

XUNIRO
ROSUVASTATINA
Comprimidos recubiertos
Caja , 14 Comprimidos recubiertos , 20 Miligramos
Caja , 14 Comprimidos recubiertos , 10 Miligramos
1 Caja, 14 Comprimidos recubiertos, 10 mg
1 Caja, 14 Comprimidos recubiertos, 20 mg
1 Caja, 14 Comprimidos recubiertos, 10 mg
Caja , 14 Comprimidos recubiertos , 10 Miligramos
COMPOSICIÓN:
Forma farmacéutica: Comprimido (tableta) recubierto.Fórmula
Rosuvastatina 10 mg:
Cada COMPRIMIDO RECUBIERTO contiene: Rosuvastatina cálcica equivalente a Rosuvastatina base10 mg. Excipientes c.s.
Rosuvastatina 20 mg: Cada COMPRIMIDO RECUBIERTO contiene: Rosuvastatina cálcica equivalente a Rosuvastatina base 20 mg. Excipientes c.s.
Forma farmacéutica y concentraciones: Comprimido (tableta) recubierta de 10 mg, circulares, de color rosa, con una de sus caras ranurada y lisa la otra. Comprimido (tableta) recubierto de 20 mg, circulares, de color rosa, con ambas caras lisas.
PRINCIPIO ACTIVO (S) / GRUPO FARMACOLÓGICO:
Descripción: La rosuvastatina cálcica es un agente reductor de lípidos sintético para la administración oral. El nombre químico de la rosuvastatina cálcica es ácido bis[(E]-7-[4-(4-fluorofenil)-6-isopropil-2[metil (metilsulfonil) amino] pirimidin-5-il-](3R, 5S)-3, 5-dihidroxihept-6-enoico] con la siguiente fórmula estructural:
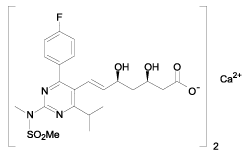
La fórmula empírica de la rosuvastatina cálcica es (C22H27FN3O6S)2Ca y el peso molecular es de 1001.14. La rosuvastatina cálcica es un polvo blanco amorfo que es rápidamente soluble en el agua y en el metanol y ligeramente soluble en etanol. La rosuvastatina cálcica es un compuesto hidrofílico con un coeficiente de partición (octanol/ agua) de 0.13 a pH de 7.0. Cada comprimido recubierto de 10 mg y 20 mg contiene los siguientes ingredientes inactivos: lactosa monohidrato, celulosa microcristalina, carbonato de calcio ligero, croscarmelosa sódica, estearato de magnesio, dióxido de silicio, talco, dióxido de titanio, alcohol polivinílico, polietilen glicol ·3000, color FD&C rojo No. 40 laca, color D&C amarillo No. 6 laca, óxido de hierro rojo agua purificada.
INDICACIONES TERAPÉUTICAS:
Indicaciones y uso
Hiperlipidemia y dislipidemia mixta: La rosuvastatina está indicada como tratamiento adyuvante a la dieta para reducir el colesterol (C) total, C-LDL, ApoB , C- no HDL y los triglicéridos elevados y aumentar el C-HDL en pacientes adultos con hiperlipidemia primaria o dislipidemia mixta. Se debe emplear agentes reductores de lípidos además de una dieta restringida en grasas saturadas y colesterol cuando la respuesta a la dieta y a las intervenciones no farmacológicas solas no haya sido adecuada. Pacientes pediátricos de 10 a 17 años de edad con hipercolesterolemia familiar heterocigota.
No procede.
Hipertrigliceridemia: La rosuvastatina está indicada como terapia adyuvante para el tratamiento de los pacientes adultos con hipertrigliceridemia.
Disbetalipoproteína primaria (hiperlipoproteinemia tipo III): La rosuvastatina está indicada como un adyuvante a la dieta para el tratamiento de los pacientes con disbetalipoproteinemia primaria (hiperlipoproteinemia tipo III).
Hipercolesterolemia familiar homocigota: La rosuvastatina está indicada como tratamiento adyuvante a otros tratamientos reductores de lípidos (por ejemplo, aféresis de LDL) o solo, si estos tratamientos no están disponibles para reducir el C-LDL, C total y ApoB en pacientes adultos con hipercolesterolemia familiar homocigota.
Retraso de la progresión de la aterosclerosis: La rosuvastatina está indicada como terapia adyuvante para retrasar la progresión de la aterosclerosis en los pacientes adultos como parte de la estrategia de tratamiento para disminuir el C-total y el C-LDL a las concentraciones objetivo.
Prevención primaria de la enfermedad cardiovascular: No procede.
Limitaciones de uso: La rosuvastatina no se ha estudiado en las dislipidemias de Fredrickson tipos I y V.
MECANISMO DE ACCIÓN:
Farmacología clínica
Mecanismo de acción: La rosuvastatina es un inhibidor selectivo y competitivo del la HMG-CoA reductasa, la enzima que limita la velocidad de conversión de la 3-hidroxi-3-metilglutaril coenzima A a mevalonato, un precursor del colesterol. Los estudios en animales in vivo y en células de animales y de humanos cultivadas in vitro han mostrado que la rosuvastatina tiene una alta captura y selectividad para actuar en el hígado, el órgano blanco para la reducción del colesterol. En los estudios in vivo e in vitro, la rosuvastatina produce sus efectos modificadores de los lípidos de dos maneras. Primero, aumenta el número hepático de receptores de LDL en la membrana celular para aumentar la captura y el catabolismo de la LDL. Segundo, la rosuvastatina inhibe la síntesis hepática de VLDL, lo que reduce el número total de partículas de VLDL y LDL.
Farmacocinética
Absorción: En los estudios clínicos en el hombre, las concentraciones plasmáticas máximas se alcanzaron de 3 a 5 horas después de la dosis oral. Tanto la Cmáx como el ABC aumentaron en proporción aproximada a la dosis de la rosuvastatina. La biodisponibilidad absoluta de la rosuvastatina es de aproximadamente el 20%. La administración de la rosuvastatina no varió después de su administración en la mañana o en la noche.
Distribución: El volumen medio de distribución en el estado de equilibrio de la rosuvastatina es de aproximadamente 134 litros. El 88% de la rosuvastatina se une a las proteínas plasmáticas, mayormente a la albúmina. Esta unión es reversible e independiente de las concentraciones plasmáticas.
Metabolismo: La rosuvastatina no se metaboliza extensamente; aproximadamente el 10% de la dosis radiomarcada se recupera como metabolito. El metabolito más importante es la N-desmetil rosuvastatina, que se forma principalmente mediante el citocromo P450 2C9 y los estudios in vitro han demostrado que la N-desmetil rosuvastatina tiene aproximadamente un sexto a un medio de la actividad inhibitoria de la HMG-CoA reductasa del compuesto original. En general, el compuesto original es responsable de más del 90% de la actividad inhibitoria del la HMG-CoA reductasa del plasma.
Excreción: Después de la administración oral, la rosuvastatina y sus metabolitos se excretan principalmente en las heces (90%). La vida media de eliminación (t1/2) de la rosuvastatina es de aproximadamente 19 horas. Después de una dosis intravenosa, aproximadamente el 28% de la depuración corporal total se efectuó por la vía renal y el 72% por la vía hepática.
Raza: Un análisis de farmacocinética de poblaciones no reveló diferencias clínicamente relevantes en la farmacocinética entre grupos caucásicos, hispanos y negros o afro-caribeños. Sin embargo, los estudios de farmacocinética, incluyendo los realizados en los EE. UU. han demostrado una elevación de aproximada de aproximadamente 2 veces la exposición mediana (ABC y Cmáx) en sujetos asiáticos cuando se comparó con un grupo control caucásico.
• Género: No hubo diferencias en las concentraciones plasmáticas de la rosuvastatina entre hombres y mujeres.
• Geriatría: No hubo diferencias en las concentraciones plasmáticas de la rosuvastatina entre poblaciones ancianas (edad ≥ 65 años) y no ancianas.
• Deterioro renal: El deterioro renal leve a moderado (CLcr ≥30 mL/min/1.73 m2) no tuvo influencia sobre las concentraciones plasmáticas de la rosuvastatina. Sin embargo, las concentraciones plasmáticas de la rosuvastatina aumentaron a un grado clínicamente significativo (cerca de 3 veces) en los pacientes con deterioro renal grave (CLcr <30 mL/min/1.73 m2 ) que no se sometían a hemodiálisis en comparación con los sujetos sanos (CLcr >80 mL/min/1.73 m2).
• Hemodiálisis: Las concentraciones plasmáticas en estado de equilibrio de la rosuvastatina en pacientes con hemodiálisis crónica fueron aproximadamente un 50% mayores en comparación con los sujetos sanos con función renal normal.
• Deterioro hepático: En pacientes con enfermedad hepática crónica alcohólica, las concentraciones plasmáticas de la rosuvastatina aumentaron modestamente. En los pacientes con enfermedad en estadio Child-Pugh A, la Cmáx y el ABC se incrementaron en un 60% y 5%, respectivamente, en comparación con los pacientes con función hepática normal. En los pacientes con enfermedad en estadio Child-Pugh B, la Cmáx y el ABC se incrementaron en un 100% y 21%, respectivamente, en comparación con los pacientes con función hepática normal.
CONTRAINDICACIONES:
La rosuvastatina está contraindicada en las siguientes afecciones:
• Pacientes con hipersensibilidad conocida a cualquiera de los componentes de la fórmula. Se han reportado reacciones de hipersensibilidad, que incluyen erupción cutánea, prurito y angioedema con la rosuvastatina [ver Reacciones adversas [6.1]).
• Pacientes con enfermedad hepática activa, que puede incluir elevaciones persistentes e inexplicables de las concentraciones de las transaminasas (ver Advertencias y precauciones [5.2]).
• Mujeres embarazadas o que se puedan embarazar. Debido a que los inhibidores de la HMG-CoA reductasa disminuyen la síntesis del colesterol y posiblemente la de otras substancias biológicamente activas derivadas del colesterol, la rosuvastatina puede causar daño fetal cuando se administra a mujeres embarazadas. Además, no parece haber un beneficio con el tratamiento durante el embarazo, y no se ha establecido la seguridad en mujeres embarazadas. Si la paciente se embaraza mientras toma este medicamento, la paciente debe ser informada sobre el peligro potencial al feto y de la falta del beneficio clínico conocido con el uso continuo durante el embarazo (ver Uso en poblaciones específicas 8.1] y Toxicología no clínica [13.2]).
• Madres que amamantan. Debido a que otro fármaco de esta clase pasa a la leche materna y debido a que los inhibidores de la HMG-CoA reductasa tienen el potencial de causar reacciones adversas graves en niños que amantan, las mujeres que requieren del tratamiento con la rosuvastatina deben ser aconsejadas de no amamantar a sus niños (ver Uso en poblaciones específicas [8.3]).
REACCIONES ADVERSAS:
Las siguientes reacciones adversas se discuten con mayor detalle en otras secciones de esta información para prescribir:
• rabdomiolisis con mioglobinuria e insuficiencia renal y miopatía (incluyendo miositis) (ver Advertencias y precauciones [5.1])
• anormalidades de las enzimas hepáticas (ver Advertencias y precauciones [5.2])
En la base de datos de los estudios controlados de la rosuvastatina (controlados con placebo o con comparador activo) de 5394 pacientes con una duración media del tratamiento de 15 semanas, el 1.4% de los pacientes descontinuaron debido a reacciones adversas. Las reacciones adversas más comunes que llevaron a la descontinuación del tratamiento fueron:
• mialgia
• dolor abdominal
• náusea
Las reacciones adversas más comúnmente reportadas (incidencia ≥2%) en la base de datos de los estudios controlados de rosuvastatina de 5394 pacientes fueron:
• dolor de cabeza
• mialgia
• dolor abdominal
• astenia
• náusea
Experiencia en estudios clínicos: Debido a que los estudios se realizan en condiciones ampliamente variables, las frecuencias de las reacciones adversas observadas en los estudios clínicos de un medicamento no se pueden comparar directamente con las frecuencias de los estudios clínicos de otro medicamento y pueden no reflejar las frecuencias observadas en lapráctica clínica. Las reacciones adversas reportadas en ≥2% de los pacientes en los estudios clínicos controlados con placebo y con una frecuencia mayor que la observada con el placebo se muestran en la tabla 1. En estos estudios el tratamiento tuvo una duración de hasta 12 semanas.
|
Tabla 1. Reacciones adversas* reportadas en ≥2% de los pacientes tratados con rosuvastatina y mayor que con el placebo en los estudios |
||||||
|
Reacciones adversas |
Rosuvas- tatina n= 291 |
Rosuvas-tatina n= 283 |
Rosuvas-tatina n= 64 |
Rosuvas-tatina n= 106 |
Total Rosuvas-tatina 5 mg-40 mg n= 744 |
Placebo n=382 |
|
Dolor de cabeza |
5.5 |
4.9 |
3.1 |
8.5 |
5.5 |
5.0 |
|
Náusea |
3.8 |
3.5 |
6.3 |
0 |
3.4 |
3.1 |
|
Mialgia |
3.1 |
2.1 |
6.3 |
1.9 |
2.8 |
1.3 |
|
Astenia |
2.4 |
3.2 |
4.7 |
0.9 |
2.7 |
2.6 |
|
Estreñimiento |
2.1 |
2.1 |
4.7 |
2.8 |
2.4 |
2.4 |
|
* Reacciones adversas por término preferido de COSTART. |
||||||
Otras reacciones adversas reportadas en los estudios clínicos fueron dolor abdominal, mareo, hipersensibilidad (incluyendo erupción cutánea, prurito, urticaria y angioedema) y pancreatitis. También se reportaron las siguientes anormalidades de laboratorio: tira reactiva positiva para proteinuria y hematuria microscópica (ver Advertencias y precauciones [5.4]; elevación de la creatina fosfoquinasa, transaminasas, glucosa, glutamiltranspeptidasa, fosfatasa alcalina y de la bilirrubina; y anormalidades de las pruebas de función tiroidea. En un estudio en el que se trató a 981 pacientes con rosuvastatina 40 mg (n=700) o placebo (n=281) con una duración media del tratamiento de 1.7 años, el 5.6% de los sujetos tratados con rosuvastatina versus el 2.8% de los sujetos tratados con placebo descontinuaron debido a reacciones adversas. Las reacciones adversas más comunes que llevaron a la descontinuación del tratamiento fueron mialgia, elevación de las enzimas pancreáticas, dolor de cabeza y náusea (ver Estudios clínicos [14.7]). Las reacciones adversas reportadas en ≥2% de los pacientes y con mayor frecuencia que con el placebo se muestran en la tabla 2.
|
Tabla 2. Reacciones adversas* reportadas en ≥2% de los pacientes tratados con rosuvastatina y mayor que con el placebo en el estudio clínico de 981 pacientes |
||
|
Reacciones adversas |
Rosuvastatina 40 mg n=700 |
Placebo n=281 |
|
Mialgia |
12.7 |
12.1 |
|
Artralgia |
10.1 |
7.1 |
|
Dolor de cabeza |
6.4 |
5.3 |
|
Mareo |
4.0 |
2.8 |
|
CPK elevada |
2.6 |
0.7 |
|
Dolor abdominal |
2.4 |
1.8 |
|
ALT >3 veces el límite normal superior† |
2.2 |
0.7 |
|
* Reacciones adversas por término preferido de MedDRA †Frecuencia registrada como valor anormal de laboratorio |
||
En otro estudio, se trató a 17,082 participantes con rosuvastatina 20 mg (n=8901) o placebo (n=8901) con una duración media de 2 años. Un mayor porcentaje de pacientes tratados con rosuvastatina versus los pacientes tratados con placebo, 6.6% y 6.2%, respectivamente, descontinuaron el medicamento del estudio debido a algún evento adverso, independientemente de la causalidad con el tratamiento. La reacción adversa más común fue la mialgia que llevó a la descontinuación del tratamiento. En este estudio, hubo una mayor frecuencia significativamente de diabetes mellitus reportada en pacientes que tomaron rosuvastatina (2.8%) versus los pacientes que tomaron placebo (2.3%). La HbA1c media se elevó significativamente en un 0.1% en los pacientes tratados con rosuvastatina en comparación con los pacientes tratados con placebo. El número de pacientes con una HbA1c >6.5% al final del estudio fue significativamente mayor en los pacientes tratados con rosuvastatina que en los tratados con placebo (ver Advertencias y precauciones [5.5] y Estudios clínicos [14.8]).Las reacciones adversas reportadas en ≥2% de los pacientes y con mayor frecuencia que con el placebo se muestran en la tabla 3.
|
Tabla 3. Reacciones adversas* reportadas en ≥2% de los pacientes tratados con rosuvastatina y mayor que con el placebo en el estudio clínico de 17,082 pacientes (% de pacientes) |
||
|
Reacciones adversas |
Rosuvastatina |
Placebo n=8901 |
|
Mialgia |
7.6 |
6.6 |
|
Artralgia |
3.8 |
3.2 |
|
Estreñimiento |
3.3 |
3.0 |
|
Náusea |
2.4 |
2.3 |
|
* Reacciones adversas surgidas del tratamiento por término preferido de MedDRA |
||
Pacientes pediátricos de 10 a 17 años de edad: No procede.
Experiencia de postcomercialización: Las siguientes reacciones adversas se han identificado durante el empleo después de la aprobación de la rosuvastatina: artralgia, insuficiencia hepática, hepatitis, ictericia, pérdida de la memoria, depresión y trastornos del sueño (incluyendo insomnio y pesadillas). Debido a que estas reacciones se reportan voluntariamente de una población de tamaño incierto, no siempre es posible calcular confiablemente su frecuencia o establecer una relación causal con la exposición al medicamento.
Toxicología no clínica
Carcinogénesis, mutagénesis y deterioro de la fertilidad: En un estudio de carcinogenicidad en ratas a dosis de 2, 20, 60 u 80 mg/Kg/día por sonda oral, la incidencia de pólipos del estroma uterino aumentó significativamente en las hembras con 80 mg/Kg/día con una exposición sistémica 20 veces la exposición humana a 40 mg/día basada en el ABC. La incidencia elevada de los pólipos no se observó con las dosis menores. En un estudio de carcinogenicidad de 107 semanas en ratones a los que se les administró 10, 60 ó 200 mg/Kg/día por sonda oral, se observó un aumento de la incidencia de adenoma/carcinoma hepatocelular con 200 mg/Kg/día con exposiciones sistémicas 20 veces la exposición humana de 40 mg/día basada en el ABC. No se observó una incidencia elevada de tumores hepatocelulares a dosis menores. La rosuvastatina no fue mutagénica ni clastogénica con o sin activación metabólica en la prueba de Ames con Salmonella typhimurium y Escherichia coli, en el ensayo de linfoma de ratón ni en el ensayo de aberraciones cromosómicas en células de pulmón de hámster chino. La rosuvastatina fue negativa en el ensayo de micronúcleo en el ratón. En los estudios de fertilidad de ratón con dosis por sonda oral de 5, 15 y 50 mg/Kg/día, se trataron a machos durante 9 semanas antes y durante el apareamiento y se trató a las hembras 2 semanas antes del apareamiento y durante el mismo hasta el día 7 de la gestación. No se observaron efectos adversos sobre la fertilidad con 50 mg/ Kg/día (exposición sistémica de hasta 10 veces la exposición humana a 40 mg/día basada en el ABC). En los testículos de perros tratados con rosuvastatina a 30 mg/Kg/día durante un mes, se observaron células espermatídicas gigantes. Las células espermatídicas gigantes se observaron en monos después del tratamiento de 6 meses con 30 mg/Kg/día además de la vacuolación del epitelio de los túbulos seminíferos. Las exposiciones en los perros fueron 20 veces y en los monos, 10 veces la exposición humana a 40 mg/día basada en el área corporal. Se observaron hallazgos similares con otros fármacos de esta clase.
Toxicología y/o farmacología animal
Desarrollo embrionario-fetal: En la rata, la rosuvastatina cruza la placenta y se encuentra en el tejido fetal y el líquido amniótico al 3% y 20%, respectivamente, de la concentración plasmática materna después de una dosis única por sonda oral de 25 mg/Kg en el día 16 de la gestación.
En conejos, se observó un mayor distribución tisular fetal (25% de la concentración plasmática materna) después de una dosis única por sonda oral de 1 mg/ Kg en el día 18 de la gestación. En las ratas hembras a las que se les administró la rosuvastatina por sonda oral a dosis de 2, 10 y 50 mg/Kg/día desde antes del apareamiento hasta el día 7 postcoito resulta en la disminución del peso corporal fetal (de los productos hembras) y retraso de la osificación con la dosis alta (exposiciones sistémicas de 10 veces la exposición humana con 40 mg/día basada en el ABC). En ratas preñadas a las que se les administró la rosuvastatina por sonda oral a dosis de 2, 10 y 50 mg/Kg/día desde el día 7 de la gestación hasta el día 21 de la lactancia (destete), se presentó una disminución de la sobrevivencia de los productos en los grupos a los que se les administró la dosis de 50 mg/Kg/día, exposiciones sistémicas ≥12 veces la exposición humana con 40 mg/día basada en superficie corporal. En conejos preñados a los que se les administró por sonda oral dosis de 0.3, 1 y 3 mg/Kg/día desde el día 6 de la gestación hasta el día 18 de lactancia (destete), exposiciones equivalentes a la exposición humana con 40 mg/día basada en superficie corporal, se observó disminución de la viabilidad fetal y mortalidad materna. La rosuvastatina no fue teratogénica en ratas a ≤25 mg/Kg/día ni en conejos a ≤3 mg/Kg/ día (exposiciones sistémicas equivalentes a la exposición humana con 40 mg/día basada en el ABC o la superficie corporal, respectivamente).
Toxicidad del sistema nervioso central (SNC)
Se han observado lesiones vasculares del SNC, caracterizadas por hemorragia perivascular, edema e infiltración de células mononucleares de los espacios perivasculares en perros tratados con otros miembros de esta clase de fármacos. Un fármaco químicamente similar de esta clase produjo degeneración del nervio óptico (degeneración walleriana de las fibras retinogeniculadas) dependiente de la dosis en perros a la dosis que produjo concentraciones plasmáticas de aproximadamente 30 veces mayores que la concentración media del fármaco en humanos que toman la dosis más alta recomendada. Se observó edema, hemorragia y necrosis parcial del intersticio del plexo coroidal en un perro hembra moribunda que se sacrificó en el día 24 con 90 mg/Kg/día por sonda oral (exposiciones sistémicas de 100 veces la exposición humana con 40 mg/día basada en el ABC). Se observó opacidad corneal en perros tratados por 52 semanas con 6 mg/Kg/día por sonda oral (exposiciones sistémicas de 20 veces la exposición humana con 40 mg/día basada en el ABC). Se observó displasia retiniana y pérdida retiniana en perros tratados durante 4 semanas con sonda oral con 90 mg/Kg/día (exposiciones sistémicas de 100 veces la exposición humana con 40 mg/día basado en el ABC). Las dosis ≤30 mg/Kg/día (exposiciones sistémicas ≤60 veces la exposición humana con 40 mg/día basada en el ABC) no revelaron hallazgos retinianos durante el tratamiento hasta por 1 año.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones medicamentosas
Ciclosporina: La ciclosporina aumentó significativamente la exposición a la rosuvastatina. Por lo tanto, en pacientes que tomen ciclosporina, el tratamiento debe limitarse a 5 mg de rosuvastatina una vez al día (ver Dosis y administración [2.5], Advertencias y precauciones [5.1] y Farmacología clínica [12.3]).
Gemfibrozilo: El gemfibrozilo aumentó significativamente la exposición a la rosuvastatina. Por lo tanto, el tratamiento combinado con la rosuvastatina y el gemfibrozilo deben evitarse. Si se emplean, no exceder de 10 mg de rosuvastatina una vez al día (ver Dosis y administración [2.6] y Farmacología clínica [12.3]).
Inhibidores de la proteasa: La coadministración de la rosuvastatina con ciertos inhibidores de la proteasa en combinación con ritonavir tiene diferentes efectos sobre la exposición a la rosuvastatina. Las combinaciones de los inhibidores de la proteasa lopinavir/ritonavir y atazanavir/ritonavir aumentan la exposición a la rosuvastatina (ABC) hasta tres veces (ver tabla 4 en Farmacología clínica [12.3]). Con estas combinaciones, la dosis de la rosuvastatina debe limitarse a 10 mg. Las combinaciones de tipranavir/ ritonavir o fosamprenavir/ritonavir producen poco o no producen cambio de la exposición a la rosuvastatina. Se debe tener precaución cuando la rosuvastatina se coadministre con los inhibidores de la proteasa administrados en combinación con el ritonavir (ver Dosis y administración [2.5], Advertencias y precauciones [5.1] y Farmacología clínica [12.3]).
Anticoagulantes cumarínicos: La rosuvastatina aumentó significativamente el INR de los pacientes que recibieron anticoagulantes cumarínicos. Por lo tanto, se debe tener precaución cuando se administren los anticoagulantes cumarínicos junto con la rosuvastatina. En los pacientes que tomen anticoagulantes cumarínicosy rosuvastatina de manera concomitante, el INR debe determinarse antes de comenzar la rosuvastatina y lo suficientemente frecuente al principio del tratamiento para asegurar que no ocurra alguna alteración significativa del INR (ver Advertencias y precauciones [5.3] y Farmacología clínica [12.3]).
Niacina: El riesgo de los efectos sobre el músculo esquelético puede aumentar cuando la rosuvastatina se emplea en combinación con la niacina. Se debe considerar una reducción de la dosis de la rosuvastatina en este escenario (ver Advertencias y precauciones [5.1]). Fenofibrato: Cuando la rosuvastatina se coadministró con el fenofibrato no se observó ninguna elevación del ABC de la rosuvastatina ni del fenofibrato clínicamente significativa. El beneficio de las alteraciones adicionales de las concentraciones de los lípidos con el uso combinado de la rosuvastatina con los fibratos debe ser ponderada cuidadosamente contra el riesgo potencial de esta combinación (ver Advertencias y precauciones [5.1] y Farmacología clínica [12.3]).
Interacciones fármaco-fármaco
Citocromo P450 3A4: La depuración de la rosuvastatina no depende del metabolismo por el citocromo P450 3A4 en grado clínicamente significativo.
|
Tabla 4. Efecto de los fármacos coadministrados sobre la exposición sistémica a la rosuvastatina |
|||
|
Fármaco coadministrado y régimen de dosificación |
Rosuvastatina |
||
|
Dosis (mg)* |
Cambio del ABC† |
Cambio de Cmáx† |
|
|
Ciclosporina-dosis estable requerida (75 mg-200 mg BID) |
10 mg QD por 10 días |
↑ 7 veces‡ |
↑ 11 veces‡ |
|
Gemfibrozilo 600 mg BID por 7 días |
80 mg |
↑ 1.9 veces‡ |
↑ 2.2 veces‡ |
|
Combinación de lopinavir/ ritonavir 400 mg/100 mg BID por 10 días |
20 mg QD por 7 días |
↑ 2 veces‡ |
↑ 5 veces‡ |
|
Combinación de atazanavir/ ritonavir 300 mg/100 mg QD por 7 días |
10 mg |
↑ 3 veces‡ |
↑ 7 veces‡ |
|
Combinación de tipranavir/ ritonavir 500 mg/200 mg BID por 11 días |
10 mg |
↑ 26% |
↑ 2 veces |
|
Fosamprenavir/ritovavir 700 mg/100 mg BID por 7 días |
10 mg |
↑ 8% |
↑ 45% |
|
Fenofibrato 67 mg TID por 7 días |
10 mg |
↑ 7% |
↑ 21% |
|
Combinación de antiácidos de hidróxido de aluminio y magnesio administrados simultáneamente con 2 horas de diferencia |
40 mg 40 mg |
↓ 54%‡ ↓ 22% |
↓ 50%‡ ↓ 16% |
|
Eritromicina 500 mg QID por 7 días |
80 mg |
↓ 20% |
↓ 31% |
|
Ketoconazol 200 mg BID por 7 días |
80 mg |
↑ 2% |
↓ 5% |
|
Itraconazol 200 mg BID por 7 días |
10 mg 80 mg |
↑ 39% ↑ 28% |
↑ 36% ↑ 15% |
|
Fluconazol 200 mg QD por 11 días |
80 mg |
↑ 14% |
↑ 9% |
|
* Dosis únicas a menos que se indique de otra manera † Razón media (con/sin el fármaco coadministrado y sin cambio = 1 vez) o % del cambio (con/sin el fármaco coadministrado = 0%); los símbolos de ↑ y ↓ indican que la exposición aumentó o disminuyó, respectivamente ‡ Clínicamente significativo (ver Dosis y administración [2] y Advertencias y precauciones [5]) |
|||
|
Tabla 5. Efecto de la coadministración de la rosuvastatina sobre la exposición sistémica a otros fármacos |
|||
|
Régimen de dosis de rosuvastatina |
Fármaco coadministrado |
||
|
Nombre y dosis |
Cambio del ABC |
Cambio de la Cmáx |
|
|
40 mg QD por 10 días |
Warfarina* 25 mg dosis única |
R-warfarina ↑ 4% S-warfarina ↑ 6% |
R-warfarina ↓1% S-warfarina 0% |
|
40 mg QD por 12 días |
Digoxina 0.5 mg dosisúnica |
↑ 4% |
↑ 4% |
|
40 mg QD por 28 días |
Anticonceptivo oral (etinil estradiol 0.035 mg & norgestrel 0.180, 0.215 y 0.250 mg) QD por 21 días |
EE ↑ 26% NG ↑ 34% |
EE ↑25% NG ↑ 23% |
|
EE = etinil estradiol, NG = norgestrel * Efectos farmacodinámicos clínicamente significativos (ver Advertencias y precauciones [5.4]) |
|||
INFORMACIÓN COMPLEMENTARIA:
Referencia: U.S. Department of Health and Human Services.
DA Online Label Repository.
SANOFI-AVENTIS
Av. De los Shyris N° 3727 y Naciones Unidas
Edificio Silva Nuñez, Piso 7
PBX: 593 (2) 299 4300, opción 7
Fax: 593 (2) 299 4349
Casilla 17-210-0244
Quito- Ecuador
RECOMENDACIONES:
Advertencias y precauciones
Efectos sobre el músculo esquelético: Se han reportado casos de miopatía y rabdomiolisis con insuficiencia renal aguda secundaria a mioglobinuria con los inhibidores de la HMG-CoA reductasa, incluyendo la rosuvastatina. Estos efectos pueden presentarse con cualquier dosis, pero se incrementa con la dosis más alta (40 mg). Se debe prescribir la rosuvastatina con precaución en pacientes con factores predisponentes de miopatía (por ejemplo, edad ≥65 años, hipotiroidismo tratado inadecuadamente, deterioro renal).
El riesgo de miopatía durante el tratamiento con la rosuvastatina puede aumentar con la administración concurrente de algunos tratamientos reductores de lípidos (fibratos o niacina), gemfibrozilo, ciclosporina, lopinavir/ritonavir o atazanavir/ ritonavir (ver Dosis y administración [2] e Interacciones medicamentosas [7]).
El tratamiento con la rosuvastatina debe descontinuarse si se presenta una elevación marcada de las concentraciones de la creatina quinasa o si se diagnostica o si se sospecha de miopatía. El tratamiento con la rosuvastatina debe detenerse temporalmente en cualquier paciente con alguna enfermedad aguda y grave sugestiva de miopatía o que predisponga al desarrollo de insuficiencia renal secundaria a rabdomiolisis (por ejemplo, sepsis, hipotensión, deshidratación, cirugía mayor, traumatismo, enfermedades endocrinas y de los electrolitos graves o convulsiones descontroladas). A todos los pacientes se les debe recomendar reportar inmediatamente dolor, hipersensibilidad o debilidad musculares inexplicables, particularmente si se acompaña de fiebre o malestar.
Anormalidades de las enzimas hepáticas y monitoreo: Se recomienda que se realicen pruebas de función hepática antes y a las 12 semanas después de iniciar el tratamiento y de cualquier elevación de la dosis y periódicamente (por ejemplo, semestralmente) a partir de entonces. Se han reportado aumento de las transaminasas séricas (AST [SGOT] o ALT [SGPT]) con el empleo de inhibidores de la HMG-CoA reductasa, incluyendo la rosuvastatina. En la mayoría de los casos, las elevaciones fueron transitorias y se resolvieron o mejoraron durante el tratamiento continuo o después de una interrupción breve del tratamiento. Hubo dos casos de ictericia cuya relación causal con la rosuvastatina no se pudo determinar y que se resolvió después de la descontinuación del tratamiento. No hubo casos de insuficiencia hepática o enfermedad hepática irreversible en esto estudios.
En un análisis acumulado de los estudios controlados con placebo, los incrementos de las transaminasas séricas >3 veces el límite superior normal (LSN) ocurrieron en el 1.1% de los pacientes que tomaron rosuvastatina versus el 0.5% de los pacientes tratados con placebo. Se debe vigilar a los pacientes que desarrollan concentraciones elevadas de las transaminasas hasta que se resuelvan la anormalidades. Si persistiera el incremento de la ALT o la AST >3 veces el LSN, se recomienda la reducción de la dosis o el retiro de la rosuvastatina. La rosuvastatina debe emplearse con precaución en pacientes que consumen cantidades importantes de alcohol y/o que tienen historia de enfermedad hepática crónica (ver Farmacología clínica [12.3]). La enfermedad hepática activa, que puede incluir la elevación persistente e inexplicable de las transaminasas, es una contraindicación al uso de la rosuvastatina (ver Contraindicaciones [4]).
Anticoagulantes cumarínicos concomitantes: Se debe tener precaución cuando se administren anticoagulantes junto con la rosuvastatina, debido a la potenciación del efecto de los anticoagulantes del tipo de la cumarina en la prolongación del tiempo de protrombina/INR. En pacientes que toman anticoagulantes cumarínicos y rosuvastatina de forma concomitante, se debe determinar el INR antes de iniciar la rosuvastatina y frecuentemente al principio del tratamiento para asegurar que no ocurra alguna alteración significativa del INR (ver Interacciones medicamentosas [7.4]).
Proteinuria y hematuria: En el programa de estudios clínicos de rosuvastatina, se observó tira reactiva positiva para proteinuria y hematuria microscópica entre los pacientes tratados con la rosuvastatina. Estos hallazgos fueron más frecuentes en pacientes que tomaron 40 mg de rosuvastatina en comparación con las dosis menores de rosuvastatina o los inhibidores de la HMG-CoA reductasa usados como comparadores, aunque generalmente fue transitoria y no se asoció al empeoramiento de la función renal. Aunque no se conoce el significado clínico de este hallazgo, se debe considerar la reducción de la dosis en pacientes en tratamiento con rosuvastatina y proteinuria y/o hematuria inexplicadas y persistentes durante el examen de orina rutinario. Efectos endocrinos: Se han reportado incrementos de la concentración de la HbA1c y de la glucosa sérica en ayuno con los inhibidores de la HMG-CoA reductasa, incluyendo la rosuvastatina (ver Reacciones adversas [6.1.]). Aunque los estudios clínicos han mostrado que la rosuvastatina sola no reduce la concentración plasmática del cortisol ni deteriora la reserva adrenal, se debe tener precaución si la rosuvastatina se administra de forma concomitante con fármacos que pueden disminuir las concentraciones o la actividad de las hormonas esteroideas endógenas como el ketoconazol, la espironolactona y la cimetidina.
Uso en poblaciones específicas
Embarazo
Efectos teratogénicos: Categoría del embarazo X La rosuvastatina está contraindicada en mujeres que están embarazadas o que puedan embarazarse. El colesterol y los triglicéridos séricos aumentan durante el embarazo normal, y los productos del colesterol son esenciales para el desarrollo fetal. La aterosclerosis es un proceso crónico y la descontinuación de los fármacos reductores de los lípidos durante el embarazo debe tener poco impacto sobre los resultados a largo plazo del tratamiento de la hiperlipidemia primaria (ver Contraindicaciones [4]). No existen estudios adecuados y bien controlados de la rosuvastatina en mujeres embarazadas. Existen reportes de anomalías congénitas después de la exposición intrauterina a los inhibidores de la HMG-CoA reductasa. En una revisión de aproximadamente 100 embarazos seguidos prospectivamente en mujeres expuestas a otros inhibidores de la HMG-CoA reductasa, la incidencia de anormalidades congénitas, abortos espontáneos y muertes fetales/óbitos no excedió la frecuencia esperada en la población general. Sin embargo, en este estudio sólo se pudo excluir un aumento del riesgo de anormalidades congénitas de 3 a 4 veces sobre la incidencia de fondo. En el 89% de estos casos, el tratamiento farmacológico comenzó antes del embarazo y se detuvo durante el primer trimestre cuando se identificó el embarazo. La rosuvastatina cruza la placenta en ratas y conejos. En las ratas, la rosuvastatina no fue teratogénica con exposiciones sistémicas equivalentes a la dosis terapéutica humana de 40 mg/día. Con 10-12 veces la dosis humana de 40 mg/día, hubo una disminución de la sobrevivencia de los productos, disminución del peso corporal fetal entre los productos hembras y retraso de la osificación. En los conejos, la viabilidad de los productos disminuyó y aumentó la mortalidad materna a dosis equivalentes a la dosis humana de 40 mg/día (ver Toxicología no clínica [13.2]). La rosuvastatina puede causar daño fetal cuando se administra a la mujer embarazada. Si la paciente se embaraza mientras toma la rosuvastatina, la paciente debe ser informada del riesgo potencial para el feto y la falta de beneficio clínico conocido con el empleo continuo durante el embarazo.
Trabajo de parto y parto: No procede.
Madres que amamantan: No se conoce si la rosuvastatina se excreta en la leche humana, pero una pequeña cantidad de otro fármaco de esta misma clase pasa a la leche humana. En las ratas, la concentración de la rosuvastatina en la leche materna es 3 veces mayor que la concentración plasmática; sin embargo, las concentraciones del fármaco en la leche materna no reflejan con precisión las concentraciones en la leche materna humana. Debido a que otro fármaco de esta misma clase pasa a la leche humana y debido a que los inhibidores del a HMG-CoA reductasa tienen el potencial para causar reacciones adversas serias en niños que amamantan, las mujeres que requieran del tratamiento con la rosuvastatina deben ser aconsejadas de no amamantar a sus bebés (ver Contraindicaciones [4]).
Uso pediátrico: No procede.
Uso geriátrico: De los 10,275 pacientes en los estudios clínicos con la rosuvastatina, 3159 (31%) tenían 65 años de edad y más y 698 (6.8%) tenían 75 años y más. No se observaron diferencias generales en la seguridad y efectividad entre estos sujetos y los sujetos más jóvenes, y en otra experiencia clínica reportada no se ha identificado ninguna diferencia en la respuesta entre los pacientes ancianos y jóvenes, pero no se puede descartar una mayor sensibilidad de algunos individuos mayores. Los pacientes ancianos tienen un riesgo mayor de miopatía y la rosuvastatina debe prescribirse con precaución en los ancianos (ver Advertencias y precauciones [5.1] y Farmacología clínica [12.3]).
Deterioro renal: La exposición a la rosuvastatina no está influida por el deterioro renal leve a moderado (CLcl ≥30 mL/min/1.73 m2); sin embargo, la exposición a la rosuvastatina se incrementa a un grado clínicamente significativo en pacientes con deterioro renal grave que no se están sometiendo a hemodiálisis (ver Dosis y administración [2.7], Advertencias y precauciones [5.1] y Farmacología clínica [12.3]).
Deterioro hepático: La rosuvastatina está contraindicada en pacientes con enfermedad hepática aguda, que puede incluir elevaciones de las concentraciones de las transaminasas hepáticas persistentes e inexplicadas. Se sabe que la enfermedad hepática crónica alcohólica aumenta la exposición a la rosuvastatina. La rosuvastatina debe ser utilizada con precaución en estos pacientes (ver Contraindicaciones [4], Advertencias y precauciones [5.2] y Farmacología clínica [12.3]).
Pacientes asiáticos: En los estudios de farmacocinética se ha demostrado un aumento de aproximadamente 2 veces la exposición mediana a la rosuvastatina en sujetos asiáticos cuando se comparó con los controles caucásicos. La dosis de la rosuvastatina puede ajustarse en pacientes asiáticos (ver Dosis y administración [2.4] y Farmacología clínica [12.3]).
Abuso y dependencia del fármaco: No procede.
Información para asesorar al paciente
Efectos sobre el músculo esquelético: Se debe recomendar a los pacientes reportar inmediatamente el dolor, hipersensibilidad o debilidad musculares inexplicadas, particularmente si se acompañan de malestar o fiebre.
Uso concomitantes de antiácidos: Cuando se administra la rosuvastatina con antiácidos que combinan el hidróxido de aluminio y magnesio, el antiácido debe tomarse cuando menos 2 horas después de la administración de la rosuvastatina.
Embarazo: Si una paciente se embaraza mientras está tomando este medicamento, la paciente debe ser informada sobre el peligro potencial al feto y la falta de beneficio clínico conocido con el empleo continuo durante el embarazo.
Enzimas hepáticas: Se recomienda que las enzimas hepáticas se revisen antes y a las 12 semanas después del inicio del tratamiento y con cualquier elevación de la dosis y periódicamente a partir de entonces.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis y administración
Información general sobre la dosificación: El rango de dosis de la rosuvastatina es de 5 a 40 mg oralmente, una vez al día. La dosis de inicio habitual es de 10-20 mg.
La rosuvastatina puede ser administrada como dosis única en cualquier momento del día, con o sin alimento. Cuando se inicie el tratamiento con la rosuvastatina o cuando se cambie de otro inhibidor de la HMG-CoA reductasa, primero se debe utilizar la dosis inicial de rosuvastatina apropiada y sólo después titular la dosis de acuerdo a la respuesta del paciente y el objetivo individualizado del tratamiento. Después del inicio o de la titulación de la rosuvastatina, se deben analizar las concentraciones de los lípidos de 2 a 4 semanas y se debe ajustar la dosis de acuerdo a esto. La dosis de 40 mg de rosuvastatina debe utilizarse solamente en los pacientes que no logren su C-LDL objetivo al utilizar la dosis de 20 mg (ver Advertencias y precauciones [5.1]).
Hipercolesterolemia familiar heterocigota en pacientes pediátricos (10 a 17 años de edad): No procede. Hipercolesterolemia familiar homocigota: Se recomienda una dosis de inicio de 20 mg una vez al día. La respuesta al tratamiento debe estimarse por las concentraciones del C-LDL.
Dosis en pacientes asiáticos: Se debe considerar el inicio del tratamiento con la rosuvastatina con 5 mg una vez al día en pacientes asiáticos (ver Uso en poblaciones especiales [8.8] y Farmacología clínica [12.3]).
Uso con ciclosporina, lopinavir/ritonavir o atazanavir/ritonavir: En pacientes que toman ciclosporina, la dosis de rosuvastatina debe limitarse a 5 mg una vez al día (ver Advertencias y precauciones [5.1] e Interacciones medicamentosas [7.1]). En pacientes que tomen una combinación de lopinavir y ritonavir o atazanavir y ritonavir, la dosis de rosuvastatina debe limitarse a 10 mg una vez al día. (ver Advertencias y precauciones [5.1] e Interacciones medicamentosas [7.3]).
Tratamiento concomitante con reductor de lípidos: El riesgo de efectos sobre los músculos esqueléticos puede aumentar cuando la rosuvastatina se utiliza en combinación con niacina o fenofibrato. Se debe considerar una reducción de la dosis de la rosuvastatina en este escenario (ver Advertencias y precauciones [5.1] e Interacciones medicamentosas [7.5, 7.6]). Se debe evitar la combinación con gemfibrozilo, debido al incremento de la exposición a la rosuvastatina con su uso concomitante. Si la rosuvastatina se emplea en combinación con el gemfibrozilo, la dosis de la rosuvastatina debe limitarse a 10 mg una vez al día (ver Advertencias y precauciones [5.1]) e Interacciones medicamentosas [7.2]).
Dosis en pacientes con deterioro renal grave: En los pacientes con deterioro renal grave (Clcr <30 mL/min/1.73 m2) sin someterse a hemodiálisis, la dosis de la rosuvastatina debe iniciarse con 5 mg una vez al día y no exceder de 10 mg una vez al día (ver Uso en poblaciones específicas [8.6] y Farmacología clínica [12.3]).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis: No existe un tratamiento específico en el caso de sobredosis. En este caso, el paciente debe ser tratado sintomáticamente y se deben instituir medidas de apoyo como se requieran. La hemodiálisis no aumenta significativamente la depuración de la rosuvastatina.
PRESENTACIÓN:
Cómo se suministra: La rosuvastatina se suministra en comprimidos (tabletas) recubiertos de 10 mg y 20 mg. Los comprimidos de 10 mg son circulares, de color rosa, con una de sus caras ranurada y lisa la otra. Los comprimidos de 20 mg son circulares, de color rosa, con ambas caras lisas. Caja con 14 comprimidos recubiertos.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a temperatura no mayor de 30ºC, protegido de la humedad y de la luz.
BIBLIOGRAFÍA:
Estudios clínicos
Hiperlipidemia y dislipidemia mixta: La rosuvastatina reduce el C total, C-LDL, ApoB, C no HDL y TG y aumenta el C-HDL en pacientes adultos con hiperlipidemia o dislipidemia mixta.
Estudio de rango de dosis: En un estudio multicéntrico, doble ciego, controlado con placebo, de rango de dosis en pacientes con hiperlipidemia, la rosuvastatina administrada como una dosis única diaria durante 6 semanas redujo significativamente el C total, C-LDL y ApoB con todo el rango de dosis (tabla 6).
|
Tabla 6. Respuesta a la dosis en pacientes con hiperlipidemia (cambio porcentual medio ajustado desde la evaluación inicial en la semana 6) |
|||||||
|
Dosis |
N |
C-total |
C-LDL |
C no HDL |
ApoB |
TG |
C-HDL |
|
Placebo |
13 |
-5 |
-7 |
-7 |
-3 |
-3 |
3 |
|
Rosuvastatina 5 mg |
17 |
-33 |
-45 |
-44 |
-38 |
-35 |
13 |
|
Rosuvastatina 10 mg |
17 |
-36 |
-52 |
-48 |
-42 |
-10 |
14 |
|
Rosuvastatina 20 mg |
17 |
-40 |
-55 |
-51 |
-46 |
-23 |
8 |
|
Rosuvastatina 40 mg |
18 |
-46 |
-63 |
-60 |
-54 |
-28 |
10 |
Estudio controlado con comparador activo: Se comparó la rosuvastatina con los inhibidores de la HMG-CoA reductasa atorvastatina, simvastatina y pravastatina en un estudio multicéntrico, abierto, de rango de dosis de 2240 pacientes con hiperlipidemia o dislipidemia mixta. Después de la aleatorización, se trató a los pacientes durante 6 semanas con una dosis única diaria con rosuvastatina, atorvastatina, simvastatina o pravastatina (figura 1 y tabla 1).
Figura 1. Cambio porcentual del C-LDL por dosis de rosuvastatina, atorvastatina, simvastatina y pravastatina en la semana 6 en pacientes con hiperlipidemia o dislipidemia mixta
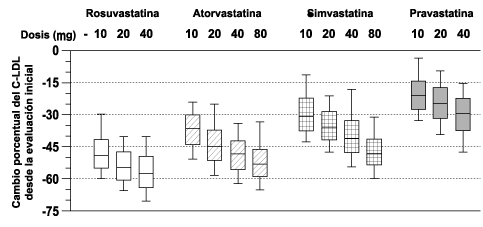
La gráfica de cajas y bigotes es una representación de los valores de las percentiles 25, 50 y 75; los bigotes representa los valores de las percentiles 10 y 90. C-LDL media de la evaluación inicial: 198 mg/dL.
|
Tabla 7. Cambio porcentual del C-LDL desde la evaluación inicial hasta la semana 6 (media LS*) por grupo de tratamiento (los tamaños de las muestras variaron de 156 a 167 pacientes por grupo) |
||||
|
Tratamiento |
10 mg |
20 mg |
40 mg |
80 mg |
|
Rosuvastatina |
-46† |
-52‡ |
-55§ |
--- |
|
Atorvastatina |
-37 |
-43 |
-48 |
-51 |
|
Simvastatina |
-28 |
-35 |
-39 |
-46 |
|
Pravastatina |
-20 |
-24 |
-30 |
--- |
|
* Los errores estándares correspondientes son de aproximadamente 1.00 † La rosuvastatina 10 mg redujo el C-LDL significativamente más que la atorvastatina 10 mg; pravastatina 10 mg, 20 mg y 40 mg; simvastatina 10 mg, 20 mg y 40 mg. (p>0.002) ‡ La rosuvastatina 20 mg redujo el C-LDL significativamente más que la atorvastatina 20 mg; pravastatina 20 mg y 40 mg; simvastatina 20 mg, 40 mg y 80 mg. (p>0.002) § La rosuvastatina 40 mg redujo el C-LDL significativamente más que la atorvastatina 40 mg; pravastatina 40 mg; simvastatina 40 mg, y 80 mg. (p>0.002) |
||||
Hipercolesterolemia familiar heterocigota: No procede.Hipertrigliceridemia
Estudio de dosis-respuesta: En un estudio doble ciego, controlado con placebo, de dosis-respuesta en pacientes con concentraciones iniciales de los triglicéridos (TG) de 273 a 817 mg/dL, la rosuvastatina administrada como dosis única diaria (5 a 40 mg) durante 6 semanas redujo significativamente las concentraciones séricas de TG (tabla 9).
|
Tabla 8. Cambio porcentual medio (mín, máx) desde la evaluación inicial de la dosis-respuesta en pacientes con hipertrigliceridemia primaria durante 6 semanas |
|||||
|
Dosis |
Placebo (n=26) |
Rosuvas-tatina 5 mg (n=25) |
Rosuvas-tatina 10 mg (n=23) |
Rosuvas-tatina 20 mg (n=27) |
Rosuvas-tatina 40 mg (n=25) |
|
TG |
1 (-40, 72) |
-21 (-58, 38) |
-37 (-65, 5) |
-37 (-72, 11) |
-43 (-80, 7) |
|
C no HDL |
2 (-13, 19) |
-29 (-43, -8) |
-49 (-59, -20) |
-43 (-74, 12) |
-51 (-62, -6) |
|
C-VLDL |
2 (-36, 53) |
-25 (-62, 49) |
-48 (-72, 14) |
-49 (-83, 20) |
-56 (-83, 10) |
|
C total |
1 (-13, 17) |
-24 (-40, -4) |
-40 (-51, -14) |
-34 (-61, -11) |
-40 (-51, -4) |
|
C- LDL |
5 (-30, 52) |
-28 (-71, 2) |
-45 (-59, 7) |
-31 (-66, 34) |
-43 (-61, -3) |
|
C-HDL |
-3 (-25, 18) |
3 (-38, 33) |
8 (-8, 24) |
22 (-5, 50) |
17 (-14, 63) |
Disbetalipoproteinemia primaria (hiperlipoproteinemia tipo III): En un estudio multicéntrico, doble ciego cruzado, 32 pacientes (27 con e2/e2 y 4 con la mutación de apo E [Arg145Cys]) con disbetalipoproteinemia (hiperlipoproteinemia tipo III) entraron en un período inicial de prueba de 6 semanas con dieta del cambio de estilo de vida terapéutico (CEVT) de la NCEP (siglas en inglés del Programa Nacional de Educación sobre el Colesterol) . Después del período de prueba con la dieta, los pacientes se aleatorizaron a una secuencia de tratamientos junto con la dieta de CEVT, cada período de la secuencia de 6 semanas: rosuvastatina 10 mg seguida de rosuvastatina 20 mg o rosuvastatina 20 mg, seguida de rosuvastatina 10 mg. La rosuvastatina redujo el C no HDL (criterio de valoración principal) y la concentración del resto de las lipoproteínas remanentes circulantes. Los resultados se muestran en la siguiente tabla.
|
Tabla 9. Efectos modificadores de los lípidos de la rosuvastatina 10 mg y 20 mg en la disbetalipoproteinemia primaria (hiperlipoproteinemia tipo III) después de 6 semanas por cambio porcentual de la mediana |
|||
|
Valor de la mediana en la evaluación inicial (mg/ dL) |
Cambio porcentual del valor de la mediana (IC 95%) desde la evaluación inicial con rosuvastatina 10 mg |
Cambio porcentual del valor de la mediana (IC 95%) desde la evaluación inicial con rosuvastatina 20 mg |
|
|
C total |
342.5 |
-43.3 (-46.9, -37.5) |
-47.6 (-51.6, -42.8) |
|
Triglicéridos |
503.5 |
-40.1 (-44.9, -33.6) |
-43.0 (-52.5, -33.1) |
|
C no HDL |
294.5 |
-48.2 (-56.7, -45.6) |
-56.4 (-61.4, -48.5) |
|
C VLDL + C-IDL |
209.5 |
-46.8 (-53.7, -39.4) |
-56.2 (-67.7, -43.7) |
|
C-LDL |
112.5 |
-54.4 (-59.1, -47.3) |
-57.3 (-59.4, -52.1) |
|
C-HDL |
35.5 |
10.2 (1.9, 12.3) |
11.2 (8.3, 20.5) |
|
C-RLP |
82.0 |
-56.4 (-67.1, -49.0) |
-64.9 (-74.0, -56.6) |
|
Apo-E |
16.0 |
-42.9 (-46.3, -33.3) |
-42.5 (-47.1, -35.6) |
Hipercolesterolemia familiar (HF) homocigota: Estudio de titulación de la dosis: en un estudio abierto, de titulación forzada, se evaluó la respuesta de pacientes con HF homocigota (n=40, 8-63 años) a la rosuvastatina 20 y 40 mg titulada en un intervalo de 6 semanas. En la población general, la reducción del C-LDL desde la evaluación inicial fue del 22%. Cerca de un tercio de los pacientes se beneficiaron del aumento de su dosis de 20 mg a 40 mg con reducción adicional mayor del 6%. En los 27 pacientes con al menos una reducción del 15% del C-LDL, la reducción media de C-LDL fue del 30% (reducción mediana del 28%). Entre los 13 pacientes con una reducción del C-LDL <15%, 3 no tuvieron ningún cambio ni un incremento del C-LDL. Se observaron reducciones del C-LDL de 15% o mayores en 3 de 5 pacientes con estado conocido negativo del receptor.
Pacientes pediátricos con hipercolesterolemia familiar heterocigota: No procede.
Retraso de la progresión de la aterosclerosis: En un estudio aleatorizado, doble ciego y controlado con placebo, se evaluó el efecto del tratamiento con la rosuvastatina sobre la aterosclerosis carotídea por ultrasonido de modo B en pacientes con C-LDL elevada, con riesgo bajo (riesgo de Framingham <10% durante 10 años) para enfermedad coronaria sintomática y con aterosclerosis subclínica evidenciada por el grosor de la capa íntima-media (IM) de la carótida. Se aleatorizaron a 984 pacientes (de los cuales se analizaron 876) a rosuvastatina 40 mg o placebo una vez al día, en una proporción 5:2.
Se utilizó el ultrasonido de las paredes carotídeas para determinar la tasa anualizada del cambio por paciente desde la evaluación inicial hasta los dos años y el grosor de la capa IM de la carótida máximo de 12 segmentos. La diferencia estimada de la tasa del cambio del grosor IM de la carótida máximo analizado en los 12 sitios de la arteria carotídea entre los pacientes tratados con la rosuvastatina y los pacientes tratados con el placebo fue de -0.0145 mm/año (IC 95% -0.0196, -0.0093; p<0.0001).
La tasa anualizada de cambio desde la evaluación inicial del grupo placebo fue de +0.0131 mm/ año (p<0.0001). La tasa anualizada de cambio desde la evaluación inicial del grupo tratado con la rosuvastatina fue de -0.0014 mm/año (p=0.32). A nivel del paciente individual, el grupo tratado con la rosuvastatina, el 52.1% de los pacientes demostraron una ausencia de progresión de la enfermedad (definida como una tasa de cambio anualizada negativa), en comparación con el 37.7% de los pacientes en el grupo del placebo.
Prevención primaria de la enfermedad cardiovascular: No procede.
LEYENDAS DE PROTECCIÓN:
Manténgase fuera del alcance de los niños.


