KOGENATE
FACTOR VIII (FACTOR ANTIHEMOFÍLICO A)
Polvo liofilizado para solución inyectable
Vial , 250 Unidades Internacionales
Vial , 500 Unidades Internacionales
Vial , 1000 Unidades Internacionales
COMPOSICIÓN:
Composición cualitativa y cuantitativa: La proteína octocog alfa (en lo sucesivo, rFVIII) es un concentrado deshidratado estéril, estable, purificado, no pirógeno, que se fabrica mediante tecnología de ADN recombinante.KOGENATE Bayer viene en forma de un vial de polvo liofilizado que contiene lo siguiente: 250, 500, 1000, 2000, 3000 UI potencia nominal.
Para ver la lista completa de excipientes, consulte la sección “Lista de excipientes”.
Lista de excipientes: Sacarosa, Histidina, Glicina, cloruro de sodio, cloruro de calcio y polisorbato KOGENATE Bayer es un polvo liofilizado para inyección que viene con un diluyente (agua para inyección).
Forma farmacéutica: KOGENATE Bayer es un polvo liofilizado inyectable que viene con un diluyente.
INDICACIONES TERAPÉUTICAS:
Indicaciones: Tratamiento de la hemofilia A y profilaxis del sangrado. Tratamiento profiláctico de rutina para prevenir o reducir la frecuencia de episodios de sangrado en adultos con hemofilia A. Tratamiento profiláctico de pacientes pediátricos para reducir la frecuencia de episodios de sangrado espontáneos en la hemofilia A y disminuir considerablemente el riesgo de daño articular en comparación con el tratamiento episódico.10 Tratamiento por inducción de tolerancia inmunitaria (ITI) de pacientes con inhibidores.
KOGENATE Bayer no contiene factor de von Willebrand y no está indicado para tratar la enfermedad de von Willebrand.
MECANISMO DE ACCIÓN:
Propiedades farmacológicas
Propiedades farmacodinámicas
Grupo farmacoterapéutico: ATC-Código: B02B D0252.Mecanismo de acción: KOGENATE Bayer constituye una manera de reemplazar temporalmente el factor de coagulación VIII que falta para lograr una hemostasia efectiva.
Efectos farmacodinámicos: El tiempo parcial de tromboplastina activada (activated partial thromboplastin time, aPTT) se prolonga en las personas con hemofilia. La determinación del aPTT es un ensayo convencional in vitro para la actividad biológica del factor VIII. El tratamiento con rFVIII normaliza el aPTT durante el período de dosificación efectiva.
Estudios Clínicos
Infusión continua: En un estudio clínico realizado con pacientes adultos con hemofilia A que se sometieron a una cirugía mayor se demostró que KOGENATE Bayer se puede usar en infusión continua durante las cirugías (en forma pre-y postoperatoria). En este estudio se usó heparina para prevenir la tromboflebitis en el lugar de infusión, tal como se hace con cualquier otra infusión intravenosa de largo plazo.
Tolerancia inmunitaria: Los datos de dos estudios han demostrado que KOGENATE Bayer se puede usar para inducir tolerancia inmunitaria en pacientes con hemofilia A que han formado inhibidores del factor VIII. Una vez establecida la tolerancia inmunitaria, es posible prevenir o controlar nuevamente los sangrados con factor VIII, y el paciente puede continuar con el tratamiento profiláctico de factor VIII como terapia de mantenimiento.
Profilaxis en adultos: Un estudio clínico en curso de 3 años de duración, multicéntrico, de etiqueta abierta, con grupos paralelos, prospectivo, aleatorizado, controlado, sobre el efecto de la profilaxis regular con KOGENATE Bayer en comparación con el uso a demanda, sobre la frecuencia de sangrado y las complicaciones articulares en adultos y adolescentes incluyó 84 PTPs con hemofilia A grave (nivel de FVIII < 1 UI/dl), de 15 a 50 años de edad. Los pacientes presentaban una buena similitud al inicio en cuanto a las características demográficas y de la enfermedad. La cantidad mediana de sangrados en el año antes de la inscripción era de 18. Los pacientes fueron aleatorizados en una relación 1:1 a recibir un régimen profiláctico estándar (25 UI/kg tres veces por semana) o uso a demanda de KOGENATE Bayer. Se permitió el aumento escalonado de la dosis profiláctica de a 5 UI/kg/infusión después de los años 1 y 2, hasta un máximo de 35 UI/kg/infusión. El criterio de valoración primario, frecuencia de sangrado, se analizó en la población según la intención de tratar tras un período de seguimiento con una mediana de alrededor de 1.4 años. La mediana de la tasa de sangrado anual en el grupo con tratamiento a demanda fue de 33 sangrados/sujeto/año en comparación con 0 sangrados/sujeto/año en el grupo con tratamiento profiláctico (p < 0.0001). La mayoría de los sangrados ocurrió en las articulaciones: hubo 24 sangrados articulares/sujeto/año en el grupo con tratamiento a demanda en comparación con 0 sangrados articulares/sujeto/año en el grupo con tratamiento profiláctico. Hubo veintidós (22) de 42 (52%) sujetos con tratamiento profiláctico que no experimentaron sangrado, y 12 de 42 (29%) sujetos con tratamiento profiláctico que experimentaron solamente 1-2 sangrados durante el período de seguimiento. Hubo una reducción del 94% en la media de la tasa de sangrado anual: 36.9 sangrados/sujeto/año en el grupo con tratamiento a demanda en comparación con 2.2 sangrados/sujeto/año en el grupo con tratamiento profiláctico. Hubo una reducción del 93% en la media de la tasa de sangrado articular anual: 30 sangrados/sujeto/año en el grupo con tratamiento a demanda en comparación con 2 sangrados/sujeto/año en el grupo con tratamiento profiláctico. El tratamiento profiláctico comparado con el tratamiento a demanda con KOGENATE Bayer tuvo como resultado reducciones importantes en los sangrados, independientemente de la característica inicial examinada, incluida la edad, el antecedente de sangrado y la presencia o ausencia de articulaciones con sangrado recurrente (Tabla 4). En particular, la mediana de la tasa de sangrado anual entre los pacientes con tratamiento a demanda >30 años de edad fue de 30.7, en comparación con 0 entre los pacientes con tratamiento profiláctico en el mismo grupo de edad. Además, la gravedad de sangrado autoinformada con mayor frecuencia entre los pacientes con tratamiento profiláctico fue el grado leve (44%), en comparación con el grado moderado (58%) en los pacientes con tratamiento a demanda (Tabla 5). Entre los pacientes con tratamiento profiláctico: la cantidad media de infusiones por dia/semana fue 2.8, y la mediana de la dosis por infusión profiláctica fue de 26 UI/kg.
No se detectaron inhibidores.
El perfil de eventos adversos fue congruente con la información que figura en la etiqueta actual.
|
Tabla 3. Sangrados anuales generales y por característica al inicio. |
|||||||
|
A demanda |
Profilaxis |
||||||
|
N |
Mediana |
% con 0 sangrados |
N |
Mediana |
% con 0 sangrados |
||
|
General |
42 |
33 |
2.4 |
42 |
0 |
52 |
|
|
Edad |
<18 |
1 |
21.7 |
0 |
2 |
0 |
100 |
|
18 a <30 |
21 |
43.8 |
4.8 |
19 |
0.7 |
42.1 |
|
|
≥30 |
20 |
30.7 |
0 |
21 |
0 |
57.1 |
|
|
Cantidad de sangrados en el año previo |
<15 ≥15 |
10 32 |
24.4 34.6 |
0 2.9 |
14 27 |
0 0.7 |
69.2 48 |
|
Presencia de articulaciones con sangrado recurrente |
No |
11 |
17.6 |
9.1 |
14 |
0 |
57.1 |
|
Sí |
31 |
43.8 |
0 |
28 |
0.3 |
50 |
|
|
Tabla 4. Gravedad del sangrado |
||
|
A demanda |
Profilaxis |
|
|
Gravedad del sangrado |
||
|
Cantidad de sangrados |
2363 (100%) |
196 (100%) |
|
Faltantes |
150 (6.3%) |
24 (12.2%) |
|
Leves |
396 (16.8%) |
87 (44.4%) |
|
Moderados |
1364 (57.7%) |
71 (36.2%) |
|
Graves |
453 (19.2%) |
14 (7.1%) |
Propiedades farmacocinéticas
Absorción: No procede. KOGENATE Bayer se administra directamente en el torrente sanguíneo por inyección intravenosa (IV).
Distribución: Aunque no se han realizado estudios de distribución específicos, tras la administración de KOGENATE Bayer, la actividad pico del factor VIII disminuye siguiendo una declinación exponencial de dos fases. Esta es similar a la del factor VIII derivado del plasma. KOGENATE Bayer se une a su portador proteínico natural, el factor de von Willebrand (von Willebrand factor, vWF) y queda confinado principalmente en el espacio vascular.Metabolismo: KOGENATE Bayer es metabolizado mientras produce su actividad biológica durante la activación de la cascada coagulatoria.
Eliminación/Excreción: Después de la administración de rFVIII, la actividad pico de factor VIII disminuye mediante una declinación exponencial de dos fases, con una vida media terminal de unas 15 horas.En esto es similar al factor VIII derivado del plasma, que tiene una vida media terminal media de aproximadamente 13 horas. Los datos de vida media de KOGENATE Bayer no cambiaron después de 24 semanas de tratamiento exclusivo, lo que indica eficacia continua sin ninguna evidencia de inhibición del factor VIII.
Datos preclínicos sobre seguridad: La administración de dosis varias veces superiores a la dosis clínica recomendada (respecto del peso corporal) no tuvo ningún efecto tóxico agudo o subagudo para KOGENATE Bayer en animales de laboratorio (ratones, ratas, conejos y perros).No se realizaron estudios específicos con administración repetida de KOGENATE Bayer, como toxicidad sobre la reproducción, toxicidad crónica y carcinogenicidad, debido a la respuesta inmunitaria a las proteínas heterólogas en todas las especies de mamíferos no humanos.
La evaluación in vitro del potencial mutagénico de KOGENATE Bayer de primera generación no logró demostrar que hubieran mutaciones inversas, ni aberraciones cromosómicas, a dosis considerablemente mayores que la dosis clínica máxima esperada. La evaluación in vivo de KOGENATE Bayer en animales con dosis que oscilaron entre 10 y 40 veces la dosis clínica máxima esperada, también indicó que KOGENATE Bayer no tiene potencial mutagénico.
CONTRAINDICACIONES:
Intolerancia o reacciones alérgicas conocidas a los ingredientes del preparado.
REACCIONES ADVERSAS:
Experiencia clínica: Durante los estudios clínicos realizados en pacientes tratados previamente (PTP), se informó de 451 eventos adversos (EA) ocurridos durante 24,936 infusiones (1.8%). Solamente 24 eventos, observados en 13 pacientes, se consideraron al menos remotamente relacionados con la administración de KOGENATE Bayer. La incidencia en el estudio de eventos adversos relacionados al menos remotamente con el medicamento fue de 0.1%, respecto de la cantidad de infusiones administradas.
|
Tabla 6. Eventos adversos en PTPs |
||||
|
Eventos adversos en PTPs que se relacionan definitivamente con KOGENATE Bayer, productos de factor VIII o proteínas de administración parenteral. |
Cantidad total de pacientes: 73 Cantidad de pacientes con AE (%). |
Cantidad total de infusiones: 24,936 Eventos adversos por infusión (%). |
||
|
SOC principal del MedDRA. |
Entidad médica. |
Término preferido. |
||
|
Trastornos generales y condiciones del lugar de la administración. |
Reacciones en el lugar de infusión. |
Reacciones en el lugar de infusión. |
3 (4.1) |
0.01 |
|
Trastornos de la piel y de los tejidos subcutáneos. |
Reacciones por hipersensibilidad relacionadas con la piel. |
Sarpullido, picazón. |
6 (8.2) |
0.02 |
En estudios clínicos con 73 PTPs (definidos como pacientes con más de 100 días de exposición), a los que se dio seguimiento durante cuatro años, no se observó nueva formación de inhibidores.
En estudios clínicos con PUPs y MTPs pediátricos, se recibieron informes de 726 eventos adversos ocurridos en el transcurso de 9,389 infusiones (7.7%). Entre estos se incluyó la complicación esperada de formación de inhibidores en 9 pacientes.
|
Tabla 7. Eventos adversos en PUPs y MTPs |
||||
|
Eventos adversos en PUPs/MTPs que se relacionan definitivamente con KOGENATE Bayer, productos de factor VIII o proteínas de administración parenteral |
Cantidad total de pacientes: 61 Cantidad de pacientes con AE (%) |
Cantidad total de infusiones: 9,389 Eventos adversos por infusión (%) |
||
|
SOC principal del MedDRA |
Entidad médica |
Término preferido |
||
|
Trastornos de la sangre y el sistema linfático. |
Formación de inhibidores. |
Inhibición del factor VIII. |
9 (15)* |
N.A. |
|
Trastornos generales y condiciones del lugar de la administración. |
Reacciones en el lugar de infusión. |
Reacciones en el lugar de infusión. |
4 (6.6) |
0.04 |
|
Trastornos de la piel y de los tejidos subcutáneos. |
Reacciones por hipersensibilidad relacionadas con la piel. |
Sarpullido, picazón. |
10 (16.4) |
0.01 |
|
* El denominador de los nuevos inhibidores es N=60, puesto que un paciente tenía un inhibidor preexistente. |
||||
Experiencia posterior a la comercialización: Las siguientes reacciones adversas se han identificado durante el uso posterior a la aprobación de KOGENATE Bayer. Puesto que el informe de tales reacciones es voluntario y proviene de una población de tamaño incierto, no siempre es posible estimar de modo confiable su frecuencia, ni establecer una relación causal con la exposición al medicamento. Entre los pacientes tratados con KOGENATE Bayer se ha informado de casos infrecuentes de reacciones alérgicas o por hipersensibilidad graves (entre otras, hinchazón facial, rubor, erupción cutánea, disminución de la presión sanguínea, náuseas, sarpullido, intranquilidad, falta de aire, taquicardia, sensación de opresión en el pecho, hormigueo, urticaria y vómito), sobre todo en pacientes de muy corta edad o que habían tenido reacciones previas a otros concentrados de factor VIII.
|
Tabla 8. Experiencia de eventos adversos posterior a la comercialización. |
||
|
Experiencia posterior a la comercialización |
||
|
SOC principal del MedDRA |
Entidad médica |
Término preferido |
|
Trastornos de la sangre y el sistema linfático. |
Formación de inhibidores. |
Inhibición del factor VIII. |
|
*Trastornos de la piel y del tejido subcutáneo. |
Reacciones por hipersensibilidad relacionadas con la piel. |
Picazón, urticaria, sarpullido. |
|
*Trastornos generales y condiciones en el lugar de administración. |
Reacción en el lugar de infusión. |
Reacción en el lugar de infusión. |
|
Reacción febril relacionada con la infusión. |
Pirexia. |
|
|
Trastornos del sistema inmunológico. |
Reacciones sistémicas por hipersensibilidad. |
Reacción anafiláctica por hipersensibilidad. |
|
Trastornos del sistema nervioso. |
Disgeusia. |
Disgeusia. |
|
*Es posible que un mismo paciente haya informado de más de un evento. |
||
Incompatibilidades: Este producto no se debe mezclar ni con otros medicamentos ni con solventes. Solamente se pueden usar los equipos de administración que se suministran, ya que el tratamiento puede fracasar debido a la adsorción del factor de coagulación VIII humano en las superficies internas de algunos equipos de infusión.
Bayer S. A.
Para mayor información, comuníquese con la
Dirección Médica de Bayer S. A.
Luxemburgo N34-359 y Av. Portugal esquina.
Edif. Cosmopolitan Parc, pisos 6 y 7
Quito-Ecuador.
Telf: (593) 2 3975200
informacionmedicaandina@bayer.com
farmacovigilancia.ecuador@bayer.com
www.andina.bayer.com
REACCIONES ADVERSAS:
Eventos adversos
Resumen del perfil de seguridad: La reacción adversa al medicamento que se informa con mayor frecuencia es la formación de anticuerpos neutralizantes (principalmente en pacientes no tratados previamente [previously untreated patients, PUPs] o mínimamente tratados [minimally treated patients, MTPs]).
Lista tabulada de reacciones adversas: Las reacciones adversas al medicamento se presentan dentro de cada agrupación por frecuencia y clases de órganos o sistemas. Los eventos que aparecen en cursivas se relacionan con la experiencia posterior a la comercialización del producto.
Muy frecuentes: ≥ 10%.
Frecuentes: ≥ 1% a < 10% (>1/100 a <1/10) .
Poco frecuentes: ≥ 0.1% a < 1% (>1/1,000 a <1/100). Infrecuentes: ≥ 0.01% a < 0.1% (>1/10,000 a <1/1,000).
|
Muy Frecuentes |
Frecuentes |
Poco frecuentes |
Infrecuentes |
Desconocida |
|
Trastornos de la sangre y el sistema linfático. |
||||
|
Desarrollo de Inhibidores al FVIII en PUPs/MTPs. |
Formación de inhibidores del factor VIII (en estudios de pacientes tratados previamente (previously treated patients, PTPs)). |
|||
|
Trastornos generales y condiciones del lugar de la administración. |
||||
|
Reacción en el lugar de infusión. |
||||
|
Reacción febril relacionada con la infusión. |
||||
|
Trastornos del sistema inmunológico. |
||||
|
Reacciones por hipersensibilidad relacionadas con la piel. |
||||
|
Reacciones por hipersensibilidad sistémicas (incluyendo anafilácticas). |
||||
|
Trastornos del sistema nervioso |
||||
|
Disgeusia. |
||||
Descripción de reacciones adversas selectas: En estudios clínicos, KOGENATE Bayer se ha usado para el tratamiento de episodios de sangrado en 60 PUPs y MTPs pediátricos (estos últimos se definen como pacientes con 4 o menos días de exposición). Nueve de los 60 PUPs/MTPs (15%) tratados con KOGENATE Bayer formaron inhibidores: En total, seis de los 60 (10%) tuvieron un título de más de 10 UB, y 3 de los 60 (5%) tuvieron un título inferior a 10 UB. La media de los días de exposición en el momento de detección del inhibidor en estos pacientes fue de 9 días (intervalo de 3 a 18 días). Para el tratamiento de pacientes que formaron inhibidores, consulte la Sección “Dosis y método de administración”.
Cuatro de los cinco pacientes que no habían llegado a 20 días de exposición al final del estudio, alcanzaron finalmente más de 20 días de exposición durante un seguimiento posterior al estudio y uno de ellos presentó un inhibidor de título bajo. El quinto paciente no pudo ser localizado para el seguimiento.
En estudios clínicos con 73 PTPs (definidos como pacientes con más de 100 días de exposición), a los que se dio seguimiento durante cuatro años, no se observó nueva formación de inhibidores.
En estudios extensos con KOGENATE Bayer posteriores al registro y en los cuales participaron más de 1000 pacientes, se observó lo siguiente: en los subgrupos de PUPs/MTPs (definidos como pacientes con menos de 20 días de exposición), menos de 11% tuvieron nueva formación de inhibidores. Menos de 0.2% de los PTPs tuvieron nueva formación de inhibidores.
Los registros disponibles, han reportado tasas de inhibidores en PUPs con Hemofilia A grave en un rango de 28 a 38% para los productos de FVIII.
Efectos sobre la capacidad de conducir y usar máquinas: No se han observado efectos sobre la capacidad para conducir vehículos o usar máquinas.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones con otros medicamentos y otras formas de interacción: No se sabe de ninguna interacción con otros medicamentos.
Interacciones farmacológicas: Aparte de las interacciones conocidas del factor VIII con otras proteínas de la coagulación, no se ha establecido ninguna otra interacción farmacológica.
Interacciones entre medicamento y alimentos: No se han establecido interacciones con alimentos.
Interacciones entre medicamento y productos a base de hierbas: No se han establecido interacciones con preparados a base de hierbas.
Interacciones entre medicamento y procedimientos de laboratorio: No se sabe de ninguna interacción con procedimientos de laboratorio.
RECOMENDACIONES:
Advertencias y precauciones especiales de empleo: Hipersensibilidad conocida a las proteínas de ratón o de hámster. Se debe alertar a los pacientes en cuanto a que la posible presencia de opresión en el pecho, mareos, hipotensión leve y náuseas durante la infusión podría ser un primer signo de hipersensibilidad y reacción anafiláctica. Se debe instituir tratamiento sintomático y terapia para la hipersensibilidad, según sea apropiado. Si se presenta una reacción alérgica o anafiláctica, la inyección/infusión se debe suspender de inmediato. En caso de choque, se deben observar los estándares médicos actuales para el tratamiento del mismo.31 La formación de anticuerpos neutralizantes en circulación contra el factor VIII puede presentarse durante el tratamiento de pacientes con hemofilia A. La formación de inhibidores es particularmente común entre los niños de corta edad con hemofilia grave durante los primeros años de tratamiento, o en los pacientes de cualquier edad que hayan recibido pocos tratamientos previos con factor VIII. No obstante, la formación de inhibidores puede ocurrir en cualquier momento durante el tratamiento de un paciente con hemofilia A. Los pacientes tratados con cualquier preparado de factor antihemofílico, incluso rFVIII, deben ser vigilados estrechamente en cuanto a la formación de anticuerpos contra KOGENATE Bayer, mediante observación clínica y pruebas de laboratorio apropiadas, según lo recomiende el centro de tratamiento de la hemofilia del paciente.
Los pacientes hemofílicos con factores de riesgo cardiovascular, pueden tener el mismo riesgo de desarrollar eventos cardiovasculares que los pacientes no hemofílicos, una vez que la coagulación ha sido normalizada con el tratamiento con FVIII.
Pueden llegar a observarse infecciones relacionadas con el catéter si la administración de KOGENATE Bayer se realiza mediante dispositivos de acceso venoso central (central venous access devices, CVADs). Estas infecciones no han sido asociadas con el producto en sí.
Embarazo y lactancia: Categoría de embarazo C en los Estados Unidos. No se sabe si KOGENATE Bayer podría causar lesiones fetales al ser administrado a una mujer embarazada o afectar la capacidad reproductiva.No se han realizado estudios de reproducción animal con rFVIII. No debe usarse KOGENATE Bayer durante el embarazo y la lactancia, a menos que los beneficios contrapesen claramente cualquier riesgo potencial.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Dosis y método de administración
Método de administración: KOGENATE Bayer se administra directamente en el torrente sanguíneo por inyección intravenosa (IV).
Pauta posológica: La cantidad de unidades de factor VIII administradas se expresa en unidades internacionales (UI), las cuales se relacionan con el estándar actual de la Organización Mundial de la Salud (OMS) para productos de factor VIII. La actividad de factor VIII en el plasma se expresa como un porcentaje (respecto al plasma humano normal) o en UI (relacionadas con el estándar internacional para el factor VIII en plasma). Una unidad internacional (UI) de actividad de factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma humano normal. El cálculo de la dosis de factor VIII necesaria se basa en el hecho empírico de que 1 UI de factor VIII por kilogramo de peso corporal aumenta la actividad de factor VIII en plasma en 1.5 % a 2.5 % respecto de la actividad normal.La dosificación y duración de la terapia de sustitución para lograr la hemostasia se deben personalizar en función de las necesidades del paciente (peso, gravedad del trastorno de la función hemostática, lugar y magnitud/gravedad del sangrado, título de inhibidores y nivel de factor VIII que se desea). El efecto clínico de rFVIII es el elemento más importante en la evaluación de la eficacia del tratamiento. Quizás deba administrarse más KOGENATE Bayer de lo que se calcule para lograr resultados clínicos satisfactorios. Si la dosis calculada no permite alcanzar las concentraciones de factor VIII esperadas o no es posible controlar el sangrado después de administrar la dosis calculada, debe sospecharse la presencia de un inhibidor circulante en el paciente. Es necesario corroborar la presencia de este y cuantificar la concentración del inhibidor mediante un análisis de laboratorio apropiado. Cuando está presente un inhibidor, el requisito posológico de KOGENATE Bayer es sumamente variable y la respuesta clínica es la única manera de determinar la dosis adecuada.
El aumento porcentual in vivo de la concentración de factor VIII se puede estimar multiplicando por 2% la dosis de KOGENATE Bayer por kilogramo de peso corporal (UI/kg).17 18 19 Cálculo 1:
Dosis necesaria (UI) = peso corporal (kg) x aumento deseado del factor VIII (% de lo normal) x 0.5 (UI/kg) Cálculo 2: Aumento esperado del factor VIII (% de lo normal) = (2%/UI/kg x UI administradas)/peso corporal (kg).
Use el cálculo anterior. Además, puede usar el enunciado general de la tabla a continuación para ver instrucciones adicionales sobre cómo calcular las dosis según la gravedad de la hemorragia.> La dosis única usual es de 10 a 30 UI/kg de peso corporal.
La dosis necesaria para lograr la hemostasia depende del tipo y la gravedad del episodio de sangrado.
|
Tabla 1. Dosis necesaria para lograr la hemostasia |
||
|
Episodio hemorrágico. |
Nivel de actividad de factor VIII en plasma terapéuticamente necesario. |
Dosis necesaria para mantener el nivel terapéutico en plasma. |
|
Hemorragia menor (superficial, sangrados iniciales, sangrados en las articulaciones). |
20-40%. |
10-20 UI/kg Repetir la dosis si persisten las evidencias de sangrado. |
|
Hemorragia moderada a mayor (sangrados musculares, sangrados en la cavidad oral, hemartrosis definidas, traumatismos conocidos). |
30-60% |
15-30 UI/kg Repetir una dosis 12 a 24 horas después si fuera necesario. |
|
Cirugía (procedimientos quirúrgicos menores). |
||
|
Hemorragia mayor a posiblemente mortal (sangrados intracraneales, intraabdominales o intratorácicos, sangrados gastrointestinales, sangrados en el sistema nervioso central, sangrado en los espacios retrofaríngeo o retroperitoneal, o en la vaina del músculo psoas ilíaco). |
80-100% |
Dosis inicial de 40-50 UI/kg Repetir la dosis de 20-25 UI/kg cada 8 a 12 horas. |
|
Fracturas Traumatismos en la cabeza. |
||
|
Cirugías (procedimientos quirúrgicos mayores). |
~100% |
a) Por infusiones en bolo Dosis preoperatoria de 50 UI/kg Verificar la actividad ~100% antes de la cirugía. Repetir según sea necesario luego de 6 a 12 horas inicialmente, y durante 10 a 14 días hasta que termine la cicatrización. b) Por infusión continua Aumente la actividad de factor VIII antes de la cirugía con una infusión en bolo inicial, seguida inmediatamente por infusión continua (en UI/h/kg), ajustada en función de la eliminación (o aclaración) diaria del paciente y de las concentraciones de factor VIII deseadas, durante 7 días por lo menos. |
Velocidad de administración: Según los datos de estudios clínicos con pacientes en edades de 0 a 68 años, la dosis completa se administra en un tiempo medio de 5 minutos.Se debe adaptar la velocidad de administración a la respuesta de cada paciente de modo individual.
El producto se debe administrar en menos de 3 horas después de la reconstitución.
Infusión continua: KOGENATE Bayer se puede infundir por infusión continua. La velocidad de infusión se debe calcular con base en la eliminación y en la concentración de factor VIII deseada. En un estudio clínico llevado a cabo en pacientes adultos con hemofilia A que se sometieron a una cirugía mayor, el intervalo de velocidades de infusión de KOGENATE Bayer fue de 0.2 a 3.6 ml/h. Ejemplo: en el caso de un paciente de 75 kg con una tasa de eliminación de 3 mL/h/kg, la velocidad de infusión inicial debería ser de 3 UI/h/kg para lograr una concentración de factor VIII de 100%. Para calcular los mL/hora, multiplique la velocidad de infusión en UI/h/kg por el peso corporal (kg)/concentración de la solución (UI/mL).
|
Tabla 2. Cálculo de la velocidad de infusión con base en la eliminación y en la concentración de factor VIII deseada. |
|||||
|
Concentración de factor VIII en plasma deseada. |
Velocidad de infusión, UI/kg. |
Velocidad de infusión para un paciente de 75 kg, mL/h. |
|||
|
Eliminación: 3 mL/h/kg |
Concentraciones de la solución de Kogenate Bayer |
||||
|
100 UI/mL |
200 UI/mL |
400 UI/mL |
|||
|
100% (1 UI/mL) |
3.0 |
2.25 |
1.125 |
0.56 |
|
|
60% (0.6 UI/mL) |
1.8 |
1.35 |
0.68 |
0.34 |
|
|
40% (0.4 UI/mL) |
1.2 |
0.9 |
0.45 |
0.225 |
|
Posiblemente se requieran velocidades de infusión más altas en afecciones con eliminación acelerada en los casos de sangrado mayor y de daños tisulares extensos durante las intervenciones quirúrgicas. Las velocidades de infusión subsiguientes se deben calcular con base en las concentraciones de factor VIII y la eliminación recalculada para cada día después de la cirugía con base en la ecuación: eliminación = velocidad de infusión/concentración real de factor VIII.
Durante la infusión continua es necesario cambiar las bolsas de infusión cada 24 horas.
Para el cálculo de la velocidad de infusión inicial, el valor de eliminación se puede obtener realizando una curva de declinación previa a la cirugía o a partir del valor promedio de la población (3.0 a 3.5 mL/h/kg), que luego se ajusta en forma apropiada. Velocidad de infusión (en UI/kg/h) = Eliminación (en mL/h/kg) x concentración de factor VIII deseada (en UI/mL) La estabilidad clínica e in vitro se ha demostrado mediante el uso de bombas ambulatorias con un depósito de cloruro de polivinilo (PVC). KOGENATE Bayer contiene una concentración baja de polisorbato 80 como excipiente, el cual, según se sabe, aumenta la velocidad de extracción del di-(2-etilhexil) ftalato (DEHP) de los materiales de PVC. Esto se debe tener en consideración en los casos de administración por infusión continua.
Inducción de tolerancia inmunitaria: Únicamente personas con experiencia en el tratamiento de pacientes con hemofilia A e inhibidores deberían iniciar y conducir la inducción de tolerancia inmunitaria (ITI). Si los pacientes forman inhibidores del factor VIII se debería considerar el tratamiento por ITI con KOGENATE Bayer en los casos pertinentes. En los casos con títulos de inhibidores >5 UB (unidades Bethesda), la dosis sugerida para la ITI oscila entre 50 UI/kg/día, tres veces por semana y 200 UI/kg/día todos los días. Se pueden usar dosis más altas, a discreción del médico tratante. Luego de una ITI con buenos resultados, la dosis de factor VIII se debe reducir y continuar como una terapia profiláctica de mantenimiento.
Información adicional sobre poblaciones especiales
Pacientes pediátricos: El uso de KOGENATE Bayer es apropiado para pacientes pediátricos. Se han realizado estudios de seguridad y eficacia en pacientes pediátricos menores de 4 años, no tratados previamente y mínimamente tratados.
Pacientes geriátricos: Los estudios clínicos con KOGENATE Bayer no han incluido cantidades suficientes de pacientes de 65 años o más, como para poder determinar si estos responden de modo distinto al de los pacientes más jóvenes. No obstante, en la experiencia clínica con KOGENATE Bayer y otros productos de factor VIII no se han identificado diferencias entre los pacientes jóvenes y de edad avanzada. Al igual que cualquier otro paciente que reciba rFVIII, la selección de la dosis para un paciente de edad avanzada se debe personalizar.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis: No se tienen informes de síntomas de sobredosis.
PRESENTACIÓN:
Naturaleza y contenido del envase: Se acuerdo con la documentación TRD/CMC. Opción 1 (“Clásico”: sobresello de aluminio y vial de diluyente). Presentación del empaque: KOGENATE Bayer viene en los siguientes viales de uso único. Se provee un volumen adecuado de agua estéril para inyección (Farmacopea Estadounidense [USP]), una aguja de transferencia estéril de doble punta, una aguja filtro estéril, y un equipo de administración estéril. Opción 2 (BIO-SET: BIO-SET y jeringa de diluyente). Presentación del empaque: KOGENATE Bayer viene en los siguientes viales de uso único. También se provee una jeringa precargada de diluyente que contiene agua estéril (cumple los requisitos químicos de la USP para el agua estéril para inyección, con excepción del pH) para reconstitución, un equipo estéril de administración, dos hisopos impregnados en alcohol, un apósito estéril, y una torunda de algodón estéril.
Opción 3 (adaptador para viales/Medimop: Adaptador para viales y jeringa de diluyente) Presentación del empaque: KOGENATE Bayer con adaptador para viales viene como frascos de uso único.76 También se provee una jeringa precargada de diluyente que contiene agua estéril (cumple los requisitos químicos de la USP para el agua estéril para inyección, con excepción del pH) para reconstitución, un adaptador para viales estéril, y un equipo estéril de administración.
Opción 4 (Mix2Vial: Adaptador para viales y vial de diluyente). Presentación del empaque: KOGENATE Bayer viene en los siguientes frascos de uso único. Se provee un volumen adecuado de agua estéril para inyección (USP) y un equipo de filtro para transferencia Mix2Vial™.
|
Tabla 5. Potencia y opciones de diluyentes. |
|
|
Actividad aproximada de FVIII (UI) |
Diluyente (ml) |
|
250 |
2.5 |
|
500 |
2.5 |
|
1000 |
2.5 |
|
2000 |
5.0 |
|
3000 |
5.0 |
El producto está a la venta en diversas presentaciones y kits. Elija las opciones apropiadas, según la Solicitud de autorización comercial nacional.
Instrucciones de uso/manejo: Las instrucciones de reconstitución apropiadas para las presentaciones aprobadas en los mercados respectivos, se deben incluir en las etiquetas locales.
“Clásico”: Sobresello de aluminio y vial de diluyente. En el caso de la infusión, el producto se debe preparar en condiciones asépticas. Si cualquier componente del empaque se encuentra abierto o dañado, no lo use. Los medicamentos parenterales deben ser inspeccionados visualmente en busca de partículas y cambios de color antes de la administración. No utilice KOGENATE Bayer si observa partículas o turbidez en la solución.
KOGENATE Bayer debe ser reconstituido y administrado con los componentes que se proporcionan con cada empaque. Tras la reconstitución, el producto se debe usar en el lapso de 3 horas. Para infusión continua en estudios in vitro se ha demostrado la estabilidad química y física en uso, durante 24 horas a 30°C, dentro de bolsas de PVC.
El producto reconstituido debe ser filtrado antes de la administración para eliminar posibles partículas en la solución. La filtración puede realizarse siguiendo los pasos para la reconstitución y/o administración tal como se describe a continuación. Es importante utilizar el equipo de administración que se proporciona con el producto para la administración ya que incorpora un filtro en línea.En los casos en que no pueda utilizarse el equipo de administración (p. ej., cuando es una infusión periférica o por línea central), se debe utilizar un filtro separado compatible con KOGENATE Bayer.
El equipo de administración que se proporcionó con el producto no debe utilizarse para la extracción de sangre ya que contiene un filtro en línea. Cuando deba extraerse sangre antes de una infusión, utilice un equipo de administración sin filtro, luego administre la infusión de KOGENATE Bayer por medio de un filtro separado. Si tiene preguntas acerca de KOGENATE Bayer y los filtros compatibles separados, comuníquese con Bayer a [detalles de contacto local].
Reconstitución: Lávese siempre las manos antes de realizar los procedimientos siguientes:

Transferencia por vacío
1. Caliente los viales cerrados de diluyente y de concentrado a una temperatura no mayor de 37°C (99°F).
2. Después de retirar las tapas abatibles de plástico (Fig. A), limpie de modo aséptico los tapones de goma de ambos viales con alcohol, teniendo cuidado de no tocar el tapón de goma.
3. Retire la cubierta protectora de un extremo del cartucho de plástico de la aguja de transferencia e inserte la aguja en el tapón del frasco de diluyente (Fig. B).
4. Retire la parte restante de la cubierta protectora, invierta el vial de diluyente y perfore el sello de goma del vial de concentrado (Fig. C) con la aguja en ángulo.
5. El vacío hará fluir el diluyente hacia el vial de concentrado. Sostenga el vial del diluyente en ángulo respecto al vial de concentrado para dirigir el chorro de diluyente hacia la pared del vial de concentrado (Fig. C). Evite que se forme demasiada espuma. Si el diluyente no fluye hacia el vial, el vacío es insuficiente y no se debe usar el producto.
6. Después de retirar el vial del diluyente y la aguja de transferencia (Fig. D), haga girar el vial hasta disolver por completo el polvo sin formar demasiada espuma (Fig. E).
7. Vuelva a limpiar con alcohol la parte superior del vial de KOGENATE Bayer reconstituido. Permita que el tapón seque al aire.
8. Una vez disuelto por completo el polvo concentrado, cargue la jeringa con la solución haciéndola pasar por la aguja filtro que viene en el empaque (Fig. F). Remueva con cuidado todo el aire llevándolo a la parte inferior del vial pero cerciórese que haya extraído toda la solución 84 Sustituya la aguja filtro con el equipo de administración suministrado e inyecte por vía intravenosa.
NOTA: vea las instrucciones anexas para el Equipo de infusión con filtro.
9. Si el mismo paciente tiene que recibir más de un vial, es posible cargar una misma jeringa con el contenido de dos viales mediante el uso de dos agujas filtro por separado, antes de conectar la aguja de punción venosa.
Los medicamentos parenterales deben ser inspeccionados en busca de partículas y cambios de color antes de la administración, siempre que la solución y el envase lo permitan.
BIO-SET®: BIO-SET y Jeringa de Diluyente
Para infusión, el producto debe prepararse en condiciones asépticas. Si cualquiera de los componentes del empaque está abierto o dañado, no lo utilice. Los medicamentos parenterales deben ser inspeccionados visualmente en busca de partículas y cambios de color antes de la administración. No use KOGENATE Bayer si observa partículas o turbidez en la solución.
KOGENATE Bayer debe ser reconstituido y administrado con los componentes que se proporcionan con cada empaque. Tras la reconstitución, el producto se debe usar en el plazo de 3 horas. Para infusiones continuas, en estudios in vitro, se ha demostrado la estabilidad química y física en uso, durante 24 horas a 30°C, dentro de bolsas de PVC.
Se debe filtrar el producto reconstituido antes de la administración para eliminar las posibles partículas en la solución. La filtración puede realizarse siguiendo los pasos para la reconstitución y/o administración tal como se describe a continuación. Es importante utilizar el equipo de administración provisto con el producto para la administración, ya que incorpora un filtro en línea. En los casos en que no pueda utilizarse el equipo de administración (p. ej., cuando es una infusión periférica o en línea central), se debe utilizar un filtro separado compatible con KOGENATE Bayer.
El equipo de administración que se proporcionó con el producto no debe utilizarse para la extracción de sangre ya que contiene un filtro en línea. Cuando deba extraerse sangre antes de una infusión, utilice un equipo de administración sin filtro, luego administre la infusión de KOGENATE Bayer por medio de un filtro para inyección. Si tiene preguntas acerca de KOGENATE Bayer y los filtros compatibles separados, comuníquese con Bayer a [detalles de contacto local].
Reconstitución
1. Caliente los viales cerrados de diluyente (si fuera necesario) y concentrado hasta una temperatura no mayor que 37°C (99°F).
2. Retire la tapa del concentrado. Saque la jeringa precargada del diluyente y retire la tapa de la punta. (Fig. A).
3. Conecte la jeringa precargada del diluyente en el vial del concentrado atornillándola suavemente en la conexión del BIO-SET® (Fig. B).
4. Coloque el vial sobre una superficie rígida, antideslizante, y sosténgalo firmemente con una mano. Con el pulgar y el dedo índice de la otra mano, presione con fuerza hacia abajo la placa ubicada cerca de la punta de la jeringa (Fig. C) hasta que la placa haga contacto con el borde superior del BIO-SET®. Esto confirma que el sistema está activado (Fig. D).
5. Sujete el émbolo por la placa superior y retírelo de la caja de cartón. Evite tocar los lados y las roscas del émbolo. Atornille de inmediato el émbolo en el tapón de goma de la jeringa (Fig. E).
6. Inyecte el diluyente en el concentrado presionando lentamente el émbolo hacia abajo (Fig. F).
7. Haga girar suavemente el vial hasta disolver por completo el polvo sin formar demasiada espuma (Fig. G).
8. Inspeccione visualmente en busca de partículas y cambios de color antes de la administración.
9. Invierta el conjunto de vial/jeringa y transfiera la solución a la jeringa que utilizó para vaciar el diluyente (Fig. H). Cerciórese de que todo el contenido del vial del producto reconstituido pase a la jeringa. Remueva con cuidado todo el aire llevándolo a la parte inferior del vial pero cerciórese que haya extraído toda la solución.
10. Destornille la jeringa cargada para desconectarla del vial de concentrado vacío (Fig. I).
11. Conecte la jeringa cargada al equipo de administración suministrado e inyecte de inmediato por vía intravenosa (Fig. J).
12. Si el mismo paciente tiene que recibir más de un frasco, se debe usar la jeringa de diluyente suministrada para reconstituir el polvo de los viales de producto tal como se explicó anteriormente. Luego, las soluciones reconstituidas se deben combinar en una jeringa de plástico de mayor volumen (no se suministra) para inyectarlas como de costumbre (Fig. J).

Para infusión, el producto debe prepararse en condiciones asépticas. Si cualquiera de los componentes del empaque está abierto o dañado, no lo utilice. Los medicamentos parenterales deben ser inspeccionados visualmente en busca de partículas y cambios de color antes de la administración. No use KOGENATE Bayer si observa partículas o turbidez en la solución.
KOGENATE Bayer debe ser reconstituido y administrado con los componentes que se proporcionan con cada empaque. Tras la reconstitución, el producto se debe usar en el plazo de 3 horas. Para infusiones continuas, en estudios in vitro se ha demostrado la estabilidad química y física en uso, durante 24 horas a 30°C, dentro de bolsas de PVC. Se debe filtrar el producto reconstituido antes de la administración para eliminar las posibles partículas en la solución. La filtración se realiza utilizando el adaptador para viales.
1. Caliente los viales cerrados y la jeringa entre las manos hasta que adquieran una temperatura confortable (no más de 37°C [99°F]).
2. Retire la tapa protectora del vial (A). Limpie de modo aséptico el tapón de goma con alcohol, teniendo cuidado de no tocar el tapón de goma.
3. Coloque el vial del producto sobre una superficie firme y antideslizante. Desprenda la cubierta de papel del recipiente de plástico del adaptador para viales. No retire el adaptador del recipiente de plástico. Sosteniendo el recipiente del adaptador, colóquelo sobre el vial del producto y presiónelo con firmeza hacia abajo (B). El adaptador entrará en la tapa del vial. No retire todavía el recipiente del adaptador.
4. Sujete el émbolo por la placa superior y retírelo de la caja de cartón. Evite tocar los lados y las roscas del émbolo. Atornille de inmediato el émbolo, haciéndolo girar con firmeza hacia la derecha, en el tapón de goma roscado de la jeringa (C).
5. Mientras sostiene la jeringa por el cilindro, desprenda la tapa de la punta de la jeringa (D). No toque la punta de la jeringa, ni con su mano ni con ninguna otra superficie. Coloque la jeringa a un lado para usarla más tarde.
6. Ahora, retire y deseche el recipiente del adaptador (E).
7. Atornille la jeringa precargada en el adaptador para viales roscado, girándola hacia la derecha (F).
8. Inyecte el diluyente presionando lentamente el émbolo hacia abajo (G).
9. Haga girar suavemente el vial hasta que se disuelva todo el material (H). No agite el vial. Asegúrese de que el polvo se disuelva por completo. No use ninguna solución que contenga partículas visibles o que se vea turbia.
10. Cargue la jeringa con la solución manteniendo el vial del extremo por encima del adaptador para viales y la jeringa (I); luego, tire lenta y suavemente del émbolo. Cerciórese de que todo el contenido del vial pase a la jeringa. Remueva la mayor cantidad de aire posible antes de quitar la jeringa del vial esto debe realizarlo lentamente moviendo el aire al fondo del vial.
11. Con el émbolo en su lugar, retire la jeringa del adaptador para viales (este último debe permanecer unido al vial). Conecte la jeringa cargada al equipo de administración suministrado e inyecte por vía intravenosa (J).
12. Si el mismo paciente tiene que recibir más de un frasco, reconstituya cada frasco con la jeringa de diluyente que se suministra; luego, combine las soluciones en una jeringa de mayor volumen (no se suministra) e inyecte como de costumbre.
Los medicamentos parenterales deben ser inspeccionados en busca de partículas y cambios de color antes de la administración, siempre que la solución y el envase lo permitan.

Mix2Vial: Adaptador para Viales y Vial de Diluyente Para infusión continua, el producto debe prepararse en condiciones asépticas. Si cualquiera de los componentes del empaque está abierto o dañado, no lo utilice. Los medicamentos parenterales deben ser inspeccionados en busca de partículas y cambios de color antes de la administración. No utilice KOGENATE Bayer si observa partículas o turbidez en la solución. KOGENATE Bayer debe ser reconstituido y administrado con los componentes que se proporcionan con cada empaque.
Tras la reconstitución, el producto se debe usar en el plazo de 3 horas. Para infusiones continuas, en estudios in vitro se ha demostrado la estabilidad química y física en uso, durante 24 horas a 30°C, dentro de bolsas de PVC. Se debe filtrar el producto reconstituido antes de la administración para eliminar las posibles partículas en la solución. La filtración se realiza utilizando un adaptador Mix2vial.
Reconstitución
1. Caliente los viales cerrados de diluyente y de concentrado a una temperatura no mayor de 37°C o 99°F.
2. Coloque el vial del producto, el vial del diluyente y el Mix2Vial™ sobre una superficie plana.
3. Asegúrese de haber retirado las tapas abatibles de los viales del producto y el diluyente, y de haber tratado los tapones con una solución aséptica y haberlos dejado secar antes de abrir el empaque del Mix2Vial.
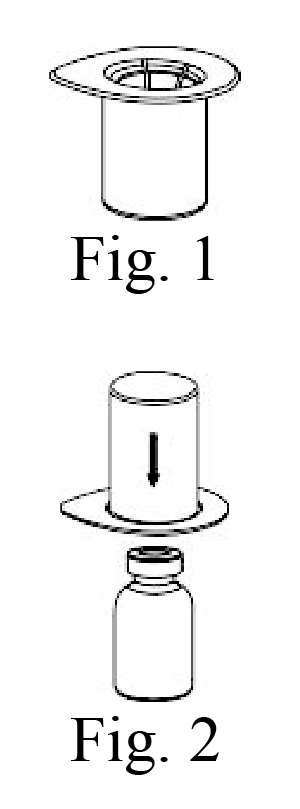
4. Abra el empaque del Mix2Vial desprendiéndolo de la tapa (Fig. 1). Deje el Mix2Vial en el empaque transparente. Coloque el vial del diluyente en una superficie plana y sosténgalo con firmeza. Sujete el Mix2Vial junto con el empaque e inserte el extremo azul en el tapón del diluyente (Fig. 2).
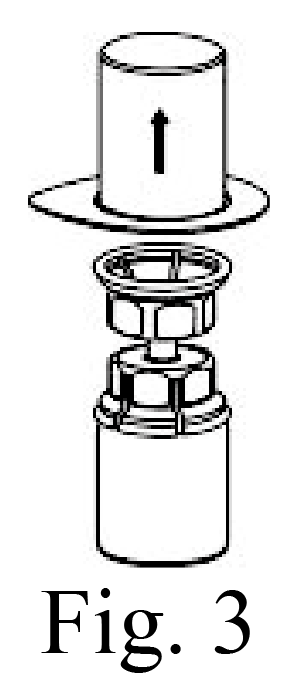
5. Retire cuidadosamente el empaque transparente del equipo Mix2Vial. Cerciórese de levantar únicamente el empaque y no el equipo Mix2Vial (Fig. 3).
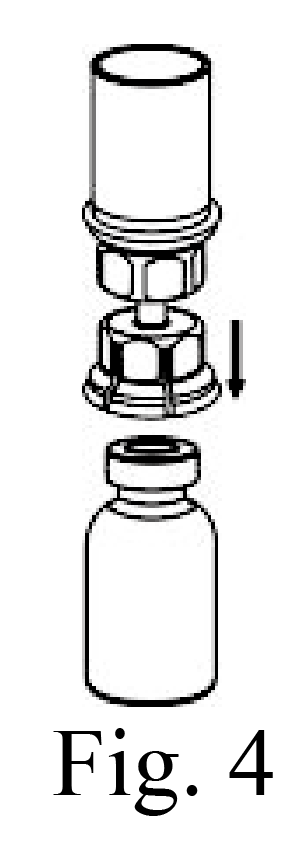
6. Con el vial del producto apoyado firmemente en una superficie, invierta el vial del diluyente con el equipo conectado, e inserte el adaptador trasparente en el tapón del vial del producto (Fig. 4). El diluyente pasará automáticamente al vial del producto.
7. Con los viales de diluyente y de producto todavía conectados, haga girar suavemente el vial del producto para asegurarse de que el polvo se disuelva por completo (Fig. 5). No agite el vial.
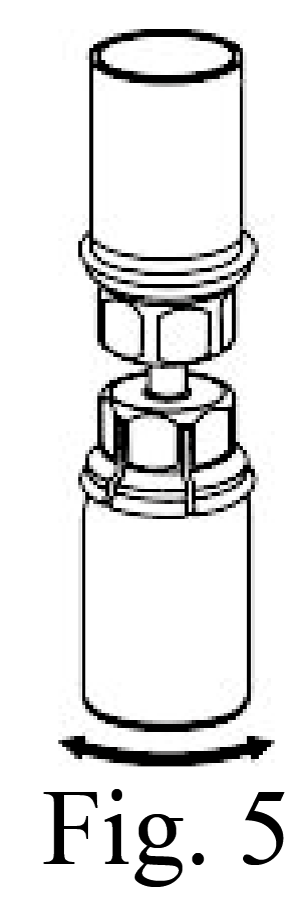
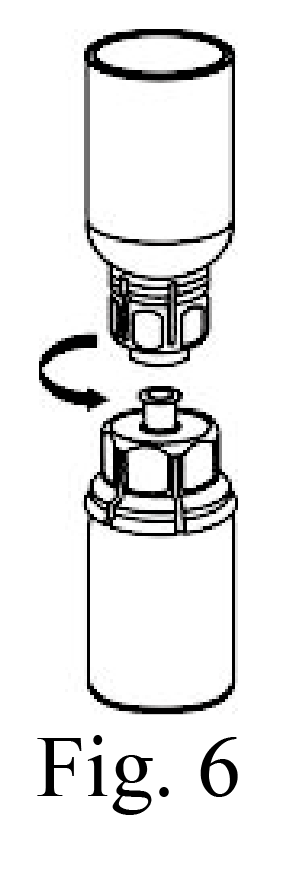
8. Sujete con una mano el lado del producto del equipo Mix2Vial y con la otra, el lado azul del diluyente del equipo Mix2Vial, y destornille el equipo para separar las dos piezas (Fig. 6). Remueva la mayor cantidad de aire posible antes de quitar la jeringa del vial esto debe realizarlo lentamente moviendo el aire al fondo del vial.
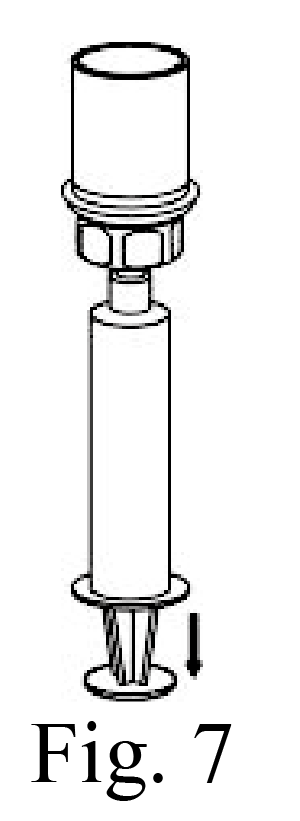
9. Llene de aire una jeringa estéril vacía. Mientras el vial del producto está en posición vertical, atornille la jeringa en el equipo Mix2Vial. Inyecte aire en el vial del producto. Mientras mantiene presionado el émbolo de la jeringa, invierta el sistema y cargue la jeringa con el concentrado tirando lentamente del émbolo hacia atrás (Fig. 7).
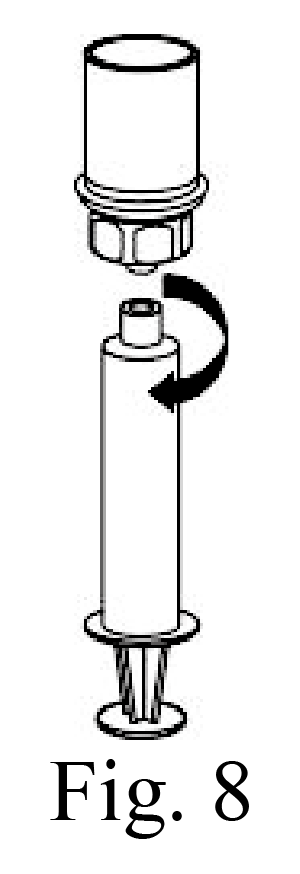
10. Ahora que el concentrado ya se encuentra en la jeringa, sujete firmemente el cilindro de la jeringa (con el émbolo de la jeringa hacia abajo) y destornille la jeringa del equipo Mix2Vial (Fig. 8). Conecte la jeringa a un equipo de administración hecho de tubos Microbore. El uso de otros equipos de administración sin tubos Microbore puede ocasionar una mayor retención de la solución dentro del equipo de administración.
11. Si el mismo paciente recibirá más de un frasco, es posible cargar una misma jeringa con el contenido de dos frascos mediante otro equipo Mix2Vial sin usar, antes de conectar la aguja de punción venosa.
12. Nombre comercial apropiado: debe ser inspeccionado visualmente en busca de partículas y cambios de color antes de la administración.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Precauciones especiales de conservación: De acuerdo con la documentación TRD/CMC.
KOGENATE Bayer se debe almacenar en refrigeración (2-8°C). No lo use después de la fecha de vencimiento que se indica en el frasco. Es posible almacenar el polvo liofilizado a una temperatura de hasta <xx>°C durante <yy> meses; por ejemplo, en situaciones de almacenamiento en el hogar. Si el producto se almacena fuera del refrigerador, anote la fecha en que fue retirado de refrigeración y anote una nueva fecha de vencimiento en la caja de cartón y el vial. La nueva fecha de vencimiento debe ser <yy> meses después de la fecha en que el producto fue retirado del refrigerador o la fecha de vencimiento previamente impresa, lo que ocurra primero. Una vez que retire el producto del refrigerador, no podrá volver a almacenarlo en refrigeración. Evite el congelamiento. Evite la exposición a condiciones de luz extremas y almacene el polvo liofilizado dentro de la caja de cartón mientras no lo use. Según la autorización comercial nacional, los períodos de almacenamiento a temperatura ambiente pueden ser diferentes. En algunos países se considera que la temperatura ambiente es de 30°C y, por lo tanto, esto se debe verificar conforme a las condiciones reales de almacenamiento aprobadas en la Solicitud de autorización comercial.
Vida útil: De acuerdo con la documentación TRD/CMC.