

EPAMIN XR 100 MG
FENITOINA
Cápsulas de liberación prolongada
1 Frasco, 100 Cápsulas de liberación prolongada,
COMPOSICIÓN:
Composición cualitativa y cuantitativa:
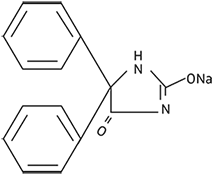
La fenitoína sódica está relacionada con barbitúricos en estructura química, pero tiene un anillo de cinco miembros. El nombre químico es 5,5-difenil-2, 4-imidazolidinediona de sodio, y presenta la siguiente fórmula estructural:
Cada cápsula de EPAMIN® XR 100 mg (fenitoína sódica CÁPSULA DE LIBERACIÓN PROLONGADA) para la administración oral contiene:
100 mg de fenitoína sódica
INDICACIONES TERAPÉUTICAS:
Detalles clínicos: EPAMIN® XR está indicada para el tratamiento de convulsiones tónico-clónicas (gran mal) y psicomotoras (lóbulo temporal) y la prevención y tratamiento de convulsiones que se producen durante o después de una neurocirugía.
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacológicas:
Propiedades farmacodinámicas:
Mecanismo de acción: El mecanismo preciso por el cual fenitoína ejerce su efecto terapéutico no se ha establecido, pero se cree que involucra el bloqueo de los canales de sodio de la membrana que dependen del voltaje, lo que produce una disminución en las descargas neuronales sostenidas de alta frecuencia.
Propiedades farmacocinéticas: El rendimiento in vivo del producto está caracterizado por una tasa lenta y extendida de absorción, con concentraciones pico en sangre esperadas entre las 4 y 12 horas, como se contrasta con fenitoína sódica cápsulas de liberación rápida, con una rápida tasa de absorción y concentraciones pico en sangre esperadas entre las 1 ½ y 3 horas.
Eliminación: La vida media plasmática en el hombre después de la administración oral de la fenitoína promedia en las 22 horas, con un rango que varía de 7 a 42 horas.
Absorción: Para EPAMIN® XR cápsulas, los niveles séricos máximos se presentan de 4 a 12 horas después de la administración. Los niveles terapéuticos del estado estacionario se alcanzan por lo menos de 7 a 10 (5 a 7 vidas medias) días después del inicio del tratamiento.
Cuando se necesiten determinaciones del nivel sérico, se deberán obtener por lo menos 5 a 7 de vida media después de iniciado el tratamiento, de un cambio de la dosis, del agregado o sustracción de otro fármaco al régimen para que se alcance un equilibrio o un estado estacionario.
Metabolismo: La fenitoína es metabolizada principalmente por la enzima CYP2C9 hepática del citrocromo P450 y en menor medida por CYP2C19. Debido a que la fenitoína se hidroliza en el hígado por un sistema de enzimas que se satura a concentraciones plasmáticas altas, pequeñas dosis incrementales pueden aumentar la vida media y producir aumentos muy sustanciales en las concentraciones séricas, cuando se encuentran en un rango superior. Al aumentar la dosis en un 10% o más, podría causar intoxicación debido a que la concentración en el estado estacionario puede aumentarse desproporcionadamente.
En la mayoría de los pacientes con una dosificación constante sostenida, se alcanzan niveles séricos de fenitoína estables. Puede haber una amplia variabilidad interpaciente con igual dosificación en los niveles séricos de fenitoína. Los pacientes con niveles inusualmente bajos pueden no cumplir con el tratamiento o hipermetabolizar la fenitoína. Los niveles inusualmente altos son el resultado de disfunción hepática, variantes alélicas CYP2C9 y CYP2C19 o interacciones de fármacos que causan interferencia metabólica. El paciente con variaciones amplias en los niveles séricos de fenitoína, a pesar de las dosis estándar, presenta un difícil problema clínico. Puede ser de gran ayuda contar con las determinaciones de nivel sérico en este tipo de pacientes. Dado que la fenitoína exhibe una alta unión proteica, los niveles de fenitoína libre pueden verse alterados en pacientes cuyas características de unión proteica difieran de lo normal.
Excreción: La mayor parte del fármaco se excreta en la bilis como metabolitos inactivos que son luego reabsorbidos desde el tracto gastrointestinal y luego eliminados en la orina. La excreción urinaria de la fenitoína y sus metabolitos ocurre en parte con la filtración glomerular, pero más importante aún, a través de la secreción tubular.
Poblaciones especiales:
Pacientes con enfermedad hepática o renal: Se ha reportado que los pacientes con enfermedad renal o hepática, o aquéllos con hipoalbuminemia, presentan una mayor fracción de fenitoína libre (consulte Posología y método de administración).
Edad:
Población geriátrica: La depuración de la fenitoína tiende a disminuir con la edad avanzada (20% menos en pacientes con más de 70 años respecto a la de pacientes entre 20 a 30 años). Ya que la depuración de fenitoína disminuye levemente en los pacientes de edad avanzada, se puede necesitar una dosificación inferior o menos frecuente (consulte Posología y método de administración).
Sexo/Raza: Ni el género ni la raza tienen un impacto importante en la farmacocinética de la fenitoína. Embarazo: En la bibliografía se ha informado que la depuración plasmática de fenitoína por lo general aumentó durante el embarazo, alcanzó el pico en el tercer trimestre y volvió al nivel previo al embarazo algunas semanas o meses después del parto.
Estudios de interacciones medicamentosas: La fenitoína es metabolizada principalmente por la enzima CYP2C9 hepática del citocromo P450 y en menor medida por CYP2C19.
La fenitoína es un inductor potente de enzimas hepáticas metabolizadoras de medicamentos (consulte Interacciones medicamentosas y otras formas de interacción).
Farmacogenómica: La actividad de CYP2C9 está disminuida en individuos con variantes genéticas como los alelos CYP2C9*2 y CYP2C9*3. Los portadores de alelos variantes, que dan como resultado un metabolismo intermedio (p. ej., *1/*3, *2/*2) o deficiente (p. ej., *2/*3, *3/*3) tienen una depuración reducida de fenitoína. Otros alelos CYP2C9 disminuidos o no funcionales también pueden resultar en una disminución de la depuración de la fenitoína (p. ej., * 5, * 6, * 8, * 11).
La prevalencia del fenotipo de metabolizador lento CYP2C9 es de aproximadamente 2-3% en la población blanca, 0,5-4% en la población asiática y <1% en la población afroamericana. La prevalencia del fenotipo intermedio CYP2C9 es de aproximadamente 35% en la población blanca, 24% en la población afroamericana y 15-36% en la población asiática (consulte Advertencias y precauciones especiales de uso y Propiedades farmacocinéticas).
PROPIEDADES FARMACÉUTICAS:
Particularidades farmacéuticas:
Lista de excipientes: Lactosa monohidratada, azúcar glas (impalpable), talco (grado 140), estearato de magnesio.
Incompatibilidades: Ninguna disponible.
Periodo de validez: No utilizar después de la fecha de vencimiento indicada en el empaque.
HALLAZGOS DE LABORATORIO CLÍNICO:
Datos de seguridad preclínicos:
Carcinogénesis: En estudios de carcinogenicidad, se administró fenitoína en la dieta a ratones (10, 25 o 45 mg/k/día) y ratas (25, 50 o 100 mg/kg/día) durante 2 años. La incidencia de tumores hepatocelulares aumentó en ratones macho y hembra bajo la dosis más alta. No se observaron aumentos en la incidencia de tumores en ratas. Las dosis más altas analizadas en estos estudios se vieron asociadas con niveles séricos máximos de fenitoína por debajo de las concentraciones terapéuticas humanas.
En estudios de carcinogenicidad informados en la bibliografía, se administró fenitoína en la dieta a ratones y ratas en dosis de hasta 600 ppm (aproximadamente 160 mg/kg/día) y 2.400 ppm (aproximadamente 120 mg/kg/día), respectivamente, durante 2 años. Las incidencias de tumores hepatocelulares aumentaron en ratones hembra, en todas las dosis analizadas, salvo la más baja. No se observaron aumentos en la incidencia de tumores en ratas.
Mutagénesis: La fenitoína fue negativa en la prueba de Ames y en el ensayo in vitro de clastogenicidad en células de ovario de hámster chino (CHO).
En estudios informados en la bibliografía, la fenitoína fue negativa en el ensayo in vitro de linfoma en ratones y en el ensayo in vivo de micronúcleo en ratones. La fenitoína fue clastogénica en el ensayo in vitro de intercambio de cromátidas hermanas en células CHO.
CONTRAINDICACIONES:
EPAMIN® XR está contraindicada en pacientes con:
• Antecedentes de hipersensibilidad a la fenitoína, a sus componentes inactivos o a otras hidantoínas (consulte Advertencias y precauciones especiales de uso). Las reacciones han incluido angioedema.
• Antecedentes de hepatotoxicidad aguda previa atribuible a la administración de fenitoína (consulte Advertencias y precauciones especiales de uso).
• La coadministración con delavirdina debido a la posible pérdida de respuesta virológica, y la posible resistencia a la delavirdina o a la clase de inhibidores de la transcriptasa inversa no nucleósidos.
No use durante o cuando se sospecha embarazo, o durante la lactancia.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad, embarazo y lactancia:
Fertilidad: No se ha evaluado adecuadamente a la fenitoína para determinar los efectos sobre la fertilidad masculina o femenina.
Lactancia:
Resumen de riesgos: La fenitoína se excreta en la leche materna. Se deben considerar los beneficios del amamantamiento para la salud y el desarrollo, junto con la necesidad clínica de la administración de EPAMIN® XR en la madre y cualquier posible efecto adverso en el lactante amamantado debido a la administración de EPAMIN® XR o a cualquier afección materna subyacente.
Uso durante el embarazo:
Consideraciones clínicas:
Riesgo materno asociado a la enfermedad: Puede producirse un aumento en la frecuencia de las convulsiones durante el embarazo debido a la alteración de la farmacocinética de la fenitoína. La medición periódica de las concentraciones séricas de fenitoína puede ser de importancia en el tratamiento de mujeres embarazadas como guía para un ajuste apropiado de la dosificación (consulte Posología y método de administración). Sin embargo, es probable que se indique la restitución de la dosificación original después del parto.
Resumen de riesgos: En seres humanos, la exposición prenatal a la fenitoína puede aumentar los riesgos de malformaciones congénitas y otros resultados adversos del desarrollo. La exposición prenatal a la fenitoína está asociada con una mayor incidencia de malformaciones, incluyendo hendiduras orofaciales y defectos cardiacos. Además, se ha descrito el síndrome fetal por hidantoína, un patrón de anomalías que incluye rasgos craneofaciales dismórficos, hipoplasia de uñas y dedos, anormalidades del crecimiento (incluida la microcefalia) y déficit cognitivo en hijos de mujeres epilépticas que estaban siendo administradas con fenitoína sola o de manera concomitante con otros fármacos antiepilépticos durante el embarazo (consulte Datos). Se han informado varios casos de neoplasias, como neuroblastoma, en niños cuyas madres estaban siendo administradas con fenitoína durante el embarazo.
La administración de fenitoína en animales preñados produjo un aumento en la incidencia de malformaciones fetales y otras manifestaciones de toxicidad del desarrollo (incluidos muerte embriofetal, deterioro del crecimiento y anomalías de la conducta) en múltiples especies en dosis de importancia clínica (consulte Datos).
En la población general de EE. UU., el riesgo de fondo estimado de presentar defectos de nacimiento importantes y abortos espontáneos en embarazos clínicamente reconocidos es de un 2 a un 4% y de un 15 a un 20%, respectivamente.
Se desconoce el riesgo de fondo de los principales defectos de nacimiento y aborto espontáneo para la población indicada.
Reacciones adversas fetales/neonatales: Puede ocurrir un trastorno de sangrado potencialmente mortal relacionado con niveles disminuidos de factores de coagulación dependientes de la vitamina K en neonatos expuestos a la fenitoína en el útero. Se puede prevenir esta afección inducida por el fármaco con la administración de vitamina K a la madre antes del parto y al neonato después del parto.
Datos:
Datos en humanos: Los metaanálisis utilizando datos publicados de estudios observacionales y registros, han estimado un riesgo aproximadamente 2,4 veces mayor de cualquier malformación importante en niños con exposición prenatal a la fenitoína en comparación con los controles. Se ha reportado un mayor riesgo de defectos cardiacos, hendiduras faciales e hipoplasia digital. El síndrome de hidantoína fetal es un patrón de anomalías congénitas, incluyendo anomalías craneofaciales, hipoplasia de uñas y dígitos, deficiencia del crecimiento prenatal y deficiencias en el desarrollo neurológico.
Datos sobre animales: La administración de fenitoína a ratas, conejos y ratones preñadas durante la organogénesis produjo muerte embriofetal, malformaciones fetales y disminución del crecimiento fetal. Se observaron malformaciones (incluidas anomalías craneofaciales, cardiovasculares, neurales, en las extremidades y los dedos) en ratas, conejos y ratones en dosis a partir de 100 mg/kg, 75 mg/kg y 12,5 mg/kg, respectivamente.
EFECTO EN LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS:
Efectos sobre la capacidad de conducir y usar maquinaria: No conduzca, opere maquinaria pesada ni realice otras actividades peligrosas hasta que sepa cómo lo afecta EPAMIN® XR. EPAMIN® XR puede lentificar sus procesos cognitivos y habilidades motoras.
REACCIONES ADVERSAS:
Las siguientes reacciones adversas serias se describen en otra parte del prospecto:
• Convulsiones precipitadas por la interrupción del medicamento, estado epiléptico (consulte Advertencias y precauciones especiales de uso)
• Conductas e ideas suicidas (consulte Advertencias y precauciones especiales de uso)
• Reacciones dermatológicas serias (consulte Advertencias y precauciones especiales de uso)
• Reacción al medicamento con eosinofilia y síntomas sistémicos (DRESS)/Hipersensibilidad multiorgánica (consulte Advertencias y precauciones especiales de uso)
• Hipersensibilidad (consulte Advertencias y precauciones especiales de uso)
• Efectos cardiacos (consulte Advertencias y precauciones especiales de uso)
• Angioedema (consulte Advertencias y precauciones especiales de uso)
• Lesión hepática (consulte Advertencias y precauciones especiales de uso)
• Complicaciones hematopoyéticas (consulte Advertencias y precauciones especiales de uso)
• Efectos sobre la vitamina D y los huesos (consulte Advertencias y precauciones especiales de uso)
• Exacerbación de la Porfiria (consulte Advertencias y precauciones especiales de uso)
• Teratogenicidad y otros daños al recién nacido (consulte Advertencias y precauciones especiales de uso)
• Hiperglucemia (consulte Advertencias y precauciones especiales de uso)
En estudios clínicos o informes de postcomercialización, se identificaron las siguientes reacciones adversas asociadas a la administración de EPAMIN® XR. Debido a que estas reacciones son informadas de manera voluntaria por una población de tamaño desconocido, no siempre es posible calcular de manera confiable su frecuencia o establecer una relación causal con la exposición al medicamento.
Cuerpo en general: Se han observado reacciones alérgicas en forma de erupción y raramente en formas más graves y el DRESS, al igual que angioedema (consulte Advertencias y precauciones especiales de uso). También se ha documentado anafilaxia.
También se ha informado tosquedad en los rasgos faciales, lupus eritematoso sistémico, periarteritis nodosa y alteraciones en la inmunoglobulina.
Sistema digestivo: Insuficiencia hepática aguda (consulte Advertencias y precauciones especiales de uso), hepatitis tóxica, daño hepático, náuseas, vómitos, estreñimiento, aumento de tamaño de los labios e hiperplasia gingival.
Sistema hematológico y linfático: Se han documentado ocasionalmente complicaciones hematopoyéticas, algunas mortales, asociadas con la administración de fenitoína. Estas incluyeron trombocitopenia, leucopenia, granulocitopenia, agranulocitosis y pancitopenia con o sin supresión de la médula ósea. Aunque ha habido casos de macrocitosis o anemia megaloblástica, estas afecciones responden al tratamiento con ácido fólico. Se ha documentado linfadenopatía que incluye hiperplasia de los ganglios linfáticos benigna, pseudolinfoma, linfoma y enfermedad de Hodgkin (consulte Advertencias y precauciones especiales de uso). También se ha documentado aplasia pura de células rojas.
Alteraciones en los resultados de las pruebas de laboratorio: La fenitoína puede disminuir las concentraciones séricas de las hormonas de la tiroides (T4 y T3), en ocasiones, con aumento de la hormona estimulante de la tiroides (TSH), pero en general en caso de ausencia de hipotiroidismo clínico. La fenitoína también puede producir valores menores a los normales para las pruebas de dexametasona o metirapona. Fenitoína puede producir aumentos en los niveles séricos de glucosa (consulte Advertencias y precauciones especiales de uso), fosfatasa alcalina y glutamiltranspeptidasa gamma (GGT).
Sistema nervioso: Las reacciones adversas más comunes encontradas con la terapia con fenitoína son reacciones del sistema nervioso y generalmente están relacionadas con la dosis. Las reacciones incluyen nistagmo, ataxia, dificultad para hablar, disminución de la coordinación, somnolencia y confusión mental. También se han observado mareos, vértigo, insomnio, nerviosismo transitorio, contracciones motoras, parestesias y dolor de cabeza. También ha habido informes poco frecuentes de discinesias inducidas por fenitoína, incluidos corea, distonía, temblor y asterixis, similares a aquellas inducidas por fenotiazina y otros medicamentos neurolépticos. Se ha informado atrofia cerebelosa, la que parece ser más probable en entornos de niveles elevados de fenitoína y/o con la administración a largo plazo de fenitoína (consulte Advertencias y precauciones especiales de uso).
Se ha observado una polineuropatía periférica sensorial predominante en los pacientes que reciben tratamiento a largo plazo con fenitoína.
Piel y apéndices: Las manifestaciones dermatológicas, a veces acompañadas por fiebre, han incluido erupciones escarlatiniformes o morbiliformes. Una erupción morbiliforme (similar al sarampión) es la más común; los otros tipos de dermatitis se presentan con más poca frecuencia. Otras formas más serias que pueden ser mortales han incluido ampollas, dermatitis exfoliativa o purpúrica, pustulosis exantemámica aguda generalizada, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica (consulte Advertencias y precauciones especiales de uso). También ha habido informes de hipertricosis y urticaria.
Sentidos especiales: El sentido alterado del gusto incluye gusto metálico.
Urogenital: Enfermedad de Peyronie.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones medicamentosas y otras formas de interacción:
Interacciones medicamentosas: La fenitoína se une ampliamente a las proteínas plasmáticas y tiende al desplazamiento competitivo. La fenitoína se metaboliza principalmente por la enzima CYP2C9 del citocromo hepático P450 y en menor medida por CYP2C19, la cual es muy susceptible a la interacción con fármacos inhibidores porque está sujeta al metabolismo saturable. La inhibición del metabolismo podría producir aumentos importantes en las concentraciones de fenitoína circulante y aumentar el riesgo de toxicidad del fármaco El monitoreo de los niveles séricos de fenitoína es recomendado cuando se sospecha que existen interacciones farmacológicas.
La fenitoína es un potente inductor de las enzimas hepáticas metabolizadoras del medicamento.
Fármacos que afectan las concentraciones de fenitoína: En la Tabla 2 se incluyen las interacciones medicamentosas que ocurren comúnmente y que afectan las concentraciones de fenitoína. Sin embargo, no está previsto que esta lista sea inclusiva o exhaustiva. Se debe consultar la información de prescripción individual sobre los medicamentos relevantes.
La adición o la interrupción de la administración de estos agentes en los pacientes en terapia con fenitoína pueden requerir un ajuste de la dosis de fenitoína para alcanzar el resultado clínico óptimo.
Tabla 2. Medicamentos que afectan las concentraciones de fenitoína
|
Agente de interacción |
Ejemplos |
|
Medicamentos que pueden aumentar los niveles séricos de fenitoína |
|
|
Medicamentos antiepilépticos |
Etosuximida, felbamato, oxcarbazepina, metsuximida, topiramato |
|
Azoles |
Fluconazol, ketoconazol, itraconazol, miconazol, voriconazol |
|
Agentes antineoplásicos |
Capecitabina, fluorouracilo |
|
Antidepresivos |
Fluoxetina, fluvoxamina, sertralina |
|
Agentes reductores del ácido gástrico |
Antagonistas de H2 (cimetidina), omeprazol |
|
Sulfonamidas |
Sulfametizol, sulfafenazol, sulfadiazina, sulfametoxazol-trimetoprima |
|
Otros |
Ingesta aguda de alcohol, amiodarona, cloranfenicol, clordiazepóxido, disulfiram, estrógeno, fluvastatina, isoniazida, metilfenidato, fenotiazinas, salicilatos, ticlopidina, tolbutamida, trazodona, warfarina |
|
Medicamentos que pueden disminuir los niveles séricos de fenitoína |
|
|
Antiácidosa |
Carbonato de calcio, hidróxido de aluminio, hidróxido de magnesio Prevención o manejo terapéutico: No se deben tomar antiácidos y fenitoína en el mismo momento del día |
|
Agentes antineoplásicos normalmente en combinación |
Bleomicina, carboplatino, cisplatino, doxorrubicina, metotrexato |
|
Agentes antivirales |
Fosamprenavir, nelfinavir, ritonavir |
|
Medicamentos antiepilépticos |
Carbamazepina, vigabatrina |
|
Otros |
Abuso crónico de alcohol, diazepam, diazóxido, ácido fólico, reserpina, rifampicina, hierba de San Juanb, sucralfato, teofilina |
|
Medicamentos que pueden aumentar o disminuir los niveles séricos de fenitoína |
|
|
Medicamentos antiepilépticos |
Fenobarbital, valproato sódicoc, ácido valproicoc |
a Los antiácidos pueden afectar la absorción de fenitoína.
b La potencia de inducción de la hierba de San Juan puede variar ampliamente según la preparación.
c El valproato sódico y el ácido valproico son medicamentos similares. El término valproato se ha utilizado para representar estos medicamentos.
Fármacos afectados por la fenitoína: En la Tabla 3 se incluyen las interacciones medicamentosas que ocurren comúnmente y que se ven afectadas por la administración de fenitoína. Sin embargo, no está previsto que esta lista sea inclusiva o exhaustiva. Se deben consultar los prospectos de empaque individuales del medicamento. La adición o la interrupción de la administración de fenitoína durante la terapia concomitante con estos agentes pueden requerir el ajuste de la dosis de estos agentes para alcanzar el resultado clínico óptimo.
Tabla 3. Medicamentos afectados por la administración de fenitoína
|
Agente de Interacción |
Ejemplos |
|
Medicamentos cuya eficacia se ve deteriorada por la administración de fenitoína |
|
|
Azoles |
Fluconazol, ketoconazol, itraconazol, posaconazol, voriconazol |
|
Agentes antineoplásicos |
Irinotecán, paclitaxel, tenipósido |
|
Delavirdina |
La administración de fenitoína puede disminuir considerablemente las concentraciones de delavirdina. Esto puede provocar la pérdida de la respuesta virológica y una posible resistencia (consulte Contraindicaciones) |
|
Agentes bloqueadores neuromusculares |
Cisatracurio, pancuronio, rocuronio y vecuronio: Se ha producido resistencia a la acción de bloqueo neuromuscular de los agentes de bloqueo neuromuscular no despolarizantes en pacientes en tratamiento crónico con fenitoína. Se desconoce si la administración de fenitoína tiene el mismo efecto sobre otros agentes no despolarizantes Prevención o manejo terapéutico: Se debe monitorear cuidadosamente a los pacientes para una recuperación del bloqueo neuromuscular más rápida que la esperada, y los requisitos de la tasa de infusión pueden ser mayores |
|
Warfarina |
Se han informado aumentos y disminuciones en las respuestas de PT/INR (tiempo de protrombina/Relación Normalizada Internacional) cuando fenitoína se administra de forma simultánea con warfarina |
|
Otros |
Corticosteroides, doxiciclina, estrógenos, furosemida, anticonceptivos orales, paroxetina, quinidina, rifampicina, sertralina, teofilina y vitamina D |
|
Medicamentos cuyo nivel disminuye por la administración de fenitoína |
|
|
Anticoagulantes |
Apixabán, dabigatrán, edoxabán, rivaroxabán |
|
Medicamentos antiepilépticosa |
Carbamazepina, felbamato, lamotrigina, topiramato, oxcarbazepina, lacosamida |
|
Antiplaquetarios |
Ticagrelor |
|
Agentes antilipémicos |
Atorvastatina, fluvastatina, simvastatina |
|
Agentes antivirales |
Efavirenz, lopinavir/ritonavir, indinavir, nelfinavir, ritonavir, saquinavir Fosamprenavir, cuando se administra fenitoína con fosamprenavir solo, puede disminuir la concentración de amprenavir, el metabolito activo Fenitoína, cuando se administra con la combinación de fosamprenavir y ritonavir, puede aumentar la concentración de amprenavir |
|
Bloqueadores de los canales de calcio |
Nifedipino, nimodipino, nisoldipino, verapamilo |
|
Otros |
Albendazol (disminuye el metabolito activo), clorpropamida, clozapina, ciclosporina, digoxina, disopiramida, ácido fólico, metadona, mexiletina, praziquantel, quetiapina |
a El efecto de la administración de fenitoína sobre los niveles séricos de fenobarbital, ácido valproico y valproato de sodio es impredecible.
Hiperamonemia con el uso concomitante de valproato: La administración concomitante de fenitoína y valproato se ha asociado con un aumento del riesgo de hiperamonemia asociada al valproato. Los pacientes tratados concomitantemente con estos dos fármacos deben ser monitorizados en cuanto a signos y síntomas de hiperamonemia.
Interacción del fármaco con preparaciones nutritivas/alimentación enteral: La bibliografía sugiere que los pacientes que han recibido preparaciones alimenticias enterales y/o suplementos nutricionales relacionados presentan menores niveles séricos de fenitoína. Por lo tanto, se sugiere que no se administre concomitantemente fenitoína con preparaciones alimenticias enterales. Podrían necesitarse controles más frecuentes de los niveles séricos de fenitoína en estos pacientes.
Interacciones con pruebas de laboratorio/medicamentosas: Se deberá tener cuidado al utilizar métodos inmunoanalíticos para medir las concentraciones séricas de fenitoína.
PRECAUCIONES Y ADVERTENCIAS:
Advertencias y precauciones especiales de uso:
Advertencias: Convulsiones precipitadas por la interrupción del medicamento, estado epiléptico.
Estado epiléptico: La interrupción abrupta del tratamiento con fenitoína en pacientes epilépticos puede precipitar status epilepticus. Cuando, a juicio del clínico, surja la necesidad de reducción de la dosificación, interrupción o sustitución por otra medicación anticonvulsivante alternativa, la implementación deberá ser gradual. Sin embargo, en el caso de una reacción alérgica o de hipersensibilidad, se podría requerir una sustitución más rápida por una terapia alternativa. En este caso, el tratamiento alternativo debe consistir en un fármaco anticonvulsivante que no pertenezca a la clase química de las hidantoínas.
Comportamiento suicida e ideación: Los fármacos antiepilépticos (FAE), incluido EPAMIN® XR, aumentan el riesgo de ideas o comportamientos suicidas en pacientes tratados con estos fármacos para cualquier indicación.
Los pacientes tratados con cualquier FAE para cualquier indicación deben ser monitoreados ante la aparición o el empeoramiento de la depresión, pensamientos o comportamientos suicidas y/o cualquier otro cambio inusual en el estado de ánimo o comportamiento.
Los análisis combinados de 199 ensayos clínicos controlados con placebo (monoterapia y terapia adyuvante) de 11 FAE diferentes mostraron que los pacientes aleatorizados a alguno de los FAE tuvieron aproximadamente el doble del riesgo (riesgo relativo ajustado 1,8, IC del 95%: 1,2; 2,7) de presentar ideación o comportamiento suicida, comparados con los pacientes aleatorizados al grupo placebo. En estos ensayos, que tenían una duración de tratamiento medio de 12 semanas, la tasa de incidencia estimada de comportamiento o ideación suicida fue del 0,43% entre 27.863 pacientes tratados con FAE, en comparación con una tasa del 0,24% entre 16.029 pacientes tratados con placebo, lo que representa el aumento de aproximadamente un caso de pensamientos o comportamiento suicidas por cada 530 pacientes tratados. Ocurrieron cuatro suicidios entre los pacientes tratados con el medicamento durante los ensayos y ninguno entre los pacientes que recibían tratamiento con placebo, pero el número es demasiado pequeño como para permitir cualquier conclusión sobre el efecto del fármaco en el suicidio.
El mayor riesgo de ideación o comportamiento suicida con FAE se observó ya en la primera semana de comenzado el tratamiento con FAE, y continuó durante el tratamiento bajo evaluación. No se pudo evaluar el riesgo de ideación o comportamiento suicida después de las 24 semanas porque la mayoría de los ensayos incluidos en los análisis no se extendían más allá de este periodo.
El riesgo de ideación o comportamiento suicida era, por lo general, coherente entre los fármacos de la información analizada. El hallazgo de un mayor riesgo con FAE de diferentes mecanismos de acción y a través de un rango de indicaciones señala que este riesgo aplica a todos los FAE administrados para cualquier indicación. El riesgo no varió en forma sustancial en relación con la edad (5 a 100 años) en los ensayos clínicos analizados.
La Tabla 1 muestra el riesgo absoluto y relativo por indicación para todos los FAE evaluados.
Tabla 1. Riesgo por indicación para fármacos antiepilépticos en el análisis combinado
|
Indicación |
Pacientes bajo placebo con eventos cada 1.000 pacientes |
Pacientes bajo fármaco con eventos cada 1.000 pacientes |
Riesgo relativo: Incidencia de eventos en pacientes bajo fármaco/Incidencia en pacientes bajo placebo |
Diferencia de riesgo: Pacientes adicionales bajo fármaco con eventos cada 1.000 pacientes |
|
Epilepsia |
1,0 |
3,4 |
3,5 |
2,4 |
|
Psiquiatría |
5,7 |
8,5 |
1,5 |
2,9 |
|
Otros |
1,0 |
1,8 |
1,9 |
0,9 |
|
Total |
2,4 |
4,3 |
1,8 |
1,9 |
El riesgo relativo de ideación o comportamiento suicida fue mayor en los ensayos clínicos de epilepsia que en los ensayos clínicos de psiquiatría u otras afecciones, pero las diferencias del riesgo absoluto fueron similares para las indicaciones de epilepsia y psiquiatría.
Quien considere recetar EPAMIN® XR u otro FAE debe hacer un balance entre el riesgo de ideación o comportamiento suicida y el riesgo de la enfermedad no tratada. La epilepsia y muchas otras enfermedades para las que se recetan FAE están asociadas a la morbilidad y mortalidad, y a un mayor riesgo de ideación o comportamiento suicida. En caso de que aparezcan ideación o comportamiento suicida durante el tratamiento, el médico debe considerar si la aparición de cualquiera de estos síntomas en cualquier paciente está relacionada con la enfermedad que se trata.
Debe informarse a los pacientes, sus cuidadores y las familias que los FAE aumentan el riesgo de presentar ideas y comportamientos suicidas y se los debe aconsejar sobre la necesidad de estar alerta ante la aparición o el empeoramiento de los signos y síntomas de depresión; cualquier cambio inusual en el estado de ánimo o comportamiento o la aparición de ideas y comportamientos suicidas o de pensamientos de autolesión. Los comportamientos alarmantes deben informarse de inmediato a los proveedores de atención médica.
Reacciones dermatológicas graves: EPAMIN® XR puede causar reacciones dermatológicas severas (SCARs, por sus siglas en inglés), que pueden ser fatales. Las reacciones que se han reportado en pacientes tratados con fenitoína han incluido necrólisis epidérmica tóxica (NET), síndrome de Stevens-Johnson (SJS), pustulosis exantemática aguda generalizada (PEAG), y reacción al fármaco con eosinofilia y síntomas sistémicos (DRESS) (consulte Advertencias y precauciones especiales de uso). La aparición de los síntomas ocurre por lo general en el plazo de 28 días, pero puede ocurrir más tarde. EPAMIN® XR debe interrumpirse al primer signo de erupción, a menos que la erupción claramente no esté relacionada con el medicamento. Si se presentan signos o síntomas de reacciones dermatológicas severas, no se debe reanudar la administración de este medicamento y se deberá considerar un tratamiento alternativo. En caso de erupción, se evaluará al paciente para determinar signos y síntomas de SCARs.
Estudios en pacientes de origen chino han detectado una fuerte asociación entre el riesgo de desarrollar SJS/NET y la presencia del HLA-B*1502, una variante alélica hereditaria del gen HLA B, en pacientes administrados con carbamazepina. Es escasa la evidencia que sugiere que el HLA-B*1502 pueda ser un factor de riesgo para el desarrollo de SJS/NET en pacientes de origen asiático que usan otros fármacos antiepilépticos asociados al SJS/NET, incluida la fenitoína. Además, estudios retrospectivos, de casos y de asociación a nivel de genoma en pacientes de ascendencia del sudeste asiático también han identificado un mayor riesgo de SCAR en portadores de la variante CYP2C9*3 de función disminuida, que también se ha asociado con una disminución del aclaramiento de fenitoína. Se debe considerar no suministrar EPAMIN® XR como tratamiento alternativo a la carbamazepina en pacientes que son positivos para el HLA-B*1502 o en portadores de CYP2C9*3 (consulte Propiedades farmacocinéticas).
El uso del genotipado del alelo HLA-B*1502 o CYP2C9 tiene limitaciones significativas y nunca deberá sustituir la vigilancia clínica y el tratamiento del paciente adecuados. No se ha estudiado el rol de otros posibles factores en el desarrollo y morbilidad del SJS/NET, como, por ejemplo, dosis de fármacos antiepilépticos (FAE), el cumplimiento, los fármacos concomitantes, las comorbilidades y el nivel de monitoreo dermatológico.
Reacción al fármaco con eosinofilia y síntomas sistémicos (DRESS)/Hipersensibilidad multiorgánica: Se han informado casos de reacción al fármaco con eosinofilia y síntomas sistémicos (DRESS), también conocida como hipersensibilidad multiorgánica, en pacientes que usan fármacos antiepilépticos, incluido EPAMIN® XR. Algunos de estos eventos han resultado mortales o potencialmente mortales. Por lo general, el síndrome DRESS se presenta, aunque no exclusivamente, con fiebre, erupción, linfadenopatía, y/o inflamación facial, en asociación con otro sistema de órganos, como hepatitis, nefritis, anormalidades hematológicas, miocarditis o miositis a veces semejantes a una infección viral aguda. A menudo se desarrolla eosinofilia. Dado que este trastorno varía en su expresión, puede que estén presentes otros sistemas de órganos involucrados que aquí no se mencionaron. Es importante destacar que se pueden presentar manifestaciones tempranas de hipersensibilidad, como fiebre y linfadenopatía, sin que se evidencie una erupción. Si se presentan estos signos o síntomas, se deberá evaluar al paciente en forma inmediata. Se debe interrumpir el tratamiento con EPAMIN® XR si no se puede determinar una etiología alternativa de los signos o síntomas.
Hipersensibilidad: EPAMIN® XR y otras hidantoínas están contraindicados en pacientes que han presentado hipersensibilidad a la fenitoína (consulte Contraindicaciones y Advertencias y precauciones especiales de uso). Además, considere alternativas a fármacos estructuralmente similares como las carboxamidas (p. ej., carbamazepina), barbitúricos, succinimidas y oxazolidindionas (p. ej., la trimetadiona) para estos mismos pacientes. De manera similar, considere alternativas al tratamiento con EPAMIN® XR si el paciente, o algún familiar directo, presentan antecedentes de reacciones de hipersensibilidad a estos fármacos de similar estructura.
Efectos cardiacos: Se han notificado casos de bradicardia y paro cardiaco en pacientes tratados con EPAMIN® XR, tanto a las dosis y niveles recomendados de fenitoína, como en asociación con la toxicidad de fenitoína (consulte Sobredosis). La mayoría de los reportes de paro cardiaco ocurrieron en pacientes con enfermedad cardiaca subyacente.
Angioedema: Se ha notificado angioedema en pacientes tratados con EPAMIN® XR en el contexto poscomercialización. Se debe suspender EPAMIN® XR de inmediato si aparecen síntomas de angioedema, como hinchazón facial, perioral o de las vías respiratorias superiores. Se debe suspender EPAMIN® XR permanentemente si no se puede establecer una etiología alternativa clara a la reacción.
Lesión hepática: Se han informado casos de hepatotoxicidad aguda con EPAMIN® XR, incluidos casos poco frecuentes de insuficiencia hepática aguda. Estos eventos pueden ser parte del espectro del DRESS o pueden ocurrir en forma aislada (consulte Advertencias y precauciones especiales de uso). Otras manifestaciones comunes incluyen ictericia, hepatomegalia, niveles elevados de transaminasas en suero, leucocitosis y eosinofilia. El curso clínico de la hepatotoxicidad aguda por fenitoína varía desde una recuperación rápida a resultados mortales. En estos pacientes con hepatotoxicidad aguda, se deberá interrumpir el tratamiento con EPAMIN® XR inmediatamente y de forma permanente.
Complicaciones hematopoyéticas: Ocasionalmente, se han informado complicaciones hematopoyéticas (algunas mortales) relacionadas con la administración de EPAMIN® XR. Algunas de estas complicaciones fueron trombocitopenia, leucopenia, granulocitopenia, agranulocitosis y pancitopenia con o sin supresión de la médula ósea.
Varios informes sugieren una relación entre la fenitoína y el desarrollo de linfadenopatía (local o generalizada), incluidos hiperplasia nodular linfoide benigna, pseudolinfoma, linfoma y enfermedad de Hodgkin. Si bien no se ha establecido una relación causa y efecto, los casos de linfadenopatías señalan la necesidad de diferenciar esta afección de otros tipos de patologías de los ganglios linfáticos. La afectación de los ganglios linfáticos puede ocurrir con o sin síntomas o signos de DRESS (consulte Advertencias y precauciones especiales de uso).
Para todos los casos de linfadenopatía, se indica un periodo extenso de seguimiento y se deberá realizar el mejor esfuerzo para lograr controlar las convulsiones a través de fármacos antiepilépticos alternativos.
Precauciones:
Hiperglucemia: Se ha documentado hiperglucemia como resultado del efecto inhibidor del fármaco en la secreción de insulina. La fenitoína también puede elevar el nivel de glucosa plasmática en pacientes diabéticos.
Niveles séricos de fenitoína por sobre el rango terapéutico: Los niveles séricos de la fenitoína sostenidos sobre el rango terapéutico pueden producir estados de confusión conocidos como “delirio”, “psicosis” o “encefalopatía” o raramente disfunción cerebelar irreversible y/o atrofia cerebelosa. En consecuencia, al primer signo de toxicidad aguda, se recomienda la determinación inmediata de los niveles séricos. Se recomienda una reducción en la dosis del tratamiento de fenitoína si los niveles séricos son excesivos; y si los síntomas persisten, se recomienda la finalización del tratamiento.
Enfermedad hepática y/o renal o hipoalbuminemia: El hígado es el sitio principal de biotransformación de la fenitoína; los pacientes con función hepática deteriorada, los de edad avanzada o los que se encuentran muy enfermos podrían presentar signos tempranos de toxicidad.
Debido a que la fracción de fenitoína no unida aumenta en pacientes con enfermedad renal o hepática, o en aquellos con hipoalbuminemia, el monitoreo de los niveles séricos de fenitoína se debe basar en la fracción no unida en esos pacientes.
Información para los pacientes: Indique a los pacientes que deben tomar EPAMIN® XR solamente como fue prescripto.
Se debe advertir a los pacientes que usan fenitoína sobre la importancia de seguir estrictamente el régimen de dosificación indicado y de informar a su médico sobre cualquier afección clínica que imposibilite tomar el fármaco por vía oral, según se indicó, p. ej., cirugía, etc.
Los pacientes deben conocer los signos de toxicidad temprana y los síntomas de posibles reacciones hematológicas, dermatológicas, de hipersensibilidad o hepáticas. Estos síntomas pueden incluir, entre otros, fiebre, dolor de garganta, erupción, úlceras en la boca, fácil formación de hematomas, linfadenopatía, hinchazón hepáticas, anorexia, náuseas/vómitos o ictericia. El paciente deberá entender que, como estos signos y síntomas pueden indicar una reacción grave, tiene que informar inmediatamente cualquier incidente al médico. Además, el paciente deberá saber que estos signos y síntomas deberán informarse aun si son leves o si ocurren después del uso prolongado.
Se debe advertir a los pacientes que deberán tener cuidado con el uso de otros medicamentos o bebidas alcohólicas sin consultar primero a su médico (consulte Interacciones medicamentosas y otras formas de interacción).
Se debe advertir a los pacientes que ciertos medicamentos sin prescripción (p. ej., antiácidos, cimetidina y omuscazol), vitaminas (p. ej., ácido fólico) y suplementos herbales (p. ej., hierba de San Juan) pueden alterar sus niveles de fenitoína (consulte Interacciones medicamentosas y otras formas de interacción).
Debe enfatizarse la importancia de la higiene dental para minimizar el desarrollo de hiperplasia gingival y sus complicaciones.
Se debe informar a los pacientes, sus cuidadores y las familias que los FAE, incluido EPAMIN® XR, aumentan el riesgo de tener ideas y comportamientos suicidas; y se les debe aconsejar sobre la necesidad de estar alerta ante la aparición o el empeoramiento de los signos y síntomas de depresión, cualquier cambio inusual en el estado de ánimo o comportamiento, o la aparición de ideas y comportamientos suicidas o de pensamientos de autolesión. Las conductas alarmantes deben informarse de inmediato a los proveedores de cuidados médicos.
Efectos sobre la vitamina D y huesos: El uso crónico de fenitoína en pacientes con epilepsia ha sido asociado con una disminución de la densidad mineral ósea (osteopenia, osteoporosis y osteomalacia) y fractura de huesos. La fenitoína induce enzimas hepáticas metabolizadoras. De esta manera, puede reforzar el metabolismo de la vitamina D y disminuir los niveles de vitamina D; y podría provocar insuficiencia de vitamina D, hipocalcemia e hipofosfatemia. Se deberá considerar evaluar con pruebas de hueso de laboratorio y exámenes radiológicos, según corresponda, e iniciar los planes de tratamiento conforme a los lineamientos establecidos.
Exacerbación de la porfiria: Dado que existen informes aislados que asocian a la fenitoína con la exacerbación de la porfiria, se deberá tener precaución al utilizar este fármaco en pacientes con esa enfermedad.
Teratogenicidad y otros daños al recién nacido: EPAMIN® XR puede causar daño fetal al administrarse en mujeres embarazadas. La exposición prenatal a fenitoína puede aumentar los riesgos de malformaciones congénitas y otros resultados desfavorables del desarrollo (consulte Propiedades farmacocinéticas).
Se ha informado un aumento de la frecuencia de malformaciones importantes (tales como hendidura orofacial y defectos cardiacos) y anormalidades características del síndrome fetal por hidantoína, que incluyen características dismórficas craneofaciales, hipoplasia de uñas y dedos, anormalidades del crecimiento (incluida microcefalia) y déficits cognitivos entre niños que nacieron de madres epilépticas a quienes se les administró fenitoína sola o en combinación con otros medicamentos antiepilépticos durante el embarazo. Se han informado varios casos de neoplasias, incluido neuroblastoma.
Un trastorno de sangrado potencialmente mortal relacionado con niveles disminuidos de factores de coagulación dependientes de la vitamina K puede ocurrir en recién nacidos expuestos a fenitoína en el útero. Esta afección inducida por el medicamento se puede prevenir con la administración de vitamina K a la madre antes del parto y al neonato después del nacimiento.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología y método de administración:
Dosificación en adultos:
Dosis diaria dividida: La dosis inicial recomendada para pacientes que no han recibido tratamiento previo es una dosis de EPAMIN® XR 100 mg (fenitoína sódica cápsulas de liberación prolongada) vía oral tres veces al día.
Ajustar la dosificación a los requisitos de cada paciente hasta un máximo de dos cápsulas tres veces al día. En la mayoría de los adultos, la dosis de mantenimiento satisfactoria será una cápsula tres a cuatro veces al día.
Dosis una vez al día: En adultos, si se establece el control de las convulsiones con dosis divididas de tres EPAMIN® XR 100 mg (fenitoína sódica cápsulas de liberación prolongada) al día, puede considerarse una dosis una vez al día con 300 mg de EPAMIN® XR (fenitoína sódica cápsulas de liberación prolongada) una vez al día. Estudios que comparan dosis indicaron que la absorción, los niveles séricos máximos, la vida media biológica, la diferencia entre valores máximos y mínimos y la cantidad de fármaco recuperado en la orina fueron equivalentes. La dosis una vez al día ofrece un beneficio a cada paciente o al personal de cuidados para los pacientes institucionalizados, y está previsto para administrarse sólo a pacientes que necesitan esta cantidad de fármaco a diario. Un importante problema al motivar a pacientes que no cumplen también podría atenuarse cuando el medicamento se debe administrar una vez al día. Sin embargo, debe advertirse a los pacientes que no se olviden ninguna dosis involuntariamente.
Sólo EPAMIN® XR (fenitoína sódica cápsulas de liberación prolongada) está recomendado para dosificación una vez al día. Las diferencias inherentes en las características de disolución y las tasas de absorción resultantes de la fenitoína, debido a distintos procedimientos de fabricación y/o formas farmacéuticas, descartan tal recomendación para otros productos con fenitoína. Cuando se receta un cambio en la forma farmacéutica o marca, debe realizarse un cuidadoso monitoreo de los niveles séricos de la fenitoína.
Dosis de carga: Algunas autoridades han propuesto la administración de una dosis de carga oral de fenitoína en adultos que requieren rápidos niveles de suero en estado de equilibrio y en casos donde la administración intravenosa no es la mejor opción. Este régimen de dosificación debe emplearse para pacientes en clínica u hospital donde pueden monitorearse de cerca los niveles séricos de fenitoína. Los pacientes con antecedentes de enfermedad renal o hepática no deben recibir el régimen de carga oral.
Al principio, un gramo de EPAMIN® XR (fenitoína sódica cápsulas de mantenimiento normal de la dosificación 24 horas después de la dosis de carga, con determinaciones frecuentes de los niveles séricos.
Dosificación en poblaciones especiales:
Pacientes con enfermedad hepática o renal: Dado que los pacientes con enfermedad renal o hepática, o aquellos con hipoalbuminemia, presentan una mayor fracción de fenitoína libre, se debería tener precaución a la hora de interpretar las concentraciones plasmáticas de fenitoína total. Las concentraciones de fenitoína libre pueden ser de mayor utilidad en estas poblaciones de pacientes.
Uso en pacientes con función CYP2C9 disminuida: Los pacientes que son metabolizadores intermedios o lentos de los sustratos del CYP2C9 (p. ej., *1/*3,*2/*2,*3/*3) pueden presentar un aumento de las concentraciones séricas de fenitoína en comparación con los pacientes que son metabolizadores normales (p. ej., *1/*1). Por lo tanto, los pacientes que se sabe que son metabolizadores intermedios o lentos pueden, en última instancia, requerir dosis más bajas de fenitoína para mantener concentraciones similares en estado estacionario en comparación con los metabolizadores normales. Si se desarrollan signos tempranos de toxicidad del sistema nervioso central (SNC) relacionada con la dosis, las concentraciones séricas deben controlarse de inmediato (consulte Propiedades farmacocinéticas).
Pacientes de edad avanzada: La depuración de la fenitoína disminuye ligeramente en pacientes de edad avanzada y puede ser necesaria una dosificación menor o menos frecuente (consulte Propiedades farmacocinéticas).
Pacientes pediátricos: La dosis inicial recomendada para pacientes pediátricos es 5 mg/kg/día vía oral en dos o tres dosis divididas por igual, y las dosis subsiguientes administradas según cada paciente hasta un máximo de 300 mg diarios en dosis divididas. Una dosis de mantenimiento diaria recomendada es, por lo general, 4 a 8 mg/kg/día en dosis divididas equitativamente. Los niños mayores de 6 años y los adolescentes pueden requerir la dosis mínima para adultos (300 mg/día).
Ajustes de la posología: La posología debe individualizarse para proporcionar el máximo beneficio. En algunos casos, puede ser necesario que se realicen determinaciones de los niveles sanguíneos séricos para realizar los ajustes óptimos de la posología. Los niveles mínimos proporcionan información sobre el rango del nivel sérico clínicamente efectivo y confirman el cumplimiento del paciente, y se obtienen justo antes de la próxima dosis programada del paciente. Los niveles máximos indican el umbral del individuo para la aparición de efectos secundarios relacionados con la dosis y se obtienen en el momento de la concentración máxima esperada. El efecto terapéutico sin signos clínicos de toxicidad ocurre más frecuentemente con concentraciones séricas totales de entre 10 y 20 mcg/mL (concentraciones de fenitoína no unida entre 1 y 2 mcg/mL), aunque algunos casos leves de epilepsia tónico-clónica (gran mal) se pueden controlar con niveles séricos de fenitoína más bajos. Para los pacientes con enfermedad renal o hepática, o aquellos con hipoalbuminemia, el monitoreo de las concentraciones de fenitoína no unida puede ser de mayor importancia.
Con la posología recomendada, se puede requerir un período de siete a diez días para alcanzar niveles sanguíneos en estado de equilibrio con fenitoína y los cambios en la posología (aumento o disminución) no se deben llevar a cabo en intervalos más cortos que siete a diez días.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis: No se conoce la dosis mortal en pacientes pediátricos. La dosis mortal en adultos se estima entre 2 y 5 gramos. Los síntomas iniciales son nistagmo, ataxia y disartria. Otros signos son temblor, hiperreflexia, letargo, habla dificultosa, visión borrosa, náuseas y vómitos. El paciente puede entrar en estado comatoso y sufrir hipotensión. Se han reportado casos de bradicardia y paro cardiaco (consulte Advertencias y precauciones especiales de uso). La causa de muerte se debe a la disminución de la respiración y circulación.
Hay variaciones marcadas entre los individuos con respecto a los niveles séricos de fenitoína en los que ocurre la toxicidad. El nistagmo, o mirada lateral, usualmente aparece con 20 mcg/mL, la ataxia con 30 mcg/mL, la disartria y el letargo aparecen cuando la concentración sérica es más de 40 mcg/mL; sin embargo, se han documentado concentraciones tan altas como 50 mcg/mL sin evidencia de toxicidad. Se han tomado cantidades de hasta 25 veces la dosis terapéutica, cuyo resultado fue concentraciones plasmáticas de más de 100 mcg/mL con una recuperación completa. Se han informado casos de disfunción y atrofia cerebelosa irreversible.
Tratamiento: El tratamiento es no específico dado que no hay antídoto conocido.
Se debe observar cuidadosamente la capacidad de los sistemas respiratorios y circulatorios y emplear medidas de apoyo apropiadas. Se puede considerar la hemodiálisis dado que la fenitoína no se liga completamente a las proteínas plasmáticas. Se ha utilizado la transfusión total de intercambio como tratamiento para la intoxicación grave en pacientes pediátricos.
En caso de sobredosificación aguda se deberán considerar otros supresores del SNC como el alcohol.
PRESENTACIÓN:
Naturaleza y contenido del envase:
Presentación comercial:
Frasco × 100 cápsulas de liberación prolongada + Inserto.
Fabricado por:
VIATRIS PHARMACEUTICALS LLC.
Vega Baja, Puerto Rico
Importado y distribuido por:
Aspenpharma S. A.
Quito, Ecuador
LLD_Ecu_USPI_LAB-0375-34.0_03/2022_v1
Versión: Ene 2024
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Precauciones especiales de almacenamiento:
Almacenar a temperatura no mayor a 30 °C.
Proteger de la luz y la humedad.
Todo medicamento debe conservarse fuera del alcance de los niños.
Precauciones especiales para el desecho y otra manipulación: Ninguna.


