMINISTRACIÓN
La dosificación depende de la intensidad de los dolores o de la fiebre y de la sensibilidad individual para reaccionar a metamizol.
En principio, debería optarse por la dosis más baja que sea capaz de controlar el dolor y la fiebre.
En lo que se refiere a la fiebre, en niños una dosis de 10 mg de metamizol por kilogramo de peso corporal es, en general, suficiente.
Un efecto notorio se puede esperar 30 a 60 minutos después de la administración oral y aproximadamente 30 minutos después de la administración parenteral.
En el caso de los niños y los adolescentes hasta los 14 años de edad, la dosis única a administrarse es de 8 a 16 mg de metamizol sódico 1 H2O por kilogramo de peso corporal. Los adultos y los adolescentes a partir de los 15 años (> 53 kg) pueden ingerir hasta 1000 mg por dosis única. Si el efecto no fuera eficaz, es posible administrar la dosis única hasta 4 veces al día, ello dependiendo de la dosis máxima diaria.
Las siguientes tablas de dosificación contienen las dosis únicas recomendadas y las dosis diarias máximas.
Tabla de dosificación para la solución inyectable: Cuando se trata de una administración parenteral, lo usual es que como dosis única se administre 6 mg hasta 16 mg de metamizol por kilogramo de peso corporal. Los lactantes a partir del tercer mes de vida hasta el año recibirán metamizol solución inyectable exclusivamente por vía intramuscular.
En vista de que es posible que las reacciones hipotensoras a la inyección sean dosis dependientes, las dosis únicas parenterales superiores a 1 g de metamizol deberán ser controladas con rigurosidad.
|
Edad (peso corporal) |
Dosis única |
|
3-11 meses (5-8 kg) |
0.1-0.2 ml de metamizol (equivalente a 50-100 mg de metamizol sódico 1 H2O) Sólo intramuscular |
|
1-3 años (9-15 kg) |
0.2-0.5 ml de Metamizol (equivalente a 100-250 mg de metamizol sódico 1 H2O) |
|
4-6 años (16-23 kg) |
0.3-0.8 ml de Metamizol (equivalente a 150-400 mg de metamizol sódico 1 H2O) |
|
7-9 años (24-30 kg) |
0.4-1 ml de Metamizol (equivalente a 200-500 mg de metamizol sódico 1 H2O) |
|
10-12 años (31-45 kg) |
0.5-1 ml de Metamizol (equivalente a 250-500 mg de metamizol sódico 1 H2O) |
|
13-14 años (46-53 kg) |
0.8-1.8 ml de Metamizol (equivalente a 400-900 mg de metamizol sódico 1 H2O) |
|
Adultos y adolescentes a partir de los 15 años (> 53 kg) |
1-2 ml(*) de Metamizol (equivalente a 500-1000 mg de metamizol sódico 1 H2O) |
|
(*) De requerirse, se puede aumentar la dosis única a 5 mL (equivalentes a 2500 mg de metamizol sódico 1 H2O) y la dosis diaria, a 10 mL (equivalentes a 5000 mg de metamizol sódico 1 H2O). |
|
Pacientes ancianos: En pacientes ancianos, se debería bajar la dosis ya que la eliminación de los productos del metabolismo de metamizol puede ocurrir con retardo.
En el caso de un estado general disminuido y de una depuración limitada de la creatinina: En pacientes con un estado general disminuido y una depuración limitada de la creatinina, se debería bajar la dosis ya que la eliminación de los productos del metabolismo de metamizol puede ocurrir con retardo.
Función renal o hepática deteriorada: Dado que cuando la función renal o hepática está deteriorada la velocidad de eliminación disminuye, debería evitarse las dosis elevadas y reiteradas. Si se va a administrar el fármaco sólo por un corto tiempo, no será necesario reducir la dosis. No hay experiencias en relación a una administración de larga duración.
Tipo de administración: El tipo de administración está supeditada al efecto terapéutico deseado y al estado del paciente. En muchos casos basta con la administración oral para conseguir un efecto satisfactorio. De requerirse que el fármaco actúe rápidamente o si la administración oral o rectal no está indicada, se recomienda la inyección intravenosa o intramuscular de metamizol. Al elegir la forma de administración hay que tener en cuenta que la administración parenteral está asociada con un elevado riesgo de reacciones anafilácticas o anafilactoides.
Metamizol solución inyectable se inyecta por vía intravenosa o intramuscular. En lactantes (3-11 meses) exclusivamente por vía intramuscular. La administración intramuscular debería realizarse siempre con la solución a la temperatura corporal.
Metamizol solución inyectable puede ser mezclada o diluida en solución de glucosa al 5%, solución salina al 0.9% o solución de lactato de Ringer. Sin embargo, como la estabilidad de estas mezclas es limitada, se deberá realizar la infusión inmediatamente.
En vista de la posibilidad de incompatibilidades, se recomienda no inyectar o infundir metamizol solución inyectable junto con otros medicamentos.
Duración de la administración: La duración de la administración está supeditada al tipo y gravedad de la enfermedad. En el caso de terapias con metamizol por plazos largos serán necesarios controles con hemograma periódicos, incluyendo el hemograma diferencial.
Medidas de seguridad para la administración de la inyección: Una dosis única superior a los 2 mL de metamizol (equivalentes a 1000 mg de metamizol sódico 1 H2O) requiere de un diagnóstico especialmente cuidadoso debido a que existe la sospecha de que la hipotensión crítica no relacionada con una alergia depende de la dosis.
La administración parenteral de metamizol deberá efectuarse con el paciente recostado y bajo cuidadosa vigilancia médica.
Para minimizar el riesgo de una reacción hipotensora y asegurarse de que se podrá interrumpir la administración al primer signo de una reacción anafiláctica o anafilactoide, la inyección intravenosa debe ser colocada muy lentamente, esto es, no exceder 1 mL (equivalente a 500 mg de metamizol sódico 1 H2O) por minuto.
TRATAMIENTO EN CASO DE SOBREDOSIS:
Síntomas: Después de una sobredosificación aguda se han observado náuseas, vómitos, dolor en la región abdominal, deterioro de la función renal/insuficiencia renal aguda (p.ej. debido a una nefritis intersticial) y -más raramente- síntomas nerviosos centrales (vértigo, somnolencia, coma, convulsiones) y descenso de la presión arterial hasta llegar incluso al shock y la taquicardia.
Después de administrar dosis muy altas, la excreción de ácido ribosómico puede causar enrojecimiento de la orina.
Medidas terapéuticas: No se conoce un antídoto específico para el metamizol. Si la ingesta de metamizol es muy reciente, se puede intentar limitar la absorción sistémica a través de medidas destinadas a reducir la absorción (p.ej. con carbón activado). El metabolito principal (4- N-metil-amino-antipirina) puede ser eliminado mediante hemodiálisis, hemofiltración, hemoperfusión o filtración del plasma.
El tratamiento de la intoxicación, al igual que la prevención de complicaciones severas, puede hacer necesario vigilancia y tratamiento intensivos generales y especiales.
Medidas de emergencia para casos de reacciones de hipersensibilidad (shock): Al primer signo (p.ej. reacciones cutáneas, tales como urticaria y enrojecimiento, intranquilidad, cefalea, transpiración, náuseas) interrumpir la inyección. Dejar la cánula en la vena o habilitar un acceso venoso. Aparte de las medidas de emergencia habituales, tales como colocar al paciente en posición decúbito dorsal, mantener libres las vías respiratorias y aplicar oxígeno, puede requerirse la administración de simpaticomiméticos o glucocorticoides.
PERÍODO DE VALIDEZ: No usar y/o administrar después de la fecha de expira indicada en los envases.
TEVA PERÚ S.A.
Telf.: 415-0500
PRESENTACIONES:
ANTALGINA® 1 g/2 mL: Caja x 1 ampolla.
ANTALGINA® R 5 ml: Caja x 1 ampolla.
TEVA PERÚ S.A.
Av. Venezuela 5415 - San Miguel
Telf.: 415-0500
Lima - Perú
CONDICIONES DE ALMACENAMIENTO:
Conservar a temperatura menor a 30 °C
RELACIÓN DE EXCIPIENTES:
Clorobutanol y Agua para inyección.


