GAZYVA®/GAZYVARO™
OBINUTUZUMAB
Concentrado en solución para infusión
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Principio activo: obinutuzumab.
GAZYVA es un líquido transparente, entre incoloro y ligeramente pardo, que se presenta como una dosis única de 1000 mg en un vial de vidrio de 50 ml con 40 ml de concentrado líquido apirógeno, estéril y sin conservantes (25 mg/ml).
Obinutuzumab es un anticuerpo monoclonal anti-CD20 humanizado tipo II de la subclase IgG1 obtenido mediante la humanización del anticuerpo murino B-Ly1 parental y producido a partir de un cultivo de células de ovario de hámster chino mediante técnicas de ADN recombinante.
Excipientes: L-Histidina, L-Histidina clorhidrato monohidrato, dihidrato de trehalosa y poloxámero 188.
FORMA FARMACÉUTICA: Concentrado para solución para infusión.
PROPIEDADES Y EFECTOS FARMACOLÓGICOS
Propiedades farmacodinámicas
Mecanismo de acción: GAZYVA es un anticuerpo monoclonal recombinante humanizado anti-CD20 de tipo II, del isotipo IgG1, obtenido mediante glicoingeniería. Actúa específicamente contra el dominio extracelular del antígeno transmembranario CD20 presente en la superficie de los linfocitos pre-B y de los linfocitos B maduros, tanto malignos como no malignos, pero no en la superficie de los hemocitoblastos, los linfocitos pro-B, los plasmocitos normales u otros tejidos normales. La modificación del fragmento Fc de GAZYVA mediante glicoingeniería determina que la afinidad de este anticuerpo por los receptores Fc?RIII presentes en células inmunitarias efectoras como los linfocitos citolíticos naturales (NK) y los macrófagos y monocitos sea mayor que la de los anticuerpos no sometidos a dicha modificación.
En estudios no clínicos, GAZYVA induce la muerte celular directa e interviene en la citotoxicidad celular dependiente de anticuerpos (CCDA) y en la fagocitosis celular dependiente de anticuerpos (FCDA) por medio del reclutamiento de células efectoras del sistema inmunitario con receptores Fc?RIII. Además, GAZYVA muestra una baja citotoxicidad dependiente del complemento (CDC). En modelos animales, GAZYVA causa una profunda depleción de los linfocitos B y muestra una gran eficacia antineoplásica. En comparación con los anticuerpos anti-CD20 de tipo I, GAZYVA, un anticuerpo de tipo II, se caracteriza por inducir una mayor actividad citocida directa con una reducción concomitante de la citotoxicidad dependiente del complemento. En comparación con los anticuerpos anti-CD20 no modificados por glicoingeniería, GAZYVA se caracteriza por una mayor citotoxicidad celular dependiente de anticuerpos y una mayor fagocitosis celular dependiente de anti-cuerpos como consecuencia de dicha modificación por glicoingeniería. Esto se traduce en una mayor depleción de linfocitos B y una eficacia antitumoral superior en modelos animales.
Efectos farmacodinámicos: en el ensayo clínico fundamental, BO21004/CLL11, el 91 % (40 de 44) de los pacientes evaluables tratados con GAZYVA presentaban depleción de los linfocitos B (definida como una cifra de linfocitos B CD19+ <0,07 × 109/l) al final del periodo de tratamiento, y se mantuvieron en ese estado durante los 6 primeros meses de seguimiento. La cifra de linfocitos B se recuperó al cabo de 12-18 meses de seguimiento en el 35 % (14 de 40) de los pacientes sin enfermedad progresiva y en el 13 % (5 de 40) de los pacientes con enfermedad progresiva.
Ensayos clínicos/eficacia
Leucemia linfocítica crónica: se llevó a cabo un ensayo clínico internacional y multicéntrico de fase III, aleatorizado, sin enmascaramiento, con tres grupos, que se desarrolló en dos etapas (BO21004/CLL11); en él se compararon la seguridad y la eficacia de GAZYVA más clorambucilo, de rituximab más clorambucilo y de clorambucilo solo en pacientes aquejados de leucemia linfocítica crónica no tratada anteriormente y de otras afecciones concomitantes.
Se requería que, antes de incorporarse al estudio, los pacientes presentaran una LLC CD20+ documentada y al menos uno de dos indicadores de afecciones concomitantes, a saber: una puntuación de comorbilidad (puntuación total en la Escala de Valoración Acumulativa de Enfermedades [Cumulative Illness Rating Scale, CIRS]) >6 y un ClCr <70 ml/min indicativo de una afectación de la función renal. Se excluyó a los pacientes con función hepática insuficiente (grado 3 de los NCI-CTC en las pruebas de la función hepática [AST, ALT más de 5 veces por encima del LSN durante >2 semanas; bilirrubina más de 3 veces por encima del LSN]) y con función renal insuficiente (ClCr <30 ml/min).
Un total de 781 pacientes fueron asignados aleatoriamente, en una proporción 2:2:1, a recibir GAZYVA más clorambucilo, rituximab más clorambucilo o clorambucilo solo. En la etapa 1 se comparó GAZYVA más clorambucilo con el clorambucilo solo en 356 pacientes, y en la etapa 2 se comparó GAZYVA más clorambucilo con el rituximab más clorambucilo en 663 pacientes. Los resultados relativos a la eficacia se resumen en la tabla 8 y en las figuras 1-3.
La mayoría de los pacientes recibieron GAZYVA por vía i.v. en una dosis inicial de 1000 mg administrada el día 1, el día 8 y el día 15 del primer ciclo de tratamiento. Para reducir la incidencia de RRI en los pacientes, se introdujo una enmienda en virtud de la cual 140 pacientes recibieron la primera dosis de GAZYVA administrada en 2 días (día 1 [100 mg] y día 2 [900 mg]) (v. 2.2 Posología y forma de administración). En cada ciclo de tratamiento posterior (ciclos 2-6), los pacientes recibieron 1000 mg de GAZYVA sólo el día 1. El clorambucilo se administró por vía oral en dosis de 0,5 mg/kg el día 1 y el día 15 de todos los ciclos de tratamiento (1-6).
Las características demográficas y las características iniciales estaban adecuadamente equilibradas entre los grupos de tratamiento. La mayoría de los pacientes reclutados eran de raza blanca (95 %) y de sexo masculino (61 %). La mediana de la edad fue de 73 años, y el 44 % de los pacientes tenían 75 años o más. Al inicio del estudio, el 22 % de los pacientes tenían una LLC en estadio A según la clasificación de Binet, el 42 % una LLC en estadio B y el 36 % una LLC en estadio C. La mediana de la puntuación de comorbilidad fue de 8, y el 76 % de los pacientes participantes tenían una puntuación de comorbilidad >6. La mediana del ClCr calculado fue de 62 ml/min, y el 66 % de todos los pacientes tenían un ClCr <70 ml/min. El 42 % de los pacientes incluidos en el estudio tenían un ClCr <70 ml/min además de una puntuación de comorbilidad >6. El 34 % de los pacientes fueron reclutados basándose solo en la puntuación de la comorbilidad, y el 23 % de los pacientes reclutados sólo tenían una disfunción renal.
Las afecciones médicas coexistentes notificadas con mayor frecuencia (utilizando un valor límite ≥30 %), por clases de órganos y sistemas del MedDRA, son: trastornos vasculares, 73 %; trastornos cardiacos, 46 %; trastornos gastrointestinales, 38 %; trastornos del metabolismo y de la nutrición, 40 %; trastornos renales y urinarios, 38 %; trastornos musculoesqueléticos y del tejido conjuntivo, 33 %.
La variable principal de valoración del estudio fue la supervivencia sin progresión evaluada por los investigadores (SSP-INV). Además, un comité de revisión independiente (CRI) valoró la progresión en todos los pacientes, y se evaluó la SSP determinada por el CRI (SSP-CRI).
Las principales variables secundarias de valoración de la eficacia fueron la tasa de respuesta al final del tratamiento, la remisión molecular al final del tratamiento (estado de enfermedad residual mínima) y variables basadas en el tiempo transcurrido hasta el evento (supervivencia sin eventos, nuevo tratamiento de la leucemia). Los datos de la supervivencia global de la etapa 1 se presentan en la figura 2. Se seguirá haciendo el seguimiento de la supervivencia global de la etapa 2, cuyos datos aún no son maduros.
|
Tabla 8. Resumen de la eficacia en el estudio BO21004 (CLL11) |
||||
|
Etapa 1 |
Etapa 2 |
|||
|
Clorambucilo N = 118 |
GAZYVA + clorambucilo N = 238 |
Rituximab + clorambucilo N = 330 |
GAZYVA + clorambucilo N = 333 |
|
|
Mediana del periodo de observación de 22,8 meses |
Mediana del periodo de observación de 18,7 meses |
|||
|
SSP evaluada por el investigador (SSP-INV)* N.º (%) de pacientes con eventos |
96 (81,4 %) |
93 (39,1 %) |
199 (60,3 %) |
104 (31,2 %) |
|
Mediana del tiempo transcurrido hasta el evento (meses) |
11,1 |
26,7 |
15,2 |
26,7 |
|
HR (IC 95 %) |
0,18 [0,13; 0,24] |
0,39 [0,31; 0,49] |
||
|
p (prueba de rangos logarítmicos estratificada†) |
<0,0001 |
<0,0001 |
||
|
SSP evaluada por el CRI (SSP-CRI)* N.º (%) de pacientes con eventos |
90 (76,3 %) |
89 (37,4 %) |
183 (55,5 %) |
103 (30,9 %) |
|
Mediana del tiempo transcurrido hasta el evento (meses) |
11,2 |
27,2 |
14,9 |
26,7 |
|
HR (IC 95 %) |
0,19 [0,14; 0,27] |
0,42 [0,33; 0,54] |
||
|
p (prueba de rangos logarítmicos estratificada†) |
<0,0001 |
<0,0001 |
||
|
Tasa de respuesta al final del tratamiento N.º de pacientes incluidos en el análisis |
118 |
238 |
329 |
333 |
|
Pacientes con respuesta (%) |
37 (31,4 %) |
184 (77,3 %) |
214 (65,0 %) |
261 (78,4 %) |
|
Pacientes sin respuesta (%) |
81 (68,6 %) |
54 (22,7 %) |
115 (35,0 %) |
72 (21,6 %) |
|
Diferencia entre las tasas de respuesta (IC 95 %) |
45,95 [35,6; 56,3] |
13,33 [6,4; 20,3] |
||
|
p (prueba de la X2) |
<0,0001 |
0,0001 |
||
|
Número de pacientes con respuesta completa‡ (%) |
0 (0,0 %) |
53 (22,3 %) |
23 (7,0 %) |
69 (20,7 %) |
|
Remisión molecular al final del tratamiento§ N.º de pacientes incluidos en el análisis |
90 |
168 |
244 |
239 |
|
ERM: resultado negativo¶ (%) |
0 (0 %) |
45 (26,8 %) |
6 (2,5 %) |
61 (25,5 %) |
|
ERM: resultado positivo\\ (%) |
90 (100 %) |
123 (73,2 %) |
238 (97,5 %) |
178 (74,5 %) |
|
Diferencia entre las tasas de ERM (IC 95 %) |
26,79 [19,5; 34,1] |
23,06 [17,0; 29,1] |
||
|
Supervivencia sin eventos N.º (%) de pacientes con eventos |
103 (87,3 %) |
104 (43,7 %) |
208 (63,0 %) |
118 (35,4 %) |
|
Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
10,8 |
26,1 |
14,3 |
26,1 |
|
HR (IC 95 %) |
0,19 [0,14; 0,25] |
0,43 [0,34; 0,54] |
||
|
p (prueba de rangos logarítmicos estratificada†) |
<0,0001 |
<0,0001 |
||
|
Tiempo transcurrido hasta el inicio de un nuevo tratamiento de la leucemia N.º (%) de pacientes con eventos |
65 (55,1 %) |
51 (21,4 %) |
86 (26,1 %) |
55 (16,5 %) |
|
Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
14,8 |
- |
30,8 |
- |
|
HR (IC 95 %) |
0,24 [0,16; 0,35] |
0,59 [0,42; 0,82] |
||
|
p (prueba de rangos logarítmicos estratificada†) |
<0,0001 |
0,0018 |
||
|
Supervivencia global N.º (%) de pacientes con eventos |
24 (20,3 %) |
22 (9,2 %) |
41 (12,4 %) |
28 (8,4 %) |
|
Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
NA |
NA |
NA** |
NA** |
|
HR (IC 95 %) |
0,41 [0,23; 0,74] |
0,66 [0,41; 1,06]** |
||
|
p (prueba de rangos logarítmicos estratificada†) |
0,0022 |
0,0849** |
||
|
CRI: Comité de revisión independiente; ERM: enfermedad residual mínima; HR: razón de riesgos instantáneos (hazard ratio); IC: intervalo de confianza; SSP: supervivencia sin progresión. * Definido como el tiempo transcurrido desde la aleatorización hasta la primera aparición de la progresión, la recidiva o el fallecimiento por cualquier causa según la evaluación del investigador. |
||||
|
† Estratificada según el estadio de la clasificación de Binet al inicio del estudio. ‡ Incluye a 11 pacientes del grupo de GAZYVA más clorambucilo con una respuesta completa y recuperación incompleta de la médula ósea. § Sangre y médula ósea en conjunto. ¶ La negatividad de la ERM se define como un resultado <0,0001. \\ Incluye a los pacientes con ERM y a los pacientes que presentaron una progresión de la enfermedad o fallecieron antes de concluir el tratamiento. NA = No alcanzada ** Los datos aún no son maduros. |
||||
Los resultados del análisis de subgrupos de la SSP (es decir, sexo, edad, estadio de la clasificación de Binet, ClCr, puntuación en la escala CIRS, microglobulina β2, estado del gen IGVH, anomalías cromosómicas, cifra de linfocitos al inicio del estudio) eran congruentes con los observados en el conjunto de la población por intención de tratar. El riesgo de progresión de la enfermedad o de muerte fue menor en el grupo de GAZYVA más clorambucilo (GClb) que en el grupo de rituximab más clorambucilo (RClb) y el grupo de clorambucilo solo (Clb) en todos los subgrupos. La hazard ratio (razón de riesgos instantáneos) osciló de 0,08-0,42 con GClb en comparación con Clb a 0,28-0,71 con GClb en comparación con RClb.
Figura 1. Curva de Kaplan-Meier de la supervivencia sin progresión evaluada por los investigadores (etapa 1)
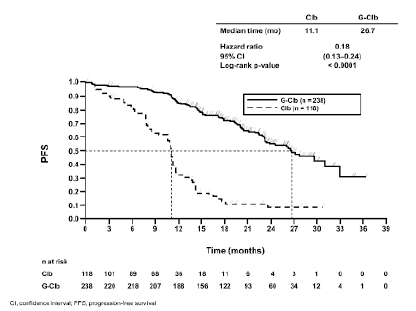
De arriba abajo: Median time (mo) = Mediana del tiempo (meses); Hazard ratio = Hazard ratio; 95% CI = (IC 95%); Log-rank p-value = p (rangos logarítmicos); PFS = SSP; Time (months) = Tiempo (meses); n at risk = n en riesgo; Clb = Clb; G-Clb = G-Clb; CI, confidence interval = IC: intervalo de confianza; PFS, progression-free survival = SSP: supervivencia sin progresión.
Figura 2. Curva de Kaplan-Meier de la supervivencia global (etapa 1)
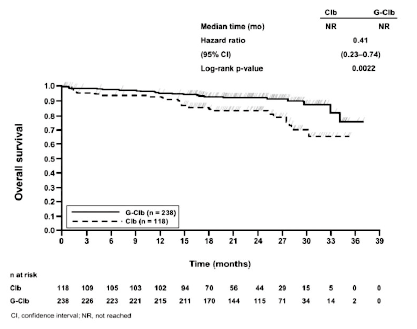
De arriba abajo: Median time (mo) = Mediana del tiempo (meses); Hazard ratio = Hazard ratio; 95% CI = (IC 95%); Log-rank p-value = p (rangos logarítmicos); Overall survival = Supervivencia global; Time (months) = Tiempo (meses); n at risk = n en riesgo; Clb = Clb; G-Clb = G-Clb; CI, confidence interval = IC: intervalo de confianza; NR, not reached = NA: no alcanzado.
Figura 3. Curva de Kaplan-Meier de la supervivencia sin progresión evaluada por los investigadores (etapa 2)
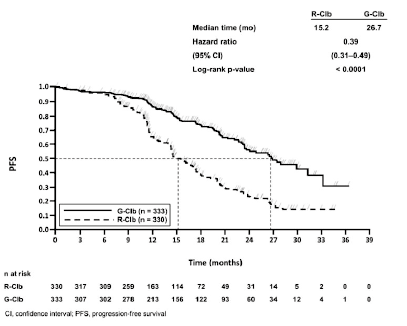
De arriba abajo: Median time (mo) = Mediana del tiempo (meses); Hazard ratio = Hazard ratio; 95% CI = (IC 95%); Log-rank p-value = p (rangos logarítmicos); PFS = SSP; Time (months) = Tiempo (meses); n at risk = n en riesgo; R-Clb = R-Clb; G-Clb = G-Clb; CI, confidence interval = IC: intervalo de confianza; PFS, progression-free survival = SSP: supervivencia sin progresión.
Resultados referidos por los pacientes: no se observaron diferencias sustanciales en ninguna de las subescalas de los cuestionarios QLQC30 y QLQ-CLL-16 administrados durante el periodo de tratamiento. Los datos del seguimiento son limitados, en especial los del grupo del clorambucilo solo. Sin embargo, hasta la fecha no se han identificado diferencias notables en la calidad de vida durante el seguimiento.
Las evaluaciones de la calidad de vida relacionada con la salud, que se refieren específicamente a la fatiga durante el periodo de tratamiento, no muestran diferen-cias estadísticamente significativas que indiquen que la adición de GAZYVA al clorambucilo aumente la fatiga que sienten los pacientes.
Linfoma no hodgkiniano (linfoma folicular)
Pacientes con linfoma folicular sin tratamiento previo: en un estudio de fase III multicéntrico, aleatorizado y sin enmascaramiento (BO21223/GALLIUM), se evaluó a 1202 pacientes con linfoma folicular en estadio II (masa tumoral voluminosa)/III/IV que no habían recibido previamente tratamiento. Los pacientes fueron asignados aleatoriamente, en una proporción 1:1, a recibir GAZYVA o rituximab en combinación con quimioterapia (CHOP, CVP o bendamustina), y a continuación tratamiento de mantenimiento con GAZYVA o rituximab en los pacientes que habían presentado una respuesta completa o parcial.
Los datos demográficos y las características iniciales de la población de pacientes con linfoma folicular estaban adecuadamente equilibrados (la mediana de la edad fue de 59 años, la mayoría de los pacientes [81 %] eran de raza blanca y de sexo femenino [53 %]). El 79 % tenían una puntuación en el índice FLIPI (Follicular Lymphoma International Prognostic Index) ≥2; el 7 % tenían una enfermedad en estadio II (masa tumoral voluminosa), el 35 % en estadio III y 57 % en estadio IV. El 57 % de los pacientes recibieron bendamustina; el 33 %, CHOP; y el 10 %, CVP. El 44 % tenían una masa tumoral voluminosa (>7 cm), el 34 % tenían al menos un síntoma B al inicio del estudio y el 97 % tenían al inicio del estudio un estado general de 0-1 según la escala del ECOG.
GAZYVA (1000 mg) se administró por vía i.v. (tal como se indica en 2.2 Posología y forma de administración) antes de la quimioterapia. La bendamustina se administró por vía i.v. los días 1 y 2 en todos los ciclos de tratamiento (ciclos 1-6) en una dosis de 90 mg/m2 al día cuando se administró en combinación con GAZYVA. Se administró la pauta posológica habitual de CHOP y CVP. Después de los ciclos 6-8, en los que GAZYVA se administró en combinación con quimioterapia, se administró tratamiento de mantenimiento con GAZYVA cada 2 meses durante 2 años en los pacientes con respuesta o hasta la progresión de la enfermedad.
Los resultados relativos a la eficacia se resumen en la tabla 9. En la figura 4 se muestran las curvas de Kaplan-Meier de la SSP.
|
Tabla 9. Resumen de la eficacia en pacientes con linfoma folicular del estudio BO21223 (GALLIUM) |
||
|
Rituximab + quimioterapia y posteriormente mantenimiento con rituximab N = 601 |
GAZYVA + quimioterapia y posteriormente mantenimiento con GAZYVA N = 601 |
|
|
Mediana del periodo de observación 34 meses |
Mediana del periodo de observación 35 meses |
|
|
Rituximab + quimioterapia y posteriormente mantenimiento con rituximab N = 601 |
GAZYVA + quimioterapia y posteriormente mantenimiento con GAZYVA N = 601 |
|
|
Variable de valoración principal |
||
|
SSP evaluada por el investigador§ (SSP-INV) N.º (%) de pacientes con eventos |
144 (24,0 %) |
101 (16,8 %) |
|
HR [IC 95 %] |
0,66 [0,51, 0,85] |
|
|
p (prueba de rangos logarítmicos estratificada*) |
0,0012 |
|
|
Estimación de la SSP a los 2 años [IC 95 %] |
80,9 [77,4, 84,0] |
87,7 [84,6, 90,1] |
|
Estimación de la SSP a los 3 años [IC 95 %] |
73,3 [68,8, 77,2] |
80,0 [75,9, 83,6] |
|
Variables de valoración fundamentales |
||
|
SSP evaluada por el CRI§ (SSP-CRI) N.º (%) de pacientes con eventos |
125 (20,8 %) |
93 (15,5 %) |
|
HR [IC 95 %] |
0,71 [0,54, 0,93] |
|
|
p (prueba de rangos logarítmicos estratificada*) |
0,0138 |
|
|
Estimación de la SSP a los 2 años [IC 95 %] |
82,0 [78,5, 85,0] |
87,2 [84,1, 89,7] |
|
Estimación de la SSP a los 3 años [IC 95 %] |
77,9 [73,8, 81,4] |
81,9 [77,9, 85,2] |
|
Tiempo transcurrido hasta la administración del siguiente tratamiento contra el linfoma N.º (%) de pacientes con eventos |
111 (18,5 %) |
80 (13,3 %) |
|
HR [IC 95 %] |
0,68 [0,51, 0,91] |
|
|
p (prueba de rangos logarítmicos estratificada*) |
0,0094 |
|
|
Supervivencia global N.º (%) de pacientes con eventos |
46 (7,7 %) |
35 (5,8 %) |
|
HR [IC 95 %] |
0,75 [0,49, 1,17]¶ |
|
|
p (prueba de rangos logarítmicos estratificada*) |
0,21¶ |
|
|
Tasa de respuesta global** al final de la inducción‡ (evaluada por el INV, TC) Pacientes con respuesta (%) (RC, RP) |
522 (86,9 %) |
532 (88,5 %) |
|
Diferencia entre las tasas de respuesta (%) [IC 95 %] |
1,7 % [-2,1 %, 5,5 %] |
|
|
p (prueba de Cochran-Mantel-Haenszel) |
0,33 |
|
|
Respuesta completa (RC) |
143 (23,8 %) 1 |
17 (19,5 %) |
|
IC 95 % (Clopper-Pearson) |
[20,4 %, 27,4 %] |
[16,4 %, 22,9 %] |
|
Respuesta parcial (RP) |
379 (63,1 %) |
[415 (69,1 %) |
|
IC 95 % (Clopper-Pearson) |
59,1 %, 66,9 %] |
[65,2 %, 72,7 %] |
|
Tasa de conversión desde el final de la inducción Pacientes con RP al final de la inducción |
222 |
271 |
|
Conversión de RP a RC |
97 (43,7 %) |
134 (49,4 %) |
|
Diferencia entre las tasas (%) [IC 95 %] |
5,7 % [-3,1 %, 14,6 %] |
|
|
Tasa de respuesta global al final del tratamiento de mantenimiento Pacientes evaluados al final del mantenimiento |
533 |
525 |
|
Pacientes con respuesta (%) (RC, RP) |
341 (64,0 %) |
371 (70,7 %) |
|
Diferencia entre las tasas de respuesta (%) [IC 95 %] |
6,7 % [1,0 %, 12,4 %] |
|
|
p (prueba de Cochran-Mantel-Haenszel) |
0,0197 |
|
|
Respuesta completa (RC) |
195 (36,6 %) |
205 (39,0 %) |
|
IC 95 % (Clopper-Pearson) |
[32,5 %, 40,8 %] |
[34,9 %, 43,4 %] |
|
Respuesta parcial (RP) |
146 (27,4 %) |
166 (31,6 %) |
|
IC 95 % (Clopper-Pearson) |
[23,7 %, 31,4 %] |
[27,7 %, 35,8 %] |
|
CRI: Comité de revisión independiente; HR: razón de riesgos instantáneos (hazard ratio); IC: intervalo de confianza; NA = no alcanzada; SSP: supervivencia sin progresión. * Los factores de estratificación fueron la pauta de quimioterapia, el grupo de riesgo según el índice FLIPI relativo al linfoma folicular y la región geográfica). ¶ Los datos aún no son maduros. La mediana no se había alcanzado en el momento del análisis. ‡ Final de la inducción = final de la fase de inducción; no incluye el mantenimiento con monoterapia. ** Evaluada según los criterios de Cheson del 2007 modificados. § Nivel de significación en este análisis provisional de la eficacia: 0,012. |
||
Se contó con datos sobre las tasas de respuesta al final de la inducción, evaluadas mediante tomografía por emisión de positrones, de 297/601 pacientes en el grupo de GAZYVA más quimioterapia y de 298/601 pacientes en el grupo del rituximab más quimioterapia del estudio. Las tasas de respuesta completa al final de la inducción, evaluadas mediante tomografía por emisión de positrones, fueron del 62,3 % en el grupo de GAZYVA más quimioterapia y del 56,7 % en el grupo del rituximab más quimioterapia. Las tasas de respuesta global fueron similares en ambos grupos, con una diferencia del 4,3 % a favor del grupo de GAZYVA más quimioterapia (85,9 % en el grupo de GAZYVA más quimioterapia frente al 81,5 % en el grupo de rituximab más quimioterapia).
Figura 4. Estimaciones de Kaplan-Meier de la supervivencia sin progresión en pacientes con linfoma folicular evaluada por los investigadores
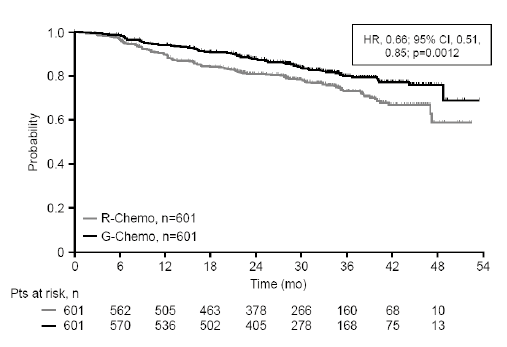
HR = HR; 95% CI = (IC 95%); Probability = Probabilidad; R-Chemo = R-quimio; G-Chemo = G-quimio; Time (months) = Tiempo (meses); Pts at risk, n = Pacientes en riesgo, n.
G-quimio: GAZYVA más quimioterapia; HR: hazard ratio; IC: intervalo de confianza; R-quimio: rituximab más quimioterapia.
Resultados de los análisis de subgrupos: en gene-ral, los resultados de los análisis de subgrupos fueron coherentes con los resultados observados en la población de pacientes con linfoma folicular, lo que respalda la robustez del resultado global. Los subgrupos evaluados incluyeron el IPI, el FLIPI, la pauta de quimioterapia, la masa tumoral voluminosa, los síntomas B al inicio del estudio, el estadio según la clasificación de Ann Arbor y el estado general según la escala del ECOG al inicio del estudio.
Linfoma folicular recidivante o resistente al tratamiento: en un estudio de fase III multicéntrico, aleatorizado y sin enmascaramiento (GAO4753g/GADOLIN), se evaluó a 396 pacientes con LNH de baja malignidad que no habían respondido o habían presentado una progresión de la enfermedad durante o hasta 6 meses después del tratamiento con rituximab o con un régimen que contuviera rituximab. Los pacientes fueron asignados aleatoriamente, en una proporción 1:1, a recibir bendamustina (B) sola (n = 202) o GAZYVA en combinación con bendamustina (G + B) (n = 194) durante 6 ciclos, cada uno de ellos de 28 días de duración. Los pacientes del grupo de G + B que no habían presentado una progresión de la enfermedad (es decir, los pacientes con una respuesta completa [RC], una respuesta parcial [RP] o una enfermedad estable [EE]) al final de la inducción siguieron recibiendo tratamiento de mantenimiento con GAZYVA hasta la progresión o hasta completar 2 años (lo que antes ocurriera).
Los datos demográficos y las características iniciales estaban adecuadamente equilibrados (la mediana de la edad fue de 63 años; la mayoría de los pacientes eran de raza blanca [88 %] y varones [58 %]). La mediana del tiempo transcurrido desde el diagnóstico inicial fue de 3 años, y la mediana del número de tratamientos previos fue de 2 (intervalo: 1-10); el 44 % de los pacientes habían recibido 1 tratamiento previo y el 34 %, 2 tratamientos previos.
GAZYVA se administró por vía i.v. en una dosis de 1000 mg los días 1, 8 y 15 del ciclo 1, el día 1 de los ciclos 2-6 y, en pacientes que no presentaban una progresión de la enfermedad, cada 2 meses hasta completar 2 años o hasta la progresión de la enfermedad. La bendamustina se administró por vía i.v. los días 1 y 2 en todos los ciclos de tratamiento (ciclos 1-6) en una dosis de 90 mg/m2 al día cuando se administró en combinación con GAZYVA o en una dosis de 120 mg/m2 al día cuando se administró sola.
El análisis principal demostró una reducción estadística y clínicamente significativa del 45 % del riesgo de progresión de la enfermedad o fallecimiento, según la evaluación del CRI, en pacientes con LNH de baja malignidad que recibieron G + B y a continuación tratamiento de mantenimiento con GAZYVA en comparación con la bendamustina sola (valor p de la prueba de rangos logarítmicos estratificada = 0,0001). Las tasas de respuesta según la evaluación del CRI al final del tratamiento de inducción y la mejor respuesta global evaluada por el CRI en un plazo de 12 meses desde el inicio del tratamiento fueron similares en los dos grupos de tratamiento. En el momento del análisis principal, la mediana del tiempo transcurrido hasta el evento en el grupo de la bendamustina fue de 14,9 meses (IC 95 %: 12,8-16,6) y la mediana no se alcanzó en el grupo de G + B (IC 95 %: 22,5-No alcanzada). Aunque los datos sobre la supervivencia global todavía no son maduros, se realizó un análisis a posteriori 8 meses después de la fecha límite para la inclusión de datos en el análisis principal. Con un perio-do de observación de 24,1 meses (mediana) en el caso de los pacientes con linfoma folicular, 48 pacientes (28,1 %) del grupo de B y 30 (18,3 %) del grupo de G + B habían fallecido. La mejoría observada en la supervivencia global en el grupo de G + B estaba respaldada por una HR estratificada de la supervivencia global de 0,62 (IC 95 %: 0,39-0,98) en este análisis a posteriori. La mediana de la supervivencia global (SG) no se había alcanzado aún en ningún grupo. Los resultados de la SSP en el análisis a posteriori son congruentes con los del análisis principal, su significación no ha cambiado, y el perfil de seguridad concuerda con el del análisis principal.
La mayoría (81,1 %) de los pacientes tenían un linfoma folicular (LF). Los resultados sobre la eficacia en la población con linfoma folicular se muestran en la tabla 10. Entre los pacientes con linfoma no folicular, el 11,6 % tenían un linfoma de la zona marginal (LZM) y el 7,1 % tenían un linfoma linfocítico bien diferenciado (linfoma linfocítico de células pequeñas [LLP]). No pueden extraerse conclusiones sobre la eficacia en el linfoma de la zona marginal y el linfoma linfocítico de células pequeñas.
|
Tabla 10. Resumen de la eficacia en pacientes con linfoma folicular del estudio GAO4753g (GADOLIN) |
||
|
Bendamustina N = 166 |
G + B y a continuación tratamiento de mantenimiento con GAZYVA N = 155 |
|
|
Mediana del periodo de observación de 20 meses |
Mediana del periodo de observación de 22 meses |
|
|
Variable de valoración principal en la población con linfoma folicular |
||
|
SSP evaluada por el CRI (SSP-CRI) N.º (%) de pacientes con eventos |
90 (54,2 %) |
54 (34,8 %) |
|
Mediana de la duración de la SSP (meses) |
13,8 |
NA |
|
HR [IC 95 %] |
0,48 [0,34, 0,68] |
|
|
p (prueba de rangos logarítmicos estratificada*) |
<0,0001 |
|
|
Variables de valoración secundarias |
||
|
SSP evaluada por el investigador (SSP-INV) N.º (%) de pacientes con eventos |
102 (61,4 %) |
62 (40,0 %) |
|
Mediana de la duración de la SSP (meses) |
13,7 |
29,2 |
|
HR [IC 95 %] |
0,48 [0,35, 0,67] |
|
|
p (prueba de rangos logarítmicos estratificada*) |
<0,0001 |
|
|
Mejor respuesta global (MRG) (evaluada por el CRI)§ N.º de pacientes incluidos en el análisis |
161 |
153 |
|
Pacientes con respuesta (%) (RC, RP) |
124 (77,0 %) |
122 (79,7 %) |
|
Diferencia entre las tasas de respuesta (%) [IC 95 %] |
2,72 [-6,74, 12,18] |
|
|
p (prueba de Cochran-Mantel-Haenszel) |
0,6142¶ |
|
|
Duración de la respuesta (evaluada por el CRI) N.º de pacientes incluidos en el análisis |
127 |
122 |
|
N.º (%) de pacientes con eventos |
74 (58,3 %) |
36 (29,5 %) |
|
Mediana de la duración de la respuesta (meses) |
11,9 |
NA |
|
HR [IC 95 %] |
0,36 [0,24, 0,54] |
|
|
Supervivencia global N.º (%) de pacientes con eventos |
36 (21,7 %) |
25 (16,1 %) |
|
Mediana del tiempo transcurrido hasta la aparición del evento (meses) |
NA¶ |
NA¶ |
|
HR [IC 95 %] |
0,71 [0,43, 1,19]¶ |
|
|
p (prueba de rangos logarítmicos estratificada*) |
0,1976¶† |
|
|
Tasa de respuesta global al final de la inducción‡ (evaluada por el CRI) Pacientes evaluados al final del tratamiento |
155 |
149 |
|
Pacientes con respuesta (%) (RC, RP) |
97 (62,6 %) |
105 (70,5 %) |
|
Diferencia entre las tasas de respuesta (%) [IC 95 %] |
7,89 [-3,05, 18,83] |
|
|
p (prueba de Cochran-Mantel-Haenszel) |
0,1713 |
|
|
Respuesta completa (RC) |
21 (13,5 %) |
14 (9,4 %) |
|
Respuesta parcial (RP) |
76 (49,0 %) |
91 (61,1 %) |
|
Enfermedad estable (EE) |
15 (9,7 %) |
12 (8,1 %) |
|
Enfermedad progresiva (EP) |
15 (9,7 %) |
15 (10,1 %) |
|
No se pudo evaluar |
4 (2,6 %) |
3 (2,0 %) |
|
No consta (NA): |
24 (15,5 %) |
14 (9,4 %) |
|
CRI: Comité de revisión independiente; HR: razón de riesgos instantáneos (hazard ratio); IC: intervalo de confianza; NA = no alcanzada; SSP: supervivencia sin progresión. * Los factores de estratificación fueron el subtipo de LNH de baja malignidad (folicular en comparación con el no folicular: no se utilizó en el análisis de los pacientes con linfoma folicular), el tipo resistente al tratamiento (monoterapia con rituximab en comparación con rituximab + quimioterapia) y las terapias previas (≤2 frente a >2). |
||
|
§ Mejor respuesta en un plazo de 12 meses desde el inicio del tratamiento. ¶ Los datos aún no son maduros. ‡ Final de la inducción: final de la fase de inducción; no incluye el tratamiento de mantenimiento con monoterapia. |
||
Figura 4.1. Curva de Kaplan-Meier de la supervivencia sin progresión evaluada por el CRI en pacientes con linfoma folicular
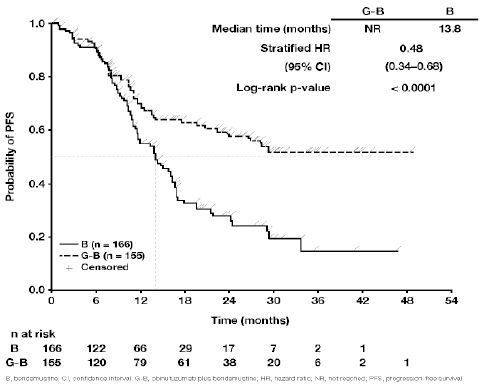
De arriba abajo: G-B = G-B; B = B; Median time (months) = Mediana del tiempo (meses); Stratified HR = HR estratificada; (95% CI) = (IC 95%); Long-rank p-value = p (rangos logarítmicos); Probability of PFS = Probabilidad de SSP; Censored = Datos bajo censura estadística; Time (months) = Tiempo (meses); n at risk = n en riesgo.
B: bendamustina; G-B: obinutuzumab más bendamustina; HR: hazard ratio; IC: intervalo de confianza; NA: no alcanzado; SSP: supervivencia sin progresión.
Resultados de los análisis de subgrupos: los resultados de los análisis de subgrupos estaban en general en consonancia con los observados en la población global de pacientes con linfoma folicular, lo que respalda la robustez del resultado global.
Figura 5. Gráfico de efectos de los análisis de subgrupos en los pacientes con linfoma folicular
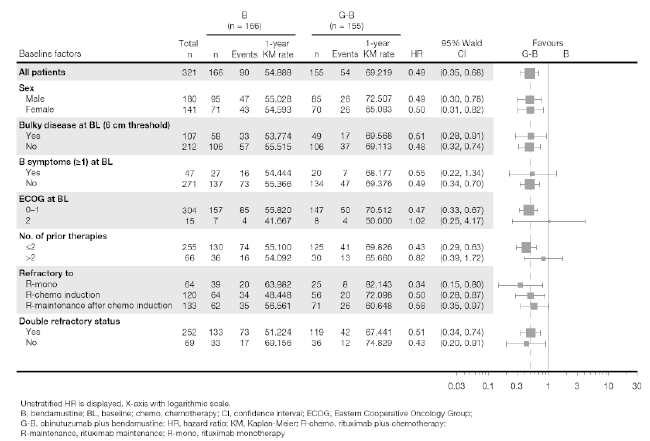
De arriba abajo: G-B = G-B; B = B; Baseline factors = Factores iniciales; Total = Total; Events = Eventos; 1-year KM rate = Tasa KM a 1 año; HR = HR; 95% Wald CI = IC 95% de Wald; Favours = A favor de; All patients = Todos los pacientes ; Sex = Sexo; Male = Masculino; Female = Femenino; Bulky disease at BL (6 cm threshold) = Masa voluminosa al IE (valor liminar: 6 cm); Yes = Sí; No = No; B symptoms (≥1) at BL = Síntomas B (≥1) al IE; ECOG at BL = Puntuación en la escala del ECOG al IE; No. of prior therapis = N.º de terapias previas; Refractory to = Resistente a; R-mono = R-mono; R-chemo induction = R-quimio inducción; R-maintenance after chemo = R-mantenimiento tras quimio inducción ; Double refractory status = Estado resistente doble.
Se muestra la HR no estratificada. Eje X con escala logarítmica. B: bendamustina; ECOG: Eastern Cooperative Oncology Group (Grupo Oncológico Cooperativo de la Costa Este de los EE.UU.); G-B: obinutuzumab más bendamustina; HR: hazard ratio; IC: intervalo de confian-za; IE: inicio del estudio; KM: Kaplan-Meier; quimio: quimioterapia; R-quimio: rituximab más quimioterapia; R-mantenimiento: mantenimiento con rituximab; R-mono: rituximab en monoterapia.
Resultados referidos por los pacientes
Pacientes con linfoma folicular sin tratamiento previo: teniendo en cuenta los resultados del cuestionario FACT-Lym obtenidos durante los periodos de tratamiento y de seguimiento, ambos grupos presentaron mejorías clínicamente significativas de los síntomas relacionados con el linfoma, que se definieron como un aumento de ≥3 puntos en la subescala del linfoma respecto al valor inicial, un aumento de ≥6 puntos en el índice de desenlaces del estudio (TOI) de FACT-Lym respecto al valor inicial y un aumento de ≥7 puntos en la puntuación total del cuestionario FACT-Lym respecto al valor inicial. Las puntuaciones de utilidad del cuestionario EQ-5D fueron similares al inicio del estudio, durante el tratamiento y el seguimiento. No se observaron diferencias significativas entre los grupos en lo que se refiere a las mediciones de la calidad de vida relacionada con la salud o del estado de salud.
Linfoma folicular recidivante o resistente al tratamiento: considerando los resultados del cuestiona-rio FACT-Lym y la escala del índice EQ-5D obtenidos durante los periodos de tratamiento y de seguimiento, la calidad de vida relacionada con la salud se mantuvo generalmente en el estudio fundamental sin que hubiera diferencias significativas entre los grupos. Sin embargo, la adición de GAZYVA a la bendamustina retrasó el tiempo transcurrido hasta el empeoramiento de la calidad de vida determinado mediante la puntuación en el índice de desenlaces del estudio (TOI) de FACT-Lym (HR = 0,83; IC 95 %: 0,60, 1,13).
Inmunogenicidad: los resultados de los ensayos de inmunogenicidad dependen en gran medida de varios factores, como la sensibilidad y la especificidad del ensayo, la metodología del ensayo, la robustez del ensayo respecto a las cantidades de GAZYVA/anticuerpo circulantes, la manipulación de las muestras, el calendario de recogida de éstas, los tratamientos farmacológicos concomitantes y la enfermedad de fondo. Por estas razones, la compara-ción de la incidencia de anticuerpos contra GAZYVA con la de anticuerpos contra otros productos puede llevar a conclusiones erróneas.
En varios momentos del ensayo fundamental en la LLC, BO21004/CLL11, se llevaron a cabo determinaciones de anticuerpos contra GAZYVA. Entre los pacientes que recibieron GAZYVA, 8 de 140 pacientes de la fase aleatorizada y 2 de 6 pacientes de la fase de preinclusión tenían anticuerpos dirigidos contra el fármaco al cabo de 12 meses de seguimiento. Ninguno de ellos presentó reacciones anafilácticas o de hipersensibilidad que se conside-raran relacionadas con los anticuerpos dirigidos contra el fármaco, y tampoco se afectó la respuesta clínica.
Ningún paciente desarrolló HAHA (anticuerpos humanos antihumanos) contra GAZYVA durante el tratamiento con GAZYVA o después del mismo, en ninguno de los estudios fundamentales en pacientes con LNH de baja malignidad.
Propiedades farmacocinéticas: se desarrolló un modelo farmacocinético poblacional para analizar los datos farmacocinéticos en 469 pacientes con LNH de baja malignidad, 342 con LLC y 130 con linfoma difuso de linfocitos B grandes de estudios de fase I, II y III que recibieron GAZYVA.
Absorción: GAZYVA se administra por vía i.v.; no se han realizado estudios clínicos con otras vías de adminis-tración. Según el modelo farmacocinético poblacional, después de la infusión del día 1 del ciclo 6 en pacientes con LLC, la mediana calculada de la Cmáx era de 465,7 µg/ml y el valor del ABC(τ) fue de 8961 µg·d/ml; en pacientes con LNH de baja malignidad, la mediana calculada de la Cmáx fue de 539,3 µg/ml y el valor del ABC(τ) fue de 10 956 µg·d/ml.
Distribución: tras la administración i.v., el volumen de distribución del compartimento central (2,72 l) se aproxima al volumen sérico, lo que indica que la distribución se limita en su mayor parte al plasma y el líquido intersticial.
Metabolismo: no se ha estudiado directamente el metabolismo de GAZYVA. Los anticuerpos se depuran principalmente por catabolismo.
Eliminación: el aclaramiento de GAZYVA fue aproximadamente de 0,11 l/día en pacientes con LLC y de 0,08 l/día en pacientes con LNH de baja malignidad; la mediana de la semivida de eliminación (t½) fue de 26,4 días en los pacientes con LLC y de 36,8 días en los pacientes con LNH de baja malignidad.
La eliminación de GAZYVA comprende dos vías paralelas que describen el aclaramiento: una vía lineal y otra no lineal que varía en función del tiempo. Durante el tratamiento inicial, predomina la vía de aclaramiento no lineal variable con el tiempo, que en consecuencia es la principal vía de aclaramiento. A medida que el tratamiento prosigue, esta vía pierde importancia y pasa a predominar la vía de aclaramiento lineal. Ello es indicativo de una disposición del fármaco mediada por la diana terapéutica (TMDD), en la que la abundancia inicial de linfocitos B CD20+ causa una rápida eliminación de GAZYVA de la circulación. Sin embargo, cuando la mayoría de esos linfocitos se han unido a GAZYVA, el impacto de la TMDD sobre la farmacocinética se minimiza.
Farmacocinética en poblaciones especiales: en el análisis farmacocinético poblacional se observó que el sexo era una covariable que explica parte de la varia-bilidad entre pacientes: en los varones, el aclaramiento en el estado de equilibrio (Clee) es un 18 % mayor y el volumen de distribución (V) es un 19 % mayor. Sin embargo, los resultados del análisis poblacional han demostrado que las diferencias en la exposición no son significativas (mediana estimada del ABC y de la Cmáx en pacientes con LLC de 11 282 µg·d/ml y de 578,9 µg/ml en las mujeres y de 8451 µg·d/ml y 432,5 µg/ml en los varones, respectivamente, en el ciclo 6; ABC y Cmáx en pacientes con LNH de baja malignidad de 13 172 µg·d/ml y 635,7 µg/ml en las mujeres y de 9769 µg·d/ml y 481,3 µg/ml en los varones, respectivamente), lo que indica que no es necesario ajustar la dosis en función del sexo.
Pacientes ancianos: el análisis farmacocinético poblacional de GAZYVA mostró que la edad no afecta a la farmacocinética de este medicamento. No se observaron diferencias significativas de la farmacocinética de GAZYVA entre los pacientes menores de 65 años (n = 454), los de 65 a 75 años (n = 317) y los mayores de 75 años (n = 190).
Pacientes pediátricos: no se han llevado a cabo estudios para investigar la farmacocinética de GAZYVA en los pacientes pediátricos.
Insuficiencia renal: el análisis farmacocinético poblacional de GAZYVA mostró que el aclaramiento de creatinina (ClCr) no afecta a su farmacocinética. En los pacientes con insuficiencia renal leve (ClCr: 50-89 ml/min, n = 464) o moderada (ClCr: 30-49 ml/min, n = 106), la farmacocinética de GAZYVA era similar a la observada en los pacientes con función renal normal (ClCr: ≥90 ml/min, n = 383). Se dispone de pocos datos farmacocinéticos (n = 8) de pacientes con insuficiencia renal grave (ClCr: 15-29 ml/min), por lo que no es posible formular recomendaciones posológicas.
Insuficiencia hepática: no se ha realizado ningún estudio farmacocinético formal ni se han obtenido datos de farmacocinética poblacional en pacientes con insuficiencia hepática.
INDICACIONES TERAPÉUTICAS
Leucemia linfocítica crónica
GAZYVA en combinación con clorambucilo está indicado para el tratamiento de pacientes con leucemia linfocítica crónica (LLC) que no han sido tratados previamente.
Linfoma folicular
GAZYVA en combinación con quimioterapia, seguido por el tratamiento de mantenimiento con GAZYVA, está indicado para el tratamiento de pacientes con linfoma folicular que no han sido tratados previamente.
GAZYVA en combinación con bendamustina, seguido por el tratamiento de mantenimiento con GAZYVA, está indicado en el tratamiento de pacientes con linfoma folicular (LF) que no han respondido o han sufrido una progresión de la enfermedad durante el tratamiento con rituximab o con un régimen que contuviera rituximab, o después del mismo.
DATOS FARMACÉUTICOS
Conservación
Viales: consérvense los viales en un refrigerador a 2-8 °C.
Este medicamento no debe usarse después de la fecha de caducidad, indicada con «EXP» en el envase.
Manténgase el vial dentro de su caja de cartón para protegerlo de la luz.
No debe congelarse. No debe agitarse.
Periodo de validez: conforme al registro local.
Periodo de validez de la solución para infusión que contiene el producto: se ha comprobado que la solución preparada para su uso mantiene la estabilidad física y química durante 24 horas a 2-8 °C, seguidas por otras 24 horas a temperatura ambiente (≤30 °C) y de una infusión que no dure más de 24 horas.
Desde el punto de vista microbiológico, la solución para infusión preparada debe usarse inmediatamente. Si no se utiliza de inmediato, los periodos y las condiciones de conservación antes usarlo son responsabilidad del usuario; normalmente no debería conservarse durante más de 24 horas a 2-8 °C, a no ser que la dilución se haya realizado en condiciones asépticas controladas y validadas.
GAZYVA no contiene conservantes antimicrobianos. Así pues, deben tomarse precauciones para que la solución para infusión no se vea comprometida desde el punto de vista microbiológico durante su preparación.
Instrucciones especiales de uso, manipulación y eliminación
Instrucciones para la dilución: la preparación de GAZYVA debe realizarla un profesional sanitario utilizando una técnica aséptica.
Para los ciclos 2-6 en la LLC y para todos los ciclos en el linfoma folicular: se extraen 40 ml del concentrado líquido de GAZYVA del vial y se diluyen en bolsas de infusión de PVC o de poliolefina sin PVC que contengan solución acuosa de cloruro de sodio al 0,9 % estéril y apirógena.
Preparación de las bolsas de infusión para la dosis del día 1 del ciclo 1 administrada en 2 días, sólo en la LLC: para distinguir convenientemente las dos bolsas de infusión para la dosis inicial de 1000 mg, se recomienda utilizar bolsas de diferentes tamaños, de modo que se puedan diferenciar la dosis de 100 mg del día 1 del ciclo 1 y la dosis de 900 mg del día 1 (continuación) o el día 2 del ciclo 1. Para preparar las 2 bolsas de infusión, se extraen 40 ml de concentrado líquido de GAZYVA del vial y se diluyen 4 ml en una bolsa de infusión de 100 ml y los 36 ml restantes en una bolsa de infusión de 250 mg de PVC o de poliolefina sin PVC que contenga solución salina acuosa al 0,9 % estéril y apirógena. Se debe rotular claramente cada bolsa de infusión.
|
Dosis de GAZYVA que debe administrarse |
Cantidad necesaria de concentrado líquido de GAZYVA |
Tamaño de la bolsa de infusión de PVC o de poliolefina sin PVC |
|
100 mg |
4 ml |
100 ml |
|
900 mg |
36 ml |
250 ml |
|
1000 mg |
40 ml |
250 ml |
No deben utilizarse otros diluyentes, como una solución de glucosa (5 %) (ver Incompatibilidades).
La bolsa debe invertirse suavemente para mezclar la solución sin que se produzca demasiada espuma.
Los medicamentos de uso parenteral deben inspeccionarse visualmente antes de su administración para descartar la presencia de partículas o cambios de color.
Incompatibilidades: no se han observado incompatibilidades entre GAZYVA y las bolsas de cloruro de polivinilo, polietileno, polipropileno o poliolefina, los equipos de infusión de cloruro de polivinilo, poliuretano o polietileno, los filtros facultativos para vía i.v. con superficies de contacto con el producto de polietersulfona, las llaves de tres vías para infusión fabricadas con policarbonato o los catéteres de poliuretano, cuando se utilizaron concentraciones de 0,4-20,0 mg/ml tras diluir GAZYVA con solución de cloruro de sodio al 0,9 %. El producto diluido no se debe agitar ni congelar.
No deben usarse otros diluyentes, como soluciones de glucosa (al 5 %), ya que no se ha estudiado su uso.
Eliminación de los medicamentos no utilizados o caducados: la emisión de productos farmacéuticos al medio ambiente debe reducirse al mínimo. Evítese tirar los medicamentos por los desagües o a la basura doméstica, y utilícense los sistemas de recogida disponibles localmente.
CONTRAINDICACIONES
GAZYVA está contraindicado en pacientes con hipersensibilidad conocida (mediada por IgE) al obinutuzumab o a cualquiera de los excipientes.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: No se han realizado estudios de los efectos de GAZYVA sobre la capacidad para conducir y utilizar máquinas. Se advertirá a los pacientes que sufran síntomas relacionados con la infusión que no deben conducir ni utilizar máquinas hasta que desaparezcan los síntomas.
REACCIONES ADVERSAS
Ensayos clínicos
Leucemia linfocítica crónica: las reacciones adversas descritas en este apartado se identificaron durante el periodo de tratamiento y el seguimiento del ensayo clínico fundamental, BO21004/CLL11, en el que GAZYVA en combinación con clorambucilo se comparó con el clorambucilo solo (etapa 1) o con rituximab más clorambucilo (etapa 2). El 81 % de los pacientes tratados con GAZYVA en asociación con clorambucilo recibieron los 6 ciclos de tratamiento, en comparación con el 89 % de los pacientes del grupo de rituximab más clorambucilo y con el 67 % de los pacientes del grupo del clorambucilo solo.
En las tablas 5 y 6 se resumen las reacciones adversas con una incidencia más elevada (diferencia ≥2 %) en los pacientes tratados con GAZYVA más clorambucilo que en los que recibieron el clorambucilo solo o rituximab más clorambucilo, respectivamente.
|
Tabla 5. Reacciones adversas notificadas con mayor frecuencia (diferencia ≥2 % en comparación con el clorambucilo solo [etapa 1]) en pacientes tratados con GAZYVA más clorambucilo* |
||
|
Reacción adversa (MedDRA) Clase de órgano, aparato o sistema |
Todos los grados (%) |
Grados 3-5† (%) |
|
GAZYVA + clorambucilo n = 241 |
||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
||
|
Reacciones relacionadas con la infusión |
68,9 |
21,2 |
|
Trastornos de la sangre y del sistema linfático |
||
|
Neutropenia |
40,7 |
34,9 |
|
Trombocitopenia |
15,4 |
11,2 |
|
Anemia |
12,4 |
4,6 |
|
Leucopenia |
7,1 |
5,4 |
|
Infecciones e infestaciones |
||
|
Infección urinaria |
6,2 |
1,7 |
|
Herpes bucal |
3,7 |
0 |
|
Rinitis‡ |
2,1 |
0 |
|
Faringitis |
2,1 |
0 |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Pirexia |
10,4 |
<1 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Tos |
9,5 |
0 |
|
Trastornos del metabolismo y de la nutrición |
||
|
Síndrome de lisis tumoral |
4,1 |
1,7 |
|
Hiperuricemia |
3,3 |
<1 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Artralgia |
4,6 |
<1 |
|
Dolor de espalda |
5,0 |
<1 |
|
Dolor torácico musculoesquelético |
2,5 |
<1 |
|
Trastornos vasculares |
||
|
Hipertensión arterial |
3,7 |
1,7 |
|
Exploraciones complementarias |
||
|
Cifra de leucocitos disminuida |
2,1 |
2,1 |
|
Cifra de neutrófilos disminuida |
2,1 |
2,1 |
|
Peso aumentado |
2,1 |
0 |
|
Trastornos cardiacos |
||
|
Fibrilación auricular |
2,1 |
<1 |
|
Neoplasias benignas, malignas y sin especificar (incluidos, quistes y pólipos) |
||
|
Carcinoma de células escamosas cutáneo |
2,1 |
1,2 |
|
Trastornos gastrointestinales |
||
|
Diarrea‡ |
10,4 |
2,5 |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Alopecia |
2,1 |
0 |
|
* En todos los grados o grados 3-5 † No se han observado reacciones adversas de grado 5 con una diferencia ≥2 % entre los grupos de tratamiento. ‡ Con la actualización de la etapa 1 y los datos de la etapa 2, ya no se notificó que la diferencia entre los grupos de tratamiento en lo que se refiere a este evento fuera ≥2 %. |
||
|
Tabla 6. Reacciones adversas notificadas con mayor frecuencia (diferencia ≥2 % en comparación con el rituximab más clorambucilo [etapa 2]) en pacientes tratados con GAZYVA más clorambucilo* |
||
|
Reacción adversa (MedDRA) Clase de órgano, aparato o sistema |
Todos los grados (%) |
Grados 3-5† (%) |
|
GAZYVA + clorambucilo n = 336 |
||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
||
|
Reacciones relacionadas con la infusión |
65,8 |
19,9 |
|
Trastornos de la sangre y del sistema linfático |
||
|
Neutropenia |
38,1 |
33,0 |
|
Trombocitopenia |
14,3 |
10,4 |
|
Leucopenia |
6,3 |
4,5 |
|
Trastornos gastrointestinales |
||
|
Diarrea |
10,1 |
2,1 |
|
Estreñimiento |
8,3 |
0 |
|
Infecciones e infestaciones |
||
|
Rinofaringitis |
5,7 |
<1 |
|
Infección urinaria |
5,4 |
1,5 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Dolor de espalda |
4,8 |
<1 |
|
Artralgia |
4,8 |
<1 |
|
Trastornos del metabolismo y de la nutrición |
||
|
Síndrome de lisis tumoral |
4,2 |
1,8 |
|
* En todos los grados o grados 3-5 † No se han observado reacciones adversas de grado 5 con una diferencia ≥2 % entre los grupos de tratamiento. |
||
Linfoma no hodgkiniano: en los pacientes con linfoma folicular, el perfil de reacciones adversas concordaba con el de la población general de pacientes con LNH de baja malignidad.
Las reacciones adversas que se describen en este apartado se identificaron durante la inducción, el manteni-miento y el seguimiento de dos estudios fundamentales en los que se investigó el uso de GAZYVA en combinación con bendamustina, CHOP o CVP, seguido por el trata-miento de mantenimiento con GAZYVA en:
- Pacientes con LNH de baja malignidad sin trata-miento previo (N = 1390; 692 pacientes en el grupo del rituximab más quimioterapia y 698 pacientes en el grupo de GAZYVA más quimioterapia), el 86 % de los cuales padecían un linfoma folicular (BO21223). De estos pacientes, el 92,7 % de los tratados con GAZYVA más quimioterapia (inducción) recibieron los 6 o los 8 ciclos de tratamiento (dependiendo de la quimioterapia) de GAZYVA.
- Pacientes con LNH de baja malignidad recidivante o resistente al tratamiento (N = 392; 198 en el grupo de la bendamustina y 194 en el grupo de GAZYVA más bendamustina), el 81 % de los cuales presentaban un linfoma folicular (GAO4753g). De estos pacientes, el 79,4 % de los tratados con GAZYVA más bendamustina recibieron los 6 ciclos de tratamiento con GAZYVA.
Las reacciones adversas se identificaron como aquellas que se registraron con una diferencia ≥2 % en compara-ción con el grupo de referencia pertinente (en lo que se refiere a las reacciones adversas de todos los grados) al menos en un estudio fundamental. Las incidencias que se presentan en la tabla 7 (reacciones adversas de todos los grados y de grados 3-5) y en todo el apartado «Reacciones adversas» en lo que se refiere al LNH de baja malignidad corresponden a la mayor incidencia de la reacción adversa en cuestión notificada en cualquiera de los estudios fundamentales. Así pues, la incidencia de reacciones adversas de grado 3-5 puede haber sido notificada en un estudio fundamental diferente al de la incidencia de reacciones adversas de todos los grados.
|
Tabla 7. Reacciones adversas notificadas en pacientes con LNH de baja malignidad tratados con GAZYVA más quimioterapia |
||
|
Reacción adversa (MedDRAa) Clase de órgano, aparato o sistema |
Todos los grados (%) |
Grados 3-5† (%) |
|
GAZYVA + Quimioterapia* |
||
|
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos |
||
|
Reacciones relacionadas con la infusión‡ |
69,6 |
12,6 |
|
Trastornos de la sangre y del sistema linfático |
||
|
Neutropenia§ |
48,1 |
44,0 |
|
Trombocitopenia |
14,4 |
10,3 |
|
Dolor en ganglio linfático |
2,1 |
0 |
|
Trastornos cardiacos |
||
|
Insuficiencia cardiaca |
2,1 |
1,0 |
|
Trastornos oculares |
||
|
Hiperemia ocular |
2,1 |
0 |
|
Trastornos gastrointestinales |
||
|
Estreñimiento |
30,8 |
0,3 |
|
Diarrea |
24,9 |
1,9 |
|
Dispepsia |
8,0 |
0 |
|
Colitis |
2,1 |
1,0 |
|
Hemorroides |
2,1 |
0,1 |
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Pirexia |
19,1 |
2,1 |
|
Astenia |
11.3 |
1,0 |
|
Dolor torácico |
4,6 |
0,6 |
|
Infecciones e infestaciones |
||
|
Infección respiratoria de vías altas |
18,3 |
2,1 |
|
Rinofaringitis |
16,8 |
0,1 |
|
Sinusitis§ |
11,9 |
1,0 |
|
Infección urinaria |
10,5 |
3,1 |
|
Herpes zóster§ |
10,2 |
1,1 |
|
Neumonía§ |
9,9 |
5,6 |
|
Rinitis |
6,7 |
0,1 |
|
Gripe |
4,3 |
0,3 |
|
Faringitis |
4,2 |
0 |
|
Infección pulmonar |
3,1 |
1,4 |
|
Exploraciones complementarias |
||
|
Hipopotasemia |
7,2 |
1,0 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Artralgia§ |
13,9 |
0 |
|
Dolor en una extremidad |
8,8 |
1,0 |
|
Dolor óseo |
5,0 |
0,1 |
|
Trastornos psiquiátricos |
||
|
Insomnio |
13,8 |
0,1 |
|
Depresión |
4,0 |
0,6 |
|
Trastornos renales y urinarios |
||
|
Disuria |
2,6 |
0,5 |
|
Incontinencia urinaria |
2,6 |
0,5 |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Tos |
27,1 |
0,1 |
|
Congestión nasal |
7,2 |
0 |
|
Rinorrea |
4,1 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Alopecia |
12,3 |
0 |
|
Prurito |
9,9 |
0,1 |
|
Sudores nocturnos |
4,2 |
0,1 |
|
Eccema |
2,6 |
0 |
|
* Y a continuación tratamiento de mantenimiento con GAZYVA. a Reacciones adversas codificadas según el MedDRA, tal como fueron notificadas por los investigadores (excluidas las reacciones relacionadas con la infusión). † No se han observado reacciones adversas de grado 5 con una diferencia ≥2 % entre los grupos de tratamiento. ‡ Definidas como cualquier evento adverso relacionado que tenga lugar durante la infusión o en las 24 horas posteriores a esta. § Observadas también durante el tratamiento de mantenimiento con una frecuencia ≥2 % superior en el grupo de GAZYVA (BO21223). |
||
En el estudio GAO4753g, los pacientes del grupo de la bendamustina recibieron 6 meses de tratamiento de inducción sólo, mientras que después del periodo de inducción los pacientes del grupo de GAZYVA más bendamustina siguieron bajo tratamiento de mantenimiento con GAZYVA. Durante el periodo de mantenimiento con GAZYVA, las reacciones adversas más frecuentes fueron las siguientes: tos (14,7 %), infecciones de las vías respiratorias altas (11,9 %), neutropenia (10,5 %), sinusitis (9,8 %), diarrea (8,4 %), reacciones relacionadas con la infusión (8,4 %), náuseas (7,7 %), fatiga (7,7 %), bronquitis (7,0 %), artralgia (7,0 %), rinofaringitis (6,3 %), infecciones urinarias (6,3 %) y pirexia (5,6 %). Las reacciones adversas de grado 3-5 más frecuentes fueron las siguientes: neutropenia (9,8 %), y anemia, neutropenia febril, trombocitopenia, septicemia, infección de las vías respiratorias altas e infección urinaria (todas ellas con una frecuencia del 1,4 %).
Información adicional sobre determinadas reacciones adversas
Reacciones relacionadas con la infusión: los síntomas asociados a RRI que se notificaron con mayor frecuencia (≥5 %) fueron: náuseas, vómitos, diarrea, cefalea, mareos, fatiga, escalofríos, pirexia, hipotensión arterial, rubefacción, hipertensión arterial, taquicardia, disnea y malestar torácico. También se han notificado casos de síntomas respiratorios, como broncoespasmo, irritación de laringe y garganta, sibilancias, edema laríngeo, y de síntomas cardiacos, como la fibrilación auricular (ver Advertencias y precauciones).
Leucemia linfocítica crónica: la incidencia de RRI fue del 65 % en la infusión de los 1000 mg iniciales de GAZYVA (20 % de los pacientes sufrieron RRI de grado 3-4). En general, el 7 % de los pacientes sufrieron una RRI que implicó la retirada de GAZYVA. La incidencia de RRI con las infusiones posteriores fue del 3 % con la segunda dosis de 1000 mg y del 1 % posteriormente. No se notificaron RRI de grado 3-5 después de las infusiones de los 1000 mg iniciales del ciclo 1.
En los pacientes en los que se aplicaron las medidas recomendadas para la prevención de las RRI —tal como se describe en el apartado 2.2 Posología y forma de administración—, se observó una disminución de la incidencia de RRI de todos los grados. Las tasas de RRI de grado 3-4 (que se basan en un número relativamente pequeño de pacientes) fueron similares antes y después de aplicar las medidas de mitigación.
Linfoma no hodgkiniano: en el ciclo 1, la incidencia global de RRI fue mayor en los pacientes que recibieron GAZYVA más quimioterapia que en los pacientes del grupo de comparación. En los pacientes que recibieron GAZYVA más quimioterapia, la mayor incidencia de RRI se registró el día 1, y disminuyó gradualmente en las infusiones posteriores. Esta tendencia decreciente se mantuvo durante el tratamiento de mantenimiento con GAZYVA solo.
En general, el 3 % de los pacientes sufrieron una RRI que implicó la suspensión del tratamiento con GAZYVA.
Neutropenia e infecciones
Leucemia linfocítica crónica: la incidencia de neutropenia fue mayor en el grupo de GAZYVA más clorambucilo que en el grupo de rituximab más clorambucilo; la neutropenia remitió espontáneamente o con la administración de factores estimulantes de las colonias de granulocitos. La incidencia de infección fue del 38 % en el grupo de GAZYVA más clorambucilo y del 37 % en el grupo de rituximab más clorambucilo (se registraron eventos de grado 3-5 en el 12 % y 14 %, respectivamente, y los eventos mortales se registraron en <1 % en ambos grupos de tratamiento). También se notificaron casos de neutropenia prolongada (2 % en el grupo de GAZYVA más clorambucilo y 4 % en el grupo de rituximab más clorambucilo) y de neutropenia de inicio tardío (16 % en el grupo de GAZYVA más clorambucilo y 12 % en el grupo de rituximab más clorambucilo; ver Advertencias y precauciones).
Linfoma no hodgkiniano: en el grupo de GAZYVA más quimioterapia, la incidencia de neutropenia fue mayor que en el grupo de comparación, con un riesgo elevado durante el periodo de inducción. La incidencia de neutropenia prolongada y de neutropenia de inicio tardío en el grupo de GAZYVA más quimioterapia fue del 3 % y del 7 %, respectivamente. La incidencia de infección fue del 78 % en el grupo de GAZYVA más quimioterapia (los eventos de grado 3-5 se registraron en el 22 % y los eventos mortales en el 3 % de los pacientes). En los pacientes que recibieron profilaxis con factores estimulantes de las colonias de granulocitos la incidencia de infecciones de grado 3-5 fue menor (verAdvertencias y precauciones).
Trombocitopenia y eventos hemorrágicos
Leucemia linfocítica crónica: la incidencia de trombocitopenia fue mayor en el grupo de GAZYVA más clorambucilo que en el grupo de rituximab más clorambucilo, especialmente durante el primer ciclo. El 4 % de los pacientes tratados con GAZYVA más clorambucilo sufrieron una trombocitopenia aguda (que tuvo lugar en las 24 horas siguientes a la infusión de GAZYVA (ver Advertencias y precauciones). La incidencia general de episodios hemorrágicos fue similar en el grupo tratado con GAZYVA y en el grupo que recibió rituximab. El número de episodios hemorrágicos mortales estaba equilibrado entre los grupos de tratamiento; sin embargo, todos los eventos en pacientes tratados con GAZYVA se notificaron en el ciclo 1. No se ha establecido una relación clara entre la trombocitopenia y los eventos hemorrágicos.
Linfoma no hodgkiniano: la trombocitopenia fue más frecuente durante el ciclo 1 en el grupo que recibió GAZYVA más quimioterapia. La trombocitopenia que tuvo lugar durante la infusión o en un plazo de hasta 24 horas después de que ésta concluyera (trombocitopenia aguda) se observó más frecuentemente en pacientes tratados con GAZYVA más quimioterapia que en el grupo de comparación pertinente. La incidencia de eventos adversos hemorrágicos fue similar en todos los grupos de tratamiento. Los eventos hemorrágicos y los eventos hemorrágicos de grado 3-5 se registraron en el 11 % y el 5 % de los pacientes, respectivamente. Aunque los eventos hemorrágicos mortales afectaron a menos del 1 % de los pacientes, ninguno de estos eventos adversos mortales tuvo lugar en el ciclo 1.
Leucoencefalopatía multifocal progresiva (LMP): se han notificado casos de leucoencefalopatía multifocal progresiva (LMP) en pacientes tratados con GAZYVA (ver Advertencias y precauciones).
Reactivación de la hepatitis B: se han notificado casos de reactivación de la hepatitis B en pacientes tratados con GAZYVA (ver Advertencias y precauciones).
Empeoramiento de afecciones cardiacas pree-xistentes: se han notificado casos de eventos cardiacos mortales en pacientes tratados con GAZYVA (ver Adver-tencias y precauciones).
Perforación gastrointestinal: se ha referido casos de perforación gastrointestinal en pacientes tratados con GAZYVA, principalmente en el LNH.
Alteraciones analíticas: poco después de la primera infusión de GAZYVA se ha observado una elevación transitoria de las enzimas hepáticas (AST, ALT y fosfatasa alcalina).
Para obtener más información, véase el apartado anterior, Información sobre determinadas reacciones adversas - Neutropenia y Trombocitopenia.
Poscomercialización: sin texto.
Alteraciones analíticas: sin texto.
INTERACCIONES CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: No se han realizado estudios formales de interacciones farmacológicas, aunque se han llevado a cabo algunos subestudios de interacciones farmacológicas de GAZYVA con bendamustina, CHOP (ciclofosfamida, doxorubicina, vincristina, prednisolona), FC (fludarabina, ciclofosfami-da) y clorambucilo. La administración concomitante de GAZYVA no tuvo ningún efecto en la farmacocinética de la bendamustina, de FC o de alguno de los componentes del régimen CHOP; por otra parte, no se observaron efectos aparentes de la bendamustina, FC, el clorambucilo o CHOP en la farmacocinética de GAZYVA. No puede descartarse el riesgo de interacciones con otros medicamentos que esté recibiendo el paciente.
DATOS PRECLÍNICOS SOBRE SEGURIDAD
Carcinogenicidad: no se han realizado estudios para evaluar el potencial carcinógeno de GAZYVA.
Mutagenicidad: no se han llevado a cabo estudios para determinar el potencial mutágeno de GAZYVA.
Trastornos de la fecundidad: no se han realizado estudios específicos en animales para evaluar los efectos de GAZYVA en la fecundidad. En estudios de toxicidad de dosis repetidas en el macaco, no se observó ningún efecto adverso sobre los órganos sexuales masculinos o femeninos.
Teratogenicidad: se llevó a cabo un estudio mejorado de la toxicidad para el desarrollo prenatal y posnatal en hembras de macaco preñadas que recibieron dosis i.v. semanales de GAZYVA —la media del ABC0-168 h en el estado de equilibrio (139 días después del apareamien-to) fue de 125 000 y 250 000 (µg·h)/ml con 25 mg/kg y 50 mg/kg, respectivamente; la media de la Cmáx fue de 1220 µg/ml y 2470 µg/ml con 25 mg/kg y 50 mg/kg, respectivamente— durante la gestación (periodo de la organogénesis; desde 20 días después del apareamiento hasta el parto). No se encontraron indicios de efectos teratógenos en la progenie expuesta, pero se observó una depleción completa de los linfocitos B 28 días después del parto. La exposición de las crías 28 días después del parto indica que GAZYVA puede atravesar la barrera placentaria. Las concentraciones en el suero de las crías 28 días después del parto estaban dentro del intervalo de concentraciones del suero materno, mientras que las concentraciones en la leche materna ese mismo día eran muy bajas (inferiores al 0,5 % de las concentraciones séricas maternas correspondientes), lo que indica que la exposición de las crías tuvo que ser prenatal. Las cifras de linfocitos B se normalizaron, y 6 meses después del parto la función inmunitaria se había restablecido.
Otros efectos: en un estudio de 26 semanas en el macaco, se registraron reacciones de hipersensibilidad y se atribuyeron al reconocimiento del anticuerpo humanizado como extraño —con la administración semanal de 5, 25 y 50 mg/kg, en el estado de equilibrio (día 176), la Cmáx fue de 377, 1530 y 2920 µg/ml, respectivamente, y el ABC0-168 h fue de 39 800, 183 000 y 344 000 (µg·h)/ml, respectivamente—. Se registraron reacciones anafilácticas o anafilactoides agudas y una mayor prevalencia de inflamación sistémica e infiltrados atribuibles a reacciones de hipersensibilidad mediadas por inmunocomplejos, como arteritis o periarteritis, glomerulonefritis e inflamación de la serosa o la adventicia. Estas reacciones determinaron la retirada no programada del estudio de 6 de los 36 animales tratados con GAZYVA durante las fases de administración y de recuperación; las alteraciones fueron parcialmente reversibles. En el ser humano no se ha observado ninguna manifestación de nefrotoxicidad relacionada causalmente con GAZYVA.
PRUEBAS DE LABORATORIO: ver Advertencias y precauciones generales.
ADVERTENCIAS Y PRECAUCIONES: Para mejorar la trazabilidad de los biomedicamentos, debe registrarse (o declararse) claramente el nombre comercial y el número de lote del producto administrado en la historia clínica del paciente.
ADVERTENCIAS Y PRECAUCIONES GENERALES
Reacciones relacionadas con la infusión (RRI)
Las reacciones adversas observadas con mayor frecuencia en pacientes que recibían GAZYVA fueron reacciones relacionadas con la infusión (RRI) que ocurrieron predominantemente durante la infusión de los 1000 mg iniciales.
En los pacientes con LLC en los que se aplicaron las medidas combinadas para la prevención de las RRI (corticosteroide adecuado, analgésico/antihistamínico por vía oral, omisión de la medicación antihipertensora) se observó una incidencia reducida de RRI de todos los grados. Las tasas de RRI de grado 3-4 (que se basaron en un número relativamente pequeño de pacientes) fueron similares antes y después de aplicar las medidas de mitigación. Se deben adoptar medidas de mitigación para reducir las RRI (ver Posología y forma de administración). La incidencia y la intensidad de los síntomas relacionados con la infusión disminuyeron sustancialmente después de infundir los 1000 mg iniciales; la mayoría de los pacientes no presentaron RRI durante las administraciones posteriores de GAZYVA (ver Reacciones adversas).
En la mayoría de los pacientes, independientemente de la indicación, las RRI fueron de leves a moderadas y pudieron tratarse reduciendo la velocidad de infusión o suspendiendo temporalmente la primera infusión; no obstante, también se han notificado casos de RRI graves y potencialmente mortales que requirieron tratamiento sintomático. Las RRI pueden ser clínicamente indistinguibles de las reacciones alérgicas mediadas por IgE (por ejemplo: anafilaxia). Los pacientes con una carga tumoral elevada o con una cifra elevada de linfocitos circulantes en la LLC (>25 × 109/l) pueden tener un riesgo elevado de sufrir RRI graves. Véase la información sobre la profilaxis en Posología y forma de administración; en la tabla 4 (Pautas para la modificación de la velocidad de infusión en caso de reacciones relacionadas con la infusión) se indica cómo tratar las RRI según el grado de la reacción.
Los pacientes no deben recibir más infusiones de GAZYVA si sufren:
- Síntomas respiratorios agudos potencialmente mortales,
- Una RRI de grado 4 (es decir, potencialmente mortal) o
- Un segundo episodio de una RRI de grado 3 (prolongada o recidivante) (tras reanudar la primera infusión o durante una infusión posterior).
Durante toda la infusión y el periodo posterior a ella, se debe vigilar estrechamente a los pacientes con afecciones cardiacas o pulmonares preexistentes. Durante las infusiones i.v. de GAZYVA los pacientes pueden presentar hipotensión arterial, por lo que se planteará la suspensión de la medicación antihipertensora en las 12 horas anteriores a la infusión, durante cada infusión de GAZYVA y en la primera hora después de concluir la administración. Si el paciente presenta un riesgo agudo de crisis hipertensiva, habrán de sopesarse los beneficios y los riesgos de suspender temporalmente la medicación antihipertensora.
Reacciones de hipersensibilidad: se han notificado reacciones de hipersensibilidad de inicio inmediato (por ejemplo, anafilaxia) y de inicio tardío (por ejemplo, enfermedad del suero) en pacientes tratados con GAZYVA. Si se sospecha una reacción de hipersensibilidad durante o después de una infusión (por ejemplo: síntomas que suelen ocurrir tras una exposición anterior y muy raras veces con la primera infusión), se detendrá la infusión y se suspenderá el tratamiento definitivamente. Los pacientes con hipersensibilidad conocida no deben recibir tratamiento con GAZYVA (ver Contraindicaciones). Clínicamente, la hipersensibilidad puede ser difícil de diferenciar de las reacciones relacionadas con la infusión.
Síndrome de lisis tumoral (SLT): se han notificado casos de síndrome de lisis tumoral (SLT) con GAZYVA. Los pacientes a los que se considera en riesgo de sufrir un síndrome de lisis tumoral —por ejemplo: los pacientes con una gran carga tumoral o una cifra de linfocitos circulantes alta (>25 x 109/l) o insuficiencia renal (ClCr <70 ml/min) o cualquier combinación de estas condiciones— deben recibir tratamiento profiláctico. La profilaxis debe consistir en la hidratación adecuada y la administración de uricostáticos (por ejemplo: alopurinol) o alguna alternativa apropiada, como una urato-oxidasa (por ejemplo: rasburicasa), antes de comenzar la infusión de GAZYVA, tal como se describe en el apartado Posología y forma de administración. Durante los primeros días del tratamien-to se vigilará estrechamente a todos los pacientes a los que se considere en riesgo, centrándose especialmente en la función renal y en los valores de potasio y de ácido úrico. Se debe seguir cualquier guía adicional conforme a las prácticas habituales. Para tratar el síndrome de lisis tumoral, es preciso corregir las alteraciones electrolíticas, controlar la función renal y el balance hídrico, así como administrar tratamiento complementario, incluida la diálisis, tal como se ha indicado.
Neutropenia: se han descrito casos de neutropenia grave y potencialmente mortal, incluida la neutropenia febril, durante el tratamiento con GAZYVA. Se debe controlar estrechamente a los pacientes que presenten neutropenia, realizando análisis regularmente hasta que se resuelva. Si fuera necesario administrar tratamiento, se hará de acuerdo con las pautas locales, y se planteará la conveniencia de administrar factores estimulantes de las colonias de granulocitos. Si hay signos de infección concomitante, se instaurará el tratamiento que proceda. También pueden darse casos de neutropenia de inicio tardío (que tuvieron lugar 28 días después de finalizar el tratamiento) o neutropenia prolongada (que persistieron más de 28 días después de haber finalizado o suspendido el tratamiento).
Trombocitopenia: durante el tratamiento con GAZYVA se han observado casos de trombocitopenia grave y potencialmente mortal, incluida la trombocitopenia aguda (que tuvieron lugar en un plazo de 24 horas después de la infusión). También se han descrito eventos hemorrá-gicos mortales en el ciclo 1 en pacientes tratados con GAZYVA. No se ha establecido una relación clara entre la trombocitopenia y los episodios hemorrágicos.
Se debe vigilar estrechamente a los pacientes para detectar la trombocitopenia, en especial durante el primer ciclo; se realizarán regularmente análisis hasta que el episodio se resuelva, y se planteará la conveniencia de retrasar la administración de la dosis en caso de trombocitopenia grave o potencialmente mortal. Se deja a discreción del médico responsable la decisión de transfundir hemoderivados (es decir, plaquetas) conforme a las prácticas del centro. También se debe tener en cuenta, sobre todo durante el primer ciclo, el uso de cualquier medicamento administrado concomitantemente que pueda empeorar los eventos relacionados con la trombocitopenia, como los antiagregantes plaquetarios y los anticoagulantes.
Empeoramiento de las afecciones cardiacas pree-xistentes: en pacientes con cardiopatías, se han registrado casos de arritmias (como la fibrilación auricular y la taquiarritmia), angina de pecho, síndrome coronario agudo, infarto de miocardio e insuficiencia cardiaca al ser tratados con GAZYVA (ver Reacciones adversas). Estos eventos pueden formar parte de una RRI y a veces son mortales, por lo que se vigilará estrechamente a los pacientes con antecedentes de cardiopatía. Además, se debe hidratar con cuidado a estos pacientes para evitar una posible hipervolemia.
Infecciones: GAZYVA no debe administrarse si existe una infección activa, y se procederá con cautela al plantear la posibilidad de usarlo en pacientes con antecedentes de infecciones recidivantes o crónicas. Pueden producirse graves infecciones bacterianas, fúngicas o víricas (en este último caso, de nueva aparición o reactivaciones) durante el tratamiento con GAZYVA y después de concluirlo. Se han notificado casos de infecciones mortales.
En los estudios en pacientes con linfoma folicular, se observó una incidencia elevada de infecciones en todas las fases de dichos estudios, incluido el seguimiento; la mayor incidencia se observó durante el mantenimiento. En la fase de seguimiento, las infecciones de grado 3-5 se observaron más en los pacientes que recibieron GAZYVA más bendamustina en la fase de inducción.
Reactivación de la hepatitis B: en pacientes tratados con anticuerpos anti-CD20, incluido GAZYVA, puede producirse la reactivación del virus de la hepatitis B (VHB), que en ocasiones da lugar a una hepatitis fulminante, insuficiencia hepática y la muerte (ver Reacciones adversas).
Antes de iniciar el tratamiento con GAZYVA, se deben realizar a todos los pacientes pruebas de detección del virus de la hepatitis B (VHB). Como mínimo se debe determinar el estado del paciente respecto al antígeno de superficie del virus de la hepatitis B (HBsAg) y respecto al anticuerpo contra el antígeno nuclear del virus de la hepatitis B (HBcAb). A estos se pueden añadir otros marcadores apropiados, según las pautas locales. No se debe tratar con GAZYVA a los pacientes con hepatitis B activa. Los pacientes con resultados positivos en las pruebas serológicas de la hepatitis B deben consultar a expertos en hepatopatías antes de iniciar el tratamiento; por otra parte, se los debe controlar y tratar conforme a las pautas médicas locales para prevenir la reactivación de la hepatitis.
Leucoencefalopatía multifocal progresiva (LMP): se han notificado casos de leucoencefalopatía multifocal progresiva (LMP) en pacientes tratados con GAZYVA (ver Reacciones adversas). Se planteará el diagnóstico de LMP en cualquier paciente que presente nuevas manifestaciones neurológicas o cambios de las manifestaciones neurológicas preexistentes. Los síntomas de LMP son inespecíficos y pueden variar dependiendo de la región del encéfalo afectada. Son comunes los síntomas motores, con signos de afectación corticoespinal (por ejemplo: debilidad muscular, parálisis y trastornos sensitivos), las anomalías sensitivas, los síntomas cerebelosos y los defectos del campo visual. Pueden producirse algunos signos y síntomas considerados como «corticales» (por ejemplo, afasia o desorientación visuoespacial). La evaluación de la LMP comprende, entre otras medidas, la consulta con un neurólogo, una exploración mediante resonancia magnética nuclear (RMN) encefálica y una punción lumbar (análisis del líquido cefalorraquídeo para detectar el ADN del virus JC). Se interrumpirá el tratamiento con GAZYVA durante la investigación de una posible LMP, y se suspenderá definitivamente si se confirma el diagnóstico. También se debe plantear la suspensión o la reducción de la dosis de cualquier quimioterapia o tratamiento inmunosupresor administrados concomitantemente. Se remitirá al paciente a un neurólogo para que éste evalúe y trate la LMP.
Vacunación: no se ha estudiado la seguridad de la inmunización con vacunas de virus vivos o atenuados después del tratamiento con GAZYVA, y no se recomienda utilizar vacunas de virus vivos durante el tratamiento hasta que no se haya normalizado la cifra de linfocitos B.
Exposición intrauterina a GAZYVA y vacunación de los lactantes con vacunas elaboradas con virus vivos:
Dada la posibilidad de depleción de los linfocitos B en lactantes cuyas madres han estado expuestas a GAZYVA durante el embarazo, se debe consultar con el médico del niño respecto a la seguridad de la inmunización con vacunas elaboradas con virus vivos y el momento de aplicarlas. Se debe plantear la posibilidad de retrasar la inmunización con vacunas elaboradas con microorganis-mos vivos de los lactantes cuyas madres hayan estado expuestas a GAZYVA durante el embarazo hasta que la cifra de linfocitos B de los lactantes se encuentre dentro del intervalo normal (ver Uso en poblaciones especiales - Embarazo).
Abuso y dependencia del fármaco: sin texto.
PAUTAS POSOLÓGICAS ESPECIALES
Niños: no se han determinado la seguridad ni la eficacia de GAZYVA en menores de 18 años.
Ancianos: no es preciso ajustar la dosis en los pacientes ancianos (ver Uso en poblaciones especiales, Uso en geriatría/Pacientes ancianos).
Insuficiencia renal: no es preciso ajustar la dosis en pacientes con insuficiencia renal leve o moderada. No se ha estudiado el uso de GAZYVA en pacientes con un ClCr <30 ml/min (ver Insuficiencia renal y Farmacocinética en poblaciones especiales).
Insuficiencia hepática
No se han determinado la seguridad ni la eficacia de GAZYVA en pacientes con insuficiencia hepática.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
Instrucciones generales: la sustitución de GAZYVA por cualquier otro biomedicamento requiere el consentimiento del médico prescriptor.
GAZYVA se administrará en infusión i.v. a través de una vía exclusiva, en un entorno con un equipo completo de reanimación inmediatamente disponible y bajo la estrecha vigilancia de un profesional sanitario con experien-cia. La infusión de GAZYVA no debe administrarse en inyección i.v. lenta o rápida (embolada). Como vehículo de la infusión se usará una solución isotónica de cloruro de sodio al 0,9 % (ver Instrucciones especiales de uso, manipulación y eliminación).
Profilaxis y premedicación para prevenir el síndrome de lisis tumoral (SLT): se considera que los pacientes con una gran carga tumoral o una cifra de linfocitos circulantes alta (>25 x 109/l) o insuficiencia renal (ClCr <70 ml/min) corren el riesgo de sufrir un síndrome de lisis tumoral y deben recibir tratamiento profiláctico. La profilaxis debe consistir en la hidratación adecuada y la administración de uricostáticos (por ejemplo: alopurinol) o alguna alternativa apropiada, como una urato-oxidasa (por ejemplo: rasburicasa), antes de comenzar la infusión de GAZYVA según la práctica habitual (ver Advertencias y precauciones). Si se considera pertinente, los pacientes seguirán recibiendo profilaxis repetida antes de cada infusión posterior.
Profilaxis y premedicación para prevenir las reacciones relacionadas con la infusión (RRI): en la tabla 1 se detalla la premedicación para reducir el riesgo de reacciones relacionadas con la infusión (ver Adverten-cias y precauciones). Se recomienda la premedicación con corticosteroides en los pacientes con linfoma folicular; dicha premedicación es obligatoria en los pacientes con LLC en la primera infusión. A continuación se describe la premedicación que debe administrarse en las siguientes infusiones y otro tipo de premedicación.
Al administrar las infusiones i.v. de GAZYVA puede producirse hipotensión arterial como síntoma de reacciones relacionadas con la infusión (RRI). En consecuencia, se planteará la suspensión de la medicación antihipertensora en las 12 horas anteriores a la infusión, durante cada infusión de GAZYVA y en la primera hora después de concluir de la administración (ver Advertencias y precauciones).
|
Tabla 1. Premedicación que debe administrarse antes de la infusión de GAZYVA para reducir el riesgo de reacciones relacionadas con la infusión |
|||
|
Día del ciclo de tratamiento |
Pacientes que necesitan premedicación |
Premedicación |
Forma de administración |
|
Ciclo 1: LLC Día 1 Día 2 LF Día 1 |
Todos los pacientes |
Corticosteroide i.v.1, 2 |
Ha de concluir al menos 1 hora antes de la infusión de GAZYVA |
|
Analgésico/antipirético por vía oral3 |
Al menos 30 minutos antes de la infusión de GAZYVA |
||
|
Antihistamínico4 |
|||
|
Todas las infusiones posteriores: LLC y LF |
Pacientes sin RRI durante la infusión anterior |
Analgésico/antipirético por vía oral3 |
Al menos 30 minutos antes de la infusión de GAZYVA |
|
Pacientes con una RRI (grado 1 o 2) con la infusión anterior |
Analgésico/antipirético por vía oral3 |
Al menos 30 minutos antes de la infusión de GAZYVA |
|
|
Antihistamínico4 |
|||
|
Pacientes con una RRI de grado 3 con la infusión anterior O |
Corticosteroide i.v.1 |
Ha de concluir al menos 1 hora antes de la infusión de GAZYVA |
|
|
Pacientes con una cifra de linfocitos >25 x 109/l antes del siguiente tratamiento |
Analgésico/antipirético por vía oral3 |
Al menos 30 minutos antes de la infusión de GAZYVA |
|
|
Antihistamínico3 |
|||
|
1 100 mg de prednisona/prednisolona o 20 mg de dexametasona o bien 80 mg de metilprednisolona. No se debe usar la hidrocortisona, dado que no ha sido efectiva en lo que respecta a la reducción de las tasas de RRI. 2 Si se administra una pauta de quimioterapia que contenga un corticosteroide el mismo día que GAZYVA, el corticosteroide puede administrarse por vía oral si se hace al menos 60 min antes de la infusión de GAZYVA, en cuyo caso no es necesaria la administración de un corticosteroide i.v. adicional como premedicación. 3 Por ejemplo, 1000 mg de paracetamol. 4 Por ejemplo, 50 mg de difenhidramina. |
|||
Posología habitual
Leucemia linfocítica crónica (en combinación con clorambucilo1)
Ciclo 1: la dosis recomendada de GAZYVA es de 1000 mg administrados durante el día 1 y el día 2, así como el día 8 y el día 15 del primer ciclo de tratamiento de 28 días, tal como se muestra en la tabla 2.
Deben prepararse dos bolsas de infusión para la primera dosis de 100 mg de la primera infusión y 900 mg para la segunda infusión. Si la administración de la dosis de 100 mg concluye sin que sea preciso modificar la velocidad de infusión o sin interrupciones, la dosis de 900 mg puede administrarse el mismo día (sin retrasar la dosis), siempre que durante toda la infusión se cuente con el tiempo, las condiciones y la supervisión médica pertinentes. Si al administrar los 100 mg iniciales fuera necesario modificar la velocidad de infusión o interrumpir la administración, la infusión de la dosis de 900 mg se administrará al día siguiente (ver tabla 2).
Ciclos 2-6: la dosis recomendada de GAZYVA es de 1000 mg, administrados el día 1 de cada ciclo de tratamiento de 28 días, tal como se muestra en la tabla 2.
|
Tabla 2. Dosis y velocidad de infusión de GAZYVA en los pacientes con LLC |
|||
|
Día del ciclo de tratamiento |
Dosis de GAZYVA |
Velocidad de infusión Consúltese en la tabla 4 el tratamiento de las RRI que se producen durante la infusión. |
|
|
Ciclo 1 |
Día 1 |
100 mg |
Se administra a una velocidad de 25 mg/h durante 4 horas. No se debe aumentar la velocidad de infusión. |
|
Día 2 o bien Día 1 (continuación) |
900 mg |
Si no se produce ninguna RRI durante la infusión previa, se debe administrar a una velocidad de 50 mg/h. La velocidad de infusión puede aumentarse a razón de 50 mg/h cada 30 minutos hasta una velocidad máxima de 400 mg/h. Si el paciente sufre una RRI durante la infusión previa, se comenzará la administración a una velocidad de 25 mg/h. La velocidad de infusión puede aumentarse a razón de hasta 50 mg/h cada 30 minutos hasta una velocidad máxima de 400 mg/h. |
|
|
Día 8 |
1000 mg |
Si no se produce ninguna RRI durante la infusión previa, cuya velocidad de infusión final fuera ≥100 mg/h, las infusiones pueden iniciarse a una velocidad de 100 mg/h y aumentarse a razón de 100 mg/h cada 30 min hasta alcanzar una velocidad máxima de 400 mg/h. Si el paciente sufre una RRI durante la infusión previa, se administrará a una velocidad de 50 mg/h. La velocidad de infusión puede aumentarse a razón de 50 mg/h cada 30 minutos hasta una velocidad máxima de 400 mg/h. |
|
|
Día 15 |
1000 mg |
||
|
Ciclos 2-6 |
Día 1 |
1000 mg |
|
|
1 Ensayos clínicos/Eficacia para obtener información sobre la dosis de clorambucilo. |
|||
Dosis diferidas u omitidas (LLC): si se omite una dosis prevista de GAZYVA, se debe administrar tan pronto como sea posible; no se esperará hasta la siguiente dosis prevista. El intervalo de tratamiento previsto para GAZYVA debe mantenerse entre las dosis.
Linfoma folicular: la dosis recomendada de GAZYVA es de 1000 mg, administrados por vía i.v. tal como se indica en la tabla 3.
Pacientes con linfoma folicular sin tratamien-to previo: en los pacientes con linfoma folicular sin tratamiento previo, GAZYVA debe administrarse con quimioterapia del siguiente modo:
- Seis ciclos de 28 días en combinación con bendamustina2 o,
- Seis ciclos de 21 días en combinación con CHOP, seguidos por 2 ciclos adicionales de GAZYVA solo o,
- Ocho ciclos de 21 días en combinación con CVP.
Los pacientes que no hayan recibido previamente trata-miento y que presenten una respuesta completa o parcial al tratamiento con GAZYVA más quimioterapia seguirán recibiendo GAZYVA (1000 mg) solo, como tratamiento de mantenimiento, cada 2 meses hasta la progresión de la enfermedad o durante un periodo de hasta 2 años.
Linfoma folicular recidivante o resistente al tratamiento: en los pacientes con linfoma folicular que han presentado una recidiva después del tratamiento con rituximab o con una pauta terapéutica que contenga rituximab, o aquellos que no responden a este tratamien-to, GAZYVA debe administrarse en 6 ciclos de 28 días en combinación con bendamustina2.
Los pacientes con linfoma folicular recidivante o resistente al tratamiento que presenten una respuesta completa o parcial o tengan una enfermedad estable deben seguir recibiendo GAZYVA solo, en dosis de 1000 mg, como tratamiento de mantenimiento una vez cada 2 meses hasta la progresión de la enfermedad o durante un periodo de hasta 2 años.
|
Tabla 3. Dosis y velocidad de infusión de GAZYVA en los pacientes con LF |
|||
|
Día del ciclo de tratamiento |
Dosis de GAZYVA |
Velocidad de infusión. Consúltese en la tabla 4 el tratamiento de las RRI que se producen durante la infusión. |
|
|
Ciclo 1 |
Día 1 |
1000 mg |
Se administra a una velocidad de 50 mg/h. La velocidad de infusión puede aumentarse a razón de hasta 50 mg/h cada 30 minutos hasta una velocidad máxima de 400 mg/h. |
|
Día 8 |
1000 mg |
Si no se produce ninguna RRI o se produce una RRI de grado 1 durante la infusión previa, cuya velocidad de infusión final fuera ≥100 mg/h, las infusiones pueden iniciarse a una velocidad de 100 mg/h y aumentarse a razón de 100 mg/h cada 30 min hasta alcanzar una velocidad máxima de 400 mg/h. Si el paciente sufre una RRI de grado 2 o superior durante la infusión previa, se administrará a una velocidad de 50 mg/h. La velocidad de infusión puede aumentarse a razón de 50 mg/h cada 30 minutos hasta una velocidad máxima de 400 mg/h. |
|
|
Día 15 |
1000 mg |
||
|
Ciclos 2-6 o 2-8 |
Día 1 |
1000 mg |
|
|
Mantenimiento de los pacientes con LF |
Cada 2 meses hasta la progresión o hasta 2 años |
1000 mg |
|
|
Véase Ensayos clínicos/Eficacia para más información sobre la dosis de bendamustina. |
|||
Dosis diferidas u omitidas (LF): si se omite una dosis prevista de GAZYVA, se debe administrar tan pronto como sea posible; no se omitirá ni se esperará hasta la siguiente dosis prevista.
Si antes del día 8 del ciclo 1 o del día 15 del ciclo 1 se producen reacciones adversas que exijan retrasar el tratamiento, dichas dosis deben administrarse tras la resolución de las reacciones adversas. En tales casos, todas las visitas posteriores y el inicio del ciclo 2 se moverán para adaptarse al retraso del ciclo 1.
Durante el mantenimiento, se debe mantener la pauta posológica original en las dosis subsiguientes.
Ajustes posológicos durante el tratamiento (todas las indicaciones): no se recomienda reducir la dosis de GAZYVA.
Para el tratamiento de los eventos adversos sintomáticos (incluidas las RRI), véanse la tabla 4, a continuación, y el apartado Advertencias y precauciones.
|
Tabla 4. Pautas para la modificación de la velocidad de infusión en caso de reacciones relacionadas con la infusión (ver Advertencias y precauciones, Reacciones relacionadas con la infusión) |
|
|
Grado 4 (potencialmente mortales) |
Se debe detener la infusión y suspender permanentemente el tratamiento. |
|
Grado 3 (graves) |
- Se debe interrumpir temporalmente la infusión y tratar los síntomas. - Cuando hayan remitido los síntomas, se reanudará la infusión a una velocidad que sea como máximo la mitad de la velocidad previa (la velocidad que se estuviera utilizando en el momento en que se produjo la RRI). - Si el paciente no sufre ningún otro síntoma de RRI, se puede reanudar el aumento escalonado de la velocidad de infusión según los incrementos e intervalos que sean apropiados para la dosis del tratamiento (ver tablas 2 y 3). o en los pacientes con LLC que reciben la dosis del día 1 del ciclo 1 dividida en 2 días, la velocidad de infusión del día 1 puede aumentarse hasta 25 mg/h al cabo de 1 hora, pero sin aumentarla más. - Se debe interrumpir la infusión y suspender permanentemente el tratamiento si el paciente sufre por segunda vez una RRI de grado 3. |
|
Grado 1-2 (leves y moderadas) |
- Se reducirá la velocidad de infusión y se tratarán los síntomas. - Cuando se hayan resuelto los síntomas, se continuará la infusión. - Si el paciente no sufre ningún otro síntoma de RRI, se puede reanudar el aumento escalonado de la velocidad de infusión según los incrementos e intervalos que sean apropiados para la dosis del tratamiento (ver tablas 2 y 3). o en los pacientes con LLC que reciben la dosis del día 1 del ciclo 1 dividida en 2 días, la velocidad de infusión del día 1 puede aumentarse hasta 25 mg/h al cabo de 1 hora, pero sin aumentarla más. |
USO EN POBLACIONES ESPECIALES
Embarazo: GAZYVA no debe administrarse durante el embarazo a no ser que el posible beneficio para la madre justifique el riesgo para el feto. Las mujeres con posibilidad de quedar embarazadas deben usar métodos anticonceptivos eficaces durante el tratamiento con GAZYVA y en los 18 meses posteriores a éste (ver Propiedades farmacocinéticas, Eliminación). Se debe considerar la posibilidad de retrasar la inmunización con vacunas elaboradas con microorganismos vivos en los lactantes cuyas madres hayan estado expuestas a GAZYVA durante el embarazo, hasta que la cifra de linfocitos B de los lactantes vuelva a estar dentro del intervalo normal. No se han realizado estudios en embarazadas. En un estudio de la reproducción en el macaco, no se evidenció toxicidad embriofetal ni efectos teratógenos, pero sí una depleción completa de los linfocitos B en las crías. Las cifras de linfocitos B se normalizaron en las crías, y a los 6 meses de edad la función inmunitaria se había restablecido (ver Datos preclínicos sobre seguridad, Teratogenicidad). Además, la concentración sérica de GAZYVA en las crías fue similar a la observada en las madres el día 28 después del parto, mientras que la concentración en la leche el mismo día fue muy baja, lo que indica que GAZYVA atraviesa la barrera placentaria.
Parto: sin texto.
Lactancia: dado que la IgG humana se segrega en la leche materna, y puesto que se desconoce el potencial de absorción y daño para el lactante, se aconsejará a las mujeres que no amamanten a sus hijos durante el tratamiento con GAZYVA y en los 18 meses posteriores a la administración de la última dosis (ver Propiedades farmacocinéticas, Eliminación). En estudios en animales se ha demostrado que GAZYVA se excreta en la leche materna (ver Datos preclínicos sobre seguridad, Teratogenicidad).
Uso en pediatría: no se han determinado la seguridad ni la eficacia de GAZYVA en menores de 18 años.
Uso en geriatría
Leucemia linfocítica crónica: en el estudio fundamental en la LLC, el 46 % (156 de 336) de los pacientes tratados con GAZYVA más clorambucilo tenían 75 o más años (mediana de la edad de 74 años). Estos pacientes sufrieron más eventos adversos graves y eventos adversos mortales que los pacientes menores de 75 años. No se observaron diferencias significativas en cuanto a la eficacia entre los pacientes de 75 o más años y los menores de 75 años (ver Ensayos clínicos/Eficacia).
Linfoma no hodgkiniano: en los estudios fundamentales en el LNH de baja malignidad, los pacientes de 65 o más años presentaron más eventos adversos graves, y eventos adversos que implicaron la retirada o eventos adversos mortales que los pacientes menores de 65 años. No se observaron diferencias clínicamente significativas en cuanto a la eficacia.
Insuficiencia renal
Leucemia linfocítica crónica: en el estudio fundamental en la LLC, el 27 % (90 de 336) de los pacientes tratados con GAZYVA más clorambucilo tenían una insuficiencia renal moderada (aclaramiento de creatinina [ClCr] <50 ml/min). Estos pacientes sufrieron más eventos adversos graves y eventos adversos mortales que los asociados a un ClCr ≥50 ml/min (ver Pautas posológicas especiales y 3.2.5 Farmacocinética en poblaciones especiales). No se observaron diferencias significativas en cuanto a la eficacia entre los pacientes con un ClCr <50 ml/min y los pacientes con un ClCr ≥50 ml/min. Se excluyó del estudio a los pacientes con un ClCr <30 ml/min (ver Ensayos clínicos/Eficacia).
Linfoma no hodgkiniano: en los estudios fundamentales en el LNH de baja malignidad, el 7,7 % de los pacientes (GAO4753g: 15 de 194) y el 5 % de los pacientes (BO21223: 35 de 698) tenían una insuficiencia renal moderada (ClCr <50 ml/min). Estos pacientes sufrieron más eventos adversos graves, eventos adversos mortales y eventos adversos que implicaran la retirada del tratamiento que los asociados a un ClCr ≥50 ml/min (ver Pautas posológicas especiales y 3.2.5 Farmacocinética en poblaciones especiales). Se excluyó de los estudios a los pacientes con un ClCr <40 ml/min (ver Ensayos clínicos/Eficacia).
Insuficiencia hepática: no se han determinado la seguridad ni la eficacia de GAZYVA en pacientes con insuficiencia hepática.
VÍA DE ADMINISTRACIÓN: Infusión intravenosa (i.v.).
Declaración de esterilidad/radiactividad: producto estéril.
SOBREDOSIS: No se dispone de experiencia en ensayos clínicos en el ser humano en lo que respecta a la sobredosis. En los ensayos clínicos con GAZYVA, se han administrado dosis de 50-2000 mg por infusión. La incidencia y la intensidad de las reacciones adversas notificadas en estos estudios no parecieron depender de la dosis. En caso de sobredosis, se procederá inmediatamente a reducir la velocidad de infusión o a interrumpir la infusión y se controlará estrechamente al paciente. Se tendrá en cuenta la necesidad de realizar regularmente hemogramas y el riesgo elevado de infecciones cuando el paciente presenta una depleción de los linfocitos B.
DESCRIPCIÓN
Clase terapéutica o farmacológica del fármaco: antineoplásico, anticuerpo monoclonal.
Código ATC: L01XC15 (obinutuzumab).
PRESENTACIÓN COMERCIAL: Caja x 1 vial de 1000 mg/40 ml + prospecto.
Medicamento biológico innovador.
Información de febrero de 2017.
Fabricado para F. Hoffmann-La Roche S.A. Basilea, Suiza por Roche Diagnostics GmbH Mannheim, Alemania.
Medicamento: manténgase fuera del alcance de los niños.
ROCHE ECUADOR S. A.
Casilla 1711-06185
Quito - Ecuador

