NEUPOGEN
FILGRASTIM
Jeringas precargadas
Caja, 1 Jeringa prellenada, 30/0.5 MU
COMPOSICIÓN:
Formas farmacéuticas y concentraciones
Neupogen es una solución transparente, incolora y libre de conservantes, disponible como:
Jeringa precargada:
• Inyección: 300 mcg/0,5 mL en una jeringa precargada de dosis única
PRINCIPIO ACTIVO (S) / GRUPO FARMACOLÓGICO:
Descripción
Filgrastim es un factor estimulante de colonias de granulocitos humanos (G-CSF) de 175 aminoácidos fabricado por tecnología de ADN recombinante. Filgrastim es producido por la bacteria Escherichia coli (E coli) en la que se ha insertado el gen del factor estimulante de colonias de granulocitos humanos. Filgrastim tiene un peso molecular de 18.800 daltons. La proteína tiene una secuencia de aminoácidos que es idéntica a la secuencia natural predicha del análisis de secuencia de ADN humano, excepto por la adición de una metionina N-terminal necesaria para la expresión en E. coli. Debido a que filgrastim se produce en E. coli, el producto no está glicosilado y, por lo tanto, difiere del G-CSF aislado de una célula humana.
La inyección de Neupogen es un líquido estéril, transparente, incoloro, sin conservantes que contiene filgrastim a una actividad específica de 1,0 ± 0.6x108 U/mg (medido por un ensayo de mitogénesis celular). El producto está disponible en viales de dosis única y jeringas prellenadas. Los viales de dosis única contienen 300 mcg/mL o 480 mcg/1,6 mL de filgrastim. Las jeringas prellenadas de dosis única contienen 300 mcg/0,5 mL o 480 mcg/0,8 mL de filgrastim. Consulte la tabla a continuación para ver la composición del producto de cada jeringa precargada.
|
300 mcg/mL (Vial) |
480 mcg/1,6 mL (Vial) |
300 mcg/0,5 mL (Jeringa) |
480 mcg/0,8 mL (Jeringa) |
|
|
Filgrastim |
300 mcg |
480 mcg |
300 mcg |
480 mcg |
|
Acetato |
0,59 mg |
0,94 mcg |
0,295 mg |
0,472 mg |
|
Polisorbato 80 |
0,04 mg |
0,064 mg |
0,02 mg |
0,032 mg |
|
Sodio |
0,035 mg |
0,056 mg |
0,0175 mg |
0,028 mg |
|
Sorbitol |
50 mg |
80 mg |
25 mg |
40 mg |
|
Agua para inyección USP c.s.* |
1 mL |
1,6 mL |
0,5 mL |
0,8 mL |
|
* Cantidad suficiente para |
||||
INDICACIONES TERAPÉUTICAS:
Indicaciones y usos
Pacientes con cáncer que reciben quimioterapia mielosupresora
Neupogen está indicado para disminuir la incidencia de infección, como se manifiesta por neutropenia febril en pacientes con neoplasias no mieloides que reciben medicamentos anticancerígenos mielosupresores asociados con una incidencia significativa de neutropenia grave con fiebre (ver Estudios clínicos).
Pacientes con leucemia mieloide aguda que reciben quimioterapia de inducción o consolidación
Neupogen está indicado para reducir el tiempo de recuperación de neutrófilos y la duración de la fiebre, después del tratamiento de quimioterapia de inducción o consolidación de pacientes con leucemia mieloide aguda (LMA) (ver Estudios clínicos).
Pacientes con cáncer sometidos a trasplante de médula ósea
Neupogen está indicado para reducir la duración de la neutropenia y las secuelas clínicas relacionadas con la neutropenia, por ejemplo, neutropenia febril, en pacientes con neoplasias no mieloides sometidas a quimioterapia mieloablativa seguida de trasplante de médula ósea (ver Estudios clínicos).
Pacientes sometidos a recolección y terapia de células progenitoras de sangre periférica autólogas
Neupogen está indicado para la movilización de células progenitoras hematopoyéticas autólogas en la sangre periférica para su recolección por leucoféresis (ver Estudios clínicos).
Pacientes con neutropenia crónica severa
Neupogen está indicado para administración crónica para reducir la incidencia y la duración de las secuelas de neutropenia (p. Ej., fiebre, infecciones, úlceras orofaríngeas) en pacientes sintomáticos con neutropenia congénita, neutropenia cíclica o neutropenia idiopática (ver Estudios clínicos).
Pacientes expuestos de forma aguda a dosis de radiación mielosupresoras (Síndrome hematopoyético del síndrome de radiación aguda)
Neupogen está indicado para aumentar la supervivencia en pacientes con exposición aguda a dosis de radiación mielosupresoras (ver Estudios clínicos).
MECANISMO DE ACCIÓN
Farmacología clínica
Grupo farmacoterapéutico: Citocinas, Código ATC: L03AA02
Mecanismo de acción
Los factores estimulantes de colonias son las glicoproteínas que actúan sobre las células hematopoyéticas uniéndose a receptores específicos de la superficie celular y estimulando la proliferación, el compromiso de diferenciación y alguna activación funcional de la célula final.
El G-CSF endógeno es un factor estimulante de colonias de linaje específico que es producido por monocitos, fibroblastos y células endoteliales. El G-CSF regula la producción de neutrófilos dentro de la médula ósea y afecta la proliferación de progenitores de neutrófilos, la diferenciación y las funciones seleccionadas de las células terminales (incluida la capacidad fagocítica mejorada, el cebado del metabolismo celular asociado con la explosión respiratoria, la muerte dependiente de anticuerpos y el aumento de la expresión de algunos antígenos de superficie celular). G-CSF no es específico de la especie y se ha demostrado que tiene efectos directos mínimos in vivo o in vitro sobre la producción o actividad de tipos de células hematopoyéticas distintas del linaje de neutrófilos.Propiedades farmacodinámicas
En los estudios de fase 1 en los que participaron 96 pacientes con diversas neoplasias malignas no mieloides, la administración de Neupogen resultó en un aumento dependiente de la dosis en los recuentos de neutrófilos circulantes en el rango de dosis de 1 a 70 mcg/kg/día. Este aumento en los recuentos de neutrófilos se observó tanto si Neupogen se administró por vía intravenosa (1 a 70 mcg/kg dos veces al día), subcutáneo (1 a 3 mcg/kg una vez al día), o por infusión subcutánea continua (3 a 11 mcg/kg/día). Con la interrupción de la terapia con Neupogen, los recuentos de neutrófilos volvieron al valor basal en la mayoría de los casos en 4 días. Los neutrófilos aislados mostraron actividad fagocítica (medida por quimioluminiscencia estimulada por zimosán) y quimiotáctica (medida por migración bajo agarosa usando N-formil-metionil-leucil-fenilalanina [fMLP] como la quimiotaxina) normal in vitro.
Se informó que el recuento absoluto de monocitos aumenta de manera dependiente de la dosis en la mayoría de los pacientes que reciben Neupogen; sin embargo, el porcentaje de monocitos en el recuento diferencial se mantuvo dentro del rango normal. Los recuentos absolutos de eosinófilos y basófilos no cambiaron y estuvieron dentro del rango normal después de la administración de Neupogen. Se han informado aumentos en los recuentos de linfocitos después de la administración de Neupogen en algunos sujetos normales y pacientes con cáncer.
Los diferenciales de glóbulos blancos (WBC) obtenidos durante los ensayos clínicos han demostrado un cambio hacia las células progenitoras de granulocitos anteriores (desplazamiento a la izquierda), incluida la aparición de promielocitos y mieloblastos, generalmente durante la recuperación de neutrófilos después del nadir inducido por quimioterapia. Además, los cuerpos de Dohle aumentaron la granulación de granulocitos y se observaron neutrófilos hipersegmentados. Tales cambios fueron transitorios y no se asociaron con secuelas clínicas, ni se asociaron necesariamente con infección.
Propiedades farmacocinéticas
Filgrastim exhibe farmacocinética no lineal. El aclaramiento depende de la concentración de filgrastim y del recuento de neutrófilos: el aclaramiento mediado por el receptor G-CSF está saturado por una alta concentración de Neupogen y disminuye por neutropenia. Además, filgrastim es eliminado por el riñón. |
La administración subcutánea de 3,45 mcg/kg y 11,5 mcg/kg de filgrastim dio como resultado concentraciones séricas máximas de 4 ng/mL y 49 ng/mL, respectivamente, dentro de 2 a 8 horas. Después de la administración intravenosa, el volumen de distribución promedió 150 mL/kg y la vida media de eliminación fue de aproximadamente 3,5 horas tanto en sujetos normales como en sujetos con cáncer. Las tasas de aclaramiento de filgrastim fueron de aproximadamente 0,5 mL/minuto/kg a 0,7 mL/minuto/kg. Las dosis parenterales únicas o las dosis intravenosas diarias, durante un período de 14 días, dieron como resultado semividas comparables. Las vidas medias fueron similares para la administración intravenosa (231 minutos, después de dosis de 34,5 mcg/kg) y para la administración subcutánea (210 minutos, después de dosis de Neupogen de 3,45 mcg/kg). Las infusiones intravenosas continuas de 24 horas de 20 mcg/kg durante un período de 11 a 20 días produjeron concentraciones séricas estables de filgrastim sin evidencia de acumulación de drogas durante el período investigado. La biodisponibilidad absoluta de filgrastim después de la administración subcutánea es del 60% al 70%.
Poblaciones específicas
Pacientes expuestos de forma aguda a dosis mielosupresoras de radiación
La farmacocinética de filgrastim no está disponible en pacientes con exposición aguda a dosis mielosupresoras de radiación. Con base en datos farmacocinéticos limitados en primates no humanos irradiados, el área bajo la curva de concentración de tiempo (AUC), que refleja la exposición a filgrastim en primates no humanos a una dosis de 10 mcg/kg de Neupogen, parece ser similar a la de los humanos a 5 mcg/kg. Las simulaciones realizadas con el modelo farmacocinético de la población indican que las exposiciones a filgrastim a una dosis de Neupogen de 10 mcg/kg en pacientes con exposición aguda a dosis mielosupresoras de radiación se espera que excedan las exposiciones a una dosis de 10 mcg/kg en primates no humanos irradiados.
Pacientes pediátricos
La farmacocinética de filgrastim en pacientes pediátricos después de la quimioterapia es similar a la de los pacientes adultos que reciben las mismas dosis normalizadas de peso, lo que sugiere que no hay diferencias relacionadas con la edad en la farmacocinética de filgrastim (ver sección Uso en poblaciones específicas).
Insuficiencia renal
En un estudio con voluntarios sanos, sujetos con insuficiencia renal moderada y sujetos con enfermedad renal en etapa terminal (n = 4 por grupo), se observaron concentraciones séricas más altas en sujetos con enfermedad renal en etapa terminal. Sin embargo, no es necesario ajustar la dosis en pacientes con insuficiencia renal.
Insuficiencia hepática
La farmacocinética y la farmacodinámica de filgrastim son similares entre sujetos con insuficiencia hepática y sujetos sanos (n = 12/grupo). El estudio incluyó a 10 sujetos con insuficiencia hepática leve (Child Pugh Clase A) y 2 sujetos con insuficiencia hepática moderada (Child Pugh Clase B). Por lo tanto, no es necesario ajustar la dosis de filgrastim para pacientes con insuficiencia hepática.
CONTRAINDICACIONES:
Neupogen está contraindicado en pacientes con antecedentes de reacciones alérgicas graves a factores estimulantes de colonias de granulocitos humanos como filgrastim o pegfilgrastim (ver Advertencias y Precauciones).
REACCIONES ADVERSAS:
Las siguientes reacciones adversas graves se analizan con mayor detalle en otras secciones del etiquetado:
• Ruptura esplénica (ver Advertencias y Precauciones).
• Síndrome de distrés respiratorio agudo (ver Advertencias y Precauciones).
• Reacciones alérgicas graves (ver Advertencias y Precauciones).
• Trastornos de células falciformes (ver Advertencias y Precauciones).
• Glomerulonefritis (ver Advertencias y Precauciones).
• Hemorragia alveolar y hemoptisis (ver Advertencias y Precauciones).
• Síndrome de fuga capilar (ver Advertencias y Precauciones).
• Trombocitopenia (ver Advertencias y Precauciones).
• Leucocitosis (ver Advertencias y Precauciones).
• Vasculitis cutánea (ver Advertencias y Precauciones).
• Aortitis (ver Advertencias y Precauciones).
Experiencia en ensayos clínicos
Debido a que los ensayos clínicos se realizan en condiciones muy variables, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no se pueden comparar directamente con las tasas en los ensayos clínicos de otro fármaco y pueden no reflejar las tasas observadas en la práctica clínica.
Reacciones adversas en pacientes con cáncer que reciben quimioterapia mielosupresora
Los siguientes datos de reacciones adversas en la Tabla 2 son de tres estudios aleatorizados, controlados con placebo en pacientes con:
• cáncer de pulmón de células pequeñas que recibe quimioterapia de dosis estándar con ciclofosfamida, doxorrubicina y etopósido (Estudio 1)
• cáncer de pulmón de células pequeñas que recibe ifosfamida, doxorrubicina y etopósido (Estudio 2), y
• Linfoma no Hodgkin (NHL) que recibe doxorrubicina, ciclofosfamida, vindesina, bleomicina, metilprednisolona y metotrexato ("ACVBP") o mitoxantrona, ifosfamida, mitoguazona, teniposido, metotrexato, ácido folínico, metilprednisolona y megtotrexato (“VIM3”) (Estudio 3).
Se aleatorizó un total de 451 pacientes para recibir Neupogen subcutáneo 230 mcg/m2 (Estudio 1), 240 mcg/m2 (Estudio 2) o 4 mcg/kg/día o 5 mcg/kg/día (Estudio 3) (n = 294) o placebo (n = 157). Los pacientes en estos estudios tenían una mediana de edad de 61 (rango 29 a 78) años y el 64% eran hombres. La etnia era 95% caucásica, 4% afroamericana y 1% asiática.
|
Tabla 2. Reacciones adversas en pacientes con cáncer que reciben quimioterapia mielosupresora (con una incidencia ≥ 5% mayor en Neupogen en comparación con placebo) |
||
|
Sistema de Clasificación de Órganos Término preferido |
Neupogen (N = 294) |
Placebo (N = 157) |
|
Transtornos de la sangre y del sistema linfático |
||
|
Trombocitopenia |
38% |
29% |
|
Trastornos gastrointestinales |
||
|
Náuseas |
43% |
32% |
|
Trastornos generales y condiciones del sitio de inyección |
||
|
Pirexia |
48% |
29% |
|
Dolor de pecho |
13% |
6% |
|
Dolor |
12% |
6% |
|
Fátiga |
20% |
10% |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||
|
Dolor de espalda |
15% |
8% |
|
Artralgia |
9% |
2% |
|
Dolor óseo |
11% |
6% |
|
Dolor en extremidades* |
7% |
3% |
|
Trastornos del sistema nervioso |
||
|
Mareos |
14% |
3% |
|
Trastornos respiratorios tóracicos y mediástinicos |
||
|
Tos |
14% |
8% |
|
Disnea |
13% |
8% |
|
Trastornos de la piel y tejido subcutáneo |
||
|
Rash |
14% |
5% |
|
Investigaciones |
||
|
Aumento de la lactato deshidrogenasa en sangre |
6% |
1% |
|
Aumento de la fosfatasa alcalina en sangre |
6% |
1% |
|
* La diferencia porcentual (Neupogen - Placebo) fue del 4%. |
||
Los eventos adversos con una incidencia ≥ 5% mayor en pacientes con Neupogen en comparación con placebo y asociados con las secuelas de la neoplasia maligna subyacente o quimioterapia citotóxica administrada incluyeron anemia, estreñimiento, diarrea, dolor oral, vómitos, astenia, malestar, edema periférico, disminución de la hemoglobina, disminución del apetito, dolor orofaríngeo y alopecia.
Reacciones adversas en pacientes con leucemia mieloide aguda
Los datos de reacciones adversas a continuación son de un estudio aleatorizado, doble ciego, controlado con placebo en pacientes con LMA (Estudio 4) que recibieron un régimen de quimioterapia de inducción de daunorrubicina intravenosa los días 1, 2 y 3; arabinósido de citosina los días 1 a 7; y etopósido los días 1 a 5 y hasta 3 cursos adicionales de terapia (inducción 2 y consolidación 1, 2) de daunorrubicina intravenosa, arabinósido de citosina y etopósido. La población de seguridad incluyó a 518 pacientes asignados al azar para recibir 5 mcg/kg /día de Neupogen (n = 257) o placebo (n = 261). La mediana de edad fue de 54 (rango 16 a 89) años y el 54% eran hombres.Las reacciones adversas con una incidencia ≥ 2% mayor en pacientes con Neupogen en comparación con placebo incluyeron epistaxis, dolor de espalda, dolor en las extremidades, eritema y erupción maculo papular. Los eventos adversos con una incidencia ≥ 2% mayor en pacientes con Neupogen en comparación con placebo y asociados con las secuelas de la neoplasia maligna subyacente o la quimioterapia citotóxica incluyeron diarrea, estreñimiento y reacción a la transfusión. Reacciones adversas en pacientes con cáncer sometidos a trasplante de médula ósea
Los siguientes datos de reacciones adversas provienen de un estudio aleatorizado sin tratamiento controlado en pacientes con leucemia linfoblástica aguda o linfoma linfoblástico que reciben quimioterapia de dosis altas (ciclofosfamida o citarabina y melfalan) e irradiación corporal total (Estudio 5) y un estudio aleatorizado sin tratamiento controlado en pacientes con enfermedad de Hodgkin (HD) y NHL sometidos a altas dosis de quimioterapia y trasplante autólogo de médula ósea (Estudio 6). Los pacientes que recibieron trasplante autólogo de médula ósea solo se incluyeron en el análisis. Un total de 100 pacientes recibieron tanto 30 mcg/kg/día como una infusión de 4 horas (Estudio 5) o 10 mcg/kg/día o 30 mcg/kg/día como una infusión de 24 horas (Estudio 6) de Neupogen (n = 72), sin control de tratamiento o placebo (n = 28). La mediana de edad fue de 30 (rango de 15 a 57) años, el 57% eran hombres.
Las reacciones adversas con una incidencia ≥ 5% mayor en pacientes con Neupogen en comparación con los pacientes que no recibieron Neupogen incluyeron erupción cutánea e hipersensibilidad.
Las reacciones adversas en pacientes que recibieron quimioterapia intensiva seguida de TMO autólogo con una incidencia ≥ 5% mayor en pacientes con Neupogen en comparación con los pacientes que no recibieron Neupogen incluyeron trombocitopenia, anemia, hipertensión, sepsis, bronquitis e insomnio.
Reacciones adversas en pacientes con cáncer sometidos a recolección de células progenitoras de sangre periférica autóloga
Los datos de reacciones adversas en la Tabla 3 provienen de una serie de 7 ensayos en pacientes con cáncer sometidos a movilización de células progenitoras de sangre periférica autólogas para su recolección por leucoféresis. Los pacientes (n = 166) en todos estos ensayos se sometieron a un régimen de movilización / recolección similar: se administró Neupogen durante 6 a 8 días, en la mayoría de los casos el procedimiento de aféresis se produjo en los días 5, 6 y 7. La dosis de Neupogen varió entre 5 mcg/kg/día a 30 mcg/kg/día y se administró por vía subcutánea mediante inyección o infusión continua. La mediana de edad fue de 39 (rango de 15 a 67) años, y el 48% eran hombres.
|
Tabla 3. Reacciones adversas en pacientes con cáncer sometidos a PBPC autólogo en la fase de movilización (≥ 5% de incidencia en pacientes con Neupogen) |
|
|
Sistema de clasificación de órganos Término preferido |
Fase de movilización (N = 166) |
|
Trastornos musculoesqueléticos y del tejido conectivo |
|
|
Dolor óseo |
30% |
|
Trastornos generales y condiciones del sitio de inyección |
|
|
Pirexia |
16% |
|
Investigaciones |
|
|
Aumento de la fosfatasa alcalina en sangre |
11% |
|
Trastornos del sistema nervioso |
|
|
Dolor de cabeza |
10% |
Reacciones adversas en pacientes con neutropenia crónica severa
Los siguientes datos de reacciones adversas se identificaron en un estudio aleatorizado y controlado en pacientes con SCN que recibieron Neupogen (Estudio 7). 123 pacientes fueron asignados al azar a un período de observación de 4 meses seguido de tratamiento con Neupogen subcutáneo o tratamiento con Neupogen subcutáneo inmediato. La mediana de edad fue de 12 años (rango de 7 meses a 76 años) y el 46% eran hombres. La dosis de Neupogen se determinó por la categoría de neutropenia. Dosis inicial de Neupogen:
• Neutropenia idiopática: 3,6 mcg/kg/día.
• Neutropenia cíclica: 6 mcg/kg/día.
• Neutropenia congénita: 6 mcg/kg/día dividido 2 veces por día.
La dosis se incrementó gradualmente a 12 mcg/kg/día dividido 2 veces por día si no hubo respuesta.
Las reacciones adversas con una incidencia ≥ 5% mayor en pacientes con Neupogen en comparación con los pacientes que no recibieron Neupogen incluyeron artralgia, dolor óseo, dolor de espalda, espasmos musculares, dolor musculoesquelético, dolor en las extremidades, esplenomegalia, anemia, infección del tracto respiratorio superior e infección del tracto urinario (la infección del tracto respiratorio superior y la infección del tracto urinario fueron mayores en el brazo de Neupogen, los eventos relacionados con la infección total fueron menores en los pacientes tratados con Neupogen), epistaxis, dolor torácico, diarrea, hipoestesia y alopecia.Inmunogenicidad
Como con todas las proteínas terapéuticas, existe un potencial de inmunogenicidad. La detección de la formación de anticuerpos depende en gran medida de la sensibilidad y la especificidad del ensayo, y la incidencia observada de positividad de anticuerpos (incluido el anticuerpo neutralizante) en un ensayo puede verse influenciada por varios factores, incluidos la metodología del ensayo, el manejo de la muestra, el momento de la recolección de la muestra, medicamentos concomitantes y enfermedad subyacente. Por estos motivos, la comparación de la incidencia de anticuerpos contra filgrastim en los estudios que se describen a continuación con la incidencia de anticuerpos en otros estudios u otros productos puede ser engañosa.
La incidencia del desarrollo de anticuerpos en pacientes que reciben Neupogen no se ha determinado adecuadamente. Si bien los datos disponibles sugieren que una pequeña proporción de pacientes desarrolló anticuerpos de unión a filgrastim, la naturaleza y especificidad de estos anticuerpos no se ha estudiado adecuadamente. En estudios clínicos con Neupogen, la incidencia de anticuerpos que se unen a filgrastim fue del 3% (11/333). En estos 11 pacientes, no se observó evidencia de una respuesta neutralizante utilizando un bioensayo basado en células.
Las citopenias resultantes de una respuesta de anticuerpos a factores de crecimiento exógenos se han informado en raras ocasiones en pacientes tratados con otros factores de crecimiento recombinantes.
Experiencia posterior a la comercialización
Se han identificado las siguientes reacciones adversas durante el uso posterior a la aprobación de Neupogen. Debido a que estas reacciones se informan voluntariamente de una población de tamaño incierto, no siempre es posible estimar de manera confiable su frecuencia o establecer una relación causal con la exposición al fármaco.
• ruptura esplénica y esplenomegalia (bazo agrandado) (ver Advertencias y Precauciones)
• síndrome de distrés respiratorio agudo (ver Advertencias y Precauciones)
• anafilaxia (ver Advertencias y Precauciones)
• trastornos de células falciformes (ver Advertencias y Precauciones)
• glomerulonefritis (ver Advertencias y Precauciones)
• hemorragia alveolar y hemoptisis (ver Advertencias y Precauciones)
• síndrome de fuga capilar (ver Advertencias y Precauciones)
• leucocitosis (ver Advertencias y Precauciones)
• vasculitis cutánea (ver Advertencias y Precauciones)
• Síndrome de Sweet (dermatosis neutrofílica febril aguda)
• disminución de la densidad ósea y osteoporosis en pacientes pediátricos que reciben tratamiento crónico con Neupogen
• aortitis (ver Advertencias y Precauciones)
Toxicología preclínica
Carcinogénesis, mutagénesis, deterioro de la fertilidad
El potencial carcinogénico de filgrastim no se ha estudiado. Filgrastim no logró inducir mutaciones genéticas bacterianas ni en presencia ni en ausencia de un sistema enzimático metabolizador de fármacos. Filgrastim no tuvo ningún efecto observado sobre la fertilidad de ratas macho o hembra en dosis de hasta 500 mcg/kg.
Toxicología y farmacología animal
Filgrastim se administró a monos, perros, hámsteres, ratas y ratones como parte de un programa de toxicología preclínica, que incluyó estudios de hasta 1 año de duración.En los estudios de dosis repetidas, los cambios observados fueron atribuibles a las acciones farmacológicas esperadas de filgrastim (es decir, aumentos dependientes de la dosis en el recuento de glóbulos blancos, aumento de neutrófilos segmentados circulantes y aumento de la relación mieloide:eritroide en la médula ósea). El examen histopatológico del hígado y el bazo reveló evidencia de granulopoyesis extramedular en curso, y se observaron aumentos relacionados con la dosis en el peso del bazo en todas las especies. Todos estos cambios se revirtieron después de la interrupción del tratamiento.
INFORMACIÓN COMPLEMENTARIA:
Fecha de referencia
Versión 4
Abril 2019
MEDICAMENTA
Casilla 17-21-027 Quito, Ecuador
RECOMENDACIONES:
Advertencias y precauciones
Ruptura esplénica
Se ha informado de ruptura esplénica, incluidos casos fatales, tras la administración de Neupogen. Evaluar a los pacientes que informan dolor en la parte superior del abdomen o el hombro izquierdo por un bazo agrandado o una ruptura esplénica.
Síndrome de distrés respiratorio agudo
El Síndrome de Distrés Respiratorio Agudo (SDRA) se ha informado en pacientes que reciben Neupogen.
Evaluar a los pacientes que desarrollan fiebre e infiltrados pulmonares o distrés respiratorio para SDRA. Interrumpir tratamiento con Neupogen en pacientes con SDRA. Reacciones alérgicas graves
Se han notificado reacciones alérgicas graves, incluida la anafilaxia, en pacientes que reciben Neupogen. La mayoría de los eventos reportados ocurrieron tras la exposición inicial. Proporcionar tratamiento sintomático para reacciones alérgicas. Las reacciones alérgicas, incluida la anafilaxia, en pacientes que reciben Neupogen pueden reaparecer dentro de los días posteriores a la interrupción del tratamiento antialérgico inicial. Suspender permanentemente Neupogen en pacientes con reacciones alérgicas graves. Neupogen está contraindicado en pacientes con antecedentes de reacciones alérgicas graves a factores estimulantes de colonias de granulocitos humanos como filgrastim o pegfilgrastim.
Trastornos de células falciformes
En pacientes con trastornos de células falciformes que reciben productos de filgrastim pueden ocurrir crisis severas y a veces fatales de células falciformes. Suspenda Neupogen si se produce una crisis drepanocítica. Glomerulonefritis
Se ha producido glomerulonefritis en pacientes que reciben Neupogen. Los diagnósticos se basaron en azotemia, hematuria (microscópica y macroscópica), proteinuria y biopsia renal. En general, los eventos de glomerulonefritis se resolvieron después de la reducción de la dosis o la interrupción de Neupogen. Si se sospecha glomerulonefritis, evalúe la causa. Si es probable la causalidad, considere la reducción de la dosis o la interrupción de Neupogen.
Hemorragia alveolar y hemoptisis
Se ha informado hemorragia alveolar que se manifiesta como infiltrados pulmonares y hemoptisis que requieren hospitalización en donantes sanos tratados con Neupogen sometidos a movilización de recolección de Células Progenitoras de Sangre Periférica (PBPC). La hemoptisis se resolvió con la interrupción de Neupogen. El uso de Neupogen para la movilización de PBPC en donantes sanos no es una indicación aprobada.
Síndrome de fuga capilar
El Síndrome de Fuga Capilar (CLS) se ha informado después de la administración de G-CSF, incluido Neupogen, y se caracteriza por hipotensión, hipoalbuminemia, edema y hemoconcentración. Los episodios varían en frecuencia, gravedad y pueden ser mortales si el tratamiento se retrasa. Los pacientes que desarrollan síntomas del síndrome de fuga capilar deben ser monitoreados de cerca y recibir un tratamiento sintomático estándar, que puede incluir la necesidad de cuidados intensivos.
Pacientes con neutropenia crónica severa
Confirme el diagnóstico de SCN antes de iniciar la terapia con Neupogen.
Se ha informado que el Síndrome Mielodisplásico (SMD) y la Leucemia Mielógena Aguda (LMA) se presentan en la historia natural de la neutropenia congénita sin terapia con citocinas. También se han observado anormalidades citogenéticas, transformación a SMD, y LMA en pacientes tratados con Neupogen para SCN. Según los datos disponibles, incluido un estudio de vigilancia posterior a la comercialización, el riesgo de desarrollar SMD y LMA parece estar limitado al subconjunto de pacientes con neutropenia congénita. La citogenética anormal y el SMD se han asociado con el desarrollo eventual de leucemia mieloide. Se desconoce el efecto de Neupogen en el desarrollo de citogenética anormal y el efecto de la administración continua de Neupogen en pacientes con citogenética anormal o SMD. Si un paciente con SCN desarrolla citogenética anormal o mielodisplasia, los riesgos y beneficios de continuar con Neupogen deben considerarse cuidadosamente.
Trombocitopenia
Se ha informado de trombocitopenia en pacientes que reciben Neupogen. Monitoreé los recuentos de plaquetas.Leucocitosis
Pacientes con cáncer que reciben quimioterapia mielosupresora
Se observaron recuentos de glóbulos blancos de 100.000/mm3 o más en aproximadamente, el 2% de los pacientes que recibieron Neupogen a dosis superiores a 5 mcg/kg /día. En pacientes con cáncer que reciben Neupogen como un complemento de la quimioterapia mielosupresora, para evitar los riesgos potenciales de leucocitosis excesiva, se recomienda suspender la terapia con Neupogen si el RAN supera los 10.000/mm3 después de que se haya producido el nadir de RAN inducido por la quimioterapia. Monitoreé los CSCs al menos dos veces por semana durante la terapia. Las dosis de Neupogen que aumentan el RAN más allá de 10.000/mm3 pueden no generar ningún beneficio clínico adicional. En pacientes con cáncer que reciben quimioterapia mielosupresora, la interrupción de la terapia con Neupogen generalmente, produjo una disminución del 50% en los neutrófilos circulantes en 1 a 2 días, con un retorno a los niveles de pretratamiento en 1 a 7 días. Recolección y terapia de células progenitoras de sangre periférica
Durante el período de administración de Neupogen para la movilización de PBPC en pacientes con cáncer, suspenda Neupogen si el recuento de leucocitos aumenta a > 100.000/mm3.
Vasculitis cutánea
Se ha informado de vasculitis cutánea en pacientes tratados con Neupogen. En la mayoría de los casos, la gravedad de la vasculitis cutánea fue moderada o grave. La mayoría de los informes incluyeron pacientes con SCN que recibieron terapia de Neupogen a largo plazo. Mantenga la terapia con Neupogen en pacientes con vasculitis cutánea. Neupogen puede iniciarse con una dosis reducida cuando los síntomas se resuelven y el RAN ha disminuido.
Posible efecto sobre las células malignas
Neupogen es un factor de crecimiento que estimula principalmente los neutrófilos. El receptor del factor estimulante de colonias de granulocitos (G-CSF) a través del cual actúa filgrastim también se ha encontrado en las líneas celulares tumorales. No se puede excluir la posibilidad de que filgrastim actúe como factor de crecimiento para cualquier tipo de tumor. No se ha establecido la seguridad de filgrastim en la Leucemia Mieloide Crónica (LMC) y la mielodisplasia.
Cuando se utiliza Neupogen para movilizar PBPC, las células tumorales pueden liberarse de la médula y posteriormente recolectarse en el producto de leucoféresis. El efecto de la reinfusión de células tumorales no se ha estudiado bien, y los datos limitados disponibles no son concluyentes.
No se recomienda el uso simultáneo con quimioterapia y radioterapia
No se ha establecido la seguridad y eficacia de Neupogen administrado simultáneamente con quimioterapia citotóxica. Debido a la posible sensibilidad de las células mieloides que se dividen rápidamente a la quimioterapia citotóxica, no use Neupogen en el período de 24 horas antes a 24 horas después de la administración de quimioterapia citotóxica (ver Dosis y Administración).La seguridad y eficacia de Neupogen no se han evaluado en pacientes que reciben radioterapia concurrente. Evite el uso simultáneo de Neupogen con quimioterapia y radioterapia.
Imágenes nucleares
El aumento de la actividad hematopoyética de la médula ósea en respuesta a la terapia con factor de crecimiento se ha asociado con cambios positivos transitorios en la imagen ósea. Esto debe tenerse en cuenta al interpretar los resultados de imágenes óseas.
Aortitis
Se ha informado de aortitis en pacientes que reciben Neupogen. Puede ocurrir tan pronto como en la primera semana después del inicio de la terapia. Las manifestaciones pueden incluir signos y síntomas generalizados como fiebre, dolor abdominal, malestar general, dolor de espalda y aumento de los marcadores inflamatorios (por ejemplo, proteína C reactiva y recuento de glóbulos blancos). Considere la aortitis en pacientes que desarrollan estos signos y síntomas sin una etiología conocida. Suspenda Neupogen si se sospecha aortitis.
Uso en poblaciones espcíficas
Embarazo
Resumen de riesgos
Los datos disponibles de los estudios publicados, incluidos varios estudios observacionales de los resultados del embarazo en mujeres expuestas a los productos de filgrastim y aquellas que no estuvieron expuestas, no han establecido una asociación con el uso de Neupogen durante el embarazo y defectos congénitos mayores, aborto espontáneo o resultados adversos maternos o fetales (ver sección de datos). Los informes en la literatura científica han descrito el paso transplacentario de Neupogen en mujeres embarazadas cuando se administra ≤ 30 horas antes del parto prematuro (≤ 30 semanas de gestación). En estudios de reproducción animal, se han estudiado los efectos de filgrastim sobre el desarrollo prenatal en ratas y conejos. No se observaron malformaciones en ninguna de las especies. No se observaron efectos maternos o fetales en ratas preñadas a dosis de hasta 58 veces las dosis en humanos. Se ha demostrado que Filgrastim tiene efectos adversos en conejas preñadas a dosis de 2 a 10 veces mayores que las dosis en humanos (ver sección Datos). Se desconoce el riesgo de fondo estimado de defectos congénitos importantes y aborto involuntario para la población indicada. Todos los embarazos tienen un riesgo de defecto de nacimiento, pérdida u otros resultados adversos. En la población general de EE. UU., Los riesgos de fondo estimados de defectos congénitos mayores y abortos involuntarios en embarazos clínicamente reconocidos son del 2% al 4% y del 15% al 20%, respectivamente.
Datos
Datos humanos
Varios estudios observacionales basados en el Registro Internacional de Neutropenia Crónica Severa (SCNIR) describieron los resultados del embarazo en mujeres con Neutropenia Crónica Severa (SCN) que estuvieron expuestas a productos de filgrastim durante el embarazo y mujeres con SCN que no estuvieron expuestas. No se observaron diferencias importantes entre las mujeres tratadas y no tratadas con respecto al resultado del embarazo (incluido el aborto espontáneo y el parto prematuro), las complicaciones del recién nacido (incluido el peso al nacer) y las infecciones. Las limitaciones metodológicas de estos estudios incluyen un tamaño de muestra pequeño y la falta de generalización debido a la condición materna subyacente.
Datos animales
Se han estudiado los efectos de filgrastim en el desarrollo prenatal en ratas y conejos. No se observaron malformaciones en ninguna de las especies. Se ha demostrado que Filgrastim tiene efectos adversos en conejas preñadas a dosis de 2 a 10 veces más altas que las dosis en humanos. En conejas preñadas que muestran signos de toxicidad materna, se observó una reducción de la supervivencia del embrión fetal (a 20 mcg / kg / día y 80 mcg / kg / día) y un aumento de los abortos (a 80 mcg / kg / día). En ratas gestantes, no se observaron efectos maternos o fetales a dosis de hasta 575 mcg / kg / día, que es aproximadamente, 58 veces mayor que la dosis humana de 10 mcg / kg / día.
Las crías de ratas a las que se les administró filgrastim durante los períodos perinatal y de lactancia mostraron un retraso en la diferenciación externa y el retraso del crecimiento (≥ 20 mcg / kg / día) y una tasa de supervivencia ligeramente reducida (100 mcg / kg / día).
Lactancia
Resumen de riesgos
Existe literatura publicada que documenta la transferencia de filgrastim a la leche humana. Hay algunos informes de casos que describen el uso de filgrastim en madres lactantes sin efectos adversos observados en los lactantes. No hay datos sobre los efectos de filgrastim en la producción de leche. Otros productos de filgrastim se secretan poco en la leche materna, y los productos de filgrastim no son absorbidos por vía oral por los recién nacidos. Los beneficios para el desarrollo y la salud de la lactancia materna deben considerarse junto con la necesidad clínica de Neupogen de la madre y cualquier posible efecto adverso en el niño amamantado por Neupogen o por la afección materna subyacente. Uso pediátrico
En pacientes con cáncer que recibieron quimioterapia mielosupresora, 15 pacientes pediátricos con una edad promedio de 2.6 (rango 1.2 a 9.4) años con neuroblastoma fueron tratados con quimioterapia mielosupresora (ciclofosfamida, cisplatino, doxorrubicina, y etopósido) seguido de Neupogen subcutáneo a dosis de 5 mcg/kg/día, 10 mcg/kg/día, o 15 mcg/kg/día durante 10 días (n = 5 / dosis) (Estudio 8). La farmacocinética de Neupogen en pacientes pediátricos después de la quimioterapia es similar a la de los adultos que reciben las mismas dosis normalizadas de peso, lo que sugiere que no hay diferencias relacionadas con la edad en la farmacocinética de Neupogen. En esta población, Neupogen fue bien tolerado. Hubo un informe de esplenomegalia palpable y un informe de hepatoesplenomegalia asociada con la terapia con Neupogen; sin embargo, el único evento adverso reportado consistentemente fue el dolor musculoesquelético, que no difiere de la experiencia en la población adulta.
La seguridad y la eficacia de Neupogen se han establecido en pacientes pediátricos con SCN (ver Estudios clínicos (12.5)). En un estudio de fase 3 (Estudio 7) para evaluar la seguridad y eficacia de Neupogen en el tratamiento de SCN, se estudiaron 123 pacientes con una mediana de edad de 12 años (rango de 7 meses a 76 años). De los 123 pacientes, 12 eran bebés (de 7 meses a 2 años), 49 eran niños (de 2 a 12 años) y 9 eran adolescentes (de 12 a 16 años). Se encuentra disponible información adicional de un estudio de vigilancia postcomercialización de SCN, que incluye el seguimiento a largo plazo de los pacientes en los estudios clínicos e información de pacientes adicionales que ingresaron directamente en el estudio de vigilancia postcomercialización. De los 731 pacientes en el estudio de vigilancia, 429 eran pacientes pediátricos <18 años de edad (rango 0.9 a 17) (ver Indicaciones y Usos, Dosis y Administración, y Estudios clínicos).
Los datos de seguimiento a largo plazo del estudio de vigilancia posterior a la comercialización sugieren que la altura y el peso no se ven afectados negativamente en pacientes que recibieron hasta 5 años de tratamiento con Neupogen. Los datos limitados de pacientes que fueron seguidos en el estudio de fase 3 durante 1,5 años no sugirieron alteraciones en la maduración sexual o la función endocrina.
Los pacientes pediátricos con neutropenia congénita (síndrome de Kostmann, agranulocitosis congénita o síndrome de Schwachman Diamond) han desarrollado anormalidades citogenéticas y se han transformado en SMD y LMA mientras reciben tratamiento crónico con Neupogen. Se desconoce la relación de estos eventos con la administración de Neupogen (ver Advertencias y Precauciones y Reacciones adversas).
El uso de Neupogen para aumentar la supervivencia en pacientes pediátricos expuestos de forma aguda a dosis mielosupresoras de radiación se basa en estudios realizados en animales y datos clínicos que respaldan el uso de Neupogen en otras indicaciones aprobadas (ver Dosis y Administración y Estudios clínicos).
Uso geriátrico
Entre 855 sujetos inscritos en 3 ensayos aleatorios controlados con placebo de pacientes tratados con Neupogen que recibieron quimioterapia mielosupresora, hubo 232 sujetos de 65 años o más, y 22 sujetos de 75 años o más. No se observaron diferencias generales en seguridad o efectividad entre estos sujetos y los sujetos más jóvenes.
Los estudios clínicos de Neupogen en otras indicaciones aprobadas (es decir, receptores de TMO, movilización de PBPC y SCN) no incluyeron un número suficiente de sujetos de 65 años o más para determinar si los sujetos de edad avanzada responden de manera diferente a los sujetos más jóvenes.
DOSIS Y VÍA DE ADMINISTRACIÓN
Posología y administración
Dosis en pacientes con cáncer que reciben quimioterapia mielosupresora o quimioterapia de inducción y/o consolidación para LMA
La dosis inicial recomendada de Neupogen es de 5 mcg/kg/día, administrada como una dosis diaria única de inyección subcutánea, por infusión intravenosa corta (15 a 30 minutos), o por infusión intravenosa continua. Obtenga un Conteo Sanguíneo Completo (CSC) y un recuento de plaquetas antes de instituir la terapia de Neupogen y controle dos veces por semana durante la terapia. Considere la escalada de dosis en incrementos de 5 mcg/kg para cada ciclo de quimioterapia, de acuerdo con la duración y la gravedad del Recuento Absoluto de Neutrófilos (RAN). Se recomienda detener la administración de Neupogen si el Recuento absoluto de neutrófilos aumenta más allá de 10.000/mm3 (ver Advertencias y precauciones).
La primera dosis de Neupogen deberá administrarse a partir de las 24 horas siguientes de finalizada la quimioterapia citotóxica. No administre Neupogen dentro de las 24 horas previas a la quimioterapia (ver Advertencias y Precauciones). Un aumento transitorio en el recuento de neutrófilos se observa típicamente de 1 a 2 días después del inicio de la terapia con Neupogen. Por lo tanto, para garantizar una respuesta terapéutica sostenida, administre Neupogen diariamente durante hasta 2 semanas o hasta que el RAN haya alcanzado 10.000/mm3 después del punto más bajo esperado de neutrófilos, inducido por quimioterapia. La duración necesaria de la terapia con Neupogen para atenuar la neutropenia inducida por quimioterapia puede depender del potencial mielosupresor del régimen de quimioterapia aplicado.
Dosis en pacientes en cáncer sometidos a trasplante de médula ósea
La dosis recomendada de Neupogen después del Trasplante de Médula Ósea (TMO) es de 10 mcg /kg/día administrada como una infusión intravenosa no más de 24 horas. La primera dosis de Neupogen debe administrarse al menos 24 horas después de la quimioterapia citotóxica y al menos 24 horas después de la infusión de la médula ósea. Monitoree los CSC y los recuentos de plaquetas con frecuencia después del trasplante de médula.
Durante el período de recuperación de neutrófilos, valorar la dosis diaria de Neupogen contra la respuesta de los neutrófilos (Ver Tabla 1).
|
Tabla 1. Ajustes de dosis recomendados durante la recuperación de neutrófilos en pacientes con cáncer después del TMO. |
|
|
Recuento Absoluto de Neutrófilos (RAN) |
Ajuste de la dosis de Neupogen |
|
> 1.000/mm3 durante 3 días consecutivos |
Disminuir a 0,5 MU (5 mcg)/kg/díaa |
|
Si la RAN permanece > 1.000/mm3 durante 3 días consecutivos más |
Suspender Neupogen |
|
Si el RAN desciende a < 1.000/mm3 durante el tratamiento |
Reanudar a 5 mcg/kg/día |
|
a Si el RAN disminuye a menos de 1.000/mm3 en cualquier momento durante la administración de 5 mcg/kg/día, aumente Neupogen a 10 mcg/kg/día, y luego siga los pasos anteriores |
|
Dosis en pacientes sometidos a recolección y terapia de células progenitoras de sangre periférica autólogas
La dosis recomendada de Neupogen para la movilización de células progenitoras de sangre periférica autólogas (PBPC) es de 10 mcg/ kg/día administrada por inyección subcutánea. Administre Neupogen durante al menos 4 días antes del primer procedimiento de leucoféresis y continúe hasta la última leucoféresis. Aunque la duración óptima de la administración de Neupogen y el programa de leucoféresis no se han establecido, la administración de Neupogen durante 6 a 7 días con leucoféresis en los días 5, 6 y 7 fue segura y efectiva (ver Estudios clínicos). Monitoreé los recuentos de neutrófilos después de 4 días de Neupogen ‚e interrumpa Neupogen si el recuento de glóbulos blancos (WBC) aumenta a más de 100.000/mm3.Dosis en pacientes con Neutropenia Crónica Severa (SCN)
Antes de comenzar con Neupogen en pacientes con sospecha de neutropenia crónica, confirme el diagnóstico de Neutropenia Crónica Severa (SCN) evaluando los CSC seriales con recuentos diferenciales y plaquetarios, y evaluando la morfología y el cariotipo de la médula ósea. El uso de Neupogen antes de la confirmación de un diagnóstico correcto de SCN puede perjudicar los esfuerzos de diagnóstico y, por lo tanto, puede perjudicar o retrasar la evaluación y el tratamiento de una afección subyacente, distinta de SCN, que causa la neutropenia. La dosis inicial recomendada en pacientes con Neutropenia Congénita es de 6 mcg/kg como una inyección subcutánea dos veces al día y la dosis inicial recomendada en pacientes con Neutropenia Idiopática o Cíclica es de 5 mcg/kg como una inyección subcutánea diaria única.
Ajustes de dosis en pacientes con Neutropenia crónica severa
Se requiere una administración diaria crónica para mantener el beneficio clínico. Individualice la dosis según el curso clínico del paciente y el RAN. En el estudio de vigilancia postcomercialización de SCN, las dosis diarias medias informadas de Neupogen fueron: 6 mcg/kg (neutropenia congénita), 2,1 mcg/kg (neutropenia cíclica) y 1,2 mcg/kg (neutropenia idiopática). En raras ocasiones, los pacientes con neutropenia congénita han requerido dosis de Neupogen mayores o iguales a 100 mcg/kg /día.Monitoreé los CSC para ajustes de dosis
Durante las primeras 4 semanas de tratamiento con Neupogen y durante las 2 semanas posteriores a cualquier ajuste de dosis, monitoree los CSC con recuentos diferenciales y plaquetarios. Una vez que un paciente está clínicamente estable, monitoree los CSC con recuentos diferenciales y de plaquetas mensualmente durante el primer año de tratamiento. Posteriormente, si el paciente está clínicamente estable, se recomienda una monitorización de rutina menos frecuente.
Dosis en pacientes con exposición aguda a dosis de radiación mielosupresoras (Síndrome hematopoyético del síndrome de radiación aguda)
La dosis recomendada de Neupogen es de 10 mcg/kg en una única inyección subcutánea diaria para pacientes expuestos a dosis de radiación mielosupresoras. Administre Neupogen lo antes posible después de la exposición sospechada o confirmada a dosis de radiación mayores a 2 gray (Gy).
Estime la dosis de radiación absorbida de un paciente (es decir, el nivel de exposición a la radiación) basándose en información de las autoridades de salud pública, biodosimetría si está disponible o hallazgos clínicos como el tiempo hasta el inicio del vómito o la cinética de agotamiento de linfocitos.
Obtenga un CSC de referencia y luego CSCs en serie aproximadamente, cada tercer día hasta que el RAN permanezca por encima de 1.000/mm3 durante 3 CSCs consecutivos. No demore la administración de Neupogen si no hay un CSC fácilmente disponible.
Continúe la administración de Neupogen hasta que el RAN permanezca mayor a 1.000/mm3 por 3 CSCs consecutivos o exceda 10.000/mm3 después de un nadir inducido por radiación.
Instrucciones importantes de administración Neupogen se suministra en jeringas precargadas de dosis única (para uso subcutáneo) (ver Formas Farmacéuticas y Concentraciones). Antes de usar, retire la jeringa precargada del refrigerador y permita que Neupogen alcance la temperatura ambiente durante un mínimo de 30 minutos y un máximo de 24 horas. Deseche cualquier jeringa precargada que quede a temperatura ambiente durante más de 24 horas. Los productos farmacéuticos parenterales deben inspeccionarse visualmente para detectar partículas y decoloración antes de la administración, siempre que la solución y el envase lo permitan (la solución es transparente e incolora). No administre Neupogen si se observan partículas o decoloración.
Deseche la porción no utilizada de Neupogen en jeringas precargadas. No guarde el medicamento no utilizado para su posterior administración.
Inyección subcutánea
Inyecte Neupogen por vía subcutánea en el área externa de la parte superior de los brazos, abdomen, muslos o áreas externas superiores de la nalga. Si los pacientes o cuidadores van a administrar Neupogen, indíqueles la técnica de inyección adecuada y pídales que sigan los procedimientos de inyección subcutánea en las Instrucciones de uso de la jeringa precargada.
La capacitación por parte del profesional de la salud debe tener como objetivo demostrar a esos pacientes y cuidadores cómo medir la dosis de Neupogen, y debe centrarse en garantizar que un paciente o cuidador pueda realizar con éxito todos los pasos en las instrucciones de uso de la jeringa precargada. Si un paciente o cuidador no es capaz de demostrar que puede medir la dosis y administrar el producto con éxito, debe considerar si el paciente es un candidato apropiado para la autoadministración de Neupogen o si el paciente se beneficiaría de una presentación de Neupogen diferente. Si un paciente o cuidador experimenta dificultades para medir la dosis requerida, especialmente si es diferente al contenido completo de la jeringa precargada de Neupogen, se puede considerar el uso del vial de Neupogen.
Si el paciente o el cuidador omite una dosis de Neupogen, indíqueles que se comuniquen con su proveedor de atención médica.
Instrucciones de administración para la jeringa precargada
Las personas con alergias al látex no deben administrar la jeringa precargada de Neupogen, porque la tapa de la aguja contiene caucho natural seco (derivado del látex). Información de asesoramiento para pacientes Aconseje al paciente que lea el etiquetado del paciente aprobado por la FDA (Información del paciente e Instrucciones de uso). Revise los pasos para la administración directa de pacientes con pacientes y cuidadores. La capacitación del profesional de la salud debe tener como objetivo garantizar que los pacientes y los cuidadores puedan realizar con éxito todos los pasos de las instrucciones de uso de la jeringa precargada, incluido mostrar al paciente o al cuidador cómo medir la dosis requerida, especialmente si un paciente debe recibir una dosis diferente a la jeringa precargada completa. Si un paciente o cuidador no puede demostrar que puede medir la dosis y administrar el producto con éxito, debe considerar si el paciente es un candidato apropiado para la autoadministración de Neupogen o si el paciente se beneficiaría de una presentación diferente de Neupogen.Informe a los pacientes sobre los siguientes riesgos y riesgos potenciales con Neupogen:
• Puede ocurrir ruptura o agrandamiento del bazo. Los síntomas incluyen dolor abdominal en el cuadrante superior izquierdo o dolor en el hombro izquierdo. Aconseje a los pacientes que informen de inmediato a su médico sobre el dolor en estas áreas [ver Advertencias y precauciones].
• Puede ocurrir disnea, con o sin fiebre, progresando a Síndrome de distrés respiratorio agudo. Aconseje a los pacientes que informen de inmediato a su médico en el caso de disnea. [ver Advertencias y precauciones].
• Pueden producirse reacciones alérgicas graves, que pueden manifestarse por erupción cutánea, edema facial, sibilancias, disnea, hipotensión o taquicardia. Aconseje a los pacientes que busquen atención médica inmediata si aparecen signos o síntomas de reacción de hipersensibilidad [ver Advertencias y precauciones].
• En pacientes con enfermedad de células falciformes, se han producido crisis de células falciformes y muerte. Analice los posibles riesgos y beneficios para los pacientes con anemia falciforme antes de la administración de factores estimulantes de colonias de granulocitos humanos [ver Advertencias y precauciones].
• La glomerulonefritis puede ocurrir. Los síntomas incluyen hinchazón de la cara o los tobillos, orina de color oscuro o sangre en la orina, o una disminución en la producción de orina. Aconseje a los pacientes que informen de inmediato a su médico sobre signos o síntomas de glomerulonefritis [ver Advertencias y precauciones].
• Puede ocurrir vasculitis cutánea, que puede manifestarse por púrpura o eritema. Aconseje a los pacientes que informen inmediatamente a su médico sobre los signos o síntomas de vasculitis [ver Advertencias y precauciones].
• La aortitis puede ocurrir. Los síntomas pueden incluir fiebre, dolor abdominal, malestar general, dolor de espalda y aumento de los marcadores inflamatorios. Aconseje a los pacientes que informen de inmediato a su médico sobre los signos y síntomas de aortitis [ver Advertencias y precauciones].
Informe a los pacientes expuestos de forma aguda a dosis de radiación mielosupresora (Síndrome hematopoyético del síndrome de radiación aguda) que los estudios de eficacia de Neupogen para esta indicación no pueden realizarse en humanos por razones éticas y de viabilidad y que, por lo tanto, la aprobación de este uso se basó en estudios de eficacia realizados en animales [ver Estudios clínicos].
Instruya a los pacientes que se autoadministran Neupogen utilizando la jeringa precargada:
• Importancia de seguir las instrucciones de uso aplicables
• Peligros de reutilizar agujas, jeringas o porciones no utilizadas de viales de dosis única.
• Importancia de seguir los requisitos locales para la eliminación adecuada de jeringas, agujas y viales no utilizados.
• Importancia de informar al profesional de la salud si se presentan dificultades al medir o administrar contenidos parciales de la jeringa precargada de Neupogen.
Instrucciones para inyectar con la jeringa precargada de Neupogen sin guarda manual
Esta sección contiene información sobre cómo administrar una inyección de Neupogen.
Importante: No intente inyectarse usted mismo a menos que su médico o enfermero le hayan enseñado cómo hacerlo.
Neupogen se inyecta en el tejido que hay justo debajo de la piel. Es lo que se llama una inyección subcutánea. Equipo necesario: Para administrarse una inyección subcutánea necesitará:
• una jeringa nueva precargada de Neupogen; y
• algodón con alcohol o similar.
¿Qué debo hacer antes de ponerme una inyección subcutánea de Neupogen?
1. Saque la jeringa de la nevera. Deje la jeringa a temperatura ambiente durante aproximadamente, 30 minutos o sostenga la jeringa precargada suavemente entre sus manos durante unos minutos. De este modo la inyección será más cómoda. No caliente Neupogen de ninguna otra forma (por ejemplo, no lo caliente en el microondas ni en agua caliente).
2. No agite la jeringa precargada.
3. No retire la cubierta de la aguja hasta que esté preparado para la inyección.
4. Compruebe la fecha de expiración de la etiqueta de la jeringa precargada (Vence). No la use si ha pasado el último día del mes indicado.
5. Compruebe el aspecto de Neupogen. Debe ser un líquido transparente e incoloro. No lo utilice si observa coloración, turbidez o partículas en el mismo.
6. Lávese cuidadosamente las manos.
7. Busque una superficie limpia, cómoda y bien iluminada y coloque todo el equipo necesario a su alcance.
¿Cómo preparo mi inyección de Neupogen?
Antes de inyectarse Neupogen, debe hacer lo siguiente:
1. Para evitar doblar la aguja, tire suavemente de la cubierta de la aguja sin girarla, como se muestra en las figuras 1 y 2.
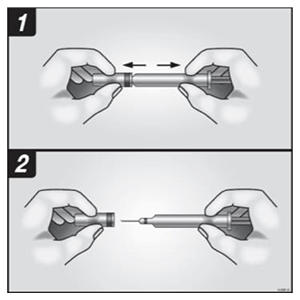
2. No toque la aguja ni empuje el émbolo.
3. Puede que observe una pequeña burbuja de aire en la jeringa precargada. Usted no debe extraer la burbuja de aire antes de la inyección. La inyección de la solución con una burbuja de aire es inofensiva.
4. Ahora ya puede usar la jeringa precargada.
¿Dónde me pongo la inyección?
Los lugares más adecuados para ponerse la inyección son la parte superior de los muslos y el abdomen. Si la inyección se la pone otra persona, también se la puede poner en la parte posterior de los brazos como muestra la imagen.
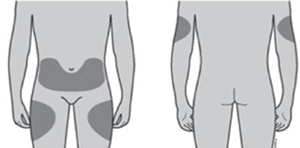
Puede cambiar de zona de inyección si observa enrojecimiento o hinchazón de la zona.
¿Cómo ponerse la inyección?
1. Desinfecte la piel usando un algodón con alcohol y pellízquela (sin apretar) entre el pulgar y el dedo índice.
2. Inserte totalmente la aguja en la piel como le ha enseñado su enfermero o médico.
3. Empuje el émbolo con una presión lenta y constante, manteniendo la piel pellizcada en todo momento hasta que la jeringa esté vacía.
4. Retire la aguja y suelte la piel.
5. Si observa una gota de sangre, puede retirarla suavemente con un poco de algodón o una gasa. No frote el lugar de inyección. Si es necesario, puede cubrir el lugar de inyección con un vendaje.
6. Utilice cada jeringa para una sola inyección. No use el resto de Neupogen que haya podido quedar en la jeringa.
Recuerde: si tiene algún problema, no dude en pedir ayuda y consejo a su médico o enfermero.
Deshacerse de las jeringas usadas
• No vuelva a poner la cubierta protectora en las agujas ya usadas, ya que podría pincharse accidentalmente.
• Mantenga las jeringas usadas fuera del alcance y de la vista de los niños.
• Las jeringas no deben tirarse en la basura del hogar. Su farmacéutico sabrá cómo deshacerse de las jeringas usadas o que ya no necesita.
Instrucciones para inyectar con la jeringa precargada de Neupogen con guarda manual
Esta sección contiene información de cómo administrarse a sí mismo una inyección de Neupogen. Es importante que no intente inyectase usted mismo a menos que su médico, enfermero o farmacéutico le hayan enseñado como hacerlo. Si tiene preguntas sobre la inyección, por favor consulte con su médico, enfermero o farmacéutico.
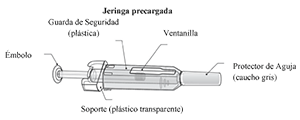
Jeringa precargada de Neupogen con guarda manual
Antes de comenzar
Lea todas las instrucciones detenidamente antes de usar la jeringa precargada
Para reducir el riesgo de pinchazos accidentales a los usuarios, cada jeringa precargada está equipada con un protector de aguja. Usted tendrá que activar manualmente el protector de la aguja (deslizar sobre la aguja) después de recibir la inyección.
NO deslice la guardia de seguridad sobre la aguja antes de administrar la inyección; se sellará en el lugar y evitará la inyección.
¿Cómo usted, o la persona inyectándolo, deben usar la jeringa precargada de Neupogen?
Su doctor le ha prescrito una jeringa precargada de Neupogen para inyección en el tejido justo debajo de la piel (subcutáneo). Su doctor, enfermero o farmacéutico puede decirle la cantidad de Neupogen que usted necesita y la frecuencia en la que se debe inyectar.
Equipo necesario: Para administrarse una inyección subcutánea necesitará:
• una jeringa nueva precargada de Neupogen; y
• algodón con alcohol o similar.
¿Qué debo hacer antes de ponerme una inyección subcutánea de Neupogen?
1. Saque la jeringa precargada de la nevera. Deje la jeringa precargada a temperatura ambiente durante aproximadamente, 30 minutos. De este modo la inyección será más cómoda. No caliente Neupogen de ninguna otra forma (por ejemplo, no lo caliente en el microondas ni en agua caliente). Adicionalmente, no deje la jeringa expuesta directamente a la luz solar.
2. No agite la jeringa precargada.
3. No retire la cubierta de la aguja hasta que esté preparado para la inyección.
4. Compruebe la fecha de expiración de la etiqueta de la jeringa precargada (Vence). No la use si ha pasado el último día del mes indicado.
5. Compruebe el aspecto de Neupogen. Debe ser un líquido transparente e incoloro. No lo utilice si observa turbidez o partículas en el mismo.
6. Lávese cuidadosamente las manos.
7. Busque una superficie limpia, cómoda y bien iluminada y coloque todo el equipo necesario a su alcance.
¿Cómo preparo mi inyección de Neupogen?
Antes de inyectarse Neupogen, debe hacer lo siguiente:
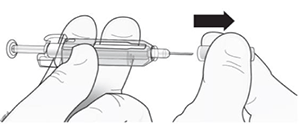
1. Para evitar doblar la aguja, tire suavemente de la cubierta de la aguja sin girarla, como se muestra en la figura.
2. No toque la aguja ni empuje el émbolo.
3. Puede que observe una pequeña burbuja de aire en la jeringa precargada. Usted no debe extraer la burbuja de aire antes de la inyección. La inyección de la solución con una burbuja de aire es inofensiva.
4. Ahora ya puede usar la jeringa precargada.
¿Dónde me pongo la inyección?
Los lugares más adecuados para ponerse la inyección son la parte superior de los muslos y el abdomen. Si la inyección se la pone otra persona, también se la puede poner en la parte posterior de los brazos como muestra la imagen.
Puede cambiar de zona de inyección si observa enrojecimiento o hinchazón de la zona.
¿Cómo ponerse la inyección?
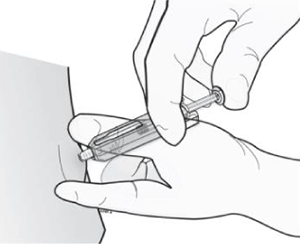
1. Desinfecte la piel usando un algodón con alcohol y pellízquela (sin apretar) entre el pulgar y el índice.
2. Inserte totalmente la aguja en la piel como le ha enseñado su médico, enfermero o farmacéutico.
3. Inyecte la dosis prescrita subcutáneamente como lo ha indicado su doctor, enfermero o farmacéutico.
4. Empuje el émbolo con una presión lenta y constante, manteniendo la piel pellizcada en todo momento hasta que la jeringa esté vacía.
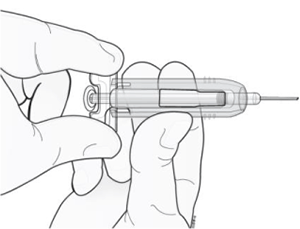
5. Retire la aguja y suelte la piel.
6. Con la aguja apuntando a la dirección contraria a usted, sostenga la jeringa por el soporte de plástico transparente con una mano.
7. Suavemente deslice la guarda de seguridad sobre la aguja y asegure que esté sellada. No agarre la guarda de seguridad muy firmemente mientras que deslice sobre la aguja.
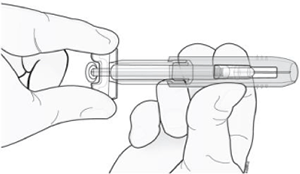
No ponga el protector de aguja sobre la aguja.
8. Si observa una gota de sangre, puede retirarla suavemente con un poco de algodón o una gasa. No frote el lugar de inyección. Si es necesario, puede cubrir el lugar de inyección con un vendaje.
9. Utilice cada jeringa para una sola inyección. No use el resto de Neupogen que haya podido quedar en la jeringa.
Recuerde: si tiene algún problema, no dude en pedir ayuda y consejo a su médico o enfermero.
Deshacerse de las jeringas usadas
• No vuelva a poner la cubierta protectora en las agujas ya usadas.
• Mantenga las jeringas usadas fuera del alcance y de la vista de los niños.
• Las jeringas usadas deben eliminarse de acuerdo con la normativa local. Pregunte a su farmacéutico como eliminar los productos que ya no se necesitan. Estas medidas ayudarán a proteger el medio ambiente.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis
No se ha determinado la dosis máxima tolerada de Neupogen. En ensayos clínicos con Neupogen de pacientes con cáncer que reciben quimioterapia mielosupresora, se han informado recuentos de leucocitos > 100.000/mm3 en menos del 5% de los pacientes, pero no se asociaron con ningún efecto clínico adverso informado. Los pacientes en los estudios de TMO recibieron hasta 138 mcg/kg/día sin efectos tóxicos, aunque hubo un aplanamiento de la curva de respuesta a la dosis por encima de las dosis diarias de más de 10 mcg/kg/día.
PRESENTACIÓN:
Caja con 1 ó 4 jeringas precargadas con 30 MU/0,5 mL de filgrastim.
Las jeringas precargadas están hechas de vidrio tipo I y tienen una aguja de acero inoxidable permanentemente incorporada en la punta. La cubierta de la aguja de la jeringa precargada contiene caucho natural (un derivado del látex).
Puede ser que no todas las presentaciones sean comercializadas.
RECOMENDACIONES SOBRE ALMACENAMIENTO
Almacenamiento y manipulación
Almacenar Neupogen entre 2 a 8°C en el cartón original para proteger de la luz. No deje Neupogen a la luz solar directa. Evitar la congelación; si está congelado, descongélelo en el refrigerador antes de la administración. Deseche Neupogen si se congeló más de una vez. Evitar la agitación. El transporte a través de un tubo neumático no ha sido estudiado.
BIBLIOGRAFÍA:
Estudios clínicos
Pacientes con cáncer que reciben quimioterapia mielosupresora
La seguridad y eficacia de Neupogen para disminuir la incidencia de infección, como se manifiesta por neutropenia febril, en pacientes con neoplasias no mieloides que reciben medicamentos anticancerígenos mielosupresores se establecieron en un ensayo aleatorizado, doble ciego, controlado con placebo realizado en pacientes con cáncer de pulmón de células pequeñas (Estudio 1).
En el Estudio 1, los pacientes recibieron hasta 6 ciclos de quimioterapia intravenosa, incluida ciclofosfamida y doxorrubicina intravenosas el día 1; y etopósido en los días 1, 2 y 3 de ciclos de 21 días. Los pacientes fueron asignados al azar para recibir Neupogen (n = 99) a una dosis de 230 mcg/m2 (4 a 8 mcg/kg/día) o placebo (n = 111). El fármaco del estudio se administró por vía subcutánea diariamente a partir del día 4, durante un máximo de 14 días. Un total de 210 pacientes fueron evaluables para eficacia y 207 fueron evaluables para seguridad. Las características demográficas y de la enfermedad se equilibraron entre los brazos con una edad media de 62 (rango 31 a 80) años; 64% hombres; 89% caucásicos; 72% de enfermedad extensa y 28% de enfermedad limitada.
La variable principal de eficacia fue la incidencia de neutropenia febril. La neutropenia febril se definió como un RAN <1.000/mm3 y una temperatura > 38.2 °C. El tratamiento con Neupogen resultó en una reducción clínica y estadísticamente significativa en la incidencia de infección, manifestada por neutropenia febril, 40% para pacientes tratados con Neupogen y 76% para pacientes tratados con placebo (p <0.001). También hubo reducciones estadísticamente significativas en la incidencia y la duración general de la infección manifestada por neutropenia febril; la incidencia, gravedad y duración de la neutropenia grave (RAN < 500/mm3); la incidencia y la duración general de los ingresos hospitalarios; y la cantidad de días reportados de uso de antibióticos. Pacientes con leucemia mieloide aguda que reciben quimioterapia de inducción o consolidación
La seguridad y eficacia de Neupogen para reducir el tiempo de recuperación de neutrófilos y la duración de la fiebre, después del tratamiento de quimioterapia de inducción o consolidación de pacientes con Leucemia Mieloide Aguda (LMA) se estableció en un ensayo aleatorizado, doble ciego, controlado con placebo, multicéntrico en pacientes con diagnóstico reciente de LMA de novo (Estudio 4).
En el Estudio 4, la terapia de inducción inicial consistió en daunorrubicina intravenosa los días 1, 2 y 3; arabinósido de citosina días 1 a 7; y etopósido los días 1 a 5. Los pacientes fueron asignados al azar para recibir Neupogen subcutáneo (n = 259) a una dosis de 5 mcg/kg/día o placebo (n = 262) desde 24 horas después de la última dosis de quimioterapia hasta la recuperación de neutrófilos (RAN ≥ 1.000/mm3 por 3 días consecutivos o ≥ 10.000/mm3 por 1 día) o por un máximo de 35 días. Las características demográficas y de la enfermedad se equilibraron entre los brazos con una edad media de 54 (rango 16 a 89) años; 54% hombres; recuento inicial de glóbulos blancos (65% < 25.000/mm3 y 27% > 100.000/mm3); 29% de citogenética desfavorable.
El criterio de valoración principal de eficacia fue la mediana de la duración de la neutropenia grave definida como recuento de neutrófilos < 500/mm3. El tratamiento con Neupogen resultó en una reducción clínica y estadísticamente significativa en la mediana del número de días de neutropenia grave, pacientes tratados con Neupogen 14 días, pacientes tratados con placebo 19 días (p = 0,0001: diferencia de 5 días (IC 95%: -6,0,-4,0)). Hubo una reducción en la mediana de la duración del uso de antibióticos por vía intravenosa, pacientes tratados con Neupogen: 15 días versus pacientes tratados con placebo: 18,5 días; una reducción en la mediana de la duración de la hospitalización, pacientes tratados con Neupogen: 20 días versus pacientes tratados con placebo: 25 días.
No hubo diferencias estadísticamente significativas entre los grupos Neupogen y placebo en la tasa de remisión completa (69% Neupogen, 68% placebo), la mediana del tiempo hasta la progresión de todos los pacientes aleatorizados (165 días Neupogen, 186 días placebo) o la mediana de supervivencia global (380 días Neupogen, 425 días placebo).
Pacientes con cáncer sometidos a trasplante de médula ósea
La seguridad y eficacia de Neupogen para reducir la duración de la neutropenia en pacientes con neoplasias no mieloides sometidas a quimioterapia mieloablativa seguida de trasplante autólogo de médula ósea se evaluó en 2 ensayos controlados aleatorios de pacientes con linfoma (Estudio 6 y Estudio 9). La seguridad y eficacia de Neupogen para reducir la duración de la neutropenia en pacientes sometidos a quimioterapia mieloablativa seguida de trasplante alogénico de médula ósea se evaluó en un ensayo aleatorizado controlado con placebo (Estudio 10).
En el Estudio 6, los pacientes con enfermedad de Hodgkin recibieron un régimen preparatorio de ciclofosfamida, etopósido y BCNU intravenoso ("CVP"), y los pacientes con linfoma no Hodgkin recibieron BCNU, etopósido, arabinósido de citosina y melfalan intravenosos ("BEAM"). Hubo 54 pacientes asignados al azar 1:1:1 para controlar, Neupogen 10 mcg/kg/día, y Neupogen 30 mcg/kg/día como una infusión continua de 24 horas comenzando 24 horas después de la infusión de médula ósea durante un máximo de 28 días. La mediana de edad fue de 33 (rango 17 a 57) años; 56% hombres; 69% de enfermedad de Hodgkin y 31% de linfoma no Hodgkin.
La variable principal de eficacia fue la duración de la neutropenia severa RAN < 500/mm3. Se produjo una reducción estadísticamente significativa en la mediana del número de días de neutropenia grave (RAN < 500/mm3) en los grupos tratados con Neupogen versus el grupo control (23 días en el grupo control, 11 días en el grupo de 10 mcg/kg/día, y 14 días en el grupo de 30 mcg/kg/día [11 días en los grupos de tratamiento combinado, p = 0,004]).En el Estudio 9, los pacientes con enfermedad de Hodgkin y linfoma no Hodgkin recibieron un régimen preparatorio de ciclofosfamida, etopósido y BCNU intravenoso ("CVP"). Hubo 43 pacientes evaluables asignados al azar a infusión subcutánea continua de Neupogen 10 mcg/kg/día (n = 19), Neupogen 30 mcg/kg/día (n = 10) y ningún tratamiento (n = 14) comenzando el día después de la infusión de médula para un máximo de 28 días. La mediana de edad fue de 33 (rango 17 a 56) años; 67% hombres; 28% de enfermedad de Hodgkin y 72% de linfoma no Hodgkin.
La variable principal de eficacia fue la duración de la neutropenia grave. Hubo una reducción estadísticamente significativa en la mediana del número de días de neutropenia severa (RAN < 500/mm3) en los grupos tratados con Neupogen versus el grupo control (21,5 días en el grupo control versus 10 días en los grupos tratados con Neupogen, p < 0,001). El número de días de neutropenia febril también se redujo significativamente en este estudio (13,5 días en el grupo control versus 5 días en los grupos tratados con Neupogen, p < 0,0001). En el Estudio 10, 70 pacientes programados para someterse a un trasplante de médula ósea por múltiples afecciones subyacentes utilizando múltiples regímenes preparatorios fueron aleatorizados para recibir Neupogen 300 mcg/m2/día (n = 33) o placebo (n = 37) días 5 a 28 después de la infusión de médula. La mediana de edad fue 18 (rango 1 a 45) años, 56% hombres. La enfermedad subyacente fue: 67% de malignidad hematológica, 24% de anemia aplásica, 9% de otros. Se produjo una reducción estadísticamente significativa en la mediana del número de días de neutropenia grave en el grupo tratado versus el grupo control (19 días en el grupo control y 15 días en el grupo de tratamiento, p < 0,001) y el tiempo de recuperación de RAN a ≥ 500/mm3 (21 días en el grupo de control y 16 días en el grupo de tratamiento, p < 0,001).Pacientes sometidos a terapia y recolección de células progenitoras de sangre periférica autólogas
La seguridad y eficacia de Neupogen para movilizar células progenitoras de sangre periférica autólogas para la recolección por leucoféresis fue respaldada por la experiencia en ensayos no controlados, y un ensayo aleatorizado que comparó el rescate de células madre hematopoyéticas utilizando células progenitoras de sangre periférica autólogas movilizadas por Neupogen para la médula ósea autóloga (Estudio 11 ). Los pacientes en todos estos ensayos se sometieron a un régimen de movilización / recolección similar: se administró Neupogen durante 6 a 7 días, en la mayoría de los casos el procedimiento de aféresis ocurrió en los días 5, 6 y 7. La dosis de Neupogen varió entre 10 a 24 mcg/kg/día y se administró por vía subcutánea mediante inyección o infusión intravenosa continua.
El injerto se evaluó en 64 pacientes que se sometieron a un trasplante utilizando células progenitoras hematopoyéticas autólogas movilizadas por Neupogen en ensayos no controlados. Dos de los 64 pacientes (3%) no alcanzaron los criterios de injerto definidos por un recuento de plaquetas ≥ 20.000/mm3 para el día 28. En ensayos clínicos de Neupogen para la movilización de células progenitoras hematopoyéticas, se administró Neupogen a los pacientes a dosis entre 5 a 24 mcg/kg/día después de la reinfusión de las células recolectadas hasta alcanzar un RAN sostenible (≥ 500/mm3). No se ha estudiado la tasa de injerto de estas células en ausencia de Neupogen después del trasplante.
El Estudio 11 fue un estudio aleatorizado y sin cegamiento de pacientes con enfermedad de Hodgkin o linfoma no Hodgkin sometidos a quimioterapia mieloablativa, 27 pacientes recibieron células progenitoras hematopoyéticas autólogas movilizadas por Neupogen y 31 pacientes recibieron médula ósea autóloga. El régimen preparatorio fue BCNU, etopósido, arabinósido de citosina y melfalan intravenosos ("BEAM"). Los pacientes recibieron Neupogen diariamente 24 horas después de la infusión de células madre a una dosis de 5 mcg/kg/día. La mediana de edad fue de 33 (rango 1 a 59) años; 64% hombres; 57% de la enfermedad de Hodgkin y 43% de linfoma no Hodgkin. El criterio de valoración principal de eficacia fue el número de días de transfusiones de plaquetas. Los pacientes asignados al azar a células progenitoras de sangre periférica autógena movilizadas por Neupogen en comparación con la médula ósea autóloga tuvieron significativamente menos días de transfusiones de plaquetas (mediana de 6 frente a 10 días).
Pacientes con neutropenia crónica severa
La seguridad y eficacia de Neupogen para reducir la incidencia y la duración de las secuelas de neutropenia (es decir, fiebre, infecciones, úlceras orofaríngeas) en pacientes adultos y pediátricos sintomáticos con neutropenia congénita, neutropenia cíclica, o neutropenia idiopática se estableció en un ensayo controlado aleatorio realizado en pacientes con neutropenia severa (Estudio 7).
Los pacientes elegibles para el Estudio 7 tenían antecedentes de neutropenia crónica severa documentada con un RAN < 500/mm3 en tres ocasiones durante un período de 6 meses, o en pacientes con neutropenia cíclica 5 días consecutivos de RAN < 500/mm3 por ciclo. Además, los pacientes deben haber experimentado una infección clínicamente significativa durante los 12 meses anteriores. Los pacientes fueron asignados al azar a un período de observación de 4 meses seguido de tratamiento con Neupogen o tratamiento inmediato con Neupogen. La mediana de edad fue de 12 años (rango de 7 meses a 76 años); 46% hombres; 34% idiopático, 17% cíclico y 49% de neutropenia congénita.
Neupogen se administró por vía subcutánea. La dosis de Neupogen se determinó por la categoría de neutropenia. Dosis inicial de Neupogen:
• Neutropenia idiopática: 3,6 mcg/kg/día.
• Neutropenia cíclica: 6 mcg/kg/día.
• Neutropenia congénita: 6 mcg/kg/día dividido 2 veces por día.
La dosis se incrementó gradualmente a 12 mcg/kg/día dividido 2 veces por día si no hubo respuesta.
El criterio de valoración principal de eficacia fue la respuesta al tratamiento con Neupogen. La respuesta RAN desde el inicio (< 500/mm3) se definió de la siguiente manera:
• Respuesta completa: mediana RAN > 1.500/mm3
• Respuesta parcial: mediana de RAN ≥ 500/mm3 y ≤ 1.500/mm3 con un aumento mínimo del 100%
• Sin respuesta: mediana RAN < 500/mm3
Hubo 112 de 123 pacientes que demostraron una respuesta completa o parcial al tratamiento con Neupogen. Los puntos finales de eficacia adicionales incluyeron una comparación entre pacientes asignados al azar a 4 meses de observación y pacientes que recibieron Neupogen de los siguientes parámetros:
• incidencia de infección
• incidencia de fiebre
• duración de la fiebre
• incidencia, duración y gravedad de las úlceras orofaríngeas
• número de días de uso de antibióticos
La incidencia para cada uno de estos 5 parámetros clínicos fue menor en el brazo de Neupogen en comparación con el brazo de control para las cohortes en cada una de las 3 categorías de diagnóstico principales. Un análisis de varianza no mostró interacción significativa entre el tratamiento y el diagnóstico, lo que sugiere que la eficacia no difirió sustancialmente en las diferentes enfermedades. Aunque Neupogen redujo sustancialmente la neutropenia en todos los grupos de pacientes, en pacientes con neutropenia cíclica, el ciclo persistió, pero el período de neutropenia se acortó a 1 día.
Pacientes expuestos de forma aguda a dosis mielosupresoras de radiación (síndrome hematopoyético del síndrome de radiación aguda) Los estudios de eficacia de Neupogen no pudieron realizarse en humanos con síndrome de radiación aguda por razones éticas y de viabilidad. La aprobación de esta indicación se basó en estudios de eficacia realizados en animales y datos que respaldan el uso de Neupogen para otras indicaciones aprobadas (ver Posología y Administración).
Debido a la incertidumbre asociada con la extrapolación de datos de eficacia animal a humanos, la selección de la dosis humana para Neupogen tiene como objetivo proporcionar exposiciones a filgrastim que excedan las observadas en estudios de eficacia animal. La dosis diaria de 10 mcg/kg se selecciona para humanos expuestos a dosis mielosupresoras de radiación porque se espera que la exposición asociada con dicha dosis exceda la exposición asociada con una dosis de 10 mcg/kg en primates no humanos (ver Propiedades farmacocinéticas). La seguridad de Neupogen a una dosis diaria de 10 mcg/kg se ha evaluado sobre la base de la experiencia clínica en indicaciones aprobadas.
La eficacia de Neupogen se estudió en un estudio aleatorizado, ciego, controlado con placebo en un modelo de lesión por radiación en primates no humanos. El tamaño de muestra planificado fue de 62 animales, pero el estudio se detuvo en el análisis intermedio con 46 animales porque se estableció la eficacia. Los macacos rhesus se asignaron al azar a un grupo control (n = 22) o tratado (n = 24). Los animales se expusieron a una irradiación corporal total de 7,4 ± 0,15 Gy administrada a 0,8 ± 0,03 Gy/min, lo que representa una dosis que sería letal en el 50% de los animales a los 60 días de seguimiento (DL50/60). A partir del día 1 después de la irradiación, los animales recibieron inyecciones subcutáneas diarias de placebo (dextrosa al 5% en agua) o filgrastim (10 mcg/kg/día). El tratamiento cegado se interrumpió cuando se cumplió uno de los siguientes criterios: RAN ≥ 1.000/mm3 por 3 días consecutivos, o RAN ≥ 10.000/mm3 por más de 2 días consecutivos dentro del día de estudio 1 a 5, o RAN ≥ 10.000/mm3 en cualquier momento después del día de estudio 5. Los animales recibieron tratamiento médico que consistía en fluidos intravenosos, antibióticos, transfusiones de sangre y otro tipo de soporte según sea necesario.
Filgrastim redujo significativamente (a un nivel de significancia de 0,023) la mortalidad a los 60 días en los primates no humanos irradiados: 21% de mortalidad (5/24) en el grupo de filgrastim en comparación con 59% de mortalidad (13/22) en el grupo control.
LEYENDAS DE PROTECCIÓN:
Venta bajo receta médica.
Mantener fuera del alcance y vista de los niños.
Contacte a su profesional de la salud o farmacéutico para cualquier aclaración sobre el uso del producto.
En el caso de omitir una dosis, contacte a su profesional de la salud.