SPIRIVA RESPIMAT
TIOTROPIO
Solución para inhalación
1 Caja, 1 Inhalador, 30 Dosis, 2.5 µg/dosis
COMPOSICIÓN:
Cada PUFF de SPIRIVA® RESPIMAT® contiene:
Tiotropio 2,5 microgramos (mcg)
La dosis suministrada es 2,5 μg de tiotropio por puff (dos puffs contienen una dosis medicinal). 2,5 μg de tiotropio equivale a 3,124 μg de bromuro de tiotropio monohidrato.
Excipientes: Cloruro de benzalconio, edetato disódico, agua purificada, ácido clorhídrico para ajuste del pH, nitrógeno.
INDICACIONES:
SPIRIVA® RESPIMAT® está indicado para:
EPOC: SPIRIVA® RESPIMAT® está indicado para el tratamiento del mantenimiento de pacientes con EPOC (incluyendo bronquitis crónica y enfisema). Se indica además para el tratamiento de la disnea asociada a la enfermedad y a la prevención de exacerbaciones.
Asma: SPIRIVA® RESPIMAT® está indicado como tratamiento de mantenimiento complementario para la mejoría de los síntomas del asma, la mejoría en la calidad de vida y la reducción de las exacerbaciones en los pacientes de 1 año de edad y mayores con asma, que continúan siendo sintomáticos con la administración de corticosteroides inhalados.
PROPIEDADES FARMACOLÓGICAS:
Grupo farmacoterapéutico: Otros fármacos para las enfermedades obstructivas de las vías aéreas, productos para inhalar, anticolinérgicos.
CÓDIGO ATC: R03BB04
Mecanismo de acción:
El bromuro de tiotropio es un antagonista de acción prolongada específico de los receptores muscarínicos, a menudo conocido en la práctica clínica con el nombre de anticolinérgico. Tiene una afinidad similar por los subtipos de los receptores muscarínicos M1 a M5. En las vías aéreas, la inhibición de los receptores M3 en la musculatura lisa produce relajación. La índole competitiva y reversible del antagonismo quedó demostrada en receptores de origen humano y animal y en preparados aislados de órganos. En estudios preclínicos in vitro e in vivo se observó que los efectos broncoprotectores eran dependientes de la dosis y duraban más de 24 horas. La prolongada duración del efecto probablemente se deba a que la disociación del tiotropio de los receptores M3 es muy lenta, con una semivida de disociación significativamente más prolongada que la observada en el caso del ipratropio. Como anticolinérgico N-cuaternario, el tiotropio es tópicamente (bronco) selectivo cuando se lo administra mediante inhalación, y presenta un rango terapéutico aceptable antes de dar lugar a efectos anticolinérgicos sistémicos. La disociación de los receptores M2 es más rápida que de los M3, lo cual en estudios in vitro funcionales, se tradujo en una selectividad (controlada cinéticamente) por el subtipo de receptores M3 mayor que por aquellos del tipo M2.
La elevada potencia y la lenta disociación de los receptores tuvieron como correlato clínico una broncodilatación significativa y prolongada en los pacientes con EPOC y asma. La broncodilatación que se produce tras la inhalación del tiotropio es primariamente un efecto localizado (sobre las vías aéreas) y no un efecto sistémico.
CONTRAINDICACIONES:
SPIRIVA® RESPIMAT® está contraindicado en pacientes con antecedentes de hipersensibilidad a la atropina o sus derivados, por ej., ipratropio y oxitropio o a cualquiera de los excipientes de este producto.
REACCIONES ADVERSAS:
Como todos los medicamentos, SPIRIVA® RESPIMAT® puede producir efectos adversos, aunque no todas las personas los sufran.
Muchos de los efectos indeseados que aquí se citan son atribuibles a las propiedades anticolinérgicas de SPIRIVA® RESPIMAT®.
Se identificaron reacciones adversas al medicamento a partir de datos obtenidos de los estudios clínicos y reportes espontáneos efectuados durante el uso posterior a la aprobación del medicamento.
Lista de reacciones adversas:
Trastornos del metabolismo y de la nutrición: Deshidratación.
Trastornos del sistema nervioso: Mareos, insomnio.
Trastornos oculares: Glaucoma, aumento de la presión intraocular, visión borrosa.
Trastornos cardiacos: Fibrilación auricular, palpitaciones, taquicardia supraventricular, taquicardia.
Trastornos respiratorios, torácicos y mediastinales: Tos, epistaxis, faringitis, disfonía, broncoespasmo, laringitis, sinusitis.
Trastornos gastrointestinales: Boca seca (usualmente leve), estreñimiento, candidiasis orofaríngea, disfagia, enfermedad por reflujo gastroesofágico, gingivitis, glositis, estomatitis, obstrucción intestinal incluyendo íleo paralítico.
Trastornos de la piel y el tejido subcutáneo, trastornos del sistema inmune: Exantema, prurito, edema angioneurótico, urticaria, infección de la piel y úlceras cutáneas, piel seca, hipersensibilidad (lo que incluye reacciones inmediatas).
Trastornos musculoesqueléticos y del tejido conectivo: Inflamación de las articulaciones.
Trastornos renales y urinarios: Retención urinaria (especialmente en hombres con factores predisponentes), disuria, infección de las vías urinarias.
Asma:
Población pediátrica: La frecuencia, el tipo y la gravedad de las reacciones adversas en los pacientes pediátricos son similares en comparación con los adultos.
Si alguno de los efectos adversos que sufre es grave o si nota cualquier efecto adverso no mencionado en este folleto, informe a su médico o farmacéutico.
INTERACCIONES:
Si bien no se han realizado estudios formales de interacciones medicamentosas, el bromuro de tiotropio se ha usado en forma concomitante con otros medicamentos que se prescriben frecuentemente para el tratamiento de la EPOC y el asma, lo que incluye broncodilatadores simpaticomiméticos, metilxantinas, esteroides orales y para inhalar, antihistamínicos, mucolíticos, modificadores del leucotrieno, cromonas y tratamiento anti-IgE, sin que se observara evidencia clínica de interacciones medicamentosas.
Se comprobó que los medicamentos que son frecuentemente usados en forma concomitante por los pacientes con EPOC (beta agonistas de acción prolongada, corticoesteroides para inhalar y sus combinaciones) no alteran la exposición al tiotropio.
La coadministración crónica de bromuro de tiotropio con otros medicamentos anticolinérgicos no ha sido estudiada. Por lo tanto, se desaconseja la coadministración crónica de otros anticolinérgicos junto con SPIRIVA® RESPIMAT®.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES:
Antes de empezar con el tratamiento de SPIRIVA® RESPIMAT® comuníquele a su médico o farmacéutico si:
• Se encuentra utilizando o ha utilizado recientemente otros medicamentos, inclusive los que no requieran de una receta para ser adquiridos, como los medicamentos herbarios.
• Si está embarazada o piensa que puede estarlo o está en periodo de lactancia.
Cuando use SPIRIVA® RESPIMAT® tenga las siguientes precauciones:
SPIRIVA® RESPIMAT®, por ser un broncodilatador de mantenimiento de una toma diaria, no debe ser usado para el tratamiento inicial de los episodios agudos de broncoespasmo ni para el alivio de los síntomas agudos. En el caso de un ataque agudo, debe usarse un agonista-beta-2 de acción rápida.
SPIRIVA® RESPIMAT® no debe usarse como tratamiento de primera línea para el asma. Debe indicarse a los pacientes asmáticos que deben continuar tomando su tratamiento antiinflamatorio, es decir, corticosteroides inhalados sin ningún cambio de dosis luego de la introducción de SPIRIVA® RESPIMAT®, incluso aunque sus síntomas mejoren.
Pueden producirse reacciones de hipersensibilidad inmediata después de la administración de SPIRIVA® RESPIMAT® solución para inhalación.
Al igual que otros medicamentos anticolinérgicos, SPIRIVA® RESPIMAT® se debe usar con precaución en los pacientes con glaucoma de ángulo estrecho, hiperplasia prostática u obstrucción del cuello de la vejiga.
Los medicamentos para inhalar pueden causar broncoespasmo inducido por la inhalación.
Como sucede con todos los medicamentos cuya vía de excreción es predominantemente renal, se deberá efectuar un estrecho control del uso de SPIRIVA® RESPIMAT® en los pacientes con insuficiencia renal de moderada a grave (depuración de la creatinina ≤ 50 mL/min).
Se deberá indicar a los pacientes la forma correcta de administrar SPIRIVA® RESPIMAT®.
Debe tenerse la precaución de evitar que la solución o la vaporización entre en contacto con los ojos. La presencia de dolor o molestia ocular, visión borrosa, halos visuales o imágenes coloreadas en asociación con enrojecimiento ocular por congestión de la conjuntiva y edema de córnea pueden ser signos de glaucoma de ángulo estrecho agudo. Si se presentara alguna combinación de estos síntomas, se deberá consultar de inmediato a un especialista.
Los colirios mióticos no son considerados un tratamiento efectivo.
SPIRIVA® RESPIMAT® no debe usarse con una frecuencia mayor a una vez al día.
Los cartuchos de SPIRIVA® RESPIMAT® se deben usar únicamente con el inhalador RESPIMAT®.
Cloruro de benzalconio:
SPIRIVA® RESPIMAT® contiene 0,0011 mg de cloruro de benzalconio en cada aplicación.
El cloruro de benzalconio puede provocar sibilancias y dificultades respiratorias. Los pacientes con asma tienen un mayor riesgo de padecer estos eventos adversos.
POSOLOGÍA Y ADMINISTRACIÓN:
Siempre use SPIRIVA® RESPIMAT® exactamente como su médico ha señalado.
El médico debe indicar la posología y el tiempo de tratamiento apropiado para su caso particular; no obstante, la dosis usual recomendada de SPIRIVA® RESPIMAT® es la inhalación de la solución pulverizada que se obtiene a partir de dos aplicaciones (puffs) una vez al día mediante el inhalador RESPIMAT® a la misma hora todos los días (ver “Instrucciones de uso”).
En el tratamiento del asma, los beneficios completos se observan luego de varias dosis de SPIRIVA® RESPIMAT®.
Poblaciones especiales: Los pacientes de edad avanzada pueden usar SPIRIVA® RESPIMAT® en la dosis recomendada.
Los pacientes con insuficiencia renal pueden usar SPIRIVA® RESPIMAT® en la dosis recomendada. No obstante, como sucede con todos los fármacos cuya excreción es predominantemente renal, se deberá efectuar un estrecho control del uso de SPIRIVA® RESPIMAT® en los pacientes con insuficiencia renal moderada a grave.
Los pacientes con insuficiencia hepática pueden usar SPIRIVA® RESPIMAT® en la dosis recomendada.
Población pediátrica:
La EPOC normalmente no se produce en los niños.
En asma, la posología recomendada de SPIRIVA® RESPIMAT® en pacientes entre 1 y 17 años de edad es la inhalación de la solución pulverizada que se obtiene a partir de dos pulsaciones (puffs) una vez al día mediante el inhalador RESPIMAT®, a la misma hora todos los días (ver “Instrucciones de uso”). En niños entre 1 y 5 años de edad, SPIRIVA® RESPIMAT® se debe utilizar con un espaciador adecuado con una mascarilla.
La eficacia y la seguridad de SPIRIVA® RESPIMAT® en pacientes pediátricos menores de 1 año de edad con asma no han sido determinadas.
Usted debe consultar con su médico o farmacéutico si tiene alguna duda sobre el uso de SPIRIVA® RESPIMAT®.
INSTRUCCIONES DE USO:
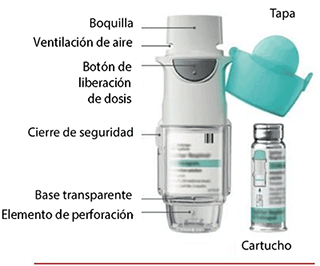
SPIRIVA® RESPIMAT® descartable:
Uso previsto: El RESPIMAT® descartable es un dispositivo inhalador que genera una bruma en dispersión lenta para inhalación.
El RESPIMAT® descartable es un dispositivo para un solo paciente previsto para uso múltiple.
Lea estas instrucciones antes de empezar a usar SPIRIVA® RESPIMAT®.
Los niños deben utilizar SPIRIVA® RESPIMAT® con la ayuda de un adulto. En pacientes entre 1 y 5 años de edad, se debe utilizar SPIRIVA® RESPIMAT® con un espaciador adecuado con una mascarilla.
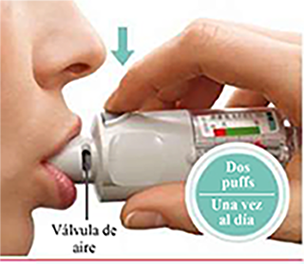
Debe utilizar este inhalador solamente UNA VEZ AL DÍA. Cada vez que lo use, inhale DOS PULSACIONES (puffs).
• Si transcurrieron más de 7 días desde la última vez que usó su inhalador, libere una pulsación con el inhalador apuntando hacia el suelo.
• Si transcurrieron más de 21 días desde la última vez que usó su inhalador, repita los pasos 4 a 6 hasta que se forme una nube visible. Luego, repita los pasos 4 a 6 tres veces más.
¿Cómo cuidar su inhalador SPIRIVA® RESPIMAT®?:
Al menos una vez a la semana debe limpiar la boquilla, incluyendo la parte metálica de su interior, únicamente con un paño o pañuelo de papel húmedo.
Las decoloraciones mínimas de la boquilla no afectan el funcionamiento de su inhalador SPIRIVA® RESPIMAT®.
¿Cuándo adquirir un nuevo inhalador SPIRIVA® RESPIMAT®?:

• El inhalador SPIRIVA® RESPIMAT® contiene 60 aplicaciones (30 dosis) si se le usa según se indica (dos aplicaciones/una vez al día).
• El indicador de dosis muestra de manera aproximada cuánta medicación queda.
• Cuando el indicador ingrese en la zona roja de la escala, usted deberá conseguir una nueva receta; quedará medicación para aproximadamente 7 días más (14 aplicaciones).
• Una vez que el indicador de dosis llegue al final de la escala roja, su SPIRIVA® RESPIMAT® se trabará automáticamente y no liberará más dosis. A partir de ese momento ya no se podrá girar más la base transparente.
• Una vez cumplidos tres meses desde el primer uso, el inhalador SPIRIVA® RESPIMAT® deberá ser desechado, aunque no haya sido utilizado.
Preparación para el primer uso:
|
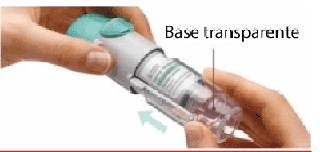
1. Retirar la base transparente • Mantenga la tapa cerrada. • Presione el cierre de seguridad con firmeza mientras retira la base transparente con la otra mano. |
|
|
2. Insertar el cartucho • Inserte el extremo estrecho del cartucho en el inhalador. • Coloque el cartucho sobre una superficie firme y empuje con firmeza hacia abajo hasta que encastre en su lugar. |
|
|
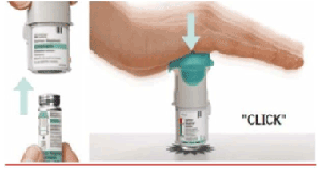
3. Volver a colocar la base transparente • Coloque la base transparente en su lugar hasta que haga click. |
|
|
4. Girar • Mantenga la tapa cerrada. • Gire la base transparente en el sentido de las flechas de la etiqueta hasta que haga click (media vuelta). |
|
|
5. Abrir • Abra la tapa hasta que quede totalmente abierta. |
|
|
6. Presionar • Apunte el inhalador hacia el suelo. • Presione el botón de liberación de dosis. • Cierre la tapa. • Repita los pasos 4 a 6 hasta que se forme una nube visible. • Cuando se haya formado una nube visible, repita los pasos 4 a 6 tres veces más. |
|
Uso diario:
|
GIRAR • Mantenga la tapa cerrada. • GIRE la base transparente en el sentido de las flechas de la etiqueta hasta que haga click (media vuelta). |
|
|
ABRIR • ABRA la tapa hasta quitarla por completo. |
|
|
PRESIONAR • Exhale en forma lenta y profunda. • Coloque sus labios alrededor de la boquilla sin cubrir las válvulas de aire. • Mientras inhala lenta y profundamente a través de la boca, PRESIONE el botón de liberación de dosis y continúe inhalando. • Contenga la respiración unos 10 segundos o durante el tiempo que le resulte cómodo. • Repita los pasos “Girar”, “Abrir” y “Presionar” hasta completar el total de 2 inhalaciones. • Cierre la tapa hasta que vuelva a usar el inhalador. |
|
Lea cuidadosamente este folleto antes de la administración de este medicamento, contiene información importante acerca de su tratamiento.
Este medicamento se le ha recetado a usted y no debe entregarlo o recomendarlo a otras personas, aunque tengan los mismos síntomas, ya que puede perjudicarles. Si considera que alguno de los efectos adversos que sufre es grave o si aprecia cualquier efecto adverso no mencionado en este folleto, pregunte a su médico o químico farmacéutico. Guarde el folleto, puede necesitar leerlo nuevamente.
USO EN POBLACIONES ESPECÍFICAS:
Fertilidad, embarazo y lactancia:
Usted no debe usar SPIRIVA® RESPIMAT® a menos que su médico lo haya recomendado para su uso.
Embarazo:
Existe una cantidad limitada de datos sobre el uso del tiotropio en las mujeres embarazadas. Los estudios preclínicos no indican efectos nocivos directos ni indirectos en lo que se refiere a la toxicidad para la reproducción en las dosis clínicamente relevantes.
Como medida de precaución, es preferible evitar el uso de SPIRIVA® RESPIMAT® durante el embarazo.
Lactancia:
No existen datos clínicos disponibles sobre mujeres que hayan sido expuestas al tiotropio durante el periodo de lactancia.
Los estudios en roedores en periodo de lactancia han indicado que una pequeña cantidad de tiotropio se excreta en la leche materna.
Por lo tanto, SPIRIVA® RESPIMAT® no debe ser usado en mujeres embarazadas o en periodo de lactancia, a menos que el beneficio previsto supere todo posible riesgo para el bebé en gestación o el lactante.
Fertilidad: No existen datos clínicos disponibles en relación con el efecto del tiotropio sobre la fertilidad.
Un estudio preclínico realizado con tiotropio no reveló indicio alguno de efectos adversos sobre la fertilidad.
Efectos sobre la capacidad de conducir vehículos y operar maquinarias: No se han realizado estudios en torno a los efectos de este producto sobre la capacidad para conducir vehículos y operar maquinarias.
El desarrollo de mareos y visión borrosa puede afectar la capacidad de conducir vehículos y operar maquinarias.
VÍA DE ADMINISTRACIÓN: Inhalatoria.
SOBREDOSIS:
Las dosis altas de SPIRIVA® RESPIMAT® pueden provocar signos y síntomas anticolinérgicos.
No se observaron efectos adversos relevantes, excepto sequedad de la boca/garganta y sequedad de la mucosa nasal, cuya incidencia fue dependiente de la dosis (10-40 μg diarios), luego de la dosificación durante 14 días de dosis de hasta 40 μg de solución para inhalar de tiotropio en voluntarios sanos, a excepción de una notable reducción de la salivación del día 7 en adelante. No se ha observado ningún efecto indeseado significativo en seis estudios a largo plazo realizados en pacientes con EPOC tratados con una dosis diaria de 10 μg de solución para inhalar de tiotropio durante 4 a 48 semanas.
Ante esta eventualidad acudir al servicio de urgencias más cercano.
PRESENTACIONES COMERCIALES:
Caja x 1 cartucho de aluminio con inhalador SPIRIVA® RESPIMAT® (válvula dosificadora) con escala medidora x 4mL solución x 60 puffs o pulsaciones (2.5mcg/pulsación) que suministra 30 dosis + folleto interno.
CONDICIONES DE CONSERVACIÓN Y ALMACENAMIENTO:
No congelar.
Consérvese a una temperatura no mayor a 30 ºC.
LEYENDAS DE PROTECCIÓN:
Manténgase fuera del alcance de los niños. Venta bajo receta médica. Producto de uso delicado. Adminístrese por prescripción y bajo vigilancia médica.
En Ecuador:
Notificación de Reacciones Adversas a Medicamentos: Es importante notificar los eventos adversos y toda información relevante para la seguridad del paciente después de la autorización del medicamento. Ello permite una supervisión continua de la relación beneficio/riesgo del medicamento. Se invita a los pacientes, así como a los profesionales de la salud a notificar eventos adversos y toda información relevante para la seguridad del paciente al siguiente e-mail: pv_local_ecuador@boehringer-ingelheim.com y al teléfono (+593) 2 397 9900. Mediante la notificación, usted puede contribuir a proporcionar mayor información sobre la seguridad del medicamento.
Chile: Importado por Boehringer Ingelheim Ltda. Isidora Goyenechea 3000, 18° Piso, Las Condes, Santiago y distribuido por Novofarma Service S.A., Av. Víctor Uribe 2280, Quilicura, Santiago por cuenta de Boehringer Ingelheim Ltda., Isidora Goyenechea 3000,° piso 18, Las Condes, Santiago. Versión 12.
Fabricado por:
Boehringer Ingelheim Pharma GmbH & Co.
KG, Ingelheim am Rhein, Alemania.
Bajo licencia de:
Boehringer Ingelheim International GmbH
Ingelheim am Rhein, Alemania,
Importado y distribuido por:
Boehringer Ingelheim del Ecuador Cía. Ltda.
Quito, Ecuador.
Reg. San. Ecuador: 29123-12-09
INCIDENTES GRAVES:
Si experimenta cualquier incidente grave en relación con el dispositivo, consulte a su médico o farmacéutico. También puede informar los incidentes graves directamente a Boehringer Ingelheim. Al informar los incidentes graves puede ayudar a suministrar más información sobre la seguridad de este dispositivo.
Preguntas frecuentes:
Es difícil insertar el cartucho lo suficientemente profundo.
¿Giró accidentalmente la base transparente antes de insertar el cartucho?
Abra la tapa, presione el botón de liberación de la dosis y luego inserte el cartucho.
¿Insertó el cartucho por el lado más grueso?
Inserte el cartucho nuevamente por el lado más delgado.
No puedo presionar el botón de liberación de la dosis.
¿Giró la base transparente?
Si no es así, gire la base transparente en un movimiento continuo hasta que haga click (media vuelta).
¿El indicador de dosis de SPIRIVA® RESPIMAT® está en cero?
El inhalador SPIRIVA® RESPIMAT® se bloquea después de 60 pulsaciones (30 dosis medicinales). Prepare y use un nuevo inhalador SPIRIVA® RESPIMAT®.
No logro girar la base transparente.
¿Ya giró la base transparente?
Si ya lo hizo, siga los pasos “ABRIR” y “PRESIONAR” indicados en “Uso diario” para administrar el medicamento.
¿El indicador de dosis de SPIRIVA® RESPIMAT® está en cero?
El inhalador SPIRIVA® RESPIMAT® se bloquea después de 60 pulsaciones (30 dosis medicinales). Prepare y use un nuevo inhalador SPIRIVA® RESPIMAT®.
El indicador de dosis de RESPIMAT® llega a cero demasiado pronto.
¿Usó SPIRIVA® RESPIMAT® de acuerdo con las indicaciones (dos pulsaciones una vez al día)?
El RESPIMAT® durará 30 días si se administran dos pulsaciones una vez al día.
¿Giró la base transparente antes de insertar el cartucho?
El indicador de dosis cuenta cada giro de la base transparente, ya sea que haya insertado o no un cartucho.
¿Accionó el dispositivo en el aire a menudo para verificar si el RESPIMAT® funcionaba?
Una vez que SPIRIVA® RESPIMAT® está preparado no es necesario efectuar pulsaciones de prueba si se lo usa diariamente.
¿Insertó el cartucho en un SPIRIVA® RESPIMAT® usado?
Siempre inserte un nuevo cartucho en un SPIRIVA® RESPIMAT® NUEVO.
Mi SPIRIVA® RESPIMAT® pulveriza automáticamente
¿La tapa estaba abierta cuando giró la base transparente?
Cierre la tapa y luego gire la base transparente.
¿Presionó el botón de liberación de la dosis al girar la base transparente?
Cierre la tapa de manera que el botón de liberación de la dosis quede tapado y luego gire la base transparente.
¿Dejó de girar la base transparente antes de que hiciera click?
Gire la base transparente en un movimiento continuo hasta que haga click (media vuelta).
No sale nada de mi SPIRIVA® RESPIMAT®.
¿Insertó un cartucho?
Si no lo hizo, inserte el cartucho.
¿Repitió los pasos “¿Gire”, “Abra” y “Presione” menos de tres veces luego de insertar el cartucho?
Repita “Gire”, “Abra”, “Presione” tres veces luego de insertar el cartucho, tal como se indica en los pasos 4 a 6 en “Preparación para el primer uso”.
¿Está en 0 el indicador de la dosis del RESPIMAT®?
Si el indicador de dosis está en 0, ya ha usado todo el medicamente y el inhalador está bloqueado. Una vez que haya ensamblado el SPIRIVA® RESPIMAT® no retire la base transparente ni el cartucho. Siempre inserte un cartucho nuevo en un SPIRIVA® RESPIMAT® NUEVO.
Para más información, consulte a su médico o farmacéutico.