
ERIVEDGE
VISMODEGIB
Cápsulas
Cápsulas , 150 Miligramos
Composición cualitativa y cuantitativa
Principio activo: Vismodegib 150.0 mg.
Excipientes de la cápsula: Celulosa microcristalina PH101, lactosa monohidrato, laurisulfato sódico, povidona K29/32, glicolato sódico de almidón, talco y estearato de magnesio (no bovino).
Forma farmacéutica: Las cápsulas de Erivedge de 150 mg son de gelatina dura y tienen el cuerpo opaco de color rosa, con la inscripción 150 mg impresa en tinta negra, y tapa opaca de color gris, con la inscripción «VISMO» impresa en tinta negra.
PROPIEDADES Y EFECTOS FARMACOLÓGICOS
Propiedades farmacodinámicas
Electrofisiología cardiaca: las dosis terapéuticas de Erivedge no tuvieron ningún efecto en el intervalo QTc. En un estudio del QTc aleatorizado, con doble enmascaramiento (doble ciego), comparativo con placebo y con un fármaco de referencia activo, con grupos paralelos, se administraron a sujetos sanos 150 mg de Erivedge cada 24 horas durante 7 días, placebo y una dosis oral única de moxifloxacino. Erivedge tampoco tuvo efectos importantes en otros parámetros electrocardiográficos (frecuencia cardiaca, intervalo PR, duración del QRS, morfología de la onda T o de la onda U).
Mecanismo de acción: el vismodegib es un inhibidor de la vía hedgehog, de baja masa molecular, que se administra por vía oral. El vismodegib se une a la proteína transmembrana smoothened y la inhibe, impidiendo así la transmisión de señales hedgehog.
Ensayos clínicos / Eficacia: se realizó un estudio fundamental internacional, multicéntrico, sin enmascara-miento, con un solo grupo y 2 cohortes (SHH4476g), en 104 pacientes con carcinoma basocelular (CBC) avanzado, tanto CBC metastásico (n = 33) como CBC localmente avanzado (n = 71). Se definió el CBC metastásico (CBCm) como un CBC diseminado desde la piel a otras partes del organismo, como los ganglios linfáticos, los pulmones, los huesos u otros órganos internos. Los pacientes con CBC localmente avanzado (CBCla) tenían lesiones cutáneas que no podían someterse a cirugía (lesiones inoperables o lesiones cuyo tratamiento quirúrgico causaría deformidades sustanciales) y en las que la radioterapia no había arrojado resultados satisfactorios o estaba contraindicada. Antes de la inclusión en el estudio se confirmó el diagnóstico de CBC mediante un examen histológico. Podían participar en el estudio los pacientes con síndrome de Gorlin que tuvieran al menos una lesión de CBC avanzado y que cumplieran los criterios de inclusión. Los pacientes recibieron una dosis oral diaria de 150 mg de Erivedge.
La mediana de la edad de todos los pacientes fue de 62 años; el 45 % de los pacientes eran mayores de 65 años. La mayoría de los pacientes eran varones (61 %) y de raza blanca (100 %); el 32 % tenían un CBCm y el 68 % presentaban un CBCla. En la cohorte del CBCm, casi todos los pacientes habían recibido anteriormente tratamiento (97 %): cirugía (97 %), radioterapia (58 %) y tratamientos sistémicos (30 %). En la cohorte del CBCla, casi todos los pacientes habían recibido anteriormente tratamiento (94 %): cirugía (89 %), radioterapia (27 %) y tratamientos sistémicos (11 %). La mediana de la duración del tratamiento de todos los pacientes fue de 12,9 meses (intervalo: 0,7-47,8 meses).
La variable de valoración principal fue la tasa de respuesta objetiva (TRO) evaluada por un centro independiente de revisión (CIR). Los resultados del análisis principal (9 meses después de la inclusión del último paciente en el estudio) y de 12 meses de seguimiento adicionales se resumen en la tabla 2.
La evaluación de la TRO por parte del investigador fue una variable de valoración secundaria. Los resultados del análisis principal (9 meses después de la inclusión del último paciente en el estudio) y de 30 meses de seguimiento adicionales se resumen en la tabla 3.
Se definió la respuesta objetiva como una respuesta completa o parcial, determinada en dos evaluaciones consecutivas realizadas al menos con 4 semanas de diferencia. En la cohorte del CBCm, la respuesta tumoral se evaluó conforme a la versión 1.0 de los criterios RECIST (criterios de evaluación de la respuesta en tumores sólidos). En la cohorte del CBCla, la respuesta tumoral se evaluó basándose en el examen visual externo del tumor y de la ulceración, las pruebas de diagnóstico por imágenes del tumor (si era pertinente) y la biopsia tumoral.
Se consideró que un paciente respondía al tratamiento si se cumplía al menos uno de los siguientes criterios y el paciente no sufría una progresión: 1) reducción ≥30 % del tamaño de las lesiones diana (suma del diámetro mayor [SDM]) respecto al tamaño inicial, determinado mediante radiografía; 2) reducción ≥30 % de la SDM respecto al valor inicial en la dimensión externamente visible de las lesiones diana; 3) resolución completa de la ulceración de todas las lesiones diana.
Otras variables de valoración secundarias fueron la duración de la respuesta (DR), la supervivencia sin progresión (SSP), la respuesta histopatológica y la supervivencia global (SG). Los resultados se muestran en las tablas 2 y 3.
|
Tabla 2. Resumen de la eficacia según la evaluación del CIR: pacientes evaluables en cuanto a la eficacia*,† |
||||
|
Resultado |
Análisis principal |
Actualización a los 12 meses |
||
|
CBCm |
CBCla (n = 63) |
CBCm (n = 33) |
CBCla (n = 63) |
|
|
Variable de valoración principal |
||||
|
Tasa de respuesta objetiva (TRO) |
||||
|
Pacientes con respuesta |
10 (30,3 %) |
27 (42,9 %) |
11 (33,3 %) |
30 (47,6 %) |
|
Respuesta completa |
0 |
13 |
0 |
14 |
|
Respuesta parcial |
10 |
14 |
11 |
16 |
|
Enfermedad estable |
21 |
24 |
20 |
22 |
|
Enfermedad progresiva ‡ |
1 |
8 |
1 |
8 |
|
IC 95 % de la respuesta global |
(15,6 %, 48,2 %) |
(30,5 %, 56,0 %) |
(19,2 %, 51,8 %) |
(35,5 %, 60,6 %) |
|
p (unilateral)†† |
0,0011 |
< 0,0001 |
No procede |
No procede |
|
Variables de valoración secundarias |
||||
|
Duración de la respuesta Mediana (meses) |
7,6 |
7,6 |
7,6 |
9,5 |
|
IC 95 % |
(5,62, NE) |
(5,7, 9,7) |
(5,5, 9,4) |
(7,4, 21,4) |
|
Supervivencia sin progresión (SSP) Mediana (meses) |
9,5 |
9,5 |
9,5 |
9,5 |
|
IC 95 % |
(7,36, NE) |
(7,39, 11,93) |
(7,4, 11,1) |
(7,4, 14,8) |
|
NE = no estimable. * La población de pacientes evaluables en cuanto a la eficacia se define como la formada por todos los pacientes inscritos en el estudio que recibieron cualquier cantidad del fármaco en estudio y en los que la interpretación de las muestras conservadas de tejido o de la biopsia inicial por parte de un anatomopatólogo independiente era compatible con un CBC. |
||||
|
† Datos inevaluables o perdidos, incluidos 1 paciente con CBCm y 4 pacientes con CBCla. ‡ La progresión en la cohorte de pacientes con CBCla se define como el cumplimiento de cualquiera de los siguientes requisitos: 1) aumento ≥20 % de la suma de las dimensiones mayores (SDM) de las lesiones diana desde el nadir (según la evaluación radiográfica o la dimensión externamente visible); 2) nuevas ulceraciones de las lesiones diana que persistan sin signos de cicatrización durante al menos 2 semanas; 3) nuevas lesiones según la evaluación radiográfica o la exploración física; 4) progresión, según los criterios RECIST, de lesiones no diana. †† Según el análisis principal realizado 9 meses después de la entrada en el estudio del último paciente. |
||||
|
Tabla 3 . Resumen de la eficacia según la evaluación del investigador: pacientes evaluables en cuanto a la eficacia*,† |
||||
|
Resultado |
Análisis principal |
Actualización a los 30 meses |
||
|
CBCm (n = 33) |
CBCla (n = 63) |
CBCm (n = 33) |
CBCla (n = 63) |
|
|
Variables de valoración secundarias |
||||
|
Tasa de respuesta objetiva (TRO) |
||||
|
Pacientes con respuesta |
15 (45,5 %) |
38 (60,3 %) |
16 |
38 (60,3 %) |
|
Respuesta completa |
0 |
20 |
0 |
20 |
|
Respuesta parcial |
15 |
18 |
16 |
18 |
|
Enfermedad estable |
15 |
15 |
14 |
15 |
|
Enfermedad progresiva‡ |
2 |
6 |
2 |
6 |
|
IC 95 % de la respuesta global |
(28,1 %, 62,2 %) |
(47,2 %, 71,7 %) |
(30,8 %, 66,2 %) |
(47,2 %, 71,7 %) |
|
p (unilateral)†† |
< 0,0001 |
< 0,0001 |
No procede |
No procede |
|
Duración de la respuesta |
||||
|
Mediana (meses) |
12,9 |
7,6 |
14,8 |
26,2 |
|
IC 95 % |
(5,55, 12,91) |
(7,43, NE) |
(5,6, 17,0) |
(9,0, 37,6) |
|
Supervivencia sin progresión (SSP) |
||||
|
Mediana (meses) |
9,2 |
11,3 |
9,3 |
12,9 |
|
IC 95 % |
(7,4, NE) |
(9,46, 16,8) |
(7,4, 16,6) |
(10,2, 28,0) |
|
Supervivencia global (SG) |
||||
|
Mediana (meses) |
No se alcanzó |
No se alcanzó |
33,4 |
No se alcanzó |
|
IC 95 % |
(13,86, NE) |
(17,6, NE) |
(18,1, NE) |
(NE) |
|
NE = no estimable. * La población de pacientes evaluables en cuanto a la eficacia se define como la formada por todos los pacientes inscritos en el estudio que recibieron cualquier cantidad del fármaco en estudio y en los que la interpretación de las muestras conservadas de tejido o de la biopsia inicial por parte de un anatomopatólogo independiente era compatible con un CBC. |
||||
|
† Datos inevaluables o perdidos, incluidos 1 paciente con CBCm y 4 pacientes con CBCla. ‡ La progresión en la cohorte de pacientes con CBCla se define como el cumplimiento de cualquiera de los siguientes requisitos: 1) aumento ≥20 % de la suma de las dimensiones mayores (SDM) de las lesiones diana desde el nadir (según la evaluación radiográfica o la dimensión externamente visible); 2) nuevas ulceraciones de las lesiones diana que persistan sin signos de cicatrización durante al menos 2 semanas; 3) nuevas lesiones según la evaluación radiográfica o la exploración física; 4) progresión, según los criterios RECIST, de lesiones no diana. †† Según el análisis principal realizado 9 meses después de la entrada en el estudio del último paciente. |
||||
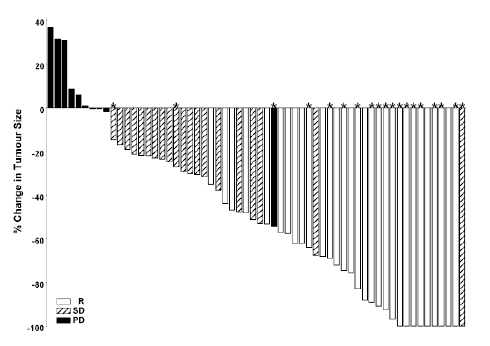
Los gráficos en cascada de las figuras 1 y 2 reflejan la evaluación del CIR tras 12 meses de seguimiento mediante la representación de la reducción máxima del tamaño de las lesiones diana de cada paciente. La mayoría de los pacientes de ambas cohortes presentaron una reducción del tamaño tumoral.
Figura 1
Cohorte del CBC metastásico

Nota: el tamaño tumoral se basa en la suma de las dimensiones mayores de las lesiones diana.
PD = enfermedad progresiva; SD = enfermedad estable; PR = respuesta parcial. En 3 pacientes el mejor cambio porcentual del tamaño tumoral fue de 0; estos pacientes están representados en la figura por barras positivas mínimas. Cuatro pacientes fueron excluidos de la figura: la evaluación de 3 pacientes con enfermedad estable se hizo únicamente en lesiones no diana, y 1 paciente no fue evaluable.
|
% Change in Tumor Size |
% de cambio del tamaño tumoral |
|
PR |
RP |
|
SD |
EE |
|
PD |
EP |
Figura 2
Cohorte del CBC localmente avanzado
Nota: el tamaño tumoral se basa en la suma de las dimensiones mayores de las lesiones diana.
PD = enfermedad progresiva; SD = enfermedad estable; R = respuesta; * = resolución completa de la(s) úlcera(s). La evaluación de la respuesta se basó en una variable de valoración compuesta definida como se indica más atrás. No se hicieron mediciones de la lesión en 4 pacientes, a los que no se incluyó en el diagrama.
|
% Change in Tumor Size |
% de cambio del tamaño tumoral |
|
R |
R |
|
SD |
EE |
|
PD |
EP |
En el momento de realizar el análisis principal del CBCm, la mayoría de las respuestas evaluadas por el CIR (6 de 10 pacientes con respuesta) tuvieron lugar en la semana 8, y se observaron respuestas adicionales en evaluaciones posteriores.
En el CBCla, la mayoría de las respuestas evaluadas por el CIR (14 de 27 pacientes con respuesta) tuvieron lugar en la semana 8, y se observaron respuestas adicionales en evaluaciones posteriores. El 54 % de los pacientes con CBCla (n = 63) tuvieron una respuesta histopatológica sin signos de CBC a las 24 semanas.
Se realizó un ensayo clínico de fase II posterior a la aprobación, multicéntrico, no comparativo, sin enmascaramiento (MO25616) en 1232 pacientes con CBC avanzado, incluidos pacientes con CBCla evaluables en lo que respecta a la seguridad y a la eficacia (n = 1119) o CBCm (n = 96). Se definió el CBC localmente avanzado (CBCla) como lesiones cutáneas que no podían someterse a cirugía (lesiones inoperables o lesiones cuyo tratamien-to quirúrgico causaría deformidades importantes) y en las que la radioterapia no había tenido resultados satisfactorios o estaba contraindicada. Se definió el CBCm como la presencia de metástasis a distancia confirmadas histológicamente. Antes de la inclusión en el estudio se confirmó el diagnóstico de CBC mediante un examen histológico. Los pacientes recibieron una dosis oral diaria de 150 mg de Erivedge.
La mediana de la edad de todos los pacientes fue de 72 años. La mayoría de los pacientes eran varones (57 %), el 8 % tenían un CBCm, mientras que el 92 % tenía un CBCla. En lo que respecta a la cohorte de pacientes con CBCm, la mayoría (91 %) de ellos se habían sometido anteriormente a cirugía o a procedimientos relacionados con el CBCm. En la cohorte del CBCla, la mayoría de los pacientes habían recibido anteriormente tratamiento: cirugía (84,5 %), radioterapia (62 %) y tratamientos sistémicos (6 %). La mediana de la duración del tratamien-to de todos los pacientes fue de 8,6 meses (intervalo: 0-44,1 meses).
Entre los pacientes de la población evaluable en lo que respecta a la eficacia que presentaban una enfermedad mensurable y confirmada histológicamente, el 68,5 % en la cohorte del CBCla y el 36,9 % en la cohorte del CBCm respondieron al tratamiento. En los pacientes con una respuesta (parcial o completa) confirmada, la mediana de la duración de la respuesta fue de 23,0 meses (IC del 95 %: 20,4-26,7) en la cohorte del CBCla y de 13,9 meses (IC 95 %: 9,2-NE) en la cohorte del CBCm. Alcanzaron una respuesta completa el 4,8 % de los pacientes en la cohorte del CBCm y el 33,4 % de los pacientes en la cohorte del CBCla.
Indicaciones terapéuticas: Erivedge está indicado para el tratamiento de pacientes adultos con carcinoma basocelular avanzado en los que no resulta adecuado el tratamiento quirúrgico.
DATOS FARMACÉUTICOS
Conservación
Periodo de validez: conforme al registro local.
Conservación: mantener a temperatura no mayor a 30°C. Manténgase el frasco bien cerrado para proteger el medicamento de la humedad.
Instrucciones especiales de uso, manipulación y eliminación: este medicamento no debe usarse después de la fecha de caducidad, indicada con «EXP» en el envase.
Eliminación de los medicamentos no utilizados o caducados: la emisión de productos farmacéuticos al medio ambiente debe reducirse al mínimo.
Evítese tirar los medicamentos por los desagües o a la basura doméstica, y utilícense los sistemas de recogida disponibles localmente.
Propiedades farmacocinéticas
Absorción: el vismodegib es un compuesto muy permeable, de baja hidrosolubilidad (clase 2 según el Sistema de Clasificación Biofarmacéutica [BCS]). La biodisponibilidad absoluta de una dosis única de vismodegib es del 31,8 %. La absorción es saturable, tal como evidencia la ausencia de un aumento de la exposición proporcional a la dosis después de administrar una dosis única de 270 mg y de 540 mg de vismodegib. En condiciones clínicamente relevantes (estado de equilibrio), la farmacocinética del vismodegib no se ve afectada por los alimentos. En consecuencia, el vismodegib puede tomarse independien-temente de las comidas.
Distribución: el volumen de distribución del vismodegib es bajo (16,4-26,6 l). La unión in vitro del vismodegib a las proteínas del plasma humano es elevada (97 %) en concentraciones clínicamente importantes. El vismodegib se une a la albúmina y a la glucoproteína ácida α1 (AAG) del suero humano. La unión in vitro a la AAG es saturable en concentraciones clínicamente relevantes. La unión ex vivo a las proteínas plasmáticas en pacientes humanos es >99 %. Las concentraciones de vismodegib se correlacionan estrechamente con las de AAG, observándose fluctuaciones paralelas de la concentración de AAG y la concentración total del fármaco a lo largo del tiempo, así como concentraciones constantemente bajas de fármaco no unido las proteínas.
Metabolismo: el vismodegib se elimina lentamente mediante una combinación de metabolismo y excreción del fármaco original. El vismodegib es predominante en el plasma, y sus concentraciones representan >98 % del total de los componentes circulantes relacionados con el fármaco. Las vías metabólicas del vismodegib en el ser humano incluyen la oxidación, la glucuronidación y una escisión infrecuente del anillo piridínico. Los dos metabolitos oxidativos más abundantes que se recuperan en las heces son producidos in vitro por CYP2C9 y CYP3A4/5 recombinantes.
Eliminación: tras administrar una dosis oral única, el vismodegib muestra un perfil farmacocinético único, con concentraciones plasmáticas sostenidas y una semivida terminal calculada de 12 días. Después de la administración diaria continua, la farmacocinética del vismodegib parece ser no lineal. Considerando la semivida de una dosis única, en los pacientes se alcanzan concentraciones plasmáticas en equilibrio antes de lo previsto (generalmente en un plazo aproximado de 7 días de administración diaria continua), con una acumulación inferior a la prevista. Se calcula que la semivida aparente del vismodegib en estado de equilibrio es de 4 días con la administración diaria continua.
Después de la administración oral del fármaco radiomarcado, el vismodegib se absorbe y se elimina lentamente mediante una combinación de metabolismo y excreción del fármaco original, la mayoría del cual se recupera en las heces (82 % de la dosis administrada); el 4,4 % de la dosis administrada se recupera en la orina. El vismode-gib y los productos metabólicos asociados se eliminan principalmente por vía hepática.
Farmacocinética en poblaciones especiales
Pacientes pediátricos: no existen datos sobre pacientes pediátricos.
Pacientes geriátricos: los datos sobre pacientes geriá-tricos son limitados. Un análisis de farmacocinética poblacional indica que la edad no tuvo una repercusión clínicamente significativa en la concentración del vismodegib en estado de equilibrio.
Insuficiencia renal: la excreción renal del vismodegib administrado por vía oral es baja (<5 %). En consecuencia, es improbable que la insuficiencia renal tenga un efecto clínicamente significativo en la farmacocinética del vismodegib. Según un análisis farmacocinético poblacional realizado en pacientes con insuficiencia renal leve (aclaramiento de creatinina [ClCr] de 50-80 ml/min indexado por superficie corporal, n = 58), moderada (ClCr de 30-50 ml/min indexado por superficie corporal, n = 16) y grave (ClCr <30 ml/min indexado por superficie corporal, n = 1), la insuficiencia renal no tuvo un efecto clínicamente significativo en la farmacocinética del vismodegib.
Insuficiencia hepática: las principales vías de eliminación del vismodegib son el metabolismo hepático y la secreción biliar e intestinal. En un estudio clínico realizado en sujetos con insuficiencia hepática (el grado de insuficiencia se basó en las concentraciones de AST y la bilirrubina total [BT] del sujeto), los resultados demostraron que en pacientes con insuficiencia hepática leve (criterios NCI-ODWG, n = 8), moderada (criterios NCIODWG, n = 6) y grave (criterios NCI-ODWG, n = 3), la farmacocinética del vismodegib fue comparable a la de sujetos con función hepática normal (n = 9).
Criterios NCI-ODWG de clasificación de la insuficiencia hepática: leve (BT ≤LSN, AST >LSN o bien LSN<BT≤1,5 × LSN, cualquier valor de AST); moderada (1,5 × LSN<BT<3 × LSN, cualquier valor de AST); grave (3 × LSN<BT<10 × LSN, cualquier valor de AST).
Contraindicaciones: Erivedge está contraindicado en las mujeres lactantes durante el tratamiento y hasta 24 meses después de recibir la última dosis, ya que podría causar graves defectos del desarrollo en los lactantes y niños amamantados (ver Advertencias y precauciones generales y Uso en poblaciones especiales).
Efectos sobre la capacidad para conducir y utilizar máquinas: No se han realizado estudios sobre los efectos de Erivedge en la capacidad para conducir y utilizar máquinas.
Pruebas de laboratorio: sin texto.
Reacciones adversas
Ensayos clínicos: la seguridad de Erivedge se ha evaluado en más de 2300 pacientes y voluntarios sanos en estudios clínicos. Los datos que se presentan a continuación corresponden a pacientes con carcinoma basocelular (CBC) avanzado que participaron en 4 ensayos de fase I y de fase II sin enmascaramiento y en un estudio posterior a la aprobación y recibieron al menos una dosis de Erivedge en monoterapia de >150 mg. Las dosis >150 mg no se tradujeron en mayores concentraciones plasmáticas en los ensayos clínicos; en el análisis se ha incluido a los pacientes tratados con dosis >150 mg.
Las reacciones adversas (RA) más frecuentes (≥10 %) notificadas en estos estudios clínicos con Erivedge se resumen en la siguiente tabla.
|
Tabla 1: Reacciones adversas que afectaron a ≥10 % de los pacientes con CBC |
|||
|
Término preferido del MedDRA |
Todos los pacientes con CBC avanzado |
||
|
Todos los grados* /%) |
Grado 3* /%) |
Grado 4* /%) |
|
|
Trastornos gastrointestinales |
|||
|
Náuseas |
48 (34,8 %) |
0 |
0- |
|
Diarrea |
46 (33,3 %) |
3 (2,2 %) |
0- |
|
Estreñimiento |
32 (23,2 %) |
0- |
0- |
|
Vómitos |
23 (16,7 %) |
0- |
0- |
|
Dispepsia |
15 (10,9 %) |
0 |
0 |
|
Trastornos generales y alteraciones en el lugar de la administración |
|||
|
Fatiga |
65 (47,1 %) |
8 (5,8 %) |
1 ( 0,7 %) |
|
Exploraciones complementarias |
|||
|
Disminución del peso |
69 (50,0 %) |
14 (10,1 %) |
0 |
|
Trastornos del metabolismo y de la nutrición |
|||
|
Disminución del apetito |
41 (29,7 %) |
3 (2,2 %) |
0 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
|||
|
Espasmos musculares |
103 (74,6 %) |
7 (5,1 %) |
0 |
|
Artralgias |
23 (16,7 %) |
1 (0,7 %) |
0 |
|
Dolor en las extremidades |
14 (10,1 %) |
1 (0,7 %) |
0 |
|
Trastornos del sistema nervioso |
|||
|
Disgeusia |
81 (58,7 %) |
0 |
0 |
|
Ageusia |
15 (10,9 %) |
0 |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
|||
|
Alopecia |
91 (65,9 %) |
0 |
0 |
|
MedDRA= Diccionario médico para las actividades de registro. |
|||
|
* Versión 3.0 de los NCI-CTCAE (criterios de toxicidad comunes del National Cancer Institute referentes a los acontecimientos adversos). |
|||
Otras reacciones adversas que afectaron a >10 % de un subgrupo de pacientes en riesgo:
Trastornos del sistema reproductor y de la mama: de los 138 pacientes con CBC avanzado, 10 eran mujeres con posibilidad de quedar embarazadas; 3 de ellas (30 %) presentaron amenorrea.
Las siguientes reacciones adversas afectaron a <10 % de los pacientes con carcinoma basocelular avanzado tratados con Erivedge:
Trastornos gastrointestinales: dolor abdominal, dolor en la parte superior del abdomen.
Trastornos generales y alteraciones en el lugar de la administración: astenia.
Exploraciones complementarias: enzimas hepáticas elevadas**, creatina-cinasa en sangre elevada.
Trastornos del metabolismo y de la nutrición: deshidratación.
Trastornos musculoesqueléticos: dolor musculoesquelético, dolor de espalda, dolor torácico musculoesquelético, mialgia, dolor de costado.
Trastornos del sistema nervioso: hipogeusia.
Trastornos de la piel y del tejido subcutáneo: madarosis, crecimiento anormal del pelo.
** El término «enzimas hepáticas elevadas» incluye los siguientes términos preferidos de eventos adversos notificados: enzimas hepáticas elevadas, aspartato-aminotransferasa (AST) elevada, resultados anormales en las pruebas de la función hepática, fosfatasa alcalina elevada en sangre, ?-glutamil transferasa (GGT) elevada y bilirrubina elevada en sangre.
En general, el perfil de seguridad observado fue constante tanto en los pacientes con CBC metastásico como en los pacientes con CBC localmente avanzado, tal como se ha descrito anteriormente.
Alteraciones analíticas: en los 138 pacientes con CBC avanzado, las anomalías de grado 3 de los parámetros analíticos después del inicio de los estudios fueron poco frecuentes (<5 %) y no se registró ninguna anomalía analítica de grado 4. Las anomalías analíticas (n >1) que supusieron un cambio desde el valor inicial hasta una RA de grado 3 fueron la concentración de sodio reducida (n = 7), la concentración de potasio reducida (n = 2) y el nitrógeno ureico sanguíneo (BUN) elevado (n = 3).
Poscomercialización: se han notificado las siguientes reacciones adversas durante el uso de Erivedge desde su aprobación:
Trastornos musculoesqueléticos y del tejido conjuntivo: fusión prematura de las epífisis (ver Adver-tencias y precauciones, Uso en poblaciones especiales y Datos preclínicos sobre seguridad).
Alteraciones analíticas: sin texto.
Interacciones con otros medicamentos y otras formas de interacción
Efectos de otros fármacos en el vismodegib
Fármacos que inhiben o inducen enzimas metabolizadoras: la eliminación del vismodegib se realiza por múltiples vías. El vismodegib se excreta principalmente como fármaco sin modificar. Múltiples enzimas del sistema del citocromo P450 (CYP450) producen varios metabolitos menores. No es previsible que se produzcan interacciones farmacocinéticas clínicamente significativas entre el vismodegib y los inhibidores del CYP450. Los resultados de un estudio clínico no evidenciaron interacciones farmacocinéticas clínicamente significativas entre el vismodegib y el fluconazol (un inhibidor moderado del CYP2C9) o el itraconazol (un inhibidor potente del CYP3A4) en voluntarios sanos.
No es previsible que los inductores del CYP3A4 alteren la exposición sistémica al vismodegib, dado que las concentraciones plasmáticas del vismodegib en equilibrio observadas en pacientes participantes en ensayos clínicos que fueron tratados concomitantemente con inductores del CYP3A4 (por ejemplo: carbamazepina, modafinilo y fenobarbital) y en los tratados concomitantemente con inhibidores del CYP3A4 (por ejemplo: eritromicina y fluconazol) fueron similares.
Fármacos que inhiben los sistemas de transporte de fármacos: no es previsible que se produzcan interacciones farmacocinéticas clínicamente significativas entre el vismodegib y los inhibidores de la glucoproteína P (P-gp). Los resultados de un estudio clínico no evidenciaron interacciones farmacocinéticas clínicamente significativas entre el vismodegib y el itraconazol (un inhibidor potente de la P-gp) en voluntarios sanos.
Fármacos que afectan al pH gástrico: no es previsi-ble que se produzcan interacciones farmacocinéticas clínicamente significativas entre el vismodegib y los fármacos que elevan el pH. Los resultados de un estudio clínico no evidenciaron interacciones farmacocinéticas clínicamente significativas entre el vismodegib y el rabeprazol (un inhibidor de la bomba de protones) en voluntarios sanos.
Efectos del vismodegib en otros fármacos: no es previsible que se produzcan interacciones farmacocinéticas clínicamente significativas entre el vismodegib y los sustratos del CYP450. Los resultados de un estudio de interacciones farmacológicas realizado en pacientes con cáncer no evidenciaron interacciones farmacocinéticas significativas entre el vismodegib y la rosiglitazona (que es sustrato del CYP2C8). Así pues, se puede descartar que el vismodegib inhiba a las enzimas del citocromo P450. Los resultados de un estudio de interacciones farmacológicas realizado en pacientes con cáncer no evidenciaron interacciones farmacocinéticas significativas entre el vismodegib y los anticonceptivos orales etinilestradiol y noretindrona.
No es previsible que se produzcan interacciones farmacocinéticas clínicamente significativas entre el vismodegib y la proteína de resistencia del cáncer de mama (BCRP). Los datos in vitro indican que el vismodegib es un inhibidor del transportador BCRP. Sin embargo, las concentraciones in vitro a las que se produjo la inhibición son significativamente mayores que las concentraciones del vismodegib no unido que se observan en los pacientes.
Uso en poblaciones especiales
Embarazo: no se han realizado estudios comparativos adecuados en mujeres embarazadas tratadas con Erivedge. Se ha demostrado que el vismodegib es embriotóxico y teratógeno en animales. Debido al papel fundamental de la vía hedgehog en la embriogénesis y a los conocidos efectos del vismodegib en el desarrollo prenatal y posnatal (ver Datos preclínicos sobre seguridad), se debe advertir a las mujeres con posibilidad de quedar embarazadas que eviten quedarse embarazadas mientras toman Erivedge y hasta 24 meses después de recibir la última dosis. Las mujeres con posibilidad de quedar embarazadas deben utilizar 2 métodos anticonceptivos aceptables (incluido un método de barrera aceptable con espermicida, cuando sea posible) durante el tratamiento y hasta 24 meses después de concluirlo (ver Advertencias y precauciones generales). Entre los métodos aceptables de anticoncepción primaria se encuentran los siguientes: anticonceptivos orales combinados, implante hormonal subcutáneo, parche hormonal, anticonceptivos hormonales (sistema intrauterino de liberación de levonor-gestrel, acetato de medroxiprogesterona depot), ligadura de trompas, vasectomía y dispositivo intrauterino (DIU). Entre las formas aceptables de anticoncepción de barrera se encuentran las siguientes: cualquier preservativo masculino (con espermicida cuando sea posible) o diafragma (con espermicida cuando sea posible).
Fecundidad: Erivedge puede afectar a la fecundidad (ver Datos preclínicos sobre seguridad - Trastornos de la fecundidad). En ensayos clínicos se han observado casos de amenorrea en mujeres con posibilidad de quedar embarazadas (ver Reacciones adversas, Ensayos clínicos). No se sabe si el trastorno de la fecundidad es reversible. En el caso de las mujeres con posibilidad de quedar embarazadas, antes de empezar el tratamiento con Erivedge es preciso comentar con ellas las estrategias de preservación de la fecundidad que existen.
Parto: sin texto.
Lactancia: no se sabe si el vismodegib se excreta en la leche humana. Erivedge está contraindicado en las mujeres lactantes, dado su potencial de causar graves defectos del desarrollo en los lactantes y niños amamantados (ver Contraindicaciones).
Uso en pediatría: no se han establecido la seguridad ni la eficacia de Erivedge en pacientes pediátricos.
Se han descrito casos de fusión prematura de las epífisis en pacientes pediátricos expuestos a Erivedge (ver Advertencias y precauciones, Reacciones adversas y Datos preclínicos sobre seguridad).
Uso en geriatría: del total de pacientes que participa-ron en estudios clínicos de Erivedge y padecían un carcinoma basocelular avanzado, cerca del 40 % tenían ≥65 años; no se observaron diferencias generales en cuanto a la seguridad y la eficacia entre esta población y pacientes más jóvenes.
Insuficiencia renal: no se han realizado estudios específicos para evaluar el efecto de la insuficiencia renal en la farmacocinética del vismodegib. Los resultados de un análisis farmacocinético poblacional no evidenciaron que la insuficiencia renal repercutiera en la farmacocinética del vismodegib. No es preciso ajustar la dosis en pacientes con insuficiencia renal.
Insuficiencia hepática: se evaluaron la farmacocinética, la seguridad y la tolerabilidad del vismodegib en pacientes con insuficiencia hepática leve, moderada o grave en un estudio clínico específico, tras administrar dosis múltiples de vismodegib. Los resultados no evidenciaron que la insuficiencia hepática repercutiera en la farmacocinética del vismodegib. No es preciso ajustar la dosis en pacientes con insuficiencia hepática leve, moderada o grave.
Datos preclínicos sobre seguridad
Carcinogenicidad: se realizaron estudios de carcinogenicidad en el ratón y la rata. La posibilidad de carcinogenicidad se identificó sólo en la rata, y se limitó a tumores benignos del folículo piloso, en concreto pilomatrixomas y queratoacantomas, respectivamente, con valores de ≥0,1 y ≥0,6 veces el ABC(0-24 h) en el estado de equilibrio con la dosis recomendada en el ser humano. No se identificaron tumores malignos en ninguna de las especies estudiadas. No se han notificado casos de tumores benignos del folículo piloso en los ensayos clínicos con el vismodegib. No se sabe qué importancia puede tener esto para los pacientes.
Mutagenicidad: el vismodegib no fue genotóxico en una serie de ensayos in vitro (prueba de mutación de Ames en Salmonella y Escherichia coli y ensayo de aberraciones cromosómicas en linfocitos periféricos humanos) en pre-sencia o ausencia de sistemas de activación metabólica.
El vismodegib no fue genotóxico en un ensayo in vivo de micronúcleos en células de médula ósea de rata en el que se utilizó una dosis única de hasta 2000 mg/kg (12 000 mg/m2; aproximadamente 120 veces superior a la dosis humana recomendada según la superficie corporal).
Trastornos de la fecundidad: en el estudio específico del vismodegib en la fecundidad de la rata, de 26 semanas de duración, no se observó ningún efecto en las variables de valoración de los órganos reproductores y la fecundidad con dosis de 100 mg/kg/día al final de la administración o de la fase de recuperación (lo que corresponde a 1,3 veces el ABC0-24 h en el estado de equilibrio con la dosis humana recomendada). Además, en los estudios de toxicidad general del vismodegib, de hasta 26 semanas de duración, realizados en ratas y pe-rros sexualmente maduros, no se observaron efectos en los órganos reproductores masculinos. La cifra elevada de células germinales degenerativas y de hipospermia en perros sexualmente inmaduros que se observó con dosis ≥50 mg/kg/día en el estudio general de la toxicidad de 4 semanas tuvo una relación incierta con el vismodegib.
En el estudio específico del vismodegib en la fecundidad de la rata, de 26 semanas de duración, los efectos relacionados con el vismodegib en los órganos reproductores femeninos se observaron con dosis de 100 mg/kg/día inmediatamente después de suspender el tratamiento, incluidos el número reducido de implantaciones, el porcentaje elevado de pérdidas previas a la implantación y el número reducido de hembras con embriones viables. No se encontraron resultados similares tras un periodo de recuperación de 16 semanas. No se observaron cambios histopatológicos correlativos. La exposición en ratas hembra a dosis de 100 mg/kg corresponde a 1,2 veces el ABC0-24 h en el estado de equilibrio con la dosis humana recomendada. Además, en el estudio general de la toxicidad del vismodegib, de 26 semanas de duración, se observó un número reducido de cuerpos lúteos con dosis de 100 mg/kg/día; el efecto no había revertido al finalizar un periodo de recuperación de 8 semanas.
Teratogenicidad: en un estudio del desarrollo embriofetal en el que se administró a ratas preñadas vismodegib diariamente durante la organogénesis, el vismodegib atravesó la barrera placentaria y fue gravemente tóxico para el producto de la concepción. Se observaron malformaciones, incluidas anomalías craneofaciales, perineo abierto y adactilia o sindactilia, en fetos de hembras que habían recibido 10 mg/kg/d (correspondientes a una exposición [ABC0-24 h] que era el 20 % de la obtenida con la dosis humana recomendada). Con la dosis de 10 mg/kg/d también aumentó la incidencia de retraso o variaciones del crecimiento fetal, así como de elementos esternales, cuerpos de las vértebras cervicales, o falanges proximales y garras incompletamente osificados o sin osificar. El vismodegib fue embrioletal en dosis ≥60 mg/kg/d (correspondiente a una exposición [ABC0-24 h] 2,8 veces superior a la que se alcanza con la dosis humana recomendada).
Otros efectos: los resultados de estudios de la toxicidad con vismodegib indicaron que existía el riesgo de efectos adversos durante el desarrollo posnatal. La administración de vismodegib a ratas causó cambios irreversibles de los dientes en crecimiento (degeneración y necrosis de los odontoblastos, formación de quistes rellenos de líquido en la pulpa dental, osificación del canal radicular y hemorragia) y el cierre de la placa epifisaria.
En los estudios de la toxicidad del vismodegib en la rata se observaron con gran frecuencia efectos neurológicos descritos como fasciculaciones o temblores corporales o de las extremidades. Estos efectos neurológicos se resolvieron completamente tras suspender la adminis-tración y no se asociaron a signos microscópicos. No se determinó si estos efectos eran de origen central o periférico; no obstante, en un estudio de autorradiografía de cuerpo entero realizado en la rata, la penetración del vismodegib en los tejidos del sistema nervioso central fue baja. En los perros no se observaron los signos clínicos correspondientes.
Advertencias y precauciones
Advertencias y precauciones generales
Muerte embriofetal o defectos congénitos graves (ver Uso en poblaciones especiales y Datos preclínicos sobre seguridad): Erivedge puede causar muerte embriofetal o defectos congénitos graves cuando se administra a mujeres embarazadas. Se ha demostrado que los inhibidores de la vía hedgehog, como Erivedge, son embriotóxicos o teratógenos en múltiples especies animales, y pueden causar graves defectos de la línea media, adactilia y otras malformaciones irreversibles en el embrión o el feto. Erivedge no debe usarse durante el embarazo, salvo en casos graves potencialmente mortales en los que el posible beneficio para la paciente supere el riesgo para el feto.
Embarazo: (ver Uso en poblaciones especiales)
Pacientes de sexo femenino: las mujeres embarazadas no deben tomar Erivedge, dado el riesgo de muerte embriofetal o de defectos congénitos graves causados por Erivedge, salvo en casos graves potencialmente mortales, en los que el beneficio previsto para la paciente supere el riesgo para el feto.
Las mujeres con posibilidad de quedar embarazadas deben utilizar 2 métodos anticonceptivos aceptables (incluido un método de barrera aceptable con espermicida, cuando sea posible) durante el tratamiento y hasta 24 meses después de concluirlo. Se asesorará a cada paciente acerca de los métodos anticonceptivos disponibles. Se considera que los siguientes métodos de anticoncepción primaria son aceptables, siempre que resulten apropiados desde el punto de vista médico: anticonceptivos orales combinados, implante hormonal subcutáneo, parche hormonal, anticonceptivos hormonales (sistema intrauterino de liberación de levonorgestrel, acetato de medroxiprogesterona depot), ligadura de trompas, vasectomía y dispositivo intrauterino (DIU). Los siguientes son métodos aceptables de anticoncepción secundaria (métodos de barrera): cualquier preservativo masculino (con espermicida cuando sea posible) o diafragma (con espermicida cuando sea posible). Se realizará una prueba de embarazo en el consultorio o en laboratorio en los 7 días anteriores al inicio del tratamiento con Erivedge y mensualmente durante el mismo.
Si la paciente queda embarazada, debe notificárselo inmediatamente al médico que la atiende para que éste evalúe la situación y la asesore convenientemente.
Pacientes de sexo masculino: Vismodegib se encuentra en el semen. Para evitar la posible exposición embrio-fetal durante el embarazo, los pacientes varones, cuando mantengan relaciones sexuales, deben usar preservativos con espermicida (cuando sea posible) mientras sigan el tratamiento con Erivedge y hasta 2 meses después de recibir la última dosis, incluso aunque se hayan sometido a una vasectomía. Los pacientes varones no deben donar semen mientras reciban tratamiento con Erivedge y hasta 2 meses después de recibir la dosis final.
Efectos sobre el desarrollo posnatal (ver Contraindicaciones, Uso en poblaciones especiales, Reacciones adversas y Datos preclínicos sobre seguridad): se han descrito casos de fusión prematura de las epífisis en pacientes expuestos a Erivedge. En ocasiones esta fusión progresó después de suspender la administración del fármaco. En ratas tratadas con vismodegib se han observado efectos adversos irreversibles en los dientes en crecimiento, así como el cierre prematuro de la placa epifisaria.
Mujeres lactantes (ver Contraindicaciones y Uso en poblaciones especiales): no se sabe en qué medida pasa el vismodegib a la leche materna. Dado que puede causar graves defectos del desarrollo, está contraindicada la lactancia en las mujeres que estén bajo tratamiento con vismodegib o que lo hayan tomado en los 24 últimos meses.
Donación de sangre: los pacientes no deben donar sangre o productos sanguíneos mientras sigan el tratamiento y durante los 24 meses posteriores a la administración de la última dosis de Erivedge.
Abuso y dependencia del fármaco: sin texto.
Posología y forma de administración
Dosis habitual: la dosis diaria recomendada de Erivedge es de 150 mg.
Erivedge debe tomarse una vez al día, con o sin alimentos. Las cápsulas deben tragarse enteras; en ningún caso deben abrirse o masticarse.
La administración de Erivedge debe mantenerse hasta la progresión de la enfermedad o hasta que aparezcan reacciones adversas inaceptables.
Dosis omitidas: se indicará a los pacientes que, si omiten la toma de alguna dosis de Erivedge, no tienen que tomar o compensar esa dosis, sino que deben proseguir la administración con la siguiente dosis programada.
Pautas posológicas especiales
Pacientes geriátricos: no es necesario ajustar la dosis en los pacientes mayores de 65 años.
Pacientes pediátricos: no se han establecido la seguridad ni la eficacia de Erivedge en pacientes pediátricos (ver Uso en poblaciones especiales).
Insuficiencia renal: no es preciso ajustar la dosis en pacientes con insuficiencia renal (ver Farmacocinética en poblaciones especiales).
Insuficiencia hepática: no es necesario ajustar la dosis en pacientes con insuficiencia hepática (ver Farmacocinética en poblaciones especiales).
Vía de administración: Vía oral.
Declaración de esterilidad/radiactividad: no procede.
Sobredosis:Erivedge se ha administrado en dosis 3,6 veces superiores a la dosis diaria recomendada de 150 mg. No se observó ningún aumento de la concentración plasmática del fármaco ni de los efectos adversos.
DESCRIPCIÓN
Clase terapéutica o farmacológica del fármaco: antineoplásico. Código ATC: L01XX43.
Presentación Comercial: Caja x 1 frasco x 28 cápsulas + inserto.
Noviembre de 2016.
Fabricado para F. Hoffmann-La Roche S.A. Basilea, Suiza por Patheon Inc., 2100.
Syntex CT, Mississauga, ON Canada L5N.
Medicamento: manténgase fuera del alcance de los niños.
ROCHE ECUADOR S.A.
Casilla 1711- 06185 CCI
Quito - Ecuador

