ATOZET
ATORVASTATINA, EZETIMIBA
Tabletas recubiertas
1 Caja, 30 Tabletas recubiertas, 10/10 mg
1 Caja, 30 Tabletas recubiertas, 10/20 mg
COMPOSICIÓN:
Forma farmacéutica y formulación: Tabletas Recubiertas
Cada TABLETA RECUBIERTA contiene:
equivalente a 500 mg y 750 mg
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
Prevención de Enfermedad Cardiovascular
ATOZET está indicado para reducir el riesgo de eventos cardiovasculares (muerte cardiovascular, infarto de miocardio no fatal, accidente cerebrovascular no fatal, hospitalización por angina inestable o necesidad de revascularización) en pacientes con enfermedad cardiaca coronaria (ECC).
Hipercolesterolemia Primaria
ATOZET está indicado como tratamiento complementario a la dieta para la reducción de niveles elevados de colesterol total (C-total), colesterol de lipoproteínas de baja densidad (C-LDL), apolipoproteína B (ApoB), triglicéridos (TG) y colesterol de lipoproteínas no de alta densidad (C-no HDL), y para aumentar el colesterol de lipoproteínas de alta densidad (C-HDL) en pacientes con hipercolesterolemia primaria (heterocigótica familiar y no familiar) o hiperlipidemia mixta.
Hipercolesterolemia Familiar Homocigótica (HFHo)
ATOZET está indicado para la reducción de los niveles elevados de C-total y C-LDL en pacientes con HFHo. Los pacientes también pueden recibir tratamientos complementarios (ej., aféresis de LDL).
MECANISMO DE ACCIÓN:
Clase Terapéutica
ATOZET (ezetimiba/atorvastatina) es un producto hipolipemiante que inhibe selectivamente la absorción
intestinal de colesterol y esteroles vegetales relacionados e inhibe la síntesis endógena de colesterol.
ATOZET
El colesterol plasmático se deriva de la absorción intestinal y la síntesis endógena. ATOZET contiene ezetimiba y atorvastatina, dos compuestos hipolipemiantes con mecanismos de acción complementarios.
ATOZET reduce los niveles elevados de C-total, C-LDL, Apo B, TG y C no-HDL, y aumenta el C-HDL mediante la inhibición dual de la absorción y la síntesis de colesterol.
Ezetimiba
Ezetimiba inhibe la absorción intestinal de colesterol. La ezetimiba es activa por vía oral y tiene un mecanismo de acción que difiere de otras clases de compuestos reductores del colesterol (por ejemplo, estatinas, secuestrantes de ácidos biliares [resinas], derivados de ácido fíbrico y estanoles vegetales). La diana molecular de ezetimiba es el transportador de esterol, Niemann-Pick C1-Like 1 (NPC1L1), que es responsable de la absorción intestinal de colesterol y de fitoesteroles.
La ezetimiba se localiza en el borde en cepillo del intestino delgado e inhibe la absorción de colesterol, induciendo una disminución en la entrega de colesterol intestinal al hígado; las estatinas reducen la síntesis de colesterol en el hígado y en conjunto, estos mecanismos distintos proporcionan una reducción complementaria de colesterol.
En un estudio clínico de 2 semanas en 18 pacientes con hipercolesterolemia, ezetimiba inhibió la absorción intestinal de colesterol en 54% en comparación con el placebo.
Se realizó una serie de estudios preclínicos para determinar la selectividad de ezetimiba en la inhibición de la absorción de colesterol. Ezetimiba inhibió la absorción de [14C]-colesterol sin ningún efecto sobre la absorción de triglicéridos, ácidos grasos, ácidos biliares, progesterona, etinil estradiol o las vitaminas liposolubles A y D.
Atorvastatina
La atorvastatina es un inhibidor selectivo y competitivo de la HMG-CoA reductasa, la enzima limitante de la velocidad que convierte 3-hidroxi-3-metil-glutaril-coenzima A en mevalonato, un precursor de los esteroles, incluido el colesterol. En modelos animales, la atorvastatina disminuye los niveles plasmáticos de colesterol y lipoproteínas al inhibir la síntesis de HMG-CoA reductasa y colesterol en el hígado y al aumentar el número de receptores hepáticos de LDL en la superficie celular para mejorar la captación y el catabolismo de la LDL; La atorvastatina también reduce la producción de LDL y el número de partículas de LDL.
Farmacocinética
ATOZET ha demostrado ser bioequivalente con la co-administración de las correspondientes dosis de las
tabletas de ezetimiba y atorvastatina.
Absorción
Los efectos de una comida rica en grasas en la farmacocinética de ezetimiba y atorvastatina cuando se
administran como ATOZET tabletas son comparables a los reportados para las tabletas individuales.
Ezetimiba
Después de la administración oral, la ezetimiba se absorbe rápidamente y se conjuga extensamente para formar un glucurónido fenólico farmacológicamente activo (ezetimiba-glucurónido). Las concentraciones plasmáticas máximas (Cmáx) medias ocurren de 1 a 2 horas para ezetimiba-glucurónido y de 4 a 12 horas para ezetimiba. La biodisponibilidad absoluta de ezetimiba no se puede determinar porque el compuesto es prácticamente insoluble en medios acuosos adecuados para inyección.
La administración concomitante de alimentos (comidas ricas en grasas o sin grasas) no tuvo efecto sobre la biodisponibilidad oral de ezetimiba administrada como ezetimiba 10 mg tabletas.
Atorvastatina
La atorvastatina se absorbe rápidamente después de la administración oral; las concentraciones plasmáticas máximas (Cmáx) ocurren dentro de 1 a 2 horas. El grado de absorción aumenta en proporción a la dosis de atorvastatina. Después de la administración oral, las tabletas recubiertas de atorvastatina son 95% a 99% biodisponibles en comparación con la solución oral. La biodisponibilidad absoluta de atorvastatina es de aproximadamente 12% y la disponibilidad sistémica de la actividad inhibidora de la HMG-CoA reductasa es de aproximadamente 30%. La baja disponibilidad sistémica se atribuye a la depuración pre-sistémica en la mucosa gastrointestinal y/o al metabolismo hepático de primer paso.
Distribución
Ezetimiba y ezetimiba-glucurónido se unen en proporciones de 99.7% y de 88 a 92% a las proteínas plasmáticas humanas, respectivamente.
Atorvastatina
El volumen promedio de distribución de atorvastatina es de aproximadamente 381 L. La atorvastatina se une ≥ 98% a las proteínas plasmáticas.
Metabolismo
Ezetimiba
La ezetimiba se metaboliza principalmente en el intestino delgado y el hígado a través de la conjugación glucurónida (una reacción fase II) con subsecuente excreción biliar. Se ha observado metabolismo oxidativo mínimo (una reacción fase I) en todas las especies evaluadas. Ezetimiba y ezetimibaglucurónido son los principales compuestos derivados del medicamento detectados en el plasma, que constituyen aproximadamente 10 a 20% y 80 a 90% del medicamento total en plasma, respectivamente. Tanto ezetimiba como ezetimiba-glucurónido se eliminan lentamente del plasma con evidencia de reciclado enterohepático significativo. La vida media de ezetimiba y de ezetimiba-glucurónido es de aproximadamente 22 horas.
Atorvastatina
La atorvastatina es metabolizada por el citocromo P450 3A4 a derivados orto- y parahidroxilados y diversos productos de la beta-oxidación. Aparte de otras rutas, estos productos también se metabolizan adicionalmente a través de la glucuronidación. In vitro, la inhibición de la HMG-CoA reductasa por los metabolitos orto- y parahidroxilados es equivalente a la de atorvastatina. Aproximadamente 70% de la actividad inhibidora circulante para la HMG-CoA reductasa se atribuye a los metabolitos activos.
Eliminación
Ezetimiba
Después de la administración oral de 14C-ezetimiba (20 mg) a sujetos humanos, ezetimiba total representó aproximadamente 93% de la radioactividad total en el plasma. Aproximadamente 78% y 11% de la radioactividad administrada se recuperó en las heces y la orina, respectivamente, durante un período de recogida de 10 días. Después de 48 horas, no hubo niveles detectables de radioactividad en el plasma.
Atorvastatina
La atorvastatina se elimina principalmente por la bilis después del metabolismo hepático y/o extrahepático. Sin embargo, el medicamento no parece sufrir una recirculación enterohepática significativa. La vida media de eliminación plasmática media de atorvastatina en los humanos es de aproximadamente 14 horas. La vida media de la actividad inhibidora para la HMG-CoA reductasa es de aproximadamente 20 a 30 horas debido a la contribución de los metabolitos activos.
Poblaciones especiales
Insuficiencia Rena
Ezetimiba
Después de una dosis única de 10 mg de ezetimiba en pacientes con enfermedad renal severa (n=8, con una CrCl media ≤30 mL/min/1.73 m2), el ABC media de ezetimiba total aumentó aproximadamente 1.5 veces, en comparación con los sujetos sanos (n=9).
Un paciente adicional en este estudio (post trasplante renal y tratado con varios medicamentos, incluyendo ciclosporina) tuvo una exposición 12 veces mayor a ezetimiba total.
Atorvastatina La enfermedad renal no tiene influencia en las concentraciones plasmáticas o los efectos lipídicos de atorvastatina y sus metabolitos activos.
Insuficiencia hepática
Ezetimiba
Después de una dosis única de 10 mg de ezetimiba, el área bajo la curva (ABC) media de ezetimiba total aumentó aproximadamente 1.7 veces en pacientes con insuficiencia hepática leve (puntuación Child- Pugh 5 o 6), en comparación con sujetos sanos. En un estudio de dosis múltiples de 14 días (10 mg al día) en pacientes con insuficiencia hepática moderada (puntuación de Child-Pugh 7 a 9), el ABC media de ezetimiba total aumentó aproximadamente 4 veces el Día 1 y el Día 14 en comparación con sujetos sanos. No es necesario ajustar la dosis para pacientes con insuficiencia hepática leve. Debido a los efectos desconocidos de la exposición elevada a ezetimiba en pacientes con insuficiencia hepática moderada o severa (puntuación de Child-Pugh > 9), no se recomienda el uso de ezetimiba en estos
pacientes.
Atorvastatina
Las concentraciones plasmáticas de atorvastatina y sus metabolitos activos se incrementan notablemente (aproximadamente 16 veces en la Cmáx y aproximadamente 11 veces en el ABC) en pacientes con enfermedad hepática alcohólica crónica (Child-Pugh B).
Uso Pediátrico
Ezetimiba
La farmacocinética de ezetimiba es similar entre niños de 6 años o más y adultos. No se dispone de datos farmacocinéticos en la población pediátrica <6 años.
Atorvastatina
El aclaramiento oral aparente de atorvastatina en sujetos pediátricos fue similar al de los adultos cuando se escaló alométricamente por peso corporal ya que el peso corporal fue la única covariable significativa en la población PK de atorvastatina con datos que incluyeron pacientes pediátricos con HeFH (edades de 10 a 17 años de edad, n = 29) en un estudio de 8 semanas de etiqueta abierta.
Uso Geriátrico
Ezetimiba
Las concentracines plasmáticas de ezetimiba total son aproximadamente 2 veces mayores en las personas de edad avanzada (≥65 años) que en los jóvenes (18 a 45 años). La reducción de C-LDL y el perfil de seguridad son similares entre personas de edad avanzada y jóvenes tratados con ezetimiba.
Atorvastatina
Las concentraciones plasmáticas de atorvastatina y sus metabolitos activos son mayores en los sujetos sanos de edad avanzada que en los adultos jóvenes, mientras que los efectos sobre los lípidos son similares a los observados en poblaciones de pacientes más jóvenes.
Raza
Basados en un meta-análisis de estudios farmacocinéticos con ezetimiba, no hubo diferencias farmacocinéticas entre los negros y los caucásicos.
Sexo
Ezetimiba
Las concentraciones plasmáticas de ezetimiba total son ligeramente superiores (<20%) en las mujeres que en los hombres. La reducción del C-LDL y el perfil de seguridad son comparables entre los hombres y las mujeres tratados con ezetimiba.
Atorvastatina
Las concentraciones de atorvastatina y sus metabolitos activos en mujeres difieren de las de los hombres (mujeres: aproximadamente 20% mayor para Cmáx y aproximadamente 10% menor para ABC). Estas diferencias no fueron clínicamente significativas, resultando en diferencias clínicamente no significativas en los efectos de lípidos entre hombres y mujeres.
Hemodiálisis
Atorvastatina
Aunque no se han realizado estudios en pacientes con enfermedad renal terminal, no se espera que la hemodiálisis mejore significativamente la depuración de atorvastatina ya que el medicamento se une ampliamente a las proteínas plasmáticas.
CONTRAINDICACIONES:
• ATOZET está contraindicado en pacientes con hipersensibilidad a ezetimiba, atorvastatina o a cualquiera de sus componentes inactivos.
• Enfermedad hepática activa o elevaciones persistentes e inexplicables de las transaminasas séricas.
• Embarazo y lactancia [ver 6. USO EN POBLACIONES ESPECÍFICAS, 6.1. Embarazo y 6.2. Madres en Período de Lactancia].
REACCIONES ADVERSAS:
Efectos en la capacidad para conducir y utilizar maquinaria:
No se han realizado estudios de los efectos en la capacidad para conducir y utilizar maquinaria. Sin embargo, ciertos efectos secundarios que se han reportado con ATOZET pueden afectar la capacidad del paciente para conducir u operar maquinaria. Las respuestas individuales a ATOZET pueden variar.
Adultos
La seguridad de ATOZET (o la co-administración de ezetimiba y atorvastatina equivalente a ATOZET) se ha evaluado en más de 2400 pacientes en 7 ensayos clínicos. ATOZET fue generalmente bien tolerado.
Las siguientes experiencias adversas frecuentes (≥ 1/100, < 1/10) o infrecuentes (≥ 1/1000, < 1/100) relacionadas con el medicamento se reportaron en pacientes que tomaban ATOZET:
Infecciones e infestaciones:
Infrecuentes: influenza.
Trastornos psiquiátricos:
Infrecuentes: depresión; insomnio; desorden del sueño.
Trastornos del sistema nervioso:
Infrecuentes: mareos; disgeusia; cefalea; parestesia.
Trastornos cardíacos:
Infrecuentes: bradicardia sinusal.
Trastornos vasculares:
Infrecuentes: sofocos.
Trastornos respiratorios, torácicos y mediastínicos:
Infrecuentes: disnea.
Trastornos gastrointestinales:
Frecuentes: diarrea.
Infrecuentes: malestar abdominal, distensión abdominal, dolor abdominal, dolor abdominal inferior, dolor abdominal superior, estreñimiento, dispepsia, flatulencia, movimientos intestinales frecuentes, gastritis, náuseas, malestar estomacal.
Trastornos de la piel y del tejido subcutáneo:
Infrecuentes: acné, urticaria.
Trastornos musculoesqueléticos y del tejido conectivo:
Frecuentes: mialgia.
Infrecuentes: artralgia, dolor de espalda, fatiga muscular, espasmos musculares, debilidad muscular, dolor en las extremidades.
Trastornos generales y condiciones en el sitio de administración:
Infrecuentes: astenia, fatiga, malestar general, edema.
Investigaciones:
Infrecuentes: ALT y/o AST elevadas, fosfatasa alcalina elevada, CK sanguínea elevada, gammaglutamiltransferasa elevada, enzimas hepáticas elevadas, prueba de función hepática anormal, aumento de peso.
En ensayos clínicos controlados, la incidencia de elevaciones clínicamente importantes de las transaminasas séricas (ALT y/o AST ≥ 3 x LSN, consecutivas) fue de 0.6% para los pacientes tratados con ATOZET. Estas elevaciones de las transaminasas fueron generalmente asintomáticas, no asociadas con colestasia y retornaron a los valores basales de forma espontánea o después de la suspensión de la terapia.
Ninguno de los pacientes tratados con ATOZET tuvo niveles de CK ≥ 10 x LSN.
Experiencia Post-comercialización y Otras Experiencias de Ensayos Clínicos
Las siguientes reacciones adversas adicionales se han reportado en el uso post-comercialización con ATOZET o en estudios clínicos o uso post-comercialización con ezetimiba o atorvastatina:
Infecciones e infestaciones: nasofaringitis.
Trastornos de la sangre y del sistema linfático: trombocitopenia.
Trastornos del sistema inmune: reacciones de hipersensibilidad, incluyendo anafilaxia, angioedema, erupción cutánea y urticaria.
Trastornos del metabolismo y de la nutrición: disminución del apetito, anorexia, hiperglucemia, hipoglucemia.
Trastornos psiquiátricos: pesadillas.
Trastornos del sistema nervioso: hipoestesia; neuropatía periférica.
Ha habido raros reportes post-comercialización de deterioro cognitivo (por ejemplo, pérdida de la memoria, olvidos, amnesia, deterioro de la memoria, confusión) asociado con el uso de estatinas. Estos problemas cognitivos se han reportado para todas las estatinas. Los reportes son generalmente no serios y reversibles con la descontinuación de la estatina, con tiempos variables para el inicio de los síntomas (1 día a años) y la resolución de los síntomas (mediana de 3 semanas).
Trastornos oculares: visión borrosa; alteración de la visión
Trastornos auditivos y del laberinto: tinnitus; pérdida de la audición.
Trastornos vasculares: hipertensión.
Trastornos respiratorios, torácicos y del mediastino: tos, dolor faringolaríngeo, epistaxis.
Trastornos gastrointestinales: boca seca, pancreatitis, enfermedad por reflujo gastroesofágico, eructos, vómitos.
Trastornos hepatobiliares: hepatitis, colelitiasis, colecistitis, colestasis.
Trastornos de la piel y del tejido subcutáneo: alopecia, prurito, erupción cutánea, eritema multiforme, edema angioneurótico, dermatitis bullosa incluyendo eritema multiforme, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica.
Trastornos musculoesqueléticos y del tejido conjuntivo: miopatía/rabdomiólisis, dolor de cuello, inflamación de las articulaciones, miositis, tendinopatía, a veces se complica por la ruptura.
Se han reportado casos muy raros de miopatía necrotizante inmuno-mediada (MNIM), una miopatía autoinmune, asociada con el uso de estatinas. La MNIM se caracteriza por: debilidad muscular proximal y creatina quinasa sérica elevada, que persiste a pesar de la descontinuación del tratamiento con estatinas; biopsia muscular que muestra miopatía necrotizante sin inflamación significativa; mejoría con agentes inmunosupresores.
Sistema reproductor y trastornos mamarios:
ginecomastia.
Trastornos generales y condiciones en el sitio de administración: dolor pectoral, dolor, edema periférico, pirexia
Investigaciones:
glóbulos blancos presentes en la orina.
Se han reportado elevaciones en los niveles de HbA1c y de glucosa sérica en ayunas con inhibidores de la HMG-CoA reductasa, incluyendo atorvastatina.
Los siguientes eventos adversos han sido reportados con algunas estatinas:
• disfunción sexual
• depresión
• casos excepcionales de enfermedad pulmonar intersticial, especialmente con la terapia a largo plazo
• diabetes mellitus: la frecuencia dependerá de la presencia o ausencia de factores de riesgo (glucosa en ayunas ≥ 5.6 mmol / L, Índice de Masa Corporal> 30 kg /m2, triglicéridos elevados antecedentes de hipertensión arterial)
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
ATOZET
No se observó interacción farmacocinética clínicamente significativa cuando ezetimiba se administró concomitantemente con atorvastatina.
Múltiples mecanismos pueden contribuir a posibles interacciones con los inhibidores de la HMG Co-A reductasa. Los medicamentos o productos naturales que inhiben ciertas vías enzimáticas (por ejemplo, CYP3A4) y/o transportadores (por ejemplo, OATP1B) pueden aumentar las concentraciones plasmáticas de atorvastatina y pueden conducir a un incremento del riesgo de miopatía/rabdomiólisis.
Consulte la información para prescribir de todos los fármacos utilizados concomitantemente para obtener más información sobre sus interacciones potenciales con atorvastatina y/o alteraciones potenciales enzimáticas o transportadoras y los posibles ajustes de la dosis y regímenes.
Interacciones CYP3A4
En estudios preclínicos se ha demostrado que ezetimiba no induce las enzimas metabolizadoras de medicamentos del citocromo P450. No se han observado interacciones farmacocinéticas clínicamente significativas entre ezetimiba y medicamentos que se sabe que son metabolizadas por los citocromos P450 1A2, 2D6, 2C8, 2C9 y 3A4, o por la N-acetiltransferasa. La atorvastatina es metabolizada por el citocromo P450 3A4. La administración concomitante de atorvastatina con inhibidores del citocromo P450 3A4 puede generar incrementos en las concentraciones plasmáticas de atorvastatina. El grado de interacción y potenciación de los efectos depende de la variabilidad del efecto sobre el citocromo P450 3A4.
Los inhibidores del citocromo P3A4 aumentan el riesgo de miopatía al reducir la eliminación del componente atorvastatina de ATOZET.
Claritromicina: El ABC de atorvastatina fue significativamente mayor con la administración concomitante de atorvastatina 80 mg con claritromicina (500 mg dos veces al día) en comparación con la de atorvastatina sola. Por tanto, en pacientes que toman claritromicina se debe tener precaución cuando la dosis de ATOZET exceda 10/20 mg.
Combinación de Inhibidores de la Proteasa: El ABC de atorvastatina fue significativamente mayor con la administración concomitante de atorvastatina con varias combinaciones de inhibidores de la proteasa del VIH, así como con el inhibidor de la proteasa de la hepatitis C telaprevir, en comparación con el de atorvastatina sola.
Por lo tanto, en pacientes que toman el inhibidor de la proteasa del VIH tipranavir más ritonavir, o el inhibidor de la proteasa de la hepatitis C telaprevir, se debe evitar el uso concomitante de ATOZET. En pacientes que toman el inhibidor de la proteasa del VIH lopinavir más ritonavir, se debe tener precaución cuando se prescriba ATOZET, y se debe usar la menor dosis necesaria. En pacientes que toman inhibidores de la proteasa del VIH saquinavir más ritonavir, darunavir más ritonavir, fosamprenavir o fosamprenavir más ritonavir, o el inhibidor de la proteasa de la hepatitis C boceprevir, la dosis de ATOZET no debe exceder de 10/20 mg y debe usarse con precaución en pacientes que toman el inhibidor de la proteasa del VIH nelfinavir, la dosis de ATOZET no debe exceder de 10/40 mg y se recomienda un estrecho monitoreo clínico.
Itraconazol: El ABC de atorvastatina fue significativamente mayor con la administración concomitante de atorvastatina 40 mg e itraconazol 200 mg. Por lo tanto, en pacientes que toman itraconazol se debe tener precaución cuando la dosis de ATOZET exceda de 10/20 mg.
Jugo de Toronja: El jugo de toronja contiene uno o más componentes que inhiben el CYP3A4 y puede aumentar las concentraciones plasmáticas de atorvastatina, especialmente con el consumo excesivo de jugo de toronja (> 1.2 litros por día).
Ciclosporina: En un estudio de ocho pacientes de trasplante renal con depuración de creatinina > 50 mL/min en una dosis estable de ciclosporina, una dosis única de 10 mg de ezetimiba produjo un aumento de 3.4 veces (rango de 2.3 a 7.9 veces) en el ABC media de ezetimiba total en comparación con una población control sana de otro estudio (n=17). En otro estudio, un paciente de trasplante renal con insuficiencia renal severa (depuración de la creatinina de 13.2 mL/min/1.73 m2) que estaba recibiendo múltiples medicamentos, incluyendo ciclosporina, demostró una exposición 12 veces mayor a ezetimiba total en comparación con controles concurrentes. En un estudio cruzado de dos períodos en doce sujetos sanos, la administración diaria de 20 mg de ezetimiba por 8 días con una dosis única de 100 mg de ciclosporina el Día 7 produjo un incremento promedio del 15% en el ABC de ciclosporina (rango 10% de disminución a 51 % de aumento) en comparación con una dosis única de 100 mg de ciclosporina sola.
La atorvastatina y los metabolitos de la atorvastatina son sustratos del transportador OATP1B1. Los inhibidores de OATP1B1 (ejemplo, ciclosporina) pueden aumentar la biodisponibilidad de atorvastatina. El ABC de atorvastatina fue significativamente mayor con la administración concomitante de atorvastatina 10 mg y ciclosporina 5.2 mg/kg/día en comparación con la de atorvastatina sola. Se debe evitar la administración concomitante de ATOZET con ciclosporina.
Otras interacciones
Antiácidos: La administración concomitante de antiácidos disminuyó la tasa de absorción de ezetimiba, pero no tuvo efecto sobre la biodisponibilidad de ezetimiba. Esta disminución de la tasa de absorción no se considera clínicamente significativa.
La administración concomitante de atorvastatina con una suspensión oral de antiácidos conteniendo hidróxidos de magnesio y aluminio disminuyó las concentraciones plasmáticas de atorvastatina y sus metabolitos activos en aproximadamente 35%; sin embargo, la reducción de C-LDL no se modificó.
Colestiramina: La administración concomitante de colestiramina disminuyó el ABC media de ezetimiba total (ezetimiba + glucurónido de ezetimiba) aproximadamente 55%. La reducción incremental de C-LDL debido a la adición de ezetimiba a colestiramina puede verse disminuida por esta interacción.
Gemfibrozilo: Se debe evitar la administración concomitante de ATOZET con gemfibrozilo.
Fenofibrato: Se debe tener precaución cuando se prescribe ATOZET y fenofibrato, dado que el fenofibrato puede causar miopatía cuando se administra solo.
Si se sospecha de colelitiasis en un paciente que recibe ATOZET y fenofibrato se indican estudios de la vesícula biliar y se debe considerar una terapia hipolipemiante alternativa [ver el texto del producto para fenofibrato y ácido fenofíbrico].
Otros fibratos: La administración concomitante de ezetimiba con otros fibratos no ha sido estudiada. Por lo tanto, no se recomienda la administración concomitante de ATOZET y otros fibratos.
Ácido Fusídico
Los pacientes tratados con ácido fusídico concomitantemente con ATOZET pueden tener un mayor riesgo de miopatía/rabdomiólisis. No se recomienda la administración concomitante con ácido fusídico. En pacientes en los que se considera esencial el uso de ácido fusídico sistémico, se debe descontinuar ATOZET durante la duración del tratamiento con ácido fusídico. En circunstancia excepcionales, donde se necesita el uso sistémico prolongado de ácido fusídico, por ejemplo para el tratamiento de infecciones severas, la necesidad de la administración concomitante de ATOZET y ácido fusídico solo debe considerarse caso por caso bajo estricta supervisión médica.
Anticoagulantes
Si se añade ATOZET a warfarina, otro anticoagulante cumarínico o fluindiona, se debe monitorear apropiadamente la Relación Normalizada Internacional (INR).
Uso en Pacientes con Accidente Cerebrovascular reciente o Ataque Isquémico Transitorio (AIT)
En un análisis post-hoc del estudio Prevención de ACV por Reducción Intensiva de los Niveles de Colesterol (SPARCL, por sus siglas en inglés), en donde se administró atorvastatina 80 mg versus placebo en 4,731 sujetos sin ECC que tuvieron un ACV o un AIT dentro de los 6 meses previos, se observó una mayor incidencia de ACV hemorrágico en el grupo de atorvastatina 80 mg en comparación con el placebo. La incidencia de ACV hemorrágico fatal fue similar en todos los grupos de tratamiento. La incidencia de ACV hemorrágico no fatal fue significativamente mayor en el grupo de atorvastatina en comparación con el grupo placebo. Algunas características basales, incluyendo ACV hemorrágico y AC lacunar en el ingreso al estudio se asociaron con una mayor incidencia de ACV hemorrágico en el grupo de atorvastatina.
INFORMACIÓN COMPLEMENTARIA:
Restricciones de uso durante el embarazo
y la lactancia:
Embarazo
La aterosclerosis es un proceso crónico y la descontinuación de los medicamentos hipolipemiantes durante el embarazo debe tener poco impacto en el desenlace de la terapia a largo plazo para la hipercolesterolemia primaria.
ATOZET
ATOZET está contraindicada durante el embarazo. Debido a que los inhibidores de la HMG-CoA reductasa disminuyen la síntesis de colesterol y posiblemente la síntesis de otras sustancias biológicamente activas derivadas del colesterol. ATOZET puede causar daño fetal cuando se administra a mujeres embarazadas. ATOZET se debe suspender tan pronto como se conozca del embarazo. Aconseje a las mujeres sobre el potencial reproductivo para que usen un método anticonceptivo eficaz durante el tratamiento con ATOZET.
Ezetimiba
No se dispone de datos clínicos sobre embarazos expuestos. Los estudios en animales de ezetimibe administrados solos no indican efectos dañinos directos o indirectos con respecto al embarazo, el desarrollo embrionario / fetal, el parto o el desarrollo postnatal.
Cuando se administró ezetimiba con estatinas, no se observaron efectos teratogénicos en los estudios de desarrollo embriofetal en ratas preñadas. En conejas preñadas, se observó una baja incidencia de malformaciones esqueléticas.
Atorvastatina
Los datos publicados son limitados, sobre atorvastatina a partir de estudios observacionales, metaanálisis
e informes de casos no han mostrado un mayor riesgo de malformaciones congénitas importantes oaborto espontáneo. Se han recibido informes poco frecuentes de anomalías congénitas después de la exposición intrauterina a otros inhibidores de la HMG-CoA reductasa. En una revisión de aproximadamente 100 embarazos seguidos prospectivamente en mujeres expuestas a simvastatina o lovastatina. La incidencia de anomalías congénitas, abortos espontáneos y muertes/muertes fetales, no superaron lo que se esperaba en la población general. El número de casos es adecuado para excluir un aumento mayor o igual a tres a cuatro veces mayor en anomalías congénitas sobre la incidencia de fondo. En el 89% de los embarazos seguidos prospectivamente, el tratamiento con medicamentos se inició antes del embarazo y se suspendió en algún momento del primer trimestre cuando se identificó el embarazo.
En estudios de reproducción en animales, ratas y conejos no hubo evidencia de toxicidad embriofetal o malformaciones congénitas en dosis de hasta 30 y 20 veces, respectivamente. La exposición humana a la dosis máxima recomendada en humanos (MRHD) de 80 mg, basada en el cuerpo. Superficie (mg/m2).
En ratas a las que se administró atorvastatina durante la gestación y la lactancia, se observó una disminución del crecimiento y el desarrollo postnatal a dosis ≥ 6 veces la MRHD.
Madres en Período de Lactancia
ATOZET está contraindicado en madres en periodo de lactancia. Debido a la posibilidad de reacciones adversas graves en el lactante amamantado. Las mujeres que están en periodo de lactancia no deben tomar ATOZET.
Ezetimiba
Los estudios en ratas han demostrado que ezetimiba y atorvastatina se excretan en la leche. No se sabe si ezetimiba o atorvastatina se excretan en la leche materna humana.
Atorvastatina
No hay información disponible sobre los efectos de la atorvastatina en el lactante amamantado o los efectos de la atorvastatina en la producción de leche. No se sabe si la atorvastatina está presente en la leche materna, pero se ha demostrado que otro medicamento de esta clase pasa a la leche humana y la atorvastatina está presente en la leche de rata.
Farmacología animal
El efecto hipocolesterolémico de ezetimiba se evaluó en monos Rhesus, un modelo para el metabolismo humano de colesterol, así como en perros. Los monos Rhesus se alimentaron con una dieta que contenía colesterol que mimetizaba una dieta occidental humana. Se encontró que la ezetimiba tiene una DE50 de 0.0005 mg/kg/día para inhibir el aumento en los niveles de colesterol plasmático (DE100 = 0.003 mg/kg/día). Se encontró que la DE50 en los perros era 0.007 mg/kg/día. Estos resultados son consistentes con ezetimiba como un inhibidor de la absorción de colesterol extremadamente potente.
En perros tratados con ezetimiba (≥ 0.03 mg/kg/día), la concentración de colesterol en la bilis de la vesícula biliar aumentó ~2 a 3 veces. Sin embargo, una dosis de 300 mg/kg/día administrada a los perros durante un año no dio lugar a la formación de cálculos biliares ni a otros efectos adversos hepatobiliares.
En ratones tratados con ezetimiba (0.3 a 5 mg/kg/día) y alimentados con una dieta normal o rica en colesterol, la concentración de colesterol en la bilis de la vesícula biliar no fue afectada o fue reducida a niveles normales, respectivamente. La relevancia de estos hallazgos preclínicos en humanos es desconocida.
Toxicología animal
Toxicidad Aguda
En animales, no se observó toxicidad después de dosis orales únicas de 5000 mg/kg de ezetimiba en ratas y ratones y 3000 mg/kg en perros.
Toxicidad Crónica
La seguridad de la administración concomitante de ezetimiba y atorvastatina se evaluó en ratas y perros.
Cuando ezetimiba se co-administró con atorvastatina, simvastatina, pravastatina o lovastatina, durante tres meses, los hallazgos toxicológicos fueron consistentes con los observados con las estatinas administradas solas
Ezetimiba
La ezetimiba fue bien tolerada por ratones, ratas y perros. No se identificaron órganos dianas de toxicidad en estudios crónicos a dosis diarias de hasta 1500 (machos) y 500 mg/kg (hembras) en ratas, hasta 500 mg/kg en ratones o hasta 300 mg/kg en perros.
Carcinogénesis
Ezetimiba
En estudios de dos años realizados en ratones y ratas, la ezetimiba no fue carcinogénica.
Atorvastatina
En un estudio de carcinogenicidad de 2 años en ratas a dosis de 10, 30 y 100 mg/kg/día, se encontraron 2 tumores raros en el músculo en las hembras con dosis altas: en una, había un rabdomiosarcoma y, en la otra, hubo un fibrosarcoma. Esta dosis representa un valor del ABC0-24 del plasma de aproximadamente 16 veces la exposición media del medicamento en plasma humano después de una dosis oral de 80 mg.
Un estudio de carcinogenicidad de 2 años en ratones que recibieron 100, 200 o 400 mg/kg/día resultó en un aumento significativo de adenomas hepáticos en los machos con dosis altas y carcinomas hepáticos en hembras con dosis altas. Estos hallazgos ocurrieron en valores del ABC0-24 del plasma de aproximadamente 6 veces la exposición media del medicamento en plasma humano después de una dosis oral de 80 mg.
Mutagénesis
La combinación de ezetimiba con atorvastatina no fue genotóxica en una serie de ensayos in vitro e in vivo.
Ezetimiba
La ezetimiba no fue genotóxica en una serie de pruebas in vivo e in vitro.
Atorvastatin
In vitro, la atorvastatina no fue mutagénica ni clastogénica en las siguientes pruebas con y sin activación metabólica: la prueba de Ames con Salmonella typhimurium y Escherichia coli, el ensayo de mutación de avance HGPRT en células pulmonares de hámster chino, y el ensayo de aberraciones cromosómicas en células pulmonares de hámster chino. La atorvastatina fue negativa en la prueba de micronúcleos de ratón in vivo.
Reproducción
Ezetimiba
La ezetimiba no afectó la fertilidad de ratas machos o hembras.
Atorvastatina
En ratas hembras, la atorvastatina a dosis de hasta 225 mg/kg (56 veces la exposición humana) no causó efectos adversos sobre la fertilidad. Los estudios en ratas macho realizados a dosis de hasta 175 mg/kg (15 veces la exposición humana) no produjeron cambios en la fertilidad. Hubo aplasia y aspermia en el epidídimo de 2 de 10 ratas tratadas con 100 mg/kg/día de atorvastatina durante 3 meses (16 veces el AUC humano en la dosis de 80 mg); los pesos de los testículos fueron significativamente más bajos a 30 y 100 mg/kg y el peso del epidídimo fue más bajo a 100 mg/kg. Las ratas machos que recibieron 100 mg/kg/día durante 11 semanas antes del apareamiento tuvieron disminución de la motilidad del esperma, la concentración de la cabeza espermátida y el aumento del esperma anormal. La atorvastatina no causó efectos adversos en los parámetros del semen ni en la histopatología de los órganos reproductivos en perros que recibieron dosis de 10, 40 o 120 mg/kg durante dos años.
Desarrollo
La administración concomitante de ezetimiba y atorvastatina no fue teratogénica en ratas. En conejas preñadas se observó una baja incidencia de malformaciones esqueléticas (esternebras fusionados y vértebras caudales fusionadas) cuando ezetimiba (1000 mg/kg; ≥146 veces la exposición humana con 10 mg diarios con base en el ABC0-24h de ezetimiba total) se administró con atorvastatina (5, 25 y 50 mg/kg). La exposición a la forma farmacológicamente activa de atorvastatina fue ≥1.4 veces la exposición humana con 10 mg diarios con base en el ABC0-24h.
Ezetimiba
La ezetimiba no fue teratogénica en ratas o conejos y no tuvo efecto sobre el desarrollo prenatal o postnatal.
Atorvastatina
La atorvastatina no fue teratogénica en ratas a dosis de hasta 300 mg/kg/día o en conejos a dosis de hasta 100 mg/kg/día. Estas dosis resultaron en múltiplos de aproximadamente 30 veces (rata) o 20 veces (conejo) la exposición humana basada en la superficie (mg/m2).
RECOMENDACIONES:
Miopatía/Rabdomiólisis
Se han reportado casos raros de rabdomiólisis con insuficiencia renal aguda secundaria a mioglobinuria con atorvastatina y con otros medicamentos de esta clase. Un antecedente de insuficiencia renal puede ser un factor de riesgo para el desarrollo de rabdomiólisis. Estos pacientes requieren un monitoreo más estrecho de los efectos sobre el músculo esquelético.
La atorvastatina, como otras estatinas, ocasionalmente causa miopatía, definida como dolores musculares o debilidad muscular en combinación con aumentos de los valores de CPK > 10 veces el LSN. Se debe considerar miopatía en cualquier paciente con mialgias difusas, sensibilidad o debilidad muscular y/o marcada elevación de CPK. Los pacientes deben ser advertidos de reportar lo más pronto posible cualquier dolor, sensibilidad o debilidad muscular inexplicables, especialmente si se acompaña de malestar o fiebre o si los signos y síntomas musculares persisten después de descontinuar ATOZET . La terapia con ATOZET se debe descontinuar si se producen niveles marcadamente elevados de CPK o si se diagnostica o sospecha miopatía.
El riesgo de miopatía durante el tratamiento con estatinas se incrementa con la administración concomitante de ciclosporina, derivados del ácido fíbrico, eritromicina, claritromicina, agentes antivirales de la hepatitis C telaprevir, elbasvir, grazoprevir, las combinaciones de inhibidores de la proteasa del VIH, incluyendo saquinavir más ritonavir, lopinavir más ritonavir, tipranavir más ritonavir, darunavir más ritonavir, fosamprenavir y fosamprenavir más ritonavir, niacina o antimicóticos azoles. Los médicos que consideran la terapia combinada con ATOZET y derivados del ácido fíbrico, eritromicina, claritromicina, elbasvir, grazoprevir, una combinación de saquinavir más ritonavir, lopinavir más ritonavir, darunavir más ritonavir, fosamprenavir o fosamprenavir más ritonavir, antimicóticos azoles o dosis hipolipemiantes de niacina deben sopesar cuidadosamente los beneficios y riesgos potenciales y deben vigilar cuidadosamente a los pacientes para detectar cualquier signo o síntoma de dolor, sensibilidad o debilidad muscular, especialmente durante los primeros meses de la terapia y durante cualquier período de ajuste ascendente de la dosis de cualquiera de los medicamentos. Se deben considerar dosis bajas iniciales y de mantenimiento de ATOZET cuando se toma concomitantemente con los medicamentos antes mencionados. En este tipo de situaciones se debe considerar la determinación periódica de la CPK, pero no hay garantía de que este monitoreo evitará la aparición de miopatía severa.
Las recomendaciones de prescripción para los agentes que interactúan se resumen en la Tabla 1.
Tabla 1 Interacciones Medicamentosas de Atorvastatina Asociadas con Mayor Riesgo de Miopatía/Rabdomiólisis
|
Agentes Interactuantes R |
ecomendaciones de Prescripción para ATOZET |
|
Ciclosporina, inhibidores de la proteasa del VIH (tipranavir más ritonavir), inhibidor de la proteasa de la hepatitis C (telaprevir), gemfibrozilo. |
Evite el uso de ATOZET. |
|
Otros fibratos (excepto fenofibrato), ácido fusídico. |
No se recomienda con ATOZET. |
|
Inhibidor de la proteasa del VIH (lopinavir más ritonavir). |
Usar con precaución y la menor dosis necesaria. |
|
Claritromicina, itraconazol, inhibidores de la proteasa del VIH (saquinavir más ritonavir*, darunavir más ritonavir, fosamprenavir, fosamprenavir más ritonavir), agentes antivirales de la hepatitis C (boceprevir, elbasvir, grazoprevir). |
No exceda de 10/20 mg al día de ATOZET. |
|
Inhibidor de la proteasa del VIH (nelfinavir) |
No exceda de 10/40 mg al día de ATOZET. |
* Utilizar con precaución y con la menor dosis necesaria
Se han reportado casos de miopatía, incluyendo rabdomiólisis, con atorvastatina co-administrada con colchicina, y se debe tener precaución cuando se prescriba ATOZET con colchicina.
Se han observado informes de miopatía y/o rabdomiólisis con inhibidores de la HMG-CoA reductasa coadministrados con daptomicina. Se debe tener precaución al prescribir inhibidores de la HMG-CoA reductasa con daptomicina, ya que cualquiera de los dos agentes puede causar miopatía y/o rabdomiólisis cuando se administra solo. Se debe considerar la posibilidad de suspender temporalmente la ATOZET en pacientes que toman daptomicina.
La terapia con ATOZET se debe interrumpir temporalmente o descontinuarla en cualquier paciente con una condición seria, aguda sugerente de una miopatía o que tenga un factor de riesgo que predisponga al desarrollo de insuficiencia renal secundaria a rabdomiólisis (p.ej., infección aguda severa, hipotensión, cirugía mayor, trauma, trastornos metabólicos, endocrinos y de electrolitos severos, y convulsiones no controladas).
Enzimas Hepáticas
En ensayos controlados de co-administración en pacientes que recibieron ezetimiba con atorvastatina, se han observado elevaciones consecutivas de las transaminasas (≥ 3 veces el límite superior normal [LSN]).
La atorvastatina, como algunas otras terapias hipolipemiantes, ha sido asociada con anormalidades bioquímicas de la función hepática.
Se recomienda que se realicen pruebas de enzimas hepáticas antes de iniciar el tratamiento con ATOZET y se repitan como sea clínicamente indicado. Ha habido raros informes post-comercialización de insuficiencia hepática fatal y no fatal en pacientes que toman estatinas, incluyendo atorvastatina. Si ocurre lesión hepática grave con síntomas clínicos y/o hiperbilirrubinemia o ictericia durante el tratamiento con ATOZET, se debe interrumpir la terapia inmediatamente. Si no se encuentra una etiología alternativa, no reinicie ATOZET.
ATOZET debe usarse con precaución en pacientes que consumen cantidades importantes de alcohol y/o que tienen antecedentes de enfermedad hepática. La enfermedad hepática activa o elevaciones persistentes, inexplicables de las transaminasas son contraindicaciones para el uso de atorvastatina.
Función Endocrina
Las estatinas interfieren con la síntesis de colesterol y teóricamente podrían atenuar la producción de esteroides adrenales y/o gonadales. Los estudios clínicos han demostrado que la atorvastatina no reduce la concentración basal de cortisol plasmático ni reduce la reserva adrenal. Los efectos de las estatinas sobre la fertilidad masculina no han sido estudiados en un número adecuado de pacientes. Se desconocen los efectos, si los hubiera, en el eje pituitario-gonadal en mujeres premenopáusicas. Se debe tener precaución si ATOZET se administra concomitantemente con medicamentos que pueden disminuir los niveles o la actividad de las hormonas esteroideas endógenas, como ketoconazol, espironolactona y cimetidina.
Insuficiencia Hepática
Debido a los efectos desconocidos de una elevada exposición a ezetimiba en pacientes con insuficiencia hepática moderada o severa, ATOZET no se recomienda en estos pacientes.
Gemfibrozilo: Debido a un aumento del riesgo de miopatía/rabdomiólisis cuando los inhibidores de la HMG-CoA reductasa se co-administran con gemfibrozilo, se debe evitar la administración concomitante de ATOZET con gemfibrozilo.
En un estudio farmacocinético, la administración concomitante de gemfibrozilo aumentó las concentraciones totales de ezetimiba aproximadamente 1.7 veces. Este incremento no se considera clínicamente significativo. No se dispone de datos clínicos.
Fenofibrato: Dado que se sabe que el riesgo de miopatía durante el tratamiento con inhibidores de la HMG-CoA reductasa se incrementa con la administración concomitante de fenofibrato, ATOZET debe administrarse con precaución cuando se utiliza concomitantemente con fenofibrato.
En un estudio farmacocinético, la administración concomitante de fenofibrato incrementó las concentraciones totales de ezetimiba aproximadamente 1.5 veces. Este aumento no se considera clínicamente significativo.
Otros fibratos: La seguridad y la eficacia de ezetimiba administrada con otros fibratos no han sido establecidas. Los fibratos pueden aumentar la excreción de colesterol en la bilis, produciendo colelitiasis. En un estudio preclínico en perros, la ezetimiba aumentó el colesterol en la bilis de la vesícula biliar. Aunque se desconoce la relevancia de este hallazgo preclínico para los humanos, no se recomienda la administración concomitante de ATOZET con otros fibratos hasta que se estudie el uso en pacientes.
Ácido Fusídico: El riesgo de miopatía/rabdomiólisis puede incrementarse por la administración.
Anticoagulantes: La administración concomitante de ezetimiba (10 mg una vez al día) no tuvo efecto significativo sobre la biodisponibilidad de warfarina y el tiempo de protrombina en un estudio de doce varones adultos sanos. Ha habido reportes post-comercialización de INR elevada en pacientes con ezetimiba añadida a warfarina o fluindiona. La mayoría de estos pacientes también estaban recibiendo otros medicamentos concomitante de ácido fusídico.
La atorvastatina no tuvo efecto clínicamente significativo en el tiempo de protrombina cuando se administró a pacientes que reciben tratamiento crónico con warfarina.
El efecto de ATOZET sobre el tiempo de protrombina no se ha estudiado.
Inhibidores de la Proteína Resistente al Cáncer de Mama (BCRP, por sus siglas en inglés): La atorvastatina es un sustrato del transportador de eflujo de BCRP. La administración concomitante de productos que son inhibidores de BCRP (ej., elbasvir y grazoprevir) puede dar lugar a un aumento de las concentraciones plasmáticas de atorvastatina y un aumento del riesgo de miopatía; por lo tanto, puede ser necesario un ajuste de la dosis de atorvastatina. La coadministración de elbasvir y grazoprevir con atorvastatina aumenta las concentraciones plasmáticas de atorvastatina en 1.9 veces debido en parte a la inhibición de CYP3A y/o BCRP; por lo tanto, la dosis de ATOZET no debe exceder de 10/20 mg al día en pacientes que reciben medicamentos concomitantes con productos que contengan elbasvir o grazoprevir.
Inductores del Citocromo P450 3A4: La administración concomitante de atorvastatina con inductores del citocromo P450 3A4 (ej., efavirenz, rifampicina) puede conducir a reducciones variables en las concentraciones plasmáticas de atorvastatina. Debido al doble mecanismo de interacción de la rifampicina, se recomienda la administración simultánea de atorvastatina con rifampicina, porque la administración retrasada de atorvastatina después de la administración de rifampicina se ha asociado con una reducción significativa en las concentraciones plasmáticas de atorvastatina.
Antipirina: Debido a que atorvastatina no afecta la farmacocinética de la antipirina, no se esperan interacciones con medicamentos metabolizados a través de las mismas isoenzimas del citocromo.
Colestipol: Las concentraciones plasmáticas de atorvastatina disminuyeron aproximadamente en 25% cuando se administraron concomitantemente colestipol y atorvastatina. Sin embargo, la reducción de CLDL fue mayor cuando atorvastatina y colestipol se administraron concomitantemente que cuando cualquier medicamento se administró solo.
Digoxina: Cuando se administraron concomitantemente dosis múltiples de atorvastatina y digoxina, las concentraciones plasmáticas de digoxina en estado de equilibrio se incrementaron en aproximadamente 20%. Los pacientes que toman digoxina deben ser monitoreados adecuadamente.
Anticonceptivos Orales: La co-administración de atorvastatina y un anticonceptivo oral aumentó los valores del ABC de noretindrona y etinilestradiol en aproximadamente 30% y 20%. Estos incrementos se deben considerar cuando se seleccione un anticonceptivo oral para una mujer que toma atorvastatina.
Amlodipina: En un estudio de interacción medicamentosa en sujetos sanos, la administración concomitante de atorvastatina 80 mg y amlodipina 10 mg resultó en un aumento del 18% en la exposición a atorvastatina, que no fue clínicamente significativa.
Niacina: El riesgo de efectos sobre el músculo esquelético puede ser aumentado cuando ATOZET se utiliza en combinación con niacina; se debe considerar una reducción en la dosis de ATOZET en este contexto.
Colchicina: Se han reportado casos de miopatía, incluyendo rabdomiólisis, con atorvastatina administrada concomitantemente con colchicina, y se debe tener precaución cuando se prescriba ATOZET con colchicina.
Daptomicina: El riesgo de miopatía y/o rabdomiólisis puede aumentar por la administración concomitante de inhibidores de la HMG-CoA reductasa y daptomicina.
DOSIS Y VÍA DE ADMINISTRACIÓN: Oral.
Generalidades
El paciente debe estar bajo una dieta hipolipemiante adecuada y debe continuar en esta dieta durante el tratamiento con ATOZET. La dosis debe ser individualizada de acuerdo con el nivel basal de C-LDL, la meta recomendada de la terapia y la respuesta del paciente. ATOZET se puede administrar como una sola dosis en cualquier momento del día, con o sin alimentos.
Adultos
Hipercolesterolemia Primaria y/o Enfermedad Cardíaca Coronaria
El rango de dosis de ATOZET es de 10/10 a 10/80 mg una vez al día. La dosis inicial recomendada de ATOZET es 10/10 o 10/20 mg una vez al día. Los pacientes que requieren una mayor reducción en CLDL (más del 55%) pueden comenzar con 10/40 mg una vez al día. Después del inicio y/o al hacer la valoración de ATOZET, los niveles de lípidos deben ser analizados dentro de 2 semanas o más y ajustar la dosis en consecuencia.
Dosis en Pacientes con Hipercolesterolemia Familiar Homocigótica
La dosis de ATOZET en pacientes con hipercolesterolemia familiar homocigótica es 10/40 o 10/80 mg al día. ATOZET debe usarse como un complemento a otros tratamientos hipolipemiantes (ej., aféresis de LDL) en estos pacientes o si esos tratamientos no están disponibles.
Pacientes Pediátricos
No se recomienda el tratamiento con ATOZET.
Pacientes Geriátricos
No se requiere ajuste de la dosis en pacientes de edad avanzada.
Insuficiencia Renal
No es necesario ajustar la dosis en pacientes con insuficiencia renal.
Coadministración con Secuestradores
de Ácidos Biliares
La dosis de ATOZET debe hacerse ya sea ≥ 2 horas antes o ≥ 4 horas después de la administración de un secuestrador de ácidos biliares.
Ciclosporina, Claritromicina, Itraconazol o Ciertos Agentes Antivirales VIH/VHC
En pacientes que toman ciclosporina o los inhibidores de proteasa del VIH tipranavir más ritonavir o el inhibidor de la proteasa de la hepatitis C telaprevir, se debe evitar la terapia con ATOZET. En los pacientes con VIH que toman lopinavir más ritonavir, se debe tener precaución cuando se prescriba ATOZET y emplear la menor dosis necesaria. En pacientes que toman claritromicina, itraconazol, o agentes antivirales de la hepatitis C boceprevir, elbasvir, grazoprevir, o en pacientes con VIH que toman una combinación de saquinavir más ritonavir, darunavir más ritonavir, fosamprenavir, o fosamprenavir más ritonavir, la terapia con ATOZET debe limitarse a 10/20 mg y se recomienda la evaluación clínica adecuada para asegurar que se usa la menor dosis necesaria de atorvastatina. En los pacientes que toman el inhibidor de la proteasa del VIH nelfinavir, la terapia con ATOZET debe limitarse a 10/40 mg y se recomienda la evaluación clínica apropiada para asegurar que se usa la menor dosis necesaria de ATOZET.
Otra Terapia Hipolipemiante Concomitante
No se recomienda la combinación de ATOZET y fibratos.
USO PEDIÁTRICO:
No hay datos suficientes para el uso seguro y eficaz de ATOZET en pacientes pediátricos.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No se puede recomendar un tratamiento específico para la sobredosis con ATOZET. En caso de una sobredosis, se deben aplicar medidas sintomáticas y de soporte.
Ezetimiba
En estudios clínicos, la administración de ezetimiba, 50 mg/día a 15 sujetos sanos hasta por 14 días, 40 mg/día a 18 pacientes con hiperlipidemia primaria hasta por 56 días y 40 mg/día a 27 pacientes con sitosterolemia homocigota por 26 semanas, fue generalmente bien tolerada.
Se han reportado unos pocos casos de sobredosis; la mayoría no se han asociado con experiencias adversas. Las experiencias adversas reportadas no han sido serias.
Atorvastatina
Debido a la unión extensa del medicamento a las proteínas plasmáticas, no se espera que la hemodiálisis mejore significativamente la depuración de atorvastatina.
PRESENTACIONES:
ATOZET 10 mg/10 mg: Caja por 30 tabletas recubiertas.
ATOZET 10 mg/20 mg: Caja por 30 tabletas recubiertas.
BIBLIOGRAFÍA:
En estudios clínicos controlados, ATOZET redujo significativamente el colesterol total (C-total), colesterol de lipoproteínas de baja densidad (C-LDL), apolipoproteína B (Apo B), triglicéridos (TG) y colesterol de lipoproteínas no de alta densidad (C no-HDL), y aumentó el colesterol de lipoproteínas de alta densidad (C-HDL) en pacientes con hipercolesterolemia.
Hipercolesterolemia Primaria
En un estudio clínico multicéntrico, doble ciego, controlado con placebo en pacientes con hiperlipidemia, 628 pacientes fueron tratados durante un máximo de 12 semanas y 246 de ellos hasta por 48 semanas adicionales. Los pacientes fueron aleatorizados para recibir placebo, ezetimiba (10 mg), atorvastatina (10 mg, 20 mg, 40 mg o 80 mg) o ezetimiba y atorvastatina co-administradas a dosis equivalentes a ATOZET (10/10, 10/20, 10/40, y 10/80) en el estudio de 12 semanas. Después de completar el estudio de 12 semanas, los pacientes elegibles fueron asignados a ezetimiba y atorvastatina co-administradas a dosis equivalentes a ATOZET (10/10-10/80) o atorvastatina (10-80 mg/día) por 48 semanas adicionales.
Los pacientes que recibieron todas las dosis de ATOZET se compararon con los que recibieron todas las dosis de atorvastatina. ATOZET redujo los valores de C total, C-LDL, Apo B, TG y C no-HDL, y aumentó el C-HDL significativamente más que la atorvastatina sola.
Tabla 2 Respuesta a Atozet en los Pacientes con Hiperlipidemia Primaria (Cambio Medioa % del Valor Basalb sin Tratamiento a las 12 semanas)
|
Tratamiento (Dosis diaria) |
N |
C-Total |
C-LDL |
APO B |
TGa |
C-HDL |
C-HDL |
|
Datos agrupados (todas las dosis de ATOZET)c |
255 |
-41 |
-56 |
-45 |
-33 |
+ 7 |
-52 |
|
Datos agrupados (todas las dosis de atorvastatina)c |
248 |
-32 |
-44 |
-36 |
-24 |
+4 |
-41 |
|
Ezetimiba 10 mg |
65 |
-14 |
-20 |
-15 |
-5 |
+4 |
-18 |
|
Placebo |
60 |
+4 |
+ 4 |
+ 3 |
-6 |
+4 |
+ 4 |
|
ATOZET por dosis |
|||||||
|
10/10 |
65 |
-38 |
-53 |
-43 |
-31 |
+ 9 |
-49 |
|
10/20 |
62 |
-39 |
-54 |
-44 |
-30 |
+ 9 |
-50 |
|
10/40 |
65 |
-42 |
-56 |
-45 |
-34 |
+ 5 |
-52 |
|
10/80 |
63 |
-46 |
-61 |
-50 |
-40 |
+ 7 |
-58 |
|
Atorvastatina por dosis |
|||||||
|
10 mg |
60 |
-26 |
-37 |
-28 |
-21 |
+ 6 |
-34 |
|
20 mg |
60 |
-30 |
-42 |
-34 |
-23 |
+ 4 |
-39 |
|
40 mg |
66 |
-32 |
-45 |
-37 |
-24 |
+ 4 |
-41 |
|
80 mg |
62 |
-40 |
-54 |
-46 |
-31 |
+ 3 |
-51 |
a Para triglicéridos, cambio Medio %del Valor Basal
b Línea base - ningún fármaco hipolipemiante
c ATOZET agrupado (10/10 - 10/80), redujo significativamente el C-total, C- LDL, Apo B, TG, C no-HDL y aumentó significativamente el C-HDL en comparación con todas las dosis de atorvastatina agrupadas (10-80 mg).
Los cambios en los criterios de valoración de lipídicos después de 48 semanas adicionales de tratamiento con ATOZET (todas las dosis) o con atorvastatina (todas las dosis) fueron generalmente consistentes con los datos de 12 semanas que se muestra arriba.
Se realizó un estudio multicéntrico, doble ciego, controlado, de 14 semanas en 621 pacientes con hipercolesterolemia familiar heterocigótica (HFHe), enfermedad cardiaca coronaria (ECC) o múltiples factores de riesgo cardiovascular (≥ 2), que se adhieren a una dieta NCEP Paso I o más estricta. Todos los pacientes recibieron atorvastatina 10 mg por un mínimo de 4 semanas antes de la aleatorización. Los pacientes fueron aleatorizados para recibir ya se ezetimiba y atorvastatina co-administradas (equivalente a ATOZET 10/10) o atorvastatina 20 mg/día en monoterapia. Los pacientes que no alcanzaron su meta de C-LDL después de 4 y/o 9 semanas de tratamiento aleatorizado se cambiaron al doble de la dosis de atorvastatina.
ATOZET 10/10 fue significativamente más eficaz que doblar la dosis de atorvastatina a 20 mg en la reducción adicional de C-total, C-LDL, TG y C no-HDL. Los resultados para C-HDL entre los dos grupos de tratamiento no fueron significativamente diferentes. (Ver Tabla 3). Además, en la Semana 4 significativamente más pacientes que recibieron ATOZET 10/10 alcanzaron un valor de C-LDL < 2.6 mmol/L (< 100 mg/dL) en comparación con los que recibieron atorvastatina 20 mg, 12% vs 2%. Los niveles basales promedios de C-LDL en los pacientes que recibieron ATOZET 10/10 y atorvastatina 20 mg fueron 186 mg/dL y 187 mg/dL, respectivamente.
Tabla 3 Respuesta de ATOZET después de 4 Semanas en Pacientes con ECC o Múltiples Factores de Riesgo Cardiovascular y un C-LDL ≥ 130 mg/dL (Cambio Medio* % del Valor Basal†
)
|
Tratamiento (Dosis diaria) |
N |
C-Total |
C-LDL |
C-HDL |
TGa |
C no-HDL |
|
ATOZET 10/10 |
305 |
-17 ‡ |
-24 ‡ |
+ 2 |
-9 ‡ |
-22 ‡ |
|
Atorvastatina 20 mg |
316 |
-6 |
-9 |
+ 1 |
-4 |
-8 |
* Para triglicéridos, cambio medio % del valor basal
† Pacientes tratados con atorvastatina 10 mg, luego cambiaron a ATOZET 10/10 o ajustados a atorvastatina 20 mg
‡ p < 0.05 diferencia con atorvastatina
El estudio de Valoración de Atorvastatina Versus Ezetimiba Añadida a Atorvastatina en Pacientes con Hipercolesterolemia (TEMPO), un estudio multicéntrico, doble ciego, controlado, de 6 semanas de duración, incluyó 184 pacientes con un nivel de C-LDL ≥ 2.6 mmol/L y ≤ 4.1 mmol/L (≥ 100 mg/dL y ≤ 160 mg/dL) y en riesgo moderado-alto de enfermedad arterial coronaria (ECC). Todos los pacientes recibieron atorvastatina 20 mg por un mínimo de 4 semanas antes de la aleatorización. Los pacientes que no estaban al nivel opcional NCEP ATP III para C-LDL (<2.6 mmol/L [< 100 mg/dL]) fueron aleatorizados para recibir ya sea ezetimiba y atorvastatina co-administradas (equivalente a ATOZET 10/20) o atorvastatina 40 mg durante 6 semanas.
ATOZET 10/20 fue significativamente más eficaz que doblar la dosis de atorvastatina a 40 mg en la reducción adicional de C-total, C-LDL, Apo B y C no-HDL. Los resultados para C-HDL y TG entre los dos grupos de tratamiento no fueron significativamente diferentes. (Ver Tabla 4). Además, significativamente más pacientes que recibieron ATOZET 10/20 alcanzaron un valor de C-LDL < 2.6 mmol/L (<100 mg/dL) en comparación con los que recibieron atorvastatina 40 mg, 84% vs 49%.
Tabla 4 Respuesta a la Atozet en los Pacientes con Hipercolesterolemia Primaria (Cambio Medioa % del Valor Basalb)
|
Tratamiento (Dosis diaria) |
N |
C-Total |
C-LDL |
Apo B |
C-HDL |
TG * |
C no-HDL |
|
ATOZET 10/10 |
92 |
20c |
-31c |
-21c |
+ 3 |
-18 |
-27 |
|
Atorvastatina 40 mg |
92 |
-7 |
-11 |
-8 |
+ 1 |
-6 |
-10 |
a Para triglicéridos, cambio medio % del valor basal
b Pacientes tratados con atorvastatina 20 mg, luego cambiaron a ATOZET 10/20 o ajustados a atorvastatina 40 mg
c p < 0.05 diferencia con atorvastatina
El estudio Ezetimiba más Atorvastatina Versus Valoración de Atorvastatina en la Consecución de Objetivos más Bajos de C-LDL en Pacientes Hipercolesterolémicos (EZ-PATH), un estudio multicéntrico, doble ciego, controlado, de 6 semanas de duración, incluyó 556 pacientes con un nivel de C-LDL ≥ 1.8 mmol/L y ≤ 4.1 mmol/L (≥ 70 mg/dL y ≤ 160 mg/dL) y en alto riesgo de enfermedad cardiaca coronaria (ECC). Todos los pacientes recibieron atorvastatina 40 mg por un mínimo de 4 semanas antes de la aleatorización. Los pacientes que no estaban al nivel opcional NCEP ATP III para C-LDL < 1.8 mmol/L (< 70 mg/dL) fueron aleatorizados para recibir ezetimiba y atorvastatina co-administradas (equivalente a ATOZET 10/40) o atorvastatina 80 mg durante 6 semanas.
ATOZET 10/40 fue significativamente más eficaz que doblar la dosis de atorvastatina a 80 mg en la reducción adicional de C-total, C-LDL, Apo B, TG y C no-HDL. Los resultados para C-HDL entre los dos grupos de tratamiento no fueron significativamente diferentes. (Ver Tabla 5). Además, significativamente más pacientes que recibieron ATOZET 10/40 alcanzaron un valor de C-LDL < 1.8 mmol/L (< 70 mg/dL) en comparación con los que recibieron atorvastatina 80 mg, 74% vs 32%.
Tabla 5 Respuesta de ATOZET en los Pacientes con Hipercolesterolemia Primaria (Cambio Medioa % del Valor Basalb)
|
Tratamiento (Dosis diaria) |
N |
C-Total |
C-LDL |
Apo B |
C-HDL |
TG * |
C no-HDL |
|
ATOZET 10/40 |
277 |
-17c |
-27c |
-81c |
0 |
-12c |
-23c |
|
Atorvastatina 80 mg |
279 |
-7 |
-11 |
-8 |
- 1 |
-6 |
-9 |
a Para triglicéridos, cambio medio % del valor basal
b Pacientes tratados con atorvastatina 40 mg, luego cambiaron a ATOZET 10/40 o ajustados a atorvastatina 80 mg
c p < 0.05 diferencia con atorvastatina
En un estudio doble ciego, controlado con placebo, de 8 semanas, 308 pacientes con hipercolesterolemia que ya recibían monoterapia con atorvastatina y que no estaban dentro de la meta de C-LDL del Programa Educacional Nacional de Colesterol (NCEP, por sus siglas en inglés) (meta de C-LDL basado en el nivel basal de C-LDL y el estatus de riesgo de ECC) se distribuyeron aleatoriamente para recibir ezetimiba 10 mg o placebo además de su terapia actual con atorvastatina.
Entre los pacientes tratados con atorvastatina no dentro de la meta de C-LDL al nivel basal (~83%), significativamente más pacientes aleatorizados a ezetimiba co-administrada con atorvastatina lograron su meta de C-LDL en el punto final del estudio en comparación con los pacientes aleatorizados a placebo co-administrado con atorvastatina, 72% vs. 27%. La ezetimiba añadida a la terapia con atorvastatina redujo el C-LDL significativamente más que el placebo añadido a la terapia con atorvastatina, 25% vs 4%. Además, ezetimiba añadida a la terapia con atorvastatina redujo significativamente el C-total, Apo B y TG en comparación con el placebo añadido a la terapia con atorvastatina.
En un estudio multicéntrico, doble ciego, controlado, de 12 semanas, de 2 fases, 1539 pacientes de alto riesgo cardiovascular, con un nivel de C-LDL entre 100 y 160 mg/dL al nivel basal con atorvastatina 10 mg al día, fueron aleatorizados a uno de tres grupos de tratamiento: atorvastatina 20 mg, rosuvastatina 10 mg o ATOZET 10/10. Después de 6 semanas de tratamiento (Fase I) siguiendo un esquema de asignación aleatoria establecido en el inicio de la Fase I, los pacientes que tomaron atorvastatina 20 mg, que no lograron alcanzar un nivel de C-LDL <100 mg/dL fueron cambiados a atorvastatina 40 mg o ATOZET 10/20 durante 6 semanas (Fase II), y pacientes similares que tomaron rosuvastatina 10 mg durante la Fase I fueron cambiados ya sea a rosuvastatina 20 mg o a ATOZET 10/20 durante la Fase II. Las reducciones de C-LDL y las comparaciones entre el grupo ATOZET y los otros grupos de tratamientoestudiados se muestran en la Tabla 6.
Tabla 6 Respuesta de ATOZET* en Pacientes de Alto Riesgo con un Nivel de C-LDL Entre 100 y 160 mg/dL en Atorvastatina 10 mg Diarios al Inicio
|
Tratamiento |
N |
Cambio Porcentual desde el Inicio† |
|||||
|
C-Total |
C-LDL |
Apo B |
TG ‡ |
C-HDL |
C no-HDL |
||
|
Fase I Cambio de atorvastatina 10 mg ATOZET 10/10 Atorvastatina 20 mg Rosuvastatina 10 mg |
120 480 939 |
-13.5 -6.4§ -7.7§ |
-22.2 -9.5§ -13.0§ |
-11.3 -6.0¶ -6.9# |
-6.0 -3.9 -1,1 |
+0.6 -1,1 + 1.1 |
-18.3 -8.1§ -10.6§ |
|
Fase II Cambio de atorvastatina 20 mg ATOZET 10/20 Atorvastatina 40 mg |
124 124 |
-10.7 -3.8 Þ |
-17.4 -6.9 Þ |
-9.8 -5.4 |
-5.9 -3,1 |
+0.7 +1.7 |
-15.1 -5.8 Þ |
|
Cambio de atorvastatina 10 mg ATOZET 10/20 Rosuvastatina 20 mg |
231 205 |
-11.8 -4,5Þ |
-17.1 -7.5 Þ |
-11.9 -4.1Þ |
-10.2 -3.2ß |
0.1 + 0,8 |
-16.2 -6.4 Þ |
* Ezetimiba y atorvastatina co-administrados equivalen a ATOZET 10/10 o 10/20
† M-estimaciones (basado en el método de Huber; el IC del 95% y valor de p se obtuvieron de ajustar un modelo de regresión robusto con términos para el tratamiento y el valor basal)
‡ Los cambios porcentuales de la media geométrica del valor basal en TG se calcularon basados en la transformación a través de la exponenciación de los modelos menos cuadrados (LS) medios y expresados como (media geométrica-1) multiplicado por 100
§ p < 0.001 versus ATOZET10/10
¶ p < 0.01 versus ATOZET10/10
# p < 0.05 versus ATOZET 10/10
Þ p < 0.001 versus ATOZET 10/20
ß p < 0.05 versus ATOZET 10/20
La Tabla 6 no contiene datos que comparan los efectos de ATOZET 10/10 o 10/20 a dosis superiores a atorvastatina 40 mg o rosuvastatina 20 mg.
Ezetimiba
En dos estudios multicéntricos, doble ciegos, controlados con placebo, de 12 semanas en 1719 pacientes con hipercolesterolemia primaria, ezetimiba redujo significativamente el C-total (–13%), C-LDL (–19%), Apo B (–14% ), TG (–8%) y C no-HDL (–17%), y aumentó el C-HDL (+3%) en comparación con el placebo. La reducción de C-LDL fue consistente en la edad, sexo y valor basal de C-LDL.
Atorvastatina
En un estudio controlado con placebo, el Ensayo Anglo-Escandinavo de Desenlaces Cardíacos (ASCOT, por sus siglas en inglés), el efecto de atorvastatina 10 mg sobre la enfermedad cardiaca coronaria fatal y no fatal se evaluó en 10,305 pacientes hipertensos, de 40 a 80 años de edad, con niveles de C-total ≤251 mg/dL (6.5 mmol/L) y al menos tres factores de riesgo cardiovasculares. Los pacientes fueron seguidos durante una media de 3.3 años. Atorvastatina 10 mg redujo significativamente (p=0.0005) la tasa de eventos coronarios (ya sea enfermedad cardíaca coronaria fatal [46 eventos en el grupo placebo versus 40 eventos en el grupo de atorvastatina] o IM no fatal [108 eventos en el grupo placebo versus 60 eventos en el grupo de atorvastatina]) en 36% (con base en la incidencia de 1.9% para atorvastatina versus 3.0% para placebo).
En un estudio controlado con placebo, el Estudio Cooperativo de Atorvastatina en Diabetes (CARDS, por sus siglas en inglés), el efecto de atorvastatina 10 mg en los criterios de valoración de enfermedades cardiovasculares (ECV) se evaluó en 2838 pacientes, de 40 a 75 años de edad, con diabetes tipo 2, uno o más factores de riesgo cardiovasculares, LDL ≤160 mg/dL y TG ≤600 mg/dL. Los pacientes fueron seguidos durante una media de 3.9 años. Atorvastatina 10 mg redujo significativamente (p<0.05) la tasa de eventos cardiovasculares mayores (ECVM) en 37%, el riesgo de ACV en 48% y el riesgo de IM en 42%.
Prevención de la Enfermedad Cardiovascular
En un estudio de ezetimiba/simvastatina, multicéntrico, aleatorizado, doble ciego, con control activo, 18,144 pacientes reclutados dentro de 10 días de hospitalización por síndrome coronario agudo (SCA, ya sea infarto agudo de miocardio [IM] o angina inestable [AI]). Todos los pacientes fueron asignados aleatoriamente en una relación 1:1 para recibir ya sea ezetimiba/simvastatina 10/40 mg (n=9067) o simvastatina 40 mg (n=9077) y fueron seguidos durante una media de 6.0 años.
Los pacientes tenían una edad media de 63.6 años; 76% eran hombres, 84% eran de raza caucásica y 27% eran diabéticos. El valor promedio de C-LDL al momento del evento calificante del estudio fue de 80 mg/dL (2.1 mmol/L) para los que estaban con terapia hipolipemiante (n=6390) y 101 mg/dL (2.6 mmol/L) para los que no estaban bajo terapia hipolipemiante previamente (n=11594). Antes de la hospitalización por el evento calificante de SCA, 34% de los pacientes estaban en tratamiento con estatinas. A un año, el valor promedio de C-LDL en los pacientes que continuaron con el tratamiento fue 53.2 mg/dL (1.4 mmol/L) para el grupo de ezetimiba/simvastatina y 69.9 mg/dL (1.8 mmol/L) para el grupo con simvastatina en monoterapia.
El criterio de valoración primario fue un compuesto que consistió en muerte cardiovascular, eventos coronarios mayores (ECM; definidos como infarto de miocardio no fatal, angina inestable documentada que requiere hospitalización o cualquier procedimiento de revascularización coronaria realizado por lo menos 30 días después de la asignación aleatoria al tratamiento) y ACV no fatal. El estudio demostró que el tratamiento con ezetimiba/simvastatina proporciona beneficio adicional en la reducción del criterio de valoración primario compuesto de muerte cardiovascular, ECM y ACV no fatal en comparación con simvastatina sola (reducción del riesgo relativo de 6.4%, p=0.016). El criterio de valoración primario ocurrió en 2572 de 9067 pacientes (tasa de Kaplan-Meier [KM] 32.72% a 7 años) en el grupo de ezetimiba/simvastatina y 2742 de 9077 pacientes (tasa de KM 34.67% a 7 años) en el grupo de simvastatina sola. (Ver Figura 1 y Tabla 7). Se espera que este beneficio adicional sea similar con la coadministración de ezetimiba y atorvastatina.
El efecto del tratamiento con ezetimiba/simvastatina fue generalmente consistente con los resultados globales para muchos subgrupos, incluyendo sexo, edad, raza, historia clínica de diabetes mellitus, niveles basales de lípidos, tratamiento previo con estatinas, ACV previo e hipertensión (ver Figura 2).
Figura 1 Efecto de ezetimiba/simvastatina en el Criterio de Valoración Primario Compuesto de Muerte Cardiovascular, Evento Coronario Mayor, o ACV no Fatal
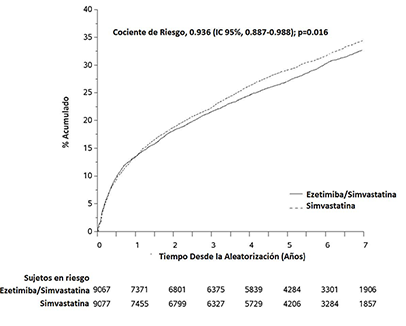
Figura 2 Análisis de Subgrupos del Criterio de Valoración Secundario Compuesto de Muerte Cardiovascular, Evento Coronario Mayor, o ACV no Fatal
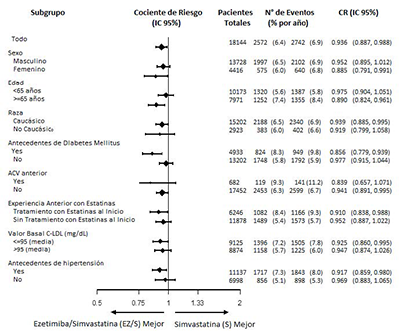
Tabla 7 Eventos Cardiovasculares Mayores por Grupo de Tratamiento en Todos los Pacientes Aleatorizados en IMPROVE-IT
|
Resultado |
Ezetimiba/ 10/40 mg * (N = 9067) |
Simvastatina 40 mg† (N = 9077) |
Cociente de Riesgo (IC 95%) |
valor p |
||
|
n |
K-M %‡ |
n |
K-M %‡ |
|||
|
Criterio de Valoración Primario de Eficacia Compuesto (Muerte CV, Eventos Coronarios Mayores y ACV no fatal) |
2572 |
32.72% |
2742 |
34.67% |
0.936 (0.887, 0.988) |
0.016 |
|
Componentes del Criterio de Valoración Primario Compuesto y Criterios de Valoración de Eficacia Seleccionados (las |
||||||
|
Muerte cardiovascular |
537 |
6.89% |
538 |
6,84% |
1.000 (0.887, 1.127) |
0.997 |
|
Evento Coronario Agudo |
||||||
|
IM no fatal |
945 |
12.77% |
1083 |
14.41% |
0.871 (0.798, 0.950) |
0.002 |
|
Angina inestable que requiere hospitalización |
156 |
2.06% |
148 |
1.92% |
1.059 (0.846, 1.326) |
0.618 |
|
Revascularización coronaria después de 30 días |
1690 |
21.84% |
1793 |
23.36% |
0.947 (0.886, 1.012) |
0.107 |
|
Accidente cerebrovascular no fatal |
245 |
3.49% |
305 |
4.24% |
0.802 (0.678, 0.949) |
0.010 |
* 6% fueron aumentado la dosis a ezetimiba/simvastatina 10/80 mg.
† 27% fueron aumentado la dosis a simvastatina 80 mg.
‡ Estimación de Kaplan-Meier a los 7 años.
En el Estudio Treating to New Targets (TNT), el efecto de atorvastatina 80 mg/día vs atorvastatina 10 mg/día en la reducción de eventos cardiovasculares se evaluó en 10,001 pacientes con evidencia clínica de ECC y un nivel de C-LDL <130 mg/dL mientras recibían atorvastatina 10 mg. Los pacientes fueron seguidos durante una media de 4.9 años. Atorvastatina 80 mg redujo significativamente (p≤0.05) la tasa de ECVM en 22%, IM no fatal no relacionado con el procedimiento en 22%, ACV fatal y no fatal en 25%, revascularización coronaria en 28%, angina en 12% y hospitalización por insuficiencia cardíaca en 26%.
En un estudio clínico multicéntrico, doble ciego, controlado con placebo, el estudio Myocardial Ischemia Reduction with Aggressive Cholesterol Lowering (MIRACL), los pacientes con un síndrome coronario agudo (IM no de onda Q o angina inestable) fueron aleatorizados para recibir atorvastatina 80 mg/día (n=1538) o placebo (n=1548). El tratamiento se inició durante la fase aguda después del ingreso hospitalario y se prolongó por un período de 16 semanas. La administración de atorvastatina 80 mg/día produjo una reducción de 16% (p=0.048) en el riesgo del criterio de valoración primario combinado, que fue definido como muerte por cualquier causa, IM no fatal, paro cardíaco resucitado o angina de pecho con evidencia de isquemia miocárdica que requirió hospitalización. Esto se basó principalmente en una reducción de 26% en la re-hospitalización por angina de pecho con evidencia de isquemia miocárdica (p=0.018).
En el Estudio Incremental Decrease in Endpoints Through Aggressive Lipid Lowering (IDEAL), el tratamiento con atorvastatina 80 mg/día se comparó con el tratamiento con simvastatina 20-40 mg/día en 8,888 pacientes de hasta 80 años de edad con antecedentes de ECC para evaluar si se podría alcanzar una reducción en el riesgo CV. En este ensayo del criterio de valoración, aleatorizado, prospectivo, abierto, ciego, los pacientes, 76% de los cuales estaban bajo tratamiento con estatinas en la aleatorización, fueron seguidos durante una media de 4.8 años. No hubo diferencia significativa entre los grupos de tratamiento para la tasa de primer evento coronario mayor (ECC fatal, IM no fatal y paro cardíaco resucitado): 411 (9.3%) en el grupo de atorvastatina 80 mg/día versus 463 (10.4%) en el grupo de simvastatina 20-40 mg/día o para la mortalidad por cualquier causa: 366 (8.2%) en el grupo de atorvastatina 80 mg/día vs 374 (8.4%) en el grupo de simvastatina 20-40 mg/día. Las proporciones de sujetos que experimentaron muerte CV o no CV fueron similares en el grupo de atorvastatina 80 mg y el grupo de simvastatina 20-40 mg.
Hipercolesterolemia Familiar Homocigótica (HFHo)
Se realizó un estudio doble ciego, aleatorizado, de 12 semanas en pacientes con un diagnóstico clínico y/o genotípico de HFHo. Se analizaron los datos de un subgrupo de pacientes (n=36) que recibían atorvastatina 40 mg al nivel basal. El aumento de la dosis de atorvastatina de 40 a 80 mg (n=12) produjo una reducción de C-LDL de 2% respecto del valor basal con atorvastatina 40 mg. La co-administración de ezetimiba y atorvastatina equivalente a ATOZET (dosis de 10/40 y 10/80 combinadas, n=24), produjo una reducción de C-LDL de 19% respecto al valor basal con atorvastatina 40 mg. En estos pacientes tratados concomitantemente con ezetimiba y atorvastatina equivalente a ATOZET (10/80, n=12) se produjo una reducción de C-LDL de 25% respecto al valor basal con atorvastatina 40 mg.
Después de completar el estudio de 12 semanas, los pacientes elegibles (n = 35), que estaban recibiendo atorvastatina 40 mg al inicio del estudio, fueron asignados a ezetimiba y atorvastatina co-administradas equivalente a ATOZET 10/40 hasta por 24 meses adicionales. Después de al menos 4 semanas de tratamiento, la dosis de atorvastatina podía ser duplicada hasta una dosis máxima de 80 mg. Al final de los 24 meses, ATOZET (dosis 10/40 y 10/80 combinadas) produjo una reducción de C-LDL que era consistente con la observada en el estudio de 12 semanas.
LEYENDAS DE PROTECCIÓN:
No se deje al alcance de los niños. Su venta requiere receta médica. Literatura exclusiva para médicos.
Hecho por:
SCHERING-PLOUGH LABO N.V.,
Heist-op-den-Berg – Bélgica.