MABTHERA
RITUXIMAB
Solución para inyección
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Principio activo: rituximab
Mabthera I.V. 100 mg/10 ml y 500 mg/50 ml
Excipientes: Citrato de sodio dihidrato, polisorbato 80, cloruro sódico, agua para inyectables c.s. y ácido clorhídrico o hidróxido de sodio (para un Ph de 6,5) c.s.
Mabthera S.C. 1400 mg/11.7ml
Excipientes: Hialurodinasa humana recombinante (rHuPH20), L-histidina, clorhidrato de L-histidina monohidrato, alfa-alfa trehalosa, dihidrato, L-metionina, polisorbato 80 y agua para preparaciones inyectables c.s.p
Formulación intravenosa
MabThera/Rituxan i.v. es un líquido límpido, incoloro, estéril, sin conservantes y apirógeno que se presenta en viales monodosis.
Viales monodosis: los viales contienen 100 mg/10 ml y 500 mg/50 ml.
Formulación subcutánea
MabThera/Rituxan s.c. es una solución entre incolora y amarilla clara y entre límpida y opalescente, estéril, sin conservantes y apirógena, que se presenta en viales monodosis.
MabThera/Rituxan s.c. contiene hialuronidasa humana recombinante (rHuPH20), una enzima que se utiliza para aumentar la dispersión y la absorción de fármacos coadministrados cuando se administran por vía s.c. (ver Uso en poblaciones especiales, Embarazo).
Los demás excipientes se describen de conformidad con los requisitos locales.
Formulación subcutánea para el linfoma no hodgkiniano: los viales monodosis contienen 1400 mg/11,7 ml (en los viales de 15 ml).
Formulación subcutánea para la leucemia linfocítica crónica: los viales monodosis contienen 1600 mg/13,4 ml (en los viales de 20 ml).
FORMA FARMACÉUTICA
Formulación intravenosa (i.v.): concentrado para solución para infusión.
Formulación subcutánea (s.c.): solución para inyección subcutánea.
PROPIEDADES Y EFECTOS FARMACOLÓGICOS
Propiedades farmacodinámicas
Mecanismo de acción
Rituximab es un anticuerpo monoclonal quimérico, murino y humano, que se une específicamente al antígeno transmembranario CD20. Este antígeno se encuentra en los linfocitos pre-B y en los linfocitos B maduros, pero no en los hemocitoblastos, los linfocitos pro-B, los plasmocitos normales u otros tejidos normales. El antígeno CD20 se expresa en >95 % de todos los LNH de linfocitos B. Este antígeno no se internaliza tras la unión al anticuer-po ni se elimina hacia al medio circundante desde la membrana celular. El CD20 no circula en el plasma como antígeno libre, de modo que no compite por la unión a los anticuerpos. El rituximab se une al antígeno CD20 en los linfocitos B y desencadena reacciones inmunitarias que intervienen en la lisis de los linfocitos B. Entre los posibles mecanismos de la lisis celular se encuentran la citotoxicidad dependiente del complemento (CDC), la citotoxicidad celular dependiente de anticuerpos (CCDA) y la inducción de la apoptosis. Por último, en estudios in vitro se ha demostrado que el rituximab sensibiliza líneas de linfoma humano de linfocitos B resistente a fármacos a los efectos citotóxicos de algunos quimioterápicos.
La cifra de linfocitos B periféricos disminuyó hasta valores inferiores a los normales después de la primera dosis de MabThera/Rituxan. En pacientes que reci-bían tratamiento de neoplasias malignas hemáticas, la recuperación de la cifra de linfocitos B comenzó en un plazo de 6 meses desde el inicio del tratamiento, aunque en algunos pacientes llevó más tiempo (ver Reacciones adversas, Ensayos clínicos, Experiencia obtenida en ensayos clínicos en hematooncología).
En pacientes con AR, la duración de la depleción de linfocitos B periféricos fue variable. La mayoría de los pacientes recibieron más tratamiento antes de que se recuperara por completo la cifra de linfocitos B. Una pequeña proporción de pacientes mostraron una deple-ción prolongada de los linfocitos B periféricos, que se prolongó 2 años o más después de la administración de la última dosis de MabThera/Rituxan i.v.
En pacientes con GPA y con PAM, la cifra de linfocitos B CD19 en sangre periférica disminuyó hasta <10 células/ìl después de las dos primeras infusiones de rituximab y se mantuvo en ese nivel en la mayoría de los pacientes hasta el mes 6.
Ninguno de los 67 pacientes en los que se evaluó la presencia de anticuerpos humanos antimurinos (HAMA) presentó resultados positivos. De los 356 pacientes con LNH en los que se evaluaron los anticuerpos humanos antiquiméricos (HACA), el 1,1 % (4 pacientes) presentaron un resultado positivo.
Ensayos clínicos / Eficacia
Formulación intravenosa
Linfoma no hodgkiniano de bajo grado o folicular.
Monoterapia con MabThera/Rituxan i.v.
Tratamiento inicial, 4 dosis a intervalos semanales
En el estudio clínico fundamental, 166 pacientes con LNH de bajo grado o folicular de linfocitos B recidivante o quimiorresistente recibieron 375 mg/m2 de MabThera/Rituxan en infusión i.v. semanal hasta completar 4 dosis. La tasa de respuesta global (TRG) en la población por intención de tratar (ITT) fue del 48 % (IC 95 %: 41-56 %), con un 6 % de respuestas completas (RC) y un 42 % de respuestas parciales (RP). La mediana proyectada del tiempo transcurrido hasta la progresión (TTP) en los pacientes que respondieron al tratamiento fue de 13,0 meses.
En un análisis de subgrupos, la TRG fue mayor en los pacientes con los subtipos histológicos B, C y D de la International Working Formulation (IWF) que en los que presentaban el subtipo A (58 % y 12 %, respectivamente); mayor en los pacientes con lesiones cuyo diámetro mayor era <5 cm en comparación con los pacientes con lesiones cuyo diámetro mayor era >7 cm (53 % y 38 %, respectivamente), y mayor en los pacientes con LNH recidivante sensible al tratamiento que en los pacientes con LNH recidivante y resistente al tratamiento (definido como una duración de la respuesta <3 meses) (50 % y 22 %, respectivamente). La TRG fue del 78 % en los pacientes que habían recibido previamente un autotrasplante de médula ósea y del 43 % en aquellos que no se sometieron a autotrasplante. No se apreció que la edad, el sexo, el grado del linfoma, el diagnóstico inicial, la presencia o ausencia de una gran masa tumoral, el valor normal o elevado de la LDH o la presencia de afectación ganglionar tuvieran un efecto estadísticamente significativo (prueba exacta de Fisher) en la respuesta a MabThera/Rituxan i.v.
Se observó una correlación estadísticamente significativa entre las tasas de respuesta y la afectación de la médula ósea. Respondieron al tratamiento el 40 % de los pacientes con afectación de la médula ósea, frente al 59 % de los pacientes sin afectación de la misma (p = 0,0186). Este dato no se vio confirmado por un análisis de regresión logística por etapas, en el que se identificaron los siguientes factores predictivos: tipo histológico, positividad inicial para bcl-2, resistencia a la última quimioterapia recibida y gran masa tumoral.
Tratamiento inicial, 8 dosis a intervalos semanales
En un estudio clínico multicéntrico con un solo grupo, 37 pacientes con LNH de bajo grado o folicular de linfocitos B recidivante o quimiorresistente recibieron 375 mg/m2 de MabThera/Rituxan en infusión i.v. semanal hasta completar 8 dosis. La TRG fue del 57 % (IC 95 %: 41-73 %; RC: 14 %; RP: 43 %), con una mediana proyectada del TTP en los pacientes con respuesta de 19,4 meses (intervalo: 5,3-38,9 meses).
Tratamiento inicial, pacientes con gran masa tumoral, 4 dosis a intervalos semanales
En los datos combinados de tres estudios, 39 pacientes con LNH de linfocitos B de bajo grado o folicular recidivante o quimiorresistente, con una gran masa tumoral (lesión única ≥10 cm de diámetro), recibieron 375 mg/m2 de MabThera/Rituxan en infusión i.v. semanal hasta completar 4 dosis.
La TRG fue del 36 % (IC 95 %: 21-51 %; RC: 3 %; RP: 33 %), con una mediana del TTP en los pacientes con respuesta de 9,6 meses (intervalo: 4,5-26,8 meses).
Retratamiento, 4 dosis a intervalos semanales
En un estudio multicéntrico con un solo grupo, 58 pacientes con LNH de linfocitos B de bajo grado o folicular recidivante o quimiorresistente que habían tenido una respuesta clínica objetiva a un ciclo anterior de Mab-Thera/Rituxan i.v. recibieron retratamiento con 375 mg/m2 de MabThera/Rituxan en infusión i.v. semanal hasta completar 4 dosis. Tres de ellos habían recibido dos ciclos de MabThera/Rituxan antes del reclutamiento, por lo que recibieron un tercer ciclo en el estudio. Dos pacientes recibieron dos veces retratamiento en el estudio. La TRG de los 60 retratamientos del estudio fue del 38 % (IC 95 %: 26-51 %; RC: 10 %; RP: 28 %), con una mediana proyectada del TTP de 17,8 meses (intervalo: 5,4-26,6 meses) en los pacientes con respuesta. Este resultado aventaja al TTP obtenido tras el ciclo anterior de MabThera/Rituxan i.v. (12,4 meses).
MabThera/Rituxan i.v. en combinación con quimioterapia
Tratamiento inicial
En un estudio aleatorizado y sin enmascaramiento, un total de 322 pacientes con linfoma folicular no tratados anteriormente recibieron o bien CVP (750 mg/m2 de ciclofosfamida, 1,4 mg/m2 de vincristina hasta un máximo de 2 mg en el día 1, y 40 mg/m2/día de prednisolona los días 1-5) cada 3 semanas durante 8 ciclos, o bien 375 mg/m2 de MabThera/Rituxan i.v. en combinación con CVP (R-CVP). MabThera/Rituxan i.v. se administró el primer día de cada ciclo de tratamiento. Un total de 321 pacientes (R-CVP: 162; CVP: 159) recibieron tratamiento y fueron evaluados para determinar la eficacia.
La mediana de seguimiento de los pacientes fue de 53 meses. En lo que respecta a la variable de valoración principal, el tiempo transcurrido hasta el fracaso terapéutico, R-CVP proporcionó un beneficio significativo en comparación con CVP (27 meses frente a 6,6 meses; p <0,0001, prueba de rangos logarítmicos). La proporción de pacientes con respuesta tumoral (RC, RC no confirmada, RP) fue significativamente mayor (p <0,0001, prueba de la ÷2) en el grupo de R-CVP (80,9 %) que en el grupo de CVP (57,2 %). El tratamiento con R-CVP prolongó significativamente el tiempo transcurrido hasta la progresión de la enfermedad o la muerte en comparación con CVP (33,6 y 14,7 meses, respectivamente; p <0,0001, prueba de rangos logarítmicos). La mediana de la duración de la respuesta fue de 37,7 meses en el grupo de R-CVP y de 13,5 meses en el grupo de CVP (p <0,0001, prueba de rangos logarítmicos). La diferencia entre los grupos de tratamiento en cuanto a la supervivencia global reveló un notable beneficio clínico (p = 0,029, prueba de rangos logarítmicos con estratificación por centros): la tasa de supervivencia a los 53 meses fue del 80,9 % en el grupo de R-CVP y del 71,1 % en el grupo de CVP.
Los resultados de otros tres ensayos aleatorizados con MabThera/Rituxan i.v. en asociación con un régimen de quimioterapia distinto a CVP (CHOP, MCP, CHVP/interferón a) también evidenciaron una mejora significativa de la tasa de respuesta, parámetros dependientes del tiempo y la supervivencia global. En la tabla 5 se resumen los resultados principales de los cuatro estudios.
|
Tabla 5. Resumen de los resultados fundamentales de cuatro estudios de fase III aleatorizados en los que se evaluaron los beneficios de MabThera/Rituxan i.v. con diferentes regímenes quimioterápicos en el linfoma folicular |
||||||
|
Estudio |
Tratamiento, n |
Mediana del seguimiento, meses |
TRG, % |
RC, % |
Mediana del TTF/SSP/SSE, meses |
Tasas de SG, % |
|
M39021 |
CVP, 159 R-CVP, 162 |
53 |
57 81 |
10 41 |
Mediana del TP: 14,7 33,6 p <0,0001 |
53 meses 71,1 80,9 p = 0,029 |
|
GLSG’00 |
CHOP, 205 R-CHOP, 223 |
18 |
90 96 |
17 20 |
Mediana del TTF: 2,6 años No alcanzada p <0,001 |
18 meses 90 95 p = 0,016 |
|
OSHO-39 |
MCP, 96 R-MCP, 105 |
45 |
75 92 |
25 50 |
Mediana de la SSP: 28.8 No alcanzada p <0,0001 |
48 meses 74 87 p = 0,0096 |
|
FL2000 |
CHVP-IFN, 183 R-CHVP-IFN, 175 |
42 |
85 94 |
49 76 |
Mediana de la SSE: 36 No alcanzada p <0,0001 |
42 meses 84 91 p = 0,029 |
|
SSE: supervivencia sin eventos; SSP: supervivencia sin progresión; tasas de SG: tasas de supervivencia en el momento de los análisis; TTF: tiempo transcurrido hasta el fracaso del tratamiento; TTP: tiempo transcurrido hasta la progresión o la muerte. |
||||||
Terapia de mantenimiento con MabThera/Rituxan i.v.
LNH folicular no tratado previamente
En un ensayo de fase III prospectivo, multicéntrico, internacional, sin enmascaramiento, 1193 pacientes con linfoma folicular avanzado no tratado previamente recibieron tratamiento de inducción con R-CHOP (n = 881), R-CVP (n = 268) o R-FCM (n = 44), según el criterio del investigador. Respondieron al tratamiento de inducción un total de 1078 pacientes, 1018 de los cuales habían sido asignados a la terapia de mantenimiento con MabThera/Rituxan i.v. (n = 505) o a la observación (n = 513). Los dos grupos de tratamiento estaban adecuadamente equilibrados en lo que respecta a las características iniciales y al estado clínico. La terapia de mantenimiento con MabThera/Rituxan i.v. consistió en una infusión única de MabThera/Rituxan i.v. de 375 mg/m2 de superficie corporal, administrada cada 2 meses hasta la progresión de la enfermedad o durante un periodo máximo de 2 años.
Tras un periodo de observación de 25 meses (mediana) desde la aleatorización, la terapia de mantenimiento con MabThera/Rituxan i.v. dio lugar a una mejoría clínica y estadísticamente significativa de la variable de valoración principal, la SSP evaluada por el investigador, en comparación con la ausencia de terapia de mantenimiento en pacientes con LNH folicular que no habían sido tratados previamente. Esta mejoría de la SSP fue confirmada por un comité de revisión independiente (CRI) (ver tabla 6).
También se observó una ventaja significativa de la terapia de mantenimiento con MabThera/Rituxan i.v. en lo que respecta a las variables de valoración secundarias: la supervivencia sin eventos (SSE), el tiempo transcurrido hasta el siguiente tratamiento contra el linfoma (TSTL) y la tasa de respuesta global (TRG) (ver tabla 6).
El análisis actualizado correspondiente a un periodo de observación de 73 meses (mediana) desde la aleatorización confirma los resultados del análisis principal (ver tabla 6).
|
Tabla 6. Cuadro general de los resultados relativos a la eficacia de la terapia de mantenimiento con MabThera/Rituxan i.v. en comparación con la observación (mediana del periodo de observación de 25 y 73 meses) |
||||
|
Parámetro de valoración de la eficacia |
Análisis principala |
Análisis actualizadob |
||
|
Observación N = 513 |
Mantenimiento con rituximab N = 505 |
Observación N = 513 |
Mantenimiento con rituximab |
|
|
Variable de valoración principal |
||||
|
Supervivencia sin progresiónc Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
NA |
NA |
49 |
NA |
|
Valor de p (prueba de rangos logarítmicos estratificada) |
p < 0,0001 |
p < 0,0001 |
||
|
HR [IC 95 %] (estratificada) |
0,50 [0,39-0,64] |
0,58 [0,48-0,69] |
||
|
Variables de valoración secundarias |
||||
|
Supervivencia global Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
NA |
NA |
NA |
NA |
|
Valor de p (prueba de rangos logarítmicos estratificada) |
p = 0,7246 |
p = 0,8959 |
||
|
HR [IC 95 %] (estratificada) |
0,89 [0,45-1,74] |
1,02 [0,71-1,47] |
||
|
Tasa de respuesta global al final del periodo de terapia de mantenimiento o del periodo de observación Pacientes evaluados al final del tratamiento |
398 |
389 |
509 |
500 |
|
Pacientes con respuesta (RC, RC no confirmada, RP) |
219/398 (55 %) |
288/389 (74 %) |
309/509 (61 %) |
395/500 (79 %) |
|
Valor de p (prueba de la X2) |
p < 0,0001 |
p < 0,0001 |
||
|
Pacientes sin respuesta |
179/398 (45 %) |
101/389 (26 %) |
200/509 (40 %) |
105/500 (21 %) |
|
Pacientes con respuesta completa (RC/RC no confirmada) |
190 (48 %) |
260 (67 %) |
268 (53 %) |
361 (72 %) |
|
Respuesta parcial (RP) |
29 (7 %) |
28 (7 %) |
41 (8 %) |
34 (7 %) |
|
Enfermedad estable (EE) |
1 (<1 %) |
0 (0 %) |
1 (<1 %) |
1 (<1 %) |
|
Enfermedad progresiva (EP) |
162 (41 %) |
79 (20 %) |
181 (36 %) |
86 (17 %) |
|
Supervivencia sin eventos |
||||
|
Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
38 |
NA |
48 |
NA |
|
Valor de p (prueba de rangos logarít-micos estratificada) |
p < 0,0001 |
p < 0,0001 |
||
|
HR [IC 95 %] (estratificada) |
0,54 [0,43-0,69] |
0,61 [0,51-0,72] |
||
|
Tiempo transcurrido hasta la administración del siguiente tratamiento contra el linfoma Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
NA |
NA |
71 |
NA |
|
Valor de p (prueba de rangos logarítmicos estratificada) |
p = 0,0003 |
p < 0,0001 |
||
|
HR [IC 95 %] (estratificada) |
0,61 [0,46-0,80] |
0,63 [0,52-0,76] |
||
|
Parámetro de valoración de la eficacia |
Análisis principala |
Análisis actualizadob |
||
|
Observación N = 513 |
Mantenimiento con rituximab N = 505 |
Observación N = 513 |
Mantenimiento con rituximab N = 505 |
|
|
Tiempo transcurrido hasta la administración del siguiente tratamiento quimioterápico Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
NA |
NA |
85 |
NA |
|
Valor de p (prueba de rangos logarítmicos estratificada) |
p = 0,0011 |
p = 0,0006 |
||
|
HR [IC 95 %] (estratificada) |
0,60 [0,44-0,82] |
0,70 [0,57-0,86] |
||
|
Tasa de transformación en la primera progresión Pacientes con progresión |
173 |
91 |
278 |
186 |
|
Pacientes con transformación |
19/513 (4 %) |
11/505 (2 %) |
24/513 (5 %) |
16/505 (3 %) |
|
HR: hazard ratio (razón de riesgos instantáneos); NA: no se alcanzó. 1 mes = 30,4375 días (es decir, 365,25 días/12 meses). Los valores de p y las HR de las variables de valoración del tipo del tiempo transcurrido hasta la aparición del evento se calcularon usando la prueba de rangos logarítmicos estratificada y la regresión de Cox estratificada, respectivamente. Los factores de estratificación fueron el tratamiento de inducción recibido y la respuesta al tratamiento de inducción. Los valores de p de las tasas de respuesta se calcularon usando la prueba de la X2, y las odds ratios (razones de posibilidades) se calcularon usando la regresión logística (los análisis de la tasa de respuesta no se ajustaron). a Fecha límite de obtención de datos clínicos: 14 de enero de 2009. Mediana del periodo de observación: 25,5 meses. b Fecha límite de obtención de datos clínicos: 31 de enero de 2013. Mediana del periodo de observación: 73 meses. c Basada en las evaluaciones de los investigadores. |
||||
El tratamiento de mantenimiento con MabThera/Rituxan i.v. aportó un beneficio constante en todos los subgrupos examinados —sexo (masculino, femenino), edad (<60 años, ≥60 años), puntuación del índice FLIPI (1, 2 o 3), terapia de inducción (R-CHOP, R-CVP o R-FCM)— e independientemente de la calidad de la respuesta al tratamiento de inducción (RC o RP).
LNH folicular recidivante o resistente al trata-miento
En un ensayo de fase III multicéntrico, internacional, prospectivo, sin enmascaramiento, 465 pacientes con LNH folicular recidivante o resistente al tratamiento fueron asignados aleatoriamente en una primera etapa a la terapia de inducción con CHOP (ciclofosfamida, doxorubicina, vincristina, prednisolona; n = 231) o a MabThera/Rituxan i.v. más CHOP (R-CHOP; n = 234). Los dos grupos de tratamiento estaban adecuadamente equilibrados en lo que respecta a las características iniciales y al estado clínico. En una segunda etapa, un total de 334 pacientes con remisión completa o parcial después de la terapia de inducción fueron asignados aleatoriamente a la terapia de mantenimiento con MabThera/Rituxan i.v. (n = 167) o a permanecer en observación (n = 167). La terapia de mantenimiento con MabThera/Rituxan i.v. consistió en una infusión única de MabThera/Rituxan i.v. de 375 mg/m2 de superficie corporal administrada cada 3 meses hasta la progresión de la enfermedad o durante un máximo de 2 años.
En el análisis final de la eficacia se incluyó a todos los pacientes aleatorizados en ambas partes del estudio. Después de un periodo de observación de 31 meses (mediana) en los pacientes aleatorizados en la fase de inducción, los resultados de los pacientes con LNH folicular recidivante o resistente al tratamiento del grupo de R-CHOP mejoraron significativamente en comparación con el grupo de CHOP (ver tabla 7).
|
Tabla 7. Fase de inducción: cuadro general de los resultados de la eficacia de CHOP y R-CHOP (mediana del periodo de observación de 31 meses) |
||||
|
CHOP |
R-CHOP |
p |
Reducción del riesgo1 |
|
|
Variables principales de valoración de la eficacia |
||||
|
TRG2 |
74 % |
87 % |
0,0003 |
ND |
|
RC2 |
16 % |
29 % |
0,0005 |
ND |
|
RP2 |
58 % |
58 % |
0,9449 |
ND |
|
Variables secundarias de valoración de la eficacia |
||||
|
SG (mediana) |
NA |
NA |
0,0508 |
32 % |
|
SSP (mediana) |
19,4 meses |
33,2 meses |
0,0001 |
38 % |
|
1 Las estimaciones se efectuaron por hazard ratios (razones de riesgos instantáneos). 2 Última respuesta tumoral según la evaluación del investigador. La prueba estadística «principal» de la «respuesta» fue la prueba de tendencias de la RC en comparación con la RP y en comparación con la ausencia de respuesta (p <0,0001) Abreviaturas: ND: no disponible; NA: no alcanzado; TRG: tasa de respuesta global; RC: respuesta completa; RP: respuesta parcial; SG: supervivencia global; SSP: supervivencia sin progresión. |
||||
En los pacientes asignados aleatoriamente a la fase de mantenimiento del ensayo, la mediana del periodo de observación fue de 28 meses a partir de la asignación aleatoria a la terapia de mantenimiento. La terapia de mantenimiento con MabThera/Rituxan i.v. se tradujo en una mejoría clínicamente importante y estadísticamente significativa de la variable de valoración principal, la SSP (tiempo transcurrido desde la asignación aleatoria a la terapia de mantenimiento hasta la recaída, la progresión de la enfermedad o la muerte), en comparación con el grupo sometido exclusivamente a observación (p <0,0001, prueba de rangos logarítmicos). La mediana de la SSP fue de 42,2 meses en el grupo de mantenimiento con MabThera/Rituxan i.v. y de 14,3 meses en el grupo de observación. Aplicando un análisis de regresión de Cox, el riesgo de enfermedad progresiva o fallecimiento disminuyó en un 61 % en el grupo de terapia de mantenimiento con MabThera/Rituxan i.v. en comparación con el grupo de observación (IC 95 %, 45-72 %). La estimación de la tasa de supervivencia sin progresión a los 12 meses, realizada según el método de Kaplan-Meier, fue del 78 % en el grupo de mantenimiento con MabThera/Rituxan i.v. y del 57 % en el grupo de observación. Un análisis de la supervivencia global confirmó los beneficios significativos del mantenimiento con MabThera/Rituxan i.v. respecto a la observación (p = 0,0039; prueba de rangos logarítmicos). La terapia de mantenimiento con MabThera/Rituxan i.v. redujo el riesgo de muerte en un 56 % (IC 95 %: 22-75 %).
La mediana del tiempo transcurrido hasta la administración de un nuevo tratamiento contra el linfoma fue significativamente mayor en el grupo de la terapia de mantenimiento con MabThera/Rituxan i.v. que en el grupo de observación (38,8 meses frente a 20,1 meses; p <0,0001, prueba de rangos logarítmicos). El riesgo de empezar un nuevo tratamiento disminuyó en un 50 % (IC del 95 %: 30-64 %). En los pacientes con una RC o una RC no confirmada como mejor respuesta durante el tratamiento de inducción, la terapia de mantenimiento con MabThera/Rituxan i.v. prolongó significativamente la mediana de la supervivencia sin enfermedad (SSE) en comparación con el grupo de observación (53,7 frente a 16,5 meses; p = 0,0003, prueba de rangos logarítmicos) (v. tabla 8). El riesgo de recidiva en los pacientes con una respuesta completa disminuyó en un 67 % (IC 95 %: 39-82 %).
|
Tabla 8. Fase de mantenimiento: cuadro general de los resultados de la eficacia de MabThera/Rituxan i.v. en comparación con la observación (mediana del periodo de observación de 28 meses) |
||||
|
Parámetro de valoración de la eficacia |
Estimación de Kaplan-Meier de la mediana del tiempo transcurrido hasta la aparición del evento (meses) |
Reducción del riesgo |
||
|
Observación (n = 167) |
MabThera/Rituxan (n = 167) |
p, prueba de rangos logarítmicos |
||
|
Supervivencia sin progresión (SSP) |
14,3 |
42,2 |
<0,0001 |
61 % |
|
Supervivencia global |
NA |
NA |
0,0039 |
56 % |
|
Tiempo transcurrido hasta iniciar un nuevo tratamiento del linfoma |
20,1 |
38,8 |
<0,0001 |
50 % |
|
Supervivencia sin enfermedad |
16,5 |
53,7 |
0,0003 |
67 % |
|
Análisis de subgrupos |
||||
|
SSP: CHOP R-CHOP RC RP |
11,6 22,1 14,3 14,3 |
37,5 51,9 52,8 37,8 |
<0,0001 0,0071 0,0008 <0,0001 |
71 % 46 % 64 % 54 % |
|
SG CHOP R-CHOP |
NA NA |
NA NA |
0,0348 0,0482 |
55 % 56 % |
|
NA: no alcanzado; a: sólo aplicable a los pacientes que presentaron una RC. |
||||
Los beneficios de la terapia de mantenimiento con MabThera/Rituxan i.v. se confirmaron en todos los subgrupos analizados, con independencia del régimen de inducción (CHOP o R-CHOP) o la calidad de la respuesta al tratamiento de inducción (RC o RP) (ver tabla 8). La terapia de mantenimiento con MabThera/Rituxan i.v. prolongó significativamente la mediana de la SSP tanto en los pacientes que respondieron al tratamiento de inducción con CHOP (mediana de la SSP: 37,5 frente a 11,6 meses, p <0,0001) como en los que respondieron al tratamiento de inducción con R-CHOP (mediana de la SSP: 51,9 frente a 22,1 meses; p <0,0071). La terapia de mantenimiento con MabThera/Rituxan i.v. también proporcionó un beneficio clínicamente significativo en cuanto a la supervivencia global de los pacientes que respondieron a CHOP y los que respondieron a R-CHOP en la fase de inducción del estudio.
La terapia de mantenimiento con MabThera/Rituxan i.v. se tradujo en beneficios constantes en todos los subgrupos de estudio —sexo (masculino, femenino), edad (≤60 años, >60 años), estadio (III, IV), estado general según la escala de la OMS (0 frente a >0), síntomas B (ausentes, presentes), afectación de la médula ósea («no» frente a «sí»), IPI (0-2 frente a 3-5), FLIPI (0-1 frente a 2 y frente a 3-5), número de localizaciones extraganglionares (0-1 frente a >1), número de localizaciones ganglionares (<5 frente a ≥5), número de regímenes anteriores (1 frente a 2), mejor respuesta al tratamiento anterior (RC/RP frente a ausencia de cambios/EP), hemoglobina (<12 g/dl frente a ≥12 g/dl), microglobulina b2 (<3 mg/l frente a ≥3 mg/l), LDH (elevada, no elevada), salvo en el pequeño subgrupo de pacientes con una gran masa tumoral.
Linfoma no hodgkiniano difuso de linfocitos B grandes
En un ensayo aleatorizado y sin enmascaramiento, un total de 399 pacientes ancianos (edad: 60-80 años) no tratados previamente que padecían linfomas difusos de linfocitos B grandes recibieron quimioterapia CHOP convencional (ciclofosfamida: 750 mg/m2; doxorubicina: 50 mg/m2; vincristina: 1,4 mg/m2 hasta un máximo de 2 mg el día 1, y prednisolona: 40 mg/m2/día los días 1-5) cada 3 semanas, durante 8 ciclos, o bien MabThera/Rituxan i.v. 375 mg/m2 + CHOP (R-CHOP). MabThera/Rituxan i.v. se administró el primer día de cada ciclo de tratamiento.
En el análisis final de la eficacia se incluyó a todos los pacientes aleatorizados (CHOP: 197; R-CHOP: 202) y la mediana de la duración del seguimiento fue de 31 meses aproximadamente. Los dos grupos de tratamiento estaban adecuadamente equilibrados en cuanto a las características iniciales y el estado clínico. En el análisis final se confirmó que en el grupo de R-CHOP había aumentado significativamente la duración de la supervivencia sin eventos (la variable principal de valoración de la eficacia, siendo los eventos la muerte, la recidiva o la progresión del linfoma, o la instauración de un nuevo tratamiento del linfoma) (p = 0,0001). Las estimaciones de Kaplan-Meier de la mediana de la supervivencia sin eventos fueron de 35 meses en el grupo de R-CHOP y de 13 meses en el grupo de CHOP, lo que representa una reducción del riesgo del 41 %. A los 24 meses, las estimaciones de la supervivencia global fueron del 68,2 % en el grupo de R-CHOP y del 57,4 % en el grupo de CHOP. Un análisis ulterior de la duración de la supervivencia global, realizado con una mediana del seguimiento de 60 meses, confirmó la ventaja de R-CHOP respecto a CHOP (p = 0,0071), lo que representa una reducción del riesgo del 32 %.
El análisis de todas las variables de valoración secundarias (tasas de respuesta, supervivencia sin progresión, supervivencia sin enfermedad, duración de la respuesta) corroboró el efecto del tratamiento con R-CHOP en comparación con CHOP. La tasa de respuesta completa después del ciclo 8 fue del 76,2 % en el grupo de R-CHOP y del 62,4 % en el grupo de CHOP (p = 0,0028). El riesgo de progresión de la enfermedad disminuyó en un 46 %, y el riesgo de recaída, en un 51 %.
El índice de riesgo para la supervivencia sin eventos y la supervivencia global (R-CHOP en comparación con CHOP) fue <0,83 y <0,95, respectivamente, en todos los subgrupos de pacientes (sexo, edad, IPI ajustado en función de la edad, estadio según la clasificación de Ann Arbor, estado general según la escala del ECOG, microglobulina b2, LDH, albúmina, síntomas B, gran masa tumoral, enfermedad extraganglionar, afectación de la médula ósea). Según el IPI ajustado en función de la edad, el régimen R-CHOP se acompañó de mejoras en los resultados en los pacientes de algo riesgo y en los de bajo riesgo.
Leucemia linfocítica crónica sin tratamiento previo y recidivante o resistente al tratamiento
En dos ensayos aleatorizados y sin enmascaramiento, un total de 817 pacientes sin tratamiento previo y 552 pacientes con LLC recidivante o resistente al tratamiento fueron asignados aleatoriamente a recibir FC (fludarabina, 25 mg/m2, y ciclofosfamida, 250 mg/m2; días 1-3) cada 4 semanas durante 6 ciclos o MabThera/Rituxan i.v. en combinación con FC (R-FC). MabThera/Rituxan i.v. se administró en una dosis de 375 mg/m2 durante el primer ciclo un día antes de la quimioterapia, y en una dosis de 500 mg/m2 el día 1 de cada ciclo de tratamiento ulterior. Se analizó la eficacia en un total de 810 pacientes (R-FC: 403, FC: 407) del estudio del tratamiento de primera línea (ver tabla 9 y tabla 10) y 552 pacientes (R-FC: 276; FC: 276) del estudio de la LLC recidivante o resistente al tratamiento (ver tabla 11).
En el estudio del tratamiento de primera línea, después de un periodo de observación con una mediana de 20,7 meses, la mediana de la supervivencia sin progresión (variable de valoración principal) fue de 40 meses en el grupo de R-FC y de 32 meses en el grupo de FC (p <0,0001, prueba de rangos logarítmicos) (ver tabla 9). El análisis de la supervivencia global mostró una mejoría de la supervivencia a favor del grupo de R-FC (p = 0,0427, prueba de rangos logarítmicos). Estos resultados se confirmaron con un seguimiento más prolongado: tras un periodo de observación de 48,1 meses (mediana), la mediana de la SSP fue de 55 meses en el grupo de R-FC y 33 meses en el grupo de FC (p <0,0001, prueba de rangos logarítmicos) y los análisis de la supervivencia global seguían mostrando un beneficio significativo del tratamiento con R-FC respecto a FC solo (p = 0,0319, prueba de rangos logarítmicos). El beneficio en lo que res-pecta a la SSP se observó constantemente en la mayoría de los subgrupos de pacientes analizados según el riesgo de enfermedad al inicio del estudio (es decir, estadios A-C según la clasificación de Binet) y se confirmó con un seguimiento más prolongado (ver tabla 10).
|
Tabla 9. Tratamiento de primera línea de la leucemia linfocítica crónica: cuadro general de los resultados de la eficacia de MabThera/Rituxan i.v. más FC en comparación con FC solo (mediana del periodo de observación de 20,7 meses) |
||||
|
Parámetro de valoración de la eficacia |
Estimación de Kaplan-Meier de la mediana del tiempo transcurrido hasta la aparición del evento (meses) |
Hazard ratio |
||
|
FC (n = 407) |
R-FC (n = 403) |
p, prueba de rangos logarítmicos |
||
|
Supervivencia sin progresión (SSP) |
32,2 (32,8)*** |
39,8 (55,3)*** |
<0,0001 (<0,0001)*** |
0,56 (0,55)*** |
|
Supervivencia global |
NA (NA)*** |
NA (NA)*** |
0,0427 (0,0319)*** |
0,64 (0,73)*** |
|
Supervivencia sin eventos |
31,1 (31,3)*** |
39,8 (51,8)*** |
<0,0001 (<0,0001)*** |
0,55 (0,56)*** |
|
Tasa de respuesta (RC, RP no confirmada, RP) |
72,7 % |
86,1 % |
<0,0001 |
NP |
|
Tasas de RC |
17,2 % |
36,0 % |
<0,0001 |
NP |
|
Duración de la respuesta* |
34,7 (36,2)*** |
40,2 (57,3)*** |
0,0040 (<0,0001)*** |
0,61 (0,56)*** |
|
Supervivencia sin enfermedad (SSE)** |
NA (48,9)*** |
NA (60,3)*** |
0,7882 (0,0520)*** |
0,93 (0,69)*** |
|
Tiempo transcurrido hasta iniciar un nuevo tratamiento de la LLC |
NA (47,2)*** |
NA (69,7)*** |
0,0052 (<0,0001)*** |
0,65 (0,58)*** |
|
Tasa de respuesta y tasas de RC analizadas usando la prueba de la X2. *** Los valores entre paréntesis corresponden a la mediana del periodo de observación de 48,1 meses (población ITT: FC: 409; R-FC: 408). NA: no alcanzado; NP: No procede. *: Sólo aplicable a pacientes con RC, RP no confirmada o RP como respuesta al final del tratamiento. **: Sólo aplicable a pacientes con RC como respuesta al final del tratamiento. |
||||
|
Tabla 10. Hazard ratios (razones de riesgos instantáneos) de la supervivencia sin progresión según el estadio de Binet (ITT) - Mediana del periodo de observación de 20,7 meses |
||||
|
Supervivencia sin progresión (SSP) |
Número de pacientes |
Hazard ratio (IC 95 %) |
p, prueba de rangos logarítmicos |
|
|
FC |
R-FC |
|||
|
Estadio A de Binet |
22 (22)* |
18 (18)* |
0,13 (0,03-0,61) (0,39 (0,15-0,98))* |
0,0025 (0,0370)* |
|
Estadio B de Binet |
257 (259)* |
259 (263)* |
0,45 (0,32-0,63) (0,52 (0,41-0,66))* |
<0,0001 (<0,0001)* |
|
Estadio C de Binet |
126 (126)* |
125 (126)* |
0,88 (0,58-1,33) (0,68 (0,49-0,95))* |
0,5341 (0,0215)* |
|
IC: intervalo de confianza. * Los valores entre paréntesis corresponden a la mediana del periodo de observación de 48,1 meses (población ITT: FC: 409; R-FC: 408). |
||||
En el estudio de la LLC recidivante o resistente al tratamiento, la mediana de la SSP (variable de valora-ción principal) fue de 30,6 meses en el grupo de R-FC y de 20,6 meses en el grupo de FC (p = 0,0002, prueba de rangos logarítmicos). El beneficio en lo que se refiere a la SSP se observó prácticamente en todos los subgrupos de pacientes analizados según el riesgo de enfermedad al inicio del estudio. Se registró un aumento leve, pero no significativo, de la supervivencia global en el grupo de R-FC en comparación con el grupo de FC.
|
Tabla 11. Tratamiento de la leucemia linfocítica crónica recidivante o resistente al tratamiento: cuadro general de los resultados de la eficacia de MabThera/Rituxan i.v. más FC en comparación con FC solo (mediana del periodo de observación de 25,3 meses) |
||||
|
Parámetro de valoración de la eficacia |
Estimación de Kaplan-Meier de la mediana del tiempo transcurrido hasta la aparición del evento (meses) |
Reducción del riesgo |
||
|
FC (n = 276) |
R-FC (n = 276) |
p, prueba de rangos logarítmicos |
||
|
Supervivencia sin progresión (SSP) |
20,6 |
30,6 |
0,0002 |
35 % |
|
Supervivencia global |
51,9 |
NA |
0,2874 |
17 % |
|
Supervivencia sin eventos |
19,3 |
28,7 |
0,0002 |
36 % |
|
Tasa de respuesta (RC, RP no confirmada, RP) |
58,0 % |
69,9 % |
0,0034 |
NP |
|
Tasas de RC |
13,0 % |
24,3 % |
0,0007 |
NP |
|
Duración de la respuesta* |
27,6 |
39,6 |
0,0252 |
31 % |
|
Supervivencia sin enfermedad (SSE)** |
42,2 |
39,6 |
0,8842 |
-6 % |
|
Tiempo transcurrido hasta iniciar un nuevo tratamiento de la LLC |
34,2 |
NA |
0,0024 |
35 % |
|
Tasa de respuesta y tasas de RC analizadas usando la prueba de la -2. NA: no alcanzado; NP: no procede. * Sólo aplicable a pacientes con RC, RP no confirmada o RP como mejor respuesta global. **: Sólo aplicable a los pacientes con RC como mejor respuesta global. |
||||
Los resultados de otros estudios de apoyo en los que usó MabThera/Rituxan i.v. en combinación con otros regímenes de quimioterapia (como CHOP, FCM, PC, PCM, bendamustina y cladribina) para el tratamiento de pacientes con LLC también han evidenciado elevadas tasas de respuesta global y prometedoras tasas de SSP sin añadir toxicidad importante al tratamiento.
Estudio de la infusión de 90 minutos de MabThera/Rituxan i.v. (U4391g)
Pacientes con linfoma no hodgkiniano folicular y linfoma no hodgkiniano difuso de linfocitos B grandes sin tratamiento previo
En un ensayo de fase III multicéntrico, prospectivo, sin enmascaramiento, con un solo grupo, 363 pacientes con linfoma difuso de linfocitos B grandes sin tratamien-to previo que recibieron 375 mg/m2 de MabThera/Rituxan i.v. más CHOP, o con LNH folicular no tratados previamente que recibieron 375 mg/m2 más CVP, fueron tratados con una infusión de 90 minutos de MabThera/Rituxan i.v. para evaluar la seguridad de la infusión de 90 minutos. Se excluyó del estudio a los pacientes con enfermedades cardiovasculares clínicamente significativas.
Los pacientes podían continuar en el estudio si no habían sufrido ninguna reacción adversa de grado 3 y 4 relacionada con la infusión en el ciclo 1 (a la velocidad de infusión habitual de MabThera/Rituxan i.v.) y tenían una cifra de linfocitos circulantes ≤5000/mm3 antes del ciclo 2. Los pacientes que continuaron en el estudio reci-bieron la infusión de MabThera/Rituxan i.v. del ciclo 2 del siguiente modo: el 20 % de la dosis total se administró en los 30 primeros minutos, y el 80 % restante en los 60 minutos siguientes. Los pacientes que toleraron la prime-ra infusión de MabThera/Rituxan i.v. de 90 minutos (ciclo 2) siguieron recibiendo las infusiones posteriores de MabThera/Rituxan i.v. de 90 minutos durante el resto del régimen de tratamiento (hasta el ciclo 6 o el ciclo 8).
La variable principal de valoración del estudio fue la aparición de reacciones adversas relacionadas con la infusión de grado 3 o 4 (evento adverso de interés) en pacientes que recibieron MabThera/Rituxan i.v. en una infusión de 90 minutos en el ciclo 2.
La tasa de RRI de grado 3 y 4 en el día de la infusión de 90 minutos de MabThera/Rituxan i.v. en el ciclo 2 o en el día siguiente fue del 1,1 % (IC 95 %: 0,3-2,8 %). La tasa de RRI de grado 3 y 4 en cualquier ciclo (ciclos 2-8) con la infusión de 90 minutos fue del 2,8 % (IC 95 %: 1,3-5,0 %) (ver tabla 12). No se observó ninguna RRI aguda mortal (ver Reacciones adversas, Ensayos clínicos).
|
Tabla 12. Cuadro general de la tasa de reacciones relacionadas con la infusión de grado 3 y 4 en el ciclo 2 y en cualquier ciclo (ciclos 2-8)* |
|||
|
N.º (%) de pacientes que presentaron |
R-CHOP n = 250 |
R-CVP n = 113 |
Total n = 363 |
|
RRI que comenzaron el día de la infusión más rápida de MabThera/Rituxan i.v. en el ciclo 2 o el día siguiente |
|||
|
- RRI de grado 3 y 4 (variable de valoración principal) |
0 (0,0 %) |
4 (3,5 %) |
4 (1,1 %) |
|
RRI que comenzaron el día de las infusiones de MabThera/Rituxan i.v. en los ciclos 2-8 o el día siguiente |
|||
|
- RRI de grado 3 y 4 |
4 (1,6 %) |
6 (5,3 %) |
10 (2,8 %) |
|
* No se resumen las reacciones adversas que tuvieron lugar antes del ciclo 2. Las RRI son las reacciones adversas relacionas con la infusión enumeradas en el plan de análisis estadístico que tuvieron lugar el día de la infusión de MabThera/Rituxan i.v. o el día siguiente. |
|||
Formulación subcutánea
Pacientes con linfoma no hodgkiniano folicular sin tratamiento previo - Estudio BO22334 (SABRINA)
Se llevó a cabo un ensayo de fase III de 2 etapas, interna-cional, multicéntrico, aleatorizado, comparativo, sin enmascaramiento, en pacientes con linfoma folicular que no habían sido tratados previamente, a fin de investigar la ausencia de inferioridad del perfil farmacocinético, junto con la eficacia y la seguridad, de MabThera/Rituxan s.c. en combinación con CHOP o CVP en comparación con MabThera/Rituxan i.v. en combinación con CHOP o CVP, y a continuación tratamiento de mantenimiento con MabThera/Rituxan.
El objetivo de la primera etapa era establecer la dosis de MabThera/Rituxan s.c. que daba lugar a Cvalle séricas de rituximab comparables a las obtenidas con MabThera/Rituxan i.v. cuando se administró como parte del tratamiento de inducción cada 3 semanas durante 8 ciclos (ver Propiedades farmacocinéticas, Distribución). En la etapa 1 participaron pacientes con linfoma folicular (LF) CD20-positivo de grado 1, 2 o 3a (n = 127) sin tratamiento previo. Los pacientes con respuesta al final de la terapia de inducción recibieron terapia de mantenimiento con la formulación correspondiente (i.v. o s.c.) utilizada en el tratamiento de inducción, cada 8 semanas durante 24 meses.
El objetivo de la etapa 2 era proporcionar datos adiciona-les sobre la eficacia y la seguridad de MabThera/Rituxan s.c. en comparación con MabThera/Rituxan i.v. usando la dosis s.c. de 1400 mg que se estableció en la etapa 1. En la etapa 2 participaron pacientes con linfoma folicular CD20-positivo de grado 1, 2 o 3a no tratados previamente (n = 283).
El diseño general del estudio fue idéntico en la etapa 1 y la etapa 2. Los pacientes fueron distribuidos aleatoriamente entre estos dos grupos de tratamiento:
• Grupo de MabThera/Rituxan s.c. (n = 205): primer ciclo de MabThera/Rituxan i.v. más 7 ciclos de MabThera/Rituxan s.c. en combinación con hasta 8 ciclos de quimioterapia CHOP o CVP administrada cada 3 semanas. Se usó MabThera/Rituxan i.v. en la dosis habitual de 375 mg/m2. MabThera/Rituxan s.c. se administró en una dosis fija de 1400 mg. Los pacientes que alcanzaron al menos una respuesta parcial al final del tratamiento de inducción pasaron a la terapia de mantenimiento con MabThera/Rituxan s.c. administrado una vez cada 8 semanas durante 24 meses.
• Grupo de MabThera/Rituxan i.v. (n = 205): 8 ciclos de MabThera/Rituxan i.v. en combinación con hasta 8 ciclos de quimioterapia CHOP o CVP administrada cada 3 semanas. Se administró MabThera/Rituxan i.v. en la dosis habitual de 375 mg/m2. Los pacientes que lograron al menos una respuesta parcial al final de la inducción pasaron a la terapia de mantenimiento con MabThera/Rituxan i.v. administrado una vez cada 8 semanas durante 24 meses.
La tasa de respuesta global (TRG, que abarca la respuesta completa [RC], la RC no confirmada y la respuesta parcial [RP]) al final del tratamiento de inducción se calculó usando la evaluación de los investigadores de la respuesta en la población ITT, que se basó en el conjunto combinado de datos de las etapas 1 y 2. Además se analizaron la TRG y la tasa de respuesta completa (TRC, que abarca la RC y la RC no confirmada) al final del tratamiento de mantenimiento y variables de valoración del tipo del tiempo transcurrido hasta el acontecimiento (la supervivencia sin progresión [SSP] y la supervivencia global [SG]). Los resultados relativos a la eficacia se presentan en la tabla 13 y se basan en un periodo de observación (mediana) de aproximadamente 37 meses.
|
Tabla 13. Resultados relativos a la eficacia en el estudio SABRINA/BO22334 |
||
|
MabThera/Rituxan s.c. n = 205 |
MabThera/Rituxan i.v. n = 205 |
|
|
Tasa de respuesta global al final de la induccióna |
||
|
N.º de pacientes con respuesta (RC, RC no confirmada, RP) |
173 |
174 |
|
Tasa de respuesta global (RC/RC no confirmada, RP) (%, [IC 95 %]) |
84,4 % [78,7; 89,1] |
84,9 % [79,2; 89,5] |
|
N.º de pacientes con respuesta completa (RC/RC no confirmada) |
66 |
66 |
|
Tasa de respuesta completa (RC/RC no confirmada) (%, [IC 95 %]) |
32,2 % [25,9; 39,1] |
32,2 % [25,9; 39,1] |
|
Tasa de respuesta global al final del mantenimiento |
||
|
Número de pacientes tratados en el mantenimiento (n) |
172 |
178 |
|
N.º de pacientes con respuesta (RC, RC no confirmada, RP) |
134 |
139 |
|
Tasa de respuesta global (RC/RC no confirmada, RP) (%, [IC 95 %]) |
77,9 % [71,0; 83,9] |
78,1 % [71,3; 83,9] |
|
N.º de pacientes con respuesta completa (RC/RC) |
87 |
100 |
|
Tasa de respuesta completa (RC/RC no confirmada) (%, [IC 95 %]) |
50,6 % [42,9; 58,3] |
56,2 % [48,6; 63,6] |
|
Supervivencia sin progresión |
||
|
Número de pacientes con el evento |
50 (24,4%) |
57 (27,8%) |
|
Hazard ratio [IC 95%] (modelo de Cox no estratificado) |
0,84 [0,57; 1,23] |
|
|
Supervivencia global |
||
|
Número de pacientes con el evento |
16 (7,8%) |
20 (9,8%) |
|
Hazard ratio [IC 95%] (modelo de Cox no estratificado) |
0,81 [0,42; 1,57] |
|
|
a La variable principal de valoración de la eficacia en la etapa 2 fue la TRG al final de la inducción, aunque los resultados combinados que se planificaron se presentan en esta tabla. Las tasas de respuesta se basan en la evaluación del investigador. Las tasas de respuesta al final del mantenimiento se basan en los pacientes que recibieron al menos un ciclo de tratamiento de mantenimiento (n). |
||
Los análisis exploratorios mostraron que las tasas de respuesta en los subgrupos de superficie corporal, quimioterapia recibida y sexo no difirieron notablemente de los valores de la población ITT global.
Leucemia linfocítica crónica - Estudio BO25341 (SAWYER)
Se realizó un estudio de fase Ib con dos partes, multicéntrico, aleatorizado, sin enmascaramiento, con grupos paralelos, en pacientes con LLC sin tratamiento previo, para investigar la ausencia de inferioridad del perfil farmacocinético, junto con la eficacia y la seguridad, de MabThera/Rituxan s.c. en combinación con quimioterapia.
El objetivo de la parte 1 era seleccionar una dosis de MabThera/Rituxan s.c. que diera lugar a Cvalle de rituximab en suero comparables a las de MabThera/Rituxan i.v. Se incluyó a pacientes con LLC sin trata-miento previo (n = 64) en cualquier momento, antes del ciclo 5, durante su tratamiento con MabThera/Rituxan i.v. en combinación con quimioterapia. Se seleccionó la dosis de 1600 mg de MabThera/Rituxan s.c. para la parte 2 del estudio.
El objetivo de la parte 2 era establecer la ausencia de inferioridad en la Cvalle observada de rituximab entre la dosis seleccionada de MabThera/Rituxan s.c. y la dosis de referencia de MabThera/Rituxan i.v.
Se distribuyó aleatoriamente a pacientes con LLC sin tratamiento previo (n = 176) en estos dos grupos de tratamiento:
- Grupo de MabThera/Rituxan s.c. (n = 88): primer ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2 en combinación con quimioterapia, más ciclos posteriores (2-6) de MabThera/Rituxan s.c. en dosis de 1600 mg en combinación con quimioterapia.
- Grupo de MabThera/Rituxan i.v. (n = 88): primer ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2 en combinación con quimioterapia y a continuación hasta 5 ciclos de MabThera/Rituxan i.v. en dosis de 500 mg/m2 en combinación con quimioterapia.
Las tasas de respuesta fueron similares en los grupos de tratamiento con MabThera/Rituxan i.v. y s.c.; la tasa de respuesta global fue del 80,7 % (IC 95 %: 70,9-88,3) y 85,2 % (IC 95 %: 76,1-91,9) en los grupos de MabThera i.v. y s.c., respectivamente. Las estimaciones puntuales de la tasa de respuesta completa fueron del 33,0 % (IC 95 %: 23,3-43,8) y 26,1 % (IC 95 %: 17,3-36,6) en los grupos de MabThera i.v. y s.c., respectivamente. En general, los resultados confirman que MabThera/Rituxan s.c. en dosis de 1600 mg tiene un perfil de beneficios y riesgos comparable al de MabThera/Rituxan i.v. en dosis de 500 mg/m2.
Formulación intravenosa
Artritis reumatoide
La eficacia y la seguridad de MabThera/Rituxan i.v. para aliviar los signos y síntomas de la artritis reumatoide (AR) quedó demostrada en tres estudios fundamentales de fase III multicéntricos, aleatorizados, comparativos con placebo y con doble enmascaramiento (doble ciego). Los pacientes aptos para participar en los estudios tenían una AR activa grave, diagnosticada conforme a los criterios del American College of Rheumatology (ACR). MabThera/Rituxan i.v. se administró en dos infusiones i.v. con un intervalo de 15 días entre ambas. Antes de cada ciclo se administró una infusión i.v. de 100 mg de metilprednisolona. Todos los pacientes recibieron concomitantemente metotrexato por vía oral. Además, todos los pacientes del estudio WA17042 recibie-ron concomitantemente glucocorticoides por vía oral los días 2-7 y los días 8-14 después de la primera infusión.
Los criterios para el retratamiento diferían entre los estudios, aplicándose una de las dos variantes siguientes: «tratamiento hasta la remisión», en la que los pacientes recibían tratamiento con una frecuencia no superior a la frecuencia semestral si no se encontraban en remisión DAS28 (es decir, DAS28-VSG ≥2,6), y «tratamiento en caso necesario», basada en la actividad de la enfermedad o la reinstauración de los síntomas clínicos (número de articulaciones inflamadas y doloridas ≥8) y con trata-miento no más frecuente que cada 16 semanas.
En el estudio WA17042 (REFLEX) participaron 517 pacientes que no habían respondido adecuadamente o no toleraron uno o más inhibidores del TNF (TNF-RI). La variable de valoración principal fue la proporción de pacientes que alcanzaron una respuesta ACR20 en la semana 24. Los pacientes recibieron 2 dosis de 1000 mg de MabThera/Rituxan i.v. o placebo. Se realizó el seguimiento de los pacientes más allá de la semana 24 en lo que respecta a las variables de valoración a largo plazo, incluida la evaluación radiográfica realizada a las 56 semanas. Durante este periodo, los pacientes podían recibir ciclos adicionales de MabThera/Rituxan i.v. según el protocolo de un estudio de extensión sin enmascaramiento. De acuerdo con el protocolo de estudio sin enmascaramiento, los pacientes recibieron ciclos adicionales basados en el criterio de «tratamiento en caso necesario».
El estudio WA17045 (SERENE) incluyó a 511 pacientes con una respuesta inadecuada al metotrexato (MTX-RI) y que no habían recibido antes tratamiento biológico. La variable de valoración principal fue la proporción de pacientes que alcanzaron una respuesta ACR20 en la semana 24. Los pacientes recibieron placebo o 2 dosis de 500 mg o 2 dosis de 1000 mg de MabThera/Rituxan i.v. El seguimiento de los pacientes se prolongó más allá de la semana 24 para evaluar las variables de valoración a largo plazo, y podían recibir más ciclos de MabThera/Rituxan i.v. conforme al criterio de «tratamiento hasta la remisión». En la semana 48 se realizó una comparación de las dosis de tratamiento activo.
Criterios de valoración de la actividad de la enfermedad
En estos estudios, la administración de 2 dosis de 1000 mg de MabThera/Rituxan i.v. incrementó significativamente la proporción de pacientes con una mejora del índice ACR de un 20 % como mínimo en comparación con los pacientes tratados con metotrexato solo (ver tabla 14). En todos los estudios del desarrollo, el beneficio terapéutico fue similar independientemente de la edad, el sexo, la superficie corporal, la raza, el número de tratamientos anteriores o el estado clínico de los pacientes. En todos los estudios, la eficacia fue sistemáticamente alta en los pacientes seropositivos para los autoanticuerpos relacionados con la enfermedad (FR, anticuerpos antipéptidos cíclicos citrulinados o ambos) en comparación con los tratados con metotrexato solo. La eficacia en los pacientes seropositivos fue mayor que la observada en los seronegativos, en los cuales fue modesta.
También se observó una mejoría clínica y estadísticamente significativa de todos los componentes de la respuesta ACR (número de articulaciones doloridas e inflamadas, evaluación global del paciente y del médico, índice HAQ [medida de la discapacidad funcional], eva-luación del dolor y proteína C reactiva (mg/dl).
|
Tabla 14. Comparación de la respuesta ACR entre estudios (población ITT) |
||||
|
Momento de valoración |
Respuesta ACR |
Placebo + MTX |
RTX + MTX (2 veces 1000 mg) |
|
|
Estudio WA17042 (TNF-RI) |
Semana 24 |
n = 201 |
n = 298 |
|
|
ACR20 |
36 (18 %) |
153 (51 %)*** |
||
|
ACR50 |
11 (5 %) |
80 (27 %)*** |
||
|
ACR70 |
3 (1 %) |
37 (12 %)*** |
||
|
Estudio WA17045 (MTX-RI) |
Semana 24 |
n = 172 |
n = 170 |
|
|
ACR20 |
40 (23 %) |
86 (51 %)*** |
||
|
ACR50 |
16 (9 %) |
44 (26 %)*** |
||
|
ACR70 |
9 (5 %) |
17 (10 %) |
||
|
Diferencia significativa respecto al placebo en el momento de valoración principal: * p ≤0,05, **p ≤0,001 ***p ≤0,0001. |
||||
El descenso del índice de actividad de la enfermedad (DAS28) fue significativamente mayor en los pacientes tratados con MabThera/Rituxan i.v. que en los que recibieron metotrexato solo. El número de pacientes que alcanzaron una respuesta EULAR de buena a moderada fue significativamente mayor con MabThera/Rituxan i.v. que con el metotrexato solo (ver tabla 15).
|
Tabla 15. Comparación entre estudios de las respuestas DAS28-VSG y EULAR (población ITT) |
||
|
Placebo + MTX |
RTX + MTX (2 veces 1000 mg) |
|
|
Estudio WA17042 (TNF-RI) |
||
|
Cambio del DAS28 en la semana 24 n Media del cambio |
n = 201 -0,4 |
n = 298 -1,9*** |
|
Respuesta EULAR (semana 24) n Moderada Buena |
n = 201 20 % 2 % |
n = 298 50 %*** 15 %*** |
|
Estudio WA17045 (MTX-RI) |
||
|
Cambio del DAS28 en la semana 24 n Media del cambio |
n = 171 –0,8 |
n = 168 –1,7*** |
|
Respuesta EULAR (semana 24) n Moderada Buena |
n = 172 29 % 5 % |
n = 170 51 %*** 12 %*** |
|
Diferencia significativa respecto al placebo en el momento de valoración principal: * p ≤0,05, **p ≤0,001 ***p ≤0,0001. |
||
Inhibición del daño articular
En los estudios WA17042 y WA17047 se evaluó radio-gráficamente el daño estructural articular y se expresó como variación del índice total de Sharp modificado y sus componentes, la puntuación de la erosión y la puntuación del estrechamiento del espacio articular.
En el estudio WA17042, realizado en pacientes con respuesta inadecuada a inhibidores del TNF (TNF-RI) tratados con MabThera/Rituxan i.v. en combinación con metotrexato, se demostró una progresión radiográfica significativamente menor a las 56 semanas en compara-ción con los pacientes tratados con metotrexato solo. También fue mayor la proporción de pacientes tratados con MabThera/Rituxan i.v. sin progresión de la erosión a las 56 semanas.
En el estudio WA17047, efectuado en pacientes que no habían recibido previamente metotrexato (755 pacientes con AR precoz de 8 semanas a 4 años de evolución), se evaluó la prevención del daño estructural articular como variable de valoración principal (ver Advertencias y precauciones). Los pacientes recibieron placebo, 2 veces 500 mg o 2 veces 1000 mg de MabThera/Rituxan i.v. en infusión. A partir de la semana 24, los pacientes podían recibir nuevas tandas de MabThera/Rituxan i.v. (o placebo hasta la semana 104) según los criterios de «tratamiento hasta remisión». La variable de valoración principal, la variación del índice total de Sharp modificado, demostró que sólo el tratamiento con MabThera/Rituxan i.v. en una dosis de 2 veces 1000 mg en combinación con metotrexato reducía significativamente la tasa de progresión del daño articular a las 52 semanas en comparación con el placebo más metotrexato (ver tabla 16). La reducción de la progresión del daño articular obedeció sobre todo a una disminución significativa en el cambio de la puntuación de la erosión.
Asimismo se observó una reducción de la tasa de daño articular progresivo a largo plazo. En el estudio WA17042, los análisis radiográficos a los 2 años demostraron una disminución significativa de la progresión del daño estructural articular en los pacientes tratados con MabThera/Rituxan i.v. (2 veces 1000 mg) más metotrexato en comparación con los que recibieron el metotrexato solo, así como una proporción significativamente mayor de pacientes sin ninguna progresión del daño articular en el periodo de 2 años.
|
Tabla 16. Resultados radiográficos al cabo de 1 año en los estudios WA17042 y WA17047 (población ITTm) |
||
|
Placebo + MTX |
RTX + MTX (2 veces 1000 mg) |
|
|
Estudio WA17042 (TNF-RI) |
n = 184 |
n = 273 |
|
Media del cambio respecto a los valores iniciales: |
||
|
Índice total de Sharp modificado |
2,30 |
1,01* |
|
Puntuación de la erosión |
1,32 |
0,60* |
|
Puntuación del estrechamiento del espacio articular |
0,98 |
0,41** |
|
Proporción de pacientes sin cambios radiográficos |
46 % |
53 % NS |
|
Proporción de pacientes sin cambios erosivos |
52 % |
60 % NS |
|
Estudio WA17047 (pacientes no tratados previamente con MTX) |
n = 232 |
n = 244 |
|
Media del cambio respecto a los valores iniciales: |
||
|
Índice total de Sharp modificado |
1,079 |
0,359** |
|
Puntuación de la erosión |
0,738 |
0,233*** |
|
Puntuación del estrechamiento del espacio articular |
0,341 |
0,126 |
|
Proporción de pacientes sin cambios radiográficos |
53 % |
64 %* |
|
Proporción de pacientes sin cambios erosivos |
55 % |
67 %* |
|
Los resultados radiográficos se evaluaron en la semana 52 en el estudio WA17047 y en la semana 56 en el estudio WA17042. 150 pacientes en principio asignados aleatoriamente al grupo de placebo + MTX en el estudio WA17042 recibieron al menos un ciclo de RTX + MTX en un año. * p <0,05; ** p <0,001; *** p <0,0001; NS: no significativo. |
||
Variables de valoración de la calidad de vida
Los pacientes tratados con MabThera/Rituxan i.v. refirieron una mejoría de todas las variables de valoración evaluadas por ellos (cuestionarios HAQ-DI, FACIT-Fatiga y SF-36). Se alcanzaron reducciones significativas en los índices de discapacidad (HAQ-DI) y fatiga (FACIT-Fatiga), así como una mejoría en los aspectos relativos a la salud física del cuestionario SF-36 en los pacientes tratados con MabThera/Rituxan i.v. en comparación con los que recibieron metotrexato solo.
|
Tabla 17. Comparación entre estudios de las respuestas a los cuestionarios HAQ-DI y FACIT-Fatiga |
||
|
Placebo + MTX1 |
RTX + MTX1 (2 veces 1000 mg) |
|
|
Estudio WA17042 (TNF-RI) |
n = 201 |
n = 298 |
|
- Media del cambio en el HAQa en la semana 24 - % de pacientes con una DMCI en el HAQ en la semana 24 - Media del cambio en el FACIT-Fatigab en la semana 24 |
-0,1 20 % -0,5 |
-0,4*** 51 % -9,1*** |
|
Estudio WA17045 (MTX-RI) |
n = 172a (170)b |
n = 170a (168)b |
|
- Media del cambio en el HAQa en la semana 24 - % de pacientes con una DMCI en el HAQ en la semana 24 - Media del cambio en el FACIT-Fatigab en la semana 24 |
-0,21 48 % 2,7 |
-0,42*** 58 %* 6,4*** |
|
a Cuestionario de evaluación de la salud (HAQ);b Evaluación funcional del tratamiento de las enfermedades crónicas (FACIT-Fatiga) Diferencia significativa respecto al placebo en el momento de valoración principal: * p <0,05; **p <0,001; ***p ≤0,0001. (Prueba de Cochran-Mantel-Haenszel del cambio cualitativo, ANOVA para la media del cambio; nótese que se muestran los cambios medios no ajustados). |
||
|
Tabla 18. Comparación entre estudios del cuestionario breve de salud SF-36 |
||
|
Placebo + MTX |
RTX + MTX (2 veces 1000 mg) |
|
|
Estudio WA17042 (TNF-RI) |
n = 197 |
n = 294 |
|
Salud física |
||
|
Media del cambio en la semana 24 |
0,9 |
5,8*** |
|
% de pacientes con una DMCI en la semana 24 |
13 % |
48 %*** |
|
Salud mental |
||
|
Media del cambio en la semana 24 |
1,3 |
4,7** |
|
% de pacientes con una DMCI en la semana 24 |
20 % |
38 %** |
|
Estudio WA17045 (MTX-RI) |
n = 147 |
n = 155 |
|
Salud física |
||
|
Media del cambio en la semana 24 |
2,7 |
5,9*** |
|
% de pacientes con una DMCI en la semana 24 |
31 % |
48 % |
|
Salud mental |
||
|
Media del cambio en la semana 24 |
2,1 |
4,4** |
|
% de pacientes con una DMCI en la semana 24 |
24 % |
35 %* |
|
DMCI = diferencia mínima clínicamente importante, definida como un aumento: >6,33 en el índice de salud mental y >5,42 en el índice de salud física; % de pacientes basado en el número de pacientes evaluables (N). Diferencia significativa respecto al placebo en el momento de valoración principal: * p ≤0,05; **p ≤0,001; ***p ≤0,0001 (prueba de Cochran-Mantel-Haenszel para el cambio cualitativo, ANOVA para la media del cambio; nótese que se muestran los cambios medios no ajustados). |
||
Evaluaciones de laboratorio
Aproximadamente el 10 % de los pacientes con artritis reumatoide mostraron resultados positivos en el análisis de HACA en los estudios clínicos. La aparición de HACA no se asoció a un deterioro clínico o a un mayor riesgo de reacciones a las infusiones posteriores en la mayoría de los pacientes. La presencia de HACA puede asociarse a un empeoramiento de las reacciones a la infusión o las reacciones alérgicas después de la segunda infusión de ciclos posteriores; en muy raras ocasiones se ha observado el fracaso de la depleción de los linfocitos B después de administrar ciclos de tratamiento adicionales.
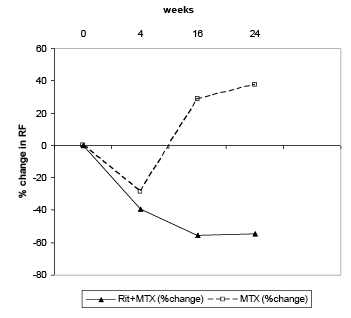
En los pacientes con un resultado positivo en el análisis del factor reumatoide (FR), se observó en los tres estudios un descenso pronunciado de la concentración de FR después del tratamiento con MabThera/Rituxan i.v. (intervalo: 45-64 %, figura 1).
Figura 1. Cambio porcentual de la concentración total de FR a lo largo del tiempo en el estudio 1 (población ITT, pacientes FR-positivos)
La concentración total de inmunoglobulinas en el plasma, así como la cifra total de linfocitos y de leucocitos, se mantuvieron generalmente dentro de los límites normales tras administrar MabThera/Rituxan i.v., salvo una caída temporal del número de leucocitos en las 4 primeras semanas después del tratamiento. Los títulos de anticuerpos IgG específicos de antígenos contra el virus de las paperas, la rubéola, la varicela, el toxoide tetánico, el virus de la gripe y Streptococcus pneumoniae permanecieron estables durante 24 horas tras la exposición a MabThera/Rituxan i.v. en pacientes con artritis reumatoide.
En los pacientes participantes en un estudio clínico se evaluaron los efectos del rituximab en diversos biomarcadores. En este subestudio se evaluó la repercusión de un solo ciclo de tratamiento con rituximab en las concentraciones de marcadores bioquímicos, como marcadores de la inflamación (interleucina 6, proteína C reactiva, proteína amiloide A sérica e isotipos A8 y A9 de la proteína S100), autoanticuerpos (FR y anticuerpos antipéptidos cíclicos citrulinados) y marcadores de la producción y el recambio óseo (osteocalcina y péptido aminoterminal del procolágeno 1 [P1NP]). El tratamiento con MabThera/Rituxan i.v., tanto en monoterapia como en combinación con metotrexato o ciclofosfamida, redujo significativamente las concentraciones de marcadores inflamatorios en comparación con el metotrexato solo, durante las primeras 24 semanas de seguimiento. Las concentraciones de marcadores del recambio óseo —osteocalcina y P1NP— aumentaron significativamente con el rituximab en comparación con el metotrexato solo.
Eficacia a largo plazo con un tratamiento de múltiples ciclos
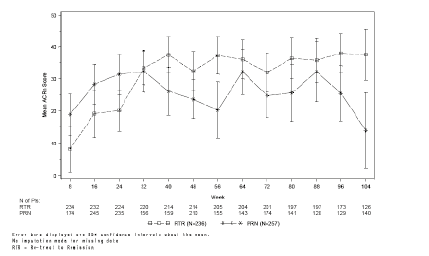
Pacientes de estudios clínicos recibieron retratamiento según una estrategia de «tratamiento hasta la remisión» o de «tratamiento en caso necesario». Ciclos repetidos de MabThera/Rituxan i.v. mantuvieron o mejoraron el beneficio terapéutico, independientemente de la estrategia de tratamiento (tratamiento hasta la remisión o tratamiento en caso necesario) (figura 2). Ahora bien, la estrategia de tratamiento hasta la remisión se tradujo generalmente en mejores respuestas y en un control más estrecho de la actividad de la enfermedad, tal como indicaron las puntuaciones ACRn, DAS28-VSG y HAQ-DI a lo largo del tiempo. En los pacientes que recibieron tratamiento en caso necesario, también reaparecieron los síntomas de la enfermedad entre los ciclos, tal como evidenciaron las puntuaciones DAS28-VSG, que se aproximaban a las cifras previas al tratamiento antes de cada ciclo (ver tabla 19).
|
Tabla 19. Valores iniciales de los parámetros de la actividad de la enfermedad antes de cada ciclo |
||||||
|
Población |
Parámetro |
C1 |
C2 |
C3 |
C4 |
C5 |
|
Tratamiento hasta la remisión |
n = 236 |
n = 218 |
n = 198 |
n = 156 |
n = 83 |
|
|
Media del valor inicial de DAS |
6,6 |
4,9 |
4,6 |
4,6 |
4,7 |
|
|
Mediana del valor inicial de ACRn |
- |
22,7 |
25,5 |
26,5 |
26,3 |
|
|
Tratamiento en caso necesario |
n = 257 |
n = 182 |
n = 139 |
n = 85 |
n = 39 |
|
|
Media del valor inicial de DAS |
6,7 |
6,2 |
6,2 |
5,9 |
6,0 |
|
|
Mediana del valor inicial de ACRn |
- |
-5,3 |
-11,1 |
-10,9 |
-4,2 |
|
|
Cambio positivo de ACRn = mejoría. |
||||||
Figura 2. Diagrama de la media de ACRn a lo largo del tiempo según los criterios de tratamiento (población MTX-RI)
Estudio con una velocidad de infusión de 120 minutos (ML25641)
En un ensayo multicéntrico, sin enmascaramiento, con un solo grupo, 351 pacientes con artritis reumatoide activa de moderada a grave que habían tenido una respuesta inadecuada al menos a un inhibidor del TNF y habían recibido metotrexato debían recibir 2 ciclos de tratamiento con MabThera/Rituxan i.v. Se consideró que podían participar en el estudio los pacientes que no hubieran recibido previamente tratamiento con MabThera/Rituxan i.v. (n = 306) y los que hubieran recibido 1-2 ciclos previos de MabThera/Rituxan i.v. 6-9 meses antes del inicio del estudio (n = 45).
Los pacientes recibieron 2 ciclos de MabThera/Rituxan i.v. en dosis de 2 × 1000 mg más tratamiento con metotrexato, administrándose el primer ciclo los días 1 y 15 y el segundo ciclo 6 meses después, los días 168 y 182. La primera infusión del primer ciclo (infusión del día 1) se administró en un periodo de 4,25 horas (255 minutos). La segunda infusión del primer ciclo (infusión del día 15) y ambas infusiones del segundo ciclo (infusiones de los días 168 y 182) tuvieron una duración de 2 horas (120 minutos). Se retiró del estudio a todos los pacientes que sufrieron con cualquier infusión una reacción grave relacionada con la misma.
El objetivo principal de este estudio era evaluar la seguridad de la administración de la segunda infusión del primer ciclo del estudio de MabThera/Rituxan i.v. con una duración de 2 horas (120 minutos).
La incidencia de RRI el día 15 fue del 6,5 % (IC 95 %: 4,1-9,7 %), cifra que concuerda con la tasa observada históricamente. No se observó ninguna RRI grave. Los datos correspondientes a las infusiones de los días 168 y 182 (infusión de 120 minutos) demuestran una baja incidencia de RRI, similar a la tasa observada históricamente, sin que se produjeran RRI graves (ver Reacciones adversas, Ensayos clínicos).
Granulomatosis con poliangitis (de Wegener) (GPA) y poliangitis microscópica (PAM)
Se incluyó a un total de 197 pacientes con granulomatosis activa grave con poliangitis de Wegener (GPA) y poliangitis microscópica (PAM) y se los trató en un estudio de ausencia de inferioridad multicéntrico, aleatorizado, con control activo y doble enmascaramiento (doble ciego). Los pacientes tenían ≥15 años, y un diagnóstico de GPA (75 % de los pacientes) o DE PAM (24 % de los pacientes) según los criterios de la conferencia de consenso de Chapel Hill (1 % de los pacientes tenían un tipo desconocido de GPA y PAM).
Los pacientes fueron asignados aleatoriamente, en una proporción 1:1, a recibir ciclofosfamida por vía oral (2 mg/kg/día) durante 3-6 meses, y a continuación azatioprina o MabThera/Rituxan i.v. (375 mg/m2) una vez por semana durante 4 semanas. Los pacientes de ambos grupos recibieron dosis altas de metilprednisolona i.v., de 1000 mg (o la dosis equivalente de otro glucocorticoide) al día, durante 1-3 días, y a continuación prednisona por vía oral (1 mg/kg/día, sin superar la dosis de 80 mg/día). La disminución progresiva de la dosis de prednisona debía concluir 6 meses después de iniciar el tratamiento del estudio.
La variable de valoración principal fue el logro de una remisión completa a los 6 meses, definida como una puntuación de actividad de la vasculitis de Birmingham para la granulomatosis de Wegener (BVAS/WG) de 0 y la suspensión del tratamiento glucocorticoide. El margen de ausencia de inferioridad preespecificado para la diferencia de tratamiento fue del 20 %. Este estudio demostró la ausencia de inferioridad de MabThera/Rituxan i.v. en comparación con la ciclofosfamida en lo que se refiere a la remisión completa a los 6 meses (ver tabla 19). Además, la tasa de remisión completa en el grupo de MabThera/Rituxan i.v. fue significativamente mayor que la tasa estimada de remisión completa en pacientes con GPA y PAM graves no tratados o tratados sólo con glucocorticoides, según los datos de referencia históricos.
Se observó la eficacia del tratamiento tanto en pacientes con GPA y PAM de reciente diagnóstico como en los pacientes con enfermedad recidivante.
|
Tabla 20. Porcentaje de pacientes que lograron una remisión completa a los 6 meses (población ITT) |
|||
|
MabThera/Rituxan (n = 99) |
Ciclofosfamida |
Diferencia entre tratamientos (MabThera/Rituxan Ciclofosfamida) |
|
|
Tasa |
63,6 % |
53,1 % |
10,6 % |
|
IC 95,1 %b |
(54,1 %, 73,2 %) |
(43,1 %, 63,0 %) |
(-3,2 %, 24,3 %)a |
|
IC = intervalo de confianza. a La ausencia de inferioridad quedó demostrada, dado que el límite inferior (-3,2 %) fue mayor que el margen de ausencia de inferioridad predeterminado (-20 %). b El nivel de confianza del 95,1 % refleja un a adicional de 0,001 para tener en cuenta un análisis intermedio de la eficacia. |
|||
Inmunogenicidad
Formulación subcutánea
Los datos del programa de desarrollo de la formulación s.c. indican que la formación de anticuerpos anti-ritu-ximab (anticuerpos humanos antiquiméricos [HACA]) después de la administración s.c. es comparable a la observada después de la administración i.v.
En el estudio SABRINA (BO22334), la incidencia de anticuerpos anti-rituximab inducida o potenciada por el tratamiento en el grupo de administración s.c. fue baja, y similar a la observada en el grupo de administración i.v. (i.v.: 1,5 %; s.c.: 2 %). La incidencia de la presencia de anticuerpos anti-rHuPH20 inducida o potenciada por el tratamiento fue del 8 % en el grupo de administración i.v. y del 13 % en el grupo de administración s.c.; ninguno de los pacientes que presentaban anticuerpos anti-rHuPH20 tenía anticuerpos neutralizantes. La proporción general de pacientes que tenían anticuerpos anti-rHuPH20 se mantuvo generalmente constante durante el periodo de seguimiento en ambas cohortes. En el estudio SAWYER (BO25341), la incidencia de anticuerpos anti-rituximab inducida o potenciada por el tratamiento fue similar en ambos grupos de tratamiento (i.v.: 6,7 %; s.c.: 2,4 %). La incidencia de la presencia de anticuerpos anti-rHuPH20 inducida o potenciada por el tratamiento, que sólo se midió en los pacientes del grupo de administración s.c., fue del 10,6 %. Ninguno de los pacientes que presentaban anticuerpos anti-rHuPH20 tenía anticuerpos neutralizantes. No se sabe qué importancia clínica puede tener la aparición de anticuerpos anti-rituximab o anti-rHuPH20 después del tratamiento con MabThera/Rituxan s.c. La presencia de anticuerpos anti-rituximab o anti-rHuPH20 no repercutió en la seguridad o en la eficacia en ninguno de estos estudios.
INDICACIONES TERAPÉUTICAS
Linfoma no hodgkiniano:
MabThera/Rituxan i.v./s.c. está indicado para el tratamiento de:
– pacientes con linfoma no hodgkiniano (LNH) de bajo grado o folicular de linfocitos B CD20+ recidivante o resistente a la quimioterapia;
– pacientes con linfoma folicular en estadio III-IV no tratado anteriormente, en combinación con quimio-terapia;
– pacientes con linfoma folicular como terapia de mantenimiento después de la respuesta al tratamiento de inducción;
– pacientes con LNH difuso de linfocitos B grandes CD20+ en combinación con el régimen de quimioterapia CHOP (ciclofosfamida, doxorubicina, vincristina y prednisona).
Leucemia linfocítica crónica:
MabThera/Rituxan i.v./s.c. en combinación con quimio-terapia está indicado en el tratamiento de pacientes con leucemia linfocítica crónica (LLC) que no han recibido previamente tratamiento y LLC recidivante o resistente al tratamiento.
Artritis reumatoide:
MabThera/Rituxan i.v. en combinación con metotrexato está indicado en pacientes adultos para:
- el tratamiento de la artritis reumatoide activa de moderada a grave cuando la respuesta a los fármacos antirreumáticos modificadores de la enfermedad (FAME), incluido el metotrexato, haya sido inade-cuada;
- el tratamiento de la artritis reumatoide activa de mo-derada a grave en pacientes con respuesta inadecuada o intolerancia a uno o más inhibidores del factor de necrosis tumoral (TNF).
Se ha demostrado que MabThera/Rituxan i.v. reduce la velocidad de progresión del daño articular determinada radiográficamente, mejora la función física e induce una respuesta clínica importante, cuando se administra con metotrexato.
Granulomatosis con poliangitis (de Wegener) (GPA) y poliangitis microscópica (PAM): MabThera/Rituxan i.v. en combinación con glucocorticoides está indicado para el tratamiento de pacientes con granulomatosis con poliangitis (GPA, también conocida como granulomatosis de Wegener) y poliangitis microscópica (PAM) sumamente activas.
PROPIEDADES FARMACOCINÉTICAS
Absorción
Formulación intravenosa
No procede.
Formulación subcutánea (1400 mg)
Estudio SparkThera (BP22333)
MabThera/Rituxan en una dosis fija de 1400 mg se administró por vía s.c. durante el mantenimiento, después de al menos un ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2, en pacientes con linfoma folicular que habían respondido previamente a MabThera/Rituxan i.v. en la inducción]. La mediana prevista de la Cmáx para la pauta de administración de MabThera/Rituxan s.c. cada 2 meses (c2m) y la de MabThera/Rituxan i.v. c2m fue comparable: 201 µg/ml y 209 µg/ml, respectivamente. De igual modo, con la pauta de administración de MabThera/Rituxan s.c. c3m y la de MabThera/Rituxan i.v. c3m, la mediana prevista de la Cmáx fue comparable: 189 µg/ml y 184 µg/ml, respectivamente. La mediana del tmáx en el grupo de MabThera/Rituxan s.c. fue aproximadamente de 3 días, en comparación con el tmáx en el grupo de MabThera/Rituxan i.v., que tuvo lugar al final o cerca del final de la infusión.
Estudio SABRINA (BO22334)
Se administró MabThera/Rituxan por vía s.c. en una dosis fija de 1400 mg durante 6 ciclos en la inducción, a intervalos de 3 semanas, después de un primer ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2, en pacientes con linfoma folicular sin tratamiento previo, en combinación con quimioterapia. La Cmáx en suero del rituximab en el ciclo 7 fue similar en los dos grupos de tratamiento, con una media geométrica (CV %) de 250,63 (19,01) µg/ml y de 236,82 (29,41) µg/ml con MabThera/Rituxan i.v. y MabThera/Rituxan s.c., respectivamente, y un índice de medias geométricas resultante (Cmáx, s.c./Cmáx, i.v.) de 0,941 (IC 90 %: 0,872-1,015).
Basándose en un análisis farmacocinético poblacional, se calculó que la biodisponibilidad absoluta era del 71 % (IC 95 %: 70,0-72,1).
Formulación subcutánea (1600 mg)
Estudio SAWYER (BO25341)
Se administró MabThera/Rituxan por vía s.c. en una dosis fija de 1600 mg durante 5 ciclos a intervalos de 4 semanas, después de un primer ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2, en pacientes con LLC sin tratamiento previo, en combinación con quimioterapia (fludarabina y ciclofosfamida [FC]). La Cmáx sérica de rituximab en el ciclo 6 fue menor en el grupo de MabThera/Rituxan s.c. que en el grupo de MabThera/Rituxan i.v. (con una dosis de 500 mg/m2 en los ciclos 2 a 6); la media geométrica (CV %) fue de 202 (36,1) µg/ml con MabThera/Rituxan i.v. y de 280 (24,6) µg/ml con MabThera/Rituxan s.c., con un índice de medias geométricas resultante (Cmáx, s.c./Cmáx, i.v.) de 0,719 (IC 90 %: 0,653-0,792). La media geométrica del tmáx en el grupo de MabThera/Rituxan s.c. fue aproximadamente de 3 días, en comparación con el tmáx en el grupo de MabThera/Rituxan i.v., que tuvo lugar al final o cerca del final de la infusión.
Distribución
Linfoma no hodgkiniano
Formulación intravenosa
De acuerdo con un análisis farmacocinético poblacional en 298 pacientes con LNH que habían recibido una infusión o múltiples infusiones de MabThera/Rituxan i.v. en monoterapia o en combinación con CHOP, las estimaciones poblacionales típicas del aclaramiento inespecífico (CL1), el aclaramiento específico (CL2) probablemente influido por los linfocitos B o la masa tumoral, y el volumen de distribución del compartimento central (V1) fueron de 0,14 l/día, 0,59 l/día y 2,7 l, respectivamente. La mediana estimada de la semivida de eliminación terminal del rituximab fue de 22 días (intervalo: 6,1-52 días). La cifra inicial de linfocitos CD19+ y el tamaño de las lesiones tumorales mensurables contribuyeron en cierta medida a la variabilidad de los datos del CL2 del rituximab en los 161 pacientes que recibieron en infusión i.v. 4 dosis de 375 mg/m2 a intervalos semanales. Los pacientes con cifras más altas de linfocitos CD19+ o con lesiones tumorales mayores presentaron un CL2 más alto. Ahora bien, gran parte de la variabilidad interindividual del CL2 se mantuvo después de la corrección en función del número de linfocitos CD19+ y del tamaño de las lesiones tumorales. V1 varió según la superficie corporal (SC) y la quimioterapia CHOP. Esta variabilidad de V1 (27,1 % y 19,0 %), a la que contribuían el intervalo de la superficie corporal (1,53-2,32 m2) y el régimen CHOP concomitante, respectivamente, fue relativamente pequeña. En la farmacocinética del rituximab no influyeron la edad, el sexo, la raza ni el estado general según la clasificación de la OMS. Este análisis indica que no es previsible que un ajuste de la dosis de rituximab con cualquiera de las covariables analizadas dé lugar a una reducción significativa de su variabilidad farmacocinética.
A 203 pacientes con LNH que no habían recibido previamente rituximab se les administraron 4 dosis de MabThera/Rituxan i.v. de 375 mg/m2 en infusión i.v. a intervalos semanales. La media de la Cmáx después de la cuarta infusión fue de 486 µg/ml (intervalo: 77,5-996,6 µg/ml). Las concentraciones séricas máxima y valle de rituximab estaban se correlacionaron inversamente con los valores iniciales del número de linfocitos B CD19+ circulantes y la masa tumoral. La mediana de las concentraciones séricas en estado de equilibrio fue mayor en los pacientes que respondieron al tratamiento que en los pacientes sin respuesta. Las concentraciones séricas fueron más altas en los pacientes con tumores de los subtipos histológicos B, C y D de la IWF (International Working Formulation) que en los pacientes con el subtipo A. El rituximab era detectable en el suero de los pacientes 3-6 meses después de concluido el último tratamiento.
A 37 pacientes con LNH se les administraron 8 dosis de MabThera/Rituxan i.v. de 375 mg/m2 en infusión i.v. a intervalos semanales. La media de la Cmáx aumentó con cada nueva infusión, desde una media de 243 µg/ml (intervalo: 16-582 µg/ml) tras la primera infusión hasta 550 µg/ml (intervalo: 171-1177 µg/ml) después de la octava.
El perfil farmacocinético del MabThera/Rituxan i.v. administrado en 6 infusiones de 375 mg/m2 en asociación con 6 ciclos de quimioterapia CHOP fue similar al registrado tras la administración de MabThera/Rituxan i.v. solo.
Formulación subcutánea (1400 mg)
Estudio SparkThera (BP22333)
Se administró por vía s.c. MabThera/Rituxan en una dosis fija de 1400 mg durante el mantenimiento, después de al menos un ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2, en pacientes con linfoma folicular que habían respondido previamente a MabThera/Rituxan i.v. en la inducción. Los valores previstos de la media y la media geométrica de la Cvalle en el ciclo 2 fueron mayores en el grupo de MabThera/Rituxan s.c. que en el grupo de MabThera/Rituxan i.v. Las medias geométricas con la pauta de MabThera/Rituxan s.c. c2m y la pauta de MabThera/Rituxan i.v. c2m fueron, respectivamente, de 32,2 µg/ml y 25,9 µg/ml; con las pautas de MabThera/Rituxan s.c. c3m y de MabThera/Rituxan i.v. c3m fueron, respectivamente, de 12,1 µg/ml y 10,9 µg/ml. De igual modo, la media y la media geométrica previstas del ABCτ en el ciclo 2 fueron mayores en el grupo de MabThera/Rituxan s.c. que en el grupo de MabThera/Rituxan i.v. Las medias geométricas con la pauta de MabThera/Rituxan s.c. c2m y la pauta de MabThera/Rituxan i.v. c2m fueron respectivamente de 5430 µg·día/ml y 4012 µg·día/ml; con la pauta de MabThera/Rituxan s.c. c3m y la pauta de MabThera/Rituxan i.v. c3m fueron, respectivamente, de 5320 µg·día/ml y 3947 µg·día/ml.
Estudio SABRINA (BO22334)
Se administró MabThera/Rituxan en una dosis fija de 1400 mg en inyección s.c., en el abdomen, a intervalos de 3 semanas. Pacientes con linfoma folicular CD20+ de grado 1, 2 o 3a que no habían recibido previamente tratamiento fueron asignados aleatoriamente, en una proporción 1:1, a recibir MabThera/Rituxan s.c. (primer ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2, seguido por 7 ciclos de MabThera/Rituxan s.c.) o MabThera/Rituxan i.v. en dosis de 375 mg/m2 (8 ciclos en total) en combinación con hasta 8 ciclos de quimioterapia CHOP o CVP administrada cada 3 semanas como parte del tratamiento de inducción. La media y la media geométrica de la Cvalle antes de la dosis en el ciclo 7 de la inducción (antes de administrar la dosis del ciclo 8) fueron mayores en el grupo de MabThera/Rituxan s.c. que en el grupo de MabThera/Rituxan i.v. La media geométrica fue de 134,6 µg/ml en el grupo de MabThera/Rituxan s.c. y de 83,1 µg/ml en el grupo de MabThera/Rituxan i.v.
De igual modo, la media y la media geométrica del ABC en el ciclo 7 de la inducción (antes de administrar la dosis del ciclo 8) fueron mayores en el grupo de MabThera/Rituxan s.c. que en el grupo de MabThera/Rituxan i.v. La media geométrica del ABC fue de 3778,9 µg·día/ml en el grupo de MabThera/Rituxan s.c. y de 2734,2 µg·día/ml en el grupo de MabThera/Rituxan i.v.
En un análisis farmacocinético poblacional en pacientes con linfoma folicular que recibieron una infusión o múltiples infusiones de MabThera/Rituxan i.v. en monoterapia o en combinación con quimioterapia, las estimaciones poblacionales del aclaramiento inespecífico (CL1), el aclaramiento específico inicial (CL2) (probablemente influido por la cifra de linfocitos B o la carga tumoral) y el volumen de distribución del compartimento central (V1) fueron de 0,194 l/día, 0,535 l/día, y 4,37 l, respectivamente. La mediana estimada de la semivida de eliminación terminal de MabThera/Rituxan s.c. fue de 29,7 días (intervalo: 9,9-91,2 días).
En el conjunto de datos del análisis final de 403 pacientes que recibieron MabThera/Rituxan s.c. o i.v. en los estudios BP22333 (277 pacientes) y BO22334 (126 pacientes), la media del peso y la superficie corporal fue de 74,4 kg (intervalo: 43,9-130 kg) y 1,83 m2 (intervalo: 1,34-2,48 m2), respectivamente. La media de la edad fue de 57,4 años (intervalo: 23-87 años). No hubo diferencias entre los parámetros demográficos y analíticos de los dos estudios. No obstante, las cifras iniciales de linfocitos B fueron notablemente inferiores en el estudio BP22333 que en el estudio BO22334, dado que los pacientes de BP22333 que participaron en el estudio habían recibido un mínimo de 4 ciclos de MabThera/Rituxan i.v. como inducción y al menos un ciclo de MabThera/Rituxan i.v. como mantenimiento, mientras que los pacientes del estudio BO22334 no habían recibido MabThera/Rituxan antes de entrar en el estudio. Sólo se dispone de los datos sobre la carga tumoral inicial de los pacientes del estudio BO22334.
Se identificó la superficie corporal como la covariable principal. Todos los parámetros del aclaramiento y el volumen aumentaron con el tamaño corporal. Entre otras dependencias de covariables, el volumen central aumentó con la edad y la constante de absorción disminuyó con la edad (para los pacientes >60 años), pero se demostró que estas dependencias se traducían en cambios insignificantes de la exposición al rituximab. Se detectaron anticuerpos contra el fármaco sólo en 13 pacientes, sin que ello diera lugar a un aumento clínicamente importante del aclaramiento.
Leucemia linfocítica crónica
Formulación intravenosa
MabThera/Rituxan se administró en infusión i.v. en una dosis de 375 mg/m2 en el primer ciclo, que aumentó hasta 500 mg/m2 en cada ciclo posterior, durante 5 dosis, en combinación con fludarabina y ciclofosfamida en pacientes con LLC. La media de la Cmáx (n = 15) fue de 408 µg/ml (intervalo: 97-764 µg/ml) después de la quinta infusión de 500 mg/m2.
Formulación subcutánea (1600 mg)
Estudio SAWYER (BO25341)
Se administró MabThera/Rituxan en una dosis fija de 1600 mg en inyección s.c., en el abdomen, a intervalos de 4 semanas. Participaron pacientes con LLC CD20+ que fueron asignados aleatoriamente, en una proporción 1:1, a recibir 6 ciclos de MabThera/Rituxan s.c. (primer ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2 y a continuación 5 ciclos de MabThera/Rituxan s.c.) o MabThera/Rituxan i.v. (primer ciclo de MabThera/Rituxan i.v. en dosis de 375 mg/m2 y a continuación 5 ciclos de MabThera/Rituxan i.v. en dosis de 500 mg/m2) en combinación con hasta 6 ciclos de quimioterapia FC administrada cada 4 semanas. La media geométrica de la Cvalle en el ciclo 5 (antes de administrar la dosis del ciclo 6) fue mayor en el grupo de MabThera/Rituxan s.c. que en el grupo de MabThera/Rituxan i.v. (97,5 µg/ml frente a 61,5 µg/ml, respectivamente). De igual modo, la media geométrica del ABC en el ciclo 6 fue mayor en el grupo de MabThera/Rituxan s.c. que en el grupo de MabThera/Rituxan i.v. (4088 µg·día/ml y 3630 µg·día/ml, respectivamente).
Artritis reumatoide
Tras la administración de dos infusiones i.v. de rituximab de 1000 mg cada una, con 2 semanas de intervalo entre ambas, la media de la semivida terminal fue de 20,8 días (intervalo: 8,58-35,9 días), la media del aclaramiento sistémico fue de 0,23 l/día (intervalo: 0,091-0,67 l/día) y la media del volumen de distribución en equilibrio fue de 4,6 l (intervalo: 1,7-7,51 l). El análisis farmacocinético poblacional de los mismos datos arrojó valores medios similares para el aclaramiento sistémico y la semivida: 0,26 l/día y 20,4 días, respectivamente. Un análisis farmacocinético poblacional reveló que la superficie corporal y el sexo eran las covariables más importantes para explicar la variabilidad interindividual de los parámetros farmacocinéticos. Tras el ajuste en función de la superficie corporal, el volumen de distribución fue mayor y el aclaramiento más rápido en los hombres que en las mujeres. Las diferencias farmacocinéticas asociadas al sexo no se consideraron clínicamente importantes, por lo que no es necesario ajustar la dosis.
La farmacocinética del rituximab se evaluó en cuatro estudios tras administrar 2 dosis i.v. de 500 mg y 1000 mg los días 1 y 15. En todos estos estudios, la farmacocinética del rituximab fue proporcional a la dosis en el intervalo limitado de las dosis evaluadas. La media de la Cmáx sérica del rituximab después de la primera infusión fue de 157-171 µg/ml al administrar 2 veces la dosis de 500 mg y de 298-341 µg/ml al administrar 2 veces la dosis de 1000 mg. Tras la segunda infusión, la media de la Cmáx fue de 183-198 µg/ml al administrar 2 veces la dosis de 500 mg y de 355-404 µg/ml al administrar 2 veces la dosis de 1000 mg. La media de la semivida de eliminación terminal fue de 15-16,5 días al administrar 2 veces la dosis de 500 mg y de 17-21 días al administrar 2 veces la dosis de 1000 mg. La media de la Cmáx fue un 16-19 % más alta después de la segunda infusión en comparación con la primera infusión con ambas dosis.
La farmacocinética del rituximab se evaluó tras adminis-trar 2 dosis i.v. de 500 mg y 1000 mg los días 1 y 15 después del retratamiento en el segundo ciclo. La media de la Cmáx sérica de rituximab después de la primera infusión fue de 170-175 µg/ml al administrar 2 veces la dosis de 500 mg y de 317-370 µg/ml al administrar 2 veces la dosis de 1000 mg. La Cmáx después de la segunda infusión fue de 207 µg /ml al administrar 2 veces la dosis de 500 mg y de 377-386 µg /ml al administrar 2 veces la dosis de 1000 mg. La media de la semivida de eliminación terminal tras la segunda infusión, después del segundo ciclo, fue de 19 días al administrar 2 veces la dosis de 500 mg y de 21-22 días al administrar 2 veces la dosis de 1000 mg. Los parámetros farmacocinéticos del rituximab fueron comparables en los dos ciclos de tratamiento.
Después de utilizar la misma pauta de administración (2 veces 1000 mg i.v., con 2 semanas de diferencia), los parámetros farmacocinéticos en los pacientes con una respuesta inadecuada al tratamiento con inhibidores del TNF fueron similares, siendo la media de la concentración sérica máxima de 369 µg/ml y la media de la semivida terminal de 19,2 días.
Granulomatosis con poliangitis (de Wegener) (GPA) y poliangitis microscópica (PAM)
De acuerdo con los resultados de un análisis farmacocinético poblacional de 97 pacientes con GPA tratados con MabThera/Rituxan i.v. en dosis de 375 mg/m2 1 vez por semana durante 4 semanas, la mediana estimada de la semivida de eliminación terminal fue de 23 días (intervalo: 9-49 días). La media del aclaramiento y del volumen de distribución del rituximab fue de 0,313 l/día (intervalo: 0,116-0,726 l/día) y 4,50 l (intervalo: 2,25-7,39 l), respectivamente. Los parámetros farmacocinéticos del rituximab en los pacientes con GPA y PAM parecen ser similares a los registrados en los pacientes con AR (ver Propiedades farmacocinéticas, Distribución).
Metabolismo
Sin texto.
Eliminación
Ver Distribución.
Farmacocinética en poblaciones especiales
No se dispone de datos farmacocinéticos en pacientes con insuficiencia hepática o renal.
Datos preclínicos sobre seguridad
Carcinogenicidad
Sin texto.
Mutagenicidad
Sin texto.
Trastornos de la fecundidad
Sin texto.
Teratogenicidad
Sin texto.
Otros efectos
Formulación subcutánea
La formulación s.c. contiene hialuronidasa humana recombinante (rHuPH20), una enzima que se utiliza para aumentar la dispersión y la absorción de fármacos coadministrados cuando se administran por vía s.c. Es improbable que se produzca una absorción sistémica de rHuPH20 tras la administración s.c. No obstante, en estudios farmacocinéticos y de toxicología realizados en animales se ha evidenciado una reducción del peso fetal y un aumento del número de resorciones después de la inyección de rHuPH20 cuando la exposición sistémica materna es la que se alcanza tras la administración accidental de una inyección i.v. rápida de un vial único de la formulación s.c. de MabThera/Rituxan en el ser humano, basándose en las presuposiciones más conservadoras posibles. No hay datos indicativos de dismorfogénesis (es decir, teratogénesis) debida a la exposición sistémica a rHuPH20.
DATOS FARMACÉUTICOS
Conservación
Formulaciones intravenosa y subcutánea
Este medicamento no debe usarse después de la fecha de caducidad, indicada con «EXP» en el envase.
Formulación intravenosa
Los viales se conservarán a 2-8 °C (en un refrigerador). No debe congelarse. El envase debe mantenerse en el acondicionamiento externo para protegerlo de la luz.
Desde el punto de vista microbiológico, la solución para infusión preparada debe usarse inmediatamente. Si no se utiliza de inmediato, los periodos y las condiciones de conservación antes usarlo son responsabilidad del usuario; normalmente no debería conservarse durante más de 24 horas a 2-8°C, a no ser que la dilución se haya realizado en condiciones asépticas controladas y validadas.
Formulación subcutánea
Debe conservarse en un refrigerador (2-8 ºC). No debe congelarse. El envase debe mantenerse en el acondiciona-miento externo para protegerlo de la luz.
Desde el punto de vista microbiológico, el producto diluido debe usarse inmediatamente. En el caso de que no se use de inmediato, la preparación debe realizarse en condiciones asépticas controladas y validadas. Los periodos y las condiciones de conservación antes usarlo son responsabilidad del usuario; normalmente no debería conservarse durante más de 48 horas a 2-8°C y 8 horas más a 30 ºC expuesto a luz diurna difusa.
Instrucciones especiales de uso, manipulación y eliminación
Formulación intravenosa
Extráigase la cantidad necesaria de MabThera/Rituxan en condiciones asépticas y dilúyase hasta una concentración calculada de rituximab de 1-4 mg/ml en una bolsa de infusión con solución salina al 0,9 %, estéril y apirógena, o una solución de glucosa al 5 %. Para mezclar la solución, invierta suavemente la bolsa de infusión para evitar que se forme espuma. Los medicamentos parenterales han de examinarse visualmente antes de su administración por si presentaran partículas o cambios de color.
Las soluciones de MabThera/Rituxan i.v. preparadas para la infusión permanecen física y químicamente estables durante 24 horas a 2-8 °C más otras 12 horas a temperatura ambiente.
Incompatibilidades
No se han descrito incompatibilidades entre MabThera/Rituxan i.v. y las bolsas o los equipos de infusión de cloruro de polivinilo o polietileno.
Formulación subcutánea
La solución de MabThera/Rituxan s.c. (una vez transferida del vial a la jeringa) es física y químicamente estable durante 48 horas a 2-8 ºC y 8 horas más a 30 ºC expuesta a la luz diurna difusa.
MabThera/Rituxan s.c. se presenta en viales monodosis con solución estéril, sin conservantes y apirógena.
Incompatibilidades
No se han observado incompatibilidades entre Mab-Thera/Rituxan s.c. y el material de las jeringas de polipropileno o policarbonato o las agujas de acero inoxidable para la transferencia e inyección.
Formulaciones intravenosa y subcutánea
Eliminación de los medicamentos no utilizados o caducados.
La emisión de productos farmacéuticos al medio ambiente debe reducirse al mínimo. Evítese tirar los medicamentos por los desagües o a la basura doméstica, y utilícense los sistemas de recogida disponibles localmente.
En lo que respecta al uso y la eliminación de las jeringas y otros objetos punzocortantes medicinales, se seguirán estrictamente las siguientes indicaciones:
- Nunca se deben reutilizar las agujas ni las jeringas.
- Todas las agujas y las jeringas deben colocarse en un recipiente desechable para objetos punzocortantes.
Los productos medicinales o el material de desecho que no se haya utilizado se eliminarán de acuerdo con las normas locales.
CONTRAINDICACIONES
MabThera/Rituxan está contraindicado en pacientes con hipersensibilidad conocida al rituximab, a cualquiera de sus excipientes o a las proteínas murinas.
Efectos sobre la capacidad para conducir y utilizar máquinas
No se han llevado a cabo estudios sobre el efecto de MabThera/Rituxan sobre la capacidad para conducir vehículos y utilizar máquinas, si bien la actividad farmacológica y los eventos adversos notificados hasta ahora no indican que sea probable tal efecto.
Pruebas de laboratorio
Sin texto.
REACCIONES ADVERSAS
Ensayos clínicos
Experiencia obtenida en ensayos clínicos de hematooncología.
Formulación intravenosa
En las tablas siguientes se resume la frecuencia de reacciones adversas (RA) notificadas en los estudios clínicos con MabThera/Rituxan i.v. en monoterapia o en asociación con quimioterapia. Estas RA se produjeron en estudios con un solo grupo o con una diferencia ≥2 % en comparación con el grupo de referencia en al menos uno de los principales estudios clínicos aleatorizados. Las RA se han categorizado en las tablas de acuerdo con la incidencia más alta registrada en cualquiera de los estudios clínicos principales. Las RA se enumeran, dentro de cada grupo de frecuencia, por orden decreciente de gravedad. Las RA se definen, según la frecuencia, del siguiente modo: muy frecuente (>1/10), frecuente (>1/100 a <1/10) y poco frecuente (>1/1000 a <1/100).
MabThera/Rituxan i.v. en monoterapia y en la terapia de mantenimiento.
Las RA de la tabla siguiente se basan en los datos de estudios con un solo grupo que incluyeron a 356 pacientes con linfoma de bajo grado o folicular que recibieron semanalmente MabThera/Rituxan i.v. en monoterapia como tratamiento o retratamiento del LNH (ver Ensayos clínicos/Eficacia). La tabla también contiene RA basadas en datos de 671 pacientes con linfoma folicular que recibieron MabThera/Rituxan i.v. como terapia de mantenimiento durante un periodo de hasta 2 años tras la respuesta al tratamiento de inducción inicial con CHOP, R-CHOP, R-CVP o R-FCM (ver Ensayos clínicos/Eficacia). Las RA se notificaron hasta 12 meses después de la monoterapia y hasta 1 mes después de la terapia de mantenimiento con MabThera/Rituxan i.v.
|
Tabla 1. Resumen de las RA notificadas en pacientes con linfoma de bajo grado o linfoma folicular que habían recibido MabThera/Rituxan i.v. en monoterapia (n = 356) o como terapia de mantenimiento (n = 671) en estudios clínicos |
|||
|
Clase de órganos y sistemas |
Muy frecuente (≥10 %) |
Frecuente (≥1 % - <10 %) |
Poco frecuente (≥0,1 % - <1 %) |
|
Infecciones e infestaciones |
Infecciones bacterianas, infecciones víricas |
Septicemia, neumonía+, infección febril+, herpes zóster+, infección respiratoria+, infecciones micóticas, infecciones de causa desconocida |
|
|
Trastornos de la sangre y del sistema linfático |
Neutropenia, leucopenia |
Anemia, trombocitopenia |
Trastornos de la coagulación, anemia aplásica transitoria, anemia hemolítica, linfadenopatía |
|
Trastornos del sistema inmunitario |
Angioedema |
Hipersensibilidad |
|
|
Trastornos del metabolismo y de la nutrición |
Hiperglucemia, disminución del peso, edema periférico, edema facial, LDH elevada, hipocalcemia |
||
|
Trastornos psiquiátricos |
Depresión, nerviosismo |
||
|
Trastornos del sistema nervioso |
Parestesias, hipoestesia, agitación, insomnio, vasodilatación, mareos, ansiedad |
Disgeusia |
|
|
Trastornos oculares |
Trastorno de la lagrimación, conjuntivitis |
||
|
Trastornos del oído y del laberinto |
Acúfenos, otalgia |
||
|
Trastornos cardiacos |
Infarto de miocardio+, arritmia, fibrilación auricular+, taquicardia, trastorno cardiaco+ |
Insuficiencia ventricular izquierda+, taquicardia supraventricular+, taquicardia ventricular+, angina de pecho+, isquemia miocárdica+, bradicardia |
|
|
Trastornos vasculares |
Hipertensión arterial, hipotensión ortostática, hipotensión arterial |
||
|
Trastornos respiratorios, torácicos y mediastínicos |
Broncoespasmo, enfermedad respiratoria, dolor torácico, disnea, tos, rinitis |
Asma, bronquiolitis obliterante, trastorno pulmonar, hipoxia |
|
|
Trastornos gastrointestinales |
Náuseas |
Vómitos, diarrea, dolor abdominal, disfagia, estomatitis, estreñimiento, dispepsia, anorexia, irritación de garganta |
Distensión abdominal |
|
Trastornos de la piel y del tejido subcutáneo |
Prurito, exantema |
Urticaria , alopecia+, sudación, sudores nocturnos |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Hipertonía, mialgias, artralgia, dolor de espalda, dolor de cuello, dolor |
||
|
Trastornos generales y alteraciones en el lugar de administración |
Fiebre, escalofríos, astenia, cefalea |
Dolor tumoral, rubefacción, malestar general, síndrome seudogripal |
Dolor en el lugar de la infusión |
|
Exploraciones complementarias |
Disminución de la concentración de IgG |
||
La frecuencia de cada término se basó en reacciones de todos los grados (de leve a grave), salvo los términos marcados con «+», en los que el recuento se basó sólo en las reacciones graves (grado ≥3 según los Criterios Comunes de Toxicidad del Instituto Nacional del Cáncer [NCI]). Sólo se indica la frecuencia más alta observada en los estudios.
MabThera/Rituxan i.v. en combinación con quimioterapia en el LNH y la LLC
Las RA enumeradas en la tabla 2 se basan en los datos del grupo de MabThera/Rituxan i.v. obtenidos en ensayos clínicos comparativos que se produjeron además de las observadas en la monoterapia y la terapia de mantenimiento o con una frecuencia mayor: de 202 pacientes con linfoma difuso de linfocitos B grandes (LDLBG) tratados con R-CHOP, así como de 234 y 162 pacientes con linfoma folicular tratados con R-CHOP o R-CVP, respectivamente, y de 397 pacientes con LLC sin tratamiento previo y 274 con LLC recidivante o resistente al tratamiento que recibieron MabThera/Rituxan i.v. en combinación con fludarabina y ciclofosfamida (R-FC) (ver Ensayos clínicos/Eficacia).
|
Tabla 2. Resumen de las RA graves notificadas en pacientes tratados con R-CHOP contra el LDLBG (n = 202), R-CHOP contra el linfoma folicular (n = 234), R-CVP contra el linfoma folicular (n = 162) o R-FC en pacientes con LLC sin tratamiento previo (n = 397) o LLC recidivante o resistente al tratamiento (n = 274) |
||
|
Clase de órganos y sistemas |
Muy frecuente (≥10 %) |
Frecuente (≥1 % - <10 %) |
|
Infecciones e infestaciones |
Bronquitis |
Bronquitis aguda, sinusitis hepatitis B* |
|
Trastornos de la sangre y del sistema linfático |
Neutropenia#, neutropenia febril, trombocitopenia |
Pancitopenia, granulocitopenia |
|
Trastornos de la piel y del tejido subcutáneo |
Alopecia |
Trastorno cutáneo |
|
Trastornos generales y alteraciones en el lugar de administración |
Fatiga, tiritona |
|
|
* Incluye la reactivación y las infecciones primarias; la frecuencia se basa en un régimen de R-FC en la LLC recidivante o resistente al tratamiento. El cálculo de la frecuencia se basó únicamente en las reacciones graves, definidas en ensayos clínicos como de grado ≥3 según los criterios comunes de toxicidad del NCI. Sólo se indica la frecuencia más alta observada en cualquiera de los ensayos. # Instauración prolongada o retardada de la neutropenia después de concluir un ciclo de R-FC en pacientes con LLC sin tratamiento previo o con LLC recidivante o resistente al tratamiento. |
||
Los términos siguientes se han notificado como eventos adversos, aunque con una incidencia similar (diferencia entre los grupos <2 %) o menor en los grupos de Mab-Thera/Rituxan i.v. que en los grupos de referencia: hematotoxicidad, infección neutropénica, infección urinaria, choque séptico, sobreinfección pulmonar, infección de un implante, septicemia estafilocócica, infección pulmonar, rinorrea, edema pulmonar, insuficiencia cardiaca, trastorno sensitivo, trombosis venosa, mucositis (sin especificar), síndrome seudogripal, edema de las extremidades inferiores, fracción de eyección anormal, pirexia, deterioro de la salud física general, caída, fracaso multiorgánico, trombosis venosa profunda de las extremidades, hemocultivo positivo, control inadecuado de la diabetes mellitus.
El perfil de seguridad de MabThera/Rituxan i.v. en combinación con otras quimioterapias (por ejemplo, MCP, CHVP-IFN) es comparable al descrito para la combinación de MabThera/Rituxan i.v. y CVP, CHOP o FC en poblaciones equivalentes.
Formulación subcutánea
Las reacciones cutáneas locales, incluidas las reacciones en el lugar de la inyección, fueron muy frecuentes (>1/10) en pacientes que recibieron MabThera s.c. En el estudio de fase III SABRINA (BO22334), se notificaron reacciones cutáneas locales hasta en el 23 % de los pacientes que recibieron MabThera/Rituxan s.c. Las reacciones cutáneas locales más frecuentes en el grupo de MabThera/Rituxan s.c. fueron el eritema en el lugar de la inyección (13 %), el dolor en el lugar de la inyección (8 %) y el edema en el lugar de la inyección (4 %). Se observaron eventos similares en el estudio SAWYER (BO25341), y se registraron hasta en el 42 % de los pacientes del grupo de MabThera/Rituxan s.c. Las reacciones cutáneas locales más frecuentes fueron el eritema en el lugar de la inyección (26 %), el dolor en el lugar de la inyección (16 %) y la tumefacción en el lugar de la inyección (5 %).
Los eventos que se observaron tras la administración s.c. fueron leves o moderados, salvo un paciente del estudio SABRINA que notificó una reacción cutánea local con una intensidad de grado 3 (exantema en el lugar de la inyección) y dos pacientes del estudio SAWYER que presentaron reacciones cutáneas locales de grado 3 (eritema en el lugar de la inyección, dolor en el lugar de la inyección y tumefacción en el lugar de la inyección). La mayor frecuencia de reacciones cutáneas locales de cualquier grado en el grupo de MabThera/Rituxan s.c. se registró en el primer ciclo s.c. (ciclo 2), y a continuación en el segundo; la incidencia disminuyó en las inyecciones posteriores.
Por lo demás, el perfil de seguridad de MabThera/Rituxan s.c. fue comparable al de la formulación i.v. No se observaron casos de anafilaxia o de reacciones de hipersensibilidad graves, síndrome de liberación de citocinas o síndrome de lisis tumoral después de la administración s.c. durante el programa de desarrollo de MabThera/Rituxan s.c.
Información adicional sobre determinadas reacciones adversas graves
Formulación intravenosa
Reacciones relacionadas con la administración
Monoterapia: tratamiento durante 4 semanas
En más del 50 % de los pacientes de los ensayos clínicos se notificaron signos y síntomas indicativos de reacciones relacionadas con la infusión, que se observaron predominantemente durante la primera infusión. En asociación con la infusión de MabThera/Rituxan i.v. se han dado casos de hipotensión arterial, fiebre, escalofríos mode-rados e intensos, urticaria, broncoespasmo, sensación de hinchazón de la lengua o la garganta (angioedema), náuseas, fatiga, cefalea, prurito, disnea, rinitis, vómitos, rubefacción y dolor en el lugar de la enfermedad, como parte de un complejo sintomático relacionado con la infusión. También se han observado algunos rasgos de síndrome de lisis tumoral.
Tratamiento de combinación (R-CVP en el LNH; R-CHOP en el LDLBG, R-FC en la LLC)
Se han observado reacciones graves relacionadas con la infusión hasta en el 12 % de todos los pacientes en el primer ciclo de tratamiento con MabThera/Rituxan i.v. en combinación con quimioterapia. La incidencia de síntomas relacionados con la infusión disminuyó sustancialmente en las infusiones ulteriores, y en el octavo ciclo fue <1 %. Se han descrito otras reacciones, como dispepsia, exantema, hipertensión arterial, taquicardia y ciertos rasgos del síndrome de lisis tumoral. También se han notificado casos aislados de infarto de miocardio, fibrilación auricular, edema pulmonar y trombocitopenia aguda reversible.
Formulación subcutánea
El riesgo de reacciones agudas relacionadas con la administración asociadas a la formulación s.c. de MabThera/Rituxan se evaluó en tres estudios clínicos.
En el estudio SparkThera (BP22333) no se notificaron reacciones graves relacionadas con la administración.
En el estudio SABRINA (BO22334) se notificaron reacciones graves relacionadas con la administración (grado ≥3) en 2 pacientes (1 %) después de administrar MabThera/Rituxan s.c. Estos eventos consistieron en erupción en el lugar de la inyección y sequedad bucal de grado 3.
En el estudio SAWYER (BO25341), se notificaron reacciones graves relacionadas con la administración (grado ≥3) en 4 pacientes (5 %) después de administrar MabThera/Rituxan s.c. Estos eventos consistieron en trombocitopenia de grado 4, y ansiedad, eritema en el lugar de la inyección y urticaria de grado 3.
Formulación intravenosa
Tratamiento de combinación con una infusión 90 minutos (R-CVP en el LNH folicular; R-CHOP en el LDLBG)
En un estudio (U4391g) realizado para caracterizar la seguridad de las infusiones de 90 minutos de MabThera/Rituxan i.v. en pacientes que toleraron bien la primera infusión convencional de MabThera/Rituxan i.v., la incidencia de RRI de grado 3 y 4 el día de la infusión de MabThera/Rituxan i.v. de 90 minutos del ciclo 2 o el día siguiente fue del 1,1 % en los 363 pacientes evaluables (IC 95 %: 0,3-2,8 %). La incidencia de RRI de grado 3 y 4 en cualquier ciclo (ciclos 2-8) con la infusión de 90 minutos fue del 2,8 % (IC 95 %: 1,3-5,0 %). No se observaron RRI agudas mortales (ver Ensayos clínicos/Eficacia).
Infecciones
Monoterapia: tratamiento durante 4 semanas
MabThera/Rituxan i.v. indujo la depleción de los linfocitos B en el 70-80 % de los pacientes, pero se asoció a una disminución de las inmunoglobulinas séricas sólo en una minoría de pacientes. El 30,3 % de 356 pacientes presentaron infecciones bacterianas, víricas, micóticas y de causa desconocida, independientemente de la eva-luación causal. En el 3,9 % de los pacientes se registraron eventos infecciosos graves (de grado 3 o 4).
Terapia de mantenimiento (LNH) hasta 2 años
Durante el tratamiento con MabThera/Rituxan i.v. se observó una mayor frecuencia de infecciones en general, incluidas las infecciones de grado 3 y 4. No se observó toxicidad acumulada en lo que respecta a las infeccio-nes notificadas durante el periodo de mantenimiento de 2 años.
Los datos de los ensayos clínicos incluyeron casos de leucoencefalopatía multifocal progresiva mortal, en pacientes con LNH, que tuvieron lugar después de la progresión de la enfermedad y el retratamiento (ver Advertencias y precauciones).
Tratamiento de combinación (R-CVP en el LNH; R-CHOP en el LDLBG, R-FC en la LLC)
No se ha observado un aumento de la frecuencia de infecciones o infestaciones. Las infecciones más frecuentes fueron las infecciones de las vías respiratorias altas, que se registraron en el 12,3 % de los pacientes tratados con R-CVP y en el 16,4 % de los que recibieron CVP. Se notificaron infecciones graves en el 4,3 % de los pacientes tratados con R-CVP y en el 4,4 % de los que recibieron CVP. No se notificó ninguna infección potencialmente mortal en este estudio.
En el estudio de R-CHOP, la incidencia global de infecciones de grado 2-4 fue del 45,5 % en el grupo de R-CHOP y del 42,3 % en el grupo de CHOP. Las infecciones micóticas de grado 2-4 fueron más frecuentes en el grupo de R-CHOP (4,5 % frente al 2,6 % en el grupo de CHOP); esta diferencia se debió a una mayor incidencia de candidiasis localizadas durante el periodo de tratamiento. La incidencia de herpes zóster de grado 2-4 fue también mayor en el grupo de R-CHOP (4,5 %) que en el grupo de CHOP (1,5 %). La proporción de pacientes con infecciones o neutropenia febril de grado 2-4 fue del 55,4 % en el grupo de R-CHOP y del 51,5 % en el grupo de CHOP.
En los pacientes con LLC, la incidencia de hepatitis B de grado 3 y 4 (reactivación e infección primaria) fue del 2 % en el grupo de R-FC y del 0 % en el grupo de FC.
Eventos hemáticos
Monoterapia: tratamiento durante 4 semanas
Se notificaron casos de neutropenia grave (grado 3 y 4) en el 4,2 % de los pacientes; de anemia grave, en el 1,1 %, y de trombocitopenia grave, en el 1,7 %.
Terapia de mantenimiento (LNH) hasta 2 años: en comparación con el grupo de observación, en el grupo de MabThera/Rituxan i.v. se encontró una mayor incidencia de leucopenia de grado 3 y 4 (2 % y 5 %, res-pectivamente) y de neutropenia de grado 3 y 4 (4 % y 10 %, respectivamente). La incidencia de trombocitopenia de grado 3 y 4 fue baja (1 % en el grupo de observación y <1 % en el grupo de MabThera/Rituxan i.v.). En aproximadamente la mitad de los pacientes con datos sobre la recuperación de los linfocitos B después del tratamiento de inducción con MabThera/Rituxan i.v. transcurrieron 12 o más meses hasta que se normalizaron las cifras de linfocitos B.
Tratamiento de combinación (R-CVP en el LNH; R-CHOP en el LDLBG, R-FC en la LLC)
Durante el tratamiento en los estudios de MabThera/Rituxan i.v. en asociación con quimioterapia, se registró generalmente mayor incidencia de leucopenia de grado 3 y 4 (R-CHOP 88 % frente a CHOP 79 %; R-FC 23 % frente a FC 12 %) y de neutropenia de grado 3 y 4 (R-CVP 24 % frente a CVP 14 %; R-CHOP 97 % frente a CHOP 88 %; R-FC 30 % frente a FC 19 % en pacientes con LLC no tratada previamente) que con la quimioterapia sola. Ahora bien, la mayor incidencia de neutropenia en los pacientes tratados con MabThera/Rituxan i.v. y quimioterapia no se asoció a una incidencia más alta de infecciones e infestaciones en comparación con los que recibieron sólo quimioterapia. En estudios de la LLC no tratada previamente y la LLC recidivante o resistente al tratamiento se ha observado que en algunos casos la neutropenia fue prolongada o se manifestó tardíamente después del tratamiento en el grupo de MabThera/Rituxan i.v. más FC.
No se observaron diferencias importantes entre los grupos por lo que respecta a la anemia o la trombocitopenia de grado 3 y 4. En el estudio sobre el tratamiento de primera línea de la LLC, se notificaron casos de anemia de grado 3 y 4 en el 4 % de los pacientes tratados con R-FC frente al 7 % de los que recibieron FC, y trombocitopenia de grado 3 y 4 en el 7 % de los pacientes del grupo de R-FC frente al 10 % en el grupo de FC. En el estudio de la LLC recidivante o resistente al tratamiento, se notificaron casos de anemia de grado 3 y 4 en el 12 % de los pacientes tratados con R-FC frente al 13 % de los que recibieron FC, y trombocitopenia de grado 3 y 4 en el 11 % de los pacientes del grupo de R-FC frente al 9 % en el grupo de FC.
Eventos cardiovasculares
Monoterapia: tratamiento durante 4 semanas
Se registraron eventos cardiovasculares en el 18,8 % de los pacientes durante el periodo de tratamiento. Los más frecuentes fueron la hipotensión y la hipertensión arterial. Se notificaron casos de arritmia de grado 3 y 4 (incluidas la taquicardia ventricular y la supraventricular) y angina de pecho de grado 3 y 4 durante una infusión de MabThera/Rituxan i.v.
Terapia de mantenimiento (LNH) hasta 2 años
La incidencia de trastornos cardiacos de grado 3 y 4 fue comparable en los dos grupos de tratamiento. Se registraron eventos cardiacos como eventos adversos graves en <1 % de los pacientes del grupo de observación y en el 3 % de los pacientes tratados con MabThera/Rituxan i.v.: fibrilación auricular (1 %), infarto de miocardio (1 %), insuficiencia ventricular izquierda (<1 %), isquemia miocárdica (<1 %).
Tratamiento de combinación (R-CVP en el LNH; R-CHOP en el LDLBG, R-FC en la LLC)
En el estudio de R-CHOP, la incidencia de arritmias cardiacas de grado 3 y 4 —sobre todo arritmias supraventriculares del tipo de la taquicardia y el aleteo auricular o la fibrilación auricular— fue mayor en el grupo de R-CHOP (6,9 % de los pacientes) que en el grupo de CHOP (1,5 % de los pacientes). Todas estas arritmias se presentaron en el contexto de una infusión de MabThera/Rituxan i.v. o se asociaron a factores predisponentes, como fiebre, infección, infarto agudo de miocardio o enfermedades respiratorias o cardiovasculares preexistentes (ver Advertencias y precauciones). No se observaron diferencias entre los grupos de R-CHOP y CHOP en la incidencia de otros eventos cardiacos de grado 3 y 4, como insuficiencia cardiaca, miocardiopatía o manifestaciones de arteriopatía coronaria.
En la LLC, la incidencia global de trastornos cardiacos de grado 3 y 4 fue baja tanto en el estudio del tratamiento de primera línea (R-FC: 4 %; FC: 3 %) como en el estudio de pacientes con LLC recidivante o resistente al tratamiento (R-FC: 4 %; FC: 4 %).
Concentraciones de IgG
Terapia de mantenimiento (LNH) hasta 2 años
Después del tratamiento de inducción, la mediana de las cifras de IgG estaba por debajo del límite inferior de la normalidad (LIN) (<7 g/l) tanto en el grupo de observación como en el grupo de MabThera/Rituxan i.v. En el grupo de observación, la mediana de las cifras de IgG aumentó después hasta valores por encima del LIN; en cambio, durante el tratamiento con MabThera/Rituxan i.v. se mantuvo constante. La proporción de pacientes con concentraciones de IgG por debajo del LIN fue de aproximadamente el 60 % en el grupo de MabThera/Rituxan i.v. durante todo el periodo de tratamiento de 2 años, mientras que en el grupo de observación disminuyó (36 % al cabo de 2 años).
Eventos neurológicos
Tratamiento de combinación (R-CVP en el LNH; R-CHOP en el LDLBG, R-FC en la LLC)
Durante el periodo de tratamiento, el 2 % de los pacientes del grupo de R-CHOP, todos ellos con factores de riesgo cardiovascular, sufrieron accidentes cerebrovasculares tromboembólicos en el primer ciclo de tratamiento. No hubo diferencias entre ambos grupos en cuanto a la incidencia de otros episodios tromboembólicos. En cambio, el 1,5 % de los pacientes del grupo de CHOP sufrieron episodios cerebrovasculares, todos ellos durante el perio-do de seguimiento.
En la LLC, la incidencia global de trastornos del sistema nervioso de grado 3 y 4 fue baja tanto en el estudio del tratamiento de primera línea (R-FC: 4 %; FC: 4 %) como en el estudio de pacientes con LLC recidivante o resisten-te al tratamiento (R-FC: 3 %; FC: 3 %).
Subpoblaciones
Monoterapia: tratamiento durante 4 semanas
Pacientes ancianos (≥65 años)
La incidencia de RA de cualquier grado y de RA de grado 3 y 4 fue similar en los ancianos (≥65 años) y en pacientes más jóvenes (88,3 % frente al 92,0 % para las RA de cualquier grado y 16,0 % frente al 18,1 % para las RA de grado 3 y 4).
Tratamiento de combinación
Pacientes ancianos (≥65 años)
En los pacientes con LLC no tratada anteriormente o con LLC recidivante o resistente al tratamiento, la incidencia de eventos adversos de la sangre y el sistema linfático de grado 3 y 4 fue mayor en los ancianos (≥65 años) que en pacientes más jóvenes.
Pacientes con gran masa tumoral
La incidencia de RA de grado 3 y 4 fue mayor en los pacientes con gran masa tumoral que en los pacientes sin una gran masa tumoral (25,6 % frente al 15,4 %). En cambio, la incidencia de RA de cualquier tipo fue similar en ambos grupos (92,3 % en los pacientes con gran masa tumoral y 89,2 % en los pacientes sin gran masa tumoral).
Retratamiento con monoterapia
El porcentaje de pacientes que notificaron RA de cual-quier grado o RA de grado 3 y 4 después del retratamiento con más ciclos de MabThera/Rituxan i.v. fue similar al descrito tras la exposición inicial (95,0 % frente al 89,7 % para las RA de cualquier grado y 13,3 % frente al 14,8 % para las RA de grado 3 y 4).
Experiencia en los ensayos clínicos en la artritis reumatoide
Formulación intravenosa
A continuación se resume el perfil de seguridad de Mab-Thera/Rituxan i.v. en el tratamiento de pacientes con AR de moderada a grave. En la población total expuesta, más de 3000 pacientes recibieron como mínimo un ciclo de tratamiento y se sometieron a seguimiento durante perio-dos que oscilaron entre 6 meses y más de 5 años, lo que equivale a una exposición global de 7198 años-paciente; aproximadamente 2300 pacientes recibieron dos o más ciclos de tratamiento durante el periodo de seguimiento.
Las RA enumeradas en la tabla 3 se basan en los datos de los periodos comparativos con placebo de cuatro ensayos clínicos multicéntricos de la AR. Las poblaciones de pacientes que recibieron MabThera/Rituxan i.v. difirieron entre los diversos estudios: desde pacientes con AR activa precoz que no habían recibido tratamiento con metotrexato (MTX), pasando por pacientes con una respuesta inadecuada al MTX (MTX-RI), hasta pacientes con respuesta inadecuada a inhibidores del TNF (TNF-RI) (ver Ensayos clínicos/Eficacia).
Se administraron 2 veces 1000 mg o 2 veces 500 mg de MabThera/Rituxan i.v., con una diferencia de 2 semanas, además de metotrexato (10-25 mg/semana) (ver Posología y forma de administración, Artritis reumatoide).
En la tabla 3 se enumeran las RA con una incidencia ≥2 %, con una diferencia ≥2 % en comparación con el grupo de referencia, y se presentan independientemente de la dosis. Las frecuencias de la tabla 3 y la nota al pie correspondiente se definen del siguiente modo: muy frecuente (≥1/10), frecuente (≥1/100 a <1/10) y poco frecuente (≥1/1000 a <1/100).
|
Tabla 3. Resumen de las RA notificadas en pacientes con artritis reumatoide en el periodo de control de los ensayos clínicos † |
||
|
Clase de órganos y sistemas |
Muy frecuente |
Frecuente |
|
Infecciones e infestaciones |
Infección de las vías respiratorias altas, infección urinaria |
Bronquitis, sinusitis, gastroenteritis, tiña de los pies |
|
Trastornos del sistema inmunitario/ Trastornos generales y alteraciones en el lugar de administración |
Reacciones relacionadas con la infusión |
Reacciones relacionadas con la infusión*: hipertensión arterial, náuseas, exantema, pirexia, prurito, urticaria, irritación de garganta, sofocos, hipotensión arterial, rinitis, escalofríos intensos, taquicardia, fatiga, dolor bucofaríngeo, edema periférico, eritema |
|
Trastornos del metabolismo y de la nutrición |
Hipercolesterolemia |
|
|
Trastornos del sistema nervioso |
Cefalea |
Parestesias, migraña, mareos, ciática |
|
Trastornos de la piel y del tejido subcutáneo |
Alopecia |
|
|
Trastornos psiquiátricos |
Depresión, ansiedad |
|
|
Trastornos gastrointestinales |
Dispepsia, diarrea, reflujo gastroesofágico, úlcera bucal, dolor en la región superior del abdomen |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
Artralgia y dolor musculoesquelético, artrosis, bursitis |
|
|
† Esta tabla incluye todos los eventos con una diferencia de incidencia ≥2 % en el grupo de MabThera/Rituxan i.v. en comparación con el placebo. * Además, entre los eventos médicamente significativos notificados como infrecuentemente asociados a RRI se encuentran los siguientes: edema generalizado, broncoespasmo, sibilancias, edema laríngeo, edema angioneurótico, prurito generalizado, anafilaxia, reacción anafilactoide. |
||
En la población total expuesta, el perfil de seguridad estaba en consonancia con el observado en el periodo comparativo de los ensayos clínicos, sin que se identificaran nuevas RA.
Múltiples ciclos
Múltiples ciclos de tratamiento se asociaron a un perfil de RA similar al observado después de la primera exposición. El perfil de seguridad mejoró en los ciclos posteriores debido al descenso de las RRI, las reagudizaciones de la AR y las infecciones, todas ellas más frecuentes en los 6 primeros meses de tratamiento.
Información adicional sobre determinadas reacciones adversas
Reacciones relacionadas con la infusión: en los estudios clínicos en la AR, las RA más frecuentes tras la administración de MabThera/Rituxan i.v. fueron las RRI. De los 3095 pacientes tratados con MabThera/Rituxan i.v., 1077 (35 %) presentaron al menos una RRI. La inmensa mayoría de las RRI fueron de grado 1 o 2 según los criterios NCI-CTC. En los estudios clínicos, <1 % (14 de 3095) de los pacientes con AR que habían recibido una infusión de MabThera/Rituxan i.v., en cualquier dosis, sufrieron una RRI grave. No hubo ninguna RRI de grado 4 según los criterios NCI-CTC ni ningún fallecimiento por RRI en los estudios clínicos (ver Reacciones adversas, Poscomercialización). La proporción de eventos de grado 3 según los criterios NCI-CTC y de RRI que implicaron la retirada disminuyó en cada ciclo, y fue raro que se produjeran del ciclo 3 en adelante.
En 720 de 3095 (23 %) pacientes se observaron signos o síntomas indicativos de RRI (es decir, náuseas, prurito, fiebre, urticaria o exantema, escalofríos moderados e intensos, pirexia, estornudos, edema angioneurótico, irritación de garganta, tos y broncoespasmo, con o sin hipotensión o hipertensión asociadas) tras la primera infusión de la primera exposición a MabThera/Rituxan i.v. La premedicación con glucocorticoides por vía i.v. redujo significativamente la incidencia y la gravedad de estos eventos (ver Advertencias y precauciones).
En un estudio diseñado para evaluar la seguridad de una infusión de MabThera/Rituxan i.v. de 120 minutos de duración en pacientes con AR, los pacientes con AR activa moderada o grave que no sufrieron ninguna RRI grave durante la primera infusión del estudio o las 24 horas siguientes a la misma podían recibir una infusión de MabThera/Rituxan i.v. de 120 minutos de duración. Se excluyó de la participación en el estudio a los pacientes que anteriormente hubieran padecido alguna RRI grave relacionada con la infusión de un tratamiento biológico para la AR. La incidencia, los tipos y la gravedad de las RRI estaban en consonancia con los observados históricamente. No se observaron RRI graves (ver Ensayos clínicos/Eficacia).
Infecciones
La tasa global de infección fue de aproximadamente 97 por 100 años-paciente en los pacientes tratados con MabThera/Rituxan i.v. Las infecciones fueron de leves a moderadas predominantemente, y consistieron en la mayoría de los casos en infecciones de las vías respiratorias altas e infecciones urinarias. La tasa de infecciones graves fue de aproximadamente 4 por 100 años-paciente; algunas de ellas fueron mortales. Además de las RA que se presentan en la tabla 3, entre los eventos clínicamente graves también se encuentra la neumonía, con una frecuencia del 1,9 %.
Neoplasias malignas
La incidencia de tumores malignos tras la exposición a MabThera/Rituxan i.v. en los estudios clínicos en la AR (0,8 por 100 años-paciente) está dentro del intervalo esperado para una población comparable en edad y sexo.
Experiencia en ensayos clínicos en la granulomatosis con poliangitis (de Wegener) (GPA) y la poliangitis microscópica (PAM)
Formulación intravenosa
En el estudio clínico de la GPA y la PAM, 99 pacientes fueron tratados con MabThera/Rituxan i.v. (375 mg/m2, 1 vez por semana durante 4 semanas) y glucocorticoides (ver Ensayos clínicos/Eficacia).
Todas las RA enumeradas en la tabla 4 fueron eventos adversos con una incidencia ≥10 % en el grupo tratado con MabThera/Rituxan i.v. Las RA de la tabla 4 fueron muy frecuentes (frecuencia ≥1/10).
|
Tabla 4. Incidencia de RA muy frecuentes (≥10 %) en pacientes con GPA y PAM tratados con MabThera/Rituxan i.v. en el estudio clínico hasta el mes 6* |
||
|
Reacciones adversas |
Rituximab n = 99 |
Ciclofosfamida n = 98 |
|
Infecciones e infestaciones Infeccionesa |
61 (61,6 %) |
46 (46,9 %) |
|
Trastornos gastrointestinales Náuseas Diarrea |
18 (18,2 %) 17 (17,2 %) |
20 (20,4 %) 12 (12,2 %) |
|
Trastornos del sistema nervioso Cefalea |
17 (17,2 %) |
19 (19,4 %) |
|
Trastornos musculoesqueléticos y del tejido conjuntivo Espasmos musculares Artralgias |
17 (17,2 %) 13 (13,1 %) |
15 (15,3 %) 9 (9,2 %) |
|
Trastornos de la sangre y del sistema linfático Anemia Leucopenia |
16 (16,2 %) 10 (10,1 %) |
20 (20,4 %) 26 (26,5 %) |
|
Trastornos generales y alteraciones en el lugar de administración Edema periférico Fatiga |
16 (16,2 %) 13 (13,1 %) |
6 (6,1 %) 21 (21,4 %) |
|
Trastornos psiquiátricos Insomnio |
14 (14,1 %) |
12 (12,2 %) |
|
Exploraciones complementarias ALT elevada |
13 (13,1 %) |
15 (15,3 %) |
|
Trastornos respiratorios, torácicos y mediastínicos Tos Epistaxis Disnea |
13 (13,1 %) 11 (11,1 %) 10 (10,1 %) |
11 (11,2 %) 6 (6,1 %) 11 (11,2 %) |
|
Trastornos vasculares Hipertensión arterial |
12 (12,1 %) |
5 (5,1 %) |
|
Trastornos del sistema inmunitario Reacciones relacionadas con la infusiónb |
12 (12,1 %) |
11 (11,2 %) |
|
Trastornos de la piel y del tejido subcutáneo Exantema |
10 (10,1 %) |
17 (17,3 %) |
|
* El diseño del estudio permitía el cambio de tratamiento o tratamiento según el criterio del médico, y 13 pacientes de cada grupo recibieron un segundo tratamiento durante el periodo de estudio de 6 meses. a Las infecciones más frecuentes en el grupo del rituximab fueron las infecciones de las vías respiratorias altas, las infecciones urinarias y el herpes zóster. b Los términos notificados más frecuentemente en el grupo del rituximab fueron el síndrome de liberación de citocinas, la rubefacción, la irritación de garganta y el temblor. |
||
Información adicional sobre determinadas reacciones adversas
Reacciones relacionadas con la infusión
Las reacciones relacionadas con la infusión (RRI) en el estudio clínico de la GPA y la PAM se definieron como cualquier evento adverso que tuviera lugar, en la población de análisis de la seguridad, en un plazo de 24 horas desde el inicio de una infusión y al que los investigadores consideraran relacionado con la infusión. Se trató con MabThera/Rituxan i.v. a 99 pacientes; el 12 % sufrieron al menos una RRI. Todas las RRI fueron de grado 1-2 según los criterios NCI-CTC. Las RRI más frecuentes fueron el síndrome de liberación de citocinas, la rubefacción, la irritación de garganta y el temblor. MabThera/Rituxan i.v. se administró en combinación con glucocorticoides i.v., que quizá hayan reducido la incidencia y la gravedad de estos eventos.
Infecciones
En los 99 pacientes tratados con MabThera/Rituxan i.v., la tasa global de infección fue aproximadamente de 210 por 100 años-paciente (IC 95 %: 173-256). Las infecciones fueron predominantemente leves o moderadas y consistieron en la mayoría de los casos en infecciones de las vías respiratorias altas, herpes zóster e infecciones urinarias. La tasa de infecciones graves fue aproximadamente de 25 por 100 años-paciente. La infección grave notificada con mayor frecuencia en el grupo de MabThera/Rituxan i.v. fue la neumonía, con una frecuencia del 4 %.
Neoplasias malignas
La incidencia de neoplasias malignas en los pacientes tratados con MabThera/Rituxan i.v. en el estudio clínico fue de 2,05 por 100 años-paciente. Considerando los índices de incidencia estandarizados, esta tasa de neoplasias malignas parece ser similar a las notificadas anteriormente en poblaciones con GPA y PAM.
Alteraciones analíticas
Formulación intravenosa
Pacientes con artritis reumatoide
En pacientes con AR tratados con MabThera/Rituxan i.v. se ha observado hipogammaglobulinemia (IgG o IgM por debajo del límite inferior de la normalidad). Tras la disminución de la IgG o la IgM, no aumentó la tasa de infecciones en general o de infecciones graves.
Los episodios de neutropenia asociados al tratamiento con MabThera/Rituxan i.v., la mayoría de los cuales fueron transitorios y de intensidad leve o moderada, se observaron en ensayos clínicos en pacientes con AR después del primer ciclo de tratamiento. La neutropenia puede presentarse varios meses después de la administración de MabThera/Rituxan i.v.
En los periodos comparativos con placebo de los ensayos clínicos, el 0,94 % (13/1382) de los pacientes tratados con MabThera/Rituxan i.v. y el 0,27 % (2/731) de los que recibieron el placebo desarrollaron neutropenia grave (grado 3 o 4). En estos estudios, las tasas de neutropenia grave fueron, respectivamente, de 1,06 y 0,53 por 100 años-paciente después del primer ciclo de tratamiento, y de 0,97 y 0,88 por 100 años-paciente después de múltiples ciclos, respectivamente. Así pues, la neutropenia puede considerarse una RA del primer ciclo exclusivamente. El momento de instauración de la neutropenia fue variable. En los ensayos clínicos, la neutropenia no se asoció a un aumento observado de las infecciones graves, y la mayoría de los pacientes siguieron recibiendo nuevos ciclos de MabThera/Rituxan i.v. después de los episodios de neutropenia.
Pacientes con granulomatosis con poliangitis (de Wegener) (GPA) y poliangitis microscópica (PAM)
Se ha observado hipogammaglobulinemia (IgA, IgG o IgM por debajo del límite inferior de la normalidad) en pacientes con GPA o PAM tratados con MabThera/Rituxan i.v.
Al cabo de 6 meses, el 27 %, 58 % y 51 % de los pacientes del grupo del MabThera/Rituxan i.v. con valores iniciales normales de inmunoglobulinas presentaban cifras bajas de IgA, IgG e IgM, respectivamente, en comparación con el 25 %, 50 % y 46 % en el grupo de la ciclofosfamida. En los pacientes con cifras bajas de IgA, IgG o IgM no aumentó la tasa de infecciones en general ni de infecciones graves.
En el estudio multicéntrico, aleatorizado, comparativo con tratamiento activo, con doble enmascaramiento (doble ciego) de la ausencia de inferioridad del MabThera/Rituxan i.v. en la GPA y la PAM, el 24 % de los pacientes del grupo del MabThera/Rituxan i.v. (ciclo único) y el 23 % de los pacientes del grupo de la ciclofosfamida desarrollaron neutropenia de grado 3 o superior según los criterios NCI-CTC. En los pacientes tratados con MabThera/Rituxan i.v., la neutropenia no se asoció a un incremento observado de las infecciones graves. No se ha estudiado en ensayos clínicos el efecto de ciclos múltiples de MabThera/Rituxan i.v. en el desarrollo de neutropenia en pacientes con GPA y PAM.
Poscomercialización
Formulación intravenosa
Pacientes con linfoma no hodgkiniano y leucemia linfocítica crónica
Las frecuencias notificadas en este apartado (reacciones raras y muy raras) se basan en estimaciones de la exposición tras la comercialización y en gran medida en datos derivados de notificaciones espontáneas.
Tras la comercialización de MabThera/Rituxan i.v. se han descrito nuevos casos de RRI graves (ver Advertencias y precauciones).
En el marco del programa de farmacovigilancia continua de MabThera/Rituxan i.v., se han observado las si-guientes RA graves:
Aparato cardiovascular
Se han registrado eventos cardiacos graves, incluidos la insuficiencia cardiaca y el infarto de miocardio, sobre todo en pacientes con trastornos cardiacos preexistentes o sometidos a tratamiento con quimioterápicos cardio-tóxicos y, la mayoría de las veces, asociados a RRI. Muy rara vez se han notificado casos de vasculitis, predominantemente cutánea, como la vasculitis leucocitoclástica.
Aparato respiratorio
Se han observado casos de fracaso o insuficiencia respiratoria e infiltración pulmonar en el contexto de RRI (ver Advertencias y precauciones). Además de eventos pulmonares asociados a la infusión, se han descrito casos de neumopatía intersticial, algunos con desenlace mortal.
Sistema hemolinfático
Se han notificado casos de trombocitopenia aguda reversi-ble relacionada con la infusión.
Piel y faneras
En raras ocasiones se han notificado casos de reacciones cutáneas ampollosas graves, incluidos algunos casos mortales de necrólisis epidérmica tóxica y síndrome de Stevens-Johnson.
Sistema nervioso
Se han descrito casos de síndrome de encefalopatía posterior reversible (SEPR) o síndrome de leucoencefalopatía posterior reversible (SLEPR). Entre los signos y síntomas se encuentran los siguientes: trastorno visual, cefalea, convulsiones y estado mental alterado, con o sin hipertensión asociada. El diagnóstico de SEPR/SLEPR debe confirmarse mediante pruebas de diagnóstico por imágenes cerebrales. Los casos notificados presentaban conocidos factores de riesgo de padecer SEPR/SLEPR, como la enfermedad de fondo, la hipertensión arterial, el tratamiento inmunosupresor y la quimioterapia.
En raras ocasiones se ha descrito casos de neuropatía craneal con o sin neuropatía periférica. Se han observado signos y síntomas de neuropatía craneal (como pérdida visual importante, pérdida auditiva, pérdida de otros sentidos y parálisis facial) en diversos momentos, hasta varios meses después de concluido el tratamiento con MabThera/Rituxan.
Trastornos generales
En raras ocasiones se han notificado reacciones del tipo de la enfermedad del suero.
Infecciones e infestaciones
Se han descrito casos de reactivación de la hepatitis B, la mayoría de las veces en pacientes que recibían MabThera/Rituxan i.v. en combinación con quimioterapia antineoplásica (ver Advertencias y precauciones). Durante el tratamiento con MabThera/Rituxan i.v. se han descrito otras infecciones víricas graves, ya fueran nuevas, reactivaciones o agudizaciones, algunas con desenlace mortal. La mayoría de los pacientes habían recibido MabThera/Rituxan i.v. en combinación con quimioterapia o como parte de un trasplante de progenitores hematopoyéticos. Ejemplos de estas graves infecciones víricas son las causadas por virus del herpes —citomegalovirus (CMV), virus de la varicela-zóster y virus del herpes simple—, el virus JC (leucoencefalopatía multifocal progresiva) (ver Advertencias y precauciones) y el virus de la hepatitis C.
En pacientes con sarcoma de Kaposi preexistente expuestos a MabThera/Rituxan i.v. se ha observado la progresión del sarcoma de Kaposi. Estos casos se produjeron en indicaciones no aprobadas y la mayoría de los pacientes eran VIH-positivos.
Aparato digestivo
Se han observado casos de perforación gastrointestinal, en ocasiones mortales, en pacientes con LNH tratados con MabThera/Rituxan i.v. en combinación con quimioterapia.
Pacientes con artritis reumatoide (AR), granulomatosis con poliangitis (de Wegener) (GPA) y poliangitis microscópica (PAM)
Como parte de la vigilancia poscomercialización continua de la seguridad de MabThera/Rituxan i.v., se han observado los siguientes eventos en el marco del tratamiento de la AR; también son previsibles, si es que no se han observado ya, en los pacientes con GPA o PAM:
Infecciones e infestaciones
Se han notificado casos de leucoencefalopatía multifocal progresiva (LMP) y reactivación de la hepatitis B.
Trastornos generales
Se han notificado casos de reacciones similares a la enfermedad del suero.
Trastornos de la piel y del tejido subcutáneo
En muy raras ocasiones se han notificado casos de necrólisis epidérmica tóxica y síndrome de Stevens-Johnson, algunos con desenlace mortal.
Trastornos de la sangre y del sistema linfático
Se han notificado casos raros de episodios de neutropenia, incluida la neutropenia grave de inicio tardío y persistente, algunos de los cuales se asociaron a infecciones mortales.
Sistema nervioso
Se han descrito casos de síndrome de encefalopatía posterior reversible (SEPR) o síndrome de leucoencefalopatía posterior reversible (SLEPR). Entre los signos y síntomas se encuentran los siguientes: trastorno visual, cefalea, convulsiones y estado mental alterado, con o sin hipertensión asociada. El diagnóstico de SEPR/SLEPR debe confirmarse mediante pruebas de diagnóstico por imágenes cerebrales. Los casos notificados presentaban conocidos factores de riesgo de padecer SEPR/SLEPR como hipertensión, tratamiento inmunosupresor u otros tratamientos concomitantes.
Trastornos generales y alteraciones en el lugar de administración
Se han registrado casos de RRI graves, algunos con desenlace mortal (ver Reacciones adversas, Ensayos clínicos).
Alteraciones analíticas
Formulación intravenosa
Linfoma no hodgkiniano
Sistema hemolinfático
En raras ocasiones la neutropenia comenzó más de 4 semanas después de la última infusión de MabThera/Rituxan i.v.
Poscomercialización
En estudios de MabThera/Rituxan i.v. en pacientes con macroglobulinemia de Waldenström se ha observado, después de iniciar el tratamiento, un aumento transitorio de la IgM que puede asociarse a hiperviscosidad y síntomas relacionados. La IgM normalmente recupera al menos su valor inicial al cabo de 4 meses.
Interacciones con otros medicamentos y otras formas de interacción
Los datos sobre posibles interacciones farmacológicas con MabThera/Rituxan de los que se dispone actualmente son limitados.
En los pacientes con LLC, la coadministración con Mab-Thera/Rituxan i.v. no pareció tener ningún efecto en la farmacocinética de la fludarabina o la ciclofosfamida; por otro lado, no se observó ningún efecto de la fludarabina ni la ciclofosfamida en la farmacocinética de MabThera/Rituxan.
La coadministración de metotrexato no tuvo ningún efecto en la farmacocinética de MabThera/Rituxan i.v. en los pacientes con AR.
Los pacientes con anticuerpos humanos antimurinos (HAMA) o anticuerpos humanos antiquiméricos (HACA) pueden sufrir reacciones alérgicas o de hipersensibilidad si reciben otros anticuerpos monoclonales diagnósticos o terapéuticos.
En el programa de estudios clínicos de la AR, 373 pacientes tratados con MabThera/Rituxan i.v. reci-bieron tratamiento ulterior con otros FAME; 240 de ellos recibieron un FAME biológico. En estos pacientes, la tasa de infecciones graves durante el tratamiento con MabThera/Rituxan i.v. (antes recibir un FAME biológico) fue de 6,1 por 100 años-paciente, frente a 4,9 por 100 años-paciente después del tratamiento con el FAME biológico.
ADVERTENCIAS Y PRECAUCIONES
Advertencias y precauciones generales
Para mejorar la trazabilidad de los biomedicamentos, debe registrarse (o declararse) claramente el nombre comercial y el número de lote del producto administrado en la historia clínica del paciente.
Pacientes con linfoma no hodgkiniano y leucemia linfocítica crónica
Reacciones relacionadas con la infusión/adminis-tración: MabThera/Rituxan se asocia a reacciones relacionadas con la infusión/administración, que pueden relacionarse con la liberación de citocinas y de otros mediadores químicos. El síndrome de liberación de citocinas puede ser indistinguible clínicamente de las reacciones de hipersensibilidad agudas.
Reacciones relacionadas con la infusión de MabThera/Rituxan i.v.: en el uso desde la comercialización, se han notificado casos de reacciones graves relacionadas con la infusión con desenlace mortal. Las reacciones graves relacionadas con la infusión, que generalmente se manifestaron en un plazo de 30 minutos a 2 horas después de iniciar la primera infusión de MabThera/Rituxan i.v., se caracterizaron por eventos pulmonares e incluyeron, en algunos casos, una lisis tumoral rápida y características del síndrome de lisis tumoral, además de fiebre, escalofríos moderados e intensos, hipotensión, urticaria, angioedema y otros síntomas (ver Reacciones adversas). Los pacientes con una gran carga tumoral o con una cifra muy elevada (>25 x 109/l) de células malignas circulantes, como los pacientes con LLC y linfoma de células del manto, pueden tener mayor riesgo de sufrir reacciones graves relacionadas con la infusión. Los síntomas de la reacción a la infusión suele revertir si se interrumpe ésta. Se recomienda tratar los síntomas relacionados con la infusión con difenhidramina y paracetamol (acetaminofeno). Puede estar indicado el tratamiento adicional con broncodilatadores o solución salina isotónica i.v. En la mayoría de los casos, la infusión puede reanudarse a una velocidad un 50 % menor (por ejemplo, de 100 mg/h a 50 mg/h) cuando los síntomas hayan desaparecido por completo. La mayoría de los pacientes que sufrieron reacciones adversas relacionadas con la infusión que no fueron potencialmente mortales pudieron concluir el ciclo entero de tratamiento con MabThera/Rituxan i.v. El tratamiento posterior de los pacientes tras la resolución completa de los signos y síntomas en raras ocasiones dio lugar a la reaparición de reacciones graves relacionadas con la infusión.
Los pacientes con una cifra elevada (>25 x 109/l) de células malignas circulantes o con una gran carga tumoral, como los pacientes con LLC y linfoma de células del manto, que pueden tener mayor riesgo de sufrir reacciones graves relacionadas con la infusión, sólo deben ser tratados con extrema precaución. Estos pacientes deben ser vigilados muy estrechamente durante todo el curso de la primera infusión. En estos pacientes, se planteará el uso de una velocidad de infusión reducida durante la primera infusión o la división de la administración en 2 días durante el primer ciclo y en cualquier ciclo ulterior si la cifra de linfocitos sigue siendo >25 × 109/l.
Reacciones de hipersensibilidad y anafilaxia: se han notificado casos de reacciones anafilácticas y otras reacciones de hipersensibilidad después de la administración i.v. de proteínas a los pacientes. Se dispondrá de epinefrina, antihistamínicos y glucocorticoides para su uso inmediato si se produjera una reacción de hipersensibilidad a MabThera/Rituxan i.v.
Reacciones relacionadas con la administración de MabThera/Rituxan s.c.: se han notificado reacciones cutáneas locales, incluidas reacciones en el lugar de inyección, en pacientes que recibieron MabThera por vía s.c. Entre los síntomas se encontraban los siguientes: dolor, hinchazón, induración, hemorragia, eritema, prurito y exantema (ver Reacciones adversas). Algunas reacciones cutáneas locales ocurrieron más de 24 horas después de la administración s.c. del fármaco. La mayoría de las reacciones cutáneas locales observadas tras la administración de la formulación s.c. fueron leves o moderadas y se resolvieron sin aplicar ningún tratamiento específico.
Todos los pacientes deben recibir siempre la primera dosis de MabThera/Rituxan mediante administración i.v. para evitar la administración irreversible de la dosis completa de MabThera/Rituxan s.c. durante el ciclo 1. En este ciclo, el paciente corre el mayor riesgo de sufrir una reacción relacionada con la infusión, que puede tratarse eficazmente reduciendo la velocidad de infusión o interrumpiendo la administración. La formulación s.c. sólo se administrará en el segundo ciclo o en los ciclos posteriores. Los pacientes que no puedan recibir la dosis completa de MabThera/Rituxan i.v. en infusión seguirán recibiendo MabThera/Rituxan i.v. hasta que se haya administrado satisfactoriamente una dosis i.v. completa. En pacientes que pueden recibir la dosis completa de MabThera/Rituxan i.v. en infusión, la segunda dosis o las dosis posteriores de MabThera/Rituxan pueden administrarse por vía s.c. usando la formulación s.c. de MabThera/Rituxan (ver Posología y forma de administración). Como ocurre con la formulación i.v., MabThera/Rituxan s.c. debe administrarse en un entorno con un equipo completo de reanimación inmediatamente disponible y bajo la estrecha vigilancia de un profesional sanitario. La premedicación, que consiste en un analgésico/antipirético y un antihistamínico, se administrará siempre antes de cada dosis de MabThera/Rituxan s.c. También debe considerarse la premedicación con glucocorticoides.
Se debe observar a los pacientes durante al menos 15 minutos después de administrar MabThera/Rituxan por vía s.c. Puede ser apropiado un periodo más prolongado en pacientes con riesgo elevado de presentar reacciones de hipersensibilidad.
Se debe indicar a los pacientes que han de ponerse inmediatamente en contacto con el médico que los trata si en cualquier momento después de la administración del fármaco aparecen síntomas que son indicativos de reacciones de hipersensibilidad graves o de un síndrome de liberación de citocinas.
Eventos pulmonares: entre los eventos pulmonares se encuentran la hipoxia, la infiltración pulmonar y la insuficiencia respiratoria aguda. Algunos de estos eventos han sido precedidos por broncoespasmo y disnea agudos. En algunos casos, los síntomas empeoraron con el tiempo, mientras que en otros se produjo una mejoría inicial seguida por un deterioro clínico. Así pues, los pacientes que sufran eventos pulmonares u otros síntomas graves relacionados con la infusión deben ser vigilados estrechamente hasta que los síntomas se resuelvan por completo. Los pacientes con antecedentes de insuficiencia pulmonar o con infiltración tumoral pulmonar son los que corren mayor riesgo de tener desenlaces desfavorables, por lo que se los tratará con gran precaución. La insuficiencia respiratoria aguda puede acompañarse de eventos como infiltración o edema intersticial pulmonar, que pueden observarse en la radiografía de tórax. El síndrome suele manifestarse 1 o 2 horas después de iniciar la primera infusión. En caso de eventos pulmonares graves, se suspenderá inmediatamente la administración de MabThera/Rituxan (ver Posología y forma de administración) y se instaurará tratamiento sintomático intensivo.
Lisis tumoral rápida: MabThera/Rituxan interviene en la lisis rápida de los linfocitos CD20+ benignos y malignos. Se ha notificado la aparición de signos y síntomas (por ejemplo: hiperuricemia, hiperpotasemia, hipocalcemia, hiperfosfatemia, insuficiencia renal aguda, LDH elevada) compatibles con un síndrome de lisis tumoral (SLT) después de la primera infusión de MabThera/Rituxan i.v. en pacientes con una cifra elevada de linfocitos malignos circulantes. Se considerará la profilaxis del síndrome de lisis tumoral en los pacientes con riesgo de presentar una lisis tumoral rápida (por ejemplo: pacientes con una gran carga tumoral o con un gran número [>25 x 109/l] de células malignas circulantes, como los pacientes con LLC o linfoma de células del manto). Se hará un seguimiento estrecho de estos pacientes y se realizará el control analítico adecuado. Se administrará el tratamiento médico apropiado a los pacientes que presenten signos y síntomas compatibles con una lisis tumoral rápida. Después del tratamiento y la resolución completa de los signos y síntomas, la terapia ulterior con MabThera/Rituxan i.v. se ha administrado junto con tratamiento profiláctico del síndrome de lisis tumoral en un número limitado de casos.
Trastornos cardiovasculares: durante la adminis-tración de MabThera/Rituxan se puede producir hipotensión arterial, por lo que se considerará la conve-niencia de retirar la medicación antihipertensora desde 12 horas antes de la administración de MabThera/Rituxan i.v./s.c. Se han descrito casos de angina de pecho y arritmias cardiacas, como aleteo y fibrilación auricular, insuficiencia cardiaca o infarto de miocardio en pacientes tratados con MabThera/Rituxan i.v./s.c. En consecuencia, se debe vigilar estrechamente a los pacientes con antecedentes de cardiopatía.
Control del hemograma: aunque MabThera/Rituxan en monoterapia no es mielosupresor, se debe actuar con cautela cuando se plantee el tratamiento de pacientes con una cifra de neutrófilos <1,5 x 109/l o una cifra de plaquetas <75 x 109/l, dado que la experiencia clínica en tales pacientes es limitada. MabThera/Rituxan i.v. se ha usado en pacientes receptores de un autotrasplante de médula ósea, así como en otros grupos de riesgo con posible hipofunción de la médula ósea, sin que indujera mielotoxicidad.
Se considerará en qué medida puede ser necesario determinar regularmente la fórmula sanguínea, incluida la cifra de plaquetas, durante la monoterapia con MabThera/Rituxan. Cuando MabThera/Rituxan se administre con CHOP o CVP, se realizarán periódicamente hemogramas de acuerdo con las prácticas médicas habituales.
Infecciones: no se debe iniciar el tratamiento con MabThera/Rituxan en pacientes con infecciones activas graves.
Hepatitis B: se han notificado casos de reactivación de la hepatitis B, incluidos casos de hepatitis fulminante, algunos de ellos mortales, en sujetos que recibían tratamiento con MabThera/Rituxan i.v., si bien la mayoría de estos sujetos estaban expuestos también a quimioterapia citotóxica. Tanto el estado de la enfermedad subyacente como la quimioterapia citotóxica constituían factores de confusión de los informes.
Antes de iniciar el tratamiento con MabThera/Rituxan, se deben realizar a todos los pacientes pruebas de detección del virus de la hepatitis B (VHB). Como mí-nimo se debe determinar el estado del paciente respecto al antígeno de superficie del virus de la hepatitis B (HBsAg) y respecto al anticuerpo contra el antígeno nuclear del virus de la hepatitis B (HBcAb). A estos se pueden añadir otros marcadores apropiados conforme a las pautas locales. No se debe tratar con MabThera/Rituxan a los pacientes con hepatitis B activa. Los pacientes con resultados positivos en las pruebas serológicas de la hepatitis B deben consultar a expertos en hepatopatías antes de iniciar el tratamiento; por otra parte, se los debe controlar y tratar conforme a las pautas médicas locales para prevenir la reactivación de la hepatitis B.
Leucoencefalopatía multifocal progresiva: se han descrito casos de leucoencefalopatía multifocal progresiva (LMP) durante el uso de MabThera/Rituxan i.v. en pacientes con LNH y LLC (ver Reacciones adversas, Poscomercialización). La mayoría de los pacientes habían recibido MabThera/Rituxan i.v. en combinación con quimioterapia o como parte de un trasplante de progenitores hematopoyéticos. Los médicos que traten a pacientes con LNH o LLC deben considerar la posibilidad de una LMP en el diagnóstico diferencial de aquellos que refieran síntomas neurológicos; la consulta con el neurólogo está clínicamente indicada.
Reacciones cutáneas: se han notificado casos de reacciones cutáneas graves, como la necrólisis epidérmica tóxica y el síndrome de Stevens-Johnson, algunos con desenlace mortal (ver Reacciones adversas, Posco-mercialización). Si se produjera un evento de este tipo presuntamente relacionado con MabThera/Rituxan, el tratamiento se suspenderá definitivamente.
Vacunación: no se ha estudiado la seguridad de la inmunización con vacunas de virus vivos después del tratamiento con MabThera/Rituxan i.v./s.c.; no se recomienda la vacunación con vacunas de virus vivos.
Los pacientes tratados con MabThera/Rituxan pueden recibir vacunas de virus no vivos, aunque las tasas de respuesta con estas vacunas pueden ser reducidas. En un estudio no aleatorizado, pacientes con LNH de bajo grado recidivante tratados con MabThera/Rituxan i.v. en monoterapia presentaron, en comparación con sujetos de referencia sanos no tratados, una menor tasa de respuesta a la vacunación con el antígeno de recuerdo del tétanos (16 % frente al 81 %) y con el neoantígeno KLH (keyhole limpet hemocyanin [hemocianina de lapa californiana]) (4 % frente al 76 % en la evaluación de un aumento del título de anticuerpos a más del doble).
La media de los títulos de anticuerpos previos al tratamiento contra una serie de antígenos (Streptococcus pneumoniae, virus de la gripe A, paperas, rubéola, varicela) se mantuvo al menos durante 6 meses después del tratamiento con MabThera/Rituxan i.v.
Pacientes con artritis reumatoide (AR), granulomatosis con poliangitis (de Wegener) (GPA) y poliangitis micros-cópica (PAM): no se han establecido la eficacia ni la seguridad de MabThera/Rituxan i.v. en el tratamiento de enfermedades autoinmunitarias aparte de la artritis reumatoide, la granulomatosis con poliangitis (de Wegener) y la poliangitis microscópica.
Reacciones relacionadas con la infusión: Mab-Thera/Rituxan i.v. se asocia a reacciones relacionadas con la infusión (RRI), que pueden relacionarse con la liberación de citocinas y de otros mediadores químicos. Antes de cada infusión de MabThera/Rituxan i.v. se administrará siempre premedicación con un analgésico/antipirético y un antihistamínico. En los pacientes con AR, la premedicación con glucocorticoides debe adminis-trarse antes de cada infusión de MabThera/Rituxan i.v., a fin de reducir la frecuencia y la gravedad de las reacciones relacionadas con la infusión (ver Posología y forma de administración y Reacciones adversas).
En los pacientes con AR, la mayoría de los eventos relacionados con la infusión que se notificaron en ensayos clínicos fueron de leves a moderados. En el marco del uso tras la comercialización se han descrito reacciones graves relacionadas con la infusión que tuvieron un desenlace mortal (ver Reacciones adversas, Poscomercialización). Se debe vigilar estrechamente a los pacientes con trastornos cardiacos preexistentes o que hayan sufrido con anterioridad reacciones cardiopulmonares adversas. Los síntomas más frecuentes fueron cefalea, prurito, irritación de garganta, rubefacción, exantema, urticaria, hipertensión arterial y pirexia. En general, la proporción de pacientes que sufrieron alguna reacción a la infusión fue mayor tras la primera infusión de cualquier ciclo de tratamiento que después de la segunda infusión. Los pacientes toleraron mejor las infusiones siguientes de MabThera/Rituxan i.v. que la infusión inicial. Menos del 1 % de los pacientes sufrieron RRI graves, y la mayoría de éstas se notificaron durante la primera infusión del primer ciclo (ver Reacciones adversas). Por lo general, las reacciones notificadas fueron reversibles tras reducir la velocidad de infusión o interrumpir la infusión de MabThera/Rituxan i.v. y administrar un antipirético, un antihistamínico y, ocasionalmente, oxígeno, suero salino isotónico i.v., broncodilatadores o glucocorticoides según las necesidades. Dependiendo de la gravedad de la reacción relacionada con la infusión y las medidas requeridas, se suspenderá MabThera/Rituxan i.v. temporal o definitivamente. En la mayoría de los casos, la infusión puede reanudarse a una velocidad un 50 % menor (por ejemplo: de 100 mg/h a 50 mg/h) cuando los síntomas hayan desaparecido por completo.
Las reacciones relacionadas con la infusión en los pacientes con GPA y PAM fueron similares a las observadas en pacientes con AR en los ensayos clínicos (ver Reacciones adversas). En los pacientes con GPA y PAM, MabThera/Rituxan i.v. se administró en combinación con dosis altas de glucocorticoides (ver Posología y forma de administración), que pueden reducir la incidencia y la gravedad de estos eventos (ver la información para los pacientes con artritis reumatoide, más atrás).
Reacciones de hipersensibilidad y anafilaxia: se han notificado casos de reacciones anafilácticas y otras reacciones de hipersensibilidad después de la adminis-tración i.v. de proteínas a los pacientes. Durante la administración de MabThera/Rituxan i.v. es preciso disponer de medicamentos para tratar inmediatamente las reacciones de hipersensibilidad (por ejemplo: epinefrina, antihistamínicos y glucocorticoides) en el caso de que sobrevengan.
Trastornos cardiovasculares: durante la infusión de MabThera/Rituxan i.v. se puede producir hipotensión arterial, por lo que debe considerarse la conveniencia de retirar la medicación antihipertensora 12 horas antes de la infusión i.v. de MabThera/Rituxan.
Se han descrito casos de angina de pecho y arritmias cardiacas, como aleteo y fibrilación auricular, insuficiencia cardiaca o infarto de miocardio en pacientes tratados con MabThera/Rituxan i.v. En consecuencia, se debe vigilar estrechamente a los pacientes con antecedentes de cardiopatía (ver el subapartado Reacciones relacionadas con la infusión, más atrás).
Infecciones: considerando el mecanismo de acción de MabThera/Rituxan y sabiendo que los linfocitos B desempeñan una función importante en el mantenimiento de la respuesta inmunitaria normal, los pacientes pueden correr mayor riesgo de infecciones después del tratamien-to con MabThera/Rituxan i.v. (ver Mecanismo de acción). MabThera/Rituxan i.v. no debe administrarse a pacientes con infección activa o inmunodeficiencia grave (por ejemplo: en caso de cifras muy bajas de linfocitos CD4 o CD8). Los médicos deben ser cautos cuando consideren la posibilidad de administrar MabThera/Rituxan i.v. a pacientes con antecedentes de infecciones recidivantes o crónicas o con enfermedades de fondo que puedan aumentar su predisposición a contraer infecciones graves (ver Reacciones adversas). A los pacientes que sufran una infección después del tratamiento con MabThera/Rituxan i.v. se los someterá a una pronta evaluación y se les administrará el tratamiento adecuado.
Hepatitis B: en pacientes con AR, granulomatosis con poliangitis y poliangitis microscópica que recibían MabThera/Rituxan i.v. se han notificado casos de reactivación de la hepatitis B, incluidos algunos con desenlace mortal.
Antes de iniciar el tratamiento con MabThera/Rituxan i.v. se deben realizar a todos los pacientes pruebas de detección del virus de la hepatitis B (VHB). Como mínimo se debe determinar el estado del paciente respecto al antígeno de superficie del virus de la hepatitis B (HBsAg) y respecto al anticuerpo contra el antígeno nuclear del virus de la hepatitis B (HBcAb). A estos se pueden añadir otros marcadores apropiados conforme a las pautas locales. No se debe tratar con MabThera/Rituxan i.v. a los pacientes con hepatitis B activa. Los pacientes con resultados positivos en las pruebas serológicas de la hepatitis B deben consultar a expertos en hepatopatías antes de iniciar el tratamiento; por otra parte, se los debe controlar y tratar conforme a las pautas médicas locales para prevenir la reactivación de la hepatitis B.
Reacciones cutáneas: se han notificado casos de reacciones cutáneas graves, como la necrólisis epidérmica tóxica y el síndrome de Stevens-Johnson, algunos con desenlace mortal (ver Reacciones adversas, Poscomerciali-zación). En el caso de que se produjera uno de estos eventos con una presunta relación con MabThera/Rituxan i.v., se suspenderá definitivamente el tratamiento.
Leucoencefalopatía multifocal progresiva: se han descrito casos de leucoencefalopatía multifocal progresiva (LMP) mortal tras utilizar MabThera/Rituxan i.v. para el tratamiento de enfermedades autoinmunitarias, incluida la AR. Varios de los casos notificados, pero no todos, presentaban posibles factores de riesgo de la LMP, como la enfermedad subyacente, el tratamiento inmunodepresor de larga duración o la quimioterapia. La LMP también se ha registrado en pacientes con enfermedades autoinmunitarias que no recibían tratamiento con Mab-Thera/Rituxan i.v. Los médicos que traten a pacientes con enfermedades autoinmunitarias deben considerar la posibilidad de una LMP en el diagnóstico diferencial de aquellos que refieran síntomas neurológicos; la consulta con el neurólogo debe considerarse clínicamente indicada.
Vacunación: no se ha estudiado la seguridad de la inmunización con vacunas de virus vivos después del tratamiento con MabThera/Rituxan i.v., por lo que no se recomienda la vacunación con este tipo de vacunas mientras dure el tratamiento con MabThera/Rituxan i.v. o la depleción de linfocitos B periféricos. Los pacientes tratados con MabThera/Rituxan i.v. pueden recibir vacunas de virus no vivos, aunque las tasas de respuesta con estas vacunas pueden ser reducidas.
En los pacientes con AR, los médicos deben evaluar el estado vacunal y aplicar las pautas de vacunación actua-les antes de iniciar el tratamiento con MabThera/Rituxan i.v. La vacunación tiene que haber finalizado al menos 4 semanas antes de la primera administración de MabThera/Rituxan i.v.
En un estudio aleatorizado, los pacientes con AR tratados con MabThera/Rituxan i.v. y metotrexato tuvieron, en comparación con los pacientes que sólo recibieron metotrexato, tasas de respuesta comparables al antígeno de recuerdo del tétanos (39 % frente al 42 %), menores tasas de respuesta a la vacuna antineumocócica de polisacáridos (43 % frente al 82 % a 2 serotipos neumocócicos por lo menos) y al neoantígeno KLH (34 % frente al 80 %) cuando se administraron al menos 6 meses después de MabThera/Rituxan i.v. Si fuera preciso utilizar vacunas elaboradas con virus no vivos durante el tratamiento con MabThera/Rituxan i.v., su aplicación debería finalizar al menos 4 semanas antes de empezar el siguiente ciclo de MabThera/Rituxan i.v.
En la experiencia global del tratamiento repetido con MabThera/Rituxan i.v. a lo largo de un año en pacientes con AR, la proporción de pacientes con títulos positivos de anticuerpos contra S. pneumoniae, los virus de la gripe, las paperas, la rubéola, la varicela y el toxoide tetánico fueron generalmente similares a las proporciones iniciales.
Pacientes con AR sin tratamiento previo con metotrexato
No se recomienda administrar MabThera/Rituxan i.v. a pacientes que no hayan recibido previamente tratamiento con metotrexato, ya que no se ha establecido que exista un balance favorable de beneficios y riesgos.
Abuso y dependencia del fármaco
Sin texto.
Pautas posológicas especiales
Niños y adolescentes: no se han establecido la seguridad ni la eficacia de MabThera/Rituxan en pacientes pediátricos.
Ancianos: no es necesario ajustar la dosis en pacientes ancianos (>65 años).
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
Instrucciones generales
Formulaciones intravenosa y subcutánea
La sustitución por cualquier otro biomedicamento re-quiere el consentimiento del médico prescriptor.
Es importante comprobar las etiquetas del producto para asegurarse de que se administra al paciente la formulación (i.v. o s.c.) y la dosis farmacéutica correctas, tal como se haya recetado.
MabThera/Rituxan debe administrarse siempre en un entorno con un equipo completo de reanimación inmediatamente disponible y bajo la estrecha vigilancia de un profesional sanitario experimentado.
Antes de cada administración de MabThera/Rituxan, se premedicará siempre al paciente con un analgésico/antipirético (por ejemplo: paracetamol [acetaminofeno]) y un antihistamínico (por ejemplo: difenhidramina).
También se planteará la premedicación con glucocorticoides, sobre todo si MabThera/Rituxan no se administra en combinación con una quimioterapia que contenga corticoesteroides (ver Advertencias y precauciones).
Ajustes posológicos durante el tratamiento
No se recomienda reducir la dosis de MabThera/Rituxan. Cuando MabThera/Rituxan se administra en combinación con quimioterapia, se debe reducir la dosis habitual de los quimioterápicos.
Formulación intravenosa
La formulación i.v. de MabThera/Rituxan no debe administrarse por vía s.c. (ver Instrucciones especiales de uso, manipulación y eliminación).
Las soluciones para infusión preparadas no deben admi-nistrarse en inyección i.v. lenta o rápida (ver Instrucciones especiales de uso, manipulación y eliminación).
Velocidad de infusión de la formulación intravenosa
Primera infusión intravenosa
La velocidad de infusión inicial recomendada es de 50 mg/h; después de los 30 minutos iniciales, la velocidad puede aumentarse a razón de 50 mg/h cada 30 minutos hasta una velocidad máxima de 400 mg/h. Esta velocidad corresponde a un periodo de administración total de 4,25 horas.
Infusiones intravenosas posteriores
Las infusiones posteriores de MabThera/Rituxan i.v. pueden iniciarse a una velocidad de 100 mg/h y aumentarse a razón de 100 mg/h cada 30 minutos hasta una velocidad máxima de 400 mg/h. Esta velocidad corresponde a un periodo de administración total de 3,25 horas.
Formulación subcutánea
La formulación s.c. de MabThera/Rituxan no debe administrarse por vía i.v. (ver Instrucciones especiales de uso, manipulación y eliminación).
MabThera/Rituxan s.c. 1400 mg debe usarse exclusivamente en el linfoma no hodgkiniano (LNH).
MabThera/Rituxan s.c. 1600 mg debe usarse exclusivamente en la leucemia linfocítica crónica (LLC).
MabThera/Rituxan s.c. debe inyectarse subcutáneamente en la pared abdominal y nunca en zonas donde la piel esté enrojecida, contusionada, dolorosa o dura o en zonas donde haya lunares o cicatrices. No se dispone de datos sobre la aplicación de la inyección en otros lugares del cuerpo, por lo que las inyecciones se limitarán a la pared abdominal.
Si durante el tratamiento con MabThera/Rituxan s.c. es preciso administrar por vía s.c. otros medicamentos, se inyectarán preferentemente en diferentes sitios.
La inyección de MabThera/Rituxan 1400 mg s.c. debe administrarse en un tiempo de aproximadamente 5 minutos.
La inyección de MabThera/Rituxan 1600 mg s.c. debe administrarse en un tiempo de aproximadamente 7 minutos.
Si se interrumpe una inyección puede reanudarse o puede usarse otro lugar, si fuera apropiado.
Dosis habitual
Linfoma no hodgkiniano de bajo grado o folicular:
Formulación intravenosa
Tratamiento inicial:
Monoterapia por vía intravenosa
La dosis recomendada de MabThera/Rituxan i.v. como monoterapia en pacientes adultos es de 375 mg/m2 de superficie corporal, administrados en infusión i.v. (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás) 1 vez por semana durante 4 semanas.
Tratamiento de combinación por vía intravenosa
La dosis recomendada de MabThera/Rituxan i.v. (R i.v.) en combinación con cualquier quimioterapia es de 375 mg/m2 de superficie corporal por ciclo, durante un total de:
- ciclos de R i.v. con CVP (21 días/ciclo);
- ciclos de R i.v. con MCP (28 días/ciclo);
- ciclos de R i.v. con CHOP (21 días/ciclo); 6 ciclos si se logra una remisión completa al cabo de 4 ciclos;
- 6 ciclos de R i.v. con CHVP-interferón (21 días/ciclo).
MabThera/Rituxan i.v. debe administrarse el día 1 de cada ciclo de quimioterapia después de la administración i.v. del componente glucocorticoide de la quimioterapia, si fuera pertinente.
Infusiones intravenosas posteriores alternativas de 90 minutos
Los pacientes que no sufran ningún evento adverso de grado 3 o 4 relacionado con la infusión en el ciclo 1 pueden recibir una infusión alternativa de 90 minutos en el ciclo 2. La infusión alternativa puede iniciarse a una velocidad de administración del 20 % de la dosis total en los 30 primeros minutos y el 80 % restante en los 60 minutos siguientes, con un tiempo de infusión total de 90 minutos. Los pacientes que toleren los 90 primeros minutos de la infusión de MabThera/Rituxan i.v. (ciclo 2) pueden seguir recibiendo las infusiones posteriores de MabThera/Rituxan i.v. de 90 minutos durante el resto del régimen de tratamiento (hasta el ciclo 6 o el ciclo 8). Los pacientes que padezcan una enfermedad cardiovascular clínicamente significativa o cuya cifra de linfocitos circulantes sea >5000/mm3 antes del ciclo 2 no deben recibir la infusión de 90 minutos (ver Reacciones adversas, Ensayos clínicos y Ensayos clínicos/Eficacia).
Retratamiento después de la recidiva
Los pacientes que hayan respondido inicialmente a MabThera/Rituxan i.v. pueden recibir MabThera/Rituxan i.v. en una dosis de 375 mg/m2 de superficie corporal, administrada en infusión i.v. 1 vez por semana durante 4 semanas (ver Ensayos clínicos/Eficacia, Retratamiento, 4 dosis a intervalos semanales).
Terapia de mantenimiento
Los pacientes que no han sido tratados previamente pueden recibir, tras la respuesta al tratamiento de inducción, terapia de mantenimiento con MabThera/Rituxan i.v. en dosis de 375 mg/m2 de superficie corporal 1 vez cada 2 meses hasta la progresión de la enfermedad o durante un periodo máximo de 2 años (12 infusiones en total).
Los pacientes con enfermedad recidivante o resistente al tratamiento pueden recibir, después de la respuesta al tratamiento de inducción, terapia de mantenimiento con MabThera/Rituxan i.v. en dosis de 375 mg/m2 de superficie corporal una vez cada 3 meses hasta la progresión de la enfermedad o durante un periodo máximo de 2 años (8 infusiones en total).
Formulación subcutánea (1400 mg)
Todos los pacientes deben recibir siempre la primera dosis de MabThera/Rituxan por vía i.v. Durante el primer ciclo, el paciente corre el mayor riesgo de sufrir una reacción relacionada con la infusión o la administración. Comenzar el tratamiento con la infusión i.v. de MabThera/Rituxan permite el manejo de las reacciones relacionadas con la infusión o la administración reduciendo la velocidad de infusión o deteniendo la infusión (ver Advertencias y precauciones). La formulación s.c. sólo se debe administrar en el segundo ciclo o en ciclos posteriores (ver los subapartados Primera adminis-tración: formulación intravenosa y Administraciones posteriores: formulación subcutánea, más adelante).
Primera administración: formulación intravenosa.
La primera administración de MabThera/Rituxan debe hacerse siempre mediante infusión i.v. en una dosis de 375 mg/m2 de superficie corporal (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás).
Administraciones posteriores: formulación subcutánea.
Los pacientes que no pueden recibir la dosis completa de MabThera/Rituxan i.v. en infusión, seguirán recibien-do MabThera/Rituxan i.v. en los ciclos posteriores hasta que se administre satisfactoriamente una dosis i.v. completa.
En los pacientes que pueden recibir la dosis completa de MabThera/Rituxan i.v. en infusión, la segunda dosis o las dosis posteriores de MabThera/Rituxan pueden administrarse por vía s.c. usando la formulación de MabThera/Rituxan s.c. (ver Advertencias y precauciones).
Tratamiento inicial:
Monoterapia por vía subcutánea
La dosis recomendada de MabThera/Rituxan s.c. usado como monoterapia en pacientes adultos es la inyección s.c. en una dosis fija de 1400 mg, independientemente de la superficie corporal del paciente, una vez por semana durante 3 semanas, tras haber recibido MabThera/Rituxan i.v. en la semana 1 (1.ª semana con MabThera /Rituxan i.v. y luego 3 semanas con MabThera/Rituxan s.c.: 4 semanas en total).
Tratamiento de combinación por vía subcutánea
MabThera/Rituxan s.c. se administrará el día 0 o el día 1 de cada ciclo de quimioterapia después de la administración del glucocorticoide de la quimioterapia, si fuera pertinente.
La dosis recomendada en combinación con cualquier quimioterapia es de 375 mg/m2 de superficie corporal de MabThera/Rituxan i.v. (R i.v.) en el primer ciclo, y posteriormente inyección s.c. de MabThera/Rituxan (R s.c.) en una dosis fija de 1400 mg, independientemente de la superficie corporal del paciente.
- Primer ciclo de R i.v. con CVP + 7 ciclos de R s.c. con CVP (21 días/ciclo)
- Primer ciclo de R i.v. con MCP + 7 ciclos de R s.c. con MCP (28 días/ciclo)
- Primer ciclo de R i.v. con CHOP + 7 ciclos de R s.c. con CHOP (21 días/ciclo); o un total de 6 ciclos (primer ciclo con R i.v. y luego 5 ciclos con R s.c.) si se logra una remisión completa al cabo de 4 ciclos
- Primer ciclo de R i.v. con CHVP-interferón + 5 ciclos de R s.c. con CHVP-interferón (21 días/ciclo).
Retratamiento después de la recidiva
Los pacientes que hayan respondido a MabThera/Rituxan i.v. o s.c. inicialmente pueden ser tratados de nuevo con MabThera/Rituxan s.c. en una dosis fija de 1400 mg, administrada en una inyección s.c. 1 vez por semana, después de una primera administración de MabThera/Rituxan en infusión i.v. en una dosis de 375 mg/m2 de superficie corporal (1.ª semana con R i.v., luego 3 semanas con R s.c.: 4 semanas en total) (ver Ensayos clínicos/Eficacia, Retratamiento, 4 dosis a intervalos semanales).
Terapia de mantenimiento
Los pacientes no tratados previamente pueden recibir, tras la respuesta al tratamiento de inducción, terapia de mantenimiento con MabThera/Rituxan s.c. en una dosis fija de 1400 mg 1 vez cada 2 meses hasta la progresión de la enfermedad o durante un periodo máximo de 2 años (12 administraciones en total).
Los pacientes que han sufrido una recidiva después del tratamiento de inducción o que no han respondido a él pueden recibir terapia de mantenimiento con MabThera/Rituxan s.c. en una dosis fija de 1400 mg 1 vez cada 3 meses hasta la progresión de la enfermedad o durante un periodo máximo de 2 años (8 administraciones en total).
Linfoma no hodgkiniano difuso de linfocitos B grandes
Formulación intravenosa
En pacientes con linfoma no hodgkiniano difuso de linfocitos B grandes, MabThera/Rituxan i.v. debe usarse en combinación con el régimen de quimioterapia CHOP (ciclofosfamida, doxorubicina, prednisona y vincristina). La dosis recomendada de MabThera/Rituxan i.v. es de 375 mg/m2 de superficie corporal, administrada el día 1 de cada ciclo de quimioterapia durante 8 ciclos, después de la administración i.v. del componente glucocorticoide de CHOP (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás).
Infusiones intravenosas posteriores alternativas de 90 minutos
Los pacientes que no sufran ningún evento adverso de grado 3 o 4 relacionado con la infusión en el ciclo 1 pueden recibir una infusión alternativa de 90 minutos en el ciclo 2. La infusión alternativa puede iniciarse a una velocidad de administración del 20 % de la dosis total en los 30 primeros minutos y el 80 % restante en los 60 minutos siguientes, con un tiempo de infusión total de 90 minutos. Los pacientes que toleren los 90 primeros minutos de la infusión de MabThera/Rituxan i.v. (ciclo 2) pueden seguir recibiendo las infusiones posteriores de MabThera/Rituxan i.v. de 90 minutos durante el resto del régimen de tratamiento (hasta el ciclo 6 o el ciclo 8). Los pacientes que padezcan una enfermedad cardiovascular clínicamente significativa o cuya cifra de linfocitos circulantes sea >5000/mm3 antes del ciclo 2 no deben recibir la infusión de 90 minutos (ver Reacciones adversas, Ensayos clínicos y Ensayos clínicos/Eficacia).
Formulación subcutánea (1400 mg)
Todos los pacientes deben recibir siempre la primera dosis de MabThera/Rituxan por vía i.v. Durante el primer ciclo, el paciente corre el mayor riesgo de sufrir una reacción relacionada con la infusión o la administración. Comenzar el tratamiento con la infusión i.v. de MabThera/Rituxan permite el manejo de las reacciones relacionadas con la infusión o la administración reduciendo la velocidad de infusión o deteniendo la infusión (ver Advertencias y precauciones). La formulación s.c. sólo se administrará en el segundo ciclo o en ciclos posteriores (ver los subapartados Primera administración: formulación intravenosa y Administraciones posteriores: formulación subcutánea, más adelante).
En pacientes con LNH difuso de linfocitos B grandes, MabThera/Rituxan s.c. 1400 mg debe usarse en combinación con el régimen de quimioterapia CHOP (ciclofosfamida, doxorubicina, prednisona y vincristina).
Primera administración: formulación intravenosa
La primera administración de MabThera/Rituxan debe hacerse siempre mediante infusión i.v. en una dosis de 375 mg/m2 de superficie corporal (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás).
Administraciones posteriores: formulación subcutánea
Los pacientes que no pueden recibir la dosis completa de MabThera/Rituxan i.v. en infusión, seguirán recibien-do MabThera/Rituxan i.v. en los ciclos posteriores hasta que se administre satisfactoriamente una dosis i.v. completa.
En los pacientes que pueden recibir la dosis completa de MabThera/Rituxan i.v. en infusión, la segunda dosis o las dosis posteriores de MabThera/Rituxan pueden administrarse por vía s.c. usando la formulación de MabThera/Rituxan s.c. (ver Advertencias y precauciones).
La dosis recomendada de MabThera/Rituxan s.c. es una dosis fija de 1400 mg, independientemente de la superficie corporal del paciente, administrada el día 1 de cada ciclo de quimioterapia durante 8 ciclos (primer ciclo de R i.v. con CHOP + 7 ciclos de R s.c. con CHOP; 8 ciclos en total) después de la administración i.v. del componente glucocorticoide de CHOP.
Leucemia linfocítica crónica
A fin de aminorar el riesgo de síndrome de lisis tumoral en los pacientes con LLC, se recomienda la profilaxis con una hidratación adecuada y la administración de uricostáticos, que comenzará 48 antes de iniciar el tratamiento. En los pacientes con LLC cuya cifra de linfocitos sea >25 ×109/l, se recomienda administrar prednisona o prednisolona, en dosis de 100 mg i.v., poco antes de la administración de MabThera/Rituxan, para reducir la incidencia y la gravedad de las reacciones agudas a la infusión y el síndrome de liberación de citosinas.
Formulación intravenosa
La dosis recomendada de MabThera/Rituxan i.v. en combinación con quimioterapia en pacientes sin trata-miento previo y pacientes con LLC recidivante o resistente al tratamiento es de 375 mg/m2 de superficie corporal administrados el día 1 del primer ciclo de tratamiento, seguidos por 500 mg/m2 de superficie corporal administrados el día 1 de cada ciclo posterior, durante 6 ciclos en total (ver Ensayos clínicos/Eficacia). La quimioterapia debe administrarse después de la infusión de MabThera/Rituxan i.v. (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás).
Formulación subcutánea (1600 mg)
Todos los pacientes deben recibir siempre la primera dosis de MabThera/Rituxan por vía i.v. Durante el primer ciclo, el paciente corre el mayor riesgo de sufrir una reacción relacionada con la infusión o la administración. Comenzar el tratamiento con la infusión i.v. de MabThera/Rituxan permite el manejo de las reacciones relacionadas con la infusión o la administración reduciendo la velocidad de infusión o deteniendo la infusión (ver Advertencias y precauciones). La formulación s.c. sólo se administrará en el segundo ciclo o en los ciclos posteriores (verlos subapartados Primera adminis-tración: formulación intravenosa y Administraciones posteriores: formulación subcutánea, más adelante).
Primera administración: formulación intravenosa
La primera administración de MabThera/Rituxan i.v. debe hacerse siempre mediante infusión i.v. en una dosis de 375 mg/m2 de superficie corporal (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás).
Administraciones posteriores: formulación subcutánea
Los pacientes que no pueden recibir la dosis completa de MabThera/Rituxan i.v., seguirán recibiendo MabThera/Rituxan i.v. en los ciclos posteriores hasta que se administre satisfactoriamente una dosis i.v. completa.
En los pacientes que pueden recibir la dosis completa de MabThera/Rituxan i.v. en infusión, la segunda dosis o las dosis posteriores de MabThera/Rituxan pueden administrarse por vía s.c. usando la formulación de MabThera/Rituxan s.c. (ver Advertencias y precauciones).
La dosis recomendada de MabThera/Rituxan s.c. es una dosis fija de 1600 mg, independientemente de la superficie corporal del paciente, administrada el día 1 de cada ciclo de quimioterapia durante 5 ciclos (primer ciclo de R i.v. con CHOP + 5 ciclos de R s.c.; 6 ciclos en total) después de la administración i.v. del componente glucocorticoide de CHOP.
Artritis reumatoide
Formulación intravenosa solamente
Se administrará premedicación con glucocorticoides para reducir la frecuencia y la gravedad de las reacciones relacionadas con la infusión. Los pacientes deben recibir 100 mg de metilprednisolona por vía i.v., cuya administración concluirá 30 minutos antes de cada infusión de MabThera/Rituxan i.v. (ver Advertencias y precauciones).
Un ciclo de MabThera/Rituxan i.v. consta de 2 infusiones i.v. de 1000 mg. La dosis recomendada de MabThera/Rituxan es de 1000 mg en infusión i.v., seguida, 2 semanas más tarde, por la segunda infusión i.v. de 1000 mg (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás).
La necesidad de administrar más ciclos se evaluará 24 semanas después del ciclo anterior; el retratamiento se administrará considerando la enfermedad residual o si la actividad de la enfermedad vuelve a un nivel superior a un DAS28-ESR de 2,6 (tratamiento hasta la remisión) (verEstudios clínicos/Eficacia, Estudios en la artritis reumatoide). Los pacientes pueden recibir ciclos adicionales no antes de que hayan transcurrido 16 semanas desde el ciclo anterior.
Infusiones alternativas posteriores de 120 minutos con la concentración de 4 mg/ml en un volumen de 250 ml: si en la infusión anterior administrada según la pauta original los pacientes no sufrieron ninguna reacción adversa grave relacionada con la infusión, se puede administrar la infusión durante 120 minutos en las infusiones posteriores. Se comienza a una velocidad de 250 mg/h durante los 30 primeros minutos y se continúa con 600 mg/h en los 90 minutos siguientes. Si la infusión de 120 minutos se tolera, puede usarse la misma velocidad de infusión alternativa de 120 minutos en las infusiones y los ciclos posteriores.
A los pacientes con una enfermedad cardiovascular clínicamente importante, incluidas las arritmias o reacciones a la infusión previas graves a cualquier biomedicamento o a MabThera/Rituxan, no se les debe administrar la infusión de 120 minutos.
Granulomatosis con poliangitis (de Wegener) (GPA) y poliangitis microscópica (PAM)
Formulación intravenosa solamente
La dosis recomendada de MabThera/Rituxan i.v. para el tratamiento de la GPA y la PAM es de 375 mg/m2 de superficie corporal, administrados en infusión i.v. (ver el subapartado Velocidad de infusión de la formulación intravenosa, más atrás) 1 vez por semana durante 4 semanas.
Se recomienda administrar metilprednisolona en dosis de 1000 mg i.v. al día durante 1-3 días, en combinación con MabThera/Rituxan i.v., a fin de tratar los síntomas de vasculitis grave; a continuación se administrará prednisona oral, en dosis de 1 mg/kg/día (no se deben superar los 80 mg/día, y se reducirá progresivamente la dosis tan pronto como sea posible desde el punto de vista clínico), durante el tratamiento con MabThera/Rituxan i.v. y después del mismo.
Se recomienda la profilaxis de la neumonía por Pneumocystis jiroveci en pacientes con GPA y PAM durante el tratamiento con MabThera/Rituxan i.v. y después del mismo, según proceda.
USO EN POBLACIONES ESPECIALES
Embarazo
Formulaciones intravenosa y subcutánea
Se sabe que las inmunoglobulinas IgG atraviesan la barrera placentaria.
Los estudios de toxicidad en el desarrollo realizados en el macaco no han revelado ningún indicio de embriotoxicidad intrauterina. En crías recién nacidas cuyas progenitoras fueron expuestas a MabThera/Rituxan, se observó una depleción de poblaciones de linfocitos B durante la fase posnatal.
No se han estudiado en ensayos clínicos las cifras de linfocitos B en neonatos humanos tras la exposición materna a MabThera/Rituxan. Aunque no hay datos adecuados y bien controlados de estudios en embarazadas, se han notificado casos de depleción transitoria de linfocitos B y linfocitopenia en algunos lactantes cuyas madres estuvieron expuestas al rituximab durante el embarazo. Por ello, MabThera/Rituxan no debe administrarse a mujeres embarazadas, salvo que los posibles beneficios superen a los riesgos.
Las mujeres en edad de concebir deben utilizar métodos anticonceptivos eficaces durante el tratamiento con MabThera/Rituxan y durante 12 meses después de su conclusión.
Formulación subcutánea
La formulación s.c. contiene hialuronidasa humana recombinante (rHuPH20) (ver Composición cualitativa y cuantitativa). Estudios farmacocinéticos y de toxicología realizados en animales evidencian una reducción del peso fetal y un aumento del número de resorciones después de la inyección de rHuPH20 cuando la exposición sistémica materna es comparable a la que se alcanza después de la administración accidental de una inyección i.v. rápida de un vial único de la formulación s.c. de MabThera/Rituxan en el ser humano, basándose en las presuposiciones más conservadoras posibles (ver Datos preclínicos sobre seguridad, Formulación subcutánea).
Para reducir el riesgo adicional de toxicidad embriofetal debido a la exposición a rHuPH20, las pacientes que se queden embarazadas mientras reciben tratamiento con MabThera/Rituxan s.c. suspenderán el tratamiento con dicha formulación.
Parto
Sin texto.
Lactancia
No se sabe si el rituximab se excreta en la leche humana. No obstante, teniendo en cuenta que la IgG materna pasa a la leche, MabThera/Rituxan no debe administrarse a mujeres lactantes.
Uso en pediatría
No se han establecido la seguridad ni la eficacia de MabThera/Rituxan en pacientes pediátricos. Se han observado casos de hipogammaglobulinemia en pacientes pediátricos tratados con MabThera/Rituxan, algunos de los cuales fueron graves y requirieron tratamiento sustitutivo con inmunoglobulinas a largo plazo. No se conocen las consecuencias de la depleción prolongada de linfocitos B en pacientes pediátricos.
Uso en geriatría
Sin texto.
Insuficiencia renal
Sin texto.
Insuficiencia hepática
Sin texto.
VÍA DE ADMINISTRACIÓN
Formulación intravenosa: infusión intravenosa.
Formulación subcutánea: inyección subcutánea.
Declaración de esterilidad/radiactividad
Estéril.
SOBREDOSIS
Formulaciones intravenosa y subcutánea
La limitada experiencia con dosis superiores a las dosis i.v. aprobadas de MabThera/Rituxan i.v. procede de ensayos clínicos en el ser humano. La mayor dosis i.v. examinada en el ser humano hasta la fecha es de 5000 mg (2250 mg/m2), que se administró en un estudio de aumento de la dosis en pacientes con LLC. No se identificó ningún problema de seguridad adicional. En caso de sobredosis, se interrumpirá de inmediato la infusión y se vigilará estrechamente al paciente.
A 3 pacientes del estudio de MabThera/Rituxan s.c. SABRINA (BO22334) se les administró involuntaria-mente la formulación s.c. por vía i.v. hasta una dosis máxima de rituximab de 2780 mg, sin observarse efectos secundarios. En caso de sobredosis o de error de medicación con MabThera/Rituxan s.c., se debe someter al paciente a estrecha vigilancia.
Se tendrá en cuenta la necesidad de realizar regularmente hemogramas y el riesgo elevado de infecciones cuando el paciente presenta una depleción de los linfocitos B.
DESCRIPCIÓN
Clase terapéutica o farmacológica del fármaco
Antineoplásico. Código ATC: L01XC02.
PRESENTACIONES COMERCIALES
Mabthera 100 mg/10 ml:
Caja x 2 viales x 100 mg/10 ml (concentrado de solución para infusión) + prospecto.
Caja x 4 viales x 100 mg/10 ml (concentrado de solución para infusión) + prospecto
Mabthera 500 mg/50 ml:
Caja x 1 vial x 500 mg/50 ml (concentrado de solución para infusión) + prospecto.
Caja x 2 viales x 500 mg/50 ml (concentrado de solución para infusión) + prospecto.
Mabthera 1400 mg/11.7 ml
Caja x 1 vial x 1400 mg/11.7 ml (solución para inyección subcutánea) + inserto.
Información de julio del 2016.
Medicamento biológico innovador
MabThera I.V. 100 mg/10 ml y MabThera 500 mg/50 ml.
Fabricante principal
F. Hoffmann-La Roche S.A. Basilea, Suiza
Fabricante alterno
Para F. Hoffmann-La Roche S.A. Basilea, Suiza por Roche Diagnostics GmbH Mannheim, Alemania.
MabThera S.C 1400mg/11.7ml
Fabricado para F. Hoffmann-La Roche Ltd. Basilea, Suiza por F. Hoffmann-La Roche Ltd. Kaiseraugst, Suiza, bajo licencia de F. Hoffmann-La Roche Ltd. Basilea, Suiza.
Medicamento: manténgase fuera del alcance de los niños.
ROCHE ECUADOR S.A.
Casilla 1711- 06185 CCI
Quito - Ecuador

