
DYSPORT
TOXINA BOTULÍNICA TIPO A
Polvo para solución inyectable
1 Caja, 1 Vial(es) unidosis, 500 U
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Ingrediente activo: Complejo de toxina tipo A de Clostridium Botulinum – hemaglutinina 500 unidades Ipsen*
* Una unidad (U) se define como la dosis mediana letal intraperitoneal en el ratón.
FORMA FARMACÉUTICA: Polvo para solución inyectable.
Indicaciones Terapéuticas: Dysport está indicado para el tratamiento de:
• Espasticidad asociada con la deformidad del pie equino en pacientes adultos después de un infarto.
• Tratamiento sintomático de la espasticidad focal de extremidades superiores en adultos.
• Deformidad dinámica del pie equino, debida a la espasticidad en pacientes con parálisis cerebral, de dos años de edad o mayores.
• Tortícolis espasmódico en adultos.
• Blefaroespasmo en adultos.
• Espasmo hemifacial en adultos.
• Hiperhidrosis axilar en adultos.
• Hiperhidrosis palmar en adultos.
• Tratamiento de líneas glabelares moderadas a severas.
• Tratamiento de líneas cantales laterales moderadas a severas.
INFORMACIÓN FARMACÉUTICA
Lista de excipientes: solución de albúmina humana, lactosa.
Incompatibilidades: ninguna conocida.
Vida útil: 24 meses.
Recomendaciones especiales de almacenamiento: almacenar a una temperatura entre 2°C - 8°C (en un refrigerador). No congelar.
Una vez reconstituido el producto y teniendo en cuenta que el mismo no contiene un agente antimicrobiano, desde el punto de vista microbiológico, se recomienda que el producto sea usado inmediatamente luego de la reconstitución. No obstante, se ha demostrado que la solución es estable durante 24 horas a 2°- 8°C posterior a su reconstitución.
Naturaleza y contenido del recipiente
Naturaleza del recipiente/tapa: viales de vidrio tipo 1 con capacidad de 3 mL, tapas de 13 mm de bromobutilo liofilizadas, selladas mediante una laminación de aluminio de 13 mm con un orificio central, plegada en los bordes.
Contenido del recipiente: polvo blanco liofilizado para reconstitución.
Instrucciones de uso, manejo y desecho: al descubrir parte central del tapón de goma, debe limpiarse con alcohol inmediatamente antes de perforar la membrana. Debe utilizarse una aguja estéril de calibre 23 o 25.
Las instrucciones para la reconstitución son específicos para cada vial el de 300 unidades y el vial 500 unidades. Estos volúmenes producen concentraciones específicas de uso para cada indicación.
|
Dosis resultante unidades por ml |
Diluente* por vial de 500 unidades |
|
500 unidades |
1 mL |
|
200 unidades |
2.5 mL |
|
100 unidades |
5 mL |
|
*Solución de Cloruro de Sodio al 0.9%, libre de preservantes |
|
Inmediatamente después del tratamiento del paciente, cualquier toxina botulínica tipo A residual que puede estar presente en cualquiera de vial o jeringa debe ser inactivada con solución diluida de hipoclorito (1% de cloro disponible). El derrame de toxina botulínica tipo A se debe limpiar con un paño absorbente empapado en una solución de hipoclorito diluido. Cualquier material utilizado o los residuos deben ser eliminados de acuerdo con las exigencias locales. Si se rompe uno de los viales, proceda como se indicó anteriormente. Recoja con cuidado los fragmentos de vidrio roto y limpie el producto, evitando cortaduras.
Si el producto entra en contacto con la piel, lave con una solución de hipoclorito de sodio (blanqueador) y enjuague con abundante agua. Si el producto entra en contacto con los ojos, enjuague con abundante agua o con una solución para enjuague ocular delicada.
En caso de lesión de la persona que maneja el material (herida o autoinyección), proceda como se indicó anteriormente y tome las medidas médicas adecuadas dependiendo de la dosis inyectada.
Recomendaciones para el desecho de materiales contaminados: las agujas, jeringas y viales – que no deben vaciarse – deben colocarse en recipientes adecuados diseñados para incineración después de su uso.
Los materiales contaminados (tela absorbente, guantes, restos de ampolletas) deben colocarse en una bolsa resistente a perforaciones para ser desechados e incinerados.
Fecha de revisión del texto: agosto 2016.
LABORATORIOS BIOPAS S. A.
PROPIEDADES FARMACOLÓGICAS
Propiedades farmacodinámicas
Grupo fármaco terapéutico: otros relajantes musculares, agentes de acción periférica.
Código ATC: M03AX01.
El complejo de toxina tipo A de Clostridium Botulinum y hemaglutinina bloquea la transmisión colinérgica periférica en las uniones neuromusculares mediante una acción presináptica en un sitio próximo al de la liberación de acetilcolina. La toxina actúa en la terminación nerviosa contrarrestando los eventos provocados por el Ca2+, que culminan en la liberación del transmisor. Esto no afecta la transmisión colinérgica postganglionar o transmisión simpática postganglionar.
La acción de la toxina implica una primera etapa de unión, en la que la toxina se une rápida y ávidamente a la membrana del nervio presináptico. A continuación se da lugar a la internalización, paso en el cual la toxina cruza la membrana presináptica sin causar parálisis. Finalmente la toxina inhibe la liberación de acetilcolina interrumpiendo el mecanismo de liberación de acetilcolina mediado por el Ca2+ disminuyendo en consecuencia el potencial de placa terminal y causando parálisis.
La recuperación de la transmisión del impulso ocurre gradualmente a medida que las nuevas terminaciones nerviosas se regeneran y se establece contacto con la placa motora post-sináptica, proceso que dura de 6 a 8 semanas en animales de experimentación.
Espasticidad focal que afecta a las extremidades superiores: la eficacia y seguridad de la toxina botulínica tipo A para el tratamiento de la espasticidad de las extremidades superiores fue evaluada en un estudio aleatorizado, multicéntrico, doble ciego, controlado con placebo que incluyó a 238 pacientes (159 toxina botulínica tipo A y 79 con placebo) con espasticidad de las extremidades superiores quienes por lo menos tenían 6 meses después del accidente cerebrovascular (90%) o una lesión cerebral postraumático (10%). El grupo muscular objetivo primario (PTMG) fueron los flexores extrínsecos de los dedos (56%), seguido por el codo (28%) y flexores de la muñeca (16%). La variable de eficacia primaria fue el tono muscular PTMG en la semana 4, según lo medido por la Escala Ashworth modificada (MAS), una escala de 5 puntos que van desde 0 (sin aumento del tono muscular) a 4 (afectado en parte[s] rígido en flexión o la extensión) y el primer criterio de valoración secundario fue la evaluación global del médico (PGA) de la respuesta al tratamiento (una escala de 9 puntos que van desde -4 [marcadamente peor], a través 0 [sin cambios], de +4 [notablemente mejorada]). Los principales resultados obtenidos en la semana 4 y la semana 12 se muestran a continuación:
|
Semana 4 |
Semana 12 |
|||||
|
Placebo (N=79) |
Toxina Botulinica Tipo A (N=80) |
Toxina Botulinica Tipo A (1000 unidades) (N=79) |
Placebo (N=79) |
Toxina Botulinica Tipo A (500 unidades) (N=80) |
Toxina Botulinica Tipo A (1000 unidades) (N=79) |
|
|
Cambio promedio MC desde el inicio del tono muscular de PTMG en el MAS |
-0.3 |
-1.2** |
-1.4** |
-0.1 n=75 |
-0.7** n=76 |
-0.8** n=76 |
|
Cambio promedio MC PGA de respuesta al tratamiento |
0.7 |
1.4* |
1.8** |
0.4 n=75 |
0.5 n=76 |
1.0* n=76 |
|
Cambio promedio MC desde el inicio del tono muscular en flexores de la muñeca en el MAS |
-0.3 n=54 |
-1.4** n=57 |
-1.6** n=58 |
-0.3 n=52 |
-0.7* n=54 |
-0.9* n=56 |
|
Cambio promedio MC desde el inicio del tono muscular en flexores de los dedos en el MAS |
-0.3 n=70 |
-0.9* n=66 |
-1.2** n=73 |
-0.1 n=67 |
-0.4* n=62 |
-0.6* n=70 |
|
Cambio promedio MC desde el inicio del tono muscular en flexores del codo en el MAS |
-0.3 n=56 |
-1.0* n=61 |
-1.2** n=48 |
-0.3 n=53 |
-0.7* n=58 |
-0.8* n=46 |
|
Cambio promedio MC desde el inicio en el hombro extensores de tono muscular en el MAS (1) |
-0.4 n=12 |
-0.6 n=7 |
-0.7 n=6 |
0.0 n=12 |
-0.9 n=7 |
0.0 n=6 |
|
*p<0.05; ** p<0.0001; MC = Mínimos Cuadrados (1) No hay pruebas estadísticas realizadas debido a la baja frecuencia de los grupos de tratamiento y placebo, ya que hay datos limitados en pacientes tratados en los músculos de los hombros. |
||||||
El objetivo principal del tratamiento (PTT) de la Escala de Evaluación de Discapacidad [DAS] se utilizó para investigar el efecto del tratamiento sobre el deterioro funcional (función pasiva). A pesar de algunas mejoras en el cambio medio desde el inicio en la semana 4 en los grupos de toxina botulínica tipo A no alcanzó significancia estadística en comparación con el placebo, la proporción de puntuación DAS en pacientes que mantuvieron la respuesta (sujetos que lograron al menos una mejora de un grado) para el PTT fue significativamente mayor a la dosis 1000U como se muestra a continuación.
|
Grupo de tratamiento |
Semana 4 % en responder |
Semana12 % en responder |
|
Toxina Boltulínica Tipo |
50.0 n=80 |
41.3 n=76 |
|
A 500U |
p = 0.13 |
p = 0.11 |
|
Toxina Boltulínica Tipo A 1000U |
62.0 n=78 p = 0.0018 |
55.7 n=76 p = 0.0004 |
|
Placebo |
39.2 n=79 |
32.9 n=75 |
|
* Los dominios incluidos en DAS son la higiene, posición de las extremidades,vestido y dolor. |
||
Se observaron además, mejoras estadísticamente significativas en la espasticidad (grado y ángulo) establecidos según la escala Tardieu, en el rango de movimiento activo de los dedos, muñeca o el codo, y en la facilidad de la aplicación de una férula por el sujeto, sobre todo en el 1000U dosis. Sin embargo, no hubo efecto del tratamiento muestra en la función activa, según la evaluación de la puntuación de Frenchay Modificado, y en los cuestionarios de calidad de vida EQ5D o SF-36.
El tiempo medio de aparición de la respuesta fue de 2 a 3 días después del tratamiento, con el efecto máximo observado en el día treinta. En ambos estudios de fase III pivotales placebo controlados, las inyecciones de Toxina Botulinica Tipo A redujo significativamente la severidad de las líneas glabelares durante un máximo de 4 meses. El efecto fue aún significativo después de 5 meses en uno de los dos estudios pivotales.
Treinta días después de la inyección, la evaluación de los investigadores mostró que el 90% (273/305) de los pacientes habían respondido al tratamiento (no exhibieron o exhibieron leves líneas glabelares con el máximo fruncimiento del ceño), frente al 3% (4/153) de los pacientes tratados con placebo. Cinco meses después de la inyección, 17% (32/190) de los pacientes tratados con toxina botulínica tipo A siguen respondiendo al tratamiento en comparación con el 1% (1/92) de los pacientes tratados con placebo en el estudio en cuestión. La propia evaluación de los pacientes con el máximo fruncimiento del ceño después de treinta días dio una tasa de respuesta del 82%df (251/305) de los pacientes tratados con la toxina botulínica tipo A y 6% (9/153) de los pacientes tratados con placebo. La proporción de pacientes que muestran una mejora de dos grados de acuerdo con la evaluación del investigador al máximo el ceño fruncido, fue del 77% (79/103) en uno de los estudio fase III pivotal donde esto se evaluó.
Un subgrupo de 177 pacientes tenía líneas moderadas o graves en reposo antes del tratamiento. La evaluación por los investigadores de esta población, treinta días después del tratamiento, mostró que el 71% (125/177) de los pacientes tipo A de toxina botulínica A tratadas se consideraron que respondieron frente a 10% (8/78) de los pacientes tratados con placebo.
El estudio a largo plazo de dosis repetidas abierto mostró que la media del tiempo hasta la aparición de la respuesta de 3 días se mantuvo en todos los ciclos de dosis repetidas. La tasa de respuesta al mimo fruncimiento del ceño tal como se determina por el investigador en el día 30 se mantuvo durante ciclos repetidos (entre el 80% y el 91% en los 5 ciclos). La tasa de respuesta en reposo durante ciclos repetidos de dosis también fue consistente con los estudios de dosis única, con un 56% a un 74% de los pacientes tratados con toxina botulínica A, fueron consideradas por los investigadores qe respondieron, treinta días después del tratamiento.
Ninguno de los criterios de valoración clínica incluyó una evaluación objetiva del impacto psicológico.
Blefaroespasmo: tres dosis toxina botulínica tipo A se investigaron por 1 ciclo de tratamiento en un estudio clínico.
La eficacia fue medida por las medianas de las diferencias de los valores del Porcentaje de la Actividad Normal (PNA) (derivado de la Escala de Incapacidad de Blefaroespasmo) entre cada grupo de tratamiento y el placebo. Una mejoría en el blefaroespasmo, dependiente de la dosis, fue evidente con dosis crecientes de Dysport, siendo en todos los grupos de tratamiento superior al placebo.
|
Diferencia entre la mediana de los cambios en los valores del PNA de la línea base en el grupo activo y la mediana de los cambios en los valores del PNA de la línea base en el grupo de placebo. Visita |
Dysport 40 Unidades (N = 30) |
Dysport 80 Unidades (N = 31) |
Dysport 120 Unidades (N = 31) |
|
Semana 4: |
31,2% |
41,3% |
48,5% |
|
Semana 8: |
36,0% |
48,3% |
55,0% |
|
Semana 12: |
36,0% |
36,3% |
50,0% |
|
Semana 16: |
10,5% [a] |
24,2% |
31,3% |
|
[a] valor p> 0,001 |
|||
Para los grupos de tratamiento de 40, 80 y 120 unidades Dysport, las medianas de los cambios en los valores del PNA en la línea base fueron estadística y significativamente mayores en comparación con el grupo placebo a las semanas 4, 8, y 12.
Una diferencia estadísticamente significativa en comparación con el grupo placebo también se observó para los grupos de tratamiento 80 unidades y 120 unidades Dysport en la semana 16, indicando una mayor duración de la respuesta en las dosis de 80 unidades y 120 unidades.
La incidencia de los eventos adversos emergentes del tratamiento (acontecimientos adversos TEAEs), específicamente ptosis, fue mayor en los grupos de tratamiento con Dysport que en el grupo de tratamiento con placebo y fue dependiente de la dosis con una mayor incidencia observada a dosis más altas de Dysport. Observar la siguiente tabla.
|
Estadística |
Placebo (N = 26) |
Dysport 40 Unidades (N = 31) |
Dysport 80 Unidades (N = 31) |
Dysport 120 Unidades (N = 31) |
|
|
Pacientes con TEAEs relacionados |
n (%) |
3 (12) |
19 (61) |
23 (74) |
26 (84) |
|
Pacientes con TEAEs oculares relacionados |
n (%) |
3 (12) |
16 (52) |
23 (74) |
26 (84) |
Líneas glabelares moderadas a severas: durante el desarrollo clínico de la toxina botulínica tipo A, más de 2600 pacientes fueron incluidos en los diferentes ensayos clínicos. En estudios clínicos, 1907 pacientes con líneas glabelares moderadas a severas han sido tratados con la dosis recomendada de 50 U de toxina botulínica tipo A.
De estos, 305 fueron tratados con 50 U en dos estudios pivotales fase III placebo controlados, doble ciego y 1200 tratados con 50U a largo plazo en un estudio fase III, abierto de dosis repetidas. El resto de pacientes fueron tratados en estudios de apoyo y de rango de dosis.
El tiempo medio de aparición de la respuesta fue de 2 a 3 días después del tratamiento, con el efecto máximo observado en el día treinta. En ambos estudios de fase III pivotales placebo controlados, las inyecciones de Toxina Botulinica Tipo A redujo significativamente la severidad de las líneas glabelares durante un máximo de 4 meses. El efecto fue aún significativo después de 5 meses en uno de los dos estudios pivotales. Treinta días después de la inyección, la evaluación de los investigadores mostró que el 90% (273/305) de los pacientes habían respondido al tratamiento (no exhibieron o exhibieron leves líneas glabelares con el máximo fruncimiento del ceño), frente al 3% (4/153) de los pacientes tratados con placebo. Cinco meses después de la inyección, 17% (32/190) de los pacientes tratados con toxina botulínica tipo A siguen respondiendo al tratamiento en comparación con el 1% (1/92) de los pacientes tratados con placebo en el estudio en cuestión. La propia evaluación de los pacientes con el máximo fruncimiento del ceño después de treinta días dio una tasa de respuesta del 82%df (251/305) de los pacientes tratados con la toxina botulínica tipo A y 6% (9/153) de los pacientes tratados con placebo. La proporción de pacientes que muestran una mejora de dos grados de acuerdo con la evaluación del investigador al máximo el ceño fruncido, fue del 77% (79/103) en uno de los estudio fase III pivotal donde esto se evaluó.
Un subgrupo de 177 pacientes tenía líneas moderadas o graves en reposo antes del tratamiento. La evaluación por los investigadores de esta población, treinta días después del tratamiento, mostró que el 71% (125/177) de los pacientes tipo A de toxina botulínica A tratadas se consideraron que respondieron frente a 10% (8/78) de los pacientes tratados con placebo. El estudio a largo plazo de dosis repetidas abierto mostró que la media del tiempo hasta la aparición de la respuesta de 3 días se mantuvo en todos los ciclos de dosis repetidas. La tasa de respuesta al máximo fruncimiento del ceño tal como se determina por el investigador en el día 30 se mantuvo durante ciclos repetidos (entre el 80% y el 91% en los 5 ciclos). La tasa de respuesta en reposo durante ciclos repetidos de dosis también fue consistente con los estudios de dosis única, con un 56% a un 74% de los pacientes tratados con toxina botulínica A, fueron consideradas por los investigadores que respondieron, treinta días después del tratamiento.
Ninguno de los criterios de valoración clínica incluyó una evaluación objetiva del impacto psicológico.
Propiedades farmacocinéticas: los estudios farmacocinéticos con la toxina botulínica en los animales tuvieron la dificultad de la elevada potencia, las pequeñas dosis que se emplean, el alto peso molecular del producto y la dificultad de marcar la toxina para obtener una actividad específica suficientemente alta. Los estudios realizados con toxina marcada con lodo 125 han demostrado que la unión al receptor es específica y saturable, y que la alta densidad de los receptores de la toxina es un factor que contribuye a la elevada potencia. Las respuestas a la dosis y a los tiempos en monos mostraron que a bajas dosis hay una demora de 2 a 3 días con efectos máximos que se aprecian a los 5 a 6 días después de la inyección. La duración de la acción, medida mediante los cambios en la alineación ocular, y la parálisis muscular variaron entre 2 semanas y 8 meses. Este patrón también se ha observado en humanos, y se atribuye al proceso de unión, internalización y los cambios en la unión neuromuscular.
Datos de seguridad preclínicos: estudios de toxicidad reproductiva en ratas y conejas preñadas, suministrando diariamente el complejo de toxina tipo A de Clostridium botulinum -hemaglutinina vía intramuscular, a dosis de 6,6 unidades/kg (79 unidades/kg totales de dosis acumulada) y 3,0 unidades/kg (42 unidades/kg totales de dosis acumulada) en ratas y conejos respectivamente, no dio lugar a toxicidad embrio/fetal. Las pérdidas de implantación a dosis tóxicas para la madre se observaron a dosis más altas en ambas especies. El complejo de toxina tipo A de Clostridium botulinum - hemaglutinina no demostró actividad teratogénica ni en ratas ni en conejos y no se observaron efectos en el estudio pre y postnatal en la generación F1 en ratas. La fertilidad de ratas machos y hembras disminuyó debido al apareamiento reducido, secundario a una parálisis muscular a dosis de 29,4 unidades/kg semanales en los machos, y se incrementó la pérdida de implantación a 20 unidades/kg semanales en las hembras.
En un estudio de toxicidad crónica llevado a cabo en ratas, de hasta 12 unidades/animal, no hubo ninguna indicación de toxicidad sistémica. Los efectos de toxicidad crónica en los estudios no clínicos se limitan a los cambios en los músculos inyectados relacionados con el mecanismo de acción del complejo de toxina tipo A de Clostridium botulinum - hemaglutinina. No hubo irritación ocular tras la administración del complejo de toxina tipo A de Clostridium botulinum -hemaglutinina en los ojos de conejos.
Contraindicaciones: Dysport está contraindicado en individuos con hipersensibilidad conocida a cualquiera de los componentes de Dysport.
Embarazo y Lactancia
Embarazo: existen datos limitados sobre el uso del complejo de toxina tipo A de Clostridium botulinum -hemaglutinina en mujeres embarazadas. Estudios en animales han mostrado toxicidad reproductiva a dosis que causan toxicidad materna. Dysport puede ser usado durante el embarazo sólo si el beneficio justifica cualquier riesgo potencial para el feto. Se debe tener precaución cuando se prescriba a mujeres embarazadas.
Lactancia: no se sabe si el complejo de toxina tipo A de Clostridium botulinum -hemaglutinina se excreta en la leche humana. La excreción del complejo no ha sido estudiada en animales, por lo que su uso no es recomendado durante la lactancia.
Efectos en la capacidad para conducir y operar máquinas: Existe un riesgo potencial de debilidad muscular o alteraciones visuales los cuales, si se experimentan, pueden afectar temporalmente la capacidad para conducir u operar maquinaria.
Efectos no deseados: La frecuencia de las reacciones adversas reportadas en los ensayos controlados con placebo después de una sola administración se define como sigue: muy frecuentes > 1/10, frecuentes > 1/100, poco frecuentes > 1/1000, <1/100; raras >1/10000, <1/1000. Se han reportado los efectos secundarios relacionados con la distribución de la toxina lejos del sitio de administración (debilidad muscular exagerada, disfagia, aspiración/neumonía por aspiración, con desenlace fatal en algunos casos muy raros). También se han reportado casos de hipersensibilidad después de su comercialización.
General: las siguientes reacciones adversas se observaron en pacientes tratados para una variedad de indicaciones que incluyen blefaroespasmo, espasmo hemifacial, tortícolis y espasticidad asociada con parálisis cerebral o accidente cerebrovascular:
Trastornos del sistema nervioso
Raros: amiotrofia neurálgica
Trastornos de la piel y tejido subcutáneo
Poco frecuentes: picazón
Raros: sarpullido en la piel
Trastornos generales y alteraciones en el lugar de administración
Frecuentes: debilidad general, fatiga, síndrome gripal, dolor/enrojecimiento en el sitio de inyección.
Frecuencia de las reacciones adversas específicas por indicación.
Adicionalmente, se reportaron las siguientes reacciones adversas específicas a las indicaciones individuales:
Espasticidad en la pierna posterior a un infarto en adultos
Trastornos gastrointestinales
Frecuentes: disfagia
Trastornos musculoesqueléticos y del tejido conectivo
Frecuentes: debilidad de los músculos de las piernas
Trastornos renales y urinarios
Frecuentes: incontinencia urinaria
Trastornos generales y alteraciones en el lugar de administración
Frecuentes: modo de andar anormal
Lesiones, intoxicaciones y complicaciones de procedimiento
Frecuentes: lesión accidental / caídas
Espasticidad focal que afecta a las extremidades superiores
|
Clasificación órganos del sistema |
Frecuencia |
Reacción adversa al medicamento |
|
Trastornos gastrointestinales |
Raro |
Disfagia |
|
Trastornos musculoesqueléticos y del tejido conectivo |
Frecuente |
Debilidad muscular |
|
Trastornos generales y alteraciones en el lugar |
Raro |
Astenia de administración |
Deformidad de pie equino debido a espasticidad focal
Trastornos gastrointestinales
Frecuentes: diarrea
Trastornos musculoesqueléticos y del tejido conectivo
Frecuentes: debilidad muscular de las piernas, dolor muscular
Trastornos renales y urinarios
Frecuentes: incontinencia urinaria
Trastornos generales y alteraciones en el lugar de administración
Frecuentes: modo de andar anormal
Lesiones traumáticas, intoxicaciones y complicaciones de procedimientos terapéuticos
Frecuentes: lesión accidental debido a caídas.
Las lesiones accidentales debido a caídas y a la marcha anormal pueden deberse a la excesiva debilidad del músculo tratado y/o la prolongación local de Dysport a otros músculos que intervienen en la deambulación y el equilibrio.
Tortícolis espasmódica
Trastornos del sistema nervioso
Frecuentes: dolor de cabeza, mareo, paresia facial
Trastornos oculares
Frecuentes: visión borrosa, agudeza visual reducida
Poco frecuentes: diplopía, ptosis
Trastornos respiratorios, torácicos y del mediastino
Frecuentes: disfonía, disnea
Raro: aspiración
Trastornos gastrointestinales
Muy frecuentes: disfagia, boca seca
Poco frecuentes: náusea
Trastornos musculoesqueléticos y del tejido conectivo
Muy frecuentes: debilidad muscular
Frecuentes: dolor de cuello, dolor musculoesquelético, mialgia, dolor en las extremidades, rigidez musculoesquelética
Poco frecuentes: atrofia muscular, trastornos en la mandíbula
La disfagia aparentemente está relacionada con la dosis y ocurre con mayor frecuencia luego de la inyección en el músculo esternocleidomastoideo. Se requerirá dieta blanda hasta que los síntomas desaparezcan.
Se espera que estos efectos secundarios desaparezcan en dos a cuatro semanas.
Blefaroespasmo y espasmo hemifacial
Trastornos del sistema nervioso
Frecuentes: paresis facial
Poco frecuentes: parálisis del nervio VII
Trastornos oculares
Muy frecuentes: ptosis
Frecuentes: diplopía, ojos secos, lagrimeo
Raras: oftalmoplejía
Trastornos de la piel y tejido subcutáneo
Frecuentes: edema de párpados
Raros: entropión
Los efectos secundarios pueden ocurrir debido a inyecciones profundas o mal aplicadas de Dysport paralizando temporalmente otros grupos de músculos cercanos.
Hiperhidrosis axilar: se reportaron las siguientes reacciones adversas en 4 estudios clínicos con 217 pacientes aproximadamente:
Trastornos de la piel y del tejido subcutáneo
Frecuente: sudoración compensatoria
Hiperhidrosis palmar: puede presentarse debilidad de los músculos adyacentes y dolor en el sitio de inyección.
Tratamiento de líneas glabelares moderadas a severas
Trastornos del sistema nervioso
Muy frecuente: dolor de cabeza
Frecuente: paresis facial (principalmente describe parálisis de frente)
Poco frecuente: mareo
Trastornos oculares
Frecuente: astenopía, ptosis, edema parpebral, aumento de lagrimeo, ojos secos, contracciones musculares (espasmos en los músculos alrededor de los ojos)
Poco frecuentes: trastornos visuales, visión borrosa, diplopía. Trastornos del movimiento de los ojos
Trastornos de la piel y tejido subcutáneo
Poco frecuentes: prurito, rash
Raras: urticaria
Trastornos generales y condiciones del sitio de administración
Muy frecuentes: reacciones en el sitio de inyección (ej. Eritema, edema, irritación, rash, prurito, parestesia, dolor, molestias, escozor y contusión)
Trastornos del sistema inmunológico
Poco frecuentes: hipersensibilidad
Tratamiento de líneas cantales laterales moderadas a severas: las siguientes reacciones adversas, usualmente de intensidad leve a moderada, fueron observadas en pacientes tratados con Dysport para la corrección de líneas cantales laterales moderadas a severas.
Trastornos oculares
Frecuente: edema ocular, ojo seco (queratoconjuntivitis sicca)
Trastornos generales y condiciones del sitio de administración
Frecuente: reacciones en el sitio de inyección (incluyendo dolor, enrojecimiento/hematoma, prurito)
Trastornos musculoesqueléticos y del tejido conectivo
Frecuente: debilidad del músculo adyacente al área de inyección. Esto puede ocasionar comúnmente ptosis en el párpado o paresis de los músculos faciales.
Trastornos del sistema nervioso
Frecuente: dolor de cabeza
Trastornos de la piel del tejido conjuntivo
Poco frecuente: prurito
Experiencia posterior a la comercialización: informar sospechas de reacciones adversas después de la autorización del medicamento es importante. Permite la supervisión continua del balance beneficio/riesgo del medicamento.
Interacciones medicamentosas y de otro género: Los efectos de la toxina botulínica pueden aumentar por fármacos que interfieren directa o indirectamente con la función neuromuscular (e.j. aminoglucósidos, bloqueadores tipo curare no despolarizantes) por lo que deben ser usados con precaución en pacientes tratados con la toxina botulínica.
Advertencias y precauciones especiales para su uso: Se han reportado efectos adversos como resultado de la distribución de la toxina en sitios alejados del sitio de administración, los cuales, en algunos casos están asociados con disfagia, neumonía y/o debilidad importante, muy rara vez, con la muerte. Los pacientes tratados con dosis terapéuticas pueden presentar debilidad muscular excesiva. El riesgo de ocurrencia de dichas reacciones adversas se puede reducir utilizando la dosis mínima efectiva y no excediendo la dosis recomendada.
Dysport solamente debe ser usado con precaución y bajo estricta supervisión médica cercana en pacientes con evidencia clínica o sub-clínica de una marcada transmisión neuromuscular deficiente (por ejemplo, miastenia gravis). Estos pacientes pueden presentar un aumento en la sensibilidad a agentes como Dysport, lo cual puede provocar una debilidad muscular excesiva con las dosis terapéuticas. Los pacientes con trastornos neurológicos subyacentes están en mayor riesgo de este efecto secundario. Raramente se han reportado casos de muerte luego del tratamiento con toxina botulínica tipo A o B; en ocasiones relacionados con disfagia, neumopatía (incluyendo pero no limitado a disnea, fallo respiratorio, paro respiratorio) y/o en pacientes con astenia importante. Pacientes con trastornos que causan defectos en la transmisión neuromuscular, dificultad para deglutir o respirar tienen un mayor riesgo de experimentar estos efectos. En estos pacientes, el tratamiento debe ser administrado bajo el control de un especialista y sólo si el beneficio del tratamiento supera el riesgo.
Dysport debe administrarse con precaución a pacientes con problemas pre-existentes para deglutir o respirar, puesto que pueden empeorar después de la distribución del efecto de la toxina en los músculos relevantes. Se ha presentado aspiración en raras ocasiones y representa un riesgo durante el tratamiento de pacientes con afección respiratoria crónica.
No se debe exceder la posología y frecuencia recomendada para la administración de Dysport.
Los pacientes y sus familiares deben ser advertidos sobre la necesidad de tratamiento médico inmediato en caso de dificultad para deglutir, hablar o respirar.
Para el tratamiento de la espasticidad asociada con parálisis cerebral en niños, Dysport sólo se debe utilizar en niños de 2 años de edad o mayores.
Dysport no debe utilizarse para tratar la espasticidad en pacientes que han desarrollado una contractura fija.
Al igual que con cualquier inyección intramuscular, Dysport sólo debe utilizarse cuando sea estrictamente necesario en pacientes con tiempos de sangrado prolongado, infección o inflamación en el sitio de la inyección.
Dysport sólo debe usarse para tratar un único paciente, durante una única sesión. Las precauciones específicas deben ser tenidas en cuenta durante la preparación y administración del producto y para la inactivación y eliminación de cualquier resto de solución reconstituida.
Se ha observado raramente la formación de anticuerpos a la toxina botulínica en pacientes que reciben Dysport. Clínicamente, los anticuerpos neutralizantes han sido detectados mediante deterioro sustancial en la respuesta a la terapia y/o la necesidad de uso constante de dosis mayores.
Cuando se tratan líneas glabelares, es esencial estudiar la anatomía facial del paciente antes de la administración. La asimetría facial, ptosis, dermatocalsia excesiva, cicatrices y cualquier alteración a esta anatomía, como resultado de intervenciones quirúrgicas previas, deben ser tenidas en consideración. Se debe tener precaución cuando el músculo objetivo muestra excesiva debilidad o atrofia.
Se debe tener especial consideración antes de la inyección de pacientes quienes hayan experimentado una reacción alérgica previa a productos que contienen toxina botulínica tipo A. El mayor riesgo de una reacción alérgica debe considerarse en relación al beneficio del tratamiento.
El efecto de administrar diferentes neurotoxinas botulinum durante el curso del tratamiento con Dysport es desconocido y debe ser evitado.
Posología y Método de Administración
Las unidades de Dysport son específicas para la preparación y no son intercambiables con otras preparaciones de toxina botulínica.
Capacitación: Dysport solamente debe ser administrado por médicos capacitados adecuadamente. Para instrucciones sobre la reconstitución del polvo para solución inyectable, manipulación y disposición de viales por favor refiérase a la sección información farmacéutica. Debe utilizarse una aguja estéril de calibre 23 o 25, o una aguja de calibre 29-30 para líneas glabelares y calibre 30 para líneas cantales laterales.
Espasticidad en la pierna posterior a un infarto en adultos
Posología: la dosis máxima administrada no debe exceder 1500 unidades, distribuidas entre los músculos gastrocnemio y sóleo, aunque deben considerarse también inyecciones en el músculo tibial posterior. El uso de electromiografía (EMG) no es una práctica clínica de rutina pero puede ayudar a identificar los músculos más activos. La dosis inicial debe reducirse si existen evidencias que sugieran que esta dosis puede provocar debilidad excesiva de los músculos objetivo, en el caso de pacientes cuyos músculos objetivo sean pequeños o pacientes que requieran inyecciones concomitantes en otros grupos de músculos . La mejoría clínica puede esperarse dentro de las primeras dos semanas posteriores a la inyección. Las inyecciones pueden repetirse aproximadamente cada 16 semanas o según se requiera para mantener la respuesta, pero con una frecuencia no menor a cada 12 semanas.
Niños: no se ha demostrado la seguridad ni la eficacia de Dysport en el tratamiento de espasticidad de la pierna después de un infarto, en niños.
Forma de administración: Dysport se reconstituye con 1.0 mL de cloruro de sodio inyectable B.P. (0.9%) para obtener una solución conteniendo 500 unidades por mL de Dysport.
Dysport se administra mediante una inyección intramuscular en los músculos recomendados descritos a detalle anteriormente cuando se trata la espasticidad en la pierna.
Espasticidad focal que afecta las extremidades superiores en adultos: la dosificación en sesiones de tratamiento iniciales y secuenciales se debe adaptar para cada individuo, con base en el tamaño, el número y el sitio de los músculos involucrados, la gravedad de la espasticidad, la presencia de debilidad muscular local, la respuesta del paciente a tratamientos anteriores, y / o la historia de eventos adversos. En los ensayos clínicos, las dosis de 500 unidades (U) y 1000 unidades se dividieron entre los músculos seleccionados como se muestra a continuación. Generalmente, no más que 1 mL debe ser administrado en el sitio como inyección única
|
Músculos inyectados |
Dosis recomendada de DYSPORT (U) |
|
Flexor carpi radialis (FCR) Flexor carpi ulnaris (FCU) |
100-200 U 100 - 200 U |
|
Flexor digitorum profundus (FDP) Flexor digitorum superficialis (FDS) Flexor Pollicis Longus Adductor Pollicis |
100 -200 U 100 -.200 U 100 -200 U 25-50 U |
|
Brachialis Brachioradialis Biceps Brachii (BB) Pronator Teres |
200 - 400 U 100 - 200 U 200 - 400 U 100 - 200 U |
|
Triceps Brachii (cabeza larga) Pectoralis Major Subscapularis Latissimus Dorsi |
150 - 300 U 150 – 300 U 150 – 300 U 150 - 300 U |
Aunque la localización real de los sitios de inyección se puede determinar por palpación, se recomienda el uso de una técnica guía para la inyección, por ejemplo, la electromiografía, la estimulación eléctrica o el ultrasonido.
La mejoría clínica se puede esperar una semana después de la inyección y puede durar hasta 20 semanas. Las inyecciones pueden repetirse cada 12-16 semanas o según sea necesario para mantener la respuesta, pero no más frecuentemente que cada 12 semanas. El grado y el patrón de la espasticidad muscular en el momento de re-inyección pueden requerir modificación en la dosis de toxina botulínica tipo A y los músculos a inyectar.
Niños: no se han demostrado ni la seguridad ni la efectividad del producto, en el tratamiento de la espasticidad focal que afecta las extremidades superiores en niños.
Pacientes mayores (≥ 65 años): la experiencia clínica no ha identificado diferencias en la respuesta entre los pacientes mayores y los adultos más jóvenes, En general, los pacientes mayores se deben observar para evaluar su tolerabilidad a Dysport, debido a la mayor frecuencia de enfermedades concomitantes y de otras terapias con medicamentos.
Forma de administración: cuando se trata la espasticidad focal sintomática de las extremidades superiores en adultos, Dysport se reconstituye con inyección de cloruro de sodio B.P, (0.9 % p/v), para lograr una solución que contenga 100 unidades / ml, o 200 unidades / ml, o 500 unidades / ml de Toxina Botulínica Tipo A la Toxina Botulínica Tipo A debe ser administrada por inyección intramuscular en los músculos como se describe anteriormente.
Deformidad del pie equino debido a espasticidad focal
Posología: la dosis inicial recomendada es de 20 unidades/kg de peso corporal administrada en una dosis dividida en ambos músculos de las dos pantorrillas. Si solamente está afectada una de las pantorrillas, debe utilizarse una dosis de 10 unidades/kg de peso corporal.
Las dosis iniciales deberán disminuirse si la evidencia sugiere que esta dosis puede provocar debilidad excesiva de los músculos objetivo, como es el caso de pacientes cuyos músculos objetivo son pequeños o pacientes que requieren inyecciones concomitantes en otros grupos musculares.
Tras evaluar la respuesta a la dosis inicial, el tratamiento subsiguiente puede valorarse en la escala de 10 unidades/kg y 30 unidades/kg dividido entre ambas piernas. La dosis máxima administrada no debe exceder de 30 unidades/kg o 1000 unidades, si este valor es menor. La administración debe dirigirse principalmente al músculo gastrocnemio, aunque también deben considerarse las inyecciones en los músculos sóleo y tibial posterior.
El uso de electromiografía (EMG) no es una práctica clínica habitual, pero puede facilitar la identificación de los músculos más activos.
La mejoría clínica puede esperarse en el transcurso de dos semanas posteriores a la inyección. Las inyecciones pueden repetirse aproximadamente cada 16 semanas o con la frecuencia necesaria para mantener la respuesta, pero no con una frecuencia menor a 12 semanas.
Forma de administración: cuando se trata la espasticidad asociada con parálisis cerebral pediátrica, reconstituir un vial de Dysport 500 U con de cloruro de sodio inyectable B.P. (0.9% p/v) para obtener una solución conteniendo 500 unidades por mL de Dysport.
Dysport se administra mediante inyección intramuscular en los músculos de la pantorrilla durante el tratamiento de espasticidad.
Tortícolis espasmódica
Posología: las dosis recomendadas para la tortícolis se aplican a los adultos de todas las edades, siempre y cuando se trate de personas de peso normal, sin evidencia de reducción de la masa muscular del cuello. Una dosis reducida puede ser apropiada en pacientes notablemente bajos de peso o en pacientes mayores cuya masa muscular puede estar reducida.
La dosis inicial recomendada para el tratamiento de tortícolis espasmódica es de 500 unidades por paciente, administrada como una dosis dividida en los dos o tres músculos más activos del cuello.
• En el caso de tortícolis rotativa, distribuir las 500 unidades administrando 350 unidades en el músculo esplenio capitis, ipsilateral a la dirección de la rotación mentón/cabeza, y 150 unidades en el músculo esternocleidomastoideo, contralateral a la rotación.
• Para laterocolis, distribuir las 500 unidades por administración de 350 unidades en el músculo esplenio capitis ipsilateral y 150 unidades en el músculo esternocleidomastoideo ipsilateral. En los casos asociados con elevación del hombro, el músculo trapecio ipsilateral o el elevador de la escápula, pueden también requerir tratamiento de acuerdo a la hipertrofia visible del músculo o a la lectura electromiográfica (EMG). Cuando sea necesario inyectar tres músculos, distribuir las 500 unidades de la siguiente manera: 300 unidades en el esplenio capitis, 100 unidades en el esternocleidomastoideo y 100 unidades en el tercer músculo.
• Para retrocolis, distribuir las 500 unidades administrando 250 unidades en cada uno de los músculos esplenio capitis. Las inyecciones bilaterales en los esplenios pueden incrementar el riesgo de debilidad muscular en el cuello.
• Todas las otras formas de tortícolis dependen en gran medida del conocimiento del especialista y de la EMG para identificar y tratar los músculos más activos. La EMG debe emplearse para el diagnóstico de todas las formas complejas de tortícolis, para una reevaluación después de inyecciones infructuosas en casos no complejos, y para guiar inyecciones en músculos profundos o en el caso de pacientes con sobrepeso cuyos músculos del cuello son difícilmente palpables.
En administraciones posteriores las dosis se pueden ajustar de acuerdo a la respuesta clínica y a los efectos secundarios observados. Se recomiendan intervalos de dosis de entre 250 y 1000 unidades; sin embargo, las dosis más altas pueden estar acompañadas por un incremento en los efectos secundarios, particularmente disfagia. La máxima dosis administrada no debe exceder de 1000 unidades. El alivio de los síntomas de tortícolis debe esperarse dentro de la primera semana después de la inyección. Las inyecciones pueden repetirse aproximadamente cada 16 semanas o según se requiera para mantener la respuesta, pero no con una frecuencia menor a 12 semanas.
Niños: no se ha demostrado la seguridad ni la eficacia de Dysport en el tratamiento de tortícolis espasmódica en niños.
Forma de administración: en el tratamiento de tortícolis espasmódica, reconstituir Dysport 500 U con cloruro de sodio inyectable B.P. (0.9%) para obtener una solución conteniendo 500 unidades por mL de Dysport.
Dysport se administra mediante inyección intramuscular para el tratamiento de tortícolis espasmódica, como se indicó anteriormente.
Blefaroespasmo y espasmo hemifacial:
Posología: en un ensayo clínico sobre el uso de Dysport para el tratamiento del blefaroespasmo esencial benigno, una dosis de 40 unidades por ojo fue significativamente eficaz. Dosis de 80 unidades y 120 unidades por ojo resultaron en una mayor duración del efecto. Sin embargo, la incidencia de eventos adversos locales, especificamente ptosis, fue relacionada con la dosis. En el tratamiento del blefaroespasmo y el espasmo hemifacial, la dosis máxima utilizada no debe exceder una dosis total de 120 unidades por ojo.
Se debe hacer una inyección de 10 unidades (0,05 ml) medialmente y 10 unidades (0,05 ml) lateralmente en la unión entre las partes preseptal y orbital de los músculos orbicular superior (3 y 4) y orbicular inferior (5 y 6) de cada ojo. Con el fin de reducir el riesgo de ptosis, se deben evitar las inyecciones cerca del elevador del párpado superior.
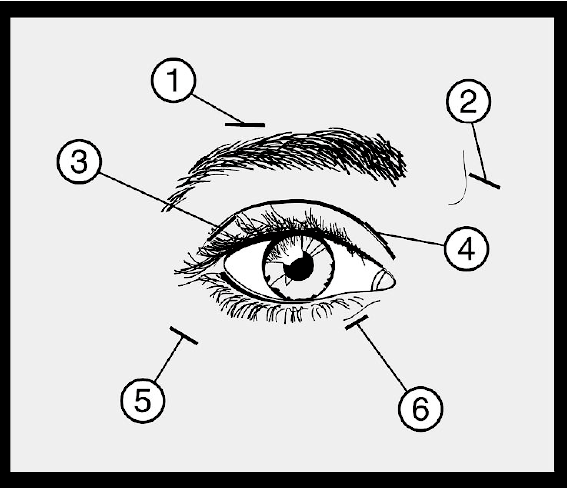
Para las inyecciones en el párpado superior, la aguja debe dirigirse lejos de su centro para evitar el músculo elevador. Se adjunta un esquema para facilitar la ejecución de dichas inyecciones. El alivio de síntomas puede esperarse al cabo de dos a cuatro días con un efecto máximo al cabo de dos semanas. Las inyecciones deben repetirse aproximadamente cada doce semanas o según se requiera para prevenir la recurrencia de los síntomas, pero no con una frecuencia menor a 12 semanas.
En las administraciones posteriores, si la respuesta del tratamiento inicial se considera insuficiente, puede ser necesario aumentar la dosis por ojo a 60 unidades: 10 unidades (0.05 mL) medialmente y 20 unidades (0.1 mL) lateralmente, 80 unidades: 20 unidades (0.1 mL) medialmente y 20 unidades (0.1 mL) lateralmente o hasta 120 unidades: 20 unidades (0.1 mL) medialmente y 40 unidades (0.2 mL) lateralmente por encima y por debajo de cada ojo en la forma descrita anteriormente. Sitios adicionales en el músculo frontal, por encima de las cejas (1 y 2) también se pueden inyectar si los espasmos interfieren con la visión.
En los casos de blefaroespasmo unilateral, las inyecciones deben limitarse al ojo afectado. Los pacientes con espasmo hemifacial deben ser tratados de la misma manera que para blefaroespasmo unilateral. Las dosis recomendadas son aplicables a adultos de todas las edades, incluyendo ancianos.
Niños: no se ha demostrado la seguridad ni la eficacia de Dysport en el tratamiento de blefaroespasmo y espasmo hemifacial en niños.
Forma de administración: durante el tratamiento de blefaroespasmo y espasmo hemifacial, reconstituir el vial de Dysport 500 U con cloruro de sodio inyectable BP (0.9% p/v) para obtener una solución conteniendo 200 unidades por mL de Dysport.
Dysport se administra mediante una inyección subcutánea medial y lateralmente en la unión de las partes preseptal y orbital de los músculos orbicular superior e inferior de cada ojo.
Hiperhidrosis axilar: la dosis máxima administrada no debe exceder 200 unidades por axila.
Posología: la dosis inicial recomendada es de 100 unidades por axila. Si no se alcanza el efecto deseado, es posible administrar hasta 200 unidades por axila para inyecciones subsiguientes. Debe determinarse previamente el área a inyectar utilizando la prueba de Yodo-almidón. Ambas axilas deben ser lavadas y desinfectadas. A continuación se administran inyecciones intradérmicas en diez sitios de 10 unidades cada una, 100 unidades por axila en total. El efecto máximo puede observarse dos semanas después de la inyección. En la mayoría de los casos, la dosis recomendada proporcionará la supresión adecuada de la secreción de sudor durante aproximadamente 48 semanas. El tiempo adecuado para una próxima aplicación se determina de manera individual, cuando la secreción de sudor del paciente ha vuelto a su nivel normal, pero con una frecuencia no menor a 12 semanas. Existe evidencia de un efecto acumulativo de dosis repetidas, de manera que el tiempo de cada tratamiento para un paciente dado debe determinarse de forma individual.
Niños: no se ha demostrado la seguridad ni la eficacia de Dysport en el tratamiento de hiperhidrosis axilar en niños.
Forma de administración: Dysport se reconstituye con 2.5 mL de solución de cloruro de sodio inyectable (0.9%) para obtener una solución conteniendo 200 unidades por mL de Dysport.
Dysport se administra mediante una inyección intradérmica en diez sitios durante el tratamiento de hiperhidrosis axilar.
Hiperhidrosis palmar
Posología
Adultos y ancianos: para hiperhidrosis palmar, la dosis total utilizada es de 120 unidades por palma, distribuida en 6 a 25 puntos de inyección subcutánea distintos, 10 unidades por punto.
Forma de administración: durante el tratamiento de hiperhidrosis palmar, el producto debe administrarse por medio de una inyección subdérmica, usualmente con una aguja de calibre 26, en las áreas hiperidróticas previamente determinadas. Algunos estudios no utilizan anestésicos, otros utilizan la congelación local de la palma o bloqueos de los nervios medial y ulnar para minimizar el dolor.
Líneas glabelares moderadas a severas
Posología y forma de administración: retirar cualquier maquillaje y desinfectar la piel con un antiséptico local.
La dosis recomendada es 50 unidades (0.25 mL de solución reconstituida) de Dysport a ser dividida en 5 sitios de inyección, 10 unidades (0.05 mL de solución reconstituida) se han de administrar intramuscularmente en cada uno de los 5 sitios: 2 inyecciones en cada músculo corrugador y una en el músculo procerus cerca al ángulo nasofrontal como se muestra a continuación:
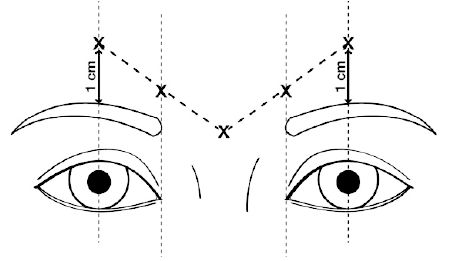
Los puntos de referencia anatómicos pueden ser más fácilmente identificados si se observa y palpa el ceño fruncido al máximo. Antes inyectar, colocar firmemente el dedo pulgar o el índice debajo del borde orbital con el fin de evitar extravasación en este lugar. Las inyecciones intramusculares deben ser llevadas a cabo en ángulo recto a la piel usando una aguja estéril de calibre 29 – 30. La aguja debe estar apuntando hacia arriba y hacia adentro durante la inyección. Con el objeto de reducir el riesgo de ptosis, evitar las inyecciones cerca al músculo elevador del parpado superior, particularmente en pacientes con gran complejo ceja-depresor (depresor superciliar). Las inyecciones en el músculo corrugador deben ser hechas en la parte central del mismo, por lo menos 1 cm por encima del borde orbital.
El intervalo de tratamiento depende de la respuesta individual del paciente después de evaluación. En estudios clínicos, un efecto óptimo fue demostrado por hasta 4 meses después de la inyección. Algunos pacientes mantuvieron la respuesta a los 5 meses. El intervalo de tratamiento no debe ser más frecuente a cada tres meses.
En caso de que el tratamiento falle o el efecto disminuya tras repetidas inyecciones, métodos alternativos de tratamiento deben ser empleados. En caso de que el tratamiento falle después de la primera sesión de tratamiento, las siguientes estrategias pueden ser consideradas:
• Análisis de las causas de la falla, por ejem. músculo inyectado incorrecto, técnica de inyección, y formación de anticuerpos neutralizantes de la toxina.
• Reevaluación de la relevancia del tratamiento con Dysport.
Niños: la seguridad y eficacia de Dysport en el tratamiento de líneas glabelares en individuos menores de 18 años, no ha sido demostrada.
Forma de administración: Dysport se reconstituye con solución de cloruro de sodio inyectable (0.9%) para obtener una solución conteniendo 200 unidades por mL de Dysport.
Dysport se administra mediante inyección intramuscular para el tratamiento de líneas glabelares moderadas a severas, como se indicó anteriormente.
Líneas cantales moderadas a severas
Posología
Pacientes de hasta 50 años de edad: la dosis recomendada en pacientes de 50 años de edad o menos es de 30 unidades (0.15 mL) de Dysport para cada ojo, dividida en 3 sitios de inyección (dosis total 60 unidades).
Pacientes mayores de 50 años de edad: la dosis recomendada en pacientes de 50 años de edad o mayores es de 45 unidades (0.15 mL) de Dysport para cada ojo, dividida en 3 sitios de inyección (dosis total 90 unidades).
Todos los pacientes: retire el maquillaje y desinfecte la piel con un antiséptico local. Las inyecciones intramusculares en el músculo orbicular del ojo deben realizarse en ángulos rectos (perpendicularmente) con respecto a la piel.
Deben administrarse 10 unidades (0.05 mL) en pacientes de 50 años o menos, o 15 unidades (0.05 mL) en pacientes de 50 años o mayores, en tres sitios de inyección. Los tres sitios de inyección se ubican en un radio de 1.5 cm del canto lateral, 1 cm fuera del borde orbital. Las ubicaciones precisas de los sitios de inyección se indican en el diagrama a continuación:
Debe solicitarse a los pacientes que sonrían. La inyección debe administrarse junto al extremo externo de las líneas cantales laterales. La inyección debe realizarse en dirección lateral, perpendicular a las líneas cantales laterales. Debe tenerse cuidado de evitar inyectar cerca del margen inferior del músculo zigomático mayor. Debe pedirse al paciente que no frote el área inyectada durante 12 horas después del tratamiento. El intervalo entre ciclos de tratamiento no debe ser menor de 12 semanas.
Niños: no se recomienda el uso de Dysport en pacientes menores de 18 años.
Forma de administración
Pacientes de hasta 50 años de edad: un vial de 500 unidades de Dysport se reconstituye con 2.5 mL de cloruro de sodio inyectable BP (0.9%) para obtener una solución conteniendo 200 unidades/mL de Dysport.
Pacientes de más de 50 años de edad: un vial de 500 unidades de Dysport se reconstituye con 1.67 mL de cloruro de sodio inyectable BP (0.9%) para obtener una solución conteniendo 300 unidades/mL de Dysport.
Sobredosis: Las dosis excesivas pueden producir parálisis neuromuscular distante y profunda. Una sobredosis puede incrementar el riesgo de que la neurotoxina ingrese al torrente sanguíneo y pueda causar complicaciones asociadas con los efectos de la intoxicación oral por botulinum (por ejemplo, disfagia y disfonía). Cuando las dosis excesivas causan parálisis de los músculos respiratorios puede necesitarse respiración asistida. No existe un antídoto específico; no se puede esperar efectos benéficos de ninguna antitoxina y se recomienda tratamiento paliativo. En caso de sobredosis, el paciente debe ser médicamente monitorizado para detectar signos y/o síntomas de debilidad muscular excesiva o parálisis muscular. Se debe instaurar tratamiento sintomático si es necesario. Los síntomas de sobredosis pueden no presentarse inmediatamente después de la inyección. Si ocurre inyección accidental o ingestión oral, el paciente debe estar bajo supervisión médica durante varias semanas en busca de signos y/o síntomas de debilidad muscular excesiva o parálisis muscular.

