KADCYLA
TRASTUZUMAB
Polvo estéril
Frasco(s) , 100 Miligramos
Frasco(s) , 160 Miligramos
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Principio activo: Trastuzumab emtansina
Forma farmacéutica: viales monodosis de 100 mg con polvo para concentrado para solución para infusión, diseñados para obtener 5 ml con 20 mg/ml de trastuzumab emtansina.
Viales monodosis de 160 mg con polvo para concentrado para solución para infusión, diseñados para obtener 8 ml con 20 mg/ml de trastuzumab emtansina.
Excipientes: Sacarosa, ácido succínico, hidróxido de sodio y polisorbato 20.
FORMA FARMACÉUTICA
Polvo estéril para concentrado para solución para infusión.
PROPIEDADES Y EFECTOS FARMACOLÓGICOS
Propiedades farmacodinámicas
Mecanismo de acción
Kadcyla (trastuzumab emtansina) es un conjugado de anticuerpo y fármaco dirigido contra HER2 que contiene trastuzumab —una IgG1 anti-HER2 humanizada— unido covalentemente a DM1 —un derivado de la maitansina inhibidor de los microtúbulos— mediante el conector tioéter estable MCC (4-[N-maleimidometil] cicloexano-1- carboxilato). Emtansina se refiere al complejo MCC-DM1. Con cada molécula de trastuzumab se conjugan por término medio 3,5 moléculas de DM1.
La conjugación de DM1 con el trastuzumab confiere la selectividad del componente citotóxico por células tumorales con sobreexpresión de HER2, aumentando así la liberación intracelular de DM1 directamente en las células malignas. Después de unirse a HER2, trastuzumab emtansina experimenta una internalización mediada por el receptor y la ulterior degradación lisosómica, lo cual da lugar a la liberación de catabolitos citotóxicos que contienen DM1 (principalmente lisina-MCC-DM1).
Kadcyla tiene los mecanismos de acción de trastuzumab y de DM1
- Trastuzumab emtansina, al igual que el trastuzumab, se une al subdominio IV del dominio extracelular de HER2 (ECD), así como a receptores de Fcã y al componente C1q del complemento. Además, Kadcyla, al igual que el trastuzumab, inhibe la liberación del dominio extracelular de HER2, inhibe la transmisión de señales a través de la vía de la fosfatidilinositol 3-cinasa (PI3-K) y participa en la citotoxicidad celular dependiente de anticuerpos (CCDA) en células de cáncer de mama humano con sobreexpresión de HER2.
- DM1, el componente citotóxico de Kadcyla, se une a la tubulina. Al inhibir la polimerización de la tubulina, tanto DM1 como Kadcyla hacen que las células se detengan en la fase G2/M del ciclo celular, lo que finalmente da lugar a la muerte celular apoptósica. Los resultados de ensayos de citotoxicidad in vitro muestran que DM1 es 20-200 veces más potente que los taxanos y los alcaloides de la vinca.
- El conector MCC ha sido diseñado para limitar la liberación sistémica y aumentar la liberación dirigida de DM1, tal como demuestra la detección de concentraciones muy bajas de DM1 libre en el plasma.
INDICACIONES TERAPÉUTICAS
Cáncer de mama
Cáncer de mama metastásico (CMM): Kadcyla, en monoterapia, está indicado para el tratamiento de pacientes con cáncer de mama HER2-positivo localmente avanzado irresecable o metastásico que han recibido previamente tratamiento con trastuzumab y un taxano.
DATOS FARMACÉUTICOS
Conservación
Viales
Consérvense los viales a 2-8 °C.
Este medicamento no debe usarse después de la fecha de caducidad, indicada con «EXP» en el envase.
Periodo de validez de la solución reconstituida
Los viales del producto reconstituidos con agua estéril para inyectables deben usarse inmediatamente después de la reconstitución. Si no se usan de inmediato, los viales reconstituidos pueden conservarse hasta 24 horas a 2-8 ºC, y se eliminarán una vez superado ese plazo.
No debe congelarse la solución reconstituida.
Periodo de validez de la solución para infusión con el producto reconstituido
La solución de trastuzumab emtansina reconstituida diluida en bolsas de cloruro de polivinilo (PVC) o de poliolefina sin PVC ni látex que contengan solución de cloruro de sodio al 0,9% o solución de cloruro de sodio al 0,45% pueden conservarse a 2-8 ºC durante ≤24 horas antes de usarlas. Durante la conservación pueden observarse partículas si se diluye en solución de cloruro de sodio al 0,9%, por lo que es necesario usar para la administración un filtro de polietersulfona (PES) de 0,2 ìm o 0,22 ìm en línea (ver Instrucciones especiales de uso, manipulación y eliminación).
No debe congelarse la solución para infusión con el producto reconstituido.
Instrucciones especiales de uso, manipulación y eliminación
Debe usarse una técnica aséptica adecuada. Se utilizarán procedimientos apropiados para la preparación de quimioterápicos.
El producto reconstituido no contiene conservantes y es para un solo uso. Se desechará la parte que no se haya utilizado.
- Con una jeringa estéril, inyéctense lentamente 5 ml de agua estéril para inyectables en un vial con 100 mg, o bien 8 ml de agua estéril para inyectables en un vial con 160 mg de trastuzumab emtansina.
- Muévase suavemente el vial en círculos hasta la total disolución. NO SE DEBE AGITAR EL VIAL.
- El vial con trastuzumab emtansina reconstituido debe conservarse a 2-8 ºC; la porción que no se haya utilizado debe eliminarse al cabo de 24 horas.
La solución reconstituida se examinará visualmente antes de su administración por si presentara partículas o cambios de color. La solución reconstituida debe carecer de partículas visibles y ser límpida o ligeramente opalescente. La solución reconstituida debe ser entre incolora y de color pardo claro. No se usará la solución reconstituida si contiene partículas visibles, está turbia o tiene un color que no es el habitual.
Instrucciones para la dilución:
Se debe determinar el volumen de solución necesario para administrar una dosis de 3,6 mg de trastuzumab emtansina/kg de peso (véase la pauta de reducción de la dosis en el apartado 2.2):
Volumen (ml) = Peso corporal (kg) x dosis (mg/kg)
20 mg/ml (concentración de la solución reconstituida)
Se extraerá la cantidad apropiada de solución del vial y se añadirá a una bolsa de infusión que contenga 250 ml de solución de cloruro de sodio al 0,45% o de solución de cloruro de sodio al 0,9%. No se debe usar solución de glucosa (5%). La solución de cloruro de sodio al 0,45% puede usarse sin filtro de polietersulfona (PES) de 0,2 o 0,22 µm en línea. Si se emplea la solución de cloruro de sodio al 0,9% para la infusión, es preciso utilizar un filtro de polietersulfona (PES) de 0,2 o 0,22 µm en línea. Una vez preparada la solución para infusión, se administrará inmediatamente. Si no se usa de inmediato, la infusión puede conservarse hasta 24 horas en el refrigerador a 2-8 ºC. Durante la conservación, la solución para infusión no debe congelarse ni agitarse.
Incompatibilidades
No debe usarse solución de glucosa (5%), dado que produce agregación de proteínas. Trastuzumab emtansina no se debe mezclar ni diluir con otros medicamentos.
Presentación
Descrita de conformidad con los requisitos locales.
Eliminación de los medicamentos no utilizados o caducados.
La emisión de productos farmacéuticos al medio ambiente debe reducirse al mínimo. Evítese tirar los medicamentos por los desagües o a la basura doméstica, y utilícense los sistemas de recogida disponibles localmente.
PROPIEDADES FARMACOCINÉTICAS
Absorción
Kadcyla se administra por vía i.v. No se han realizado estudios con otras vías de administración.
Distribución
Kadcyla, cuando se administra por vía i.v. cada 3 semanas, muestra una farmacocinética lineal con dosis de 2,4-4,8 mg/kg; el aclaramiento es más rápido en los pacientes que reciben dosis ≥1,2 mg/kg.
En los pacientes del estudio TDM4370g/BO21977 que recibieron 3,6 mg/kg de Kadcyla por vía i.v. cada 3 semanas, la media de la concentración sérica máxima (Cmáx) de trastuzumab emtansina fue de 83,4 (± 16,5) μg/ml. Según un análisis farmacocinético poblacional, después de la administración i.v., el volumen de distribución central de trastuzumab emtansina fue de 3,13 l, aproximadamente igual al volumen plasmático.
Metabolismo
Se supone que trastuzumab emtansina se cataboliza por proteólisis en los lisosomas celulares, sin que haya una participación importante de las isoenzimas del citocromo P450. En el plasma humano se han detectado catabolitos —como Lys-MCC-DM1, MCC- DM1 y DM1— en bajas concentraciones. En el estudio TDM4370g/BO21977, la media de la concentración máxima de DM1 en el ciclo 1 después de la administración de Kadcyla fue constantemente baja y, por término medio, de 4,61 ±1,61 ng/ml.
Los estudios del metabolismo in vitro en microsomas hepáticos humanos indican que DM1, un componente de trastuzumab emtansina, es metabolizado principalmente por el CYP3A4 y, en menor grado, por el CYP3A5.
Eliminación
Según el análisis farmacocinético poblacional, después de la administración i.v. de Kadcyla a pacientes con cáncer de mama metastásico HER2-positivo, el aclaramiento de Kadcyla fue de 0,68 l/día, y la semivida de eliminación (t1/2) fue de aproximadamente 4 días. No se observó ninguna acumulación de Kadcyla después de la administración repetida de infusiones i.v. cada 3 semanas.
De acuerdo con el análisis farmacocinético poblacional (n = 671), el peso corporal, la albúmina, la suma del diámetro mayor de las lesiones diana según los criterios de evaluación de la respuesta en tumores sólidos (RECIST), el dominio extracelular de HER2, las concentraciones iniciales de trastuzumab y la AST se conside-raron covariables estadísticamente significativas de los parámetros farmacocinéticos de trastuzumab emtansina. Sin embargo, la magnitud del efecto de estas covariables sobre la exposición a trastuzumab emtansina indica que, con la excepción del peso corporal, es improbable que estas covariables tengan algún efecto clínicamente significativo en la exposición a Kadcyla. Así pues, se considera apropiada la dosis basada en el peso corporal de 3,6 mg/kg cada 3 semanas sin corrección en función de otras covariables. En estudios no clínicos, los catabolitos de trastuzumab emtansina —como DM1, Lys-MCC-DM1 y MCC-DM1— se excretaron principalmente en la bilis, con eliminación mínima por la orina.
Farmacocinética en poblaciones especiales
El análisis farmacocinético poblacional de Kadcyla mostró que la raza no parecía influir en su farmacocinética. Dado que la mayoría de los pacientes de los estudios clínicos de Kadcyla eran mujeres, no se evaluó formalmente el efecto del sexo en la farmacocinética de Kadcyla.
Uso en geriatría
El análisis farmacocinético poblacional de Kadcyla mostró que la edad no afecta a la farmacocinética de este medicamento. No se observaron diferencias significativas en la farmacocinética de Kadcyla entre los pacientes menores de 65 años (n = 577), los de 65- 75 años (n = 78) y los mayores de 75 años (n = 16).
Insuficiencia renal
El análisis farmacocinético poblacional de Kadcyla mostró que el aclaramiento de creatinina (ClCr) no afecta a su farmacocinética. En los pacientes con insuficiencia renal leve (ClCr: 60-89 ml/min, n = 254) o moderada (ClCr: 30-59 ml/min, n = 53), la farmacocinética de Kadcyla era similar a la observada en los pacientes con función renal normal (ClCr: >90 ml/min, n = 361). Los datos farmacocinéticos de pacientes con insuficiencia renal grave (ClCr: 15-29 ml/min) son limitados (n = 1), por lo que no es posible formular recomendaciones posológicas.
Insuficiencia hepática
El hígado es un órgano primordial para la eliminación de DM1 y de catabolitos que contienen DM1. Se evaluó la farmacocinética de trastuzumab emtansina y de cata-bolitos que contienen DM1 después de la administración de 3,6 mg/kg de Kadcyla a pacientes con cáncer de mama metastásico HER2-positivo con función hepática normal (n = 10), insuficiencia hepática leve (clase A según la clasificación de Child-Pugh; n = 10) y moderada (clase B según la clasificación de Child-Pugh; n = 8).
- Las concentraciones plasmáticas de DM1 y de cata-bolitos que contienen DM1 (Lys-MCC-DM1 y MCC-DM1) fueron bajas, y comparables en los pacientes con insuficiencia hepática y los pacientes con función hepática normal.
- La exposición sistémica (ABC) a trastuzumab emtansina en el ciclo 1 en pacientes con insuficiencia hepática leve y moderada fue aproximadamente un 38% y un 67% menor que en los pacientes con función hepática normal, respectivamente. La exposición a trastuzumab emtansina (ABC) en el ciclo 3 después de la administración repetida en pacientes con disfunción hepática leve o moderada se encontraba dentro del intervalo observado en pacientes con función hepática normal.
No se ha estudiado el uso de Kadcyla en pacientes con insuficiencia hepática grave (clase C según la clasificación de Child-Pugh).
Ensayos clínicos / Eficacia
Eficacia
Cáncer de mama metastásico
Se realizó un estudio de fase III, aleatorizado, multicéntrico, internacional, sin enmascaramiento (TDM4370g/BO21977) en pacientes con cáncer de mama HER2- positivo localmente avanzado irresecable o metastásico que habían recibido anteriormente tratamiento con un taxano y trastuzumab, incluidos pacientes que recibieron tratamiento previo con trastuzumab y un taxano en el marco del tratamiento adyuvante y que presentaron una recidiva 6 meses después de su conclusión. Antes de la inclusión en el ensayo, se exigía que los pacientes contaran con muestras de los tumores de mama para confirmar en un laboratorio central que eran tumores HER2-positivos, definidos como aquellos con una puntuación de 3+ en la IHQ o con amplificación génica determinada mediante ISH. Las características iniciales de los pacientes y los tumores estaban adecuadamente equilibradas entre los grupos de tratamiento. En lo que se refiere a los pacientes asignados aleatoriamente al tratamiento con Kadcyla, la mediana de la edad fue de 53 años, la mayoría eran mujeres (99,8%), la mayoría eran de raza blanca (72%) y el 57% tenían tumores con receptores de estrógenos, de progesterona o de ambos tipos. El estudio comparó la seguridad y la eficacia de Kadcyla con la de lapatinib más capecitabina. Un total de 991 pacientes fueron asignados aleatoriamente al tratamiento con Kadcyla o bien a lapatinib más capecitabina, que se administraron según las pautas que se indican a continuación:
- Grupo de Kadcyla: Kadcyla en dosis de 3,6 mg/kg por vía i.v. durante 30-90 minutos el día 1 de un ciclo de 21 días.
- Grupo de referencia (lapatinib más capecitabina): lapatinib en dosis de 1.250 mg/día por vía oral 1 vez al día en ciclos de 21 días más capecitabina en dosis de 1.000 mg/m2 por vía oral 2 veces al día los días 1-14 de un ciclo de 21 días.
Las variables principales de valoración de la eficacia del estudio fueron la supervivencia sin progresión (SSP) según la evaluación de un comité de revisión independiente (CRI), la supervivencia global (SG) y las tasas de supervivencia al cabo de 1 año y 2 años.
También se evaluó durante el ensayo clínico el tiempo transcurrido hasta la progresión de los síntomas, definido por una disminución de 5 puntos en la puntuación obtenida en la subescala Trials Outcome Index-Breast (TOI-B) del cuestionario de evaluación funcional de la calidad de vida FACT-B (Functional Assessment of Cancer Therapy-Breast). Se considera que un cambio de 5 puntos en el índice TOI-B es clínicamente significativo.
|
Tabla 8. Resumen de la eficacia del estudio TDM4370g/BO21977 (EMILIA) |
||
|
Lapatinib + Capecitabina n = 496 |
Trastuzumab emtansina N = 495 |
|
|
Variables de valoración principales |
||
|
SSP evaluada por el CRI N.º (%) de pacientes en los que se registró algún evento |
304 (61,3%) |
265 (53,5%) |
|
Mediana de la duración de la SSP (meses) |
6,4 |
9,6 |
|
HR (estratificada*) |
0,650 |
|
|
IC 95% de la HR |
(0,549 , 0,771) |
|
|
p (prueba de rangos logarítmicos estratificada*) |
<0,0001 |
|
|
Supervivencia global** |
||
|
N.º (%) de pacientes que fallecieron |
182 (36,7%) |
149 (30,1%) |
|
Mediana de la duración de la supervivencia (meses) |
25,1 |
30,9 |
|
HR (estratificada*) |
0,682 |
|
|
IC 95% de la HR |
(0,548, 0,849) |
|
|
p (prueba de rangos logarítmicos*) |
0,0006 |
|
|
Tasa de supervivencia a 1 año (IC 95%) |
78,4% (74,62, 82,26) |
85,2% (81,99, 88,49) |
|
Tasa de supervivencia a 2 años (IC 95%) |
51,8% (45,92, 57,73) |
64,7% (59,31, 70,19) |
|
Variables de valoración secundarias fundamentales |
||
|
SSP evaluada por el investigador N.º (%) de pacientes en los que se registró algún evento |
335 (67,5%) |
287 (58,0%) |
|
Mediana de la duración de la SSP (meses) |
5,8 |
9,4 |
|
HR (IC 95%) |
0,658 (0,560, 0,774) |
|
|
p (prueba de rangos logarítmicos*) |
<0,0001 |
|
|
Tasa de respuesta objetiva |
||
|
Pacientes con cáncer mensurable |
389 |
397 |
|
N.º de pacientes con RO (%) |
120 (30,8%) |
173 (43,6%) |
|
Dif. (IC 95%) |
12,7% (6,0%, 19,4%) |
|
|
p (prueba de la -2 de Mantel-Haenszel*) |
0,0002 |
|
|
Duración de la respuesta objetiva (meses) N.º de pacientes con RO |
120 |
173 |
|
Mediana (IC 95%) |
6,5 (5,45, 7,16) |
12,6 (8,38, 20,76) |
|
Tiempo transcurrido hasta la progresión de los síntomas N.º de pacientes evaluables |
445 |
450 |
|
N.º (%) de pacientes en los que se registró algún evento |
257 (57,8%) |
246 (54,7%) |
|
Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
4,6 |
7,1 |
|
HR (IC 95%) |
0,796 (0,667, 0,951) |
|
|
p (prueba de rangos logarítmicos*) |
0.0121 |
|
|
SSP: supervivencia sin progresión; RO = respuesta objetiva; HR: hazard ratio (razón de riesgos instantáneos). * Estratificada por: región del mundo (EE.UU., Europa occidental, otras), número de regímenes de quimioterapia previos para el cáncer localmente avanzado o metastásico (0-1 frente a >1), y enfermedad visceral o no visceral. ** El primer análisis provisional de la supervivencia global (SG) se realizó en el momento del análisis principal de la SSP. Se observó un notable efecto del tratamiento, pero no se superó el límite de eficacia preespecificado. Se llevó a cabo un segundo análisis provisional de la SG cuando se observaron 331 eventos de SG; los resultados se presentan en esta tabla. |
||
Se observó un beneficio del tratamiento en el subgrupo de pacientes que no habían recibido antes ningún tratamiento anticanceroso sistémico en el marco del cáncer metastásico (n = 118); las HR de la SSP y la SG fueron de 0,51 (IC 95%: 0,30-0,85) y 0,61 (IC 95%: 0,32-1,16), respectivamente. Las medianas de la SSP y la SG en el grupo de Kadcyla fueron de 10,8 meses y «no alcanzada», respectivamente, en comparación con 5,7 meses y 27,9 meses, respectivamente, en el grupo de lapatinib más capecitabina.
Figura 1. Curva de Kaplan-Meier de la supervivencia sin progresión evaluada por el CRI

De arriba abajo y de izquierda a derecha: Median time (mo): mediana del tiempo (meses); Hazard Ratio: Hazard ratio; (95% CI): (IC 95 %); Log-rank p-value: p (prueba de rangos logarítmicos); Progression-Free Survival Rate: tasa de supervivencia sin progresión; Lap+Cap (n=496): Lap + Cap (n = 496); T-DM1 (n=495): T-DM1 (n = 495); Time (months): tiempo (meses); Number at Risk: N.º de pacientes en riesgo.
T-DM1: trastuzumab emtansina; Lap: lapatinib; Cap: capecitabina; CRI: Comité de revisión independiente.
La hazard ratio se calcula basándose en un modelo de Cox estratificado; p se calcula basándose en una prueba de rangos logarítmicos estratificada.
Figura 2. Curva de Kaplan-Meier de la supervivencia global
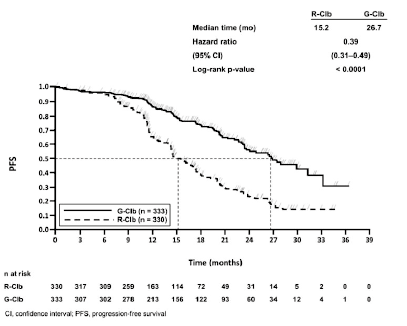
De arriba abajo y de izquierda a derecha: Median time (mo): mediana del tiempo (meses); Hazard Ratio: Hazard ratio; (95% CI): (IC 95 %); Log-rank p-value: p (prueba de rangos logarítmicos); Proportion Surviving: proporción de pacientes que sobrevivieron; Lap+Cap (n=496): Lap + Cap (n = 496); T-DM1 (n=495): T-DM1 (n = 495); Duration of Survival (months): duración de la supervivencia (meses); Number at Risk: N.º de pacientes en riesgo.
T-DM1: trastuzumab emtansina; Lap: lapatinib; Cap: capecitabina.
La hazard ratio se calcula basándose en un modelo de Cox estratificado; p se calcula basándose en una prueba de rangos logarítmicos estratificada.
En un estudio de fase II aleatorizado, multicéntrico, sin enmascaramiento (TDM4450g/BO21976) se evaluaron los efectos de Kadcyla, en comparación con trastuzumab más docetaxel, en pacientes con cáncer de mama metastásico HER2-positivo que no habían recibido previamente quimioterapia para el tratamiento de la enfermedad metastásica. Se asignó aleatoriamente a los pacientes a recibir Kadcyla en dosis de 3,6 mg/kg i.v. cada 3 semanas (n = 67) o trastuzumab en una dosis de carga de 8 mg/kg i.v. seguida por 6 mg/kg i.v. cada 3 semanas más docetaxel en dosis de 75-100 mg/m2 i.v. cada 3 semanas (n = 70).
La variable de valoración principal fue la SSP según la evaluación del investigador. La mediana de la SSP fue de 9,2 meses en el grupo de trastuzumab más docetaxel y de 14,2 meses en el grupo de Kadcyla (HR = 0,59; p = 0,035), siendo la mediana del seguimiento de aproximadamente 14 meses en ambos grupos. La TRG fue del 58,0% con trastuzumab más docetaxel y del 64,2% con Kadcyla. La mediana de la duración de la respuesta no se alcanzó con Kadcyla y fue de 9,5 meses en el grupo de referencia.
El empeoramiento de las puntuaciones FACT-B TOI fue más tardío en el grupo de Kadcyla que en el grupo de referencia (mediana del tiempo transcurrido hasta la progresión de los síntomas de 7,5 meses y de 3,5 meses, respectivamente; HR = 0,58; p = 0,022).
En un estudio de fase II, con un solo grupo y sin enmascaramiento (TDM4374g), se evaluaron los efectos de Kadcyla en pacientes con cáncer de mama HER2-positivo localmente avanzado incurable o metastásico. Todos los pacientes habían recibido previamente trata-miento con terapias dirigidas a HER2 (trastuzumab y lapatinib), y quimioterapia (una antraciclina, un taxano y capecitabina) en el marco de tratamiento neoadyuvante, adyuvante, del cáncer localmente avanzado o del cáncer metastásico. La mediana del número de antineoplásicos que habían recibido los pacientes en cualquiera de estos marcos de tratamiento fue de 8,5 (intervalo: 5-19) y en el contexto del cáncer metastásico fue de 7,0 (intervalo: 3-17), incluidos todos los fármacos destinados al trata-miento del cáncer de mama.
Los pacientes (n = 110) recibieron 3,6 mg/kg de Kadcyla por vía i.v. cada 3 semanas hasta la progresión de la enfermedad o la aparición de efectos secundarios inaceptables.
Las variables fundamentales de valoración de la eficacia fueron la TRG basada en una revisión radiológica independiente y la duración de la respuesta objetiva. La TRG fue del 32,7% (IC 95%: 24,1, 42,1), n = 36 pacientes con respuesta, tanto según la revisión del CRI como según la evaluación del investigador. La mediana de la duración de la respuesta según la evaluación del CRI no se alcanzó (IC 95%: 4,6 meses-no calculable).
Inmunogenicidad
Como ocurre con todas las proteínas terapéuticas, existe la posibilidad de una respuesta inmunitaria a trastuzumab emtansina. De los 836 pacientes de 6 estudios clínicos analizados en múltiples momentos de valora-ción para descubrir respuestas de anticuerpos contra Kadcyla, 44 pacientes (5,3%) presentaron anticuerpos contra Kadcyla en uno o más momentos de valoración posteriores a la administración; en 28 de estos pacientes las muestras iniciales eran negativas. No se sabe todavía qué importancia clínica pueden tener los anticuerpos contra trastuzumab emtansina.
Los resultados de los ensayos de inmunogenicidad dependen en gran medida de varios factores, como la sensibilidad y la especificidad del ensayo, la metodología de análisis, la manipulación de las muestras, el calendario de recogida de éstas, los tratamientos farmacológicos concomitantes y la enfermedad de fondo. Por estas razones, la comparación de la incidencia de anticuerpos contra Kadcyla con la de anticuerpos contra otros productos puede llevar a conclusiones erróneas.
CONTRAINDICACIONES: Kadcyla está contraindicado en pacientes con hipersensibilidad conocida al principio activo o a cualquier de los excipientes.
Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios de los efectos sobre la capacidad para conducir y utilizar máquinas.
Pruebas de laboratorio (ver Advertencias y precauciones generales - Hepatotoxicidad, Trombocitopenia.)
REACCIONES ADVERSAS
Ensayos clínicos
La seguridad de Kadcyla se ha evaluado en 1871 pacientes en ensayos clínicos. La tabla 6 resume las reacciones adversas que se han notificado en asociación al uso de Kadcyla en ensayos clínicos.
En este apartado, se han utilizado las siguientes cate-gorías de frecuencia: muy frecuente (>1/10), frecuente (>1/100 a <1/10) y poco frecuente (≥1/1.000 a <1/100).
|
Tabla 6. Resumen de las reacciones adversas en pacientes tratados con Kadcyla |
|||
|
Reacción adversa (MedDRA) |
Kadcyla |
||
|
Clase de órganos y sistemas |
Todos los grados (%) n = 1871 |
Grado 3-5 (%) n = 1871 |
Categoría de frecuencia |
|
Trastornos de la sangre y del sistema linfático |
|||
|
Trombocitopenia |
24,9 |
8,7 |
Muy frecuente |
|
Anemia |
14,6 |
3,8 |
Muy frecuente |
|
Neutropenia |
8,1 |
2,6 |
Frecuente |
|
Trastornos cardiacos |
|||
|
Disfunción del ventrículo izquierdo |
2,2 |
0,4 |
Frecuente |
|
Trastornos oculares |
|||
|
Sequedad ocular |
5,7 |
0,0 |
Frecuente |
|
Lagrimación aumentada |
4,1 |
0,0 |
Frecuente |
|
Visión borrosa |
4,0 |
0,0 |
Frecuente |
|
Conjuntivitis |
3,8 |
0,0 |
Frecuente |
|
Trastornos gastrointestinales |
|||
|
Náuseas |
40,0 |
0,8 |
Muy frecuente |
|
Estreñimiento |
23,7 |
0,4 |
Muy frecuente |
|
Vómitos |
19,9 |
1,0 |
Muy frecuente |
|
Diarrea |
19,2 |
0,7 |
Muy frecuente |
|
Sequedad de boca |
16,0 |
<0,1 |
Muy frecuente |
|
Dolor abdominal |
15,9 |
0,9 |
Muy frecuente |
|
Estomatitis |
15,4 |
0,1 |
Muy frecuente |
|
Dispepsia |
8,0 |
0,1 |
Frecuente |
|
Trastornos generales y alteraciones en el lugar de la administración |
|||
|
Fatiga |
36,8 |
2,5 |
Muy frecuente |
|
Pirexia |
23,0 |
0,2 |
Muy frecuente |
|
Astenia |
16,3 |
1,1 |
Muy frecuente |
|
Escalofríos |
10,3 |
≤0,1 |
Muy frecuente |
|
Edema periférico |
8,1 |
0,1 |
Frecuente |
|
Trastornos hepatobiliares |
|||
|
Insuficiencia hepática |
0,1 |
0,1 |
Poco frecuente |
|
Hiperplasia regenerativa nodular |
0,1 |
0,0 |
Poco frecuente |
|
Hipertensión portal |
0,3 |
0,1 |
Poco frecuente |
|
Trastornos del sistema inmunitario |
|||
|
Hipersensibilidad a fármacos |
2,6 |
0,1 |
Frecuente |
|
Infecciones e infestaciones |
|||
|
Infección urinaria |
11,9 |
0,4 |
Muy frecuente |
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
|||
|
Reacción relacionada con la infusión |
4,0 |
0,3 |
Frecuente |
|
Exploraciones complementarias |
|||
|
Aminotransferasas elevadas |
24,2 |
7,2 |
Muy frecuente |
|
Fosfatasa alcalina en sangre elevada |
5,3 |
0,5 |
Frecuente |
|
Trastornos del metabolismo y de la nutrición |
|||
|
Hipopotasemia |
11,0 |
2,4 |
Muy frecuente |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|||
|
Dolor musculoesquelético |
35,5 |
2,4 |
Muy frecuente |
|
Artralgias |
18,9 |
0,6 |
Muy frecuente |
|
Mialgias |
12,9 |
0,3 |
Muy frecuente |
|
Trastornos del sistema nervioso |
|||
|
Cefalea |
28,1 |
0,6 |
Muy frecuente |
|
Neuropatía periférica |
22,8 |
1,3 |
Muy frecuente |
|
Mareos |
9,5 |
0,2 |
Frecuente |
|
Disgeusia |
6,4 |
0,0 |
Frecuente |
|
Trastornos psiquiátricos |
|||
|
Insomnio |
11,7 |
0,2 |
Muy frecuente |
|
Trastornos respiratorios, torácicos y mediastínicos |
|||
|
Epistaxis |
24,3 |
0,4 |
Muy frecuente |
|
Tos |
19,5 |
0,1 |
Muy frecuente |
|
Disnea |
13,4 |
1,5 |
Muy frecuente |
|
Neumonitis |
0,7 |
0,1 |
Poco frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
|||
|
Exantema |
12,4 |
0,3 |
Muy frecuente |
|
Prurito |
6,0 |
≤0,1 |
Frecuente |
|
Trastornos vasculares |
|||
|
Hemorragia |
34,8 |
2,2 |
Muy frecuente |
|
Hipertensión arterial |
6,5 |
1,7 |
Frecuente |
Alteraciones analíticas
La siguiente tabla muestra las alteraciones analíticas observadas en pacientes tratados con Kadcyla en el ensayo clínico TDM4370g/BO21977.
|
Tabla 7. Alteraciones analíticas en los pacientes del estudio TDM4370g/BO21977 |
|||
|
Parámetro |
Trastuzumab emtansina |
||
|
Todos los grados (%) |
Grado 3 % |
Grado 4 % |
|
|
Hepáticos |
|||
|
Bilirrubina elevada |
21 |
<1 |
0 |
|
AST elevada |
98 |
8 |
<1 |
|
ALT elevada |
82 |
5 |
<1 |
|
Hemáticos |
|||
|
Cifra de plaquetas reducida |
85 |
14 |
3 |
|
Hemoglobina reducida |
63 |
54 |
1 |
|
Cifra de neutrófilos reducida |
41 |
4 |
<1 |
|
Potasio |
|||
|
Potasio reducido |
35 |
3 |
<1 |
Poscomercialización
Sin texto.
Interacciones con otros medicamentos y otras formas de interacción
No se han llevado a cabo estudios formales de interacciones farmacológicas con Kadcyla en el ser humano. Los estudios del metabolismo in vitro en microsomas hepáticos humanos indican que DM1, un componente de trastuzumab emtansina, es metabolizado principalmente por el CYP3A4 y, en menor grado, por el CYP3A5. In vitro, DM1 no induce ni inhibe el metabolismo mediado por el citocromo P450. Se debe tener precaución al administrar Kadcyla junto con inhibidores potentes del CYP3A.
DATOS PRECLÍNICOS SOBRE SEGURIDAD
Carcinogenicidad
No se han hecho estudios de carcinogenicidad con trastuzumab emtansina.
Mutagenicidad
No se ha observado ningún indicio de actividad mutagénica en un ensayo in vitro de mutación inversa en bacterias de DM1. En un ensayo de micronúcleos in vivo de trastuzumab emtansina en el macaco, no se observaron signos de daño cromosómico en las células de la médula ósea. Sin embargo, en un ensayo de micronúcleos en médula ósea de rata, DM1 mostró resultados positivos en la formación de micronúcleos después de administrar una dosis baja única en el intervalo de concentración de DM1 en el ser humano tras la administración de trastuzumab emtansina, lo que confirma que Kadcyla es aneugénico, clastogénico o ambas cosas.
Trastornos de la fecundidad
No se han realizado estudios específicos de la fecundidad con trastuzumab emtansina. No obstante, considerando los resultados de los estudios generales de toxicología en animales, cabe prever que su uso tenga efectos adversos en la fecundidad.
Teratogenicidad
No se han realizado estudios específicos del desarrollo embriofetal en animales con trastuzumab emtansina. En el marco clínico se han identificado efectos secundarios de trastuzumab en el desarrollo, aunque no se había previsto en el programa no clínico. Además, en estudios no clínicos se han detectado efectos adversos de la maitansina en el desarrollo, lo que indica que DM1 —el componente maitansinoide citotóxico inhibidor de los microtúbulos de trastuzumab emtansina— tendrá un efecto similar teratógeno y potencialmente embriotóxico.
ADVERTENCIAS Y PRECAUCIONES
Advertencias y precauciones generales
Los pacientes tratados con Kadcyla deben tener un estado tumoral HER2-positivo, evaluado mediante la determinación de la sobreexpresión de la proteína HER2 o por amplificación génica.
Toxicidad pulmonar
En ensayos clínicos con Kadcyla se han notificado casos de neumopatía intersticial, incluida la neumonía, algunos de los cuales dieron lugar a un síndrome de dificultad respiratoria aguda o tuvieron un desenlace mortal (ver Reacciones adversas). Entre los signos y síntomas se encuentran la disnea, la tos, la fatiga y los infiltrados pulmonares. Se recomienda suspender permanentemente el tratamiento con Kadcyla en pacientes con diagnóstico de neumopatía intersticial o neumonía.
Los pacientes que padecen disnea en reposo debido a complicaciones del cáncer avanzado o a enfermedades concomitantes pueden tener mayor riesgo de sufrir eventos pulmonares.
Hepatotoxicidad
Se han observado casos de hepatotoxicidad, predominantemente en forma de aumentos asintomáticos de la concentración de las aminotransferasas séricas (ele-vación de grado 1- 4), bajo tratamiento con Kadcyla en ensayos clínicos (ver Reacciones adversas). Las elevaciones de las aminotransferasas fueron generalmente transitorias; la elevación máxima se registró el día 8 después de iniciar el tratamiento, y los valores se recuperaron posteriormente, hasta alcanzar un grado ≤1 antes del siguiente ciclo. También se ha observado un efecto acumulativo de Kadcyla en las aminotransferasas. En la mayoría de los pacientes con concentración elevada de aminotransferasas, la concentración mejoró hasta alcanzar un grado 1 o un valor normal en un plazo de 30 días desde la administración de la última dosis de Kadcyla. En pacientes tratados con Kadcyla en ensayos clínicos, se observaron trastornos hepatobiliares graves, entre otros la hiperplasia regenerativa nodular del hígado y algunos trastornos con desenlace mortal debido a una lesión hepática inducida por fármacos. En los casos observados quizá existieron factores de confusión como enfermedades concomitantes o medicamentos administrados concomitantemente con conocido potencial hepatotóxico.
La función hepática debe controlarse antes de iniciar el tratamiento y antes de administrar cada dosis de Kadcyla. Las reducciones de la dosis o la suspensión del tratamiento por elevación de las aminotransferasas séricas y la bilirrubina total se especifican.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
Para impedir que se produzcan errores de medicación, es importante comprobar las etiquetas de los viales para asegurarse de que el medicamento que se está preparando y administrando es Kadcyla (trastuzumab emtansina) y no trastuzumab.
Kadcyla debe administrarse únicamente bajo la supervisión de un profesional sanitario con experiencia en el tratamiento de pacientes con cáncer.
Los pacientes tratados con Kadcyla deben presentar un estado tumoral HER2-positivo, definido como una puntuación 3+ en un análisis validado de inmunohistoquímica (IHQ) o un índice ≥2,0 en un análisis validado de hibridación in situ (ISH).
Para mejorar la trazabilidad de los biomedicamentos, debe registrarse (o declararse) claramente el nombre comercial y el número de lote del producto administrado en la historia clínica del paciente.
La sustitución por cualquier otro biomedicamento requie-re el consentimiento del médico prescriptor.
Kadcyla debe ser reconstituido y diluido por un profesional sanitario y administrado en infusión intravenosa (i.v.) (ver Instrucciones especiales de uso, manipulación y eliminación). No se debe administrar en inyección i.v. lenta o rápida.
Pauta posológica
La dosis recomendada de Kadcyla es de 3,6 mg/kg administrados en infusión i.v. cada 3 semanas (ciclo de 21 días) hasta la progresión de la enfermedad o hasta la aparición de efectos secundarios inaceptables.
La dosis inicial se administrará en una infusión i.v. con una duración de 90 minutos. Se debe observar a los pacientes durante la infusión y durante al menos 90 minutos después de la dosis inicial para detectar la aparición de fiebre, escalofríos u otras reacciones relacionadas con la infusión. Se debe vigilar estrechamente el lugar de la infusión para descubrir una posible infiltración subcutánea durante la administración del fármaco (ver Advertencias y precauciones generales - Extravasación).
Si el paciente toleró bien las infusiones anteriores, las dosis posteriores de Kadcyla pueden administrarse en infusiones de 30 minutos; se observará al paciente durante las infusiones y hasta al menos 30 minutos después de su conclusión.
La velocidad de infusión de Kadcyla debe reducirse, o se interrumpirá la infusión, si el paciente desarrolla síntomas relacionados con la infusión (ver Advertencias y precauciones generales). Se suspenderá Kadcyla en caso de reacciones a la infusión potencialmente mortales.
Dosis diferidas u omitidas
Si se omite una dosis prevista, se debe administrar tan pronto como sea posible; no se esperará hasta el siguiente ciclo previsto. La pauta de administración debe ajustarse para mantener un intervalo de 3 semanas entre las dosis. La infusión puede administrarse a la velocidad que toleró el paciente en la infusión más reciente.
Modificación de la dosis
El tratamiento de los eventos adversos sintomáticos puede requerir la interrupción temporal, la reducción de la dosis o la suspensión del tratamiento con Kadcyla, según las pautas que se presentan en las tablas 1-5.
La dosis de Kadcyla no debe aumentarse de nuevo después de haber hecho una reducción de la dosis.
|
Tabla 1. Pauta de reducción de la dosis |
|
|
Pauta de reducción de la dosis |
Nivel de dosis |
|
Dosis inicial |
3,6 mg/kg |
|
Primera reducción de la dosis |
3 mg/kg |
|
Segunda reducción de la dosis |
2,4 mg/kg |
|
Necesidad de reducir más la dosis |
Suspender el tratamiento |
|
Tabla 2. Pautas de modificación de la dosis en caso de elevación de las aminotransferasas (AST y ALT) |
||
|
Grado 2 (>2,5 a ≤5 xLSN) |
Grado 3 (>5 a ≤20 x LSN) |
Grado 4 (> 20 X LSN) |
|
Tratar con el mismo nivel de dosis. |
No administrar Kadcyla hasta que los valores de AST y ALT se recuperen, hasta alcanzar un grado ≤2, y entonces reducir un nivel de dosis. |
Suspender el tratamiento con Kadcyla. |
|
ALT = alanina-aminotransferasa; AST = aspartato-aminotransferasa; LSN = límite superior de la normalidad. |
||
|
Tabla 3. Pautas de modificación de la dosis en caso de hiperbilirrubinemia (Ver Advertencias y precauciones generales - Hepatotoxicidad) |
||
|
Grado 2 |
Grado 3 (>3 a ≤10 x LSN) |
Grado 4 |
|
No administrar Kadcyla hasta que la bilirrubina total alcance un grado ≤1, y luego tratar con el mismo nivel de dosis. |
No administrar Kadcyla hasta que la bilirrubina total alcance un grado ≤1 y luego reducir un nivel de dosis. |
Suspender el tratamiento con Kadcyla. |
|
Tabla 4 Pautas de modificación de la dosis en caso de trombocitopenia |
|
|
Grado 3 |
Grado 4 |
|
25.000 a <50.000/mm3 |
<25.000/mm3 |
|
No administrar Kadcyla hasta que la cifra de plaquetas alcance un grado ≤1 (≥75.000/mm3), y luego tratar con el mismo nivel de dosis. |
No administrar Kadcyla hasta que la cifra de plaquetas alcance un grado ≤1 (≥75.000/mm3), y luego reducir un nivel de dosis. |
|
Tabla 5 Modificaciones de la dosis en caso de disfunción del ventrículo izquierdo (Ver Advertencias y precauciones generales - Disfunción del ventrículo izquierdo.) |
||||
|
ICC sintomática |
FEVI <40% |
FEVI de 40% a ≤45% y disminución ≥10% respecto al valor inicial |
FEVI de 40% a ≤45% y disminución <10% respecto al valor inicial |
FEVI >45% |
|
Suspender el tratamiento con Kadcyla. |
No administrar Kadcyla. Repetir la determinación de la FEVI en 3 semanas. Si se confirma que la FEVI es <40%, suspender el tratamiento con Kadcyla. |
No administrar Kadcyla. Repetir la determinación de la FEVI en 3 semanas. Si la FEVI no se ha recuperado hasta valores en un margen del 10% respecto a los valores iniciales, suspender el tratamiento con Kadcyla. |
Continuar el tratamiento con Kadcyla. Repetir la determinación de la FEVI en 3 semanas. |
Continuar el tratamiento con Kadcyla. |
Pautas posológicas especiales
Ancianos
No es necesario ajustar la dosis de Kadcyla en pacientes ≥65 años (ver Uso en geriatría).
Niños y adolescentes
No se han establecido la seguridad ni la eficacia de Kadcyla en pacientes pediátricos.
Insuficiencia renal
No es necesario ajustar la dosis inicial de Kadcyla en pacientes con insuficiencia renal leve o moderada (ver Farmacocinética en poblaciones especiales). No hay datos suficientes para determinar si sería necesario ajustar la dosis en pacientes con insuficiencia renal grave.
Insuficiencia hepática
No es necesario ajustar la dosis inicial en pacientes con insuficiencia hepática leve o moderada (ver Farmacocinética en poblaciones especiales). Kadcyla no se ha estudiado en pacientes con insuficiencia hepática grave. El tratamiento de pacientes con insuficiencia hepática debe instaurarse con precaución, debido a la conocida hepatotoxicidad observada con Kadcyla (ver Adverten-cias y precauciones - Advertencias y precauciones genera-les - Hepatotoxicidad).
USO EN POBLACIONES ESPECIALES
Embarazo
No se han realizado estudios clínicos de Kadcyla en embarazadas. Tampoco se han realizado estudios de toxicología en la reproducción y el desarrollo con Kadcyla.
El trastuzumab, un componente de Kadcyla, puede producir daño o muerte fetal si se administra a mujeres embarazadas. En el marco del uso después de la comercialización, se han notificado casos de oligohidramnios, algunos asociados a hipoplasia pulmonar mortal, en embarazadas que recibieron trastuzumab. Estudios en animales de la maitansina —un compuesto químico estrechamente relacionado, de la misma clase de maitansinoides que DM1— indican que es previsible que DM1, el componente citotóxico de Kadcyla que inhibe los microtúbulos, sea teratógeno y potencialmente embriotóxico.
No se recomienda administrar Kadcyla a las embarazadas. Se indicará a las mujeres que se queden embarazadas que se pongan en contacto con su médico, y se les debe advertir de la posibilidad de daño fetal. Si una mujer embarazada es tratada con Kadcyla, se recomienda la vigilancia estrecha por parte de un equipo multidisciplinario.
Mujeres en edad de concebir
Se advertirá a las pacientes que deben utilizar métodos anticonceptivos eficaces durante el tratamiento con Kadcyla y al menos durante los 7 meses posteriores a su conclusión.
Parto
Sin texto.
Lactancia
Se ignora si Kadcyla se excreta en la leche materna humana. Debido a que muchos fármacos se excretan en la leche humana y a la posibilidad de que los lactantes amamantados cuyas madres reciben Kadcyla sufran reacciones adversas graves, las pacientes deben dejar de amamantar a sus hijos antes de iniciar el tratamiento con Kadcyla; 7 meses después de concluir el tratamiento, las pacientes pueden amamantar a sus hijos.
Uso en pediatría
No se han determinado la seguridad ni la eficacia de Kadcyla en menores de 18 años.
Uso en geriatría
No hay suficientes datos para establecer la seguridad y la eficacia de Kadcyla en pacientes ≥75 años.
Insuficiencia renal
Ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales.
Insuficiencia hepática
Ver Pautas posológicas especiales y Farmacocinética en poblaciones especiales.
VÍA DE ADMINISTRACIÓN
Infusión intravenosa.
Declaración de esterilidad/radiactividad: producto estéril.
Posología y forma de administración - Modificaciones de la dosis
Kadcyla no se ha estudiado en pacientes con concentración de aminotransferasas séricas más de 2,5 veces por encima del LSN o con una bilirrubina total más de 1,5 veces por encima del LSN antes de iniciar el tratamien-to. Se suspenderá permanentemente el tratamiento con Kadcyla en pacientes con una concentración de aminotransferasas séricas más de 3 veces por encima del LSN y concomitantemente una concentración de bilirrubina total más de 2 veces por encima del LSN.
Se han identificado casos de hiperplasia regenerativa nodular del hígado en biopsias hepáticas de pacientes tratados con Kadcyla. La hiperplasia regenerativa nodular es una afección rara, caracterizada por una transformación benigna diseminada del parénquima hepático con formación de pequeños nódulos regenerativos, que puede dar lugar a una hipertensión portal no cirrótica. Su diagnóstico sólo puede confirmarse mediante un examen histopatológico. Se debe sospechar una hiperplasia regenerativa nodular en todos los pacientes con síntomas clínicos de hipertensión portal o un patrón de tipo cirrótico en la tomografía computarizada del hígado, pero que tengan una concentración de aminotransferasas normal y no presenten otras manifestaciones de cirrosis. Se suspenderá permanentemente el tratamiento con Kadcyla si se diagnostica una hiperplasia regenerativa nodular.
Disfunción del ventrículo izquierdo
Los pacientes tratados con Kadcyla tienen mayor riesgo de desarrollar una disfunción del ventrículo iz-quierdo. Se han dado casos de una fracción de eyección del ventrículo izquierdo (FEVI) <40% en pacientes tratados con Kadcyla, por lo que la insuficiencia cardiaca congestiva (ICC) sintomática es un riesgo potencial. Se deben realizar pruebas de evaluación de la función cardiaca (ecocardiograma o ventriculografía isotópica [MUGA] antes de iniciar el tratamiento y a intervalos regulares durante el mismo (por ejemplo, cada 3 meses). No se ha estudiado el uso de Kadcyla en pacientes con una FEVI <50% antes de iniciar el tratamiento. En Posología y forma de administración - Modificaciones de la dosis, se presentan pautas específicas sobre la modificación de la dosis y la suspensión del tratamiento.
Reacciones relacionadas con la infusión
No se ha estudiado el uso de Kadcyla en pacientes en los que se ha suspendido permanentemente el tratamien-to con trastuzumab debido a reacciones relacionadas con la infusión (RRI); no se recomienda el tratamiento con Kadcyla en estos pacientes.
En los ensayos clínicos de Kadcyla se han notificado RRI, caracterizadas por uno o más de los siguientes síntomas: rubefacción, escalofríos, pirexia, disnea, hipotensión arterial, sibilancias, broncoespasmo y taquicardia. En general, estos síntomas no fueron graves (ver Reacciones adversas). En la mayoría de los pacientes, estas reacciones se resolvieron en un plazo de unas horas a un día después de finalizar la infusión. El tratamiento con Kadcyla debe interrumpirse en pacientes que sufran RRI graves. El tratamiento con Kadcyla debe suspenderse permanentemente en caso de RRI potencialmente mortales (ver Posología y forma de administración - Modificaciones de la dosis).
Reacciones de hipersensibilidad
Se debe vigilar estrechamente a los pacientes para detectar reacciones de hipersensibilidad, especialmente durante la primera infusión. En ensayos clínicos del tratamiento con Kadcyla se han observado casos de hipersensibilidad, incluidas reacciones graves de tipo anafiláctico. Se debe disponer de medicamentos para tratar tales reacciones, así como de equipo de emergencia, para usarlo de inmediato si fuera preciso.
Hemorragia
Con el tratamiento con Kadcyla se han notificado casos de eventos hemorrágicos, incluidas hemorragias en el sistema nervioso central, respiratorias y gastrointestinales. Algunos de estos eventos hemorrágicos tuvieron un desenlace mortal. En algunos de los casos observados, los pacientes también estaban recibiendo tratamiento anticoagulante o tratamiento antiplaquetario o presentaban trombocitopenia; en otros casos, no había otros factores de riesgo conocidos. Cuando, desde el punto de vista médico, sea preciso el uso concomitante estos fármacos, deberán usarse con precaución y considerarse la necesidad de un control adicional.
Trombocitopenia
En ensayos clínicos de Kadcyla se han notificado casos de trombocitopenia. La mayoría de estos pacientes presentaron eventos de grado 1 o 2 (≥50.000 plaquetas/mm3), el valor mínimo se alcanzó el día 8 y generalmente la cifra de plaquetas mejoró hasta alcanzar un grado 0 o 1 (≥75.000 plaquetas/mm3) en el momento de la siguiente dosis programada. En ensayos clínicos, la incidencia y la gravedad de la trombocitopenia fueron mayores en los pacientes asiáticos.
Durante el tratamiento con Kadcyla se debe controlar estrechamente a los pacientes con trombocitopenia (<100.000 plaquetas/mm3) y a los pacientes bajo tratamiento anticoagulante. Antes de administrar cada dosis de Kadcyla se recomienda controlar la cifra de plaquetas. No se ha estudiado el uso de Kadcyla en pacientes con una cifra de plaquetas <100.000/mm3 antes de iniciar el tratamiento. Si el paciente presenta una cifra de plaquetas reducida hasta un grado ≥3 (<50.000/mm3), no se debe administrar Kadcyla hasta que el número de plaquetas recupere un grado 1 (≥75.000/mm3). ver Posología y forma de administración - Modificaciones de la dosis.
Neurotoxicidad
En ensayos clínicos de Kadcyla se han referido casos de neuropatía periférica, principalmente de grado 1 y predominantemente sensitivas. En pacientes que sufran una neuropatía periférica de grado 3 o 4, el tratamiento con Kadcyla se suspenderá temporalmente hasta que los síntomas se resuelvan o mejoren hasta alcanzar un grado ≤2. Se controlará clínicamente a los pacientes de manera regular para detectar signos y síntomas de neurotoxicidad.
Extravasación
En los estudios clínicos de Kadcyla se han observado reacciones secundarias a la extravasación. Estas reacciones —eritema, dolor a la palpación, irritación, dolor o tumefacción de la piel en el lugar de la infusión— fueron en general leves, y se observaron más frecuentemente en las 24 horas posteriores a la infusión. Actualmente no se conoce el tratamiento específico de la extravasación de Kadcyla. Se debe vigilar minuciosamente el lugar de la infusión para descubrir cualquier posible infiltración subcutánea durante la administración del fármaco.
Abuso y dependencia del fármaco
Sin texto.
SOBREDOSIS
No existe ningún antídoto conocido para la sobredosis de trastuzumab emtansina. En caso de sobredosis, se debe vigilar estrechamente al paciente. Se han notificado casos de sobredosis con trastuzumab emtansina, la mayoría de ellos asociados a trombocitopenia; un caso fue mortal. En este caso, el paciente había recibido incorrectamente trastuzumab emtansina en dosis de 6 mg/kg y murió unas 3 semanas después de la sobredosis; no se determinó una causa de la muerte ni una relación causal con Kadcyla.
DESCRIPCIÓN
Clase terapéutica o farmacológica del fármaco: antineoplásico, conjugado de anticuerpo y fármaco.
Código ATC: no disponible aún.
PRESENTACIONES COMERCIALES
Caja x 1 vial con 100 mg de trastuzumab emtansina + prospecto.
Caja x 1 vial con 160 mg de trastuzumab emtansina + prospecto.
Medicamento biotecnológico innovador.
Información de marzo de 2017.
Fabricado para F. Hoffmann-La Roche S.A. Basilea, Suiza por F. Hoffmann-La Roche Ltd, Kaiseraugst, Suiza.
Medicamento: manténgase fuera del alcance de los niños.
ROCHE ECUADOR S.A.
Casilla 1711- 06185 CCI
Quito - Ecuador