
XTANDI
ENZALUTAMIDA
Cápsula blanda
1 Envase(s), 40 /120 Cápsulas blandas, 40 Miligramos
COMPOSICIÓN:
Fórmula cualicuantitativa
Cada CÁPSULA de 40 mg contiene:
Enzalutamida 40 mg
Excipientes: Macrogolglicéridos de Caprilocaproílo, Butilhidroxianisol, Butilhidroxitolueno.
INDICACIONES:
XTANDI está indicado para el tratamiento de pacientes (hombres adultos) con cáncer de próstata metastásico resistente a la castración (CPRC).
Acción terapéutica: Tratamiento antineoplásico endocrino. Código ATC L02BB.
MECANISMO DE ACCIÓN:
Características farmacológicas
Mecanismo de acción: Se sabe que el cáncer de próstata es sensible a los andrógenos y responde a la inhibición de la señalización de los receptores androgénicos. La señalización de los receptores androgénicos sigue favoreciendo la progresión de la enfermedad aunque las concentraciones plasmáticas de andrógenos sean bajas o incluso indetectables. La estimulación del crecimiento de las células tumorales a través de los receptores androgénicos requiere la localización nuclear y la unión al ADN. Enzalutamida es un inhibidor potente de la señalización de los receptores androgénicos que bloquea varios pasos en la vía de señalización del receptor androgénico. Enzalutamida inhibe de manera competitiva la unión de los andrógenos a los receptores androgénicos, inhibe la translocación nuclear de los receptores activados e inhibe la asociación del receptor androgénico activado al ADN, incluso en situación de sobreexpresión del receptor androgénico y de células de cáncer de próstata resistentes a los anti-andrógenos. El tratamiento con Enzalutamida reduce el crecimiento de las células de cáncer de próstata, y puede provocar la muerte de las células cancerosas y la regresión del tumor. En estudios preclínicos, Enzalutamida carece de actividad agonista de los receptores androgénicos.
Acción farmacocinética: Se evaluó la farmacocinética de la Enzalutamida en pacientes con cáncer de próstata y en sujetos sanos de sexo masculino. La semivida terminal media (t112) para la Enzalutamida en pacientes luego de una única dosis oral es de 5,8 días (intervalo de 2,8 a 10,2 días), y en aproximadamente un mes se alcanza un estado de equilibrio. Con la administración oral diaria, la Enzalutamida se acumula aproximadamente 8,3 veces en relación con una dosis única. Las fluctuaciones diarias en las concentraciones plasmáticas son bajas (cociente pico-valle: 1,25).
El aclaramiento de la Enzalutamida se realiza principalmente a través del metabolismo hepático, mediante la producción de un metabolito activo que circula en aproximadamente la misma concentración plasmática que la Enzalutamida.
Absorción: Las concentraciones plasmáticas máximas (Cmáx) de Enzalutamida en pacientes se observan entre 1 y 2 horas después de la administración. Según un estudio de equilibrio de masa en seres humanos, se calcula que la absorción oral de la Enzalutamida es, como mínimo, del 84,2%. La Enzalutamida no es un sustrato de los transportadores de eflujo gp-P o BCRP. En estado de equilibrio, los valores medios de Cmáx para la Enzalutamida y su metabolito activo son de 16,6 μg/ml (coeficiente de variación [CV] del 23% y 12,7 μg/ml (CV del 30%), respectivamente.
Los alimentos no tienen un efecto de importancia clínica sobre el grado de absorción. En ensayos clínicos, Enzalutamida se administró independientemente de los alimentos.
Distribución: El volumen aparente de distribución (V/F) medio de la Enzalutamida en pacientes luego de una dosis única oral es de 110 L (CV del 29%). El volumen de distribución de Enzalutamida es mayor que el volumen de agua corporal total, lo que indica una amplia distribución extravascular. Los estudios realizados en roedores indican que la Enzalutamida y su metabolito activo pueden atravesar la barrera hemato-encefálica.
Entre el 97% y el 98% de la Enzalutamida se une a las proteínas plasmáticas, principalmente la albúmina. El metabolito activo se une a las proteínas plasmáticas en un 95%. No hubo un desplazamiento del enlace proteínico entre Enzalutamida y otros medicamentos altamente enlazados (Warfarina, Ibuprofeno y Ácido Salicílico) in vitro.Metabolismo: La Enzalutamida se metaboliza ampliamente. En el plasma humano hay dos metabolitos principales: N-desmetil Enzalutamida (activo) y un derivado de Ácido Carboxílico (inactivo). La Enzalutamida es metabolizada por el CYP2C8 y, en menor grado, por el CYP3A4/5, los cuales participan en la formación del metabolito activo.
In vitro, N-desmetil Enzalutamida es metabolizado en el metabolito de ácido carboxilíco por la carboxilesterasa 1, la cual juega también un papel menor en el metabolismo de Enzalutamida en el metabolito de Ácido Carboxílico. La N- desmetil Enzalutamida no fue metabolizada por CYP en vitro. En condiciones de uso clínico, XTANDI es un inductor potente del CYP3A4 y un inductor moderado del CYP2C9 y CYP2Cl9, y carece de efectos de importancia clínica sobre el CYP2C8.
Eliminación y excreción: El aclaramiento aparente (CL/F) medio de la Enzalutamida en pacientes oscila entre 0,520 y 0,564 Vh. Luego de la administración oral de 14C-Enzalutamida, el 84,6% de la radiactividad se recupera 77 días después de la administración de la dosis: el 71,0% se recupera en la orina (principalmente en forma de metabolito inactivo, con cantidades mínimas de Enzalutamid y del metabolito activo) y el 13,6% en las heces (0,39% de la dosis en forma de Enzalutamida inalterada).
Los datos in vitro indican que la Enzalutamida no es un sustrato para el polipéptido transportador de aniones orgánicos lBl (OATPlBl), OATP1B3, ni del transportador de cationes orgánicos 1 (OCTI); y N-desmetil Enzalutamida no es un substrato para P-gp o BCPRP.
Los datos in vitro indican que la Enzalutamida y sus metabolitos principales no inhiben los siguientes transportadores en concentraciones clínicamente pertinentes: OATPIBI, OATP1B3, OCT2 o OATI.
Linealidad: No se observaron desviaciones importantes de la proporcionalidad a la dosis en el rango de dosis de 40 a 160 mg. Los valores de Cmín para el estado estacionario de Enzalutamida y el metabolito activo en pacientes individuales permanecieron constantes durante más de un año de terapia crónica, demostrando la farmacocinética lineal con respecto al tiempo una vez alcanzado el estado estacionario.
Poblaciones especiales
Ancianos: De los 1671 pacientes del ensayo fase 3 que recibieron Enzalutamida, 1261 pacientes (75%) tenían 65 o más años de edad y 516 (31 %) tenían 7 5 o más años. No se observó ningún efecto clínicamente relevante de la edad sobre la farmacocinética de Enzalutamida en el análisis farmacocinético de la población. No se observaron diferencias generales respecto a la seguridad o la eficacia entre estos pacientes ancianos y los pacientes más jóvenes.
Población pediátrica: No hay uso relevante de Enzalutamida en la población pediátrica en la indicación de tratamiento de hombres adultos con cáncer de próstata metastásico resistente a la castración.
Género: La Enzalutamida no está indicada para su uso en mujeres. No se ha evaluado la farmacocinética de la Enzalutamida en mujeres.
Raza: La mayoría de los pacientes del ensayo clínico aleatorizado eran de raza blanca (> 84%). En base a los datos farmacocinéticos de un estudio en pacientes japoneses con cáncer de próstata, no hubo diferencias clínicamente relevantes en la exposición entre japoneses y caucásicos. Los datos son insuficientes para evaluar las posibles diferencias en la fannacocinética de la Enzalutamida en otras razas.
Insuficiencia renal: No se han realizado estudios formales de Enzalutamida en pacientes con insuficiencia renal. Se excluyó de los ensayos clínicos a los pacientes con un valor de creatinina sérica > 177 μmol/l (2 mg/dl). Según un análisis de farmacocinética poblacional, no es necesario ajustar la dosis en los pacientes con valores calculados de aclaramiento de creatinina (CrCL) 2: 30 ml/min (estimados mediante la fórmula de Cockcroft y Gault). XTANDI no se ha evaluado en pacientes con insuficiencia renal grave (CrCL < 30 ml/min) o enfermedad renal terminal, y se aconseja precaución al tratar a estos pacientes. Es poco probable que la Enzalutamida se elimine significativamente mediante hemodiálisis intermitente o diálisis peritoneal ambulatoria continua.Insuficiencia hepática: La farmacocinética de la Enzalutamida se evaluó en sujetos con insuficiencia hepática inicial leve (N =6) o moderada (N =8) (clase A y B de Child-Pugh, respectivamente) comparados con 14 sujetos de control con una función hepática normal. Luego de administrar una dosis oral única de 160 mg de Enzalutamida, el AUC y la Cmáx de la Enzalutamida en sujetos con insuficiencia leve aumentaron un 5% y un 24%, respectivamente, y el AUC y la Cmáx de la Enzalutamida en sujetos con insuficiencia moderada aumentó un 29% y disminuyó 11 %, respectivamente, en comparación con los sujetos de control sanos. Para la suma de Enzalutamida libre más el metabolito activo libre, el AUC y la Cmáx en sujetos con insuficiencia leve aumentaron un 14% y un 19%, respectivamente, y el AUC y la Cmáx en sujetos con insuficiencia moderada aumentaron un 14% y disminuyeron un 17%, respectivamente, en comparación con los sujetos de control sanos. En general, los resultados indican que no es necesario un ajuste de dosis para pacientes con insuficiencia hepática inicial leve o moderada.
Se excluyó de los ensayos clínicos a los pacientes con insuficiencia hepática grave (clase C de Child-Pugh). No se recomienda administrar XTANDI con pacientes con insuficiencia hepática grave (clase C de Child-Pugh). Efectos farmacodinámicos: En un ensayo clínico en fase 3 de pacientes en quienes fracasó la quimioterapia previa con Docetaxel, el 54% de los pacientes tratados con Enzalutamida, en comparación con el 1,5% de los pacientes que recibieron placebo, presentó una disminución de las concentraciones de PSA de al menos un 50% con respecto a los valores iniciales.
Eficacia clínica y seguridad: La eficacia de Enzalutamida fue establecida en dos estudios clínicos fase 3, multicéntricos, controlados con placebo y aleatorizados [CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)] de pacientes con cáncer prostático metastásico progresivo que fracasaron con la terapia de privación de andrógenos [análogo de la hormona liberadora de la hormona luteinizante (LHRH) o después de orquiectomía bilateral]. El estudio PREV AIL reclutó a pacientes que nunca habían recibido quimioterapia; mientras que el estudio AFFIRM reclutó a pacientes que habían recibido Docetaxel previamente. Todos los acientes continuaron con un análogo de LHRH o tuvieron una orquiectomía bilateral previa. En el grupo con tratamiento activo, Enzalutamida fue administrada oralment a una dosis de 160 mg diarios en ambas pruebas clínicas, los pacientes recibieron placebo en el grupo de control y los pacientes podían, aunque no era requerido, tomar Prednisona (dosis diaria máxima permitida fue 10 mg de Prednisona o equivalente). Los cambios en la concentración sérica de PSA no siempre predicen independientemente un beneficio clínico. Por tanto, en ambos estudios fue recomendado que los pacientes fueran mantenidos con sus tratamientos de estudio hasta que los criterios de escontinuación fueran satisfechos tal como se especificó abajo para cada estudio.
Estudio MDV3100-03 (PREVAIL) (pacientes sin quimioterapia previa)
Un total de 1.717 pacientes sin quimioterapia previa, asintomáticos o levemente sintomáticos fueron aleatorizados 1:1 para recibir Enzalutamidavía oral a una dosis de 160 mg una vez al día (n = 872) o placebo por vía oral una vez al día (n= 845). Se admitieron pacientes con enfermedad visceral, pacientes con antecedentes de insuficiencia cardíaca leve a moderada (NYHA Clase 1 o 2), y pacientes que toman medicamentos asociados con la reducción del umbral de convulsiones. Al menos 1 medicación concomitante conocida por disminuir el umbral convulsivo fue recibida por el 52,5% de los pacientes en el grupo tratado con Enzalutamida en comparación con el 45,9% de los pacientes del grupo tratado con placebo. Se excluyeron los pacientes con antecedentes de convulsiones o una condición que pudiera predisponer a convulsiones y pacientes con dolor moderado o grave de cáncer de próstata. El tratamiento del estudio continuó hasta la progresión de la enfermedad (evidencia de la progresión radiográfica, un evento relacionado con el esqueleto, o la progresión clínica) y la iniciación de una quimioterapia citotóxica o de un agente en investigación, o hasta que apareciera una toxicidad inaceptable. Las características demográficas del paciente y las características de la enfermedad de línea de base fueron balanceadas entre los grupos de tratamiento. La mediana de la edad fue de 71 años (rango 42-93) y la distribución racial fue de 77% caucásicos, 10% asiáticos, 2% negros y 11 % de otras razas o razas desconocidas. El 68% de los pacientes tenían una puntuación de estado funcional ECOG de O y 32% de los pacientes tenía un estado funcional ECOG 1. La evaluación del dolor basal era 0-1 (asintomático) en el 67% de los pacientes y 2-3 (ligeramente sintomáticos) en el 32% de los pacientes, tal como se define por el Brief Pain Inventory Short Form (el peor dolor en las últimas 24 horas sobre una escala de 0 a 10). Aproximadamente el 45% de los pacientes tenían enfermedad de los tejidos blandos medible al inicio del estudio, y el 12% de los pacientes tenían metástasis visceral (pulmonar y/o hepáticas). Las variables de eficacia co-primarias fueron la supervivencia global y la supervivencia libre de progresión radiográfica (PRSA). Además de los criterios de valoración principales, el beneficio también se evaluó mediante el momento de inicio de la quimioterapia citotóxica, la mejor respuesta global de los tejidos blandos, el tiempo hasta el primer evento relacionado con el esqueleto, la respuesta del PSA (~ 50% de disminución respecto al valor inicial), el tiempo hasta la progresión del PSA, y el tiempo hasta la degradación total de la puntuación FACT-P. La progresión radiográfica se evaluó con el uso de estudios de imágenes secuenciales tal como se define en los criterios del Grupo de Trabajo 2 sobre Ensayos Clínicos de Cáncer de Próstata (PCWG2) (para las lesiones óseas) y / o en los Criterios de Evaluación de la Respuesta en Tumores Sólidos (RECIST v 1.1) (para las lesiones de tejidos blandos). El análisis del rPFS utilizó la evaluación radiográfica revisada centralmente de la progresión. En el análisis intermedio pre-especificado para la supervivencia general, el tratamiento con Enzalutamida demostró una mejoría estadísticamente significativa en la supervivencia global en comparación con el tratamiento con placebo, con una reducción del 29,4% en el riesgo de muerte [HR = 0,706, (95% CI: 0,596; ,837), p <0,0001]. En el análisis intermedio, 27,6% (241 de 872) de los pacientes tratados con Enzalutamida, en comparación con 35,4% (299 de 845) de los pacientes tratados con placebo, había muerto. La mediana de supervivencia global estimada fue 32,4 meses (95% CI:30.1, no alcanzado) en los pacientes tratados con Enzalutamida y fue de 30,2 meses (95% CI: 28.0, no alcanzado) en los pacientes tratados con placebo (Tabla 1 y Figuras 1 y 2). Además, el 40,3% de los pacientes tratados con Enzalutamida y 70,6% de los pacientes tratados con placebo recibieron terapias subsecuentes con un beneficio demostrado de la supervivencia. Los resultados de un análisis exploratorio actualizado de la supervivencia global realizado antes del cambio del paciente a un tratamiento abierto con Enzalutamida, que incluyó 116 eventos adicionales, fueron consistentes con el análisis intermedio (Tabla 1 y Figura 1 ).
|
Tabla1: Supervivencia global de los pacientes tratados con Enzalutamidao |
||
|
Placebo en el estudio PREVAIL (análisis por intención de tratamiento) |
Enzalutamida (N = 872) |
Placebo (N = 845) |
|
Análisis intermedio pre-especificado |
||
|
Número de muertes(%) |
241 (27.6% |
299 (35.4%) |
|
Mediana, meses (IC de 95%) |
32.4 (30.1, NR) |
30.2 (28.0, NR) |
|
Valor pª |
< 0.0001 |
|
|
Cociente de riesgo (IC de 95%) b |
0.71 (0.60, 0.84) |
|
|
ªEl valor p se deriva de una prueba no estratificada de rango logarítmico. h El cociente de riesgo se deriva de un modelo de riegos proporcionales no estratificado. El cociente de riego < 1 favorece a Enzalutamida. NR, no alcanzado. |
||
Figura 1: Curvas de Supervivencia Global de Kaplan-Meier en el estudio PREVAIL (Análisis por intención de tratamiento)
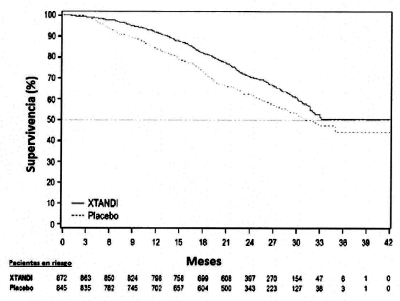
Figura 2: Supervivencia global por Subgrupo: Cociente de Riesgo e Intervalo de Confianza de 95% en el estudio PREVAIL (Análisis por Intención de Tratamiento)

En el análisis rPFS pre-especificado, una mejora estadísticamente significativa fue demostrada entre los grupos de tratamiento con una reducción del 81,4% en el riesgo de progresión radiográfica o muerte [HR = 0.186 (IC del 95%: 0.149, 0.231), p <0,0001]. Ciento dieciocho (14%) de los pacientes tratados con Enzalutamida y 321 (40%) de los pacientes tratados con placebo tuvo un evento. La mediana del rPFS no se alcanzó (IC del 95%: 13,8, no alcanzado) en el grupo tratado con Enzalutamida y fue de 3,9 meses (IC del 95%: 3,7, 5,4) en el grupo tratado con placebo (Figura 3). Se observó un beneficio consistente en el rPFS en todos los subgrupos pre-especificados de pacientes (por ejemplo, edad, rendimiento ECOG basal, PSA y LDH basales, puntuación Gleason al momento del diagnóstico, y enfermedad visceral en la selección). Un análisis rPFS pre- especificado de seguimiento basado en la evaluación del investigador de la progresión radiográfica demostró una mejoría estadísticamente significativa entre los grupos de tratamiento con una reducción del 69,3% en el riesgo de progresión radiográfica o muerte [HR CI = 0,307 (IC de 95%: 0.267, 0.353), p <0,0001]. La mediana de los rPFS fue de 19,7 meses en el grupo con Enzalutamida y 5,4 meses en el grupo placebo.
Figura 3: Curvas de supervivencia sin progresión radiográfica de Kaplan- Meier en el Estudio PREVAIL (Análisis por intención de tratamiento)
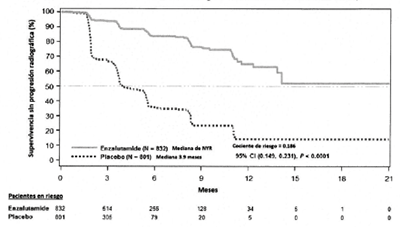
En el momento del análisis primario había 1633 pacientes aleatorizados.
Además de las variables co-primarias de eficacia, mejoras estadísticamente significativas también se demostraron en los siguientes criterios de valoración definidos de forma prospectiva.
La mediana de tiempo hasta el inicio de la quimioterapia citotóxica fue 28,0 meses para los pacientes que recibieron Enzalutamida y 10,8 meses para los pacientes que recibieron placebo (HR = 0,350, IC 95%: [0.303, 0.403], p<0,0001).
La proporción de pacientes tratados con Enzalutamida con enfermedad medible al inicio del estudio que tuvo una respuesta objetiva de tejidos blandos fue 58,8% (IC del 95%: 53,8, 63,7) en comparación con 5,0% (IC del 95%: 3,0, 7,7) de los pacientes que recibieron placebo. La diferencia absoluta en la respuesta objetivo del tejido blando entre Enzalutamida y placebo fue 53,9% (IC del 95%: 48,5%, 59,1%, p <0,0001). Respuestas completas fueron reportadas en 19,7% de los pacientes tratados con Enzalutamida en comparación con 1,0% de los pacientes tratados con placebo, y respuestas parciales se registraron en el 39, 1 % de los pacientes tratados con Enzalutamida frente a 3,9% de los pacientes tratados con placebo.
Enzalutamida disminuyó significativamente el riesgo del primer evento relacionado con el esqueleto en un 28% [HR = 0.718 (IC del 95%: 0.610, 0.844) valor p <0,0001]. Un evento relacionado con el esqueleto se definió como radioterapia o cirugía ósea por cáncer de próstata, fractura ósea patológica, compresión de la médula espinal, o cambio de la terapia antineoplásica para tratar el dolor de huesos. El análisis incluyó 587 eventos esqueléticos relacionados, de los cuales 389 eventos (66.3%) eran radiación al hueso, 79 eventos (13.5%) fueron compresión de la médula espinal, 70 eventos (11,9%) eran fractura ósea patológica, 45 eventos (7,6%) fueron cambio en la terapia antineoplásica para tratar el dolor de hueso, y 22 eventos (3,7%) fueron cirugía ósea.
Los pacientes que reciben Enzalutamida demostraron una tasa de respuesta PSA total significativamente mayor (definida como una reducción 2: 50% del valor inicial), en comparación con los pacientes que recibieron placebo, 78,0% frente a 3,5% (diferencia= 74,5%, p <0,0001). La mediana del tiempo hasta la progresión del PSA por los criterios del PCWG2 fue de 11,2 meses para los pacientes tratados con Enzalutamida y 2,8 meses para los pacientes que recibieron placebo [HR = 0,169, (IC del 95%: 0.147, 0.195), p<0,0001].
El tratamiento con Enzalutamida disminuyó el riesgo de degradación FACT-P en un 37,5% en comparación con placebo (p <0,001). La mediana del tiempo hasta la degradación en FACT-P fue de 11,3 meses en el grupo con Enzalutamida y de 5,6 meses en el grupo placebo.Estudio CRPC2 (AFFIRM) (pacientes que recibieron quimioterapia previamente).
En un ensayo clínico en fase 3, multicéntrico, aleatorizado y controlado con placebo, se evaluaron la eficacia y la seguridad de Enzalutamida en pacientes con cáncer de próstata metastásico resistente a la castración que habían recibido Docetaxel y un análogo de LHRH o habían sido sometidos a orquiectomía. En total, se aleatorizó a 1199 pacientes en una proporción 2: 1 para recibir Enzalutamida por vía oral en dosis de 160 mg una vez al día (N = 800) o placebo por vía oral una vez al día (N = 399). Los pacientes podían utilizar Prednisona, aunque esto no era un requisito. Los pacientes aleatorizados a cualquiera de los grupos continuaron el tratamiento hasta la progresión de la enfermedad (definida como confirmación radiográfica de la progresión o la aparición de un evento relacionado con el sistema óseo) y el inicio de un nuevo tratamiento antineoplásico sistémico, la aparición de una toxicidad inaceptable o el retiro.
Los siguientes datos emográficos y características basales de la enfermedad de los pacientes estaban equilibrados entre los dos grupos de tratamiento. La mediana de la edad fue de 69 años (intervalo: 41-92), y la distribución racial fue: 92,7% de raza blanca, 3,9% de raza negra, 1,1% de raza asiática y 2,1% otra. La puntuación del estado funcional del ECOG (Eastem Cooperative Oncology Group) fue de 0-1 en el 91,5% de los pacientes y de 2 en el 8,5% de los pacientes; el 28,4% tuvo una puntuación media en el Brief Pain lnventory (Cuestionario breve de dolor) 2: 4 (media del peor dolor comunicado por el paciente durante las 24 horas previas calculado durante los siete días previos a la aleatorización). La mayoría de los pacientes (91,2%) tenía metástasis en los huesos y el 23,2% tenía afectación visceral, pulmonar y/o hepática. Al momento del ingreso al estudio, 41 % de los pacientes aleatorizados tenían progresión PSA solamente, mientras que el 59% de los pacientes tuvieron progresión radiográfica. Cincuenta y un porciento (51 %) de los pacientes tomaban bifosfonatos en la línea de base.
El análisis intermedio pre-especificado en el protocolo mostró, luego de 520 muertes, una superioridad estadísticamente significativa en la mediana de la supervivencia global en los pacientes tratados con Enzalutamida en comparación con los que recibieron el placebo (Tabla 2 y Figura 4).
|
Tabla 2. Supervivencia global de pacientes tratados con Enzalutamida o placebo en el estudio AFFIRM (Análisis por intención de tratamiento) |
||
|
Enzalutamida (N = 800) |
Placebo |
|
|
Muertes(%) |
308 (38,5%) |
212 (53,1%) |
|
Mediana de la supervivencia (meses) (IC del 95%) |
18,4 (17,3; NA) |
13,6 (11,3; 15,8) |
|
valor pª |
< 0,000 |
1 |
|
Cociente de riesgo (IC del 95%)b |
0,631 (0,529; |
0,752) |
|
ªEl valor p se obtiene a partir de una prueba de rangos logarítmicos estratificada de acuerdo con la puntuación del estado funcional de ECOG (0-1 frente a 2) y la puntuación media para el dolor (puntuación< 4 frente a 2: 4). b El cociente de riesgo se obtiene a partir de un modelo de riesgos proporcionales estratificado. Un cociente de riesgo < 1 favorece a Enzalutamida. |
||
Figura 4: Curvas de Kaplan-Meier para la supervivencia global (Análisis por intención de tratamiento)
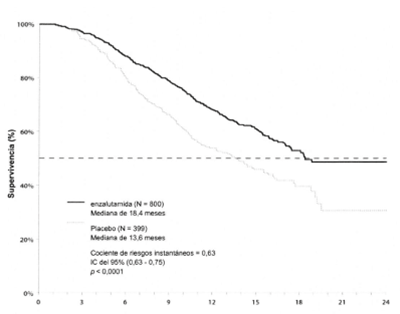
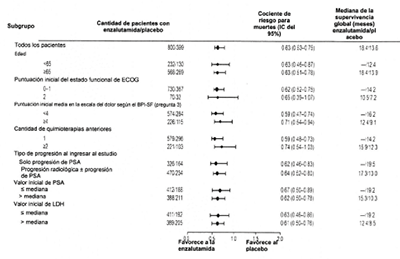
Figura 5: Supervivencia Global por subgrupos en el estudio AFFIRM: Cociente de riesgo e intervalo de confianza de 95%
Además de la mejoría observada en la supervivencia global, los criterios secundarios claves (progresión PSA, supervivencia sin progresión radiográfica y el tiempo hasta el primer evento relacionado con el esqueleto) favorecieron a Enzalutamida y fueron estadísticamente significativos luego de realizar los ajustes necesarios para múltiples análisis, como se muestra a continuación: La supervivencia sin progresión radiológica evaluada por el investigador usando RECIST v 1.1 para tejido blando y aparición de 2 o más lesiones óseas en la exploración ósea fue de 8,3 meses para pacientes tratados con XTANDI y de 2,9 meses para pacientes que recibieron placebo (cociente de riesgo = 0,404; IC del95% : [0,350; 0,466]); p < 0,0001). El análisis involucró 216 muertes sin progresión documentada y 645 eventos documentados de progresión, de los cuales 303 (47%) se debieron a progresión del tejido blando, 268 (42%) se debieron a progresión de lesión ósea y 74 (11 %) se debieron a lesiones óseas y del tejido blando. La disminución confirmada del antígeno prostático específico (PSA) del 50% o 90% fue del 54,0% y el 24,8% en pacientes tratados con Enzalutamida y del 1,5% y 0,9% en pacientes que recibieron el placebo (p < 0,0001). La mediana del tiempo hasta la progresión del PSA fue de 8,3 meses para pacientes tratados con Enzalutamida y de 3,0 meses para pacientes que recibieron placebo (cociente de riesgo= 0,248; IC del 95%: [0,204, 0,303];p < 0,0001).
La mediana de tiempo hasta el primer evento relacionado con el sistema óseo fue de 16,7 meses para pacientes tratados con Enzalutamida y de 13,3 meses para pacientes que recibieron placebo (cociente de riesgo = 0,688; IC del 95%: [0,566,0,835]; p < 0,0001). Se definió evento relacionado con el sistema óseo como radioterapia o cirugía ósea, fractura ósea patológica, compresión medular o cambio de tratamiento antineoplásico para tratar el dolor óseo. El análisis involucró 448 eventos relacionados con el sistema óseo, de los cuales 277 eventos (62%) fueron radiación al hueso, 95 eventos (21 %) fueron compresión de la médula espinal, 47 eventos (10%) fueron fractura ósea patológica, 36 eventos (8%) fueron cambio en la terapia antineoplásica para tratar el dolor óseo y 7 eventos (2 %) fueron cirugía ósea.
La eficacia de Enzalutamida en pacientes que han recibido previamente abiraterona acetato no se ha estudiado.
La tasa de respuesta para calidad de vida (Evaluación Funcional de la Terapia del cáncer: Próstata; FACT-P) fue del 43,2% para pacientes tratados con Enzalutamida y del 18,3% para pacientes que recibieron placebo (p < 0,0001).
La tasa de respuesta radiográfica objetiva evaluada por el investigador (definida como la suma de respuestas completas y parciales) entre pacientes tratados con Enzalutamida fue del 28,9% comparada con una tasa de respuesta objetiva del 3,8% para pacientes que recibieron el placebo (p < 0,0001).
El riesgo de progresión del dolor se redujo en un 44% en los pacientes tratados con Enzalutamida en comparación con los pacientes que recibieron el placebo (cociente de riesgo= 0,56; IC del 95%: [0,41, 0,78]; p = 0,0004). La mediana del tiempo hasta la progresión del dolor fue de 13,8 meses para los pacientes que recibieron el placebo y no se alcanzó para los pacientes tratados con Enzalutamida. La progresión del dolor se definió como un aumento por encima del valor inicial en la evaluación del dolor FACT-P, lo que se confirmó mediante una segunda evaluación consecutiva obtenida 3 o más semanas después.
CONTRAINDICACIONES
XTANDI está contraindicado en pacientes con hipersensibilidad al principio activo o a cualquiera de los excipientes incluidos en la composición y en mujeres que están o puedan quedar embarazadas.
Este medicamento está contraindicado en personas menores de 18 años.
REACCIONES ADVERSAS
Las reacciones adversas más comunes son astenia I fatiga, sofocos, dolor de cabeza, y la hipertensión. Otras reacciones adversas importantes incluyen caídas, fracturas no patológicas, trastorno cognitivo y neutropenia.
Las convulsiones se produjeron en el 0,4% de los pacientes tratados con Enzalutamida y en el 0, 1 % de los pacientes tratados con placebo.
Casos raros de síndrome de encefalopatía posterior reversible han sido reportados en pacientes tratados con Enzalutamida.
Las reacciones adversas observadas durante los ensayos clínicos se enumeran a continuación por categoría de frecuencia. Las categorías de frecuencia se definen de la siguiente forma: Muy frecuentes (≥1/10); frecuentes (≥: 1/100 a <1110); poco frecuentes (≥: 1 / l.000 a < 1/ 100); poco frecuentes (≥: 1 /10.000 a < 1 /1.000); muy raras (<l / 10.000). Dentro de cada grupo de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
|
Tabla 3: Reacciones adversas identificadas en las pruebas clínicas Fase 3 |
|
|
Trastornos sanguíneos y del sistema |
Poco frecuentes: Leucopenia linfático |
|
Trastornos generales |
Muy frecuentes: Astenia/fatiga |
|
Trastornos psiquiátricos |
Frecuentes: Ansiedad |
|
Poco frecuentes: Alucinaciones visuales |
|
|
Trastornos del sistema nervioso |
Muy frecuentes: Cefalea |
|
Frecuentes: Deterioro de la memoria, amnesia, perturbación de la atención, síndrome de piernas inquietas |
|
|
Poco frecuentes: Trastorno cognitivo, convulsiones, |
|
|
Infrecuentes*: Síndrome de encefalopatía posterior reversible |
|
|
Trastornos del sistema reproductivo y mamarios |
Frecuentes: Ginecomastia |
|
Trastornos vasculares |
Muy frecuentes: Sofocos, hipertensión |
|
Trastornos de la piel y del tejido |
Frecuentes: Piel seca, prurito subcutáneo |
|
Trastornos músculo-esqueléticos y del tejido conectivo |
Frecuentes: Fracturas** |
|
Lesiones, intoxicación y complicaciones de los procedimientos |
Frecuentes: Caídas |
|
* Informes espontáneos procedentes de la experiencia posterior a la comercialización ** Incluyen todas las fracturas excepto las fracturas patológicas |
|
Convulsiones: En los estudios clínicos fase 3,7 pacientes (0,4%) experimentaron una convulsión de 1.671 pacientes tratados con una dosis diaria de 160 mg Enzalutamida, mientras que un paciente (<0, 1 % ) que recibió placebo experimentó una convulsión. La dosis parece ser un predictor importante del riesgo de convulsiones, como se refleja en los datos preclínicos, y en los datos de un estudio de escalada de la dosis. En ambos estudios fase 3, se excluyeron los pacientes con factores de riesgo de convulsiones o con convulsiones previas.
En el ensayo AFFIRM, seis pacientes (0,8%) experimentaron un ataque de 800 pacientes post-quimioterapia tratados con una dosis diaria de 160 mg de Enzalutamida, mientras que ningún ataque ocurrió en pacientes que recibieron placebo. Factores potencialmente contribuyentes estuvieron presentes en varios de estos pacientes que pueden haber aumentado independientemente su riesgo de convulsiones. En el ensayo PREVAIL, un paciente (0,1 %) de los 871 pacientes sin quimioterapia previa tratados con una dosis diaria de 160 mg de Enzalutamida, y un paciente (0, 1 %) que recibió placebo, experimentaron una convulsión.
El mecanismo por el cual Enzalutamida puede disminuir el umbral de convulsiones no se conoce, pero podría estar relacionado con datos de estudios in vitro que muestran que Enzalutamida y su metabolito activo se unen al canal de cloruro cerrado por GABA y pueden inhibir su actividad.Efectos sobre la capacidad para manejar vehículos y máquinas: Debido al riesgo de convulsiones asociado al uso de XTANDI, se debe advertir a los pacientes sobre el riesgo de manejar vehículos o usar cualquier herramienta o máquina en que la pérdida repentina del conocimiento pudiera causar un daño grave a ellos mismos o a los demás.
INTERACCIONES MEDICAMENTOSAS:
Posibilidad de que otros medicamentos modifiquen las exposiciones a la Enzalutamida.
Inhibidores e inductores del CYP2C8: El CYP2C8 desempeña una función importante en Enzalutamida y en la formación de su metabolito la eliminación de activo. Luego de la administración oral a hombres sanos de gemfibrozilo (600 mg dos veces al día),un inhibidor potente del CYP2C8, el AUC de la Enzalutamida aumentó un 326%, mientras que la Cmáx de la Enzalutamida disminuyó un 18%. Para la suma de Enzalutamida enlazada más el metabolito activo no enlazado, el AUC se incrementó en 77%, mientras que la Cmáx disminuyó en 19%. Se recomienda evitar o usar con precaución los inhibidores potentes (p. ej., gemfibrozilo) o inductores potentes (p. ej., Rifampicina) del CYP2C8 durante el tratamiento con XTANDI. Si se debe administrar de manera concomitante a los pacientes un inhibidor potente del CYP2C8, la dosis de Enzalutamida se debe reducir a 80 mg una vez al día.
Inhibidores e inductores del CYP3A4: El CYP3A4 tiene una función secundaria en el metabolismo de la Enzalutamida. Luego de la administración oral a hombres sanos de Itraconazol (200 mg una vez al día), un inhibidor potente del CYP3A4, el AUC de la Enzalutamida aumentó 41 % mientras que la Cmáx se mantuvo inalterada. Para la suma de Enzalutamida no enlazada más el metabolito activo no enlazado, el AUC aumentó en 27% mientras que la Cmáx quedó nuevamente inalterada. No es necesario ajustar la dosis al administrar Enzalutamida de manera concomitante con inhibidores o inductores del CYP3A4.
Posibilidad de que Xtandi modifique las exposiciones a otros medicamentos
Inducción enzimática: Enzalutamida es un potente inductor enzimático e incrementa la síntesis de muchas enzimas y transportadores; por tanto, es de esperarse la interacción con muchos productos medicinales comunes que son substratos de enzimas o transportadores. La reducción de las concentraciones plasmáticas pueden ser substanciales, y conducir a la pérdida del efecto clínico o a su reducción. Existe también un riesgo de formación incrementada de metabolitos activos. Las enzimas que pudieran ser inducidas incluyen CYP3A en el hígado e instestino, CYP2B6, CYP2C9, CYP2Cl 9, y uridina 5-difosfo-glucuronosiltransferasa (enzimas conjugantes de UGTs - glucurónido). La proteína de transporte P-gp podría también ser inducida, y probablemente otros transportadores también, por ejemplo, la proteína 2 asociada a la Resistencia multidroga (MRP2), proteína de resistencia al cáncer de mamas (BCRP) y el polipéptido de transporte de aniones orgánicos lBl (OATPlBl).
Enzalutamida es un fuerte inhibidor de CYP3A4 y un inductor moderado de CYPC9 y CYP2C 19. La adminis-tración concomitante de Enzalutamida (160 mg una vez al día) con dosis orales únicas de sustratos sensibles del CYP a pacientes con cáncer de próstata produjo una disminución de un 86% del AUC del midazolam (sustrato del CYP3A4), 56% del AUC de la S-Warfarina (sustrato del CYP2C9) y un 70% de disminución del AUC del Omeprazol (sustrato del CYP2C 19). También es posible que se haya producido la inducción de la uridina 5 "-difosfo-glucuronosiltransferasa (UGT 1A1). Tomados en conjunto, estos resultados sugieren que Enzalutamida causa inducción enzimática vía activación del receptor nuclear de pregnano (PXR).
En un estudio clínico en pacientes con CRPC metastásico, Enzalutamida ( 160 mg una vez al día) no tuvo un efecto clínicamente relevante sobre la farmacocinética de doce-taxel administrado intravenosamente (75 mg/m2 por infusión cada 3 semanas). El AUC de Docetaxel disminuyó en 12% [cociente medio geométrico (GMR) =0.882 (IC de 90%: 0.767, 1.02)] mientras que Cmáx disminuyó en 4% [GMR = 0.963 (IC de 90%: 0.834, 1.11 )].
Son de esperarse interacciones con ciertos productos medicinales que son eliminados a través del metabolismo o transporte activo. Si su efecto terapéutico es de gran importancia para el paciente, y los ajustas de la dosis no son realizados con facilidad en base a la monitorización de la eficacia o de las concentraciones en plasma, estos productos medicinales deben ser evitados o usados con precaución. El riesgo de daño hepático después de admi-nistración de Paracetamol se sospecha que es superior en pacientes tratados concomitantemente con inductores enzimáticos.
Los grupos de productos medicinales que pueden ser afectados incluyen, pero no se limitan a los siguientes:
- Analgésicos (p. ej., Fentanilo, Trarnadol).
- Antibióticos (p. ej., laritromicina, Doxiciclina).
- Agentes anticancerígenos (p. ej., Cabazitaxel).
- Anticoagulantes (p. ej., Acenocumarol, Warfarina).
- Antiepilépticos (p. ej., Carbamazepina, Clonazepam, Fenitoína, Primidona, Ácido Valproico).
- Antipsicóticos (p. ej., Haloperidol).
- Betabloqueadores (p. ej., Bisoprolol, Propanolol).
- Benzodiacepinas (p.ej., Diazepam, Midazolam, Zolpidem).
- Bloqueadores del canal de calcio (p.ej., Diltiazem, Felodipina, Nicardipina, Nifedipina, Varapamil).
- Glicósidos cardíacos (p. ej., Digoxina).
- Corticosteroides (p. ej., Dexametasona, Prednisolona).
- Antivirales para el VIH (p. ej., Indinavir, Ritonavir).
- Estatinas metabolizadas por CYP3A4 (p. ej., Atorvastatina, Simvastatina).
- Agentes tiroideos (p.ej., Levotiroxina).
El potencial de inducción completa de Enzalutamida no puede producirse hasta aproximadamente 1 mes después del inicio del tratamiento, cuando se alcanzan las concentraciones plasmáticas de estado estacionario de Enzalutamida, aunque algunos efectos de inducción pueden ser evidentes antes. Los pacientes que toman medicamentos que son sustratos de CYP2B6, CYP3A4, CYP2C9, CYP2Cl9, CYP1A2 o UGTlAl deben ser evaluados respecto a una posible pérdida de efectos farmacológicos (o aumento de los efectos en los casos en que se forman metabolitos activos) durante el primer mes de tratamiento con Enzalutamida, y el ajuste de la dosis debe ser considerado como apropiado. En consideración de la larga vida media de Enzalutamida, los efectos sobre las enzimas pueden persistir durante un mes o más después de dejar Enzalutamida. Una reducción gradual de la dosis del medicamento concomitante puede ser necesaria cuando se interrumpe el tratamiento de Enzalutamida.
Sustratos del CYP2C8: Enzalutamida (160 mg una vez al día) no causó variaciones clínicamente significativas en el AUC de la pioglitazona (sustrato del CYP2C8) y no está indicado ningún ajuste de la dosis al administrar un sustrato del CYP2C8 de manera concomitante con Enzalutamida.
Sustratos de la gp-P: Los datos in vitro indican que la Enzalutamida puede ser un inhibidor del transportador de eflujo gp-P. El efecto de Enzalutamida sobre sustratos de la gp-P no se ha evaluado in vivo; sin embargo, bajo condiciones de uso clínico Enzalutamida puede ser un inductor de la gp-P mediante la activación del receptor nuclear de pregnano (PXR). Los medicamentos con un estrecho margen terapéutico que sean sustratos de la gp-P (p. ej., Colchicina, Dabigatrán Etexilato o Digoxina) se deben usar con precaución cuando se administran de manera concomitante con Enzalutamida, y puede ser necesario ajustar la dosis para mantenerconcentraciones plasmáticas óptimas.
Sustratos de BCRP, MRP2, OAT3 y OCTJ: Según los datos obtenidos in vitro, no se puede descartar la inhibición de BCRP y MRP2 (en el intestino) ni del transportador de aniones orgánicos 3 (OAT3) o el transportador de cationes orgánicos 1 (OCTl) (sistémicamente). En teoría, la inducción de estos transportadores también es posible, y el efecto neto se desconoce actualmente.
Efecto de los alimentos sobre la exposición a la Enzalutamida: Los alimentos no tienen un efecto de importancia clínica sobre el grado de exposición a la Enzalutamida. En ensayos clínicos, XTANDI se administró independientemente de los alimentos.
RECOMENDACIONES:
Fertilidad, embarazo y lactancia
Anticoncepción en hombres y mujeres: Se desconoce si XTANDI o sus metabolitos están presentes en el semen. Si el paciente mantiene relaciones sexuales con una mujer embarazada, deberá utilizar un preservativo durante el tratamiento con XTANDI y en los 3 meses posteriores. Si el paciente mantiene relaciones sexuales con una mujer que pueda tener hijos, debe utilizar un preservativo y otro método anticonceptivo durante el tratamiento y en los 3 meses posteriores. Los estudios en animales han mostrado toxicidad reproductiva.
Embarazo: XTANDI no está indicado para su uso en mujeres. XTANDI está contraindicado en mujeres embarazadas o que puedan quedar embarazadas. No hay datos en seres humanos sobre el uso de Enzalutamida en mujeres embarazadas.
Lactancia: XTANDI no está indicado para su uso en mujeres. Se desconoce si XTANDI o sus metabolitos se excretan en la leche materna.
Fertilidad: El tratamiento con Enzalutamida de ratones preñados resultó en una mayor incidencia de muertes embriofetales y cambios externos y esqueléticos. Estudios de toxicología reproductiva no se llevaron a cabo con Enzalutamida, pero en los estudios en ratas (4 y 26 semanas) y perros (4,13 y 39 semanas), la atrofia, aspermia / hipospermia, e hipertrofia / hiperplasia se observaron en el sistema reproductivo, en consonancia con la actividad farmacológica de Enzalutamida. En estudios en ratones (4 semanas), ratas (4 y 26 semanas) y perros (4, 13 y 39 semanas), los cambios en los órganos reproductivos asociados con Enzalutamida fueron disminución en el peso de los órganos con atrofia de la próstata y epidídimo. Cambios adicionales en los tejidos reproductivos incluyen hipertrofia / hiperplasia de la glándula pituitaria y atrofia en las vesículas seminales en ratas y hipospermia testicular y la degeneración de los túbulos seminíferos en perros. No se observaron diferencias de género en las glándulas mamarias de rata (atrofia en el macho e hiperplasia lobular en la hembra). Los cambios en los órganos reproductores en ambas especies fueron consistentes con la actividad farmacológica de Enzalutamida y se revertieron o se resolvieron parcialmente después de un período de recuperación de 8 semanas. No hubo otros cambios importantes en la patología clínica o histopatología en ningún otro sistema de órganos, incluyendo el hígado, en cualquiera de las especies.
Los estudios en animales mostraron que Enzalutamida afectó el sistema reproductivo de ratas y perros machos.
Carcinogenia, mutagénesis y fototoxicidad: No se han llevado a cabo estudios a largo plazo en animales para evaluar la posible acción cancerígena de la Enzalutamida.Enzalutamida no indujo mutaciones en el ensayo de mutagénesis microbiana (Ames) y no fue clastogénico ni en el ensayo citogenético in vitro con células de linfoma de ratón ni en el ensayo in vivo de micronúcleos de ratón. No se han realizado estudios en animales a largo plazo para evaluar el potencial carcinogénico de Enzalutamida. Enzalutamida no era fototóxico in vitro.
ADVERTENCIAS Y PRECAUCIONES:
Riesgo de convulsiones: Se debe tener precaución al administrar XTANDI a pacientes con antecedentes de convulsiones u otros factores predisponentes incluyendo, pero no limitado a, lesión cerebral subyacente, accidente cerebrovascular, tumores cerebrales primarios, metástasis cerebrales o alcoholismo. Además, el riesgo de convulsiones puede ser mayor en los pacientes que reciben medicamentos concomitantes que reducen el umbral convulsivo.
Síndrome de encefalopatía posterior reversible: Rara vez se han reportado casos del síndrome de encefalopatía posterior reversible (SEPR) en pacientes tratados con XTANDI. El SEPR es un trastorno neurológico reversible e infrecuente que puede manifestarse con síntomas de rápida evolución como convulsiones, cefalea, confusión, ceguera y otras alteraciones visuales y neurológicas, con o sin hipertensión asociada. El diagnóstico de SEPR requiere una confirmación mediante estudios por imágenes cerebrales, preferiblemente por resonancia magnética (RM). Se recomienda suspender la administración de XTANDI en pacientes que presenten SEPR.
Administración concomitante con cumarinas: Se debe evitar la administración concomitante con Warfarina y anticoagulantes de tipo cumarínico. En caso de que XTANDI se administre de manera concomitante con un anticoagulante metabolizado por el CYP2C9 (como Warfarina o acenocumarol), se deben realizar determinaciones adicionales del Índice Internacional Normalizado (IIN).
Insuficiencia renal: Se exige precaución en pacientes con insuficiencia renal grave, ya que XTANDI no se ha estudiado en esta población de pacientes.
Insuficiencia hepática: Puesto que no hay datos en pacientes con insuficiencia hepática severa y la Enzalutamida se elimina principalmente a través del hígado, no se recomienda administrar XTANDI a pacientes con insuficiencia hepática grave (clase C de Child-Pugh).
Excipientes: XTANDI contiene Sorbitol (E420). Los pacientes con problemas hereditarios raros de intolerancia a la fructosa no deben tomar XTANDI.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología y administración
Posología: La dosis recomendada de XTAND1 es de 160 mg (cuatro cápsulas de 40 mg) en una dosis oral única diaria. XTANDI puede tomarse con o sin alimentos. La castración médica con un análogo de LHRH debería continuarse durante el tratamiento de pacientes no castrados quirúrgicamente.
Si un paciente olvida tomar XTANDI a la hora habitual, debe tomar la dosis recetada lo más cerca posible de la hora habitual. Si un paciente olvida la dosis durante un día entero, el tratamiento se debe reanudar al día siguiente con la dosis diaria habitual.
Si un paciente experimenta una toxicidad ≥ Grado 3 o una reacción adversa intolerable, la dosificación debería retirarse por una semana o hasta que los síntomas mejoren hasta ≤ Grado 2, y luego debería reiniciarse a la mismo dosis o a una dosis reducida (120 mg o 80 mg) si es necesario.
Uso concomitante con inhibidores potentes del CYP2C8
Se debe evitar en lo posible el uso concomitante de inhibidores potentes de CYP2C8. Si se debe administrar de manera concomitante a pacientes un inhibidor potente del CYP2C8, la dosis de Enzalutamida se debe reducir a 80 mg una vez al día. Si se suspende la administración concomitante del inhibidor potente del CYP2C8, se debe volver a la dosis de Enzalutamida que se administraba antes de empezar el tratamiento con el inhibidor potente del CYP2C8.
Poblaciones especiales
Pacientes ancianos
No es necesario ajustar la dosis en pacientes ancianos.
Pacientes con insuficiencia hepática
No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve o moderada (clase A o B de Child-Pugh; ver sección 3). No se recomienda administrar XTANDI a pacientes con insuficiencia hepática grave (clase C de Child-Pugh).
Pacientes con insuficiencia renal
No es necesario ajustar la dosis en pacientes con insuficiencia renal leve o moderada. Se recomienda precaución en el caso de pacientes con insuficiencia renal grave o enfermedad renal terminal.
Sexo
La Enzalutamida no está indicada para su uso en mujeres.
Población pediátrica
La Enzalutamida no está indicada para su uso en niños. Forma de administración
Las cápsulas blandas de XTANDI se deben tragar enteras con agua y se pueden tomar con o sin alimentos. Este medicamento no debe partirse, abrirse ni masticarse.
SOBREDOSIS:
No existe ningún antídoto para XTANDI. En caso de sobredosis, se debe interrumpir el tratamiento con XTANDI e iniciar medidas de apoyo general teniendo en cuenta su semivida de 5,8 días. Es posible que los pacientes tengan un mayor riesgo de convulsiones luego de una sobredosis.
PRESENTACIÓN:
XTANDI se suministra en cápsulas blandas para administración oral, disponible en las siguientes presentaciones: 40 y 120 cápsulas blandas.
Cápsulas blandas oblongas de color blanco con el texto "ENZ" impreso en tinta negra en uno de los lados. Se presentan en envases que contienen 10 y 30 sobres; cada sobre contiene un blíster de dosis unitaria con 4 cápsulas blandas.
Antes de usar, observe el aspecto del medicamento.
Este medicamento solo debe utilizarse bajo estricta supervisión y vigilancia médica y no puede repetirse sin una nueva receta.
Mantener fuera del alcance de los niños.
Fecha de la última actualización: Agosto 2015.
MEDICAMENTA
Casilla 17-21-027
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Conservación: Las cápsulas blandas de XTANDI® se deben conservar a temperatura ambiente no mayor de 30 ºC. Se deben proteger de la humedad. La vida útil es de 24 meses.
No utilice este medicamento después de la fecha de vencimiento que se indica en la caja. La fecha de vencimiento hace referencia al último día de ese mes.
No tome ninguna cápsula que esté dañada o que presente pérdidas o indicios de manipulación.
Número de lote y fecha de vencimiento: Ver envase. No tome el medicamento si la fecha está vencida. Mantenga el medicamento en su envase original. Antes de tomarlo, observe el aspecto del medicamento. Si observa algún cambio en su aspecto y el medicamento aún está en el período de vida útil, consulte con el farmacéutico para determinar si puede usarlo.

