REVLIMID
LENALIDOMIDA
Cápsulas
Envase(s) , 21 Cápsulas
COMPOSICIÓN:
Fórmula cualicuantitativa
Las CÁPSULAS de 5 mg contienen:
Lenalidomida 5 mg
Excipientes: lactosa anhidra 147 mg, celulosa microcristalina 40 mg, croscarmelosa sódica 6 mg, estearato de magnesio 2 mg, gelatina 57,5 mg, dióxido de titanio (E 171) 3,5 mg
Las CÁPSULAS de 10 mg contienen:
Lenalidomida 10 mg
Excipientes: lactosa anhidra 294 mg, celulosa microcristalina 80 mg, croscarmelosa sódica 12 mg, estearato de magnesio 4 mg, gelatina 91,9 mg, dióxido de titanio (E 171) 3,70 mg, índigo carmín (E 132) 0,018 mg, óxido de hierro amarillo (E 172) 0,33 mg
Las CÁPSULAS de 15 mg contienen:
Lenalidomida 15 mg
Excipientes: lactosa anhidra 289 mg, celulosa microcristalina 80 mg, croscarmelosa sódica 12 mg, estearato de magnesio 4 mg, gelatina 91,3 mg, dióxido de titanio (E 171) 4,7 mg, índigo carmín (E 132) 0,016 mg
Las CÁPSULAS de 20 mg contienen:
Lenalidomida 20 mg
Excipientes: lactosa anhidra 244.5 mg, celulosa microcristalina 120,5 mg, croscarmelosa sódica 12 mg, estearato de magnesio 3 mg, índigo carmín (E 132) 0,0426mg, gelatina 94,12 mg, dióxido de titanio (E 171) 1,7555 mg, óxido de hierro amarillo 0,0811 mg
Las CÁPSULAS de 25 mg contienen:
Lenalidomida 25 mg
Excipientes: lactosa anhidra 200 mg, celulosa microcristalina 159 mg, croscarmelosa sódica 12 mg, estearato de magnesio 4 mg, gelatina 93,2 mg, dióxido de titanio (E 171) 2,8 mg
PRINCIPIO ACTIVO (S) / GRUPO FARMACOLÓGICO:
Descripción
REVLIMID, un análogo de talidomida, es un agente inmunomodulador con propiedades antiangiogénicas y antineoplásicas. El nombre químico es 3-(4-amino-1-oxo 1,3-dihidro-2H-isoindol-2-yl) piperidina-2,6-diona y tiene la siguiente estructura química:
Estructura Química de Lenalidomida
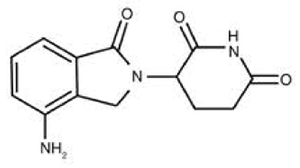
La fórmula empírica para lenalidomida es C13H13N3O3, y el peso molecular en gramos es 259,3.
Lenalidomida es un polvo sólido de color blancuzco a amarillo pálido. Es soluble en mezclas de solvente orgánico/agua, y solventes acuosos buffer. Lenalidomida es más soluble en solventes orgánicos y soluciones de pH bajo. La solubilidad fue significativamente menor en buffers menos ácidos, variando entre 0,4 y 0,5 mg/mL. Lenalidomida tiene un átomo de carbono asimétrico y puede existir como las formas óptimamente activas S(-) y R(+), y se produce como una mezcla racémica con una rotación óptica neta de cero.
REVLIMID® está disponible en cápsulas de 5 mg, 10 mg, 15 mg, 20 mg y 25 mg para administración oral. Cada cápsula contiene lenalidomida como ingrediente activo y los siguientes ingredientes inactivos: lactosa anhidra, celulosa microcristalina, croscarmelosa sódica, y estearato de magnesio. La carcasa de las cápsulas de 5 mg y 25 mg contiene gelatina, dióxido de titanio y tinta negra. La carcasa de la cápsula de 10 mg contiene gelatina, dióxido de titanio, índigo carmín, óxido de hierro amarillo, y tinta negra. La carcasa de la cápsula de 15 mg contiene gelatina, dióxido de titanio, índigo carmín, y tinta negra. La carcasa de la cápsula de 20 mg contiene gelatina, dióxido de titanio, índigo carmín, óxido de hierro amarillo, y tinta negra.
INDICACIONES TERAPÉUTICAS:
Indicaciones
Mieloma múltiple
REVLIMID en combinación con dexametasona está indicado para el tratamiento de pacientes con Mieloma Múltiple (MM).
REVLIMID está indicado como terapia de mantenimiento en pacientes con MM luego del trasplante autólogo de células madre hematopoyéticas (auto-HSCT).
REVLIMID en combinación con bortezomib y dexametasona está indicado para el tratamiento de pacientes adultos con mieloma múltiple no tratado previamente.
Síndromes mielodisplásicos
REVLIMID está indicado para el tratamiento de pacientes con anemia dependiente de transfusión debido a Síndromes Mielodisplásicos (SMD) de riesgo bajo o intermedio-1 vinculados con una anomalía citogenética por deleción del 5q con o sin anomalías citogenéticas adicionales.
Linfoma de células del manto
REVLIMID está indicado para el tratamiento de pacientes con Linfoma de Células del Manto (LCM) cuya enfermedad ha recidivado o progresado luego de dos terapias previas, una de las cuales incluyó bortezomib.
Linfoma folicular
En combinación con rituximab, está indicado para el tratamiento de pacientes con Linfoma Folicular (LF) tratado anteriormente.
Linfoma de la zona marginal
En combinación con rituximab, está indicado para el tratamiento de pacientes con Linfoma de la Zona Marginal (LZM) tratado anteriormente.
Limitaciones de uso
REVLIMID no está indicado y no está recomendado para el tratamiento de pacientes con Leucemia Linfocítica Crónica (LLC) por fuera de los estudios clínicos controlados.
MECANISMO DE ACCIÓN:
Accion farmacológica
Mecanismo de acción: La lenalidomida es un análogo de la talidomida con propiedades inmunomodulatorias, antiangiogénicas y antineoplásicas. Las actividades celulares de lenalidomida están mediadas a través de su cereblon objetivo, un componente de un complejo enzimático de ubiquitina ligasa E3 anillo cullin. In vitro, en presencia de la droga, las proteínas sustrato (incluyendo Aiolos, Ikaros y CK1α) están dirigidas a la ubiquitinación y posterior degradación que conduce a efectos citotóxicos directos e inmunomoduladores. La lenalidomida inhibe la proliferación e induce la apoptosis de determinadas células tumorales hematopoyéticas incluyendo MM, linfoma de células de manto, síndromes mielodisplásicos, linfoma folicular y linfoma de la zona marginal in vitro por deleción del 5q. La lenalidomida provoca una demora en el crecimiento del tumor en algunos modelos de tumores hematopoyéticos no clínicos in vivo, que incluyen el MM. Las propiedades inmunomoduladores de lenalidomida incluyen una cantidad incrementada y la activación de linfocitos T y de las células asesinas naturales (NK), el aumento de números de células NKT dando lugar a la citotoxicidad celular directa y reforzada dependiente de anticuerpos mediados por células (ADCC) a través de la secreción aumentada de interleukina-2 e interferón gamma, cantidades incrementadas de células NKT y la inhibición de citocinas proinflamatorias (por ejemplo, TNF-α e IL-6) por monocitos. En las células de MM, la combinación de lenalidomida y dexametasona sinergiza la inhibición de la proliferación de células y la inducción de apoptosis.
En vitro, la combinación de lenalidomida y rituximab aumenta la ADCC y la apoptosis tumoral directa en las células del linfoma folicular y aumenta el ADCC en el linfoma de la zona marginal en comparación con rituximab solo.
Farmacodinamia
Electrofisiología cardíaca
Se evaluó el efecto de lenalidomida en el intervalo QTc en 60 hombres sanos en un estudio riguroso de QT. A una dosis dos veces superior a la dosis máxima recomendada, lenalidomida no prolongó el intervalo QTc. El límite superior más grande del IC bilateral del 90 % de las diferencias entre lenalidomida y el placebo fue menor a 10 ms.
Farmacocinética
Absorción
Lenalidomida se absorbe rápidamente después de la administración oral. Después de dosis únicas y múltiples de REVLIMID en pacientes con MM o SMD, las concentraciones plasmáticas máximas se produjeron entre 0,5 y 6 horas después de la dosis.
La disposición farmacocinética de dosis únicas y múltiples de lenalidomida es lineal con los valores de AUC y Cmáx que se incrementan proporcionalmente con la dosis. La administración de dosis múltiples de REVLIMID a la dosis recomendada no tiene como resultado acumulación del fármaco.
La administración de una dosis única de 25 mg de REVLIMID con una comida rica en materia grasa en sujetos sanos reduce el grado de absorción, con una disminución aproximada, del 20% en el AUC y una disminución del 50% en la Cmáx. En los ensayos clínicos en los que se establecieron la eficacia y la seguridad de REVLIMID, el fármaco se administró sin considerar la ingesta de alimentos. REVLIMID puede administrarse con o sin alimentos.
La absorción oral de lenalidomida en pacientes con LCM es similar a la observada en pacientes con MM o SMD. Distribución
La unión in vitro de [14C]-lenalidomida a las proteínas plasmáticas es de aproximadamente un 30%.
Lenalidomida está presente en el semen a las 2 horas (1379 ng/eyaculado) y a las 24 horas (35 ng/eyaculado) de la administración de REVLIMID 25 mg diarios.
Eliminación
La semivida media de lenalidomida es de 3 horas en sujetos sanos y de 3 a 5 horas en pacientes con MM, MDS o LCM.Metabolismo
Lenalidomida tiene un metabolismo limitado. Lenalidomida sin metabolizar es el componente predominante en circulación en humanos. Dos son los metabolitos identificados, 5-hidroxi-lenalidomida y N-acetil-lenalidomida; cada uno constituye menos del 5% de los niveles circulantes totales de droga.
Excreción
La eliminación es principalmente renal. Después de la administración única por vía oral de [14C]-lenalidomida (25 mg) a sujetos sanos, aproximadamente, el 90% y el 4% de la dosis radioactiva se eliminó dentro de los diez días en la orina y las heces, respectivamente. Aproximadamente, el 82% de la dosis radioactiva se excretó como lenalidomida en la orina dentro de las 24 horas. Hidroxilenalidomida y N-acetil-lenalidomida representó el 4,6% y el 1,8% de la dosis excretada, respectivamente. El aclaramiento renal de lenalidomida supera la velocidad de filtración glomerular.
Poblaciones específicas
Insuficiencia renal: Ocho sujetos con insuficiencia renal leve (aclaramiento de creatinina (CLCr) de 50 a 79 ml / min calculados con Cockcroft-Gault), 9 sujetos con insuficiencia renal moderada (CLCr de 30 a 49 ml / min), 4 sujetos con insuficiencia renal grave (CLCr <30 ml / min), y 6 pacientes con Enfermedad Renal Terminal (ESRD) que requirieron diálisis se les administró una sola dosis de 25 mg de REVLIMID. Tres sujetos sanos de edad similar con función renal normal (CLCr> 80 ml / min) también recibieron una dosis única de 25 mg de REVLIMID. Cuando la CLCr disminuyó, la vida media aumentó y el aclaramiento de la droga disminuyó linealmente. Los pacientes con insuficiencia moderada y grave tuvieron un aumento de 3 veces la vida media y una disminución de 66% a 75% en el aclaramiento de la droga en comparación con sujetos sanos. Los pacientes en hemodiálisis (n=6) tuvieron un aumento aproximado de 4,5 veces en la vida media y una disminución del 80% en el aclaramiento de la droga en comparación con sujetos sanos. Aproximadamente, el 30% de la droga en el cuerpo se eliminó durante una sesión de hemodiálisis de 4 horas.
Ajuste la dosis inicial de REVLIMID en pacientes con insuficiencia renal basándose en el valor CLCr (ver Posología y administración).
Insuficiencia hepática: La insuficiencia hepática leve (definida como bilirrubina total > 1 a 1,5 veces el Límite Superior Normal (ULN) o cualquier aspartato transaminasa mayor que la ULN) no influyó en la disposición de la lenalidomida. No hay datos farmacocinéticos disponibles para pacientes con insuficiencia hepática moderada a grave.
Otros factores intrínsecos: La edad (39 a 85 años), el peso corporal (33 a 135 kg), el sexo, la raza y el tipo de neoplasias hematológicas (MM, MDS o LCM) no tuvieron un efecto clínicamente relevante en el aclaramiento de lenalidomida en pacientes adultos.
Acción terapéutica: Agente inmunomodulador con propiedades antiangiogénicas y antineoplásicas, análogo de la talidomida Código ATC: L04 AX04.
CONTRAINDICACIONES:
Embarazo: REVLIMID puede provocar daños al feto si se administra a una mujer embarazada. Se observaron anomalías en las extremidades de las crías de monas que recibieron lenalidomida durante la organogénesis. Este efecto se observó en todas las dosis analizadas. Debido a los resultados de este estudio del desarrollo en monos y la semejanza estructural de lenalidomida con talidomida, un teratógeno humano conocido, lenalidomida está contraindicado en mujeres embarazadas. Si este fármaco se utiliza durante el embarazo o si la paciente queda embarazada durante el tratamiento con esta medicación, el paciente debe ser notificado respecto del riesgo potencial para el feto.
Reacciones severas de hipersensibilidad: REVLIMID está contraindicado en pacientes que demostraron hipersensibilidad (por ejemplo angioedema, síndrome de Stevents-Johnson, necrólisis epidérmica tóxica) a lenalidomida o alguno de sus excipientes.
REACCIONES ADVERSAS:
Las siguientes reacciones adversas se describen en detalle en otras secciones de la información de prescripción:
- Toxicidad embrionaria y fetal. (Ver Advertencias y precauciones)
- Toxicidad hematológica. (Ver Advertencias y precauciones)
- Tromboembolia venosa y arterial. (Ver Advertencias y precauciones)
- Aumento de la mortalidad en pacientes con LLC. (Ver Advertencias y precauciones)
- Segundas neoplasias primarias. (Ver Advertencias y precauciones)
- Aumento de la mortalidad en pacientes con MM cuando se agrega Pembrolizumab a un análogo de la Talidomida y Dexametasona (ver Advertencias y precauciones)
- Hepatotoxicidad. (Ver Advertencias y precauciones)
- Reacciones cutáneas severas que incluyen hipersensibilidad. (Ver Advertencias y precauciones)
- Síndrome de lisis tumoral. (Ver Advertencias y precauciones)
- Reacciones de llamarada en el tumor. (Ver Advertencias y precauciones)
- Alteración de la movilización de células madre. (Ver Advertencias y precauciones)
- Trastornos tiroideos. (Ver Advertencias y precauciones)
- Mortalidad prematura en pacientes con MCK. (Ver Advertencias y precauciones)
Experiencia en ensayos clínicos
Dado que los ensayos clínicos se llevan a cabo bajo condiciones muy diversas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las tasas de ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica.
MM recientemente diagnosticado – Terapia de combinación con REVLIMID
Se evaluaron datos de 1.613 pacientes en un estudio fase 3 de gran volumen que recibieron por lo menos una dosis de REVLIMID con dosis baja de dexametasona (Rd) administrada en 2 duraciones de tiempo diferentes (es decir, hasta enfermedad progresiva [Rama Rd Continuo; N = 532] o por hasta dieciocho ciclos de 28 días [72 semanas, Rama Rd18; N = 540] o que recibieron melfalán, prednisona y talidomida (Rama MPT; N = 541) por un máximo de doce ciclos de 42 días (72 semanas). La mediana de la duración del tratamiento en la rama Rd Continuo fue de 80,2 semanas (rango 0,7 a 246,7) o 18,4 meses (rango 0,16 a 56,7).
En general, las reacciones adversas informadas con más frecuencia fueron comparables en la rama Rd Continuo y en la rama Rd18, e incluyeron diarrea, anemia, constipación, edema periférico, neutropenia, fatiga, dolor de espalda, náuseas, astenia e insomnio. Las reacciones de grado 3 o 4 informadas con más frecuencia incluyeron neutropenia, anemia, trombocitopenia, neumonía, astenia, fatiga, dolor de espalda, hipocalemia, erupción cutánea, cataratas, linfopenia, disnea, TVP, hiperglucemia y leucopenia. La mayor frecuencia de infecciones se produjo en la rama Rd Continuo (75%) en comparación con la rama MPT (56%). Hubo más reacciones adversas por infecciones grado 3,4 y serias en la rama Rd Continuo que en la rama MPT o en la Rd18.
En la rama Rd Continuo, las reacciones adversas más frecuentes que conducen a la interrupción de la dosis de REVLIMID fueron eventos de infección (28,8%); en total, la mediana del tiempo hasta la primera interrupción de la dosis de REVLIMID fue de 7 semanas. Las reacciones adversas más frecuentes que condujeron a reducción de la dosis de Revlimid en la rama Rd Continuo fueron los eventos hematológicos (10,7%); en total, la mediana del tiempo hasta la primera reducción de la dosis de REVLIMID fue de 16 semanas. En la rama Rd Continuo, las reacciones adversas más frecuentes que condujeron a la discontinuación de REVLIMID fueron los eventos de infección (3,4%).
En ambas ramas Rd, las frecuencias de la aparición de reacciones adversas fueron en general, mayores en los primeros 6 meses de tratamiento, y luego las frecuencias disminuyeron a lo largo del tiempo o se mantuvieron estables durante todo el tratamiento, excepto por las cataratas. La frecuencia de la aparición de cataratas aumentó a lo largo del tiempo con 0,7% durante los primeros 6 meses y hasta 9,6% hacia el segundo año de tratamiento con Rd Continuo.
La Tabla 14 resume las reacciones adversas informadas para las ramas de tratamiento Rd Continuo, Rd18 y MPT.
|
Tabla 14: Todas las reacciones adversas en ≥ 5,0% y reacciones adversas de grado 3/4 en ≥ 1,0% de los pacientes en las ramas Rd Continuo o Rd18* |
||||||
|
Sistema corporal Reacción adversa |
Todas las reacciones adversasa |
Reacciones adversas grado 3/4b |
||||
|
Rd Continuo (N = 532) |
Rd18 (N = 540) |
MPT (N = 541) |
Rd Continuo (N = 532) |
Rd18 (N = 540) |
MPT (N = 541) |
|
|
Trastornos generales y alteraciones en el lugar de administración |
||||||
|
Fatiga% |
173 (32,5) |
177 (32,8) |
154 (28,5) |
39 (7,3) |
46 (8,5) |
31 (5,7) |
|
Astenia |
50 (28,2) |
123 (22,8) |
124 (22,9) |
41 (7,7) |
33 (6,1) |
32 (5,9) |
|
Pirexiac |
114 (21,4) |
102 (18,9) |
76 (14,0) |
13 (2,4) |
7 (1,3) |
7 (1,3) |
|
Dolor de tórax no cardíaco f |
29 (5,5) |
31 (5,7) |
18 (3,3) |
<1% |
<1% |
<1% |
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
242 (45,5) |
208 (38,5) |
89 (16,5) |
21 (3,9) |
18 (3,3) |
8 (1,5) |
|
Dolor abdominal%f |
109 (20,5) |
78 (14,4) |
60 (11,1) |
7 (1,3) |
9 (1,7) |
< 1% |
|
Dispepsia f |
57 (10,7) |
28 (5,2) |
36 (6,7) |
<1% |
< 1% |
0 (0,0) |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||||
|
Dolor de espaldac |
170 ( 32,0) |
145 (26,9) |
116 (21,4) |
37 (7,0) |
34 (6,3) |
28 (5,2) |
|
Espasmos musculares f |
109 (20,5) |
102 (18,9) |
61 (11,3) |
< 1% |
< 1% |
< 1% |
|
Artralgia f |
101 (19,0) |
71 (13,1) |
66 (12,2) |
9 (1,7) |
8 (1,5) |
8 (1,5) |
|
Dolor óseo f |
87 (16,4) |
77 (14,3) |
62 (11,5) |
16 (3,0) |
15 (2,8) |
14 (2,6) |
|
Dolor en las extremidades f |
79 (14,8) |
66 (12,2) |
61 (11,3) |
8 (1,5) |
8 (1,5) |
7 (1,3) |
|
Dolor musculoesquelético f |
67 (12,6) |
59 (10,9) |
36 (6,7) |
< 1% |
< 1% |
< 1% |
|
Dolor de tórax musculoesquelético f |
60 (11,3) |
51 (9,4) |
39 (7,2) |
6 (1,1) |
< 1% |
< 1% |
|
Debilidad muscular f |
43 (8,1) |
35 (6,5) |
29 (5,4) |
< 1% |
8 (1,5) |
< 1% |
|
Dolor de cuello f |
40 (7,5) |
19 (3,5) |
10 (1,8) |
< 1% |
< 1% |
< 1% |
|
Infecciones e infestaciones |
||||||
|
Bronquitisc |
90 (16,9) |
59 (10,9) |
43 (7,9) |
9 (1,7) |
6 (1,1) |
3 (0,6) |
|
Nasofaringitis f |
80 (15,0) |
54 (10,0) |
33 (6,1) |
0 (0,0) |
0 (0,0) |
0 (0,0) |
|
Infección del tracto urinario f |
76 (14,3) |
63 (11,7) |
41 (7,6) |
8 (1,5) |
8 (1,5) |
< 1% |
|
Infección del tracto respiratorio superiorc%f |
69 (13,0) |
53 (9,8) |
31 (5,7) |
< 1% |
8 (1,5) |
< 1% |
|
Neumoníac@ |
93 (17,5) |
87 (16,1) |
56 (10,4) |
60 (11,3) |
57 (10,5) |
41 (7,6) |
|
Infección del tracto respiratorio% |
35 (6,6) |
25 (4,6) |
21 (3,9) |
7 (1,3) |
4 ( 0,7) |
1 (0,2) |
|
Gripe f |
33 (6,2) |
23 (4,3) |
15 (2,8) |
< 1% |
< 1% |
0 (0,0) |
|
Gastroenteritis f |
32 (6,0) |
17 (3,1) |
13 (2,4) |
0 (0,0) |
< 1% |
< 1% |
|
Infección del tracto respiratorio inferior |
29 (5,5) |
14 (2,6) |
16 (3,0) |
10 (1,9) |
3 (0,6) |
3 (0,6) |
|
Rinitis f |
29 ( 5,5) |
24 ( 4,4) |
14 (2,6) |
0 (0,0) |
0 (0,0) |
0 (0,0) |
|
Celulitisc |
< 5% |
< 5% |
< 5% |
8 (1,5) |
3 (0,6) |
2 (0,4) |
|
Sepsisc@ |
33 (6,2) |
26 (4,8) |
18 (3,3) |
26 (4,9) |
20 (3,7) |
13 (2,4) |
|
Trastornos del sistema nervioso |
||||||
|
Dolor de cabeza f |
75 (14,1) |
52 (9,6) |
56 (10,4) |
< 1% |
< 1% |
< 1% |
|
Disgeusia f |
39 (7,3) |
45 (8,3) |
22 (4,1) |
< 1% |
0 (0,0) |
< 1% |
|
Trastornos del sistema linfático y de la sangred |
||||||
|
Anemia |
233 (43,8) |
193 (35,7) |
229 (42,3) |
97 (18,2) |
85 (15,7) |
102 (18,9) |
|
Neutropenia |
186 (35,0) |
178 (33,0) |
328 (60,6) |
148 (27,8) |
143 (26,5) |
243 (44,9) |
|
Trombocitopenia |
104 (19,5) |
100 (18,5) |
135 (25,0) |
44 (8,3) |
43 (8,0) |
60 (11,1) |
|
Neutropenia febril |
7 (1,3) |
17 (3,1) |
15 (2,8) |
6 (1,1) |
16 (3,0) |
14 (2,6) |
|
Pancitopenia |
5 (0,9) |
6 (1,1) |
7 (1,3) |
1 (0,2) |
3 (0,6) |
5 (0,9) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||
|
Tos f |
121 (22,7) |
94 (17,4) |
68 (12,6) |
< 1% |
< 1% |
< 1% |
|
Disneac,e |
117 (22,0) |
89 (16,5) |
113 (20,9) |
30 (5,6) |
22 (4,1) |
18 (3,3) |
|
Epistaxis f |
32 (6,0) |
31 (5,7) |
17 (3,1) |
< 1% |
< 1% |
0 (0,0) |
|
Dolor orofaríngeo f |
30 (5,6) |
22 (4,1) |
14 (2,6) |
0 (0,0) |
0 (0,0) |
0 (0,0) |
|
Disnea por esfuerzo e |
27 (5,1) |
29 (5,4) |
< 5% |
6 (1,1) |
2 (0,4) |
0 (0,0) |
|
Trastornos del metabolismo y de la nutrición |
||||||
|
Disminución del apetito |
123 (23,1) |
115 (21,3) |
72 (13,3) |
14 (2,6) |
7 (1,3) |
5 (0,9) |
|
Hipocalemia% |
91 ( 17,1) |
62 (11,5) |
38 (7) |
35 (6,6) |
20 (3,7) |
11 (2,0) |
|
Hiperglucemia |
62 (11,7) |
52 (9,6) |
19 (3,5) |
28 (5,3) |
23 (4,3) |
9 (1,7) |
|
Hipocalcemia |
57 (10,7) |
56 (10,4) |
31 (5,7) |
23 (4,3) |
19 (3,5) |
8 (1,5) |
|
Deshidratación% |
25 ( 4,7) |
29 ( 5,4) |
17 ( 3,1) |
8 (1,5) |
13 (2,4) |
9 (1,7) |
|
Gota e |
< 5% |
< 5% |
< 5% |
8 (1,5) |
0 (0,0) |
0 (0,0) |
|
Diabetes mellitus% e |
< 5% |
< 5% |
< 5% |
8 (1,5) |
4 (0,7) |
2 (0,4) |
|
Hipofosfatemia e |
< 5% |
< 5% |
< 5% |
7 (1,3) |
3 (0,6) |
1 (0,2) |
|
Hiponatremia% e |
< 5% |
< 5% |
< 5% |
7 (1,3) |
13 (2,4) |
6 (1,1) |
|
Trastornos de la piel y del tejido subcutáneo |
||||||
|
Erupción cutánea |
139 (26,1) |
151 (28,0) |
105 (19,4) |
39 (7,3) |
38 (7,0) |
33 (6,1) |
|
Prurito f |
47 (8,8) |
49 (9,1) |
24 (4,4) |
< 1% |
< 1% |
< 1% |
|
Trastornos psiquiátricos |
||||||
|
Insomnio |
147 (27,6) |
127 (23,5) |
53 (9,8) |
4 (0,8) |
6 (1,1) |
0 (0,0) |
|
Depresión |
58 (10,9) |
46 (8,5) |
30 (5,5) |
10 (1,9) |
4 (0,7) |
1 (0,2) |
|
Trastornos vasculares |
||||||
|
Trombosis venosa profundac% |
55 (10,3) |
39 (7,2) |
22 (4,1) |
30 (5,6) |
20 (3,7) |
15 (2,8) |
|
Hipotensiónc% |
51 (9,6) |
35 (6,5) |
36 (6,7) |
11 (2,1) |
8 (1,5) |
6 (1,1) |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
||||||
|
Caída f |
43 (8,1) |
25 (4,6) |
25 (4,6) |
< 1% |
6 (1,1) |
6 (1,1) |
|
Contusión f |
33 (6,2) |
24 (4,4) |
15 (2,8) |
< 1% |
< 1% |
0 (0,0) |
|
Trastornos oculares |
||||||
|
Cataratas |
73 (13,7) |
31 (5,7) |
5 (0,9) |
31 (5,8) |
14 (2,6) |
3 (0,6) |
|
Cataratas subcapsulares e |
< 5% |
< 5% |
< 5% |
7 (1,3) |
0 (0,0) |
0 (0,0) |
|
Exploraciones complementarias |
||||||
|
Disminución del peso |
72 (13,5) |
78 (14,4) |
48 (8,9) |
11 (2,1) |
4 (0,7) |
4 (0,7) |
|
Trastornos cardíacos |
||||||
|
Fibrilación auricularc |
37 (7,0) |
25 (4,6) |
25 (4,6) |
13 (2,4) |
9 (1,7) |
6 (1,1) |
|
Infarto de miocardio (incluido el agudo)c ,e |
< 5% |
< 5% |
< 5% |
10 (1,9) |
3 (0,6) |
5 (0,9) |
|
Trastornos renales y urinarios |
||||||
|
Insuficiencia renal (incluida la aguda)c@,f |
49 (9,2) |
54 (10,0) |
37 (6,8) |
28 (5,3) |
33 (6,1) |
29 (5,4) |
|
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos) |
||||||
|
Carcinoma de células escamosasc e |
< 5% |
< 5% |
< 5% |
8 (1,5) |
4 (0,7) |
0 (0,0) |
|
Carcinoma de células basalesc e,f |
< 5% |
< 5% |
< 5% |
< 1% |
< 1% |
0 (0,0) |
|
Nota: Un sujeto con múltiples ocurrencias de una reacción adversa es contabilizado una sola vez bajo el SOC/PT aplicable. Sistema corporal / Reacción adversa. a Todas las reacciones adversas emergentes del tratamiento en por lo menos 5,0% de los sujetos en las ramas Rd Continuo o Rd18 y una frecuencia por lo menos 2,0% mayor (%) en las ramas Rd Continuo o Rd18 en comparación con la rama MPT. b Todas las reacciones adversas emergentes del tratamiento de grado 3 o 4 en por lo menos 1,0% de los sujetos en las ramas Rd Continuo o Rd18 y una frecuencia por lo menos 1,0% mayor (%) en cualquiera de las ramas Rd Continuo o Rd18 en comparación con la rama MPT. c Reacciones adversas serias emergentes del tratamiento en por lo menos 1,0% de los sujetos en las ramas Rd Continuo o Rd18 y una frecuencia por lo menos 1,0% mayor (%) en cualquiera de las ramas Rd Continuo o Rd18 en comparación con la rama MPT. d Los términos preferentes para el Sistema corporal y trastornos de la sangre y del sistema linfático se incluyeron por criterio médico como reacciones adversas conocidas para Rd Continuo/Rd18, y también se han informado como serias. e Nota a pie de página “a” no aplicable f Nota a pie de página “b” no aplicable. @ - reacciones adversas en las cuales por lo menos una resultó fatal % - reacciones adversas en las cuales por lo menos una se consideró de amenaza de vida (si el resultado de la reacción fue la muerte, se incluyó con los casos de muerte) |
||||||
*Las reacciones adversas incluidas en los términos de reacciones adversas combinadas:
Dolor abdominal: dolor abdominal, dolor abdominal superior, dolor abdominal inferior, dolor gastrointestinal.Neumonías: neumonía, neumonía lobar, neumonía neumocócica, bronconeumonía, neumonía por pneumocystis jiroveci, neumonía por legionella, neumonía estafilocócica, neumonía por klebsiella, neumonía atípica, neumonía bacteriana, neumonía por escherichia, neumonía estreptocócica, neumonía viral.
Sepsis: Sepsis, shock séptico, urosepsis, sepsis por escherichia, sepsis neutropénica, sepsis neumocócica, sepsis estafilocócica, sepsis bacteriana, sepsis meningocócica, sepsis enterocócica, sepis por klebsiella, sepsis por pseudomonas.
Erupción cutánea: erupción cutánea, erupción cutánea pruriginosa, erupción cutánea eritematosa, erupción cutánea maculopapular, erupción cutánea generalizada, erupción cutánea papular, erupción cutánea exfoliativa, erupción cutánea folicular, erupción cutánea macular, erupción por drogas con eosinofilia y síntomas sistémicos, eritema multiforme, erupción cutánea pustular.
Trombosis venosa profunda: trombosis venosa profunda, trombosis venosa de las extremidades, trombosis venosa.
MM recientemente diagnosticada - Terapia de mantenimiento con Revlimid después de Auto-HSCT: Los datos fueron evaluados en 1.018 pacientes en dos ensayos aleatorios quienes recibieron al menos una dosis de REVLIMID 10 mg diarios como terapia de mantenimiento después de auto-HSCT hasta la enfermedad progresiva o toxicidad inaceptable. La duración media del tratamiento con REVLIMID fue de 30,3 meses para el Estudio de Mantenimiento 1 y 24,0 meses para el Estudio de Mantenimiento 2 (rango general, en ambos estudios de 0,1 a 108 meses). A partir de la fecha de corte del 1 de marzo de 2015, 48 pacientes (21%) en el grupo del Estudio de Mantenimiento 1 de REVLIMID estaban todavía en tratamiento y ninguno de los pacientes en el grupo del Estudio de Mantenimiento 2 de REVLIMID estaba aún en tratamiento en la misma fecha de corte.
Las reacciones adversas incluidas en el Estudio de Mantenimiento 1 incluyeron los eventos reportados después del trasplante (conclusión de altas dosis de melfalán / auto-HSCT) y el período de tratamiento de mantenimiento. En el Estudio de Mantenimiento 2, las reacciones adversas fueron sólo del período del tratamiento de mantenimiento. En general, las reacciones adversas más frecuentes (más del 20% en el grupo REVLIMID) en ambos estudios fueron neutropenia, trombocitopenia, leucopenia, anemia, infección del tracto respiratorio superior, bronquitis, nasofaringitis, tos, gastroenteritis, diarrea, rash, fatiga, astenia, espasmo muscular y pirexia. Las reacciones de grado 3 o 4 más frecuentes (más del 20% en el grupo REVLIMID) incluyeron neutropenia, trombocitopenia y leucopenia. Las reacciones adversas graves de infección pulmonar y neutropenia (más del 4,5%) se produjeron en el grupo de REVLIMID. Para REVLIMID, las reacciones adversas más comunes que condujeron a la interrupción de la dosis fueron eventos hematológicos (29,7%, datos disponibles sólo en el Estudio de Mantenimiento 2). Las reacciones adversas más frecuente que condujeron a la reducción de la dosis de REVLIMID fueron eventos hematológicos (17,7%, datos disponibles sólo en el Estudio de Mantenimiento 2). Las reacciones adversas más frecuentes que condujeron a la discontinuación de REVLIMID fueron trombocitopenia (2,7%) en el Estudio de Mantenimiento 1 y neutropenia (2,4%) en el Estudio de Mantenimiento 2.
Las frecuencias de la aparición de reacciones adversas fueron generalmente, más altas en los primeros 6 meses de tratamiento y luego las frecuencias disminuyeron con el tiempo o se mantuvieron estables durante el tratamiento.
La Tabla 15 resume las reacciones adversas notificadas para los grupos de tratamiento de REVLIMID y de mantenimiento con placebo.
|
Tabla 15: Todas las reacciones adversas en el ≥5.0% y de grado 3/4 en el ≥ 1,0% de los pacientes en los grupos de REVLIMID frente a Placebo * |
||||||||
|
Estudio de Mantenimiento 1 |
Estudio de Mantenimiento 2 |
|||||||
|
Reacción adversa Sistema corporal |
Todas las reacciones adversas [a] |
Reacciones adversas Grado 3/4 [b] |
Todas las reacciones adversas [a] |
Grado 3/4 Reacciones |
||||
|
REVLIMID (N=224) n (%) |
Placebo (N=221) n (%) |
REVLIMID (N=224) n (%) |
Placebo (N=221) n (%) |
REVLIMID (N=293) n (%) |
Placebo (N=280) n (%) |
REVLIMID (N=293) n (%) |
Placebo (N=280) n (%) |
|
|
Trastornos sanguíneos y linfáticos |
||||||||
|
Neutropenia c % |
177 ( 79.0) |
94 ( 42.5) |
133 ( 59.4) |
73 ( 33.0) |
178 ( 60.8) |
33 ( 11.8) |
158 ( 53.9) |
21 ( 7.5) |
|
Trombocitopenia c % |
162 ( 72.3) |
101 ( 45.7) |
84 ( 37.5) |
67 ( 30.3) |
69 ( 23.5) |
29 ( 10.4) |
38 ( 13.0) |
8 ( 2.9) |
|
Leucopenia c |
51 (22.8) |
25 ( 11.3) |
45 ( 20.1) |
22 ( 10.0) |
93 ( 31.7) |
21 ( 7.5) |
71 ( 24.2) |
5 ( 1.8) |
|
Anemia |
47 ( 21.0) |
27 ( 12.2) |
23 ( 10.3) |
18 ( 8.1) |
26 ( 8.9) |
15 ( 5.4) |
11 ( 3.8) |
3 ( 1.1) |
|
Linfopenia |
40 ( 17.9) |
29 ( 13.1) |
37 ( 16.5) |
26 ( 11.8) |
13 ( 4.4) |
3 ( 1.1) |
11 ( 3.8) |
2 ( 0.7) |
|
Pancitopenia c d % |
1 ( 0.4) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
12 ( 4.1) |
1 ( 0.4) |
7 ( 2.4) |
1 ( 0.4) |
|
Neutropenia febril c |
39 ( 17.4) |
34 ( 15.4) |
39 ( 17.4) |
34 ( 15.4) |
7 ( 2.4) |
1 ( 0.4) |
5 ( 1.7) |
1 ( 0.4) |
|
Infecciones e Infestaciones# |
||||||||
|
Infección del tracto respiratorio superior e |
60 ( 26.8) |
35 ( 15.8) |
7 ( 3.1) |
9 ( 4.1) |
32 ( 10.9) |
18 ( 6.4) |
1 ( 0.3) |
0 ( 0.0) |
|
Infección neutropénica |
40 ( 17.9) |
19 ( 8.6) |
27 ( 12.1) |
14 ( 6.3) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
|
Neumonías* c % |
31 ( 13.8) |
15 ( 6.8) |
23 ( 10.3) |
7 ( 3.2) |
50 ( 17.1) |
13 ( 4.6) |
27 ( 9.2) |
5 ( 1.8) |
|
Bronquitisc |
10 ( 4.5) |
9 ( 4.1) |
1 ( 0.4) |
5 ( 2.3) |
139 ( 47.4) |
104 ( 37.1) |
4 ( 1.4) |
1 ( 0.4) |
|
Nasofaringitis e |
5 ( 2.2) |
2 ( 0.9) |
0 ( 0.0) |
0 ( 0.0) |
102 ( 34.8) |
84 ( 30.0) |
1 ( 0.3) |
0 ( 0.0) |
|
Gastroenteritis c |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
66 ( 22.5) |
55 ( 19.6) |
6 ( 2.0) |
0 ( 0.0) |
|
Rinitis e |
2 ( 0.9) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
44 ( 15.0) |
19 ( 6.8) |
0 ( 0.0) |
0 ( 0.0) |
|
Sinusitis e |
8 ( 3.6) |
3 ( 1.4) |
0 ( 0.0) |
0 ( 0.0) |
41 ( 14.0) |
26 ( 9.3) |
0 ( 0.0) |
1 ( 0.4) |
|
Gripe c |
8 ( 3.6) |
5 ( 2.3) |
2 ( 0.9) |
1 ( 0.5) |
39 ( 13.3) |
19 ( 6.8) |
3 ( 1.0) |
0 ( 0.0) |
|
Infección pulmonar c |
21 ( 9.4) |
2 ( 0.9) |
19 ( 8.5) |
2 ( 0.9) |
9 ( 3.1) |
4 ( 1.4) |
1 ( 0.3) |
0 ( 0.0) |
|
Infección del tracto respitario inferior e |
13 ( 5.8) |
5 ( 2.3) |
6 ( 2.7) |
4 ( 1.8) |
4 ( 1.4) |
4 ( 1.4) |
0 ( 0.0) |
2 ( 0.7) |
|
Infección c |
12 ( 5.4) |
6 ( 2.7) |
9 ( 4.0) |
5 ( 2.3) |
17 ( 5.8) |
5 ( 1.8) |
0 ( 0.0) |
0 ( 0.0) |
|
Infección del tracto urinario c d e |
9 ( 4.0) |
5 ( 2.3) |
4 ( 1.8) |
4 ( 1.8) |
22 ( 7.5) |
17 ( 6.1) |
1 ( 0.3) |
0 ( 0.0) |
|
Infección bacteriana del tracto respiratorio inferior d |
6 ( 2.7) |
1 ( 0.5) |
4 ( 1.8) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
|
Bacteriemia d |
5 ( 2.2) |
0 ( 0.0) |
4 ( 1.8) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
|
Infección de herpes c d |
11 ( 4.9) |
10 ( 4.5) |
3 ( 1.3) |
2 ( 0.9) |
29 ( 9.9) |
25 ( 8.9) |
6 ( 2.0) |
2 ( 0.7) |
|
Sepsis* c d @ |
2 ( 0.9) |
1 ( 0.5) |
0 ( 0.0) |
0 ( 0.0) |
6 ( 2.0) |
1 ( 0.4) |
4 ( 1.4) |
1 ( 0.4) |
|
Trastornos gastrointestinales |
||||||||
|
Diarrea |
122 ( 54.5) |
83 ( 37.6) |
22 ( 9.8) |
17 ( 7.7) |
114 ( 38.9) |
34 ( 12.1) |
7 ( 2.4) |
0 ( 0.0) |
|
Náuseas e |
33 ( 14.7) |
22 ( 10.0) |
16 ( 7.1) |
10 ( 4.5) |
31 ( 10.6) |
28 ( 10.0) |
0 ( 0.0) |
0 ( 0.0) |
|
Vómitos |
17 ( 7.6) |
12 ( 5.4) |
8 ( 3.6) |
5 ( 2.3) |
16 ( 5.5) |
15 ( 5.4) |
1 ( 0.3) |
0 ( 0.0) |
|
Constipación e |
12 ( 5.4) |
8 ( 3.6) |
0 ( 0.0) |
0 ( 0.0) |
37 ( 12.6) |
25 ( 8.9) |
2 ( 0.7) |
0 ( 0.0) |
|
Dolor abdominal e |
8 ( 3.6) |
7 ( 3.2) |
1 ( 0.4) |
4 ( 1.8) |
31 ( 10.6) |
15 ( 5.4) |
1 ( 0.3) |
1 ( 0.4) |
|
Dolor abdominal superior e |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
20 ( 6.8) |
12 ( 4.3) |
1 ( 0.3) |
0 ( 0.0) |
|
Trastornos generales y alteraciones en el lugar de la administración |
||||||||
|
Astenia |
0 ( 0.0) |
1 ( 0.5) |
0 ( 0.0) |
0 ( 0.0) |
87 ( 29.7) |
53 ( 18.9) |
10 ( 3.4) |
2 ( 0.7) |
|
Fatiga |
51 ( 22.8) |
30 ( 13.6) |
21 ( 9.4) |
9 ( 4.1) |
31 ( 10.6) |
15 ( 5.4) |
3 ( 1.0) |
0 ( 0.0) |
|
Pirexia e |
17 ( 7.6) |
10 ( 4.5) |
2 ( 0.9) |
2 ( 0.9) |
60 ( 20.5) |
26 ( 9.3) |
1 ( 0.3) |
0 ( 0.0) |
|
Trastornos de la piel y del tejido subcutáneo |
||||||||
|
Piel seca e |
9 ( 4.0) |
4 ( 1.8) |
0 ( 0.0) |
0 ( 0.0) |
31 ( 10.6) |
21 ( 7.5) |
0 ( 0.0) |
0 ( 0.0) |
|
Erupción cutánea |
71 ( 31.7) |
48 ( 21.7) |
11 ( 4.9) |
5 ( 2.3) |
22 ( 7.5) |
17 ( 6.1) |
3 ( 1.0) |
0 ( 0.0) |
|
Prurito |
9 ( 4.0) |
4 ( 1.8) |
3 ( 1.3) |
0 ( 0.0) |
21 ( 7.2) |
25 ( 8.9) |
2 ( 0.7) |
0 ( 0.0) |
|
Trastornos del Sistema nervioso |
||||||||
|
Parestesia e |
2 ( 0.9) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
39 ( 13.3) |
30 ( 10.7) |
1 ( 0.3) |
0 ( 0.0) |
|
Neuropatía Periférica* e |
34 ( 15.2) |
30 ( 13.6) |
8 ( 3.6) |
8 ( 3.6) |
29 ( 9.9) |
15 ( 5.4) |
4 ( 1.4) |
2 ( 0.7) |
|
Dolor de cabeza d |
11 ( 4.9) |
8 ( 3.6) |
5 (2) |
1 ( 0.5) |
25 ( 8.5) |
21 ( 7.5) |
0 ( 0.0) |
0 ( 0.0) |
|
Investigaciones |
||||||||
|
Aumento de la alanina aminotransferasa |
16 ( 7.1) |
3 ( 1.4) |
8 ( 3.6) |
0 ( 0.0) |
5 ( 1.7) |
5 ( 1.8) |
0 ( 0.0) |
1 ( 0.4) |
|
Aumento de la aspartato aminotransferasa d |
13 ( 5.8) |
5 ( 2.3) |
6 ( 2.7) |
0 ( 0.0) |
2 ( 0.7) |
5 ( 1.8) |
0 ( 0.0) |
0 ( 0.0) |
|
Trastornos del metabolismo y la nutrición |
||||||||
|
Hipopotasemia |
24 ( 10.7) |
13 ( 5.9) |
16 ( 7.1) |
12 ( 5.4) |
12 ( 4.1) |
1 ( 0.4) |
2 ( 0.7) |
0 ( 0.0) |
|
Deshidratación |
9 ( 4.0 ) |
5 ( 2.3) |
7 ( 3.1) |
3 ( 1.4) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
|
Hipofosfatemia d |
16 ( 7.1) |
15 ( 6.8) |
13 ( 5.8) |
14 ( 6.3) |
0 ( 0.0) |
1 ( 0.4) |
0 ( 0.0) |
0 ( 0.0) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||||||
|
Espasmos musculares e |
0 ( 0.0) |
1 ( 0.5) |
0 ( 0.0) |
0 ( 0.0) |
98 ( 33.4) |
43 ( 15.4) |
1 ( 0.3) |
0 ( 0.0) |
|
Mialgia e |
7 ( 3.1) |
8 ( 3.6) |
3 ( 1.3) |
5 ( 2.3) |
19 ( 6.5) |
12 ( 4.3) |
2 ( 0.7) |
1 ( 0.4) |
|
Dolor musculoesquelético e |
1 ( 0.4) |
1 ( 0.5) |
0 ( 0.0) |
0 ( 0.0) |
19 ( 6.5) |
11 ( 3.9) |
0 ( 0.0) |
0 ( 0.0) |
|
Trastornos Hepatobiliares |
||||||||
|
Hiperbilirrubinemia e |
34 ( 15.2) |
19 ( 8.6) |
4 ( 1.8) |
2 ( 0.9) |
4 ( 1.4) |
1 ( 0.4) |
2 ( 0.7) |
0 ( 0.0) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||||||
|
Tos e |
23 ( 10.3) |
12 ( 5.4) |
3 ( 1.3) |
1 ( 0.5) |
80 ( 27.3) |
56 ( 20.0) |
0 ( 0.0) |
0 ( 0.0) |
|
Disnea c e |
15 ( 6.7) |
9 ( 4.1) |
8 ( 3.6) |
4 ( 1.8) |
17 ( 5.8) |
9 ( 3.2) |
2 ( 0.7) |
0 ( 0.0) |
|
Rinorrea e |
0 ( 0.0) |
3 ( 1.4) |
0 ( 0.0) |
0 ( 0.0) |
15 ( 5.1) |
6 ( 2.1) |
0 ( 0.0) |
0 ( 0.0) |
|
Embolia pulmonar c d e |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
0 ( 0.0) |
3 ( 1.0) |
0 ( 0.0) |
2 ( 0.7) |
0 ( 0.0) |
|
Trastornos vasculares |
||||||||
|
Trombosis venosa profnda*c d % |
8 ( 3.6) |
2 ( 0.9) |
5 ( 2.2) |
2 ( 0.9) |
7 ( 2.4) |
1 ( 0.4) |
4 ( 1.4) |
1 ( 0.4) |
|
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos) |
||||||||
|
Síndrome mielodisplásico c d e |
5 ( 2.2) |
0 ( 0.0) |
2 ( 0.9) |
0 ( 0.0) |
3 ( 1.0) |
0 ( 0.0) |
1 ( 0.3) |
0 ( 0.0) |
|
Nota: Los AE se codifican en el sistema corporal / reacción adversa usando MedDRA v15.1. Un sujeto con múltiples apariciones de un AE se cuenta sólo una vez en cada categoría de AE. a. Todos los AEs emergentes del tratamiento en al menos el 5% de los pacientes del grupo de mantenimiento con Lenalidomida y al menos 2% más frecuentes (%) que el grupo de mantenimiento con placebo. b. Todos los AEs de grado 3 o 4 emergentes en el tratamiento en al menos el 1% de los pacientes del grupo de mantenimiento con Lenalidomida y al menos el 1% más frecuentes (%) que el grupo de mantenimiento con placebo. c. Todos los AEs graves emergentes del tratamiento en al menos el 1% de los pacientes del grupo de mantenimiento con Lenalidomida y al menos el 1% más frecuentes (%) que el grupo de mantenimiento con placebo. d. Nota al pie de página "a" no aplicable para ninguno de los estudios. e. Nota al pie de página "b" no aplicable para ningún estudio. @ ADRs donde al menos uno tuvo resultado fatal. % ADRs donde por lo menos uno fue considerado como una amenaza para la vida (si el resultado del evento fue la muerte, se incluye con los casos de muerte). # Todas las reacciones adversas bajo el Sistema corporal de infecciones e infestaciones excepto las infecciones raras de interés para la Salud Pública serán consideradas catalogadas. |
||||||||
|
* Reacciones adversas para términos de ADR combinados (basados en TEAE PTs relevantes incluidos en los Estudios de Mantenimiento 1 y 2 [según MedDRA v 15.1]): Neumonías: Bronconeumonía, Neumonía de Lobar, Neumonía por Pneumocystis jiroveci, Pneumonia klebsiella, Pneumonia legionella, Neumonía micoplasmal, Neumonía neumocócica, Neumonía estreptocócica, Neumonía viral, Trastorno pulmonar, Neumonitis Sepsis: sepsis bacteriana, sepsis neumocócica, sepsis, choque séptico, sepsis estafilocócica Neuropatía periférica: Neuropatía periférica, Neuropatía motora periférica, Neuropatía sensitiva periférica, Polineuropatía Trombosis venosa profunda: Trombosis venosa profunda, Trombosis, Trombosis venosa |
||||||||
Después de por lo menos una terapia previa para MM
En dos estudios, se evaluaron datos de 703 pacientes, quienes recibieron por lo menos una dosis de REVLIMID/dexametasona (353 pacientes) o placebo/dexametasona (350 pacientes).
En el grupo de tratamiento REVLIMID/dexametasona, 269 pacientes (76%) tuvieron por lo menos una interrupción de la dosis con o sin reducción de la dosis de REVLIMID en comparación con 199 pacientes (57%) en el grupo de tratamiento placebo/dexametasona. De estos pacientes que tuvieron una interrupción de la dosis con o sin reducción de la dosis, el 50% del grupo de tratamiento REVLIMID/dexametasona tuvo por lo menos una interrupción adicional de la dosis con o sin reducción de la dosis en comparación con el 21% en el grupo de tratamiento placebo/dexametasona. La mayoría de las reacciones adversas y las reacciones adversos grados 3/4 fueron más frecuentes en los pacientes que recibieron la combinación de REVLIMID/dexametasona en comparación con placebo/dexametasona.
Las tablas 16, 17 y 18 resumen las reacciones adversas informadas para los grupos REVLIMID/dexametasona y placebo/dexametasona.
|
Tabla 16: Reacciones adversas informadas en ≥ 5% de los pacientes y con una diferencia ≥ 2% en el porcentaje de pacientes entre los grupos REVLIMID/dexametasona y placebo/dexametasona |
||
|
Sistema corporal Reacciones adversas |
REVLIMID/Dex* (N = 353) n (%) |
Placebo/Dex* (N = 350) n (%) |
|
Trastornos del sistema linfático y de la sangre |
||
|
Neutropenia% |
149 (42,2) |
22 (6,3) |
|
Anemia@ |
111 (31,4) |
83 (23,7) |
|
Trombocitopenia@ |
76 (21,5) |
37 (10,6) |
|
Leucopenia |
28 (7,94) |
4 (1,1) |
|
Linfopenia |
19 (5,4) |
5 (1,4) |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Fatiga |
155 (43,9) |
146 (41,7) |
|
Pirexia |
97 (27,5) |
82 (23,4) |
|
Edema periférico |
93 (26,3) |
74 (21,1) |
|
Dolor en el pecho |
29 (8,2) |
20 (5,7) |
|
Letargia |
24 (6,8) |
8 (2,3) |
|
Trastornos gastrointestinales |
||
|
Constipación |
143 (40,5) |
74 (21,1) |
|
Diarrea@ |
136 (38,5) |
96 (27,4) |
|
Náuseas@ |
92 (26,1) |
75 (21,4) |
|
Vómitos@ |
43 (12,2) |
33 (9,4) |
|
Dolor abdominal@ |
35 (9,9) |
22 (6,3) |
|
Boca seca |
25 (7,1) |
13 (3,7) |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||
|
Calambre muscular |
118 (33,4) |
74 (21,1) |
|
Dolor de espalda |
91 (25,8) |
65 (18,6) |
|
Dolor en los huesos |
48 (13,6) |
39 (11,1) |
|
Dolor en las extremidades |
42 (11,9) |
32 (9,1) |
|
Trastornos del sistema nervioso |
||
|
Mareos |
82 (23,2) |
59 (16,9) |
|
Temblor |
75 (21,2) |
26 (7,4) |
|
Disgeusia |
54 (15,3) |
34 (9,7) |
|
Hipoaestesia |
36 (10,2) |
25 (7,1) |
|
Neuropatíaa |
23 (6,5) |
13 (3,7) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Disnea |
83 (23,5) |
60 (17,1) |
|
Nasofaringitis |
62 (17,6) |
31 (8,9) |
|
Faringitis |
48 (13,6) |
33 (9,4) |
|
Bronquitis |
40 (11,3) |
30 (8,6) |
|
Infeccionesb e infestaciones |
||
|
Infección del tracto respiratorio superior |
87 (24,6) |
55 (15,7) |
|
Neumonía@ |
48 (13,6) |
29 (8,3) |
|
Infección del tracto urinario |
30 (8,5) |
19 (5,4) |
|
Sinusitis |
26 (7,4) |
16 (4,6) |
|
Trastornos cutáneos y del tejido subcutáneo |
||
|
Erupción cutáneac |
75 (21,2) |
33 (9,4) |
|
Incremento de la transpiración |
35 (9,9) |
25 (7,1) |
|
Piel seca |
33 (9,3) |
14 (4,0) |
|
Prurito |
27 (7,6) |
18 (5,1) |
|
Trastornos del metabolismo y nutrición |
||
|
Anorexia |
55 (15,6) |
34 (9,7) |
|
Hipocalemia |
48 (13,6) |
21 (6,0) |
|
Hipocalcemia |
31 (8,8) |
10 (2,9) |
|
Disminución del apetito |
24 (6,8) |
14 (4,0) |
|
Deshidratación |
23 (6,5) |
15 (4,3) |
|
Hipomagnesemia |
24 (6,8) |
10 (2,9) |
|
Exploraciones complementarias |
||
|
Disminución de peso |
69 (19,5) |
52 (14,9) |
|
Trastornos oculares |
||
|
Visión borrosa |
61 (17,3) |
40 (11,4) |
|
Trastornos vasculares |
||
|
Trombosis venosa profunda% |
33 (9,3) |
15 (4,3) |
|
Hipertensión |
28 (7,9) |
20 (5,7) |
|
Hipotensión |
25 (7,1) |
15 (4,3) |
|
Tabla 17: Reacciones adversas grado 3/4 informadas en ≥ 2% de los pacientes y con una diferencia ≥ 1% en el porcentaje de pacientes entre los grupos REVLIMID/dexametasona y placebo/dexametasona |
||
|
Sistema corporal Reacción adversa |
REVLIMID/Dex# (N = 353) n (%) |
Placebo/Dex# (N = 350) |
|
Trastornos del sistema linfático y de la sangre |
||
|
Neutropenia% |
118 (33,4) |
12 (3,4) |
|
Trombocitopenia@ |
43 (12,2) |
22 (6,3) |
|
Anemia@ |
35 (9,9) |
20 (5,7) |
|
Leucopenia |
14 (4,0) |
1 (0,3) |
|
Linfopenia |
10 (2,8) |
4 (1,1) |
|
Neutropenia febril% |
8 (2,3) |
0 (0,0) |
|
Trastornos generales y alteraciones del lugar de administración |
||
|
Fatiga |
23 (6,5) |
17 (4,9) |
|
Trastornos vasculares |
||
|
Trombosis venosa profunda% |
29 (8,2) |
12 (3,4) |
|
Infeccionesb e infestaciones |
||
|
Neumonía@ |
30 (8,5) |
19 (5,4) |
|
Infección del tracto urinario |
5 (1,4) |
1 (0,3) |
|
Trastornos del metabolismo y la nutrición |
||
|
Hipocalemia |
17 (4,8) |
5 (1,4) |
|
Hipocalcemia |
13 (3,7) |
6 (1,7) |
|
Hipofosfatemia |
9 (2,5) |
0 (0,0) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Embolia pulmonar@ |
14 (4,0) |
3 (0,9) |
|
Distrés respiratorio@ |
4 (1,1) |
0 (0,0) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Debilidad muscular |
20 (5,7) |
10 (2,9) |
|
Trastornos gastrointestinales |
||
|
Diarrea@ |
11 (3,1) |
4 (1,1) |
|
Constipación |
7 (2,0) |
1 (0,3) |
|
Náuseas@ |
6 (1,7) |
2 (0,6) |
|
Trastornos cardíacos |
||
|
Fibrilación auricular@ |
13 (3,7) |
4 (1,1) |
|
Taquicardia |
6 (1,7) |
1 (0,3) |
|
Insuficiencia cardíaca congestiva@ |
5 (1,4) |
1 (0,3) |
|
Trastornos del sistema nervioso |
||
|
Síncope |
10 (2,8) |
3 (0,9) |
|
Mareos |
7 (2,0) |
3 (0,9) |
|
Trastornos oculares |
||
|
Cataratas |
6 (1,7) |
1 (0,3) |
|
Catarata unilateral |
5 (1,4) |
0 (0,0) |
|
Trastorno psiquiátrico |
||
|
Depresión |
10 (2,8) |
6 (1,7) |
|
Tabla 18: Reacciones adversas graves reportadas en ≥ 1% de los pacientes y con una diferencia ≥ 1% en el porcentaje de pacientes entre los grupos REVLIMID/dexametasona y placebo/dexametasona |
||
|
Sistema corporal Reacción adversa |
REVLIMID/Dex& (N = 353) n (%) |
Placebo/Dex& (N = 350) n (%) |
|
Trastornos del sistema linfático y de la sangre |
||
|
Neutropenia febril% |
6 (1,7) |
0 (0,0) |
|
Trastornos vasculares |
||
|
Trombosis venosa profunda% |
26 (7,4) |
11 (3,1) |
|
Infeccionesb e infestaciones |
||
|
Neumonía |
33 (9,3) |
21 (6,0) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Embolia pulmonar@ |
13 (3,7) |
3 (0,9) |
|
Trastornos cardíacos |
||
|
Fibrilación auricular@ |
11 (3,1) |
2 (0,6) |
|
Insuficiencia cardíaca congestiva@ |
5 (1,4) |
0 (0,00) |
|
Trastornos del sistema nervioso |
||
|
Accidente cerebrovascular@ |
7 (2,0) |
3 (0,9) |
|
Trastornos gastrointestinales |
||
|
Diarrea@ |
6 (1,7) |
2 (0,6) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Dolor de hueso |
4 (1,1) |
0 (0,0) |
|
Para las tablas 16, 17 y 18 de arriba: @ reacciones adversas en las cuales por lo menos una resultó fatal. % reacciones adversas en las cuales por lo menos una se consideró que puso en peligro la vida (si el resultado de la reacción fue muerte, se incluye en los casos de muerte) . |
||
La mediana de duración de exposición entre los pacientes tratados con REVLIMID/dexametasona fue 44 semanas, mientras que la mediana de duración de exposición entre los pacientes tratados con placebo/dexametasona fue 23 semanas. Esto debería considerarse cuando se compara la frecuencia de reacciones adversas entre los dos grupos de tratamiento REVLIMID/dexametasona vs. placebo/dexametasona.
Tromboembolia venosa y arterial
TEV y TEA están aumentados en pacientes tratados con REVLIMID
La trombosis venosa profunda se informó como una reacción adversa grave (7,4%) o severa (8,2%) en una mayor proporción en el grupo REVLIMID/dexametasona en comparación con el 3,1% y el 3,4% en el grupo placebo/dexametasona, respectivamente, en los 2 estudios en pacientes con por lo menos 1 terapia previa, con discontinuación debido a reacciones adversas relacionadas con la trombosis venosa profunda informadas en proporciones comparables entre los grupos. En el estudio NDMM, se informó trombosis venosa profunda como una reacción adversa (todos los grados: 10,3%, 7,2%, 4,1%), como una reacción adversa seria (3,6%, 2,0%, 1,7%), y como una reacción adversa de grado 3/4 (5,6%, 3,7%, 2,8%) en las ramas Rd Continuo, Rd18 y MPT, respectivamente. Las discontinuaciones y las reducciones de la dosis debido a reacciones adversas de trombosis venosa profunda se informaron a tasas comparables entre las ramas Rd Continuo y Rd18 (ambas < 1%). La interrupción del tratamiento de REVLIMID por reacciones adversas de trombosis venosa profunda se informó a tasas comparables entre las ramas Rd Continuo (2,3%) y Rd18 (1,5%).
La Embolia Pulmonar (EP) se informó como una reacción adversa seria (3,7%) o de grados 3/4 (4,0%) en una proporción mayor en el grupo REVLIMID/dexametasona en comparación con el 0,9% en el grupo placebo/dexametasona en los 2 estudios en pacientes con por lo menos 1 terapia previa, con discontinuación debido a reacciones adversas relacionadas con la trombosis venosa profunda informadas en proporciones comparables entre los grupos. En el estudio NDMM, se informó trombosis venosa profunda como una reacción adversa (todos los grados: 10,3%, 7,2%, 4,1%), como una reacción adversa seria (3,6%, 2,0%, 1,7%), y como una reacción adversa de grado 3/4 (5,6%, 3,7%, 2,8%) en las ramas Rd Continuo, Rd18 y MPT, respectivamente.
El infarto de miocardio se informó como una reacción adversa seria (1,7%) o severa (1,7%) en una tasa más alta en el grupo de REVLIMID/dexametasona en comparación con 0,6% y 0,6%, respectivamente, en el grupo placebo/dexametasona. La discontinuación debido a reacciones adversas de infarto de miocardio (incluido el agudo) fue de 0,8% en el grupo de REVLIMID/dexametasona y ninguna en el grupo placebo/dexametasona. En el estudio NDMM, se informó infarto de miocardio (incluido el agudo) como una reacción adversa (todos los grados: 2,4%, 0,6% y 1,1%), como una reacción adversa seria (2,3%, 0,6% y 1,1%), o como una reacción adversa severa (1,9%, 0,6% y 0,9%) en las ramas Rd Continuo, Rd18 y MPT, respectivamente. Se informó ACV como una reacción adversa seria (2,3%) o severa (2,0%) en el grupo REVLIMID/dexametasona en comparación con 0,9% y 0,9%, respectivamente, en el grupo placebo/dexametasona. La discontinuación por ACV fue del 1,4% en el grupo REVLIMID/ dexametasona y del 0,3% en el grupo placebo/dexametasona. En el estudio NDMM, se informó ACV como una reacción adversa (todos los grados: 0,8%, 0,6% y 0,6%), como una reacción adversa seria (0,8%, 0,6 % y 0,6%) o como una reacción adversa severa (0,6%, 0,6%, 0,2%) en las ramas Rd Continuo, Rd18 y MPT, respectivamente.
Otras reacciones adversas después de por lo menos una terapia previa para MM : En estos dos estudios, se informaron las siguientes reacciones adversas no descriptas más arriba que se produjeron en una proporción del ≥ 1% y de por lo menos dos veces el porcentaje de placebo
Trastornos del sistema linfático y de la sangre: pancitopenia, anemia hemolítica autoinmune.
Trastornos cardíacos: bradicardia, infarto de miocardio, angina de pecho.
Trastornos endocrinos: hirsutismo.
Trastornos oculares: ceguera, hipertensión ocular. Trastornos gastrointestinales: hemorragia gastrointestinal, glosodinia.
Trastornos generales y alteraciones en el lugar de administración: malestar.
Investigaciones: pruebas de la función hepática con resultados anormales, incremento de la alanina aminotransferasa.
Trastornos del sistema nervioso: isquemia cerebral. Trastornos psiquiátricos: cambios del estado de ánimo, alucinaciones, pérdida de la libido.
Trastorno del aparato reproductor y de la mama: disfunción eréctil.
Trastornos respiratorios, torácicos y mediastínicos: tos, ronquera.
Trastornos de la piel y del tejido subcutáneo: exantema, hiperpigmentación de la piel.
Pacientes con mieloma múltiple no tratado previamente que son elegibles para trasplante que recibieron lenalidomida en combinación con bortezomib y dexametasona
En los estudios PETHEMA GEM2012 (Grupos combinados A y B (RVd), n = 458) e IFM 2009 (Grupo A (RVd), n = 356), la reacción adversa grave observada con más frecuencia (≥ 5%) con lenalidomida en combinación con bortezomib y dexametasona fue:
• Neumonía (5,9%) del PETHEMA GEM2012 En el estudio PETHEMA GEM2012. Las reacciones adversas observadas con mayor frecuencia con lenalidomida en combinación con bortezomib subcutáneo y dexametasona fueron: neuropatía periférica (35,2%), neutropenia (31,9%), trombocitopenia (25,3%).
En el estudio IFM 2009, las reacciones adversas observadas con mayor frecuencia con lenalidomida en combinación con bortezomib intravenoso y dexametasona fueron: neuropatía periférica (54,8%), linfopenia (52,2%). Pacientes con mieloma múltiple no tratado previamente que no son elegibles para trasplante que recibieron lenalidomida en combinación con bortezomib y dexametasona
En el estudio SWOG S0777 (Grupo B (RVd), n = 262), las reacciones adversas graves observadas con más frecuencia (≥ 5%) con lenalidomida en combinación con bortezomib intravenoso y dexametasona que con lenalidomida en combinación con dexametasona fueron:
Hipotensión (6,5%), infección pulmonar (5,7%), deshidratación (5,0%).
Las reacciones adversas observadas con mayor frecuencia con lenalidomida en combinación con bortezomib y dexametasona que con lenalidomida en combinación con dexametasona fueron: Fatiga (73,7%), neuropatía periférica (71,8%), trombocitopenia (57,6%), estreñimiento (56,1%), hipocalcemia 50,0%).
Síndromes mielodisplásicos
Un total de 148 pacientes recibieron por lo menos 1 dosis de 10 mg de REVLIMID en el estudio clínico de SMD relacionado con deleción del 5q. Por lo menos un evento adverso se informó en todos los 148 pacientes que fueron tratados con la dosis inicial de 10 mg de REVLIMID. Los eventos adversos informados con mayor frecuencia estuvieron relacionados con trastornos del sistema linfático y de la sangre, trastornos de la piel y del tejido subcutáneo, trastornos gastrointestinales, y trastornos generales y alteraciones en el lugar de administración. Los eventos adversos observados informados con mayor frecuencia fueron trombocitopenia (61,5%; 91/148) y neutropenia (58,8%; 87/148). Le siguieron diarrea (48,6%; 72/148), prurito (41,9%; 62/148), erupción cutánea (35,8%; 53/148) y fatiga (31,1%; 46/148). La tabla 19 resume los eventos adversos que fueron informados en ≥5% de los pacientes tratados con REVLIMID en el estudio clínico de SMD relacionado con deleción del 5q. La tabla 20 resume las reacciones adversas grado 3 y grado 4 observadas con mayor frecuencia independientemente de la relación con el tratamiento con REVLIMID. En los estudios de rama única realizados, a menudo no es posible distinguir eventos adversos que están relacionados con el fármaco y aquellos que reflejan la enfermedad subyacente del paciente.
|
Tabla 19: Resumen de eventos adversos informados en ≥5% de los pacientes tratados con REVLIMID® en el estudio clínico de SMD relacionado con deleción del 5q |
||
|
Sistema corporal/ |
10 mg general (N=148) |
|
|
Pacientes con por lo menos un evento adverso |
148 |
(100,0) |
|
Trastornos del sistema linfático y de la sangre |
||
|
Trombocitopenia |
91 |
(61,5) |
|
Neutropenia |
87 |
(58,8) |
|
Anemia |
17 |
(11,5) |
|
Leucopenia |
12 |
(8,1) |
|
Neutropenia febril |
8 |
(5,4) |
|
Trastornos del tejido cutáneo y subcutáneo |
||
|
Prurito |
62 |
(41,9) |
|
Erupción cutánea |
53 |
(35,8) |
|
Piel seca |
21 |
(14,2) |
|
Contusión |
12 |
(8,1) |
|
Sudor nocturno |
12 |
(8,1) |
|
Mayor transpiración |
10 |
(6,8) |
|
Equimosis |
8 |
(5,4) |
|
Eritema |
8 |
(5,4) |
|
Trastornos gastrointestinales |
||
|
Diarrea |
72 |
(48,6) |
|
Constipación |
35 |
(23,6) |
|
Náuseas |
35 |
(23,6) |
|
Dolor abdominal |
18 |
(12,2) |
|
Vómitos |
15 |
(10,1) |
|
Dolor abdominal superior |
12 |
(8,1) |
|
Boca seca |
10 |
(6,8) |
|
Heces blandas |
9 |
(6,1) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Nasofaringitis |
34 |
(23,0) |
|
Tos |
29 |
(19,6) |
|
Disnea |
25 |
(16,9) |
|
Faringitis |
23 |
(15,5) |
|
Epistaxis |
22 |
(14,9) |
|
Disnea por esfuerzo |
10 |
(6,8) |
|
Rinitis |
10 |
(6,8) |
|
Bronquitis |
9 |
(6,1) |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Fatiga |
46 |
(31,1) |
|
Pirexia |
31 |
(20,9) |
|
Edema periférico |
30 |
(20,3) |
|
Astenia |
22 |
(14,9) |
|
Edema |
15 |
(10,1) |
|
Dolor |
10 |
(6,8) |
|
Escalofríos |
9 |
(6,1) |
|
Dolor de tórax |
8 |
(5,4) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Artralgia |
32 |
(21,6) |
|
Dolor de espalda |
31 |
(20,9) |
|
Calambre muscular |
27 |
(18,2) |
|
Dolor en las extremidades |
16 |
(10,8) |
|
Mialgia |
13 |
(8,8) |
|
Hinchazón periférica |
12 |
(8,1) |
|
Trastornos del sistema nervioso |
||
|
Mareos |
29 |
(19,6) |
|
Dolor de cabeza |
29 |
(19,6) |
|
Hipoestesia |
10 |
(6,8) |
|
Disgeusia |
9 |
(6,1) |
|
Neuropatía periférica |
8 |
(5,4) |
|
Infecciones e infestaciones |
||
|
Infección del tracto respiratorio superior |
22 |
(14,9) |
|
Neumonía |
17 |
(11,5) |
|
Infección del tracto urinario |
16 |
(10,8) |
|
Sinusitis |
12 |
(8,1) |
|
Celulitis |
8 |
(5,4) |
|
Trastornos del metabolismo y la nutrición |
||
|
Hipocalemia |
16 |
(10,8) |
|
Anorexia |
15 |
(10,1) |
|
Hipomagnesemia |
9 |
(6,1) |
|
Investigaciones |
||
|
Aumento de alanina aminotransferasa |
12 |
(8,1) |
|
Trastornos psiquiátricos |
||
|
Insomnio |
15 |
(10,1) |
|
Depresión |
8 |
(5,4) |
|
Trastornos renales y urinarios |
||
|
Disuria |
10 |
(6,8) |
|
Trastornos vasculares |
||
|
Hipertensión |
9 |
(6,1) |
|
Trastornos endocrinos |
||
|
Hipotiroidismo adquirido |
10 |
(6,8) |
|
Trastornos cardíacos |
||
|
Palpitaciones |
8 |
(5,4) |
|
[a] El sistema corporal y los eventos adversos están codificados usando el diccionario MedDRA. El sistema corporal y los eventos adversos se enumeran en orden descendente de frecuencia para la columna general. Un paciente con manifestaciones múltiples de un EA se cuenta sólo una vez en la categoría de EA. |
||
|
Tabla 20: Eventos adversos grado 3 y 4 observados con mayor frecuencia[1] independientemente de la relación con el tratamiento del estudio |
||
|
Eventos adversos[2] |
10 mg (N=148) |
|
|
Pacientes con por lo menos un EA grados 3/4 |
131 |
(88,5) |
|
Neutropenia |
79 |
(53,4) |
|
Trombocitopenia |
74 |
(50,0) |
|
Neumonía |
11 |
(7,4) |
|
Erupción cutánea |
10 |
(6,8) |
|
Anemia |
9 |
(6,1) |
|
Leucopenia |
8 |
(5,4) |
|
Fatiga |
7 |
(4,7) |
|
Disnea |
7 |
(4,7) |
|
Dolor de espalda |
7 |
(4,7) |
|
Neutropenia febril |
6 |
(4,1) |
|
Náuseas |
6 |
(4,1) |
|
Diarrea |
5 |
(3,4) |
|
Pirexia |
5 |
(3,4) |
|
Sepsis |
4 |
(2,7) |
|
Mareos |
4 |
(2,7) |
|
Granulocitopenia |
3 |
(2,0) |
|
Dolor de tórax |
3 |
(2,0) |
|
Embolia pulmonar |
3 |
(2,0) |
|
Dificultad respiratoria |
3 |
(2,0) |
|
Prurito |
3 |
(2,0) |
|
Pancitopenia |
3 |
(2,0) |
|
Calambre muscular |
3 |
(2,0) |
|
Infección del tracto respiratorio |
2 |
(1,4) |
|
Infección del tracto respiratorio superior |
2 |
(1,4) |
|
Astenia |
2 |
(1,4) |
|
Insuficiencia multiorgánica |
2 |
(1,4) |
|
Epistaxis |
2 |
(1,4) |
|
Hipoxia |
2 |
(1,4) |
|
Efusión pleural |
2 |
(1,4) |
|
Neumonitis |
2 |
(1,4) |
|
Hipertensión pulmonar |
2 |
(1,4) |
|
Vómitos |
2 |
(1,4) |
|
Aumento de la transpiración |
2 |
(1,4) |
|
Artralgia |
2 |
(1,4) |
|
Dolor en las extremidades |
2 |
(1,4) |
|
Dolor de cabeza |
2 |
(1,4) |
|
Síncope |
2 |
(1,4) |
|
[1] Eventos adversos con frecuencia ≥1% en el grupo general de 10 mg. Los grados 3 y 4 se basan en los Criterios de Toxicidad Común del Instituto Nacional del Cáncer (NCI CTC, National Cancer Institute Common Toxicity Criteria) versión 2. [2] Los eventos adversos están codificados usando el diccionario MedDRA. Un paciente con manifestaciones múltiples de un EA se cuenta sólo una vez en la categoría del evento adverso. |
||
En otros estudios clínicos de REVLIMID en pacientes con SMD, se informaron los siguientes eventos adversos graves (independientemente de la relación con el tratamiento con el fármaco experimental) no descriptos en las tablas 19 ó 20:
Trastornos del sistema linfático y de la sangre: anemia hemolítica tipo cálida, infarto esplénico, depresión de la médula ósea, coagulopatía, hemólisis, anemia hemolítica, anemia refractaria.
Trastornos cardíacos: insuficiencia cardíaca congestiva, fibrilación auricular, angina de pecho, paro cardíaco, insuficiencia cardíaca, paro cardiorrespiratorio, cardiomiopatía, infarto de miocardio, isquemia miocárdica, fibrilación auricular agravada, bradicardia, shock cardiogénico, edema pulmonar, arritmia supraventricular, taquiarritmia, disfunción ventricular.
Trastornos del oído y del laberinto: vértigo.
Trastornos endocrinos: enfermedad de Basedow. Trastornos gastrointestinales: hemorragia gastrointestinal, colitis isquémica, perforación intestinal, hemorragia rectal, pólipos del colon, diverticulitis, disfagia, gastritis, gastroenteritis, enfermedad de reflujo gastroesofágico, hernia inguinal obstructiva, síndrome de intestino irritable, melena, pancreatitis por obstrucción biliar, pancreatitis, absceso perirrectal, obstrucción del intestino delgado, hemorragia gastrointestinal superior.Trastornos generales y alteraciones en el lugar de administración: progresión de la enfermedad, caída, marcha anormal, pirexia intermitente, nódulo, escalofríos, muerte súbita.
Trastornos hepatobiliares: hiperbilirrubinemia, colecistitis aguda, colecistitis, insuficiencia hepática.
Trastornos del sistema inmune: hipersensibilidad.
Infecciones e infestaciones: infección, bacteremia, infección de línea central, infección por Clostridium, infección de oído, sepsis por Enterobacter, infección por hongos, infección viral por herpes, gripe, infección del riñón, sepsis por Klebsiella, neumonía lobar, infección localizada, infección oral, infección por Pseudomonas, shock séptico, sinusitis aguda, sinusitis, infección estafilocócica, urosepsis.
Lesión, envenenamiento y complicaciones de procedimiento: fractura de fémur, reacción a la transfusión, fractura de vértebra cervical, fractura de cuello femoral, fractura de pelvis, fractura de cadera, sobredosis, hemorragia post-procedimiento, fractura de costilla, accidente de tránsito, fractura con compresión de médula espinal.
Investigaciones: aumento de creatinina en sangre, cultivo negativo, disminución de hemoglobina, pruebas de función hepática con resultados anormales, aumento de troponina I.
Trastornos del metabolismo y de la nutrición: deshidratación, gota, hipernatremia, hipoglucemia. Trastornos musculoesqueléticos y del tejido conjuntivo: artritis, artritis agravada, artritis gotosa, dolor de cuello, pirofosfato condrocalcinosis.
Neoplasias benignas, malignas y no especificadas: leucemia aguda, leucemia mieloide aguda, carcinoma bronquioloalveolar, cáncer de pulmón con metástasis, linfoma, cáncer de próstata con metástasis.
Trastornos del sistema nervioso: accidente cerebrovascular, afasia, infarto cerebeloso, infarto cerebral, disminución del nivel de conciencia, disartria, migraña, compresión de médula espinal, hemorragia subaracnoidea, accidente isquémico transitorio.
Trastornos psiquiátricos: estado de confusión. Trastornos renales y urinarios: insuficiencia renal, hematuria, insuficiencia renal aguda, azotemia, cálculos uretrales, masa renal.
Trastornos del sistema reproductivo y de la mama: dolor pélvico.
Trastornos respiratorios, torácicos y mediastínicos: bronquitis, enfermedad obstructiva crónica de las vías respiratorias exacerbada, insuficiencia respiratoria, disnea exacerbada, enfermedad pulmonar intersticial, infiltración pulmonar, sibilancia.
Trastornos de la piel y del tejido subcutáneo: dermatosis neutrofílica febril aguda.
Trastornos del sistema vascular: trombosis venosa profunda, hipotensión, trastorno de la arteria aorta, isquemia, tromboflebitis superficial, trombosis.
Linfoma de Células del Manto (LCM)
En el ensayo de LCM, un total de 134 pacientes recibieron al menos 1 dosis de REVLIMID. La mediana de edad fue de 67 años (rango 43-83 años), 128/134 (96 %) eran caucásicos, 108/134 (81 %) eran hombres y 82/134 (61 %) tenía una duración de LCM de al menos 3 años.
La Tabla 21 resumen las reacciones adversas más frecuentemente observadas independientemente de la relación con el tratamiento con REVLIMID. En los 134 pacientes tratados en este estudio, la mediana de duración del tratamiento fue de 95 días (1-1.002 días). Setenta y ocho pacientes (58 %) recibieron 3 o más ciclos de terapia, 53 pacientes (40 %) recibieron 6 o más ciclos y 26 pacientes (19 %) recibieron 12 o más ciclos. Setenta y seis pacientes (57 %) experimentó al menos una interrupción de dosis debido a eventos adversos, y 51 pacientes (38 %) experimentaron al menos una reducción de dosis debido a eventos adversos. Veintiséis pacientes (19 %) discontinuaron el tratamiento debido a eventos adversos.
|
Tabla 21: Incidencia de reacciones adversas (≥10%) o Eventos Adversos (EA) grados 3/4 (en al menos 2 pacientes) con linfoma de células de manto |
||
|
Sistema corporal/ |
Todos los EAs1 (N=134) n (%) |
EAs2 grados 3/4 (N=134) n (%) |
|
Trastornos generales y condiciones del lugar de administración de la inyección |
||
|
Fatiga |
45 (34) |
9 (7) |
|
Pirexia$ |
31 (23) |
3 (2) |
|
Edema periférico |
21 (16) |
0 |
|
Astenia$ |
19 (14) |
4 (3) |
|
Deterioro general de la salud física |
3 (2) |
2 (1) |
|
Trastornos gastrointestinales |
||
|
Diarrea$ |
42 (31) |
8 (6) |
|
Náuseas |
40 (30) |
1 (<1) |
|
Constipación |
21 (16) |
1 (<1) |
|
Vómitos$ |
16 (12) |
1 (<1) |
|
Dolor abdominal$ |
13 (10) |
5 (4) |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||
|
Dolor de espalda |
18 (13) |
2 (1) |
|
Espasmos musculares |
17 (13) |
1 (<1) |
|
Artralgia |
11 (8) |
2 (1) |
|
Debilidad muscular$ |
8 (6) |
2 (1) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Tos |
38 (28) |
1 (<1) |
|
Disnea$ |
24 (18) |
8 (6) |
|
Efusión pleural |
10 (7) |
2 (1) |
|
Hipoxia |
3 (2) |
2 (1) |
|
Embolia pulmonar |
3 (2) |
3 (2) |
|
Insuficiencia respiratoria$ |
2 (1) |
2 (1) |
|
Dolor orofaríngeo |
13 (10) |
0 |
|
Infecciones e infestaciones |
||
|
Neumonía@ $ |
19 (14) |
12 (9) |
|
Infección respiratoria superior |
17 (13) |
0 |
|
Celulitis$ |
3 (2) |
2 (1) |
|
Bacteriemia$ |
2 (1) |
2 (1) |
|
Sepsis estafilocócica$ |
2 (1) |
2 (1) |
|
Infección del tracto urinario$ |
5 (4) |
2 (1) |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Rash + |
30 (22) |
2 (1) |
|
Prurito |
23 (17) |
1 (<1) |
|
Trastornos del sistema linfático y de la sangre |
||
|
Neutropenia |
65 (49) |
58 (43) |
|
Thrombocitopenia% $ |
48 (36) |
37 (28) |
|
Anemia$ |
41 (31) |
15 (11) |
|
Leucopenia$ |
20 (15) |
9 (7) |
|
Linfopenia |
10 (7) |
5 (4) |
|
Neutropenia febril$ |
8 (6) |
8 (6) |
|
Trastornos del metabolismo y de la nutrición |
||
|
Disminución del apetito |
19 (14) |
1 (<1) |
|
Hipocalemia |
17 (13) |
3 (2) |
|
Deshidratación$ |
10 (7) |
4 (3) |
|
Hipocalcemia |
4 (3) |
2 (1) |
|
Hiponatremia |
3 (2) |
3 (2) |
|
Trastornos renales y urinarios |
||
|
Insuficiencia renal$ |
5 (4) |
2 (1) |
|
Trastornos vasculares |
||
|
Hipotensión@ $ |
9 (7) |
4 (3) |
|
Trombosis venosa profunda$ |
5 (4) |
5 (4) |
|
Neoplasias benignas, malignas o no especificadas (incluidos quistes y pólipos) |
||
|
Exacerbación tumoral |
13 (10) |
0 |
|
Carcinoma de células escamosas de la piel$ |
4 (3) |
4 (3) |
|
Exploraciones complementarias |
||
|
Disminución de peso |
17 (13) |
0 |
|
1-Eventos adversos del ensayo de LCM – Todos los eventos adversos emergentes del tratamiento con > 10 % de pacientes 2-Eventos adversos grado 3/4 del ensayo de LCM – Todos los eventos adversos emergente de grado 3/4 emergentes del tratamiento en 2 o más pacientes $-Eventos adversos serios del ensayo de LCM – Todos los eventos adversos serios emergentes del tratamiento en 2 o más pacientes @ - Eventos adversos en los que al menos uno provocó un resultado fatal % - Eventos adversos en los que al menos uno se consideró con riesgo de muerte (si el resultado fue la muerte, se incluyó con casos de muerte) # - Todos los eventos adversos bajo el Sistema Corporal de Infecciones a excepción de infecciones raras de interés para la Salud Pública se considerará enumerado + - Todos los eventos adversos bajo términos del nivel alto (HLT) de erupción cutánea se considerarán enumerados |
||
Las siguientes reacciones adversas que se manifestaron en otras indicaciones , que incluyen otro estudio sobre LCM y no se describen anteriormente han sido reportados (5%-10%) en pacientes tratados con monoterapia de REVLIMID para linfoma de células de manto.
Trastorno cardíaco: insuficiencia cardíaca
Trastornos del oído y del laberinto: vértigo.
Trastornos generales y alteraciones en la zona de la administración: escalofríos.
Trastornos musculoesqueléticos y del tejido conectivo: Dolor en las extremidades.
Infecciones e infestaciones: Infección del tracto respiratorio, sinusitis, nasofaringitis, herpes oral. Trastornos del sistema nervioso: disgeusia, dolor de cabeza, neuropatía periférica, letargo.
Trastornos psiquiátricos: insomnio.
Trastornos de la piel y del tejido subcutáneo: Piel seca, sudoración nocturna.
Los siguientes eventos adversos serios no descriptos anteriormente y reportados en 2 o más pacientes tratados con monoterapia de REVLIMID para linfoma de células de manto.
Trastornos sanguíneos y linfáticos: Neutropenia. Trastornos cardíacos: infarto de miocardio (incluido infarto agudo de miocardio –MI-), taquicardia supraventricular.
Infecciones e infestaciones: Colitis por clostridium difficile, sepsis.
Neoplasias benignas, malignas o no especificadas (incluidos quistes y pólipos): Carcinoma de células basales.
Trastornos respiratorios, torácicos y mediastínicos: enfermedad pulmonar obstructiva crónica, embolia pulmonar.
Linfoma folicular o linfoma de zona marginal La seguridad de REVLIMID/rituximab se evaluó en dos estudios clínicos con 398 pacientes que ya habían sido tratados por linfoma folicular o linfoma de la zona marginal; AUGMENT (N=176) y MAGNIFY (N=222) [véase Estudios clínicos]. Los sujetos del estudio tenían más de 18 años, presentaban ≤2 ECOG PS, RAN ≥1.000 células/mm3 y un recuento de plaquetas ≥ 75.000/mm3 (salvo que exista compromiso secundario medular por linfoma), hemoglobina ≥ 8g/dL, AST y ALT ≤ 3 x ULN (salvo compromiso hepático con linfoma documentado) y clearance de creatinina ≥ 30 mL/min. Los sujetos con VIH, hepatitis B o C activos no fueron elegibles.
En el estudio AUGMENT, los pacientes recibieron 20 mg diarios de REVLIMID por vía oral durante los días 1-21 de cada ciclo de 28 días con 375 mg/m2 de rituximab semanal (días 1, 8, 15 y 22 del ciclo 1), luego el día 1 de los ciclos 2 a 5 (n=176) o placebo con 375 mg/m2 de rituximab semanal (días 1, 8, 15 y 22 del ciclo 1), luego el día 1 de los ciclos 2 a 5 (n=180) durante hasta 12 ciclos. En el estudio MAGNIFY los pacientes recibieron 20 mg diarios de REVLIMID por vía oral durante los días 1-21 de cada ciclo de 28 días con 375 mg/m2 de rituximab semanal (días 1, 8, 15 y 22 del ciclo 1), luego el día 1 de los ciclos 3, 7, 9 y 11 de la fase de inducción del estudio (n=222). En el estudio AUGMENT, 88,1% de los pacientes completaron al menos 6 ciclos con REVLIMID/rituximab y 71% de los pacientes completaron 12 ciclos. En el estudio en curso MAGNIFY, al 1 de mayo de 2017, 62,2% de los pacientes habían completado 6 ciclos con REVLIMID/rituximab y 30,6% de los pacientes los 12 ciclos.
Entre ambos estudios (AUGMENT y MAGNIFY) los pacientes presentaron una mediana etaria de 64,5 años (26 a 91); 49% fueron hombres y 81% blancos.
En 6 pacientes tratados con REVLIMID/rituximab se registraron reacciones adversas mortales (1,5%). Estas reacciones adversas mortales (1 por paciente) incluyeron paro cardiorrespiratorio, arritmia, insuficiencia cardiopulmonar, síndrome de disfunción multiorgánica, sepsis y daño renal agudo. Las reacciones adversas graves se registraron en un 26% de pacientes tratados con REVLIMID/rituximab en AUGMENT y en un 29% en MAGNIFY. La reacción adversa grave más frecuente que se registró en ≥ 2,5% de los pacientes en el brazo REVLIMID/rituximab fue neutropenia febril (3%). La suspensión permanente de REVLIMID o rituximab a causa de una reacción adversa se registró en 14,6% de los pacientes del brazo REVLIMID/rituximab. La reacción adversa más común (en al menos 1%) por la cual se debió suspender REVLIMID o rituximab fue neutropenia (4,8%).Las reacciones adversas más comunes que aparecieron en al menos 20% de los sujetos fueron: neutropenia (48%), fatiga (37%), diarrea (32%), constipación (27%), náuseas (21%) y tos (20%).
|
Tabla 22: Reacciones adversas de cualquier grado (≥5%) o Reacciones adversas grado 3/4 (≥1%) en pacientes con LF y LZM con una diferencia entre brazos >1% al compararse con el brazo control en el Estudio AUGMENT |
||||
|
Sistema de clasificación de órganos |
Todas las reacciones adversas 1 |
Reacciones adversas |
||
|
Brazo REVLIMID + Rituximab (N=176) n (%) |
Rituximab + Placebo (brazo control) (N=180) n (%) |
Brazo REVLIMID + Rituximab (N=176) n (%) |
Rituximab + Placebo (brazo control) (N=180) n (%) |
|
|
Infecciones e infestaciones |
||||
|
Infección del tracto respiratorio superior |
32 (18) |
23 (13) |
2 (1.1) |
4 (2.2) |
|
Gripe % |
17 (10) |
8 (4.4) |
1 (< 1) |
0 (0) |
|
Neumonía 3,$,% |
13 (7) |
6 (3.3) |
6 (3.4) |
4 (2.2) |
|
Sinusitis |
13 (7) |
5 (2.8) |
0 (0) |
0 (0) |
|
Infección del tracto urinario$ |
13 (7) |
7 (3.9) |
1 (< 1) |
1 (< 1) |
|
Bronquitis |
8 (4,5) |
6 (3,3) |
2 (1,1) |
0 (0) |
|
Gastroenteritis $ |
6 (3,4) |
4 (2,2) |
2 (1,1) |
0 (0) |
|
Neoplasias benignas, malignas o no especificadas (incluidos quistes y pólipos) |
||||
|
Exacerbación tumoral $ |
19 (11) |
1 (< 1) |
1 (< 1) |
0 (0) |
|
Trastornos del sistema circulatorio y linfático |
||||
|
Neutropenia 3,$, % |
102 (58) |
40 (22) |
88 (50) |
23 (13) |
|
Leucopenia $,% |
36 (20) |
17 (9) |
12 (7) |
3 (1,7) |
|
Anemia 3,$ |
28 (16) |
8 (4,4) |
8 (4,5) |
1 (< 1) |
|
Trombocitopenia 3,$,% |
26 (15) |
8 (4,4) |
4 (2,3) |
2 (1,1) |
|
Linfopenia |
8 (4.5) |
14 (8) |
5 (2,8) |
2 (1,1) |
|
Neutropenia febril 3,$,% |
5 (2,8) |
1 (< 1) |
5 (2,8) |
1 (< 1) |
|
Trastornos del metabolismo y de la nutrición |
||||
|
Disminución del apetito |
23 (13) |
11 (6) |
2 (1,1) |
0 (0) |
|
Hipokalemia % |
14 (8) |
5 (2.8) |
4 (2.3) |
0 (0) |
|
Hiperuricemia |
10 (6) |
8 (4,4) |
1 (< 1) |
1 (< 1) |
|
Trastornos del sistema nervioso |
||||
|
Dolor de cabeza |
26 (15) |
17 (9) |
1 (< 1) |
0 (0) |
|
Mareos |
15 (9) |
9 (5) |
0 (0) |
0 (0) |
|
Trastornos vasculares |
||||
|
Hipotensión % |
9 (5) |
1 (< 1) |
1 (< 1) |
0 (0) |
|
Eventos tromboembólicos a,$ |
8 (4.5) |
2 (1.1) |
4 (2.3) |
2 (1.1) |
|
Trastornos respiratorios, torácicos y mediastínicos |
||||
|
Tos b |
43 (24) |
35 (19) |
1 (< 1) |
0 (0) |
|
Disnea $ |
19 (11) |
8 (4,4) |
2 (1,1) |
1 (< 1) |
|
Dolor orofaríngeo |
10 (6) |
8 (4,4) |
0 (0) |
0 (0) |
|
Embolia pulmonar 3,$ |
4 (2.3) |
1 (< 1) |
4 (2,3) |
1 (< 1) |
|
Enfermedad obstructiva crónica pulmonar $ |
3 (1,7) |
0 (0) |
2 (1,1) |
0 (0) |
|
Insuficiencia respiratoria 3,$ |
2 (1,1) |
1 (< 1) |
2 (1,1) |
0 (0) |
|
Trastornos gastrointestinales |
||||
|
Diarrea $,% |
55 (31) |
41 (23) |
5 (2.8) |
0 (0) |
|
Constipación |
46 (26) |
25 (14) |
0 (0) |
0 (0) |
|
Dolor abdominal c ,$ |
32 (18) |
20 (11) |
2 (1,1) |
0 (0) |
|
Vómitos $ |
17 (10) |
13 (7) |
0 (0) |
0 (0) |
|
Dispepsia |
16 (9) |
5 (2.8) |
0 (0) |
0 (0) |
|
Stomatitis |
9 (5) |
7 (3.9) |
0 (0) |
0 (0) |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Erupción cutánea $,d |
39 (22) |
14 (8) |
5 (2,8) |
2 (1.1) |
|
Prurito $,e |
36 (20) |
9 (5) |
2 (1,1) |
0 (0) |
|
Piel seca |
9 (5) |
6 (3,3) |
0 (0) |
0 (0) |
|
Dermatitis acneiforme |
8 (4,5) |
0 (0) |
2 (1,1) |
0 (0) |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||||
|
Espasmos musculares |
23 (13) |
9 (5) |
1 (< 1) |
1 (< 1) |
|
Dolor en extremidades $ |
8 (4,5) |
9 (5) |
2 (1) |
0 (0) |
|
Trastornos renales Insuficiencia renal aguda 3,$,@,% |
3 (1,7) |
0 (0) |
2 (1,1) |
0 (0) |
|
Trastornos cardíacos |
||||
|
Taquicardia supraventricular 3,$ |
2 (1,1) |
0 (0) |
2 (1,1) |
0 (0) |
|
Trastornos generales y alteraciones en la zona de la administración |
||||
|
Fatiga |
38 (22) |
33 (18) |
2 (1.1) |
1 (< 1) |
|
Pirexia 3,$ |
37 (21) |
27 (15) |
1 (< 1) |
3 (1,7) |
|
Astenia $,% |
24 (14) |
19 (11) |
2 (1,1) |
1 (< 1) |
|
Edema Periférico$ |
23 (13) |
16 (9) |
0 (0) |
0 (0) |
|
Escalofríos |
14 (8) |
8 (4,4) |
0 (0) |
0 (0) |
|
Malestar |
13 (7) |
10 (6) |
0 (0) |
0 (0) |
|
Malestar de tipo gripal |
9 (5) |
7 (3,9) |
0 (0) |
0 (0) |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
14 (8) |
11 (6) |
0 (0) |
0 (0) |
|
Exploraciones complementarias |
||||
|
Aumento de la alanina aminotransferasa |
18 (10) |
15 (8) |
3 (1,7) |
1 (< 1) |
|
Disminución en recuento de glóbulos blancos |
16 (9) |
13 (7) |
5 (2,8) |
2 (1,1) |
|
Disminución en recuento de linfocitos |
12 (7) |
12 (7) |
6 (3,4) |
2 (1,1) |
|
Aumento de bilirrubina |
10 (6) |
0 (0) |
0 (0) |
0 (0) |
|
Descenso de peso |
12 (7) |
2 (1,1) |
0 (0) |
0 (0) |
|
Nota: El Sistema de clasificación de órganos y las reacciones adversas están codificados usando el diccionario MedDRA. Un paciente con manifestaciones múltiples de una reacción adversa se cuenta sólo una vez en la categoría de Sistema corportal/Reacción adversa. 1 Todos los EAs emergentes del tratamiento en al menos 5% de los pacientes en el grupo REVLIMID+rituximab y con al menos 1% de mayor frecuencia que el grupo rituximab +placebo (brazo control). 2 Todos los EAs emergentes del tratamiento de grado 3 o 4 en al menos 1% de los pacientes en el grupo REVLIMID+rituximab y con al menos 1% de mayor frecuencia que el grupo rituximab +placebo (brazo control). 3 Todos los EAs graves emergentes del tratamiento en al menos 1% de los pacientes en el grupo REVLIMID+rituximab y con al menos 1% de mayor frecuencia que el grupo rituximab +placebo (brazo control). $-Reacciones adversas graves informadas. @ - Reacciones adversas en los que al menos una provocó un resultado fatal % - Reacciones adversas en los que al menos una se consideró con riesgo de vida (si el resultado fue la muerte, se incluyó con casos de muerte) * Reacciones adversas para términos de RAM (Reacciones Adversas Medicamentosas) combinados (basados en TEAE PTs relevantes [según MedDRA v 21.0]): a El término combinado “Eventos tromboembólicos” incluye los siguientes TPs: embolia pulmonar, trombosis venosa profunda, accidente cerebrovascular, embolia y trombosis. b El término combinado de EA “Tos” incluye los siguientes TPs: tos y tos productiva. c El término combinado de EA “Dolor abdominal” incluye los siguientes TPs: dolor abdominal y dolor abdominal superior d El término combinado de EA “Erupción cutánea” incluye los siguientes TPs: erupción cutánea maculopapular, erupción cutánea eritematosa, erupción cutánea papular, erupción cutánea pruriginosa, erupción cutánea generalizada. e El término combinado de EA “Prurito” incluye los siguientes TPs: prurito, prurito generalizado, erupción cutánea pruriginosa y prurito alérgico. |
||||
Experiencia poscomercialización
Las siguientes reacciones adversas han sido identificadas a partir de experiencia poscomercialización a escala mundial con REVLIMID. Debido a que una población de tamaño incierto reporta voluntariamente estas reacciones , no siempre es posible calcular la frecuencia de manera confiable o establecer una relación causal con la exposición al fármaco.
Trastornos endocrinos: Hipotiroidismo, hipertiroidismo.Trastornos hepatobiliares: Insuficiencia hepática (incluyendo fatalidad), hepatitis tóxica, hepatitis citolítica, hepatitis colestásica, hepatitis citolítica / colestática mixta, pruebas de laboratorio hepáticas, transitorias anormales.Trastornos del sistema inmunológico: Angioedema, enfermedad aguda de injerto contra el huésped
(trasplante hematopoyético alogénico); rechazo de trasplante de órgano sólido.
Infecciones e infestaciones: Reactivación viral (como el virus de la hepatitis B y el herpes zóster [culebrilla]). Se han reportado casos de leucoencefalopatía progresiva multifocal.
Neoplasias benignas, malignas y no especificadas (incluidos los quistes y los pólipos): Síndrome de lisis tumoral, reacción de exacerbación tumoral. Trastornos respiratorios, torácicos y mediastínicos: neumonitis.
Trastornos de la piel y del tejido subcutáneo: Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS).
Para más información, llame al Departamento Científico: Teléfono PBX 5005005, E.mail: farmacovigilancia@medicamenta.com.ec
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones medicamentosas
La coadministración de una dosis individual o múltiple de dexametasona (40 mg) no tuvo un efecto clínico en la farmacocinética de dosis múltiple de REVLIMID (25 mg).
La coadministración de REVLIMID (25 mg) después de dosis múltiples de un inhibidor de la P-gp tal como quinidina (600 mg dos veces/día) no aumentó significativamente la Cmáx o la AUC de lenalidomida.
La coadministración del inhibidor de la P-gp y del sustrato temsirolimus (25 mg) con REVLIMID (25 mg) no alteró significativamente la farmacocinética de lenalidomida, temsirolimus o sirolimus (metabolito del temsirolimus).Estudios in vitro demostraron que REVLIMID es un sustrato de la P-glicoproteína (P-gp). REVLIMID no es un sustrato de la proteína de resistencia del cáncer de mama humano (BCRP), de los transportadores de proteína de Resistencia a Múltiples Fármacos (MRP) MRP1, MRP2 o MRP3, de transportadores de anión orgánico (OAT) OAT1 y OAT3, polipéptido 1B1 de transporte de anión orgánico (OATP1B1), transportadores de catión orgánico (OCT) OCT1 y OCT2, proteína de toxina de extrusión y múltiples fármacos (MATE) MATE1, y transportadores nuevos de catión orgánico (OCTN) OCTN1 y OCTN2. Lenalidomida no es un inhibidor de la bomba exportadora de P-GP, bomba exportadora de sales biliares (BSEP), BCRP, MRP2, OAT1, OAT3, OATP1B1, OATP1B3, u OCT2. Lenalidomida no inhibe o induce las isoenzimas CYP450. La lenalidomida tampoco inhibe la formación de la glucuronidación de bilirrubina en los microsomas de hígado humano con UGT1A1 genotipado como UGT1A1*1/*1, UGT1A1*1/*28 y UGT1A1*28/*28.
Digoxina
Al co-administrar digoxina con dosis múltiples de REVLIMID (10 mg/día), la Cmáx y el AUC0-∞ de digoxina se incrementaron en un 14%. Durante la administración de REVLIMID, se recomienda el monitoreo periódico de los niveles plasmáticos de digoxina de acuerdo con el criterio clínico y sobre la base de la práctica clínica estándar en pacientes que reciben este medicamento.
Terapias concomitantes que pueden incrementar el riesgo de trombosis
Los agentes eritropoyéticos u otros agentes que pueden incrementar el riesgo de trombosis, como terapias que contienen estrógenos, deberían utilizarse con precaución luego de hacer una evaluación de riesgo-beneficio en pacientes con mieloma múltiple que reciben REVLIMID.
Warfarina
La co-administración de dosis múltiples de REVLIMID (10 mg/día) con una dosis única de warfarina (25 mg) no tuvo efecto sobre la farmacocinética de lenalidomida y R- y S-warfarina. Se observaron cambios esperados en las evaluaciones de laboratorio de tiempo de protrombina (PT, Prothrombin Time) y rango internacional normalizado (INR, International Normalized Ratio) luego de la administración de warfarina, pero estos cambios no se vieron afectados por la administración concomitante de REVLIMID. Se desconoce si hay interacción entre dexametasona y warfarina. En pacientes con MM que reciben warfarina en forma concomitante, se recomienda controlar el PT y el INR.
INFORMACIÓN COMPLEMENTARIA:
Fecha de última revisión del texto de referencia: Mayo 2019.
Fecha última revisión local: Abril 2020. Elaborado por: CELGENE INTERNATIONAL SARL (Route de Perreux I, 2017, Boudry, Suiza)
Acondicionado en Uruguay por: ADIUM PHARMA S.A, Ruta 8, Km 17500 Zonamérica, Montevideo, República Oriental del Uruguay.
Distribuído por: MEDICAMENTA ECUATORIANA S.A. QUITO – ECUADOR
MEDICAMENTA
Casilla 17-21-027 Quito, Ecuador
RECOMENDACIONES:
Advertencias y precauciones
Toxicidad embrionaria y fetal: REVLIMID es un análogo de talidomida y está contraindicado su uso durante el embarazo. Talidomida es un conocido teratógeno humano que provoca defectos congénitos que pueden poner en peligro la vida. Un estudio de desarrollo embriofetal en monos indica que lenalidomida produjo malformaciones en la cría de monas que recibieron el medicamento durante el embarazo, lo cual se asemeja a los defectos congénitos observados en humanos tras la exposición a talidomida durante el embarazo.
REVLIMID solo está disponible por medio del Programa de Minimizacion de Riesgo.
Pacientes femeninas en edad reproductiva: Las mujeres en edad reproductiva deben evitar el embarazo por al menos 4 semanas antes de comenzar la terapia con REVLIMID, durante la terapia, durante las interrupciones de la dosis y por 4 semanas luego de completar el tratamiento.
Las pacientes femeninas deben comprometerse a mantener una abstinencia sexual heterosexual o a usar dos métodos anticonceptivos confiables,a partir de 4 semanas antes de iniciar el tratamiento con REVLIMID, durante la terapia, durante las interrupciones de dosis y continuar por 4 semanas luego de la discontinuación de la terapia con REVLIMID.
Se debe obtener una prueba de embarazo negativa antes de iniciar el tratamiento. La prueba de embarazo debe realizarse dentro de las 72 horas previas a la prescripción de la terapia de REVLIMID, y luego en forma mensual en mujeres con ciclos menstruales regulares o cada 2 semanas en mujeres con ciclos menstruales irregulares [Ver Uso en poblaciones específicas].
Pacientes masculinos: Lenalidomida está presente en el semen de pacientes que reciben el fármaco. Por tal motivo, los pacientes masculinos siempre deben usar preservativo de látex o sintético durante cualquier contacto sexual con mujeres en edad reproductiva mientras reciben REVLIMID y por hasta 4 semanas después de discontinuar REVLIMID, incluso si se han sometido a una vasectomía exitosa. Los pacientes masculinos que toman REVLIMID no deben donar esperma [Ver Uso en poblaciones específicas]. Donación de sangre: Los pacientes no deben donar sangre durante el tratamiento con REVLIMID y durante 4 semanas tras la discontinuación del medicamento ya que la sangre podría darse a una paciente embarazada cuyo feto no debe exponerse a REVLIMID.
Riesgo reproductivo y requisitos especiales de prescripción
(Programa de Minimización de Riesgo)
Debido al riesgo embrionario y fetal [Ver Uso en poblaciones específicas], REVLIMID sólo está disponible por medio de un programa restringido bajo un Plan de Minimización de Riesgo.
Los componentes requeridos del Programa son los siguientes:
• Los médicos deben estar registrados por el programa al inscribirse y cumplir con los requerimientos.
• Los pacientes deben firmar un consentimiento informado y cumplir con los requerimientos. En particular, las pacientes en edad reproductiva que no están embarazadas deben cumplir con los requerimientos en relación con las pruebas de embarazo y los métodos anticonceptivos.
Solo debe entregase REVLIMID a pacientes que están autorizados para recibir este producto y cumplen con los requerimientos del Programa de Minimización de Riesgo.Para mayor información acerca del programa, comunicarse con MEDICAMENTA ECUATORIANA S.A., Quito. PBX 5005005 . E-mail: farmacovigilancia@medicamenta.com.ec
Si se produce un embarazo durante el tratamiento, debe suspenderse de inmediato el tratamiento con REVLIMID.Toda sospecha de exposición del feto a REVLIMID debe ser informada a MEDICAMENTA ECUATORIANA S.A., Edificio Medicamenta Ecuatoriana S.A., Quito. PBX 5005005 . E-mail: farmacovigilancia@medicamenta.com.ec
Toxicidad hematológica
REVLIMID puede causar neutropenia y trombocitopenia significativas.
Controlar a los pacientes con neutropenia para detectar signos de infección. Indicar a los pacientes que observen si hay sangrado o hematomas, particularmente, con el uso de medicamentos concomitantes que pueden aumentar el riesgo de sangrado. Los pacientes que toman REVLIMID deben realizarse recuentos sanguíneos completos de forma periódica según se describe a continuación.
Los pacientes que toman REVLIMID en combinación con dexametasona o como terapia de mantenimiento de REVLIMID para MM deben realizarse recuentos sanguíneos completos cada 7 días (semanalmente) por los primeros 2 ciclos, en los días 1 y 15 del ciclo 3, y cada 28 días (4 semanas) de ahí en adelante. Puede ser necesaria una interrupción y/o reducción de dosis. En estudios clínicos sobre terapia de mantenimiento para MM, se informó neutropenia grado 3 o 4 en hasta el 59% de pacientes tratados con REVLIMID y trombocitopenia grado 3 o 4 en hasta el 38% de los pacientes tratados con REVLIMID. En pacientes con MM no tratados previamente que son elegibles para trasplante, que toman Revlimid en combinación con bortezomib y dexametasona, se debe evaluar el recuento sanguíneo completo cada 7 días (semanalmente) durante el primer ciclo, posteriormente antes del comienzo de cada ciclo subsiguiente. Con el uso continuado de Revlimid en combinación con dexametasona, monitoree mensualmente (cada 4 semanas).
Los pacientes que toman REVLIMID para el Síndrome Mielodisplásico (SMD) deben realizarse recuentos sanguíneos completos semanalmente durante las primeras 8 semanas y por lo menos mensualmente de ahí en adelante. Los pacientes que toman REVLIMID para mieloma múltiple deben realizarse recuentos sanguíneos completos cada dos semanas durante las primeras 12 semanas y luego mensualmente de ahí en adelante. Se observó toxicidad hematológica grado 3 o 4 en el 80% de los pacientes incluidos en el estudio sobre síndrome mielodisplásico. En el 48% de los pacientes que desarrollaron neutropenia grado 3 o 4, la mediana de tiempo hasta la manifestación fue de 42 días (rango, 14-411 días) y la mediana de tiempo hasta la recuperación documentada fue de 17 días (rango, 2-170 días). En el 54% de los pacientes que desarrollaron trombocitopenia grado 3 o 4, la mediana de tiempo hasta la manifestación fue de 28 días (rango, 8-290 días) y la mediana de tiempo hasta la recuperación documentada fue de 22 días (rango, 5-224 días).
Los pacientes que toman REVLIMID para LCM deben realizarse recuentos sanguíneos completos semanalmente durante el primer ciclo (28 días), cada 2 semanas durante los ciclos 2-4, y luego mensualmente de ahí en adelante. Los pacientes pueden requerir la interrupción de la dosis y/o la reducción de la dosis.
En el ensayo de LCM, se informó neutropenia grado 3 y 4 en el 43 % de los pacientes. Se informó trombocitopenia grado 3 o 4 en el 28 % de los pacientes.
Los pacientes que toman REVLIMID para el tratamento de LF o LZM deben realizarse recuentos sanguíneos completos semanalmente durante las primeras 3 semanas del ciclo 1 (28 días) y cada 2 semanas durante los ciclos 2-4, y luego mensualmente de ahí en adelante. Los pacientes pueden requerir la interrupción de la dosis y/o la reducción de la dosis. En los estudios AUGMENT y MAGNIFY se informó neutropenia grado 3 y 4 en el 50 % y en el 33% de los pacientes, respectivamente, del grupo de pacientes REVLIMID/rituximab.
Trombocitopenia grado 3 o 4 fue informado en el 2% y 8% de los pacientes, respectivamente, del grupo de pacientes REVLIMID/rituximab (ver Reacciones adversas).
Tromboembolia venosa y arterial
Los eventos tromboembólicos venosos [ETV (TVP y TEP)], y eventos tromboembólicos arteriales (TEA, infarto de miocardio y accidente cerebrovascular)aumentan en pacientes tratados con REVLIMID. Un riesgo significativamente aumentado de trombosis venosa profunda (7,4%) y de embolia pulmonar (3,7%) surgió en pacientes con MM, después de por lo menos una terapia previa, que fueron tratados con REVLIMID y terapia de dexametasona en comparación con pacientes tratados en el grupo con placebo y dexametasona (3,1% y 0,9%) en ensayos clínicos con uso variable de terapias anticoagulantes. En el estudio de mieloma múltiple recientemente diagnosticado (NDMM) en el cual casi todos los pacientes recibieron profilaxis antitrombótica, se informó TVP como reacción adversa seria (3,6%; 2,0% y 1,7%) en las ramas Rd Continuo, Rd18 y MPT, respectivamente. La frecuencia de reacciones adversas serias de EP fue similar entre las ramas Rd Continuo, Rd18 y MPT (3,8%; 2,8% y 3,7%, respectivamente).
El infarto de miocardio (1,7%) y el Accidente Cerebrovascular ACV (2,3%) aumentan en pacientes con MM después de por lo menos una terapia previa en la que fueron tratados con REVLIMID y dexametasona en comparación con pacientes tratados con placebo y dexametasona (0,6% y 0,9%) en los estudios clínicos. En el estudio NDMM, se informó infarto de miocardio (incluido el agudo) como una reacción adversa seria (2,3%; 0,6% y 1,1%) en las ramas Rd Continuo, Rd18 y MPT, respectivamente. La frecuencia de reacciones adversas serias de ACV fue similar entre las ramas Rd Continuo, Rd18 y MPT (0,8%; 0,6% y 0,6%, respectivamente). Los pacientes con factores de riesgo conocidos, incluyendo trombosis previas, pueden estar en un riesgo mayor y se deben tomar acciones para tratar de minimizar todos los factores modificables (por ejemplo, hiperlipidemias, hipertensión, tabaquismo).
En ensayos clínicos controlados que no utilizaron tromboprofilaxis concomitante, el 21,5% de los eventos trombóticos globales (eventos embólicos y trombóticos de consulta estándar en el Diccionario Médico para Actividades Regulatorias) ocurrió en pacientes con MM refractario y en recaída que fueron tratados con REVLIMID y dexametasona en comparación con 8,3% de trombosis en pacientes tratados con placebo y dexametasona. La mediana de tiempo hasta el primer evento de trombosis fue 2,8 meses. En el estudio NDMM, en el cual casi todos los pacientes recibieron profilaxis antitrombótica, la frecuencia global de eventos trombóticos fue del 17,4% en pacientes en las ramas Rd Continuo y Rd18 combinadas, y del 11,6% en la rama MPT. La mediana del tiempo hasta el primer evento de trombosis fue de 4,3 meses en las ramas Rd Continuo y Rd18 combinadas. Se recomienda tromboprofilaxis. El régimen de tromboprofilaxis se debe hacer sobre la base de una evaluación de los riesgos subyacentes del paciente. Instruya a los pacientes para que informen de inmediato cualquier signo o síntoma que podría sugerir eventos trombóticos. Los Agentes Estimuladores de la Eritropoyesis (ESA) y los estrógenos podrían, además, aumentar el riesgo de trombosis y su uso se debe basar en una decisión sobre el riesgo-beneficio en pacientes que reciben REVLIMID.
En el estudio AUGMENT, la incidencia de ETV (incluyendo TVP y TEP) en pacientes con LF o LZM fue de 3,4% en el grupo de pacientes tratados con REVLIMID/ rituximab (ver Reacciones adversas).
En el estudio AUGMENT, la incidencia de TEA (incluyendo IM) en pacientes con LF o LZM fue de 0,6% en el grupo de pacientes tratados con REVLIMID/ rituximab (ver Reacciones adversas).
Se recomienda tromboprofilaxis. El régimen de tromboprofilaxis se debe hacer sobre la base de una evaluación de los riesgos subyacentes del paciente. Instruya a los pacientes para que informen de inmediato cualquier signo o síntoma que podría sugerir eventos trombóticos. Los Agentes Estimuladores de la Eritropoyesis (ESA) y los estrógenos podrían, además, aumentar el riesgo de trombosis y su uso se debe basar en una decisión sobre el riesgo-beneficio en pacientes que reciben REVLIMID.
Aumento de mortalidad en pacientes con LLC En un ensayo clínico prospectivo aleatorizado (1:1) en el tratamiento de primera línea de pacientes con leucemia linfocítica crónica, el tratamiento con el agente único REVLIMID aumentó el riesgo de muerte en comparación con el agente único clorambucilo. En un análisis intermedio, hubo 34 muertes entre 210 pacientes en la rama de tratamiento con REVLIMID en comparación con 18 muertes entre 211 pacientes en la rama de tratamiento con clorambucilo, y el hazard ratio para la supervivencia global fue de 1,92 [IC del 95%: 1,08 – 3,41], consistente con un aumento del 92% en el riesgo de muerte. El ensayo se suspendió por cuestiones de seguridad en julio de 2013.Las reacciones adversas cardiovasculares serias, incluidos fibrilación auricular, infarto de miocardio y falla cardíaca, ocurrieron más frecuentemente en la rama de tratamiento de REVLIMID. REVLIMID no está indicado ni tampoco recomendado para LLC fuera de los ensayos clínicos controlados.
Segundas neoplasias primarias
En ensayos clínicos en pacientes con MM que recibían REVLIMID, se observó un aumento de segundas neoplasias primarias hematológicas más tumor sólido (SPM, por sus siglas en inglés), particularmente LMA y SMD. El aumento SPM hematológicas, incluyendo de casos de LMA y SMD se produjo en 5,3% de los pacientes con NDMM que recibían REVLIMID en combinación con melfalán oral, en comparación con 1,3% de pacientes que recibían melfalán sin REVLIMID. La frecuencia de casos de LMA y SMD en pacientes con NDMM tratados con REVLIMID en combinación con dexamtasona sin melfalán se observó en 0,4% de los casos.
En pacientes que recibieron terapia de mantenimiento con REVLIMID seguida de dosis alta de melfalán intravenoso y auto-HSCT, se produjeron SPM hematológicas en 7,5 % de los casos, en comparación con 3,3 % de pacientes que recibieron placebo. La incidencia de SPM de tumores hematológicos además de tumores sólidos (excluyendo carcinoma de células escamosas y carcinoma de células basales) fue de 14,9 % en comparación con 8,8 % en pacientes que recibieron placebo con un seguimiento promedio de 91,5 meses. La SPM de cáncer de piel no melanoma, incluyendo carcinoma de células escamosas y carcinoma de células basales se produjo en 3,9 % de los pacientes que recibían terapia de mantenimiento con REVLIMID, en comparación con 2,6 % de la rama placebo.En pacientes con MM recurrente o refractario tratados con REVLIMID/dexametasona, la incidencia de SPM de tumores hematológicos, además de tumores sólidos (excluyendo carcinoma de células escamosas y carcinoma de células basales) fue de 2,3 % versus 0,6 % en la rama de dexametasona sola. La SPM de cáncer de piel no melanoma, incluyendo carcinoma de células escamosas y carcinoma de células basales se produjo en 3,1 % de los pacientes que recibían terapia de mantenimiento con REVLIMID/dexametasona, en comparación con 0,6 % de la rama dexametasona sola.
Los pacientes que recibieron terapia con REVLIMID hasta la progresión de la enfermedad no mostraron una mayor incidencia de segundas neoplasias primarias invasivas que los pacientes tratados en las ramas con REVLIMID de duración fija. Seguir de cerca a los pacientes a fin de detectar el desarrollo de segundas neoplasias primarias. Tener en cuenta tanto el posible beneficio de REVLIMID como el riesgo de segundas neoplasias primarias cuando se considera el tratamiento con REVLIMID.
En pacientes con mieloma múltiple recién diagnosticado que recibieron lenalidomida en combinación con bortezomib y dexametasona, la frecuencia de SPM hematológica fue de 0,0% a 0,8% y la frecuencia de SPMs de tumores sólidos fue del 0,4% al 4,5%.
En el ensayo clínico AUGMENT en pacientes con LF o LZM que recibían REVLIMID/rituximab, se observó SPM hematológicas más tumor sólido, particularmente LMA.En el ensayo clínico AUGMENT las SPM hematológicas de LMA ocurrió en el 0,6% de los pacientes con LF o LZM que recibían tratamiento con REVLIMID/rituximab.
La incidencia de las SPMs hematológicas de tumor sólido (excluyendo cánceres de piel no melanoma) fue de 1,7% en el grupo de RIVLIMID/rituximab con seguimiento promedio de 29,8 meses (rango de 0,5 a 51,3 meses)(ver Reacciones adversas).
Seguir de cerca a los pacientes a fin de detectar el desarrollo de segundas neoplasias primarias. Tener en cuenta tanto el posible beneficio de REVLIMID como el riesgo de segundas neoplasias primarias cuando se considera el tratamiento con REVLIMID.
Aumento de la mortalidad en pacientes con MM cuando se agrega Pembrolizumab a un análogo de talidomida y dexametasona
En dos ensayos clínicos aleatorizados en pacientes con MM, la adición de pembrolizumab a un análogo de la talidomida más dexametasona, un uso para el que no está indicado el anticuerpo bloqueante PD-1 o PD L1, dio como resultado un aumento de la mortalidad. El tratamiento de pacientes con MM con un anticuerpo bloqueante PD-1 o PD-L1 en combinación con un análogo de talidomida más dexametasona no se recomienda fuera del ensayo clínico controlado.
Hepatotoxicidad
Se han presentado casos de insuficiencia hepática, incluyendo casos fatales, en los pacientes tratados con lenalidomida en combinación con dexametasona. En estudios clínicos el 15 % de los pacientes experimentó hepatotoxicidad (con características hepatocelulares, colestáticas y mixtas); 2 % de los pacientes con MM y el 1 % de los pacientes con Mielodisplasia tuvieron eventos serios de hepatotoxicidad. Se desconoce el mecanismo de la hepatotoxicidad inducida por el medicamento. Entre los posibles factores de riesgo destacan las enfermedades virales hepáticas preexistentes, los elevados niveles iniciales de enzimas hepáticas y las medicaciones concomitantes. Se debe realizar un control periódico de las enzimas hepáticas e interrumpir la administración de Revlimid si se detecta elevación de dichas enzimas. Una vez alcanzados nuevamente los valores iniciales, se puede considerar la reanudación del tratamiento con una dosis más baja.
Lenalidomida se excreta por los riñones. Es importante ajustar la dosis a los pacientes con insuficiencia renal para evitar niveles plasmáticos que pueden aumentar el riesgo de efectos secundarios hematológicos o hepatotoxicidad más elevados. Se recomienda el control de la función hepática, especialmente cuando existe un historial de infección viral hepática o concurrente o cuando la lenalidomida se combina con medicamentos que se sabe están asociados con disfunción hepática.
Reacciones cutáneas severas que incluyen reacciones de hipersensibilidad
Se informaron angioedema y reacciones cutáneas severas, incluso síndrome de Stevens-Johnson necrólisis epidérmica tóxica (TEN) y reacción farmacológica con eosinofilia y síntomas sistémicos (DRESS). DRESS puede presentarse con una reacción cutánea (como erupción cutánea o dermatitis exfoliativa), eosinofilia, fiebre y / o linfadenopatía con complicaciones sistémicas como hepatitis, nefritis, neumonitis, miocarditis y / o pericarditis. Estos eventos pueden ser fatales. Los pacientes con antecedentes de erupción cutánea grado 4 asociada con el tratamiento con talidomida no deben recibir REVLIMID. Debe considerarse la interrupción o suspensión de REVLIMID por erupción cutánea grado 2-3. REVLIMID debe suspenderse en caso de angioedema, erupción cutánea grado 4, erupción cutánea exfoliativa o bullosa o si hay sospechas de síndrome de Stevens-Johnson, TEN o DRESS y no debe reanudarse después de la suspensión debido a estas reacciones.
Síndrome de lisis tumoral
Se informaron instancias fatales de síndrome de lisis tumoral durante el tratamiento con lenalidomida. Los pacientes en riesgo de síndrome de lisis tumoral son aquellos con alta carga tumoral antes del tratamiento. Estos pacientes deben seguirse de cerca y se deben tomar las precauciones debidas.
Reacción de llamarada en el tumor
Se produjo la reacción de llamarada en el tumor durante el uso de lenalidomida en la fase de investigación para tratar la Leucemia Linfocítica Crónica (CCL, Chronic Lymphocytic Leukemia) y linfoma, y se caracteriza por una inflamación de los ganglios linfáticos, febrícula, dolor y erupción cutánea. REVLIMID no está indicado ni tampoco recomendado para LLC fuera de los ensayos clínicos controlados.
Se recomienda el monitoreo y la evaluación de las reacciones de llamarada en el tumor en pacientes con LCM. La reacción de llamarada en el tumor puede imitar la Progresión de la Enfermedad (PE). En el ensayo de LCM, 13/134 (10%) de los pacientes experimentaron reacciones de llamarada; todos los casos notificados fueron de grado 1 o 2. Todos los episodios tuvieron lugar en el ciclo 1 y un paciente desarrolló reacciones de llamarada nuevamente en el ciclo 11. Se puede continuar la administración de lenalidomida en pacientes con reacciones de llamarada de grados 1 y 2 sin interrupción o modificación, según el criterio del médico. Los pacientes con reacciones de llamarada de grado 1 o 2 también pueden recibir tratamiento con corticoides, Antiinflamatorios No Esteroides (AINE) y/o analgésicos narcóticos para el manejo de los síntomas de la reacción de llamarada. En los pacientes con reacción de llamarada grado 3 o 4, se recomienda suspender el tratamiento con lenalidomida hasta que se resuelva la reacción de llamarada a ≤ grado 1. Los pacientes con reacciones de llamarada de grado 3 o 4 pueden recibir tratamiento para el manejo de los síntomas según las directrices para el tratamiento de las reacciones de llamarada de grados 1 y 2.
Alteración de la movilización de células madre Se informó una disminución de la cantidad de células CD34+ recolectadas después del tratamiento (> 4 ciclos) con REVLIMID. En pacientes que son candidatos al auto-HSCT, la derivación a un centro de transplantes se debe realizar al principio del tratamiento para optimizar el momento de la recolección de células madre. En pacientes que recibieron más de 4 ciclos de un tratamiento de REVLIMID o para quienes se ha recolectado una cantidad inadecuada de células CD34+ con G-CSF solo, se puede considerar G-CSF con ciclofosfamida o la combinación de G-CSF con un inhibidor CXCR4.
Trastornos tiroideos
Se han reportado hipotiroidismo e hipertiroidismo. Medir la función tiroidea antes del inicio del tratamiento con REVLIMID y durante la terapia.
Mortalidad prematura en pacientes con LCM
En otro estudio LCM, hubo un aumento en las muertes prematuras (dentro de las 20 semanas), 12,9% en el grupo REVLIMID versus 7,1% en el grupo control. En el análisis exploratorio multivariable, los factores de riesgo de muerte prematura incluyen una carga tumoral alta, una puntuación MIPI en el momento del diagnóstico y un recuento alto de leucocitos (WBC) al inicio (≥ 10 x 109 / l). Neuropatía periférica: [Ver Reacciones adversas]. Electrofisiología cardíaca
Se ha observado la prolongación del intervalo QTc en el ECG durante el tratamiento con lenalidomida. El tratamiento concomitante con drogas que prolongan el intervalo QT y el tratamiento de pacientes con síndrome de QT largo solo deben realizarse con gran precaución y con un control de ECG regular (consulte Propiedades / Efectos).
Efecto inmunosupresor
Lenalidomida tiene un fuerte efecto inmunosupresor. Por lo tanto, el tratamiento concomitante con otros agentes inmunomoduladores debe realizarse solo con precaución. El efecto de las vacunas puede verse afectado. No deben administrarse vacunas con organismos vivos durante el tratamiento con lenalidomida debido al riesgo de infección.
Intolerancia a la lactosa
Las cápsulas de Revlimid contienen lactosa. Los pacientes con una rara intolerancia hereditaria a la galactosa, deficiencia de lactasa de Lapp o malabsorción de glucosa-galactosa no deben tomar este medicamento.
Terapia combinada
Para otros medicamentos administrados en combinación con lenalidomida, consulte la información de prescripción correspondiente al producto.
Fertilidad, embarazo y lactancia
Registro de exposición
Embarazo
Hay un registro de exposición del embarazo que supervisa los resultados del embarazo en las mujeres expuestas a REVLIMID durante el embarazo, así como las parejas femeninas de los pacientes varones que están expuestos a REVLIMID. Este registro también se utiliza para entender la causa raíz para el embarazo.
Resumen de riesgo
Basándose en el mecanismo de acción y hallazgos de estudios en animales [ver Datos en animales], REVLIMID puede causar daño embrionario y fetal cuando se administra a una mujer embarazada y está contraindicado durante el embarazo. REVLIMID es un análogo de la talidomida.
Talidomida es un teratógeno humano, que induce una frecuencia alta de daños congénitos severos y con riesgo de vida, tales como amelia (ausencia de miembros), focomelia (miembros cortos), hipoplasia de los huesos, ausencia de huesos, anormalidades externas del oído (incluidas anotía, micropinna, canales auditivos externos pequeños o ausentes), parálisis facial, anormalidades del ojo (anoftalmía, microftalmía) y defectos cardíacos congénitos. También se han documentado malformaciones del tracto alimentario, urinario y de los genitales, y se ha reportado mortalidad en el nacimiento o inmediatamente después en aproximadamente, el 40 % de los recién nacidos.
Lenalidomida provocó defectos en las extremidades similares a los provocados por talidomida en las crías de monas. La lenalidomida cruzó la placenta después de la administración a conejas embarazadas y ratas preñadas (ver Datos en animales). Si este fármaco se utiliza durante el embarazo o si la paciente queda embarazada mientras toma este fármaco, la paciente debe estar advertida respecto del posible riesgo para el feto.
Si el embarazo se produce durante el tratamiento, suspender el fármaco inmediatamente. Bajo estas condiciones, derive el paciente a un obstetra/ginecólogo experto en toxicidad reproductiva para mayor evaluación y asesoramiento.
Cualquier sospecha de exposición fetal a REVLIMID debe informarse a la ARCSA y al Laboratorio MEDICAMENTA al teléfono: 02-500 5005 y /o por e-mail: farmacovigilancia@medicamenta.com.ec Se desconoce el riesgo de fondo estimado de los principales defectos congénitos y abortos espontáneos para la población indicada. El riesgo de fondo estimado en la población general de los Estados Unidos de los principales defectos de nacimiento es del 2% -4% y de aborto espontáneo es del 15%-20% de los embarazos clínicamente reconocidos.
Datos en animales
En un estudio de toxicidad del desarrollo embriofetal en monos, se produjo teratogenicidad, incluso defectos en las extremidades similares a los causados por talidomida, en la cría cuando monas embarazadas recibieron lenalidomida oral durante la organogénesis. La exposición (AUC) en monos en la dosis más baja fue de 0,17 veces la exposición a la máxima dosis recomendada en humanos (MRHD, Maximum Recommended Human Dose, por sus siglas en inglés) de 25 mg. Estudios similares en conejas y ratas embarazadas a 20 veces y 200 veces la dosis Máxima Recomendada en Humanos (MRHD), respectivamente, evidenciaron embrioletalidad en conejas y ningún efecto adverso relacionado con la reproducción en ratas. En un estudio de desarrollo pre- y posnatal en ratas, los animales recibieron lenalidomida desde la organogénesis hasta la lactancia. El estudio reveló algunos efectos adversos en las crías de las ratas hembra tratadas con lenalidomida en dosis de hasta 500 mg/kg (aproximadamente, 200 veces la dosis humana de 25 mg.sobre la base del área de superficie corporal). Las crías macho exhibieron un leve retraso en la madurez sexual y las crías hembra tuvieron aumentos de peso corporal levemente más bajos durante la gestación que las crías macho. Al igual que con talidomida, es posible que el modelo en ratas no aborde, de manera adecuada, todo el espectro de posibles efectos de lenalidomida sobre el desarrollo embriofetal en humanos.
Después de la administración oral diaria de lenalidomida desde el día 7 de la gestación hasta el día 20 de la gestación en conejas embarazadas, las concentraciones de lenalidomida en plasma fetal fueron de aproximadamente, el 20-40% de la Cmáx materna. Después de una sola dosis oral a ratas embarazadas, se detectó lenalidomida en el plasma fetal y tejidos; las concentraciones de radiactividad en los tejidos fetales fueron generalmente, inferiores a las de los tejidos maternos. Estos datos indicaron que la lenalidomida atravesaba la placenta.
Mujeres y varones de potencial reproductivo
Pruebas de embarazo
REVLIMID puede causar daño fetal cuando se administra durante el embarazo (ver Uso en poblaciones específicas). Verifique el estado del embarazo de las mujeres con potencial reproductivo antes de iniciar el tratamiento con REVLIMID y durante el tratamiento. Aconseje a las mujeres con potencial reproductivo que deben evitar el embarazo 4 semanas antes del tratamiento, mientras están tomando REVLIMID, durante las interrupciones de la dosis y durante al menos 4 semanas después de completar el tratamiento.Las mujeres con potencial reproductivo deben tener una (1) prueba de embarazo negativa antes de iniciar el tratamiento. La prueba de embarazo debe realizarse dentro de las 72 horas previas a la prescripción de la terapia de REVLIMID, y luego en forma mensual en mujeres con ciclos menstruales regulares o cada 2 semanas en mujeres con ciclos menstruales irregulares [Ver Uso en poblaciones específicas].
Una vez que el tratamiento ha comenzado y durante las interrupciones de la dosis, las pruebas de embarazo deben repetirse cada 4 semanas en mujeres con ciclos menstruales regulares. Si los ciclos menstruales son irregulares, la prueba de embarazo debe realizarse cada 2 semanas. Se debe realizar la prueba de embarazo y brindar asesoramiento si una paciente no tiene su período o si hay alguna anormalidad en su sangrado menstrual. El tratamiento con REVLIMID debe ser descontinuado durante esta evaluación.
Anticoncepción
Mujeres
Las mujeres con potencial reproductivo deben comprometerse a la abstención de forma continua de tener relaciones sexuales heterosexuales o al uso de 2 métodos de control de natalidad confiable simultáneamente: una forma altamente efectiva de anticoncepción: ligadura de trompas, DIU, hormonal (píldoras anticonceptivas, inyecciones, parches hormonales, anillos vaginales o implantes) y 1 método anticonceptivo eficaz adicional: látex masculino o preservativo sintético, diafragma o capuchón cervical. La anticoncepción debe comenzar 4 semanas antes de iniciar el tratamiento con REVLIMID, durante la terapia, durante las interrupciones de la dosis y continuar durante 4 semanas después de discontinuar el tratamiento con REVLIMID. La anticoncepción fiable está indicada incluso cuando ha habido un historial de infertilidad, a menos que sea consecuencia de una histerectomía. Las mujeres con potencial reproductivo deben ser referenciadas a un proveedor calificado de métodos anticonceptivos de ser necesario.
Varones
La lenalidomida está presente en el semen de los hombres que toman REVLIMID. Por lo tanto, los hombres deben usar siempre un condón de látex o sintético durante cualquier contacto sexual con las mujeres con potencial reproductivo mientras toman REVLIMID y hasta 4 semanas después de suspender REVLIMID, incluso si se han sometido a una vasectomía exitosa. Los pacientes varones que toman REVLIMID no deben donar esperma.
Lactancia
Resumen del riesgo
No hay información sobre la presencia de lenalidomida en la leche humana, los efectos de REVLIMID en el lactante amamantado, o los efectos de REVLIMID sobre la producción de leche. Debido a que muchos fármacos se excretan en la leche humana y debido a la posibilidad de reacciones adversas en lactantes producidas por REVLIMID, aconseje a las mujeres a que no amamanten durante el tratamiento con REVLIMID.
Toxicología no clínica
Carcinogénesis, mutagénesis, deterioro de la fertilidad
No se han realizado estudios de carcinogenicidad con lenalidomida.
Lenalidomida no fue mutagénica en la prueba de Ames de mutación bacteriana inversa y no indujo aberraciones cromosómicas en linfocitos de sangre periférica humana cultivados, o mutación en el locus timidina kinasa (tk) de células de linfoma de ratón L5178Y. Lenalidomida no incrementó la transformación morfológica en ensayo de embrión de hámster sirio ni indujo los micronúcleos en los eritrocitos policromáticos de médula ósea de ratas macho.
Un estudio de fertilidad y desarrollo embrionario temprano en ratas, con administración de lenalidomida hasta 500 mg/kg (aproximadamente, 200 veces la dosis humana de 25 mg sobre la base del área de superficie corporal) no produjo toxicidad parental ni efectos adversos sobre la fertilidad.
En un estudio de toxicidad del desarrollo embriofetal en monos, se produjo teratogenicidad, incluso defectos en las extremidades similares a los causados por talidomida, en la cría cuando monas embarazadas recibieron lenalidomida oral durante la organogénesis. La exposición (AUC) en monos en la dosis más baja fue de 0,17 veces la exposición a la máxima dosis recomendada en humanos (MRHD, Maximum Recommended Human Dose, por sus siglas en inglés) de 25 mg. Estudios similares en conejas y ratas embarazadas a 20 veces y 200 veces la dosis Máxima Recomendada en Humanos (MRHD) respectivamente, evidenciaron embrioletalidad en conejas y ningún efecto adverso relacionado con la reproducción en ratas. En un estudio de desarrollo pre- y posnatal en ratas, los animales recibieron lenalidomida desde la organogénesis hasta la lactancia. El estudio reveló algunos efectos adversos en las crías de las ratas hembra tratadas con lenalidomida en dosis de hasta 500 mg/kg (aproximadamente, 200 veces la dosis humana de 25 mg.sobre la base del área de superficie corporal). Las crías macho exhibieron un leve retraso en la madurez sexual y las crías hembra tuvieron aumentos de peso corporal levemente más bajos durante la gestación que las crías macho. Al igual que con talidomida, es posible que el modelo en ratas no aborde de manera adecuada, todo el espectro de posibles efectos de lenalidomida sobre el desarrollo embriofetal en humanos.
Uso en poblaciones específicas
Uso pediátrico
No se ha establecido la seguridad y eficacia en pacientes pediátricos.
Uso geriátrico
MM en combinación: En general, de los 1613 pacientes del estudio de NDMM que recibieron tratamiento en el estudio, el 94% (1521/1613) tenía 65 años de edad o más, mientras que el 35% (561/1613) tenía más de 75 años de edad. El porcentaje de pacientes mayores de 75 años fue similar entre los grupos del estudio (Rd continuo: 33%, Rd18: 34%, MPT: 33%). En general, en todos los grupos del tratamiento, la frecuencia en la mayoría de las categorías de AEs (por ejemplo, todos los AEs, AEs de grado 3/4 y AEs graves) fue más elevada en los mayores (> 75 años) que en los sujetos más jóvenes de (≤ 75 años de edad). Los AEs de grado 3 o 4 en el sistema corporal de los trastornos generales y condiciones del sitio de la administración se informaron de forma consistente a una mayor frecuencia (con una diferencia de al menos el 5%) en los sujetos mayores que en los sujetos más jóvenes en todos los grupos de tratamiento. Los TEAEs de grado 3 o 4 de los sistema corporales en las infecciones e infestaciones, los trastornos cardíacos (incluyendo insuficiencia cardíaca e insuficiencia cardíaca congestiva), los trastornos de la piel y de los tejidos subcutáneos y los trastornos renales y urinarios (incluyendo la insuficiencia renal) también se informaron ligeramente, pero consistentemente, con más frecuencia (<5% de diferencia), en sujetos mayores que en sujetos más jóvenes en todos los grupos de tratamiento. Para otros sistemas corporales (por ej., trastornos del sistema sanguíneo y linfático, infecciones e infestaciones, trastornos cardíacos y trastornos vasculares), hubo una tendencia menos consistente de la frecuencia incrementada de los AEs de grado 3/4 en sujetos mayores frente a sujetos más jóvenes en todos los grupos de tratamiento. Los AEs graves se informaron generalmente, con una frecuencia más alta en los sujetos mayores que en los sujetos más jóvenes en todos los grupos de tratamiento.
Terapia de mantenimiento para MM: En general, el 10% (106/1018) de los pacientes tenía 65 años de edad o más, mientras que ningún paciente tenía más de 75 años de edad. Los AEs de grado 3 o 4 fueron más altos en el grupo REVLIMID (más del 5% más alto) en los pacientes de 65 años de edad o más en comparación con los pacientes más jóvenes. La frecuencia de los AEs de grado 3 o 4 en los trastornos de la sangre y del sistema linfático fueron mayores en el grupo de REVLIMID (más alto del 5% más alto) en los pacientes de 65 años de edad o más en comparación con los pacientes más jóvenes. No hubo un número suficiente de pacientes de 65 años de edad o mayores en los estudios de mantenimiento de REVLIMID que experimentaran un AE grave o que discontinuaran la terapia debido a una AE para determinar si los pacientes ancianos responden de manera diferente a la seguridad, que los pacientes más jóvenes.
El MM después de por lo menos una terapia previa: De los 703 pacientes con MM que recibieron el tratamiento experimental en los estudios 1 y 2, el 45% tenían 65 años o más, mientras que el 12% de los pacientes tenían 75 años o más. El porcentaje de pacientes de 65 años o más no fue significativamente diferente entre los grupos con REVLIMID/dexametasona y placebo/dexametasona. De los 353 pacientes que recibieron REVLIMID/dexametasona, el 46% tenía 65 años o más. En ambos estudios, los pacientes >65 años de edad fueron más propensos que los pacientes ≤65 años a experimentar trombosis venosa profunda, embolia pulmonar, fibrilación auricular e insuficiencia renal luego del uso de REVLIMID. No se observaron diferencias en cuanto a la eficacia entre los pacientes mayores de 65 años de edad y los pacientes más jóvenes.
De los 148 pacientes con SMD relacionado con deleción del 5q reclutados en el estudio principal, el 38% tenían 65 años o más, mientras que el 33% tenía 75 años o más. Aunque la frecuencia global de eventos adversos (100%) fue la misma en pacientes mayores de 65 años de edad que en pacientes más jóvenes, la frecuencia de eventos adversos graves fue mayor en pacientes mayores de 65 años de edad que en pacientes más jóvenes (54% vs. 33%). Una mayor proporción de pacientes de más de 65 años de edad discontinuaron los estudios clínicos debido a eventos adversos en comparación con la proporción de pacientes más jóvenes (27% vs. 16%). No se observaron diferencias en cuanto a la eficacia entre los pacientes mayores de 65 años de edad y los pacientes más jóvenes.
De los 134 pacientes con LCM inscriptos en el ensayo de LCM, el 63 % tenía 65 años o más, mientras que el 22 % de los pacientes tenía 75 años o más. La frecuencia global de eventos adversos fue similar en pacientes de más de 65 años de edad y en pacientes más jóvenes (98 % vs. 100 %). La incidencia global de los eventos adversos de grado 3 y 4 también fue similar en estos 2 grupos de pacientes (79 % vs. 78 %, respectivamente). La frecuencia de eventos adversos fue más alta en pacientes de más de 65 años de edad que en pacientes más jóvenes (55 % vs. 41 %). No se observaron diferencias en la eficacia entre pacientes de más de 65 años de edad y pacientes más jóvenes. LF o LZM en combinación: En general, 48% (282/590) de pacientes tenían más de 65 años mientras que 14% (82/590) tenían más de 75 años. La frecuencia general de reacciones adversas fue similar en pacientes de más de 65 años y en los más jovenes en los dos estudios encuestados (98%). Las reacciones adversas de grado 3 o 4 fueron más en el brazo REVLIMID (más del 5%) en los pacientes de más de 65 años en comparación con los pacientes mas jóvenes (71 % contra 59%). La frecuencia de reacciones adversas grado 3 o 4 fue mayor en el brazo con REVLIMID (mayor del 5%) en los pacientes de más de 65 años en comparación con los pacientes mas jóvenes en las categorías trastornos del sistema circulatorio y linfático (47 % contra 40%) e infecciones e infestaciones (16% contra 11%). Las reacciones adversas graves fueron más en el brazo con REVLIMID (más del 5%) en los pacientes de más de 65 años en comparación con los pacientes mas jóvenes (37% contra 18%). La frecuencia de reacciones adversas grado 3 o 4 fue mayor en el brazo con REVLIMID (mayor del 5%) en los pacientes de más de 65 años en comparación con los pacientes mas jóvenes en la categoría infecciones e infestaciones (15% contra 6%).
Como es más probable que en los pacientes de edad avanzada disminuya la función renal, se debe tener cuidado al elegir la dosis. Controlar la función renal.
Insuficiencia renal
Ajuste la dosis inicial de REVLIMID basándose en el valor de aclaramiento de creatinina y para los pacientes en diálisis (ver Posología y administración ).
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología y administración: REVLIMID debe tomarse por vía oral a aproximadamente, la misma hora todos los días, con o sin la ingesta de alimentos. Las cápsulas de REVLIMID deben tragarse enteras con agua. Las cápsulas no deben abrirse, romperse o masticarse. Mieloma múltiple Terapia de combinación con REVLIMID La dosis inicial recomendada de REVLIMID es 25 mg una vez por día por vía oral los días 1-21 de ciclos repetidos de 28 días en combinación con dexametasona. En el caso de pacientes > 75 años de edad, puede reducirse la dosis inicial de dexametasona. El tratamiento se debe continuar hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. En los pacientes que no son elegibles para auto-HSCT el tratamiento debe continuarse hasta la progresión de enfermedad o la aparición de toxicidad inaceptable. Para pacientes que son elegibles para auto-HSCT la movilización de celulas madre hematopoyéticas debe ocurrir dentro de los cuatro ciclos de tratamiento que contengan de Revlimid (Ver Advertencias y precauciones).
Revlimid en combinación con bortezomib y dexametasona en pacientes con mieloma múltiple no tratado previamente
Tratamiento inicial: Revlimid en combinación con bortezomib y dexametasona.
El tratamiento con Revlimid en combinación con bortezomib y dexametasona no debe iniciarse si el recuento absoluto de neutrófilos (ANC) es <1000/mcL , y / o los recuentos de plaquetas son <50000/mcL.
La dosis inicial recomendada de Revlimid es de 25 mg por vía oral una vez al día, ya sea:
a) los días 1-14 de cada ciclo de 21 días, ó
b) los días 1-21 de cada ciclo de 28 días.
Bortezomib debe administrarse mediante inyección subcutánea (1,3 mg / m2 de superficie corporal) dos veces por semana en los días 1, 4, 8 y 11 de cada ciclo de 21 días o 28 días.
La dosis recomendada de dexametasona es
a) 20 mg por vía oral una vez al día en los días 1, 2, 4, 5, 8, 9, 11 y 12, ó
b) 40 mg por vía oral una vez al día en los días 1 a 4 y 9 a 12 de cada ciclo.
Se recomiendan hasta ocho ciclos de tratamiento de 21 días o seis ciclos de 28 días (24 semanas de tratamiento inicial).
Esquema de dosificación recomendado para Revlimid en combinación con bortezomib y dexametasona
|
Hasta 8 ciclos |
Día (del ciclo de 21 días) |
||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15-21 |
|
|
Revlimid (25 mg) |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
|
|
Bortezomib (1,3 mg/m2) |
• |
• |
• |
• |
|||||||||||
|
Dexametasona (20 mg) |
• |
• |
• |
• |
• |
• |
• |
• |
|||||||
o
|
Hasta 6 ciclos |
Día (del ciclo de 28 días) |
|||||||||||||||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 |
16 |
17 |
18 |
19 |
20 |
21 |
22-28 |
|
|
Revlimid (25 mg) |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
• |
|
|
Bortezomib (1,3 mg/m2) |
• |
• |
• |
• |
||||||||||||||||||
|
Dexametasona (40 mg) |
• |
• |
• |
• |
• |
• |
• |
|||||||||||||||
Ajustes de dosis por toxicidades hematológicas durante el tratamiento del MM
Se recomiendan los lineamientos de modificación de dosis, tal como se resumen en la Tabla 11 a continuación, para manejar la neutropenia o trombocitopenia grados 3 o 4 u otra toxicidad grado 3 o 4 que se considere en relación con REVLIMID.
|
Tabla 11: Ajustes de dosis por toxicidades |
|
|
Recuentos plaquetarios |
|
|
Trombocitopenia en MM |
|
|
Cuando las plaquetas: |
Acción recomendada: Días 1-21 de ciclos repetidos de 28 días |
|
Caen a < 30.000/mcL |
Interrumpir el tratamiento con REVLIMID, seguir con un hemograma completo semanal |
|
Regresan a ≥ 30.000/mcL |
Reanudar REVLIMID con una reducción de 5mg de la dosis anterior. |
|
Para cada caída posterior < 30.000/mcL |
Interrumpir el tratamiento con REVLIMID Regresan a ≥ 30.000/mcL Reanudar REVLIMID con una reducción de 5mg de la dosis anterior. No dosificar por debajo de 5 mg día por medio. |
|
Recuento Absoluto de Neutrófilos (ANC) |
|
|
Neutropenia en MM |
|
|
Cuando los neutrófilos |
Acción recomendada: Días 1-21 de ciclos repetidos de 28 días |
|
Caen a < 1000/mcL |
Interrumpir el tratamiento con REVLIMID, seguir con un CBC semanal |
|
Regresan a ≥ 1000/mcL y neutropenia es la única toxicidad |
Reanudar REVLIMID en 25 mg diarios o en la dosis inicial |
|
Regresan a ≥ 1000/mcL y hay otra toxicidad |
Reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg día por medio. Para cada caída posterior < 1000/mcL Interrumpir el tratamiento con REVLIMID Regresan a ≥ 1000/mcL. Reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg día por medio. |
Revlimid en combinación con bortezomib y dexametasona en pacientes con mieloma múltiple no tratados previamente
|
Etapas de la reducción de la dosis |
|
|
Lenalidomida |
|
|
Dosis inicial |
25 mg |
|
Nivel de dosis -1 |
20 mg |
|
Nivel de dosis -2 |
15 mg |
|
Nivel de dosis -3 |
10 mg |
|
Nivel de dosis -4 |
5 mg |
|
Nivel de dosis -5 |
5 mg día por medio |
|
Trombocitopenia |
|
|
Cuando las plaquetas |
Acción recomendada |
|
Descienden a < 30000 /mcL |
Interrumpa el tratamiento con lenalidomida y siga el CBC semanalmente. |
|
Vuelven a ≥ 50000 mc/L |
Reanudar lenalidomida a nivel de dosis -1 |
|
Por cada caída posterior por debajo de 30000/mcL |
Interrumpa el tratamiento con lenalidomida |
|
Vuelven a ≥ 50000 /mcL |
Reanude lenalidomida en el siguiente nivel de dosis más bajo. No administre por debajo de 5 mg día por medio. |
|
Neutropenia |
|
|
Cuando los neutrófilos |
Acción recomendadaa |
|
Primero descienden a < 500/mcL o neutropenia febril (fiebre ≥ 38.5°C y ANC < 1000/mcL) |
Interrumpa el tratamiento con lenalidomida y realice el CBC semanalmente |
|
Vuelven a ≥ 1000/mcL |
Reanude lenalidomida a nivel de dosis -1 |
|
Por cada caída posterior por debajo de 500/ mcL o neutropenia febril |
Interrumpa el tratamiento con lenalidomida |
|
Vuelven a ≥ 1000/mcL |
Reanude lenalidomida en el siguiente nivel de dosis más bajo. No administre por debajo de 5 mg día por medio. |
|
a A criterio del médico, si la neutropenia es la única toxicidad en cualquier nivel de dosis, agregue el factor estimulante de colonias de granulocitos (G-CSF) y mantenga el nivel de dosis de lenalidomida. |
|
Terapia de mantenimiento con REVLIMID luego del auto-HSCT
Después de realizar el auto-HSCT, iniciar el tratamiento de mantenimiento con REVLIMID luego de una adecuada recuperación hematológica (ANC ≥ 1000/mcl y/o recuento de plaquetas ≥ 75.000/mcl). La dosis inicial de REVLIMID recomendada es 10 mg diarios en forma continua (días 1-28 de ciclos repetidos de 28 días) hasta la progresión de la enfermedad o la aparición de toxicidad inaceptable. Después de 3 ciclos de terapia de mantenimiento, la dosis puede aumentarse a 15 mg diarios si es tolerada. Ajuste de dosis para toxicidades hematológicas durante el tratamiento de MM
Para manejar la neutropenia o la trombocitopenia de grado 3 o 4 u otra toxicidad de grado 3 o 4 que se considere relacionada con REVLIMID, se recomienda seguir las directrices para la modificación de dosis resumidas en la Tabla 12 que está a continuación.
|
Tabla 12: Ajustes de dosis para toxicidades hematológicas para MM |
|
|
Recuentos plaquetarios |
|
|
Trombocitopenia en MM |
|
|
Cuando las plaquetas |
Acción recomendada |
|
Caen a < 30.000/mcL |
Interrumpir el tratamiento con REVLIMID, realizar un hemograma completo semanal |
|
Regresan a ≥ 30.000/mcL |
Reanudar REVLIMID en la dosis inmediatamente inferior, en forma continua los días 1-28 de ciclos repetidos de 28 días. |
|
Si la dosis diaria es de 5 mg, |
|
|
Para una caída posterior < 30.000/mcL |
Interrumpir el tratamiento con REVLIMID. No dosificar por debajo de 5 mg diarios los días 1 a 21 del ciclo de 28 días. |
|
Regresan a ≥ 30.000/mcL |
Reanudar REVLIMID con 5 mg diarios los días 1 a 21 del ciclo de 28 días. No dosificar por debajo de 5 mg diarios los días 1 a 21 del ciclo de 28 días. |
|
Recuento Absoluto de Neutrófilos (ANC) |
|
|
Neutropenia en MM |
|
|
Cuando los neutrófilos |
Acción recomendada |
|
Caen a < 500/mcL |
Interrumpir el tratamiento con REVLIMID, hacer un hemograma completo semanal |
|
Regresan a ≥ 500/mcL |
Reanudar REVLIMID con la dosis inmediatamente inferior, en forma continua los días 1-28 de ciclos repetidos de 28 días. |
|
Si la dosis diaria es de 5 mg |
|
|
Para cada caída posterior < 1000/mcL |
Interrumpir el tratamiento con REVLIMID. No dosificar por debajo de 5 mg diarios los días 1 a 21 del ciclo de 28 días. |
|
Regresan a ≥ 500/mcL |
Reanudar REVLIMID con 5 mg diarios los días 1 a 21 del ciclo de 28 días. No dosificar por debajo de 5 mg diarios los días 1 a 21 del ciclo de 28 días. |
Otras toxicidades en MM
En cuanto a otras toxicidades grados 3/4 que se consideran relacionadas con REVLIMID, suspender el tratamiento y reiniciar, según el criterio del médico, en el próximo nivel de dosis más bajo cuando se resuelva la toxicidad a ≤ grado 2.
Ajuste de la dosis inicial para insuficiencia renal en mieloma múltiple
(ver Tabla 13) Síndromes mielodisplásicos. La dosis inicial recomendada de REVLIMID es 10 mg diarios. El tratamiento se continúa o modifica sobre la base de resultados clínicos y de laboratorio.
Ajustes de dosis por toxicidades hematológicas durante el tratamiento del síndrome mielodisplásico
Los pacientes que reciben una dosis inicial de 10 mg y que experimentan trombocitopenia deben tener un ajuste de dosis de la siguiente manera:
|
Recuento de plaquetas |
|
|
Si se manifiesta trombocitopenia DENTRO de las 4 semanas de iniciar el tratamiento con 10 mg diarios en SMD: |
|
|
Si el basal es ≥ 100.000/mcL |
|
|
Cuando las plaquetas |
Acción recomendada |
|
Caen a < 50.000/mcL Regresan a ≥ 50.000/mcL |
Interrumpir tratamiento con REVLIMID Reanudar REVLIMID en 5 mg diarios |
|
Si el basal es < 100.000/mcL |
|
|
Cuando las plaquetas |
Acción recomendada |
|
Caen a 50% del valor basal Si el basal es ≥ 60.000/mcL y regresa a ≥ 50.000/mcL Si el basal es < 60.000/mcL y regresa a ≥ 30.000/mcL- |
Interrumpir tratamiento con REVLIMID Reanudar REVLIMID en 5 mg diarios Reanudar REVLIMID en 5 mg diarios |
|
Si se manifiesta trombocitopenia DESPUÉS de 4 semanas de iniciar el tratamiento en 10 mg diarios en SMD: |
|
|
Cuando las plaquetas |
Acción recomendada |
|
< 30.000/mcL ó < 50.000/mcL y transfusiones plaquetarias |
Interrumpir tratamiento con REVLIMID |
|
Regresan a ≥ 30.000/mcL (sin insuficiencia hemostática) |
Reanudar REVLIMID en 5 mg diarios |
Los pacientes que experimentan trombocitopenia con 5 mg diarios deben tener un ajuste de dosis de la siguiente manera:
|
Si se manifiesta trombocitopenia durante el tratamiento con 5 mg diarios en SMD: |
|
|
Cuando las plaquetas |
Acción recomendada |
|
<30.000/mcL ó < 50.000/mcL y transfusiones plaquetarias Regresan a ≥ 30.000/mcL (sin insuficiencia hemostática) |
Interrumpir tratamiento con REVLIMID Reanudar REVLIMID en 5 mg día por medio |
Los pacientes que reciben una dosis inicial de 10 mg y que experimentan neutropenia deben tener un ajuste de dosis de la siguiente manera:
|
Recuento Absoluto de Neutrófilos (ANC) |
|
|
Si se manifiesta neutropenia DENTRO de las 4 semanas de iniciar el tratamiento en 10 mg diarios en SMD: |
|
|
Si el ANC basal es ≥ 1000/mcL |
|
|
Cuando los neutrófilos |
Acción recomendada |
|
Caen a < 750/mcL Regresan a ≥ 1000/mcL |
Interrumpir tratamiento con REVLIMID Reanudar REVLIMID en 5 mg diarios |
|
Si el ANC basal < 1000/mcL |
|
|
Cuando los neutrófilos |
Acción recomendada |
|
Caen a < 500/mcL Regresan a ≥ 500/mcL |
Interrumpir tratamiento con REVLIMID Reanudar REVLIMID en 5 mg diarios |
|
Si se manifiesta neutropenia DESPUÉS de 4 semanas de iniciar tratamiento en 10 mg diarios en SMD: |
|
|
Cuando los neutrófilos |
Acción recomendada |
|
< 500/mcL por ≥ 7 días ó < 500/mcL asociado con fiebre (≥ 38,5ºC) Regresan a ≥ 500/mcL |
Interrumpir tratamiento con REVLIMID Reanudar REVLIMID en 5 mg diarios |
Los pacientes que experimentan neutropenia en 5 mg diarios deben tener un ajuste de dosis de la siguiente manera:
|
Si se manifiesta neutropenia durante el tratamiento con 5 mg diarios en SMD: |
|
|
Cuando los neutrófilos |
Acción recomendada |
|
< 500/mcL por ≥ 7 días ó < 500/mcL asociado con fiebre (≥ 38,5ºC) Regresan a ≥ 500/mcL |
Interrumpir tratamiento con REVLIMID Reanudar REVLIMID en 5 mg día por medio |
Otras toxicidades grados 3/4 en SMD
En cuanto a otras toxicidades grados 3/4 que se consideran relacionadas con REVLIMID, suspender el tratamiento y reiniciar, según el criterio del médico, en el próximo nivel de dosis más bajo cuando la toxicidad se haya resuelto a ≤ grado 2.
Ajuste de la dosis inicial para insuficiencia renal en SMD:
(ver Tabla 13)
Linfoma de células del manto
La dosis inicial recomendada de REVLIMID es de 25 mg/día por vía oral en los días 1-21 de ciclos repetidos de 28 días para el linfoma de células del manto en recidiva o refractario. El tratamiento debe continuar hasta la progresión de la enfermedad o hasta que se manifieste una toxicidad inaceptable. El tratamiento continúa, se modifica o se discontinúa sobre la base de los hallazgos clínicos o de laboratorio.
Ajustes de dosis por toxicidades hematológicas durante el tratamiento de LCM
Se recomiendan los lineamientos de modificación de dosis, tal como se resumen a continuación, para manejar la neutropenia o trombocitopenia grados 3 o 4 u otras toxicidades grado 3 o 4 que se consideren en relación con REVLIMID.
|
Recuento de plaquetas |
|
|
Si se manifiesta trombocitopenia durante el tratamiento en LCM: |
|
|
Cuando las plaquetas |
Acción recomendada |
|
Caen a < 50.000/mcL Regresan a ≥50.000/mcL |
Interrumpir tratamiento con REVLIMID y realizar un hemograma completo de forma semanal Reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg diarios. |
|
Recuento Absoluto de Neutrófilos (ANC) |
|
|
Neutropenia durante el tratamiento en LCM: |
|
|
Cuando los neutrófilos |
Acción recomendada |
|
Caen a <1000/mcL por al menos 7 días o caen a <1.000/mcL con una temperatura asociada ≥ 38,5º C o caen a <500/mcL |
Interrumpir tratamiento con REVLIMID y realizar un hemograma completo de forma semanal |
|
Regresan a ≥1.000/mcL |
Reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg diarios. |
Otras toxicidades grados 3/4 en LCM
En cuanto a otras toxicidades grados 3/4 que se consideran relacionadas con REVLIMID, suspender el tratamiento y reiniciar, según el criterio del médico, en el próximo nivel de dosis más bajo cuando la toxicidad se haya resuelto a ≤ grado 2.
Ajuste de dosis inicial para insuficiencia renal en LCM:
(Ver Tabla 13)
Posología recomendada para linfoma folicular o linfoma de la zona marginal
La dosis inicial recomendada de REVLIMID es de 20 mg/día por vía oral en los días 1-21 de ciclos repetidos de 28 días por un máximo de 12 ciclos en combinación con rituximab. Véase sección de estudios clínicos para linfoma folicular y linfoma de la zona marginal para la posología específica del estudio AUGMENT. Para ajustar la dosis debido a toxicidad ocasionada por rituximab véase la información sobre prescripcion del producto. Ajustes de dosis por toxicidades hematológicas durante el tratamiento de LF o LZM.
Para controlar la neutropenia o trombocitopenia grados 3 o 4 u otras toxicidades grado 3 o 4 que se consideren en relación con REVLIMID se recomienda seguir los lineamientos de modificación de dosis, tal como se resumen a continuación:
|
Recuento de plaquetas |
|
|
Si se manifiesta trombocitopenia durante el tratamiento en LF o LZM: |
|
|
Cuando las plaquetas |
Acción recomendada |
|
Disminuyen a menos de 50.000/mcL |
Interrumpir tratamiento con REVLIMID y realizar un hemograma completo de forma semanal |
|
Vuelven por lo menos a 50.000/mcL |
Si la dosis inicial fue de 20 mg diarios, reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg diarios. Si la dosis inicial fue de 10 mg diarios, reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg día por medio. |
|
Recuento Absoluto de Neutrófilos (RAN) |
|
|
Neutropenia durante el tratamiento en LCM: |
|
|
Cuando los neutrófilos |
Acción recomendada |
|
Disminuyen a menos de 1.000/mcL por al menos 7 días o Disminuyen a menos de 1.000/mcL asociado con fiebre de al menos 38,5º C o Disminuyen a menos de 500/mcL |
Interrumpir tratamiento con REVLIMID y realizar un hemograma completo de forma semanal |
|
Vuelven por lo menos a 1.000/mcL |
Si la dosis inicial fue de 20 mg diarios, reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg diarios. Si la dosis inicial fue de 10 mg diarios, reanudar REVLIMID en 5 mg menos que la dosis anterior. No dosificar por debajo de 5 mg día por medio. |
Otras toxicidades grados 3/4 en LF o LZM
En cuanto a otras toxicidades grados 3/4 que se consideran relacionadas con REVLIMID, suspender el tratamiento y reiniciar, según el criterio del profesional en salud, en el próximo nivel de dosis más bajo cuando la toxicidad se haya resuelto a grado 2 o menor.
Ajuste de dosis inicial para insuficiencia renal en LF o LZM:
(Ver Tabla 13)
Dosis inicial para insuficiencia renal
Las recomendaciones para las dosis iniciales para pacientes con insuficiencia renal se indican en la siguiente tabla (ver Poblaciones específicas).
|
Tabla 13. Ajuste de dosis inicial para insuficiencia renal. |
|||
|
Función renal (Cockcroft-Gault) |
Dosis en terapia de combinación con REVLIMID para MM y para LCM |
Dosis en terapia de combinación con REVLIMID para LF y LZM |
Dosis en terapia de mantenimiento con REVLIMID después de auto-HSCT para MM y para SMD |
|
CLCr 30 a 60 mL/min |
10 mg una vez por día |
10 mg una vez por día |
5 mg una vez por día |
|
CLCr < 30 mL/min (no requiere diálisis) |
15 mg día por medio |
5 mg una vez por día |
5 mg día por medio |
|
CLCr < 30 mL/min (requiere diálisis) |
5 mg una vez por día. Los días de diálisis, administrar la dosis después de la diálisis. |
5 mg una vez por día. Los días de diálisis, administrar la dosis después de la diálisis. |
5 mg tres veces por semana, administrar luego de la diálisis |
Terapia de combinación con REVLIMID para MM: para CLCr 30 a 60 mL/min, considerar la escalada de la dosis a 15 mg después de 2 ciclos si el paciente tolera la dosis de 10 mg de lenalidomida sin toxicidad limitante de la dosis. Terapia de mantenimiento con REVLIMID después del auto-HSCT para MM y para LCM, y SMD: basar el aumento o la disminución subsiguiente de la dosis de REVLIMID en la tolerancia al tratamiento de cada paciente(ver Posología y administración).
Terapia de combinación con REVLIMID para LF o para LZM: para pacientes con CLcr 30 a 60 mL/min, considerar la escalada de la dosis de REVLIMID a 15 mg después de 2 ciclos si el paciente tolera la terapia.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis
No hay experiencia específica en el manejo de sobredosis de lenalidomida en pacientes con MM, SMD, LCM, LF o LZM. En estudios de búsqueda de dosis en sujetos sanos, algunos sujetos fueron expuestos hasta 200 mg (administrados como 100 mg dos veces al día) y, en estudios de dosis únicas, algunos sujetos fueron expuestos hasta 400 mg.
Los principales efectos adversos informados fueron prurito, urticaria, erupción cutánea, y transaminasas hepáticas elevadas. En ensayos clínicos, la toxicidad limitante de la dosis fue la neutropenia y la trombocitopenia.ANTE LA EVENTUALIDAD DE UNA SOBREDOSIFICACIÓN, CONCURRIR AL HOSPITAL MÁS CERCANO O COMUNICARSE AL TELÉFONO: MEDICAMENTA ECUATORIANA S.A., Quito. PBX 5005005 . E-mail: farmacovigilancia@medicamenta.com.ec
PRESENTACIÓN:
Cápsulas REVLIMID 5 mg: Cápsulas opacas blancas que tienen impreso en tinta negra “REV” en una mitad y “5 mg” en la otra mitad. Caja de 14, 21 y 28 cápsulas.
Cápsulas REVLIMID 10 mg: Cápsulas opacas azul/verde y amarillo pálido que tienen impreso en tinta negra “REV” en una mitad y “10 mg” en la otra mitad. Caja de 14, 21 y 28 cápsulas.
Cápsulas REVLIMID 15 mg: Cápsulas opacas azul pálido y blanco que tienen impreso en tinta negra “REV” en una mitad y “15 mg” en la otra mitad. Caja de 14, 21 y 28 cápsulas.
Cápsulas REVLIMID 20 mg: Cápsulas opacas azul y azul-verde que tienen impreso en tinta negra “REV” en una mitad y “20 mg” en la otra mitad. Caja de 14, 21 y 28 cápsulas.
Cápsulas REVLIMID 25 mg: Cápsulas opacas blancas que tienen impreso en tinta negra “REV” en una mitad y “25 mg” en la otra mitad. Caja de 14, 21 y 28 cápsulas.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Conservación: Almacenar a temperatura no mayor a 30°C.Manejo y eliminación: Se debe tener cuidado con el manejo de REVLIMID. Las cápsulas de REVLIMID no deben abrirse ni romperse. Si el polvo de la cápsula de REVLIMID toma contacto con la piel, lávela inmediata y cuidadosamente con agua y jabón. Si REVLIMID toma contacto con las membranas mucosas, limpie bien con agua. Se deben considerar procedimientos para el manejo y el desecho adecuados de fármacos anticancerígenos. Se han publicado diversas pautas sobre el tema.
Recetar no más de un suministro para 28 días.
BIBLIOGRAFÍA:
Estudios clínicos
Mieloma múltiple
Ensayo clínico abierto, aleatorizado, en pacientes con MM recientemente diagnosticado: Se realizó un ensayo aleatorizado, de centros múltiples, abierto, de 3 ramas de 1.623 pacientes para comparar la eficacia y la seguridad de REVLIMID y dosis baja de dexametasona (Rd) administrado por 2 duraciones de tiempo diferentes a melfalán, prednisona y talidomida (MPT) en pacientes con MM recientemente diagnosticado que no eran candidatos para transplante de células madre (SCT). En la primera rama del estudio, se administró Rd de forma continua hasta progresión de la enfermedad [rama Rd Continuo]. En la segunda rama, se administró Rd por hasta dieciocho ciclos de 28 días [72 semanas, rama Rd18]). En la tercera rama, se administró melfalán, prednisona y talidomida (MPT) por un máximo de doce ciclos de 42 días (72 semanas). A los fines de este estudio, un paciente que tenía < 65 años de edad no era candidato para un SCT si el paciente se negaba a someterse a terapia de SCT o el paciente no tenía acceso al SCT debido al costo o a otros motivos. Los pacientes fueron estratificados en la aleatorización por edad (≤ 75 vs. >75 años), etapa (etapas I y II de ISS vs. etapa III) y país.
Los pacientes en las ramas Rd Continuo y Rd18 recibieron REVLIMID 25 mg una vez por día los días 1 a 21 de ciclos de 28 días. Se administró dexametasona 40 mg una vez por día los días 1, 8, 15 y 22 de cada ciclo de 28 días. Para los pacientes > 75 años de edad, la dosis inicial de dexametasona fue de 20 mg por vía oral una vez por día los días 1, 8, 15 y 22 de ciclos repetidos de 28 días. La dosis inicial y los regimenes para Rd Continuo y Rd18 se ajustaron de acuerdo con la edad y la función renal. Todos los pacientes recibieron anticoagulación profiláctica, siendo la aspirina la utilizada con mayor frecuencia.
Las características basales demográficas y relacionadas con la enfermedad de los pacientes estaban balanceadas entre las 3 ramas. En general, los pacientes del estudio tenían enfermedad en etapa avanzada. De la población total del estudio, la mediana de la edad fue 73 en las 3 ramas, con el 35% de los pacientes totales > 75 años de edad; el 59% tenía etapa ISS I/II; el 41% tenía etapa ISS III; el 9% tenía insuficiencia renal severa (clearance de creatinina [CLCr] < 30 ml/min); 23% tenía insuficiencia renal moderada (CLCr > 30 a 50 ml/min; 44% tenía insuficiencia renal leve (CLCr > 50 a 80 ml/min). En cuanto al estado funcional según ECOG, el 29% tenía grado 0, el 49% tenía grado 1, el 21% grado 2, y 0,4% ≥ grado 3.
El criterio de valoración de eficacia primaria, Supervivencia Libre de Progresión (PFS), se definió como el tiempo desde la aleatorización hasta la primera documentación de la progresión de la enfermedad según determinó el Comité Independiente de Adjudicación de Respuesta (IRAC), en base a los criterios del Grupo de Trabajo Internacional sobre Mieloma Múltiple (IMWG) o muerte por cualquier causa, lo que se produjera primero durante el estudio hasta el final de la fase de seguimiento de PFS. Para el análisis de eficacia de todos los criterios de valoración, la comparación primaria fue entre las ramas Rd Continuo y MPT. Los resultados de eficacia se resumen en la tabla a continuación. La PFS fue significativamente más prolongada con Rd Continuo que con MPT: HR 0,72 (IC del 95%: 0,61-0,85 p <0,0001). Un menor porcentaje de sujetos en la rama Rd Continuo en comparación con la rama MPT tuvo eventos de PFS (52% vs. 61%, respectivamente). La mejoría en la mediana del tiempo de PFS en la rama Rd Continuo en comparación con la rama MPT fue de 4,3 meses. La tasa de respuesta del mieloma fue mayor con Rd Continuo en comparación con MPT (75,1% vs. 62,3%); con una respuesta completa en 15,1% de los pacientes en la rama Rd Continuo vs. 9,3% en la rama MPT. La mediana del tiempo hasta la primera respuesta fue de 1,8 meses en la rama Rd Continuovs. 2,8 meses en la rama MPT.
Para el análisis intermedio de supervivencia total (OS), con corte de datos el 3 de marzo de 2014, la mediana del tiempo de seguimiento para todos los pacientes sobrevivientes es de 45,5 meses, con 697 eventos de muerte, lo que representa el 78% de los eventos preespecificados necesarios para el análisis final planificado de supervivenci total (697/896 de los eventos de OS finales). La HR de la OS observada fue 0,75 para Rd Continuo vs. MPT (IC del 95% = 0,62; 0,90).
|
Tabla 1: Resumen de resultados clínicos – Estudio MM-020 (población con intención de tratar- ITT-) |
|||
|
Rd Continuo (N = 535) |
Rd18 |
MPT |
|
|
PFS - IRAC (meses)g |
|||
|
Cantidad de eventos PFS |
278 (52,0) |
348 (64,3) |
334 (61,1) |
|
Medianaa del tiempo PFS, meses (IC 95%)b |
25,5 (20,7, 29,4) |
20,7 (19,4, 22,0) |
21,2 (19,3, 23,2) |
|
HR [IC 95%]c; valor de pd |
|||
|
Rd Continuo vs. MPT |
0,72 (0,61, 0,85); <0,0001 |
||
|
Rd Continuo vs. Rd18 |
0,70 (0,60, 0,82) |
||
|
Rd18 vs. MPT |
1,03 (0,89, 1,20) |
||
|
Supervivencia total (meses)h |
|||
|
Cantidad de eventos de muerte |
208 (38,9) |
228 (42,1) |
261 (47,7) |
|
Medianaa del tiempo OS, meses (95% CI)b |
58,9 (56,0, NE)f |
56,7 (50,1, NE) |
48,5 (44,2, 52,0 ) |
|
HR [IC 95%]c |
|||
|
Rd Continuo vs. MPT |
0,75 (0,62, 0,90) |
||
|
Rd Continuo vs. Rd18 |
0,91 (0,75, 1,09) |
||
|
Rd18 vs. MPT |
0,83 (0,69, 0,99) |
||
|
Tasa de respuestae IRAC, n (%)g |
|||
|
CR |
81 (15,1) |
77 (14,2) |
51 (9,3) |
|
VGPR |
152 (28,4) |
154 (28,5) |
103 (18,8) |
|
PR |
169 (31,6) |
166 (30,7) |
187 (34,2) |
|
Respuesta total: CR, VGPR, o PR |
402 (75,1) |
397 (73,4) |
341 (62,3) |
|
CR = Respuesta Completa; d = Dosis Baja de Dexametasona; HR = Hazard Ratio; IRAC = Comité Independiente de Adjudicación de Respuesta; M = Melfalán; NE = No Estimable; OS = Supervivencia Total; P = Prednisona; PFS = Supervivencia Libre de Progresión; PR = Respuesta Parcial; R = lenalidomida; Rd Continuo = Rd Administrada Hasta Documentación de Enfermedad Progresiva; Rd18 = Rd Administrada por ≤ 18 ciclos; T = Talidomida; VGPR = Respuesta Parcial Muy Buena; vs = Versus. a La mediana se basa en el estimado de Kaplan-Meier. b El Intervalo de Confianza del 95% (IC) acerca de la mediana. c En base al modelo de riesgos proporcionales de Cox que compara las funciones de riesgo relacionadas con las ramas de tratamiento indicadas. d El valor de p se basa en la prueba de log-rank no estratificada de las diferencias de la curva de Kaplan-Meier entre las ramas de tratamiento indicadas. e Mejor evaluación de respuesta durante la fase de tratamiento del estudio f Incluye pacientes sin datos de evaluación de respuesta o cuya única evaluación fue “respuesta no evaluable”. g Fecha de corte de datos = 24 de mayo de 2013. h Fecha de corte de datos = 3 de marzo de 2014. |
|||
Curvas de Kaplan-Meier de supervivencia libre de progresión según evaluación del IRAC (población ITT) entre ramas Rd Continuo, Rd18 y MPT
Fecha de corte: 24 de mayo de 2013
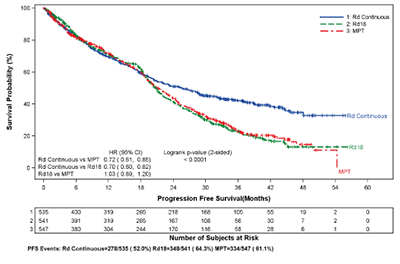
IC = Intervalo de Confianza; d = dosis baja de dexametasona; HR = Hazard Tatio; IRAC = Comité Independiente de Adjudicación de Respuesta; M = Melfalán; P = Prednisona; R = Lenalidomida; Rd Continuo = Rd Administrada Hasta Documentación de Enfermedad Progresiva; Rd18 = Rd Administrada por ≤ 18 Ciclos; T = Talidomida
Curvas de Kaplan-Meier de supervivencia total (población ITT) entre ramas
Rd Continuo, Rd18 y MPT
Fecha de corte: 3 de mayo de 2014
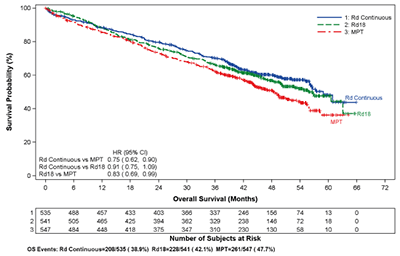
IC = Intervalo de Confianza; d = Dosis Baja de Dexametasona; HR = Hazard Ratio; M = Melfalán; P = Prednisona; R = Lenalidomida; Rd Continuo = Rd Administrada Hasta Documentación de Enfermedad Progresiva; Rd18 = Rd Administrada por ≤ 18 ciclos; T = Talidomida.
La experiencia clínica con lenalidomida en combinación con bortezomib y dexametasona en pacientes no tratados previamente con mieloma múltiple elegibles para trasplante.
La eficacia (según los criterios de respuesta del Grupo de Trabajo Internacional sobre Mieloma (IMWG)) y la seguridad de la lenalidomida en combinación con bortezomib y dexametasona (RVd) se evaluaron en dos estudios multicéntricos de fase 3: PETHEMA GEM2012 e IFM 2009.
PETHEMA GEM2012
El estudio PETHEMA GEM2012 fue un estudio multicéntrico de fase 3, aleatorizado, controlado, abierto que comparó 2 regímenes de acondicionamiento antes del trasplante (busulfan-melfalán y MEL200) en sujetos que habían recibido RVd como terapia inicial. RVd se administró en seis ciclos de 4 semanas (24 semanas). Los pacientes recibieron lenalidomida 25 mg / día por vía oral en los días 1-21, bortezomib subcutáneo 1,3 mg / m2 en los días 1, 4, 8 y 11, y dexametasona 40 mg / día por vía oral en los días 1-4, 9-12 de ciclos repetidos de 28 días. Después del tratamiento inicial, los pacientes recibieron un régimen de acondicionamiento con busulfán-melfalán o MEL200 (aleatorización 1: 1) y ASCT. Los pacientes también recibieron dos ciclos adicionales (8 semanas) de RVd después de la ASCT. En total 458 pacientes fueron incluidos en el estudio.
En el estudio PETHEMA GEM2012, al final del tratamiento inicial con RVd, la tasa ≥ VGPR fue del 67% y la tasa de CR fue del 33% con el 47% (217/458) de sujetos con MRD negativa (sensibilidad 10-4). De los sujetos con ≥ VGPR, el 64% (196/305) fue MRD negativo (sensibilidad 10-4). La tasa ≥ VGPR postrasplante fue del 75% y la tasa de CR fue del 44% con 63% (287/458) de sujetos con MRD negativo (sensibilidad 10-4). De los sujetos con ≥ VGPR, el 79% (271/344) fue MRD negativo (sensibilidad 10-4).
IFM 2009
El estudio IFM 2009 fue un estudio multicéntrico de fase 3, aleatorizado, controlado, abierto que comparó RVd con y sin ASCT como tratamiento inicial para pacientes con mieloma múltiple no tratados previamente que son elegibles para trasplante. Los pacientes recibieron lenalidomida 25 mg / día por vía oral los días 1 a 14, bortezomib intravenoso 1,3 mg / m2 en los días 1, 4, 8 y 11 y dexametasona 20 mg / día por vía oral los días 1, 2, 4, 5, 8, 9, 11 y 12 de ciclos repetidos de 21 días. RVd se administró en 8 ciclos de 3 semanas (24 semanas) sin ASCT inmediato (grupo A) o tres ciclos de 3 semanas (9 semanas) antes de ASCT (grupo B). Los pacientes en el grupo B también recibieron dos ciclos adicionales de RVd de 3 semanas después de la ASCT. En total 700 pacientes fueron incluidos en el estudio.
En el estudio IFM 2009, al final del tratamiento inicial, la tasa ≥ VGPR fue del 68%, con una tasa de CR del 31%. De los sujetos con ≥ VGPR, el 57% (136/237) fue MRD negativo (sensibilidad 10-4).
Experiencia clínica con lenalidomida en combinación con bortezomib y dexametasona en pacientes no tratados previamente con mieloma múltiple que no son elegibles para trasplante
El estudio SWOG S0777 evaluó la adición de bortezomib a una base de lenalidomida y dexametasona, como tratamiento inicial, seguido de Rd continuado hasta la progresión de la enfermedad, en pacientes con mieloma múltiple no tratados previamente que no estaban destinados para el trasplante de células madre.
Los pacientes en el grupo de lenalidomida, bortezomib y dexametasona (RVd) recibieron lenalidomida por vía oral los días 1-14, bortezomib intravenoso 1,3 mg / m2 los días 1, 4, 8 y 11, y dexametasona 20 mg / día por vía oral. días 1, 2, 4, 5, 8, 9, 11 y 12 de ciclos repetidos de 21 días hasta ocho ciclos de 21 días (24 semanas). Los pacientes del grupo de lenalidomida y dexametasona (Rd) recibieron lenalidomida 25 mg / día por vía oral los días 1-21 y la dexametasona 40 mg / día por vía oral los días 1, 8, 15 y 22 de ciclos repetidos de 28 días hasta seis ciclos de 21 días (24 semanas). Los pacientes en ambos grupos tomaron Rd: lenalidomida 25 mg / día por vía oral en los días 1-21 y dexametasona 40 mg / día por vía oral en los días 1, 8, 15 y 22 de ciclos repetidos de 28 días. El tratamiento debía continuar hasta la progresión de la enfermedad.
La variable principal de eficacia en el estudio fue la Supervivencia Libre de Progresión (PFS). En total, se reclutaron 523 pacientes en el estudio, con 263 pacientes aleatorizados a RVd y 260 pacientes aleatorizados a Rd. La demografía y las características basales relacionadas con la enfermedad de los pacientes estaban bien equilibradas entre los grupos.
Los resultados de la PFS (revisión de IRAC, reglas de censura de la EMA) con un corte al 1º de diciembre de 2016, donde el tiempo de seguimiento promedio para los sujetos supervivientes fue de 60.6 meses, mostraron una reducción del 24% en el riesgo de progresión de la enfermedad o muerte a favor de RVd (HR = 0,76; CI del 95%: 0,62 a 0,94). La mediana de la PFS general fue de 41,7 meses (CI del 95%: 33,1, 51,5) en el grupo de RVd frente a 29,7 meses (IC del 95%: 24,2, 37,8) en el grupo Rd. Se observó una reducción del 28% en el riesgo de muerte para los sujetos en el grupo RVd en comparación con los del grupo Rd (HR = 0,72; IC del 95% = 0.56 a 0,94). La mediana de OS general fue de 89,1 meses (IC del 95% 76,1, NE) en el grupo RVd frente a 67,2 meses (CI del 95% 58,4, 90,8) en el grupo Rd. De manera similar, la tasa ≥ VGPR fue mayor en el grupo RVd (58%) en comparación con el grupo Rd (32%).
Ensayos clínicos aleatorizados, controlados con placebo - Mantenimiento después del auto-HSCT: Se realizaron dos estudios multicéntricos, aleatorizados, doble ciego, de grupos paralelos y controlados con placebo para evaluar la eficacia y la seguridad de la terapia de mantenimiento con REVLIMID en el tratamiento de pacientes con MM después del auto-HSCT. En el estudio de mantenimiento 1, los pacientes entre 18 y 70 años de edad que habían sido sometidos a terapia de inducción seguida de auto-HSCT, fueron elegibles. La terapia de inducción debe haber ocurrido dentro de los 12 meses. Dentro de 90 a 100 días después del auto-HSCT, los pacientes con al menos una respuesta estable de la enfermedad fueron aleatorizados 1: 1 para recibir REVLIMID o mantenimiento con placebo. En el Estudio de Mantenimiento 2, los pacientes con edades <65 años al momento del diagnóstico que habían sido sometidos a terapia de inducción seguida de auto-HSCT y habían logrado al menos una respuesta estable de la enfermedad en el momento de la recuperación hematológica, fueron elegibles. Dentro de los 6 meses después del auto-HSCT, los pacientes fueron aleatorizados 1: 1 para recibir REVLIMID o mantenimiento con placebo. Los pacientes elegibles para ambos ensayos tuvieron CLcr de ≥ 30 ml / minuto.
En ambos estudios, la dosis de mantenimiento de REVLIMID fue de 10 mg una vez al día, los días 1-28 de ciclos repetidos de 28 días, podría aumentarse a 15 mg una vez al día, después de 3 meses en ausencia de toxicidad limitante de la dosis y el tratamiento debía ser continuado hasta la progresión de la enfermedad o la retirada del paciente por otra razón. La dosis se redujo, o el tratamiento se interrumpió temporalmente o se detuvo, según fue necesario para manejar la toxicidad. Un aumento de dosis a 15 mg una vez al día se realizó para 135 pacientes (58%) en el estudio de mantenimiento 1 y en 185 pacientes (60%) en el estudio de mantenimiento 2.
La demografía y las características basales relacionadas con la enfermedad de los pacientes fueron similares en los dos estudios y reflejaron una población típica de MM después del auto-HSCT (ver Tabla 2).
|
Tabla 2: Características demográficas basales y relacionadas con la enfermedad - Estudios de mantenimiento 1 y 2 |
||||
|
Estudio de mantenimiento 1 |
Estudio de mantenimiento 2 |
|||
|
REVLIMID N=231 |
Placebo N=229 |
REVLIMID N=307 |
Placebo N=307 |
|
|
Edad (años): |
||||
|
Promedio |
58,0 |
58,0 |
57,5 |
58,1 |
|
(Min, max) |
(29,0; 71.0) |
(39,0; 71.0) |
(22,7; 68.3) |
(32,3; 67.0) |
|
Sexo, n(%) |
||||
|
Masculino |
121 (52) |
129 (56) |
169 (55) |
181 (59) |
|
Femenido |
110 (48) |
138 (45) |
138 (45) |
126 (41) |
|
Etapa ISS al momentodel diagnóstico, n(%) |
||||
|
Etapa I oII |
120 (52) |
131 (57) |
232 (76) |
250 (81) |
|
Etapa I |
62 (27) |
85 (37) |
128 (42) |
143 (47) |
|
Etapa II |
58 (25) |
46 (20) |
104 (34) |
107 (35) |
|
Etapa III |
39 (17) |
35 (15) |
66 (21) |
46 (15) |
|
Faltante |
72 (31) |
63 (28) |
9 (3) |
11 (4) |
|
CrCl en el Post-auto-HSCT, n(%) |
||||
|
<50 mL/min |
23 (10) |
16 (7) |
10 (3) |
9 (3) |
|
≥50 mL/min |
201 (87) |
204 (89) |
178 (58) |
200 (65) |
|
Faltante |
7 (3) |
9 (4) |
119 (39) |
98 (32) |
|
Fecha de corte de datos = 1 de marzo de 2015. |
||||
El criterio principal de valoración de la eficacia de ambos estudios fue la PFS definida desde la aleatorización a la fecha de progresión o muerte, lo que tuvo lugar primero; los estudios individuales no fueron impulsados para un criterio de valoración de la supervivencia general. Ambos estudios fueron desenmascarados por las recomendaciones de sus respectivos comités de monitoreo de datos y después de superar los umbrales respectivos para los análisis provisionales preestablecidos de PFS. Después del desenmascaramiento, los pacientes siguieron siendo monitoreados como antes. Se permitió a los pacientes en el grupo placebo del Estudio de Mantenimiento 1 cruzarse para recibir REVLIMID antes de la progresión de la enfermedad (76 pacientes [33%] cruzaron a REVLIMID); a los pacientes en el Estudio de Mantenimiento 2 no se les recomendó cruzar. Los resultados de la eficacia se resumen en la siguiente tabla. En ambos estudios, el análisis primario de la PFS al momento de desenmascaramiento fue significativamente más largo con REVLIMID comparado con placebo: el Estudio de Mantenimiento 1 HR 0,38 (IC del 95%: 0,27-0,54 p <0,001) y el Estudio de Mantenimiento 2 HR 0,50 (IC del 95%: 0,39- 0,64 p <0,001). Para ambos estudios, la PFS se actualizó con una fecha de corte el 1º de marzo de 2015, como se muestra en la tabla y los siguientes gráficos de Kaplan Meier. Con un seguimiento más prolongado (promedio 72,4 y 86,0 meses, respectivamente), los análisis actualizados de la PFS para ambos estudios continúan mostrando una ventaja de la PFS para REVLIMID comparado con placebo: el Estudio de Mantenimiento 1 HR 0,38 (IC del 95%: 0,28-0,50) con la PFS promedio de 68,6 meses y el Estudio de Mantenimiento 2 HR 0,53 (IC del 95%: 0,44-0,64) con una PFS promedio de 46,3 meses.El análisis descriptivo de los datos de la OS (supervivencia general, por sus siglas en inglés) con fecha de corte el 1º de febrero de 2016 se proporciona en la Tabla 3. El tiempo promedio de seguimiento fue de 81,6 y 96,7 meses para el Estudio de Mantenimiento 1 y el Estudio de Mantenimiento 2, respectivamente. La OS promedio fue de 111,0 y 84,2 meses para REVLIMID y placebo, respectivamente, para el Estudio de Mantenimiento 1 y 105,9 y 88,1 meses para REVLIMID y placebo, respectivamente, para el Estudio de Mantenimiento 2.
|
Tabla 3: Supervivencia libre de progresión y supervivencia general de la aleatorización en los Estudios de Mantenimiento 1 y 2 (población ITT post-auto-HSCT) |
||||
|
Estudio de Mantenimiento 1 |
Estudio de Mantenimiento 2 |
|||
|
REVLIMID N = 231 |
Placebo N = 229 |
REVLIMID N = 307 |
Placebo N = 307 |
|
|
PFS en el desenmascaramiento |
||||
|
Eventos PFS n (%) |
46 (20) |
98 (43) |
103 (34) |
160 (52) |
|
Promedio en meses [IC del 95%]] |
33,9 [NE, NE] |
19,0 [16,2; 25.6] |
41,2 [38,3; NE] |
23,0 [21,2; 28,0] |
|
Cociente de riesgo [IC del 95%] |
0.38 [0,27; 0,54] |
0.50 [0,39; 0,64] |
||
|
Prueba del orden logarítmico valor de p 3 |
<0,001 |
<0,001 |
||
|
PFS en el análisis de actualización 1 Marzo 2015 (Estudios 1 y 2) |
||||
|
Eventos PFS n (%) |
97 (42) |
116 (51) |
191 (62) |
248 (81) |
|
Promedio en meses [IC del 95%]] |
68,6 [52,8; NE] |
22,5 [18.8; 30,0] |
46,3 [40,1; 56,6] |
23,8 [21,0; 27,3] |
|
Cociente de riesgo [IC del 95%] |
0,38 [0,28; 0,50] |
0,53 [0,44; 0,64] |
||
|
OS en el análisis de actualización 1 Feb 2016 (Estudios 1 y 2) |
||||
|
Eventos OS n (%) |
82 (35) |
114 (50) |
143 (47) |
160 (52) |
|
Promedio en meses [IC del 95%]] |
111.0 [101,8; NE] |
84.2 [71,0; 102,7] |
105.9 [88,8; NE] |
88.1 [80,7; 108,4] |
|
Cociente de riesgo [IC del 95%] |
0,59 [0,44; 0,78] |
0,90 [0,72; 1,13] |
||
|
Fecha del desenmascaramiento en el Estudio de Mantenimiento 1 y 2 = 17 de diciembre de 2009 y 7 de julio de 2010, respectivamente. Auto-HSCT = Trasplante Autólogo de Células Madre Hematopoyéticas; IC = Intervalo de Confianza; |
||||
Curvas Kaplan-Meier de supervivencia libre de progresión de la aleatorización (Población ITT Post-Auto-HSCT) en el Estudio de Mantenimiento 1 entre los grupos REVLIMID y Placebo (Fecha de corte de datos actualizada el 1 de marzo de 2015)
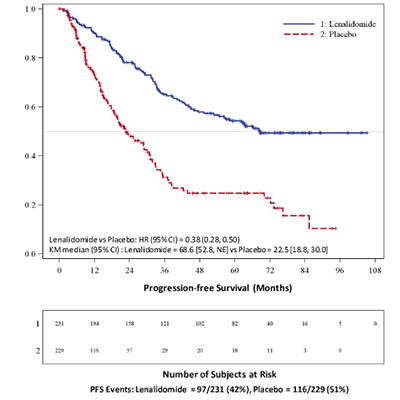
Auto-HSCT = Trasplante Autólogo de Células Madre Hematopoyéticas; IC = Intervalo de Confianza; HR = Cociente de Riesgo; ITT =Intención de Tratar; KM = Kaplan - Meier; PFS = Supervivencia Libre de Progresión; Vs = Versus.
Curvas Kaplan-Meier de supervivencia libre de progresión de la aleatorización (Población ITT Post-Auto-HSCT población) en el Estudio de Mantenimiento 2 entre los grupos REVLIMID y Placebo (Fecha de corte de datos actualizada el 1 de marzo de 2015)
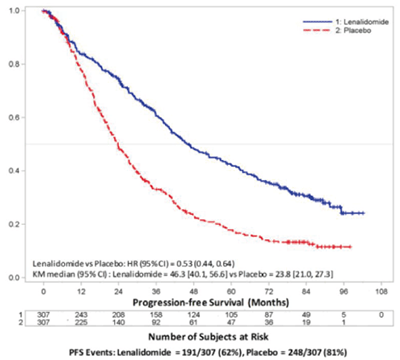
Auto-HSCT = Trasplante Autólogo de Células Madre Hematopoyéticas; IC = Intervalo de Confianza; HR = Cociente de Riesgo; ITT =Intención de Tratar; KM = Kaplan - Meier; PFS = Supervivencia Libre de Progresión; Vs = Versus.
Estudios clínicos abiertos, aleatorizados en pacientes con MM después de al menos una terapia previa. Se realizaron dos estudios aleatorizados (Estudios 1 y 2) para evaluar la eficacia y seguridad de REVLIMID. Estos estudios de centros múltiples, multinacionales, a doble-ciego, controlados con placebo compararon REVLIMID más terapia de pulsos con dexametasona en dosis alta por vía oral con la terapia con dexametasona sola en pacientes con mieloma múltiple que habían recibido por lo menos un tratamiento anterior. Estos estudios incluyeron a pacientes con recuentos absolutos de neutrófilos (ANC) ≥ 1000/mm3, recuento de plaquetas ≥ 75.000/mm3, creatinina sérica ≤ 2,5 mg/dL, SGOT/AST o SGT/ALT en sangre ≤ 3 x límite superior de lo normal (ULN) y bilirrubina directa sérica ≤ 2 mg/dL.
En ambos estudios, los pacientes en el grupo REVLIMID/dexametasona tomaron 25 mg de REVLIMID por vía oral una vez por día en los días 1 a 21 y una cápsula de placebo similar una vez por día en los días 22 a 28 de cada ciclo de 28 días. Los pacientes en el grupo placebo/dexametasona tomaron 1 cápsula de placebo en los días 1 a 28 de cada ciclo de 28 días. Los pacientes en ambos grupos de tratamiento tomaron 40 mg de dexametasona por vía oral una vez por día en los días 1 a 4, 9 a 12, y 17 a 20 de cada ciclo de 28 días durante los primeros 4 ciclos de terapia.La dosis de dexametasona se redujo a 40 mg por vía oral una vez por día en los días 1 a 4 de cada ciclo de 28 días después de los primeros 4 ciclos de terapia. En ambos estudios, el tratamiento continuó hasta la progresión de la enfermedad.
En ambos estudios, se permitieron ajustes de dosis en base a resultados clínicos y de laboratorio. Se permitieron reducciones secuenciales de dosis a 15 mg diarios, 10 mg diarios y 5 mg diarios, por toxicidad [ver Posología y administración].
La tabla 4 resume las características basales del paciente y de la enfermedad en los dos estudios. En ambos estudios, las características basales demográficas y relacionadas con la enfermedad fueron comparables entre los grupos REVLIMID/dexametasona y placebo/dexametasona.
|
Tabla 4: Características basales demográficas y |
||||
|
Estudio 1 |
Estudio 2 |
|||
|
Revlimid/Dex N=177 |
Placebo/Dex N=176 |
Revlimid/Dex N=176 |
Placebo/Dex N=175 |
|
|
Características del paciente |
||||
|
Edad (años) |
||||
|
Mediana |
64 |
62 |
63 |
64 |
|
Min - Máx |
36-86 |
37-85 |
33-84 |
40-82 |
|
Sexo |
||||
|
Hombres |
106 (60 %) |
104 (59 %) |
104 (59 %) |
103 (59 %) |
|
Mujeres |
71 (40 %) |
72 (41 %) |
72 (41 %) |
72 (41 %) |
|
Raza/etnicidad |
||||
|
Blanca |
141 (80%) |
148 (84 %) |
172 (98 %) |
175 (100 %) |
|
Otra |
36 (20 %) |
28 (16 %) |
4 (2 %) |
0 (0 %) |
|
Desempeño ECOG |
||||
|
Estado 0-1 |
157 (89 %) |
163 (95 %) |
150 (85 %) |
144 (82 %) |
|
Características de la enfermedad |
||||
|
Estadio basal de mieloma múltiple (Durie-Salmon) |
||||
|
I |
3 % |
3 % |
6 % |
5 % |
|
II |
32 % |
31 % |
28 % |
33 % |
|
III |
64 % |
66 % |
65 % |
63 % |
|
B2-microglobulina (mg/L) |
||||
|
≤ 2,5 mg/L |
52 (29%) |
51 (29%) |
51 (29%) |
48 (27%) |
|
> 2,5 mg/L |
125 (71%) |
125 (71%) |
125 (71%) |
127 (73%) |
|
Número de terapias anteriores |
||||
|
Nro. de terapias anteriores antimieloma |
||||
|
1 |
38 % |
38 % |
32 % |
33 % |
|
≥2 |
62 % |
62 % |
68 % |
67 % |
|
Tipos de terapias anteriores |
||||
|
Transplante de célula madre |
62 % |
61 % |
55 % |
54 % |
|
Talidomida |
42 % |
46 % |
30 % |
38 % |
|
Dexametasona |
81 % |
71 % |
66 % |
69 % |
|
Bortezomib |
11 % |
11 % |
5 % |
4 % |
|
Melfalán |
33 % |
31 % |
56 % |
52 % |
|
Doxorubicina |
55 % |
51 % |
56 % |
57 % |
El objetivo primario de eficacia en ambos estudios fue el tiempo hasta la progresión (TTP, Time To Progression). TTP se definió como el tiempo desde la aleatorización hasta la primera manifestación de enfermedad progresiva.
Los análisis intermedios previamente planificados de ambos estudios mostraron que la combinación de REVLIMID/dexametasona fue significativamente superior a dexametasona sola para TTP. Los estudios fueron subsiguientemente abiertos para permitir que los pacientes del grupo placebo/dexametasona aún en estudio recibieran tratamiento con la combinación REVLIMID/dexametasona. Para ambos estudios, se analizaron los datos de seguimiento prolongado acerca de la supervivencia con entrecruzamientos. En el estudio 1, la mediana del tiempo de supervivencia fue 39,4 meses (IC 95%: 32,9; 47,4) en el grupo REVLIMID/dexametasona y 31,6 meses (IC 95%: 24,1; 40,9) en el grupo placebo/dexametasona con un cociente de riesgo de 0,79 (IC 95%: 0,61 -1,03). En el estudio 2, la mediana del tiempo de supervivencia fue 37,5 meses (IC 95%: 29,9; 46,6) en el grupo REVLIMID/dexametasona y 30,8 meses (IC 95%: 23,5-40,3) en el grupo placebo/dexametasona, con un cociente de riesgo de 0,86 (IC 95%: 0,65-1,14).
|
Tabla 5: Resultados del TTP en el estudio 1 y en el estudio 2 |
||||
|
Estudio 1 |
Estudio 2 |
|||
|
REVLIMID/Dex N=177 |
Placebo/Dex N=176 |
REVLIMID/Dex N=176 |
Placebo/Dex N=175 |
|
|
TTP |
||||
|
Eventos n (%) |
73 (41) |
120 (68) |
68 (39) |
130 (74) |
|
TTP mediano en meses [IC 95%] |
13,9 [9,5; 18,5] |
4,7 [3,7; 4,9] |
12,1 [9,5; NE] |
4,7 [3,8; 4,8] |
|
Cociente de riesgos instantáneos (hazard ratio) [IC 95%] |
0,285 [0,210; 0,386] |
0,324 [0,240: 0,438] |
||
|
Prueba del orden logarítmico valor de p 3 |
<0,001 |
<0,001 |
||
|
Respuesta |
||||
|
Respuesta Completa (CR) n (%) |
23 (13) |
1 (1) |
27 (15) |
7 (4) |
|
Respuesta Parcial (RR/PR) n (%) |
84 (48) |
33 (19) |
77 (44) |
34 (19) |
|
Respuesta global n (%) |
||||
|
Respuesta global n (%) |
107 (61) |
34 (19) |
104 (59) |
41 (23) |
|
Valor de p |
<0,001 |
<0,001 |
||
|
Cociente de posibilidades [IC 95%] |
6,38 [3,95; 10,32] |
4,72 [2,98; 7,49] |
||
Valor estimado según Kaplan-Meier del tiempo hasta la progresión – Estudio 1
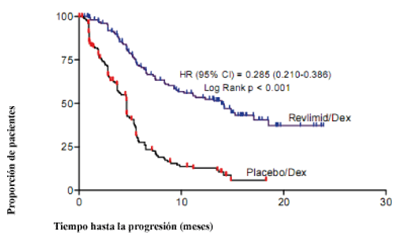
Valor estimado según Kaplan-Meier del tiempo hasta la progresión – Estudio 2
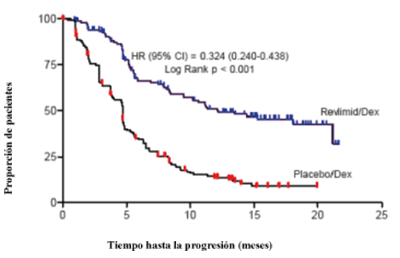
Síndromes mielodisplásicos con anomalía citogenética por deleción del 5q
Se evaluó la eficacia y seguridad de REVLIMID en pacientes con anemia dependiente de transfusión en SMD (Síndrome Mielodisplásico) de riesgo bajo o Intermedio 1 con una anomalía citogenética por deleción del 5q (q31-33) en aislamiento o con anomalías citogenéticas adicionales, en una dosis de 10 mg una vez por día o 10 mg una vez por día durante 21 días cada 28 días en un estudio abierto, de grupo único, en múltiples centros. El estudio principal no fue diseñado ni tenía la potencia para comparar de forma prospectiva la eficacia de los dos regímenes de administración. Se permitieron reducciones secuenciales de dosis a 5 mg diarios y 5 mg día por medio, así como retrasos de dosis, por toxicidad (Posología y administración).
Este estudio principal reclutó a 148 pacientes que tenían anemia dependiente de transfusión de glóbulos rojos. La dependencia de la transfusión de glóbulos rojos se definió como haber recibido ≥2 unidades de glóbulos rojos dentro de las 8 semanas anteriores al tratamiento del estudio. El estudio incorporó a pacientes con recuentos absolutos de neutrófilos (ANC) ≥500 células/mm3, recuentos plaquetarios ≥ 50.000/mm3, creatinina sérica ≤ 2,5 mg/dL, SGOT/AST o SGPT/ALT séricos ≤ 3 x límite superior del normal (ULN), y bilirrubina directa sérica ≤ 2,0 mg/dL. Se permitió el factor estimulante de colonias de granulocitos para los pacientes que manifestaron neutropenia o fiebre asociada con neutropenia. Las características basales de los pacientes y relacionadas con la enfermedad se resumen en la tabla 6.
|
Tabla 6: Características basales demográficas y relacionadas con la enfermedad en el estudio sobre SMD |
||
|
Global (N=148) |
||
|
Edad (años) |
||
|
Mediana |
71,0 |
|
|
Min, Máx |
37,0 |
95,0 |
|
Género |
n |
(%) |
|
Hombres |
51 |
(34,5) |
|
Mujeres |
97 |
(65,5) |
|
Raza |
n |
(%) |
|
Blanca |
143 |
(96,6) |
|
Otra |
5 |
(3,4) |
|
Duración de SMD (años) |
||
|
Mediana |
2,5 |
|
|
Min, Máx |
0,1 |
20,7 |
|
Anomalía citogenética Del 5 (q31-33) |
n |
(%) |
|
Sí |
148 |
(100,0) |
|
Otras anomalías citogenéticas |
37 |
(25,2) |
|
Puntaje IPSS [a] |
n |
(%) |
|
Bajo (0) |
55 |
(37,2) |
|
Intermedio-1 (0,5-1,0) |
65 |
(43,9) |
|
Intermedio-2 (1,5-2,0) |
6 |
(4,1) |
|
Alto (≥2,5) |
2 |
(1,4) |
|
Faltante |
20 |
(13,5) |
|
Clasificación FAB [b] de revisión central |
n |
(%) |
|
RA |
77 |
(52,0) |
|
RARS |
16 |
(10,8) |
|
RAEB |
30 |
(20,3) |
|
CMML |
3 |
(2,0) |
|
[a] Categoría de riesgo IPSS: Bajo (puntaje combinado = 0), Intermedio-1 (puntaje combinado = 0,5 a 1,0), Intermedio-2 (puntaje combinado = 1,5 a 2,0), Alto (puntaje combinado ≥2,5); Puntaje combinado = (puntaje de blastos en médula + puntaje cariotipo + puntaje citopenia). [b] Clasificación francesa-norteamericana-británica (FAB) del SMD. |
||
La frecuencia de independencia de transfusión de glóbulos rojos se modificó a partir de los criterios de respuesta del Grupo de Trabajo Internacional (IWG, International Working Group) para SMD. La independencia de transfusión de glóbulos rojos se definió como la ausencia de toda transfusión de glóbulos rojos durante 56 días (8 semanas) consecutivos “sucesivos” durante el período de tratamiento.
La independencia de transfusión se observó en 99/148 (67%) pacientes (IC 95% [59, 74]). La mediana de la duración desde la fecha cuando se declaró por primera vez la independencia de transfusión de glóbulos rojos (es decir, el último día del período de 56 días libre de transfusión) hasta la fecha cuando se recibió una transfusión adicional luego del período de 56 días libre de transfusión entre los 99 respondedores fue de 44 semanas (rango de 0 a >67 semanas). El 90% de los pacientes que lograron un beneficio en sus transfusiones lo hicieron al completar tres meses en el estudio.
Las tasas de independencia de transfusión de glóbulos rojos no se vieron afectadas por la edad o el género.
La dosis de REVLIMID se redujo o se interrumpió por lo menos una vez debido a un evento adverso en 118 (79,7%) de los 148 pacientes; la mediana de tiempo hasta la primera reducción o interrupción de dosis fue de 21 días (media, 35,1 días; rango, 2-253 días), y la mediana de la duración de la primera interrupción de dosis fue de 22 días (media, 28,5 días; rango, 2-265 días). Una segunda reducción o interrupción de dosis debido a eventos adversos fue necesaria en 50 (33,8%) de los 148 pacientes. La mediana del intervalo entre la primera y la segunda reducción o interrupción de dosis fue de 51 días (media, 59,7 días; rango, 15-205 días) y la mediana de la duración de la segunda interrupción de dosis fue de 21 días (media, 26 días; rango, 2-148 días).
Linfoma de células del manto
Se realizó un ensayo multicéntrico, de un único grupo de tratamiento, abierto sobre lenalidomida como agente único para evaluar la seguridad y eficacia de esta droga en pacientes con linfoma de células de manto que han sufrido una recaída o fueron resistentes a bortezomib o al régimen que contiene bortezomib. A los pacientes con clearance de creatinina ≥ 60 mL/min se les administró lenalidomida a una dosis de 25 mg una vez por día durante 21 días cada 28 días. A los pacientes con clearance de creatinina ≥30 mL/min y < 60 mL/min se les administró lenalidomida a una dosis de 10 mg una vez por día durante 21 días cada 28 días. El tratamiento continuó hasta las siguientes situaciones: progresión de la enfermedad, toxicidad inaceptable o suspensión del consentimiento.
El ensayo incluyó pacientes que tenían al menos 18 años de edad con LCM demostrada con biopsia, con enfermedad medible por medio de una tomografía computarizada. Se les requirió a los pacientes que hayan recibido tratamiento previo con antraciclina o mitoxantrona, ciclofosfamida, rituximab y bortezomib, solo o en combinación. A los pacientes se les requirió que tengan una enfermedad resistente documentada (definida como sin respuesta de PR o de mejoría durante el tratamiento con bortezomib o con un régimen que contiene bortezomib), o enfermedad en recaída (definida como progresión dentro de un año posterior al tratamiento con bortezomib o con un régimen que contiene bortezomib). Al momento de la inscripción, los pacientes tuvieran recuentos absolutos de neutrófilos (ANC) ≥ 1500/ mm3, recuentos de plaquetas ≥ 60,000/mm3, límite superior normal de SGOT/AST o SGPT/ALT ≤ 3x séricos a menos que existiera evidencia documentada de implicación del hígado por el linfoma, bilirrubina total sérica ≤1,5 x ULN excepto en casos del síndrome de Gilbert o compromiso documentado del hígado por el linfoma y clearance de creatinina calculado (fórmula Cockcroft-Gault) >30 mL/min.
La mediana de la edad era de 67 años (43-83), el 81 % eran hombres y el 96 % eran caucásicos. La tabla a continuación resume las características basales relacionadas con la enfermedad y la terapia previa contra el linfoma en el ensayo del linfoma de células de manto.
|
Tabla 7: Características basales relacionadas con la enfermedad y terapia previa contra el linfoma en el ensayo del linfoma de células de manto |
|
|
Características basales de la enfermedad y terapia previa contra el linfoma |
Total de pacientes (N=134) |
|
ECOG Estado funcionala – n (%) |
|
|
0 |
43 (32) |
|
1 |
73 (54) |
|
2 |
17 (13) |
|
3 |
1 (<1) |
|
Etapa avanzada de LCM, n (%) |
|
|
III |
27 (20) |
|
IV |
97 (72) |
|
Puntaje MIPI alto o intermediob, n (%) |
90 (67) |
|
Carga tumoral altac, n (%) |
77 (57) |
|
Enfermedad voluminosad, n (%) |
44 (33) |
|
Enfermedad extranodal, n (%) |
101 (75) |
|
Número de terapias sistemáticas anti-linfoma previas, n (%) |
|
|
Mediana (rango) |
4 (2, 10) |
|
1 |
0 (0) |
|
2 |
29 (22) |
|
3 |
34 (25) |
|
≥ 4 |
71 (53) |
|
Número de sujetos que recibieron una terapia previa conteniendo, n (%): |
|
|
Antraciclina/mitoxantrona |
133 (99) |
|
Ciclofosfamida |
133 (99) |
|
Rituximab |
134 (100) |
|
Bortezomib |
134 (100) |
|
Resistente a terapia previa con bortezomib, n (%) |
81 (60) |
|
Resistente a la última terapia, n (%) |
74 (55) |
|
Trasplante autólogo previo de células madre o médula ósea |
39 (29) |
|
a) ECOG = Eastern Cooperative Oncology Group b) MIPI = LCM International Prognostic Index c) La carga tumoral alta se define como al menos una lesión que es ≥5 cm de diámetro o 3 lesiones que son ≥3 cm de diámetro d) La enfermedad voluminosa se define como al menos una lesión que es ≥7cm del diámetro más largo. |
|
Los criterios de valoración en el ensayo de LCM fueron índice de respuesta global (ORR) y duración de la respuesta (DOR). Un comité de revisión independiente determinó la respuesta sobre la base de la revisión de los escaneos radiográficos de acuerdo con una versión modificada de los Criterios de respuesta a los linfomas del taller internacional (Cheson, 1999). La DOR se define como el tiempo desde la respuesta inicial (al menos PR) hasta la progresión documentada de la enfermedad. Los resultados de eficacia de la población de LCM se basaron en todos los pacientes evaluables que recibieron al menos una dosis de la droga en estudio y se presentan en la Tabla 8. La mediana de tiempo hasta la respuesta fue de 2,2 meses (rango 1,8 a 13 meses).
|
Tabla 8: Resultados de respuesta en el ensayo pivotal del linfoma de células de manto |
||
|
Análisis de respuesta (N = 133) |
N (%) |
CI 95% |
|
Índice de respuesta global (IWRC) (CR + CRu +PR) |
34 (26) |
(18,4 ; 33,9) |
|
Respuesta completa (CR + CRu) |
9 (7) |
(3,1; 12,5) |
|
CR |
1 (1) |
|
|
CRu |
8 (6) |
|
|
Respuesta Parcial (PR) |
25 (19) |
|
|
Duración de la respuesta (meses) |
Mediana |
IC 95% |
|
Duración de la respuesta global (CR + CRu + PR) (N = 34) |
16,6 |
(7,7; 26,7) |
Linfoma folicular y de zona marginal
La eficacia de REVLIMID con rituximab fue evaluada en pacientes con linfoma folicular y de zona marginal en recaída o refractario en los estudios AUGMENT ((NCT01938001) y MAGNIFY (NCT01996865).
AUGMENT es un estudio aleatorizado, doble ciego, multicéntrico (n=358) en el cual los pacientes con linfoma folicular y de zona marginal en recaída o refractario se aleatorizaron 1:1 para administrárseles REVLIMID y rituximab o rituximab y placebo. Este estudio incluyó pacientes con diagnóstico de linfoma folicular de grado 1, 2 o 3 que se habían sometido al menos a 1 tratamiento sistémico previo, de tipo refractario o en recaída, no refractario a rituximab, que presentaban al menos una lesión nodular o extra nodular medible y confirmada por TC o resonancia magnética y cuya función medular, hepática y renal era adecuada. La aleatorización se estratificó según linfoma folicular comparado con linfoma de la zona marginal, tratamiento previo con rituximab, y tiempo desde otro tratamiento contra el linfoma. En AUGMENT, REVLIMID se administró una vez al día en una dosis de 20 mg una vez al día durante los días 1 a 21 de ciclos repetidos de 28 días durante un máximo de 12 ciclos o hasta toxicidad inaceptable. La dosis de rituximab fue 375 mg/m2 por semana en el Ciclo 1 (días 1, 8, 15 y 22) y el día 1 de cada uno de los ciclos 2 a 5 de 28 días. Los cálculos de la dosis de rituximab se hicieron según el Área de Superficie Corporal (ASC) del paciente con el peso actual del paciente. Los ajustes de dosis de REVLIMID se permitieron según los hallazgos clínicos y de laboratorio. A un paciente con insuficiencia renal moderada (≥30 to <60 mL/minuto) se le administró una dosis inicial más baja de REVLIMID de 10 mg por día en el mismo esquema. Luego de 2 ciclos, la dosis de REVLIMID se podía aumentar a 15 mg al día en los días 1 a 21 de cada ciclo de 28 días si el paciente toleraba la medicación.
MAGNIFY es un estudio abierto, multicéntrico (n=232) en el cual, a pacientes con linfoma folicular, de la zona marginal o de células manto en recaída o refractario se les administraron 12 ciclos de inducción de REVLIMID y rituximab. El estudio incluyó pacientes con diagnóstico de linfoma folicular grado 1, 2, 3a y 3b (incluyéndose el transformado), de la zona marginal o linfoma de células del manto Etapa I a IV, tratados previamente para dichas enfermedades, que luego del último tratamiento su enfermedad sea refractaria o esté en recaída, que presentaran al menos una lesión nodular o extra nodular medible magnética y cuya función medular, hepática y renal era adecuada. También se incluyeron pacientes refractarios a rituximab. La información de los sujetos a quienes se les administró al menos 1 dosis del tratamiento inicial en los primeros 12 ciclos de inducción (n=222) en el estudio MAGNIFY se incluyó en la evaluación de eficacia de REVLIMID/rituximab en pacientes con linfoma folicular o de la zona marginal en recaída o refractario. En MAGNIFY, se administró una dosis de 20 mg los días 1-21 de los ciclos repetidos de 28 días durante hasta 12 ciclos o toxicidad inaceptable, progresión de la enfermedad o retiro del consentimiento. La dosis de rituximab fue 375 mg/m2 por semana en el Ciclo 1 (días 1, 8, 15 y 22) y el día 1 de ciclo por medio de 28 días (ciclos 3, 5, 7, 9 y 11) hasta completar 12 ciclos. Los cálculos de la dosis de rituximab se hicieron según el Área de Superficie Corporal (ASC) del paciente con el peso actual del paciente. Los ajustes de dosis de REVLIMID se permitieron según los hallazgos clínicos y de laboratorio. La Tabla 9 resume las características basales del paciente y de la enfermedad en los estudios AUGMENT y MAGNIFY.
|
Tabla 9: Características basales demográficas y de la enfermedad en pacientes con LF y LZM en los estudios AUGMENT y MAGNIFY |
|||
|
Parámetro |
Estudio AUGMENT |
Estudio MAGNIFY |
|
|
REVLIMID + Rituximab (N=178) |
Rituximab + Placebo (N=180) |
REVLIMID + Rituximab (N=222) |
|
|
Edad (años) |
|||
|
Mediana (Máx, Mín) |
64 (26, 86) |
62 (35, 88) |
65 (35, 91) |
|
Distribución etaria, n (%) |
|||
|
<65 años |
96 (54) |
107 (59) |
103 (46) |
|
≥65 años |
82 (46) |
73 (41) |
119 (54) |
|
Sexo, n (%) |
|||
|
Hombres |
75 (42) |
97 (54) |
122 (55) |
|
Mujeres |
103 (58) |
83 (46) |
100 (45) |
|
Raza |
|||
|
Blancos |
118 (66) |
115 (64) |
206 (93) |
|
Otras razas |
54 (30) |
64 (36) |
14 (6) |
|
No recabada o informada |
6 (3) |
1 (0,6) |
2 (1) |
|
Área de superficie corporal (ASC, m2) |
|||
|
Mediana (Máx, Mín) |
1,8 (1,4; 3,1) |
1,8 (1,3; 2,7) |
2 (1,3; 2,6) |
|
Tipo de enfermedad LF o LZM |
|||
|
Linfoma folicular |
147 (83) |
148 (82) |
177 (80) |
|
Linfoma de zona marginal |
31 (17) |
32 (18) |
45 (20) |
|
Subtipo LZM en el diagnóstico (investigador), n (%) |
|||
|
MALT |
14 (45) |
16 (50) |
10 (22) |
|
Nodal |
8 (26) |
10 (31) |
25 (56) |
|
Esplénico |
9 (29) |
6 (19) |
10 (22) |
|
Etapa de LF en el diagnóstico (investigador), n (%) |
|||
|
Grado 1-2 |
125 (85) |
123 (83) |
149 (84) |
|
Grado 3a |
22 (15) |
25 (17) |
28 (16) |
|
Indice FLIPI basal (calculado), n (%) |
No recabado |
||
|
Riesgo bajo (0,1) |
52 (29) |
67 (37) |
|
|
Riesgo intermedio (2) |
55 (31) |
58 (32) |
|
|
Riesgo alto (≥3) |
69 (39) |
54 (30) |
|
|
Ausencia de datos |
2 (1) |
1 (0,6) |
|
|
ECOG basal, n (%) |
|||
|
0 |
116 (65) |
128 (71) |
102 (46) |
|
1 |
60 (34) |
50 (28) |
113 (51) |
|
2 |
2 (1) |
2 (1) |
7 (3) |
|
Carga tumoral altab basal, n (%) |
|||
|
Si |
97 (54) |
86 (48) |
148 (67) |
|
No |
81 (46) |
94 (52) |
74 (33) |
|
Cantidad de tratamientos previos sistémicos contra el linfoma |
|||
|
1 |
102 (57) |
97 (54) |
94 (42) |
|
>1 |
76 (43) |
83 (46) |
128 (58) |
|
Fecha de corte de datos: 22 de junio de 2018 (AUGMENT) y 1 de mayo de 2017 (MAGNIFY) a El paciente presentó 0 (n=2) o 1 tratamiento sistémico previo. b Definido por criterios GELF ECOG: Eastern Cooperative Oncology Group. FLIPI: Índice de Pronóstico Internacional Específico Para el Linfoma Folicular. |
|||
En AUGMENT, un Comité de Revisión Independiente (IRC) midió la eficacia en la población por Intención de Tratar (ITT) en base a la supervivencia libre de progresión mediante los criterios de respuesta del International Working Group del año 2007. Los resultados de eficacia se resumen en la Tabla 10.
|
Tabla 10: Resultados de eficacia en pacientes del estudio AUGMENT (Población ITT de LF y LZM) |
||
|
Parámetro |
REVLIMID + Rituximab (N=178) |
Rituximab + Placebo (N=180) |
|
SLP |
||
|
Pacientes con evento, n (%) |
68 (38,2) |
115 (63,9) |
|
Muerte |
6 (8,8) |
2 (1,7) |
|
Progresión de la enfermedad |
62 (91,2) |
113 (98,3) |
|
SLP, mediana a [95% CI] (meses) |
39,4 [ 22,9; NE] |
14,1 [11,4; 16,7] |
|
HR b [95% CI] |
0,46 [ 0,34; 0,62] |
|
|
Valor p c |
<0,0001 |
|
|
Respuesta objetiva (RC+RP), n(%) [95% CI] d |
138 (77,5) |
96 (53,3) |
|
a Mediana calculada a partir del análisis Kaplan-Meier b Cociente de riesgo y su IC calculados mediante el modelo de riesgos proporcionales de Cox ajustado a las 3 estratificaciones: tratamiento previo con rituximab (si, no), tiempo dese el último tratamiento contra el linfoma (≤ 2, >2 años) e histología de la enfermedad (LF, LZM). c valor p basado en la prueba del orden logarítmico estratificado según los 3 factores mencionados: tratamiento previo con rituximab (si, no), tiempo dese el último tratamiento contra el linfoma (≤ 2, >2 años) e histología de la enfermedad (LF, LZM). d Intervalo de confianza exacto para distribución binomial. |
||
Curvas de Kaplan-Meier de Supervivencia libre de progresión según evaluación del IRC entre los brazos del estudio AUGMENT ((Población ITT de LF y LZM)
A = Factores de estratificación: tratamiento previo con rituximab (si, no), tiempo dese el último tratamiento contra el linfoma (≤ 2, >2 años) e histología de la enfermedad (LF, LZM).
CI: intervalo de confianza; HR= cociente de riesgo; KM= Kaplan-Meier; PFS: supervivencia libre de progresión. Linfoma folicular
En AUGMENT, la respuesta objetiva según evaluación del IRC en pacientes con linfoma folicular fue 80% (118/147) [IC 95%: 73%, 86%)] en el brazo REVLIMID y rituximab comparado con 55% (82/148) [IC 95%: 47%, 64%)] en el brazo control.
En MAGNIFY, la respuesta global según evaluación del investigador fue 59% (104/177) [IC 95%: 51,66] en pacientes con linfoma folicular. La mediana de duración de la respuesta no se alcanzó con una mediana de tiempo de seguimiento de 7,9 meses [IC 95%: 4,6; 9,2).
Linfoma de la zona marginal
En AUGMENT, la respuesta objetiva según evaluación del IRC en pacientes con linfoma de la zona marginal fue 65% (20/31) [IC 95%: 45%, 81%)] en el brazo REVLIMID y rituximab comparado con 44% (14/32) [IC 95%: 26%, 62%)] en el brazo control.
En MAGNIFY, la respuesta global según evaluación del investigador fue 51% (23/45) [IC 95%: 36,66] en pacientes con linfoma de la zona marginal. La mediana de duración de la respuesta no se alcanzó con una mediana de tiempo de seguimiento de 11,5 meses [IC 95%: 8,0; 18,9).
LEYENDAS DE PROTECCIÓN:
Mantener fuera del alcance de los niños.
Este medicamento se encuentra dentro del plan de farmacovigilancia activa, y presenta plan de gestión de riesgo.
Este medicamento solo debe utilizarse bajo estricto control y vigilancia médica y no puede repetirse sin nueva receta médica.
Especialidad medicinal autorizada por el Ministerio de Salud.