

VIDAZA
AZACITIDINA
Polvo para suspensión inyectable
Caja , 1 Frasco ampolla
COMPOSICIÓN:
Fórmula cualicuantitativa
Cada VIAL contiene:
Azacitidina 100 mg
Excipientes: Manitol 100mg
INDICACIONES TERAPÉUTICAS:
Indicaciones
VIDAZA está indicado para el tratamiento de pacientes adultos, que no están aptos para recibir un trasplante de células madre hematopoyéticas (HSCT, por sus siglas en inglés), con:
• Síndromes Mielodisplásicos (SMD) intermedio 2 y de alto riesgo según el Sistema Internacional de Puntaje Pronóstico (IPSS),
• Leucemia Mielomonocítica Crónica (CMML) con entre 10-29% de blastos en la médula sin trastorno mieloproliferativo,
• Leucemia Mieloide Aguda (LMA) con entre 20-30 % de blastos y displasia multilineal de acuerdo con la clasificación de la Organización Mundial de la Salud (OMS)
• En LMA con >30 % de blastos en la médula de acuerdo con la clasificación de la OMS.
MECANISMO DE ACCIÓN:
Acción farmacológica
Acción terapéutica
Agente antineoplásico
Código ATC: L01BC07
Propiedades farmacodinámicas
Mecanismo de acción: Se cree que la Azacitidina ejerce sus efectos antineoplásicos a través de múltiples mecanismos, incluyendo citotoxicidad sobre células hematopoyéticas anormales en la médula ósea e hipometilación del ADN. Los efectos citotóxicos de azacitidina podrían originarse de mecanismos múltiples, incluyendo inhibición del ADN, síntesis del ARN y de proteínas, incorporación al ARN y ADN, y activación de las vías de daño del ADN. Las células no proliferativas son relativamente insensibles a azacitidina. La incorporación de azacitidina al ADN resulta en la inactivación de metiltransferasas del ADN que genera hipometilación del ADN. La hipometilación del ADN de genes metilados de forma aberrante involucrados en la regulación del ciclo de células normales, las vías de diferenciación y de muerte podrían dar como resultado una reexpresión de genes y restauración de las funciones de supresión del cáncer para las células cancerígenas. No se ha establecido la importancia relativa de la hipometilación del ADN versus la citotoxicidad u otras actividades de azacitidina para los resultados clínicos.
Eficacia y seguridad clínica
Población adulta (SMD, LMMC y LMA [20-30 % de blastos en la médula])
La eficacia y seguridad de VIDAZA se estudiaron en un estudio comparativo internacional, multicéntrico, controlado, abierto, randomizado, de grupo paralelo, de fase 3 (AZA PH GL 2003 CL 001) en pacientes adultos con: SMD intermedio-2 y de alto riesgo según el Sistema Internacional de Puntaje Pronóstico (IPSS), Anemia Refractaria con blastos en exceso (RAEB), Anemia Refractaria con Blastos en Exceso en Transformación (RAEB-T) y leucemia mielomonocítica crónica modificada (mCMML) de acuerdo con el sistema francés-estadounidense-británico (FAB) de clasificación. Los pacientes con RAEB-T (21-30 % de blastos) hoy en día están considerados como pacientes con LMA bajo el sistema de clasificación actual de la OMS. La azacitidina junto con los mejores cuidados paliativos (BSC, por su sigla en inglés) (n = 179) fue comparada con los Regímenes Convencionales de Cuidado (CCR).
Los CCR consistían en únicamente BSC (n = 105), citarabina de dosis bajas más BSC (n = 49) o quimioterapia de inducción estándar junto con BSC (n = 25). Los pacientes fueron preseleccionados por sus médicos a 1 de 3 CCR previos a la randomización. Los pacientes recibieron este régimen preseleccionado si no fueron randomizados a VIDAZA. Como parte de los criterios de inclusión, se requería que los pacientes tengan un estado funcional del Grupo Cooperativo de Oncólogos del Este (ECOG, Eastern Cooperative Oncology Group) de 0-2. Los pacientes con SMD secundario fueron excluidos del estudio. El criterio de evaluación primario del estudio fue la sobrevida global. VIDAZA fue administrado en una dosis subcutánea de 75 mg/m2 diarios durante 7 días, seguidos de un período de descanso de 21 días (ciclo de tratamiento de 28 días) por una mediana de 9 ciclos (rango = 1-39) y un promedio de 10,2 ciclos. Dentro de la población con Intención de Tratar (ITT), la edad promedio fue de 69 años (rango: entre 38 y 88 años).
En el análisis ITT de 358 pacientes (179 con azacitidina y 179 con CCR), el tratamiento con VIDAZA fue asociado con la mediana de sobrevida de 24,46 meses versus la de 15,02 meses para aquellos que reciben tratamiento CCR, una diferencia de 9,4 meses con un valor p de log rank estratificado de 0,0001.
El índice de riesgo para el efecto del tratamiento fue de 0,58 (IC del 95 %: 0,43; 0,77). Los índices de sobrevida de dos años fueron de 50,8% en pacientes que recibieron azacitidina versus un 26,2 % en pacientes que recibieron CCR (p < 0,0001).
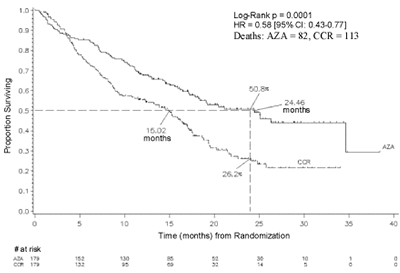
REFERENCIAS = AZA = Azacitidina; CCR = Tegímenes Convencionales de Cuidado; CI = Intervalo de Confianza; HR = Índice de Riesgo.
Los beneficios de sobrevida de VIDAZA fueron consistentes independientemente de la opción de tratamiento CCR (BSC solo, citarabina en dosis bajas más BSC o quimioterapia estándar de inducción más BSC) utilizada en el grupo de control.
Cuando los subgrupos citogenéticos de IPSS fueron analizados, se observaron hallazgos similares en términos de la mediana de sobrevida general en todos los grupos (citogenética buena, intermedia, mala, incluida monosomía 7).
En análisis de subgrupos de edad, se observó un aumento en la mediana de sobrevida general para todos los grupos (< 65 años, ≥ 65 años y ≥ 75 años).
El tratamiento con VIDAZA se asoció con una mediana de tiempo transcurrido hasta la muerte o transformación a LMA de 13,0 meses versus de 7,6 meses para aquellos que reciben tratamiento CCR, una mejora de 5,4 meses con un valor p estratificado de log-rank de 0,0025.
El tratamiento con VIDAZA también estuvo asociado con una reducción en citopenias y sus síntomas relacionados. El tratamiento con VIDAZA llevó a una disminución de transfusiones de glóbulos rojos y plaquetas. De los pacientes en el grupo de azacitidina que fueron dependientes de las transfusiones de glóbulos rojos al inicio, el 45,0% de estos pacientes se volvieron independientes de las transfusiones de glóbulos rojos durante el período de tratamiento, en comparación con el 11,4% de los pacientes en los grupos combinados de CCR (una diferencia estadísticamente significativa (p < 0,0001) de 33,6% (IC del 95 %: 22,4; 44,6). En pacientes que fueron dependientes de las transfusiones de glóbulos rojos en la línea de base y se volvieron independientes, la mediana de duración de la independencia de transfusiones de glóbulos rojos fue de 13 meses en el grupo de azacitidina.La respuesta fue evaluada por el investigador o por el Comité Independiente de Revisión (IRC, por sus siglas en inglés). La respuesta total (remisión completa [RC] + remisión parcial [RP]), según se determinó el investigador, fue del 29% en el grupo de azacitidina y del 12% en el grupo combinado de CCR (p = 0,0001). La respuesta total (RC + RP), según determinó el IRC en AZA PH GL 2003 CL 001, fue del 7 % (12/179) en el grupo de azacitidina en comparación con el 1 % (2/179) en el grupo CCR combinado (p = 0,0113).
Las diferencias entre las evaluaciones de respuesta del IRC y del investigador fueron una consecuencia de los criterios del Grupo de Trabajo Internacional (IWG, por sus siglas en inglés) que requirieron una mejora en los recuentos en sangre periférica y en el mantenimiento de estas mejoras por un mínimo de 56 días. También se demostró un beneficio de sobrevida en pacientes que no habían adquirido una respuesta completa/parcial luego del tratamiento con azacitidina. La mejora hematológica (mayor o menor), según determinó el IRC, se logró en el 49% de los pacientes que recibieron azacitidina en comparación con el 29% de los pacientes tratados con CCR combinados (p < 0,0001).
En pacientes con una o más anormalidades citogenéticas en la línea de base, el porcentaje de pacientes con una respuesta citogenética mayor fue similar en los grupos de azacitidina y de CCR combinados. La respuesta citogenética menor fue más alta y estadísticamente significativa (p = 0,0015) en el grupo de azacitidina (34%) en comparación con el grupo CCR (10%).
Población adulta de 65 años o más con LMA con > 30% blastos en la médula
Los resultados que se presentan más abajo representan la población de análisis con Intención de Tratar (ITT, por sus siglas en inglés) estudiada en AZAAML-001 (ver la indicación aprobada en la sección 4.1).
La eficacia y la seguridad de VIDAZA se estudiaron en un estudio en fase 3 internacional, multicéntrico, controlado, abierto y en grupos paralelos en pacientes de 65 años o más, a quienes se había diagnosticado LMA de novo o secundaria con >30 % de blastos en la médula, de acuerdo con la clasificación de la OMS,y que no eran elegibles para un HSCT. VIDAZA más BSC (n = 241) se comparó con las PCC. Las PPC consistieron en BSC solo (n = 45), citarabina en dosis baja más BSC (n = 158) o quimioterapia intensiva estándar con citarabina y antraciclina más BSC (n = 44). Los pacientes fueron preseleccionados por su médico a 1 de las 3 PCC antes de la aleatorización.
Los pacientes recibieron este régimen preseleccionado si no eran aleatorizados a VIDAZA. Como parte de los criterios de inclusión, se requería que los pacientes tuvieran un estado funcional según la escala del ECOG de 0-2 y anomalías citogenéticas de riesgo bajo o intermedio. El criterio de valoración principal del estudio fue la sobrevida global.
VIDAZA se administró como una dosis SC de 75 mg/m2/día durante 7 días, seguida de un período de descanso de 21 días (ciclo de tratamiento de 28 días), durante una mediana de 6 ciclos (rango: de 1 a 28) y una media de 8,8 ciclos, los pacientes con solo BSC durante una mediana de 3 ciclos (rango: de 1 a 20), pacientes con citarabina en dosis baja durante una mediana de 4 ciclos (rango de 1 a 25) y pacientes con quimioterapia intensiva estándar durante una mediana de 2 ciclos (rango: de 1 a 3, ciclo de inducción más 1 o 2 ciclos de consolidación).
Dentro de la población ITT, los grupos VIDAZA y CCR fueron comparables en cuanto a parámetros basales. La mediana de edad de los sujetos fue 75,0 años (rango: de 64 a 91 años); el 75,2 % de los pacientes eran caucásicos y el 59,0 %, hombres. En el basal, el 60,7 % se clasificó como LMA no especificada de otra manera; el 32,4 % como LMA con cambios relacionados con la mielodisplasia; el 4,1 % como neoplasias mieloides relacionadas con la terapia y el 2,9 % como LMA con anomalías genéticas recurrentes, según la clasificación de la OMS.
En el análisis ITT de 488 pacientes (241 VIDAZA y 247 PCC), el tratamiento con VIDAZA se asoció con una mediana de sobrevida de 10,4 meses versus 6,5 meses en los que recibieron tratamiento con PCC, una diferencia clínicamente significativa de 3,8 meses, con un valor de p con la prueba de rango logarítmico estratificado de 0,1009 (bilateral). La razón de riesgos con respecto al efecto del tratamiento fue 0,85 (IC 95 % = 0,69; 1,03). Las tasas de sobrevida de un año fueron 46,5 % en pacientes que recibieron VIDAZA versus 34,3 % en pacientes que recibieron PCC.
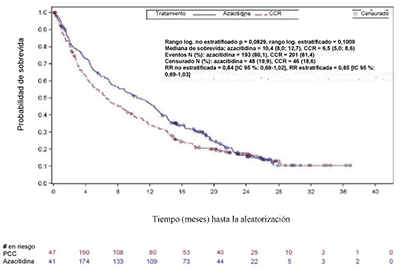
El modelo de Cox PH ajustado en cuanto a factores pronósticos preespecificados definió una RR para VIDAZA versus PCC de 0,80 (IC 95%=0,66; 0,99; p = 0,0355). Además, si bien el estudio no tenía suficiente poder para demostrar una diferencia estadísticamente significativa al comparar azacitidina con los grupos de tratamiento con PCC preseleccionados, la sobrevida de los pacientes tratados con VIDAZA fue más prolongada en comparación con la sobrevida lograda con las opciones de tratamiento de PCC BSC solo, citarabina en dosis bajas más BSC, y fue similar en comparación con la sobrevida alcanzada con la quimioterapia intensiva estándar más BSC.
En todos los subgrupos preespecificados de edad ([ <75 años y ≥75 años], sexo, raza, estado funcional del ECOG [0 o 1 y 2], riesgo citogenético basal [escaso e intermedio], región geográfica, clasificación de la OMS de la LMA [incluyendo LMA con cambios relacionados con la mielodisplasia], recuento de glóbulos blancos basales [≤5 x 109/l y >5 x 109/l], blastos en la médula ósea basales [≤50 % y >50 %] y antecedentes de SMD), hubo una tendencia el beneficio de sobrevida global a favor de VIDAZA. En algunos subgrupos preespecificados, la RR de sobrevida global alcanzó significancia estadística que incluyendo pacientes con riesgo citogenético bajo, pacientes con LMA con cambios relacionados con la mielodisplasia, pacientes < 75 años, pacientes femeninas y pacientes blancos. Las respuestas hematológicas y citogenéticas fueron evaluadas por el investigador y por el CRI con resultados similares. La tasa de respuesta global (remisión completa [RC] + remisión completa con recuperación incompleta del Recuento Sanguíneo [RCi], según fue determinada por el CRI, fue 27,8 % en el grupo VIDAZA y 25,1 % en el grupo PCC combinado (p = 0,5384). En pacientes que lograron la RC o la RCi, la mediana de duración de la remisión fue 1,4 meses (IC 95 % = 7,2; 15,2) en los sujetos tratados con VIDAZA y 12,3 meses (IC 95 % = 9,0; 17,0) en los sujetos tratados con PCC. Asimismo, se demostró un beneficio de sobrevida en pacientes que no habían logrado una respuesta completa con VIDAZA en comparación con las PCC. El tratamiento con VIDAZA mejoró el recuento de sangre periférica y llevó a una reducción en la necesidad de transfusiones de glóbulos rojos y plaquetas.
Se consideraba que un paciente era dependiente de transfusiones de glóbulos rojos o plaquetas en el basal si el sujeto recibía una transfusión de glóbulos rojos o plaquetas o más durante los 56 días (8 semanas) de aleatorización o antes de esta, respectivamente. Se consideraba que un paciente era independiente de transfusiones de glóbulos rojos o plaquetas durante el período de tratamiento si el sujeto no recibía transfusiones de glóbulos rojos o plaquetas durante cualquiera de los 56 días consecutivos durante el período de informe, respectivamente.
De los pacientes del grupo VIDAZA que eran dependientes de transfusiones de glóbulos rojos en el basal, el 38,5 % (IC 95 % = 31,1; 46,2) dejó de ser dependiente de estas durante el período de tratamiento en comparación con el 27,6 % (IC 95 % = 20,9; 35,1) de los pacientes de los grupos PCC combinadas. En los pacientes que eran dependientes de transfusiones de glóbulos rojos en basal y lograron la independencia de transfusiones durante el tratamiento, la mediana de duración de la independencia de transfusiones de glóbulos rojos fue 13,9 meses en el grupo VIDAZA y no se alcanzó en el grupo PCC.
De los pacientes del grupo VIDAZA que eran dependientes de transfusiones de plaquetas en el basal, el 40,6 % (IC 95 % = 30,9; 50,8) dejaron de ser dependientes de estas durante el período de tratamiento en comparación con el 29,3 % (IC 95 % = 19,7; 40,4) de los pacientes de los grupos PCC combinadas. En los pacientes que eran dependientes de transfusiones de plaquetas en el basal y lograron la independencia de transfusiones durante el tratamiento, la mediana de duración de la independencia de transfusión de plaquetas fue 10,8 meses en el grupo VIDAZA y 19,2 meses en el grupo CCR. La calidad de vida relacionada con la salud (HRQoL, por sus siglas en inglés) se evaluó usando el Cuestionario de Calidad de Vida de la Organización Europea para la Investigación y el Tratamiento del Cáncer (EORTC QLQ-C 30, por sus siglas en inglés). Los datos de calidad de vida relacionada con la salud pudieron analizarse en un subconjunto de toda la población del ensayo. Si bien hay limitaciones en el análisis, los datos disponibles sugieren que los pacientes no experimentan un deterioro significativo de la calidad de vida durante el tratamiento con VIDAZA.
Farmacocinética
Propiedades farmacocinéticas
Absorción
Tras la administración por vía subcutánea de una dosis única de 75 mg/m2, azacitidina se absorbió rápidamente; produciéndose concentraciones plasmáticas máximas de 750 ± 403 ng/ml a las 0,5 horas después de la administración (el primer punto de extracción de muestras). La biodisponibilidad absoluta de azacitidina después de la administración por vía subcutánea en relación con la intravenosa (dosis únicas de 75 mg/m2) fue de aproximadamente, el 89 %, basado en el área bajo la curva (AUC).
El área bajo la curva y la Concentración Plasmática Máxima (Cmáx) de la administración de azacitidina por vía subcutánea fueron aproximadamente, proporcionales en el intervalo de dosis de 25 a 100 mg/m2.
Distribución
Luego de la administración intravenosa, el volumen promedio de distribución fue de 76 ± 26 l, y el clearance sistémico fue de 147 ± 47 l/h.
Biotransformación
A partir de la información obtenida in vitro, el metabolismo de azacitidina no está mediado por las isoenzimas del citocromo P450 (CYPs, por sus siglas en inglés), UDP-Glucuronosiltransferasas (UGTs), Sulfotransferasas (SULTs) y Glutatión-Transferasas (GSTs, por sus siglas en inglés).Azacitidina sufre hidrólisis espontánea y deaminación mediada por citidina deaminasa. En las fracciones S9 del hígado humano, la formación de metabolitos fue independiente del NADPH, lo cual implica que el metabolismo de azacitidina no fue mediado por las isoenzimas del citocromo P450. Un estudio in vitro de azacitidina con hepatocitos humanos cultivados indica que a concentraciones de 1,0 μM a 100 μM (esto es, aproximadamente, 30 veces más que las concentraciones que se pueden alcanzar clínicamente), azacitidina no induce los citocromos CYP 1A2, 2C19, o 3A4 o 3A5. En estudios para evaluar la inhibición de una serie de isoenzimas P450 (CYP 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4), la azacitidina hasta 100 μM no produce inhibición. Por lo tanto, es poco probable la inducción o la inhibición de la enzima CYP por azacitidina a concentraciones plasmáticas clínicamente alcanzables. Eliminación
La azacitidina se aclara rápidamente del plasma con una vida media promedio de eliminación (t½) luego de la administración subcutánea de 41 ± 8 minutos. No ocurre ninguna acumulación luego de la administración subcutánea de 75 mg/m2 de azacitidina una vez por día durante 7 días. La excreción urinaria es la vía primaria de eliminación de azacitidina y/o sus metabolitos. Luego de la administración intravenosa y subcutánea de 14C-azacitidina, se recuperó el 85 y 50% de la radioactividad administrada en la orina, respectivamente, mientras que el < 1% se recuperó en las heces.
Poblaciones especiales
No se han estudiado formalmente los efectos de la insuficiencia hepática (ver sección Posología dosificación y administración), género, edad o raza en la farmacocinética de azacitidina.
Insuficiencia renal
La insuficiencia renal no tiene un efecto importante en la exposición farmacocinética de azacitidina tras administraciones únicas y múltiples por vía subcutánea. Tras la administración por vía subcutánea de una dosis única de 75 mg/m2, los valores de exposición medios (AUC y Cmáx) de los sujetos con insuficiencia renal leve, moderada y grave aumentaron en un 11-21 %, un 15-27 % y un 41-66 %, respectivamente, en comparación con los sujetos con la función renal normal. Sin embargo, la exposición estaba dentro del mismo rango general de exposiciones observado para los sujetos con la función renal normal. Azacitidina puede administrarse a pacientes con insuficiencia renal sin la necesidad de realizar un ajuste de la dosis inicial siempre que se monitorice la toxicidad en estos pacientes, puesto que azacitidina y/o sus metabolitos se excretan principalmente por el riñón. Farmacogenómica
El efecto de los polimorfismos de citidina deaminasa sobre el metabolismo deazacitidina no se ha estudiado formalmente.
CONTRAINDICACIONES:
Hipersensibilidad conocida a la sustancia activa o a cualquiera de los excipientes.
Tumores hepáticos malignos y avanzados (ver sección Advertencias y precauciones especiales para su uso). Lactancia (ver sección Fertilidad, embarazo y lactancia).
REACCIONES ADVERSAS:
Resumen de perfil de seguridad
Población adulta con SMD, LMMC y LMA (20-30 % de blastos en la médula). Las reacciones adversas posible o probablemente relacionadas con la administración de VIDAZA se manifestaron en el 97% de los pacientes. Las reacciones adversas graves más frecuentes observadas en el ensayo pivotal (AZA PH GL 2003 CL 001) fueron neutropenia febril (8,0 %) y anemia (2,3 %), que también se reportaron en los estudios complementarios (CALGB 9221 y CALGB 8921). Otras reacciones adversas graves de estos 3 estudios incluyeron infecciones tales como: sepsis neutropénica (0,8%) y neumonía (2,5%) (algunas con desenlace mortal), trombocitopenia (3,5 %), reacciones de hipersensibilidad (0,25 %) y reacciones hemorrágicas (por ejemplo, hemorragia cerebral) [0,5 %], hemorragia gastrointestinal [0,8 %] y hemorragia intracraneal [0,5 %]).Las reacciones adversas más comúnmente informadas con el tratamiento de azacitidina fueron reacciones hematológicas (71,4 %) que incluyen trombocitopenia, neutropenia y leucopenia (generalmente, de grado 3-4), eventos gastrointestinales (60,6 %) que incluyen náuseas, vómitos (generalmente, grado 1-2) o reacciones en la zona de aplicación de la inyección (77,1 %; generalmente, grado 1-2).
Población adulta de 65 años o más con LMA con > 30 % de blastos en la médula
Las reacciones adversas graves más frecuentes (≥10 %) observadas en el estudio AZA-AML-001 en el grupo de tratamiento con azacitidina fueron neutropenia febril (25,0 %), neumonía (20,3 %) y pirexia (10,6 %). Otras reacciones adversas graves reportadas con menos frecuencia en el grupo de tratamiento con azacitidina incluyeron: sepsis (5,1 %), anemia (4,2 %), sepsis neutropénica (3,0 %), infección de las vías urinarias (3,0 %), trombocitopenia (2,5 %), neutropenia (2,1 %), celulitis (2,1 %), mareos (2,1 %) y disnea (2,1 %).
Las reacciones adversas reportadas con más frecuencia (≥30 %) con el tratamiento con azacitidina fueron eventos gastrointestinales, incluyendo constipación (41,9 %), náuseas (39,8 %) y diarrea (36,9 %), (por lo general, grado 1-2), trastornos generales y alteraciones en el lugar de administración, incluyendo pirexia (37,7 %; por lo general, grado 1-2) y eventos hematológicos, incluyendo neutropenia febril (32,2 %) y neutropenia (30,1 %), (por lo general, grado 3-4).
Las reacciones adversas serias más comunes que figuran en el estudio pivotal (AZA PH GL 2003 CL 001) y que también se informaron en los estudios de soporte (CALGB 9221 y CALGB 8921) incluyeron neutropenia febril (8,0 %) y anemia (2,3 %). Otras reacciones adversas serias informadas de estos 3 estudios incluyeron infecciones como: sepsis neutropénica (0,8%) y neumonía (2,5%) (algunas con resultado fatal), trombocitopenia (3,5%), reacciones de hipersensibilidad (0,25 %) y reacciones hemorrágicas (por ejemplo, hemorragia cerebral [0,5 %], hemorragia gastrointestinal [0,8 %] y hemorragia intracraneal [0,5 %]).
Listado tabulado de las reacciones adversas
La Tabla 1 debajo contiene las reacciones adversas asociadas al tratamiento con azacitidina, obtenidas de los principales ensayos clínicos sobre SMD y LMA y de la experiencia postcomercialización.
Las frecuencias se definen como: muy frecuente (≥1/10), frecuente (de ≥1/100 a <1/10), poco frecuente (de ≥1/1.000 a <1/100), rara (de ≥1/10.000 a <1/1.000), muy rara (<1/10.000) y desconocida (no se puede estimar con la información disponible). Dentro de cada agrupamiento de frecuencia, se presentan los efectos no deseados según la seriedad que va decreciendo. Las reacciones adversas se presentan en la tabla que se encuentra a continuación conforme a la mayor frecuencia observada en cualquiera de los principales estudios clínicos.
|
Tabla 1: Reacciones adversas al medicamento en pacientes con SMD o LMA |
|||||
|
Clasificación de órganos y sistemas |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
Raras |
Desconocidas |
|
Infecciones e infestaciones |
Neumonía*, (incluyendo bacterial, viral y fúngica), nasofaringitis |
Sepsis* (incluyendo bacterial, viral y fúngica), sepsis neutropénica*, infección de las vías respiratorias (incluye vías respiratorias superiores y bronquitis), infección de las vías urinarias, celulitis, diverticulitis, infección fúngica de la cavidad bucal, sinusitis, faringitis, rinitis, herpes simple, infección cutánea |
Fascitis necrotizante* |
||
|
Trastornos de la sangre y del sistema linfático |
Neutropenia febril*, neutropenia, leucopenia, trombocitopenia, anemia |
Pancitopenia*, Insuficiencia medular |
|||
|
Trastornos del sistema inmunológico |
Reacciones de hipersensibilidad |
||||
|
Trastornos del metabolismo y de la nutrición |
Anorexia, disminución del apetito, hipokalemia |
Deshidratación |
Síndrome de lisis tumoral |
||
|
Trastornos psiquiátricos |
Insomnio |
Estado de confusión, ansiedad |
|||
|
Trastornos del sistema nervioso |
Mareos, cefalea |
Hemorragia intracraneal *, síncope, somnolencia, letargo |
|||
|
Trastornos oculares |
Hemorragia ocular, hemorragia conjuntival |
||||
|
Trastornos cardíacos |
Derrame pericárdico |
Pericarditis |
|||
|
Trastornos vasculares |
Hipotensión*, hipertensión, hipotensión ortostática, hematoma |
||||
|
Trastornos respiratorios, torácicos y mediastínicos |
Disnea, epistaxis |
Efusión pleural, disnea de esfuerzo, dolor faringolaríngeo |
Enfermedad pulmonar intersticial |
||
|
Trastornos gastrointestinales |
Diarrea, vómitos, estreñimiento, náuseas, dolor abdominal (incluye dolor en la parte superior del abdomen y malestar abdominal) |
Hemorragia gastrointestinal* (incluye hemorragia bucal), hemorragia hemorroidal, estomatitis, hemorragia gingival, dispepsia |
|||
|
Trastornos hepatobiliares |
Insuficiencia hepática*, coma hepático progresivo |
||||
|
Trastornos de la piel y del tejido subcutáneo |
Petequias, prurito (incluye generalizado), exantema, equimosis |
Púrpura, alopecia, urticaria, eritema, exantema macular |
Dermatosis neutrofílica febril aguda |
||
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia, dolor musculoesquelético (incluye dolor de espalda, huesos y en las extremidades) |
Espasmos musculares, mialgia |
|||
|
Trastornos renales y urinarios |
Insuficiencia renal*, hematuria, elevación de la creatinina sérica |
Acidosis tubular renal |
|||
|
Trastornos generales y alteraciones en el lugar de administración |
Pirexia*, fatiga, astenia, dolor torácico, eritema en el lugar de la inyección, dolor en el lugar de la inyección, reacción (no especificada) en el lugar de la inyección |
Equimosis, hematoma, induración, exantema, prurito, inflamación, decoloración, nódulo y hemorragia (en el lugar de la inyección), malestar, escalofríos, hemorragia en el lugar del catéter |
Necrosis en el lugar de inyección (en el lugar de la inyección) |
||
|
Exploraciones complementarias |
Disminución del peso |
||||
|
*= se han informado muy pocos casos fatales Descripción de las reacciones adversas seleccionadas |
|||||
Reacciones adversas hematológicas
Las reacciones adversas hematológicas más comúnmente informadas (≥10%) que se asociaron con el tratamiento con azacitidina incluyeron anemia, trombocitopenia, neutropenia, neutropenia febril y leucopenia, y en general, fueron de grado 3 o 4. Existe un mayor riesgo de que estos eventos ocurran dentro de los primeros 2 ciclos, y pasados estos ciclos, los eventos ocurren con menor frecuencia en pacientes con restauración de la función hematológica.La mayoría de las reacciones adversas hematológicas se controlaron por medio de un monitoreo de rutina de hemogramas completos y al retrasar la administración de azacitidina en el siguiente ciclo, con el uso de antibióticos profilácticos y/o el soporte del factor de crecimiento (por ejemplo, G-CSF) para neutropenia y transfusiones para tratar anemia y trombocitopenia, según sean necesarias.Infecciones
La mielosupresión podría causar neutropenia y un riesgo aumentado de infección. En los pacientes que han recibido azacitidina se han notificado reacciones adversas graves, como sepsis neutropénica y neumonía, algunas con desenlace mortal. Las infecciones pueden tratarse con el empleo de un antiinfeccioso y refuerzo con factor del crecimiento (p. ej., G-CSF) para la neutropenia.
Sangrado
El sangrado podría ocurrir en pacientes que reciben azacitidina. Se han informado reacciones adversas serias, tales como: hemorragia gastrointestinal y hemorragia intracraneal. Los pacientes deben ser monitoreados por signos y síntomas de sangrado, particularmente, aquellos con trombocitopenia preexistente o relacionada con el tratamiento.
Hipersensibilidad
Las reacciones serias de hipersensibilidad se han reportado en pacientes que reciben azacitidina. En caso de una reacción del tipo anafiláctica, el tratamiento con azacitidina debe interrumpirse de inmediato e iniciarse el tratamiento sintomático adecuado.
Reacciones adversas de la piel y del tejido subcutáneo
La mayoría de las reacciones adversas de la piel y del tejido subcutáneo se asociaron con la zona de aplicación de la inyección. Ninguna de estas reacciones adversas produjo una interrupción de azacitidina, ni la reducción de la dosis de azacitidina en los estudios pivotales. La mayoría de las reacciones adversas ocurrieron durante los dos primeros ciclos y tendieron a disminuir con los ciclos posteriores. Las reacciones adversas subcutáneas, tales como, rash, inflamación, prurito, eritema y lesiones de la piel en la zona de la inyección podrían requerir el uso de medicamentos concomitantes, tales como: antihistamínicos, corticosteroides y Antiinflamatorios No Esteroides (AINES).
Estas reacciones cutáneas deben diferenciarse de las infecciones del tejido blando, que a veces se producen en el lugar de la inyección. Se reportaron infecciones del tejido blando, incluso celulitis y fascitis necrotizante que en casos raros lleva a la muerte con azacitidina en el entorno de postcomercialización. Para obtener información sobre el manejo clínico de las reacciones adversas infecciosas, ver sección Infecciones.
Reacciones adversas gastrointestinales
Las reacciones adversas gastrointestinales más frecuentemente informadas que se asociaron con el tratamiento con azacitidina incluyeron constipación, diarrea, náuseas y vómitos. Estas reacciones adversas se controlaron sintomáticamente con antieméticos para las náuseas y los vómitos; antidiarreicos para la diarrea, y laxantes y/o productos para ablandar las heces en caso de constipación.
Reacciones adversas renales
Las anormalidades renales, desde creatinina sérica elevada y hematuria, a acidosis renal tubular, insuficiencia renal y muerte, fueron reportadas en pacientes tratados con azacitidina (ver sección Advertencias y precauciones especiales para su uso).
Reacciones adversas hepáticas
Se ha informado que los pacientes con gran carga tumoral debido a la metástasis, experimentaron insuficiencia hepática, coma hepático progresivo y la muerte durante el tratamiento con azacitidina (ver sección Advertencias y precauciones especiales para su uso).
Eventos cardíacos
Los datos de un estudio clínico que permitió el enrolamiento de pacientes con antecedentes conocidos de enfermedad cardiovascular o pulmonar mostraron un aumento estadísticamente significativo de los eventos cardíacos en pacientes con LMA recientemente diagnosticada tratados con (ver sección Advertencias y precauciones especiales para su uso).
Población de edad avanzada
Hay poca información de seguridad disponible respecto de azacitidina administrada a pacientes ≥85 años (con 14 [5,9%] pacientes ≥85 años en el estudio AZA-AML-001).Notificación de sospechas de reacciones adversas Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas.
Efectos sobre la capacidad para conducir y utilizar máquinas
La influencia de azacitidina sobre la capacidad para conducir y utilizar máquinas es pequeña o moderada. Se ha notificado fatiga con el uso de azacitidina. Por lo tanto, se recomienda precaución al conducir un vehículo o utilizar máquinas.
Incompatibilidades
Este producto farmacéutico no se debe mezclar con otros medicamentos a excepción de aquellos mencionados en las secciones: Vida útil y Precauciones especiales para la eliminación y otro manejo.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones
Interacciones con otros medicamentos y otras formas de interacción en base a los datos in vitro, el metabolismo de azacitidina no parece estar mediado por las isoenzimas del citocromo P450 (CYPs, por sus siglas en inglés), UDP-Glucuronosiltransferasas (UGTs), Sulfotransferasas (SULTs) y Glutatión-Transferasas (GSTs, por sus siglas en inglés); por lo tanto, las interacciones in vivo relacionadas con estas enzimas de metabolización no son comunes. Son poco probables los efectos inhibitorios o inductivos clínicamente significativos de azacitidina sobre las enzimas del citocromo P450 (ver sección Propiedades farmacocinéticas). No se han realizado estudios clínicos formales de interacción de drogas con azacitidina.
INFORMACIÓN COMPLEMENTARIA:
Elaborado en: Baxter Oncology GmbH
Kanstrasse 2
33790 Halle/Westfallen, Germany
Acondicionado en Uruguay por: ADIUM PHARMA S.A, Ruta 8, Km 17500, Zonamérica, Montevideo, República Oriental del Uruguay
Distribuido por: MEDICAMENTA ECUATORIANA S.A.
AV.6 DE DICIEMBRE 3144 Y JUAN BOUSSINGAULT, EDIFICIO TORRE 6 , PISO 12 170518 . QUITO – ECUADOR.
Fecha de última revisión: 31/01/2019
MEDICAMENTA
Casilla 17-21-027
RECOMENDACIONES:
Advertencias y precauciones
Datos de seguridad preclínica
La azacitidina induce ambas mutaciones genéticas y aberraciones cromosómicas en sistemas in vitro celulares de bacterias y de mamíferos. El potencial carcinogenético de azacitidina fue evaluado en ratones y ratas. La azacitidina indujo tumores del sistema hematopoyético en ratones hembra, cuando se lo administró de forma intraperitoneal 3 veces por semana durante 52 semanas. Se observó una incidencia aumentada de tumores en el sistema linforreticular, los pulmones, las glándulas mamarias y la piel en ratones tratados con azacitidina administrada intraperitonealmente durante 50 semanas. Un estudio de tumorigenicidad en ratas reveló una incidencia aumentada de tumores testiculares.
Los primeros estudios de embriotoxicidad en ratones revelaron una frecuencia del 44% de muerte embrionaria intrauterina (reabsorción aumentada) luego de una única inyección intraperitoneal de azacitidina durante la organogénesis. Se han detectado anormalidades de desarrollo en el cerebro de los ratones cuando se administró azacitidina durante o antes del cierre del paladar duro. En ratas, azacitidina no causó reacciones adversas cuando se la administró antes de la implantación, pero fue claramente embriotóxico cuando se administró durante la organogénesis. Las anormalidades fetales durante la organogénesis en ratas incluyeron: Anormalidades del SNC (exencefalia/encefalocele), anormalidades en las extremidades (micromelia, pie equino, sindactilia, oligodactilia) y otros (microftalmia, micronagtia, gastrosquisis, edema y anormalidades en las costillas).
La administración de azacitidina a ratones macho antes del apareamiento con ratones hembra que no están tratados, provocó una deficiencia en la fertilidad y la pérdida de las crías durante el posterior desarrollo embrionario y posnatal.
El tratamiento de las ratas macho trajo como resultado un descenso en el peso de los testículos y del epidídimo, recuentos de espermatozoides disminuidos, índices bajos de embarazo, aumento de embriones anormales y pérdida aumentada de embriones en hembras apareadas (ver sección Advertencias y precauciones especiales para su uso).
Advertencias y precauciones especiales para su uso Toxicidad hematológica
El tratamiento con Azacitidina está asociado con la anemia, neutropenia y trombocitopenia, especialmente durante los 2 primeros ciclos (ver sección Efectos adversos). Se deben realizar hemogramas completos para controlar la respuesta y toxicidad tal como se requiera, pero al menos antes de cada ciclo de tratamiento. Luego de la administración de la dosis recomendada para el primer ciclo, la dosis para los ciclos posteriores se debe reducir o retrasar su administración en base a los recuentos de nadir y a la respuesta hematológica (ver sección Posología dosificación y administración). Se les debe recomendar a los pacientes que informen a la brevedad episodios febriles. A los pacientes y a los médicos también se les recomienda que estén atentos y observen los signos y síntomas de hemorragia.
Insuficiencia hepática
No se realizaron estudios formales en pacientes con insuficiencia hepática. Se ha informado que los pacientes con carga tumoral extensiva debido a la metástasis experimentaron coma hepático progresivo y la muerte durante el tratamiento con azacitidina, especialmente en los pacientes con albúmina sérica de línea de base < 30 g/l. Azacitidina está contraindicada en pacientes con tumores hepáticos malignos avanzados (ver sección Contraindicaciones).
Insuficiencia renal
Se informaron anormalidades renales que variaron desde creatinina sérica elevada a insuficiencia renal y muerte en pacientes tratados con azacitidina intravenosa en combinación con otros agentes quimioterapéuticos. Además, la acidosis tubular renal, definida como una caída del bicarbonato sérico a < 20 mmol/l en asociación con orina alcalina e hipocalemia (potasio sérico < 3 mmol/l) se desarrolló en 5 pacientes con Leucemia Mieloide Crónica (LMC) tratada con azacitidina y etopósido. Si se presentan disminuciones del bicarbonato sérico (<20 mmol/l) o aumentos de la creatinina sérica o nitrógeno ureico en sangre (BUN, por sus siglas en inglés) de origen desconocido, se debe reducir la dosis o retrasar la administración (ver sección Posología dosificación y administración). Se les debe recomendar a los pacientes que informen inmediatamente a los profesionales de la salud si presentan oliguria o anuria. Aunque no se observaron diferencias clínicamente relevantes en la frecuencia de las reacciones adversas entre los sujetos con la función renal normal en comparación con los que presentaban insuficiencia renal, se debe monitorear atentamente la toxicidad en los pacientes con insuficiencia renal, puesto que azacitidina y/o sus metabolitos se excretan principalmente por el riñón (ver sección Posología dosificación y administración).
Análisis de laboratorio
Las pruebas de funcionamiento del hígado, la creatinina y el bicarbonato séricos se deben determinar antes de comenzar con la terapia y antes de cada ciclo de tratamiento. Los recuentos completos de sangre se deben realizar antes de iniciar la terapia y según sea necesario para monitorear la respuesta y la toxicidad, pero como mínimo, antes de cada ciclo de tratamiento (ver también sección Efectos adversos).
Enfermedad cardíaca y pulmonar
Los pacientes con antecedentes de insuficiencia cardíaca congestiva severa, con enfermedad cardíaca clínicamente no estable o con enfermedad pulmonar fueron excluidos de los estudios pivotales para registro (AZA PH GL 2003 CL 001 y AZA-AML-001) y, por lo tanto, no se ha establecido la seguridad y eficacia de VIDAZA en estos pacientes. Datos recientes de un estudio clínico en pacientes con antecedentes conocidos de enfermedad cardiovascular o pulmonar mostraron una incidencia significativamente mayor de eventos cardíacos con VIDAZA (ver sección Efectos adversos). Por lo tanto, se recomienda tener cuidado al prescribir VIDAZA a estos pacientes. Se debe considerar una evaluación cardiopulmonar antes y durante el tratamiento con VIDAZA.
Fascitis necrotizante
Fascitis necrotizante, incluyendo casos fatales, ha sido reportada en pacientes tratados con VIDAZA. El tratamiento con VIDAZA debe discontinuarse en pacientes que desarrollan fascitis necrotizante y un tratamiento apropiado debe ser iniciado rápidamente.
Síndrome de lisis tumoral
Los pacientes con riesgo de síndrome de lisis tumoral son aquellos con carga tumoral alta previa al tratamiento. Estos pacientes deben ser monitoreados de cerca y precauciones apropiadas deben ser tomadas.
Fertilidad, embarazo y lactancia
Mujeres en edad fértil / Anticoncepción en varones y mujeres
Los varones y mujeres en edad fértil tienen que utilizar un método anticonceptivo efectivo durante y hasta 3 meses después del tratamiento.
Embarazo
No se dispone de datos suficientes de la utilización de azacitidina en mujeres embarazadas. Los estudios en ratones muestran toxicidad reproductiva (ver sección Datos de seguridad preclínica). Se desconoce el riesgo potencial en humanos. En base a resultados de estudios en animales y su mecanismo de acción, no se debe utilizar azacitidina durante el embarazo, especialmente, durante el primer trimestre, a menos que sea estrictamente necesario. Las ventajas del tratamiento se deben comparar con el posible riesgo en el que puede estar el feto en cada caso individual.
Lactancia
Se desconoce si azacitidina / metabolitos se excretan en la leche humana. Debido a las potenciales reacciones adversas serias en el lactante, está contraindicado amamantar durante la terapia con azacitidina.
Fertilidad
No existen datos en humanos sobre el efecto de azacitidina en la fertilidad. En los animales, se han documentado reacciones adversas de azacitidina sobre la fecundidad masculina (ver sección Datos de seguridad preclínica). Se les recomienda a los pacientes hombres no tener hijos durante el tratamiento y deben utilizar una anticoncepción efectiva durante y hasta 3 meses después del tratamiento. Antes de comenzar el tratamiento, se les debe recomendar a los pacientes hombres que se asesoren respecto de la conservación de esperma.
DOSIS Y VÍA DE ADMINISTRACIÓN
Posología, dosificación y administración
El tratamiento con VIDAZA debe comenzar y ser monitoreado bajo la supervisión de un médico experimentado en el uso de agentes quimioterapéuticos. Los pacientes deben ser medicados previamente con antieméticos para las náuseas y vómitos.
Posología
La dosis inicial recomendada para el primer ciclo del tratamiento, para todos los pacientes independientemente de los valores hematológicos iniciales de laboratorio, es de 75 mg/m2 del área de la superficie corporal, diariamente durante 7 días, seguida de un período de descanso de 21 días (ciclo de tratamiento de 28 días).
Se recomienda tratar a los pacientes por un mínimo de 6 ciclos. El tratamiento debe continuar siempre y cuando el paciente se siga beneficiando o hasta que progrese la enfermedad. Los pacientes deben ser monitoreados con respecto a la respuesta/toxicidad hematológica y toxicidades renales (ver sección Advertencias y precauciones especiales para su uso); podría necesitarse un retraso en el comienzo del próximo ciclo o una reducción de dosis, tal como se describe a continuación.Análisis de laboratorio
Antes de iniciar el tratamiento y antes de cada ciclo de tratamiento, deben realizarse pruebas de función hepática y determinarse la creatinina y el bicarbonato séricos. Deben efectuarse recuentos sanguíneos completos antes del inicio del tratamiento y cuando sea necesario para monitorizar la respuesta y la toxicidad, pero como mínimo, antes de cada ciclo de tratamiento.
Ajuste de dosis debido a toxicidad hematológica La toxicidad hematológica se define como el recuento más bajo alcanzado en un ciclo determinado (nadir) si las plaquetas ≤ 50,0 x 109/l y/o el recuento absoluto de neutrófilos (ANC) ≤ 1 x 109/l.
La recuperación se define como un aumento de la(s) línea(s) celular(es) en las que se observó toxicidad hematológica en al menos la mitad de la diferencia entre el nadir y el recuento basal más el recuento nadir (por ejemplo, hemograma en la recuperación ≥ recuento nadir + (0,5 x [recuento basal –recuento nadir]).
Pacientes sin disminución de los recuentos sanguíneos iniciales (es decir, glóbulos blancos (WBC) ≥ 3,0 x 109/l y RAN ≥ 1,5 x 109/l, y plaquetas ≥ 75,0 x 109/l) previo al primer tratamiento.
Si se observa toxicidad hematológica luego del tratamiento con VIDAZA, el próximo ciclo de terapia de VIDAZA se debe retrasar hasta que el recuento plaquetario y el RAN se hayan recuperado. Si la recuperación se logra en 14 días, no es necesario un ajuste de dosis. Sin embargo, si no se logra la recuperación dentro de los 14 días, se debe reducir la dosis de acuerdo con la siguiente tabla. Después de las modificaciones de dosis, la duración del ciclo debe volver a ser de 28 días.
|
Recuentos nadir |
% de la dosis en el siguiente ciclo, si la recuperación* no se alcanza en un plazo de 14 días |
|
|
RAN (x 109/l) |
Plaquetas |
|
|
< 1,0 |
< 50,0 |
50% |
|
> 1,0 |
> 50,0 |
100% |
|
*Recuperación = recuentos ≥ Recuento Nadir + (0,5 x [recuento inicial – recuento Nadir]). |
||
Pacientes con recuentos sanguíneos iniciales reducidos (es decir, leucocitos < 3,0 × 109/l o RAN < 1,5 × 109/l o recuento plaquetario < 75,0 × 109/l) antes del primer tratamiento
Luego del tratamiento con VIDAZA, si la disminución en los glóbulos blancos, RAN o plaquetas desde antes del tratamiento es ≤ 50% o mayor de 50% pero con una mejora en cualquier diferenciación de línea celular, el siguiente ciclo no se debe retrasar y no se debe realizar un ajuste de dosis.
Si la reducción en los glóbulos blancos o el RAN o las plaquetas es mayor al 50% con respecto al de antes del tratamiento y no hay mejoras en la diferenciación de la línea celular, el siguiente ciclo de la terapia de VIDAZA se debe retrasar hasta que el recuento de plaquetas y el RAN se hayan recuperado. Si la recuperación se logra en 14 días, no es necesario un ajuste de dosis. No obstante, si no se logra una recuperación en 14 días, se debe determinar la celularidad de la médula ósea. Si la celularidad de la médula ósea es > 50 %, no se deben realizar ajustes de dosis. Si la celularidad de la médula ósea es ≤ 50 %, el tratamiento se debe retrasar y reducir la dosis de acuerdo con la siguiente tabla:
|
Celularidad de la médula ósea |
% de la dosis en el siguiente ciclo, si la recuperación no se alcanza en un plazo de 14 días |
|
|
Recuperación* ≤ 21 días |
Recuperación* > 21 días |
|
|
15-50% |
100% |
50% |
|
< 15% |
100% |
33% |
|
*Recuperación = recuentos ≥ recuento nadir + (0,5 x [recuento inicial – recuento nadir]) |
||
Después de las modificaciones de dosis, la duración del ciclo debe volver a ser de 28 días.
Poblaciones especiales
Personas de edad avanzada: No se recomiendan ajustes de dosis específicos para los pacientes de edad avanzada. Dado que los pacientes de edad avanzada son más propensos a tener una función renal disminuida, puede ser conveniente monitorear la función renal.
Pacientes con insuficiencia renal: Se puede administrar azacitidina a pacientes con insuficiencia renal sin la necesidad de ajustar la dosis inicial (ver sección Propiedades farmacocinéticas). Si se producen disminuciones inexplicadas de los niveles de bicarbonato sérico a menos de 20 mmol/l, la dosis deberá disminuirse en un 50 % en el siguiente ciclo. Si se producen aumentos inexplicados de la creatinina sérica o del nitrógeno ureico en sangre (NUS) a ≥ 2 veces superiores a los valores iniciales y superiores al Límite Superior de la Normalidad (LSN), el siguiente ciclo deberá retrasarse hasta que los valores vuelvan a la normalidad o a los valores iniciales, y la dosis deberá disminuirse en un 50 % en el siguiente ciclo de tratamiento. (ver sección Advertencias y precauciones especiales para su uso).
Pacientes con Insuficiencia hepática: No se realizaron estudios formales en pacientes con insuficiencia hepática (ver sección Advertencias y precauciones especiales para su uso). Los pacientes con deficiencia hepática severa deben ser monitoreados cuidadosamente con respecto a los efectos adversos. No se recomienda ninguna modificación específica a la dosis inicial en pacientes con disfunción hepática previo al tratamiento inicial; las posteriores modificaciones de dosis se deben basar en valores hematológicos de laboratorio. VIDAZA está contraindicado en pacientes con tumores hepáticos malignos avanzados (ver secciones Contraindicaciones y Advertencias y precauciones especiales para su uso).
Personas de edad avanzada: No se recomiendan ajustes de dosis específicos para los pacientes de edad avanzada. Dado que los pacientes de edad avanzada son más propensos a tener una función renal disminuida, puede ser conveniente monitorear la función renal.
Población pediátrica: No se recomienda utilizar VIDAZA en niños de menos de 18 años debido a datos de seguridad y eficacia insuficientes.
Forma de administración
Vía subcutánea: VIDAZA reconstituido debe inyectarse de manera subcutánea en la parte superior del brazo, el muslo o el abdomen. Las zonas de aplicación de las inyecciones deben rotar. Las nuevas inyecciones se deben aplicar al menos a 2,5 cm de la zona anterior y nunca en áreas sensibles, con equimosis, enrojecidas o endurecidas.
Procedimiento de reconstitución
VIDAZA debe reconstituirse con agua para inyectables. La vida útil del medicamento reconstituido puede extenderse al reconstituirlo con agua para inyectables refrigerada (2°C a 8°C). Los detalles sobre el almacenamiento del producto reconstituido figuran a continuación.
1. Se deben reunir los siguientes suministros:
• Frasco(s) ampolla de azacitidina; frasco(s) ampolla de agua para inyectables; guantes quirúrgicos no estériles.
• Paños embebidos en alcohol; jeringas para inyección de 5 ml con aguja.
2. Se deben extraer 4 ml de agua para inyectables dentro de la jeringa, asegurándose de purgar cualquier aire atrapado dentro de la jeringa.
3. La aguja de la jeringa que contiene 4 ml de agua para inyectables debe insertarse a través de la tapa de goma del frasco ampolla de azacitidina y luego inyectar el agua para inyectables dentro del frasco ampolla.
4. Luego de retirar la jeringa y la aguja, el frasco ampolla debe agitarse enérgicamente hasta lograr una suspensión turbia uniforme. Luego de la reconstitución, cada ml de la suspensión contendrá 25 mg de azacitidina (100 mg/4 mL). El producto reconstituido es una suspensión homogénea, turbia, libre de aglomerados. La suspensión debe desecharse si contiene partículas grandes o aglomerados. No filtrar la suspensión luego de la reconstitución, dado que esto podría retirar el principio activo. Se debe tener en cuenta que los filtros están presentes en algunos adaptadores, picos y sistemas cerrados; por lo tanto, tales sistemas no se deben utilizar para la administración de la droga luego de la reconstitución.
5. El tapón de goma se debe limpiar y se debe insertar una nueva jeringa con aguja. Invierta el frasco ampolla asegurándose de que la punta de la aguja esté debajo del nivel de líquido. Tire el émbolo hacia atrás para extraer la cantidad de fármaco requerido para la dosis adecuada, asegurándose de eliminar el aire atrapado dentro de la jeringa. Luego, se debe retirar la jeringa con la aguja del frasco ampolla y se debe tirar la aguja.
6. Posteriormente, se debe ajustar con firmeza a la jeringa una aguja subcutánea nueva (se recomienda de calibre 25). No se debe purgar la aguja antes de la inyección, con el objetivo de reducir la incidencia de las reacciones en la zona de aplicación local de la inyección.
7. Cuando se necesita más de un frasco ampolla deben repetirse todos los pasos anteriores para la preparación de la suspensión. Para dosis que requieren más de un frasco ampolla, la dosis debe divididirse en partes iguales (por ejemplo, la dosis de 150 mg = 6 ml, dos jeringas con 3 ml en cada jeringa).Debido a la retención en el vial y la aguja, puede no ser factible retirar toda la suspensión del vial.
8. El contenido de la jeringa de administración debe volver a suspenderse inmediatamente antes de la administración. La temperatura de la suspensión en el momento de la inyección debe ser de aproximadamente, entre 20ºC - 25ºC. Para volver a suspender, haga rodar la jeringa fuertemente entre las palmas hasta lograr una suspensión uniforme y turbia. La suspensión debe desecharse si contiene partículas grandes o conglomerados.
Almacenamiento del producto reconstituido
Para uso inmediato: La suspensión de VIDAZA puede prepararse inmediatamente antes de su uso y la suspensión reconstituida debe administrarse dentro de los 45 minutos. Si el tiempo transcurrido es mayor de 45 minutos, la suspensión reconstituida se debe desechar de forma adecuada y se debe preparar una dosis nueva. Para un uso posterior: Cuando se reconstituyen con agua para inyectables que no se ha refrigerado, la suspensión reconstituida debe colocarse en un refrigerador (entre 2ºC y 8ºC) inmediatamente después de la reconstitución, y mantenerse en el refrigerador por un máximo de 8 horas. Si el tiempo transcurrido en el refrigerador es mayor de 8 horas, la suspensión se debe descartar de forma adecuada y se debe preparar una nueva dosis.
Cuando se reconstituyen con agua para inyectables refrigerada (2ºC a 8º C), la suspensión reconstituida debe colocarse en un refrigerador (entre 2ºC y 8ºC) inmediatamente después de la reconstitución, y mantenerse en el refrigerador por un máximo de 22 horas. Si el tiempo transcurrido es mayor de 22 horas, la suspensión se debe desechar de forma adecuada y se debe preparar una dosis nueva.
La jeringa llenada con suspensión reconstituida debe dejarse reposar por hasta 30 minutos antes de la administración para alcanzar una temperatura de aproximadamente, 20°C - 25°C, durante un tiempo máximo de 30 minutos antes de la administración. Si el tiempo transcurrido es mayor de 30 minutos, la suspensión se debe desechar de forma adecuada y se debe preparar una nueva dosis.
Cálculo de la dosis individual
La dosis total, de acuerdo con el área de superficie corporal (SC, superficie corporal ), puede calcularse de la siguiente manera: Dosis total (mg) = Dosis (mg/m2) x SC (m2) La siguiente tabla se provee sólo como un ejemplo de cómo calcular las dosis individuales de azacitidina en base al valor promedio de SC de 1,8 m2.
|
Dosis, mg/m2 (% de la dosis inicial recomendada) |
Dosis total basada en un valor de SC de 1,8 m2 |
Cantidad de frascos ampolla requeridos |
Volumen total de suspensión reconstituida requerida |
|
75 mg/m2 (100%) |
135 mg |
2 frascos ampolla |
5,4 ml |
|
37,5 mg/m2 (50%) |
67,5 mg |
1 frasco ampolla |
2,7 ml |
|
25 mg/m2 (33%) |
45 mg |
1 frasco ampolla |
1,8 ml |
Forma de administración
El producto VIDAZA reconstituido debe inyectarse de forma subcutánea (insertar la aguja a un ángulo 45-90°) con una aguja calibre 25 en el brazo, abdomen o muslo. Las dosis mayores de 4 ml deben inyectarse en dos sitios diferentes. Las zonas de aplicación de las inyecciones deben rotar. Las nuevas inyecciones se deben aplicar al menos a 2,5 cm de la zona anterior y nunca en áreas sensibles, con hematomas, enrojecidas o endurecidas. Cualquier producto sin utilizar o material residual se debe desechar de acuerdo con los requerimientos locales. Vía administración endovenosa: Reconstituir el número necesario de viales de VIDAZA hasta alcanzar la dosis necesaria. Reconstituir cada vial de VIDAZA, con 10 ml. de agua estéril para inyección. Agitar o hacer girar en forma vigorosa hasta que todas las partículas sólidas desaparezcan. La solución resultante de esta reconstitución contiene 10mg/ml de Azacitidina. La solución debe ser clara. Las drogas de administración parenteral deben ser inspeccionadas para descartar la presencia de partículas sólidas y decoloración antes de la administración, siempre que la solución y los contenedores lo permitan.
Retire la cantidad necesaria de la solución de VIDAZA para administrar la dosis necesaria e inyecte en una bolsa de infusión de 50-100 mL de cloruro de sodio al 0,9% o solución de Ringer Lactato.
Incompatibilidad de las soluciones intravenosas VIDAZA es incompatible con soluciones de dextrosa al 5%, Hespan o soluciones que contengan bicarbonato. Estas soluciones tienen el riesgo potencial de aumentar la tasa de degradación de VIDAZA y por lo tanto, deben ser evitadas.
Administración intravenosa
La solución de VIDAZA se administra por vía intravenosa. Administre la dosis total en un período de 10 a 40 minutos. La administración debe completarse en un período de una hora luego de la reconstitución del vial de VIDAZA. Estabilidad de la solución: VIDAZA reconstituido para administración intravenosa puede almacenarse a 25ºC, pero su administración debe completarse en la siguiente hora.
Precauciones especiales para la eliminación y otro manejo
Recomendaciones para el manejo seguro
VIDAZA es un producto citotóxico y, como sucede con otros compuestos potencialmente tóxicos, se debe tener precaución en la manipulación y preparación de suspensiones de azacitidina. Se deben aplicar procedimientos adecuados para la manipulación y eliminación de los productos medicinales para el cáncer. Si la azacitidina reconstituida entra en contacto con la piel, lavar inmediatamente y con cuidado la piel con agua y jabón. Si entra en contacto con las membranas mucosas, limpiar con mucha agua.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis
Se informó un caso de sobredosis con azacitidina durante los estudios clínicos. Un paciente experimentó diarrea, náuseas y vómitos luego de recibir una única dosis intravenosa de aproximadamente 290 mg/, casi 4 veces la dosis de inicio recomendada.
En el caso de sobredosis, el paciente debe ser monitoreado con hemogramas adecuados y debe recibir tratamiento de soporte, según sea necesario. No existe un antídoto específico para la sobredosis de azacitidina.
Ante la eventualidad de una sobredosificación, concurrir al hospital más cercano o comunicarse al teléfono: MEDICAMENTA ECUATORIANA S.A., Edificio"Medicamenta Ecuatoriana S.A. Avda. Juan Diguja No. Oe1-82 y Vozandes en la ciudad de Quito. PBX 2277-032. FAX 2272-506. E-mail: farmacovigilanciavidaza@medicamenta.com.ec
PRESENTACIÓN:
Inyectable liofilizado 100 mg: Envase conteniendo 1 frasco ampolla.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Conservación
Luego de la reconstitución (para administración subcutánea): Cuando VIDAZA se reconstituye con agua para inyectables que no se ha refrigerado, la estabilidad química y física del medicamento reconstituido se ha demostrado a 25º C durante 45 minutos y entre 2°C y 8°C durante 8 horas.La vida útil del medicamento reconstituido se puede extender por medio de la reconstitución con agua para inyectables refrigerada (2°C a 8°C). Cuando VIDAZA se reconstituye con agua para inyectables refrigerada (2°C a 8°C), la estabilidad química y física del medicamento reconstituido se ha demostrado entre 2°C y 8°C durante 22 horas.
Desde el punto de vista microbiológico, el producto reconstituido se debería usar de inmediato. Si no se usa de inmediato, los tiempos de almacenamiento y las condiciones previas al uso son responsabilidad del usuario, y no deberían extenderse por más de 8 horas entre 2ºC y 8ºC cuando se lo reconstituye con agua para inyectables que no se ha refrigerado o por más de 22 horas cuando se reconstituyó con agua para inyectables refrigerada (2ºC a 8ºC).
Luego de la reconstitución (para administración endovenosa): VIDAZA reconstituido para administración endovenosa puede ser almacenado a 25°C (77°F), pero la administración debe completarse dentro de la siguiente hora luego de la reconstitución.
Precauciones especiales para el almacenamiento
Viales sin abrir: Este producto farmacéutico no requiere ninguna condición especial de almacenamiento.
Suspensión reconstituida: Para las condiciones de almacenamiento del producto medicinal reconstituido ver el item Vida útil. Luego de la recontitución.
LEYENDAS DE PROTECCIÓN:
Todo medicamento debe conservarse fuera del alcance de los niños.
Este medicamento solo debe utilizarse bajo estricto control y vigilancia médica y no puede repetirse sin nueva receta. Especialidad medicinal autorizada por el Ministerio de Salud.


