
VARGATEF
NINTEDANIB
Cápsulas
Caja, 120 Cápsulas, 100 mg
Caja, 60 Cápsulas, 150 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN: VARGATEF® 100 mg: 1 CÁPSULA blanda contiene 100 mg de nintedanib (= base libre) correspondiente a 120,4 mg de 1H-Indol-6-ácido carboxílico, 2,3-dihidro-3-[[[4-[metil[(4-metil-1-piperazinil)acetil] amino]fenil] amino]fenilmetilen]-2-oxo-, metil éster, (3Z)-, etanosulfonato (1:1) (= nintedanib esilato).
VARGATEF® 150 mg: 1 CÁPSULA blanda contiene 150 mg nintedanib (= base libre) correspondiente a 180,6 mg de 1H-Indol-6-ácido carboxílico, 2,3-dihidro-3-[[[4-[metil[(4-metil-1-piperazinil)acetil] amino]fenil]amino]fenilmetilen]-2-oxo-, metil éster, (3Z)-, etanosulfonato (1:1) (= nintedanib esilato).
USO EN POBLACIONES ESPECÍFICAS:
• Embarazo, lactancia y fertilidad:
Anticoncepción: Nintedanib puede causar daño fetal (véase la sección Toxicología). Debe advertirse a las mujeres con potencial para procrear que eviten quedar embarazadas mientras reciben tratamiento con VARGATEF® y que deben usar métodos anticonceptivos altamente eficaces durante el tratamiento con VARGATEF® y hasta cumplidos al menos 3 meses desde la última dosis de este fármaco. Actualmente se desconoce si nintedanib puede reducir la eficacia de los anticonceptivos hormonales y, por ende, las mujeres que usen tales anticonceptivos deben agregar un método de barrera.
Embarazo: No existe información sobre el uso de VARGATEF® en las mujeres embarazadas; sin embargo, los estudios preclínicos en animales han confirmado la toxicidad para la reproducción con este fármaco (véase la sección Toxicología). Dado que nintedanib puede causar daño fetal también en los seres humanos, este fármaco no debe ser utilizado durante el embarazo (véase la sección Contraindicaciones), y deben realizarse pruebas de embarazo antes del inicio del tratamiento con VARGATEF® y durante el tratamiento, según corresponda.
Se debe indicar a las pacientes que deben notificar a su médico o farmacéutico si quedaran embarazadas durante el tratamiento con VARGATEF®.
Si la paciente quedara embarazada mientras está recibiendo VARFATEF®, debe interrumpirse el tratamiento y la paciente deberá ser asesorada sobre el potencial riesgo para el feto.
Lactancia: No existe información sobre la eliminación de nintedanib y sus metabolitos en la leche humana.
Los estudios preclínicos indicaron que se segregaron pequeñas cantidades de nintedanib y sus metabolitos (≤ 0,5 % de la dosis administrada) en la leche de las ratas en período de lactancia.
No se puede excluir la posibilidad de un riesgo para los neonatos/lactantes. Debe suspenderse la lactancia durante el tratamiento con VARGATEF®.
Fertilidad: Sobre la base de las investigaciones preclínicas, no existe evidencia de que este fármaco afecte la fertilidad masculina (véase la sección Toxicología). A partir de los estudios de toxicidad de administración crónica y subcrónica, no existe evidencia de que la fertilidad en las ratas hembras se vea afectada en un nivel de exposición sistémica comparable al observado con la dosis recomendada máxima en los seres humanos (MRHD), de 200 mg dos veces al día (véase la sección Toxicología).
Para obtener información de docetaxel relacionada con la fertilidad, el embarazo y la lactancia, sírvase consultar la información del producto correspondiente de docetaxel.
PROPIEDADES FARMACOLÓGICAS: Grupo farmacoterapéutico: Agentes antineoplásicos - Inhibidores de la proteína cinasa de tirosina.
Código ATC: L01EX09
Mecanismo de acción: Nintedanib es un triple inhibidor de la angiocinasa que actúa bloqueando la actividad de la cinasa de los receptores del factor de crecimiento endotelial vascular (VEGFR 1-3), de los receptores de factor de crecimiento derivado de plaquetas (PDGFR α y ß) y de los receptores de factor de crecimiento de fibroblastos (FGFR 1-3). Nintedanib se une competitivamente al sitio de unión a la adenosina trifosfato (ATP) de estos receptores y bloquea la señalización intracelular que es crucial para la proliferación y la supervivencia de las células endoteliales y perivasculares (pericitos y células de la musculatura lisa vascular). También resultan inhibidas la proteína cinasa de tirosina pseudo Fms (Flt)-3, la proteína cinasa de tirosina específica de linfocitos (Lck) y la proteína cinasa de tirosina del proto-oncogén Src (Src).
Efectos farmacodinámicos: La angiogénesis tumoral es un factor esencial que contribuye al crecimiento tumoral, la progresión y la formación de metástasis, y es desencadenada principalmente por la liberación de factores proangiogénicos secretados por las células tumorales (es decir, VEGF y bFGF) para atraer a las células endoteliales y también perivasculares del huésped, para promover el suministro de oxígeno y nutrientes a través del sistema vascular del huésped. En los modelos preclínicos de la enfermedad, nintedanib, como agente único, interfirió efectivamente con la formación y el mantenimiento del sistema vascular tumoral, lo que condujo a la inhibición del crecimiento tumoral y la estasis tumoral. En particular, el tratamiento de xenoinjertos tumorales con nintedanib condujo a una rápida reducción de la densidad microvascular del tumor, la cobertura vascular de los pericitos y la perfusión tumoral.
Las mediciones realizadas mediante resonancia magnética dinámica con realce de contraste (DCE-MRI) indicaron un efecto antiangiogénico de nintedanib en los seres humanos. Dicho efecto no fue claramente dependiente de la dosis, pero la mayoría de las respuestas se observaron con dosis de ≥ 200 mg. El análisis de regresión logística reveló una asociación estadísticamente significativa del efecto antiangiogénico con la exposición a nintedanib. Los efectos detectados mediante DCE-MRI se observaron 24-48 hs después de la primera toma del medicamento y se mantuvieron o incluso se incrementaron luego del tratamiento continuado a lo largo de varias semanas. No se determinó ninguna correlación de la respuesta de DCE-MRI y la posterior reducción clínicamente significativa observada en el tamaño de la lesión objetivo, pero la respuesta de DCE-MRI estuvo asociada con la estabilización de la enfermedad.
Ensayos Clínicos:
— Eficacia en el estudio pivote de fase III LUME-Lung 1
La eficacia y la seguridad de VARGATEF® se investigaron en 1314 pacientes con NSCLC localmente avanzado, metastásico o recurrente tras una línea de quimioterapia previa. El estudio incluyó 658 pacientes (50,1 %) con adenocarcinoma, 555 pacientes (42,2%) con carcinoma escamocelular y 101 pacientes (7,7%) con otras histologías tumorales.
Los pacientes fueron aleatorizados (1:1) a recibir VARGATEF® 200 mg por vía oral dos veces al día en combinación con 75 mg/m2 de docetaxel i.v. cada 21 días (n = 655), o bien placebo por vía oral dos veces al día en combinación con 75 mg/m2 de docetaxel cada 21 días (n = 659). La aleatorización se estratificó de acuerdo con el estado del Grupo Oncológico Cooperativo del Este (Eastern Cooperative Oncology Group, ECOG) (0 vs. 1), pretratamiento con bevacizumab (sí vs. no), metástasis cerebrales (sí vs. no) e histología tumoral (histología escamosa vs. no escamosa).
Las características de los pacientes estuvieron equilibradas entre los grupos de tratamiento dentro de la población general del estudio y dentro del subgrupo de pacientes con adenocarcinoma. En la población general, el 72,7% de los pacientes eran de sexo masculino. La mayoría de los pacientes eran de raza no asiática (81,6%), la mediana de la edad fue 60,0 años, la categoría de estado funcional ECOG inicial fue 0 (28,6%) o 1 (71,3%); uno de los pacientes tenía estado funcional ECOG 2 en el nivel inicial. El 5,8% de los pacientes tenían metástasis cerebral estable al momento del ingreso al estudio y el 3,8% había sido tratado anteriormente con bevacizumab.
El estadio de la enfermedad se determinó al momento del diagnóstico sobre la base de los criterios de la Unión Internacional Contra el Cáncer (Union Internationale Contre le Cancer, UICC) / Comité Conjunto de Cáncer de Estados Unidos (American Joint Committee on Cancer, AJCC) Edición 6 o Edición 7. En la población general, el 16,0 % de los pacientes tenía enfermedad en estadio < IIIB/IV, el 22,4 % tenía enfermedad en estadio IIIB y el 61,6% tenía enfermedad en estadio IV. El 9,2% de los pacientes ingresó al estudio con estadio de enfermedad localmente recurrente tal como habían sido evaluados en el nivel inicial. De los pacientes con histología tumoral de adenocarcinoma, el 15,8% tenía enfermedad en estadio < IIIB/IV, el 15,2% tenía enfermedad en estadio IIIB y el 69,0% tenía enfermedad en estadio IV. El 5,8% de los pacientes con adenocarcinoma ingresó al estudio con estadio de enfermedad localmente recurrente tal como habían sido evaluados en el nivel inicial. “Localmente recurrente” se definió como la reaparición local del tumor sin metástasis al momento del ingreso en el estudio.
El criterio de valoración primario fue la supervivencia libre de progresión (PFS), según lo evaluado por un comité de revisión independiente (IRC), basada en la población por intención de tratar (ITT) y determinada mediante histología. La supervivencia general (OS) fue el criterio de valoración secundario clave. Otros parámetros de eficacia fueron la respuesta objetiva, el control de la enfermedad, el cambio en el tamaño del tumor y la calidad de vida relacionada con la salud.
Tal como se puede apreciar en la Tabla 5, la adición de VARGATEF® al docetaxel condujo a una reducción estadísticamente significativa del 21% en el riesgo de progresión o muerte para la población general del estudio (HR: 0,79; IC del 95 %: 0,68 - 0,92; p = 0,0019) según lo determinado por el IRC. Este resultado fue confirmado en el análisis de PFS de seguimiento (HR: 0,85, IC del 95%: 0,75 - 0,96; p = 0,0070), el cual incluyó todos los eventos obtenidos al momento del análisis de OS final. El análisis de supervivencia general realizado en la población general no alcanzó la significancia estadística (HR 0,94; IC 95 %: 0,83 - 1,05).
Es digno de mención el hecho de que los análisis previamente planificados en función de la histología indicaron una diferencia estadísticamente significativa en la OS entre los grupos de tratamiento únicamente en la población de pacientes con adenocarcinoma.
La adición de VARGATEF® al docetaxel condujo a una reducción estadísticamente significativa del 23 % en el riesgo de progresión o muerte para la población con adenocarcinoma (HR: 0,77; IC del 95 %: 0,62 - 0,96). En concordancia con estas observaciones, los criterios de valoración relacionados del estudio, tales como el control de la enfermedad y el cambio en el tamaño tumoral, evidenciaron mejorías significativas.
|
Tabla 5. Resultados de eficacia para el estudio LUME-Lung 1 para todos los pacientes y para los pacientes con histología tumoral de tipo adenocarcinoma |
||||
|
Todos los pacientes |
Histología tumoral de tipo adenocarcinoma |
|||
|
Vargatef (n = 565) |
Placebo (n = 569) |
Vargatef (n = 277) |
Placebo (n = 285) |
|
|
Supervivencia libre de progresión (PFS)* |
||||
|
Cantidad de muertes o progresiones, n (%) |
339 (60,0) |
375 (65,9) |
152 (54,9) |
180 (63,2) |
|
Mediana de PFS [meses] |
3,4 |
2,7 |
4,0 |
2,8 |
|
HR (IC del 95 %) ** |
0,79 (0,68; 0,92) |
0,77 (0,62; 0,96) |
||
|
Valor p de la prueba de rangos logarítmicos estratificada** |
0,0019 |
0,0193 |
||
|
Control de la enfermedad [%] |
48,5 |
37,6 |
60,6 |
43,9 |
|
Cociente de probabilidades (IC 95 %)+ |
1,56 (1,23; 1,98) |
1,98 (1,41; 2,77) |
||
|
Valor p+ |
0,0002 |
<0,0001 |
||
|
Respuesta objetiva [%] |
3,4 |
1,9 |
4,3 |
3,5 |
|
Cociente de probabilidades (IC 95 %)+ |
1,77 (0,85; 3,89) |
1,25 (0,53; 3,01) |
||
|
Valor p+ |
0,1283 |
0,6122 |
||
|
Media ajustada del mejor cambio porcentual del tamaño tumoral respecto del nivel inicial [%] |
-3,93 |
1,15 |
-7,38 |
-0,28 |
|
Valor p° |
0,0002 |
0,0002 |
||
|
Supervivencia general (OS)*** |
(n = 655) |
(n = 659) |
(n = 322) |
(n = 336) |
|
Cantidad de eventos de OS, n (%) |
564 (86,1) |
557 (84,5) |
259 (80,4) |
276 (82,1) |
|
Mediana de OS [meses] |
10,1 |
9,1 |
12,6 |
10,3 |
|
HR (IC del 95 %) |
0,94 (0,83; 1,05) |
0,83 (0,70; 0,99) |
||
|
Valor p de la prueba de rangos logarítmicos estratificada* |
0,2720 |
0,0359 |
||
|
* Análisis de PFS primario basado en un total de 713 eventos de PFS en la población general del estudio. ** Estratificado por puntaje de PS de ECOG inicial (0 versus 1), metástasis cerebrales en el nivel inicial (sí versus no) y tratamiento previo con bevacizumab (sí versus no), y en la población de todos los pacientes también se aplicó estratificación por histología tumoral (escamoso versus no escamoso). *** Análisis de OS basado en un total de 1121 muertes en la población general del estudio. + El cociente de probabilidades y el valor p se obtienen a partir de un modelo de regresión logística con ajuste para el puntaje de desempeño de ECOG inicial (0 versus 1) y en la población de todos los pacientes se aplica un ajuste adicional para histología tumoral (escamoso versus no escamoso). ° La media ajustada del mejor cambio porcentual respecto del nivel inicial y valor p se generan a partir de un modelo ANOVA con ajuste para PS de ECOG inicial (0 versus 1), metástasis cerebrales en el nivel inicial (sí versus no) y tratamiento previo con bevacizumab (sí versus no). En la población de todos los pacientes además se aplicó un ajuste por histología tumoral (escamoso versus no escamoso). Un paciente (135301) tuvo una PS de ECOG inicial de 2. |
||||
Se demostró una mejoría estadísticamente significativa en la OS a favor del tratamiento con VARGATEF® más docetaxel en los pacientes con adenocarcinoma con una reducción del 17% en el riesgo de muerte (HR 0,83, p = 0,0359) y una mediana de mejoría de la OS de 2,3 meses (10,3 versus 12,6 meses, Figura 1).
Figura 1. Curva de Kaplan-Meier para la supervivencia general para los pacientes con histología tumoral de adenocarcinoma, por grupo de tratamiento, en el estudio LUME-Lung 1
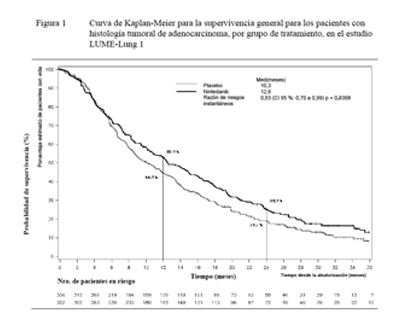
Se llevó a cabo una evaluación previamente especificada en la población de pacientes con adenocarcinoma que habían ingresado en el estudio con una prognosis de tratamiento particularmente desfavorable, es decir, pacientes que tuvieron progresión durante la terapia de primera línea o poco después de ella, antes de su ingreso al estudio. Esta población incluyó a los pacientes con adenocarcinoma identificados en el nivel inicial como pacientes que habían tenido progresión y que habían ingresado al estudio menos de 9 meses después del inicio de su terapia de primera línea. El tratamiento de estos pacientes con VARGATEF® en combinación con docetaxel redujo el riesgo de muerte en un 25%, en comparación con el tratamiento de placebo más docetaxel (HR 0,75; IC del 95 %: 0,60 - 0,92; p = 0,0073). La mediana de OS mejoró a razón de 3 meses (Vargatef®: 10,9 meses; placebo: 7,9 meses).
En un análisis post-hoc en pacientes con adenocarcinoma que tuvieron progresión e ingresaron al estudio ≥ 9 meses después del inicio de su terapia de primera línea, la diferencia no alcanzó una significancia estadística (HR para OS: 0,89, IC del 95%: 0,66 - 1,19).
La proporción de pacientes con adenocarcinoma en estadio < IIIB/IV al momento del diagnóstico fue reducida y estuvo equilibrada entre los grupos de tratamiento (placebo: 54 pacientes (16,1%); VARGATEF®: 50 pacientes, (15,5 %)). La HR para estos pacientes para la PFS y la OS fue 1,24 (IC del 95 %: 0,68; 2,28) y 1,09 (IC del 95 %: 0,70; 1,70), respectivamente. Sin embargo, el tamaño de la muestra fue reducido, no hubo ninguna interacción significativa y el intervalo de confianza fue amplio e incluyó la HR para la OS de la población general de pacientes con adenocarcinoma.
Calidad de vida: El tratamiento con VARGATEF® no modificó significativamente el tiempo hasta el deterioro de los síntomas previamente especificados de tos, disnea y dolor, pero condujo a un deterioro significativo en la escala de síntomas de diarrea. No obstante ello, se observó un beneficio general del tratamiento con nintedanib sin que se viera afectada negativamente la calidad de vida autoinformada.
Efecto sobre el intervalo QT: Se efectuaron y se analizaron mediciones de QT/QTc a partir de un estudio específico en el cual se comparó la monoterapia de nintedanib frente a la monoterapia de sunitinib en pacientes con carcinoma renal. En este estudio, dosis únicas orales de 200 mg de nintedanib y dosis múltiples orales de 200 mg de nintedanib administradas dos veces al día durante 15 días no prolongaron el intervalo QTcF.
Sin embargo, no se llevó a cabo ningún estudio exhaustivo en relación con el intervalo QT para nintedanib administrado en combinación con docetaxel.
Población pediátrica: No se han llevado a cabo estudios clínicos en niños ni adolescentes.
• Farmacocinética:
La farmacocinética (PK) de nintedanib puede considerarse lineal en relación con el tiempo (es decir, los datos de las dosis únicas pueden extrapolarse a datos de dosis múltiples). La acumulación observada tras la administración de dosis múltiples fue de 1,04 veces para la Cmáx y de 1,38 veces para el AUCτ. Las concentraciones valle de nintedanib se mantuvieron estables durante más de un año.
Absorción: Nintedanib alcanzó las concentraciones plasmáticas máximas aproximadamente 2 - 4 horas después de la administración por vía oral como cápsulas de gelatina blanda en estado posprandial (rango: 0,5 - 8 horas. La biodisponibilidad absoluta de una dosis de 100 mg fue 4,69 % (IC del 90 %: 3,615 - 6,078) en voluntarios sanos. La absorción y la biodisponibilidad se ven reducidas por los efectos de los transportadores y por un grado sustancial de metabolismo de primer paso.
La proporcionalidad a la dosis se demostró a través del incremento de la exposición a nintedanib (rango de dosis de 50 - 450 mg una vez al día y de 150 - 300 mg dos veces al día). Las concentraciones plasmáticas en estado de equilibrio dinámico se lograron dentro de un lapso de administración de una semana como máximo.
Tras la ingesta de alimentos, la exposición a nintedanib se incrementó aproximadamente un 20% en comparación con la administración en ayunas (IC: 95,3 - 152,5 %) y la absorción fue más lenta (mediana de tmáx; en ayunas: 2,00 horas; en estado posprandial: 3,98 horas).
Distribución: Nintedanib sigue una cinética de disposición como mínimo bifásica. Tras la infusión intravenosa, se observó un importante volumen de distribución (Vss: 1050 L, 45,0 % gCV).
El grado de unión a las proteínas de nintedanib observado in vitro en el plasma humano fue elevado, con una fracción ligada del 97,8 %. Se considera que la albúmina sérica es la principal proteína de unión. Nintedanib se distribuye preferentemente en el plasma, con una relación sangre: plasma de 0,869.
Biotransformación: La reacción metabólica prevalente en el caso de nintedanib es la escisión hidrolítica por esterasas, que conduce a la formación de la fracción ácido libre BIBF 1202. BIBF 1202 luego es glucuronizado por las enzimas UGT (a saber, UGT 1A1, UGT 1A7, UGT 1A8 y UGT 1A10) con la consecuente transformación en el BIBF 1202 glucurónido.
Sólo un grado mínimo de la biotransformación de nintedanib estuvo relacionada con las vías del CYP, siendo CYP 3A4 la enzima predominante en dicho proceso. El principal metabolito dependiente de CYP no pudo ser detectado en el plasma en el estudio de absorción, distribución, metabolismo y eliminación (ADME) en humanos. In vitro, el metabolismo dependiente de CYP representó aproximadamente un 5 % en comparación con alrededor de un 25 % en el caso de la escisión de ésteres.
En los experimentos in vivo preclínicos, BIBF 1202 no evidenció eficacia a pesar de su actividad en los receptores que son el blanco de acción del fármaco.
Eliminación: La depuración plasmática total tras la administración por infusión intravenosa fue elevada (CL: 1390 ml/min, 28,8 % gCV). La eliminación urinaria del principio activo inalterado dentro de las 48 horas fue de aproximadamente el 0,05 % de la dosis (gCV 31,5 %) tras la administración por vía oral, y de aproximadamente el 1,4 % de la dosis (gCV 24,2 %) tras la administración por vía intravenosa; la depuración renal fue 20 ml/min (gCV 32,6 %). La principal vía de eliminación de la radioactividad relacionada con el fármaco tras la administración por vía oral de [14C] nintedanib fue la excreción fecal/biliar (93,4% de la dosis, gCV 2,61%). La contribución de la eliminación renal a la depuración total fue baja (0,649 % de la dosis, gCV 26,3%). La recuperación total se consideró completa (superior al 90%) dentro de los 4 días subsiguientes a la administración. La vida media terminal de nintedanib fue de entre 10 y 15 horas (% gCV aprox. 50%;).
Relación exposición-respuesta: En los análisis exploratorios de la relación farmacocinética (PK)-eventos adversos, una mayor exposición a nintedanib tendió a estar asociada con elevaciones en las enzimas hepáticas, pero no con eventos adversos gastrointestinales.
No se efectuaron análisis de farmacocinética-eficacia para los criterios de valoración clínicos. El análisis de regresión logística reveló una asociación estadísticamente significativa entre la exposición a nintedanib y la respuesta de DCE-MRI.
Factores intrínsecos y extrínsecos. Poblaciones especiales: Las propiedades farmacocinéticas de nintedanib fueron similares en los voluntarios sanos, en los pacientes con FPI, pacientes con SSc-ILD y en los pacientes oncológicos. Sobre la base de los resultados de los análisis de farmacocinética poblacional y las investigaciones descriptivas, la exposición a nintedanib no se vio influenciada por el sexo (con corrección para peso corporal), la existencia de una insuficiencia renal leve o moderada (estimada sobre la base de la depuración de creatinina), la presencia de metástasis hepáticas, el puntaje de estado funcional ECOG, el consumo de alcohol ni el genotipo de P-gp. Los análisis de farmacocinética poblacional indicaron efectos moderados sobre la exposición a nintedanib dependientes de la edad, el peso corporal y la raza (véase a continuación). Sobre la base de la elevada variabilidad entre individuos de la exposición que se observó en los estudios clínicos, estos efectos no se consideran clínicamente relevantes. (véase la sección Advertencias y precauciones especiales).
Edad: La exposición a nintedanib se incrementó en forma lineal en función de la edad. Los valores de AUCτ,ss evidenciaron una reducción del 16 % para un paciente de 45 años (percentilo 5) y se incrementaron a razón de un 13 % para un paciente de 76 años (percentilo 95) respecto de un paciente con una mediana de edad de 62 años. El rango de edad cubierto por el análisis fue de 29 a 85 años; aproximadamente el 5 % de la población era mayor de 75 años.
No se han efectuado estudios en poblaciones pediátricas.
Peso corporal: Se observó una correlación inversa entre el peso corporal y la exposición a nintedanib. Los valores de AUCτ,ss se incrementaron a razón de un 25% para un paciente de 50 kg (percentilo 5) y se redujeron a razón de un 19 % para un paciente de 100 kg (percentilo 95) respecto de un paciente con una mediana de peso de 71,5 kg.
Raza: La exposición media de la población a nintedanib fue 33 50 % más alta en los pacientes procedentes de China, Taiwán e India y un 16 % más alta en los pacientes japoneses, en tanto que fue 16 22 % más baja en los pacientes de Corea, en comparación con los caucásicos (con corrección para peso corporal).
Los datos obtenidos a partir de sujetos de raza negra fueron muy limitados, pero se ubicaron dentro del mismo rango que aquellos de los sujetos caucásicos.
Insuficiencia hepática: En un estudio específico de dosis única de fase I en el que se tomó como blanco a sujetos sanos, la exposición a nintedanib, considerando tanto la Cmáx como el AUC, fue 2,2 veces mayor en voluntarios con insuficiencia hepática leve (Child Pugh A; IC del 90 % de la Cmáx: 1,3 - 3,7; IC del 90 % del AUC: 1,2 - 3,8, respectivamente). En voluntarios con insuficiencia hepática moderada (Child Pugh B), la exposición fue 7,6 veces mayor en términos de la Cmáx (IC del 90 %: 4,4 - 13,2) y 8,7 veces mayor en términos del AUC (IC del 90 %: 5,7 - 13,1), respectivamente, en comparación con pacientes sanos. No se estudiaron sujetos con insuficiencia hepática grave (Child Pugh C).
• Potencial de interacciones medicamentosas:
Metabolismo: No es dable esperar que se produzcan interacciones medicamentosas entre nintedanib y los sustratos del CYP, los inhibidores del CYP o los inductores del CYP, ya que nintedanib, BIBF 1202 y el glucurónido BIBF 1202 no evidenciaron efectos de inhibición ni de inducción de las enzimas del CYP en los ensayos preclínicos y nintedanib no fue metabolizado en un grado relevante por las enzimas del CYP.
Transporte: Nintedanib es un sustrato de la P-gp. Para el potencial de interacción de nintedanib con este transportador, véase la sección Interacciones. Se ha comprobado que nintedanib no es un sustrato ni un inhibidor de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 o MRP-2 in vitro. Nintedanib tampoco fue un sustrato de la proteína de resistencia al cáncer de mama (BCRP). Sólo se observó un débil potencial inhibidor sobre OCT-1, BCRP y P-gp in vitro, el cual se considera de escasa relevancia clínica. Lo mismo aplica a nintedanib en cuanto a ser un sustrato de OCT-1.
CONTRAINDICACIONES: VARGATEF® está contraindicado en pacientes con hipersensibilidad conocida a nintedanib, al maní o a la soja o a cualquiera de sus excipientes.
VARGATEF® está contraindicado durante el embarazo (véanse las secciones Embarazo, Lactancia y fertilidad, Toxicología).
Para obtener información sobre las contraindicaciones de docetaxel, sírvase consultar la información del producto correspondiente de docetaxel.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR VEHÍCULOS Y OPERAR MAQUINARIA: No se han llevado a cabo estudios de los efectos de este fármaco sobre la capacidad para conducir vehículos y operar maquinaria.
Debe indicarse a los pacientes que deben tener precaución al conducir vehículos u operar maquinaria durante el tratamiento con VARGATEF®.
REACCIONES ADVERSAS: Resumen del perfil de seguridad: Los datos de seguridad que se brindan a continuación están basados en un estudio global, doble ciego, aleatorizado, pivote, de fase III, el estudio 1199.13 (LUME-Lung 1), en el cual se comparó el tratamiento con VARGATEF® más docetaxel frente a un placebo más docetaxel en pacientes con NSCLC localmente avanzado o metastásico o recurrente tras la quimioterapia de primera línea y también se basan en los datos observados durante el período posterior a la comercialización de nintedanib. Las reacciones adversas específicas de VARGATEF® informadas con mayor frecuencia fueron diarrea, elevación de los valores de las enzimas hepáticas (ALT y AST) y vómitos. En la Tabla 3 se brinda un resumen de las reacciones adversas, ordenadas por Clasificación por sistema y órgano (SOC).
Para el manejo de las reacciones adversas seleccionadas, véase la sección Advertencias y precauciones especiales.
Resumen tabulado de reacciones adversas:
|
Tabla 3. Resumen de reacciones adversas |
|
|
Terminología de la clasificación por sistema y órgano del MedDRA |
Reacciones adversas de nintedanib |
|
Trastornos gastrointestinales |
Diarrea Vómitos Náuseas Dolor abdominal Perforación2) Pancreatitis3) |
|
Trastornos hepatobiliares |
Lesión hepática producida por medicamentos Elevación de las enzimas hepáticas Alanina aminotransferasa (ALT) Aspartato aminotransferasa (AST) Fosfatasa alcalina (ALKP) Gamma glutamiltransferasa (GGT) Hiperbilirrubinemia |
|
Trastornos vasculares |
Hipertensión Tromboembolia venosa Sangrado2) |
|
Trastornos de la sangre y del sistema linfático |
Neutropenia1) Trombocitopenia |
|
Infecciones e infestaciones |
Septicemia1) Neutropenia febril1) Abscesos |
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito Deshidratación Desequilibrio electrolítico Descenso de peso |
|
Trastornos del sistema nervioso |
Neuropatía periférica1) |
|
Trastornos de la piel y del tejido subcutáneo |
Mucositis1), incl. estomatitis Exantema, Prurito |
|
1) Sírvase consultar también la información del producto de docetaxel. 2) La frecuencia no resultó incrementada en los pacientes tratados con nintedanib más docetaxel en comparación con placebo más docetaxel. 3) Se han informado eventos de pancreatitis en pacientes que toman nintedanib para el tratamiento de la FPI y NSCLC. La mayoría de estos eventos se informó en los pacientes tratados por la indicación FPI. |
|
INTERACCIONES: Glicoproteína P (P-gp): Nintedanib es un sustrato de la P-gp (véase la sección Farmacocinética). La coadministración con ketoconazol, un inhibidor potente de la P-gp, incrementó la exposición a nintedanib por un factor de 1,61 sobre la base del AUC y por un factor de 1,83 sobre la base de la Cmáx en un estudio de interacciones medicamentosas específico.
En un estudio de interacciones medicamentosas realizado con rifampicina, un potente inductor de la P-gp, la exposición a nintedanib se redujo a un 50,3% sobre la base del AUC y a un 60,3% sobre la base de la Cmáx ante la coadministración con rifampicina en comparación con la administración de nintedanib solo.
Si se coadministran junto con VARGATEF®, los inhibidores potentes de la P-gp (p. ej., ketoconazol o eritromicina) pueden incrementar la exposición a nintedanib. En tales casos, debe implementarse un control estrecho de los pacientes a fin de determinar la tolerabilidad a nintedanib. El manejo de las reacciones adversas puede requerir la reducción de la dosis o bien la suspensión temporaria o definitiva del tratamiento con VARGATEF® (véase la sección Posología y administración).
Los inductores potentes de la P-gp (p. ej., rifampicina, carbamazepina, fenitoína y hierba de San Juan) pueden reducir la exposición a nintedanib. Debe considerarse la elección de otra medicación concomitante alternativa que tenga un potencial de inducción de la P-gp nulo o mínimo.
Alimentos: Se recomienda que VARGATEF® se administre con alimentos (véase la sección Farmacocinética).
Enzimas del citocromo (CYP): Sólo una pequeña parte de la biotransformación de nintedanib involucró las vías del CYP. Nintedanib y sus metabolitos, la fracción ácido libre BIBF 1202 y su glucurónido BIBF 1202 glucurónido, no inhibieron ni indujeron las enzimas del CYP en los estudios preclínicos, (véase la sección Farmacocinética). Por lo tanto, se considera que la probabilidad de que se produzcan interacciones medicamentosas con nintedanib basadas en el metabolismo del CYP es baja.
Coadministración con otros fármacos: La coadministración de nintedanib junto con docetaxel (75 mg/m²) no alteró la farmacocinética de estos fármacos en ningún grado relevante.
No se exploró el potencial de interacciones de nintedanib con los anticonceptivos hormonales.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES:
• Trastornos gastrointestinales:
— Diarrea: La diarrea fue el evento gastrointestinal informado con mayor frecuencia y se presentó en estrecha asociación temporal con la administración de docetaxel (véase la sección Reacciones adversas). En el estudio clínico LUME-Lung 1 (véase la sección Estudios clínicos), la mayoría de los pacientes tuvieron diarrea de leve a moderada. El 6,3% de los pacientes tuvieron diarrea de grado ≥3 en el caso del tratamiento combinado en comparación con un 3,6% en los pacientes tratados con docetaxel solo. La diarrea debe tratarse ante la aparición de los primeros signos con hidratación adecuada y con medicamentos antidiarreicos, p. ej., loperamida, y puede requerir la reducción de la dosis o la interrupción temporaria o definitiva del tratamiento con VARGATEF® (véase la sección Posología y administración).
— Náuseas y vómitos: Las náuseas y los vómitos, en la mayoría de los casos de gravedad leve a moderada, fueron eventos adversos gastrointestinales informados con frecuencia (véase la sección Reacciones adversas). Si los síntomas persisten a pesar de la instauración de atención médica de soporte adecuada (lo que incluye tratamiento antiemético), puede ser necesaria una reducción de la dosis o bien la interrupción temporaria o definitiva del tratamiento con VARGATEF® (véase la sección Posología y administración).
La diarrea y los vómitos pueden producir deshidratación con o sin desequilibrio electrolítico, lo que podría conducir a un deterioro de la función renal. En el caso de deshidratación, se requiere la administración de electrolitos y líquidos. Deben controlarse los niveles plasmáticos de electrolitos en el caso de producirse eventos adversos gastrointestinales relevantes.
• Neutropenia y septicemia:
Se observó una mayor frecuencia de neutropenia de grado CTCAE > 3 en los pacientes tratados con VARGATEF® en combinación con docetaxel en comparación con los pacientes que recibieron tratamiento con docetaxel solo. Se han observado complicaciones posteriores de dicho cuadro, como septicemia o neutropenia febril.
Deben controlarse los recuentos hematológicos durante el tratamiento, en particular durante el tratamiento combinado con docetaxel. Debe efectuarse un control frecuente mediante hemogramas completos al comienzo de cada ciclo de tratamiento y alrededor del nadir en los pacientes que reciban tratamiento con nintedanib en combinación con docetaxel, y según esté clínicamente indicado tras la administración del último ciclo de la combinación.
• Función hepática:
La seguridad y la eficacia de VARGATEF® no han sido estudiadas en pacientes con insuficiencia hepática moderada (Child Pugh B) o grave (Child Pugh C). Por lo tanto, no se recomienda el tratamiento con VARGATEF® en dichos pacientes.
Sobre la base de que existe una mayor exposición, es posible que los pacientes con insuficiencia hepática leve (Child Pugh A) corran más riesgos de sufrir eventos adversos (véanse las secciones Posología y administración, Farmacocinética).
Se han observado casos de lesión hepática producida por medicamentos con el tratamiento con nintedanib. En el período posterior a la comercialización, se ha informado lesión hepática grave con desenlace mortal. Las elevaciones de las enzimas hepáticas (ALT, AST, ALKP, gamma glutamiltransferasa (GGT)) y de los valores de bilirrubina fueron reversibles con la reducción de la dosis o la interrupción del tratamiento, en la mayoría de los casos.
Deben investigarse los niveles de transaminasas, ALKP y bilirrubina al iniciar un tratamiento combinado de VARGATEF® más docetaxel. Los valores de tales parámetros deben controlarse según esté clínicamente indicado o bien a intervalos periódicos durante el tratamiento, es decir, en la fase de combinación con docetaxel al comienzo de cada ciclo de tratamiento y con una frecuencia mensual en el caso de que VARGATEF® se continúe como monoterapia tras la interrupción del docetaxel.
Si se detectaran elevaciones relevantes de las enzimas hepáticas, podría ser necesario reducir la dosis o interrumpir temporaria o definitivamente el tratamiento con VARGATEF® (véase la sección Posología y administración/Tabla 2). Deben investigarse las causas alternativas de las elevaciones de las enzimas hepáticas y deben tomarse las medidas necesarias según sea pertinente.
En el caso de cambios específicos en los valores de los parámetros hepáticos (AST/ALT > 3 veces el ULN en combinación con bilirrubina ≥ 2 veces el ULN y ALKP < 2 veces el ULN), debe suspenderse el tratamiento con VARGATEF®. A menos que exista una causa alternativa confirmada, VARGATEF® debe interrumpirse definitivamente (véase la sección Posología y administración/Tabla 2).
Las mujeres y los pacientes de raza asiática tienen un mayor riesgo de elevaciones de las enzimas hepáticas.
La exposición a nintedanib se incrementó de manera lineal en función de la edad de los pacientes y tuvo una correlación inversa con el peso corporal, lo que también puede dar lugar a un mayor riesgo de desarrollar elevaciones de las enzimas hepáticas (véase la sección Farmacocinética).
Se recomienda un monitoreo estrecho en los pacientes que presenten estos factores de riesgo.
• Hemorragia:
La inhibición del VEGFR podría estar asociada con un mayor riesgo de sangrado. En el estudio clínico (LUME-Lung 1) con VARGATEF®, la frecuencia de sangrado en ambos grupos de tratamiento resultó comparable. La epistaxis leve a moderada representó el evento hemorrágico más frecuente. No hubo desequilibrios en los sangrados respiratorios o mortales, y no se informó ningún caso de sangrado intracerebral. La mayoría de los eventos hemorrágicos mortales estuvieron asociados con tumores.
En el período posterior a la comercialización, se han observado eventos hemorrágicos serios y no serios, algunos de los cuales resultaron mortales. En los pacientes que presentan eventos hemorrágicos de grado 3/4, deben evaluarse cuidadosamente los riesgos y beneficios de continuar el tratamiento con VARGATEF® y puede considerarse la interrupción de VARGATEF®. Si se reanuda el tratamiento con VARGATEF®, se recomienda una dosis diaria reducida (véase la sección Posología y administración/Tabla 1).
Los pacientes con sangrado pulmonar reciente (> 2,5 ml de sangre roja) y los pacientes con tumores de ubicación anatómica central con evidencia radiográfica de invasión local de grandes vasos sanguíneos o evidencia radiográfica de tumores necróticos o cavitarios han sido excluidos de los estudios clínicos. Por lo tanto, no se recomienda el tratamiento con VARGATEF® en dichos pacientes.
• Metástasis cerebrales:
— Metástasis cerebrales estables: No se observó ningún incremento de la frecuencia de sangrado cerebral en los pacientes con metástasis cerebrales pretratadas adecuadamente que habían estado estables durante ≥ 4 semanas antes del inicio del tratamiento con VARGATEF®. No obstante ello, dichos pacientes deben ser sometidos a un estrecho control en pos de signos y síntomas de sangrado cerebral.
— Metástasis cerebrales activas: Los pacientes con metástasis cerebrales activas fueron excluidos de los estudios clínicos, y no se recomienda el tratamiento con VARGATEF® en dichos pacientes.
• Anticoagulación terapéutica: No existen datos disponibles en relación con los pacientes afectados por una predisposición hereditaria al sangrado o los pacientes que están recibiendo una dosis completa de tratamiento anticoagulante previo al inicio del tratamiento con VARGATEF®. En los pacientes que están recibiendo un tratamiento crónico con dosis bajas de heparinas de bajo peso molecular o ácido acetilsalicílico, no se observó ningún incremento en la frecuencia de sangrado. Se permitió la continuación de la toma de VARGATEF® en los pacientes que desarrollaron eventos tromboembólicos durante el tratamiento y que requirieron tratamiento anticoagulante, y no se observó una mayor frecuencia de eventos hemorrágicos en dichos pacientes. Los pacientes que estén recibiendo terapia concomitante con anticoagulantes, tales como warfarina o fenprocoumona, deben ser sometidos a controles regulares para detectar posibles cambios en los valores de tiempo de protrombina, o de RIN, o bien episodios de sangrado clínico.
Eventos tromboembólicos arteriales: La frecuencia de eventos tromboembólicos arteriales fue comparable entre los dos grupos de tratamiento en el estudio 1199.13, de fase III (LUME-Lung 1). Los pacientes con antecedentes recientes de infarto de miocardio o accidente cerebrovascular fueron excluidos de este estudio. No obstante ello, se observó una frecuencia incrementada de eventos tromboembólicos arteriales en los pacientes con fibrosis pulmonar idiopática (FPI) tratados con monoterapia de nintedanib.
Debe tenerse precaución al tratar pacientes con riesgo cardiovascular incrementado, lo que incluye enfermedad coronaria conocida. Debe considerarse la interrupción del tratamiento en los pacientes que desarrollen signos o síntomas de isquemia de miocardio aguda.
Tromboembolia venosa: Los pacientes tratados con VARGATEF® tienen un riesgo incrementado de desarrollar tromboembolia venosa, lo que incluye trombosis venosa profunda. Debe realizarse un control estrecho de estos pacientes en pos de la detección de posibles eventos tromboembólicos. VARGATEF® debe ser interrumpido en los pacientes que tengan reacciones de tromboembolia venosa potencialmente mortales.
Perforaciones gastrointestinales: La frecuencia de perforaciones gastrointestinales fue comparable entre los grupos de tratamiento en el estudio LUME-Lung 1. Debido al mecanismo de acción de nintedanib, los pacientes podrían tener un mayor riesgo de padecer eventos de perforación gastrointestinal. Se han informado casos de perforaciones gastrointestinales, algunas de los cuales fueron mortales, en el período posterior a la comercialización. Debe tenerse especial cuidado al tratar a pacientes con una cirugía abdominal previa o antecedentes recientes de perforación de un órgano hueco. Por lo tanto, debe dejarse transcurrir un mínimo de 4 semanas luego de una cirugía mayor, lo que incluye una cirugía abdominal, antes de iniciar la administración de VARGATEF®. El tratamiento con VARGATEF® debe suspenderse definitivamente en los pacientes que desarrollen una perforación gastrointestinal.
Complicaciones de la cicatrización de las heridas: Sobre la base de su mecanismo de acción, nintedanib podría dificultar la normal cicatrización de las heridas. No se observó un aumento de la frecuencia de problemas de cicatrización de las heridas en los estudios clínicos. No se llevó a cabo ningún estudio específico en el que se investigara el efecto de nintedanib sobre la cicatrización de las heridas. Por lo tanto, el tratamiento con VARGATEF® debe ser iniciado, o reanudado en el caso de haber sido suspendido por una intervención quirúrgica, tras la confirmación de una correcta cicatrización de las heridas sobre la base del criterio clínico.
Poblaciones especiales: En el estudio 1199.13 (LUME-Lung 1), hubo una frecuencia más alta de eventos adversos serios en los pacientes tratados con VARGATEF® más docetaxel con un peso corporal de menos de 50 kg en comparación con los pacientes con un peso corporal ≥ 50 kg; sin embargo, la cantidad de pacientes con un peso corporal inferior a 50 kg fue reducida. Por lo tanto, se recomienda un control estrecho en los pacientes cuyo peso corporal sea < 50 kg.
POSOLOGÍA Y ADMINISTRACIÓN:
Posología: El tratamiento con VARGATEF® debe ser iniciado y supervisado por un médico con experiencia en el uso de terapias oncológicas.
Para obtener información sobre la posología, el modo de administración y las modificaciones de la dosis de docetaxel, sírvase consultar la información del producto correspondiente de docetaxel.
La dosis recomendada de VARGATEF® es 200 mg dos veces al día administrada con un intervalo de aproximadamente 12 horas entre sí, en los días 2 a 21 de un ciclo de tratamiento estándar de docetaxel de 21 días.
VARGATEF® no debe ser administrado el mismo día de la administración de la quimioterapia de docetaxel (= día 1).
NO DEBE EXCEDERSE LA DOSIS DIARIA MÁXIMA RECOMENDADA DE 400 MG.
Tras la interrupción de docetaxel, los pacientes pueden continuar el tratamiento con VARGATEF® mientras se observe un beneficio clínico o bien hasta que se produzca una toxicidad inaceptable.
Ajustes de la dosis: Como medida inicial para el manejo de las reacciones adversas (véanse Tabla 1 y 2), el tratamiento con VARGATEF® debe suspenderse temporariamente hasta que la reacción adversa específica se haya resuelto a un nivel que permita la continuación del tratamiento (a grado 1 o al valor inicial). El tratamiento con VARGATEF® puede reanudarse en una dosis menor. Se recomienda realizar ajustes escalonados de la dosis a razón de 100 mg por día (es decir, una reducción de 50 mg por cada toma diaria) sobre la base del perfil individual de seguridad y tolerabilidad del paciente, según se describe en la Tabla 1 y en la Tabla 2.
En el caso de que la(s) reacción(ones) adversa(s) persista(n), es decir, si el paciente no tolera el régimen de dos tomas diarias de 100 mg cada una, deberá interrumpirse definitivamente el tratamiento con VARGATEF®.
En el caso de elevaciones específicas de la aspartato aminotransferasa (AST) / alanina aminotransferasa (ALT) a valores equivalentes a > 3 veces el límite normal superior (ULN) en combinación con un incremento de la bilirrubina total a ≥ 2 veces el ULN y un nivel de fosfatasa alcalina (ALKP) < 2 veces el ULN (véase la Tabla 2), debe suspenderse el tratamiento con VARGATEF®. A menos que exista una causa alternativa confirmada, VARGATEF® debe interrumpirse definitivamente (véase la sección Advertencias y precauciones especiales).
|
Tabla 1. Ajustes de dosis recomendados para VARGATEF® en caso de diarrea, vómitos y otras reacciones adversas no hematológicas o hematológicas, excepto elevaciones de las enzimas hepáticas (véase la Tabla 2) |
|
|
Grado CTCAE* de las reacciones adversas |
Ajuste de dosis |
|
Diarrea igual a grado 2 durante más de 7 días consecutivos a pesar de la administración de tratamiento antidiarreico** O BIEN Diarrea > grado 3 a pesar de la administración de tratamiento antidiarreico** |
Tras la interrupción del tratamiento y la recuperación a grado 1 o valor inicial, reducción de la dosis de 200 mg dos veces al día a 150 mg dos veces al día y, en el caso de considerarse necesaria una segunda reducción de la dosis, de 150 mg dos veces al día a 100 mg dos veces al día. |
|
Vómitos ** > grado 2 Y/O Náuseas > grado 3 a pesar de la administración de tratamiento antiemético** |
|
|
Otra reacción adversa hematológica o no hematológica > grado 3 |
|
|
* CTCAE: Criterios Terminológicos Comunes para Eventos Adversos ** Véase también la sección Advertencias y precauciones especiales |
|
|
Tabla 2. Ajustes de dosis recomendados para VARGATEF® en caso de elevaciones de AST y/o ALT y de la bilirrubina |
|
|
Elevaciones de la AST / ALT y de la bilirrubina |
Ajuste de dosis |
|
Elevación de los valores de AST y/o ALT a > 2,5 veces el ULN en combinación con elevación de la bilirrubina total a ≥ 1,5 veces el ULN O BIEN Elevación de los valores de AST y/o ALT a > 5 veces el ULN |
Tras la interrupción del tratamiento y la recuperación de los valores de las transaminasas a un nivel ≤ 2,5 veces el ULN en combinación con el retorno de la bilirrubina al rango normal, reducción de la dosis de 200 mg dos veces al día a 150 mg dos veces al día y, en el caso de que se considere necesaria una 2da. reducción de la dosis, de 150 mg dos veces al día a 100 mg dos veces al día. |
|
Elevación de los valores de AST y/o ALT a > 3 veces el ULN en combinación con un aumento de la bilirrubina total a ≥ 2 veces el ULN y ALKP < 2 veces el ULN |
A menos que exista una causa alternativa confirmada, VARGATEF® debe interrumpirse definitivamente. |
|
AST: Aspartato aminotransferasa; ALT: Alanina aminotransferasa; ALKP: Fosfatasa alcalina; ULN: Límite normal superior |
|
• Poblaciones especiales:
Población pediátrica: La seguridad y la eficacia de VARGATEF® en pacientes pediátricos no han sido estudiadas en estudios clínicos.
Pacientes de edad avanzada (≥ 65 años): No se observaron diferencias en general en lo que respecta a la seguridad y la eficacia en los pacientes de edad avanzada en comparación con los pacientes menores de 65 años. No se requiere ajuste de la dosis inicial en función de la edad del paciente (véase la sección Farmacocinética).
Raza: Sobre la base de los análisis de farmacocinética poblacional, no se requiere a priori ningún ajuste de la dosis de VARGATEF® (véanse las secciones Poblaciones especiales, Advertencias y precauciones especiales, Farmacocinética). Son limitados los datos de seguridad disponibles en relación con los pacientes de raza negra.
Peso corporal: Sobre la base de los análisis de farmacocinética poblacional, no se requiere a priori ningún ajuste de la dosis de VARGATEF® (véase la sección Farmacocinética).
Insuficiencia renal: Menos del 1 % de una dosis única de nintedanib se elimina a través de los riñones (véase la sección Farmacocinética). No se requiere un ajuste de la dosis inicial en los pacientes con insuficiencia renal leve a moderada. La seguridad, la eficacia y la farmacocinética de nintedanib no han sido estudiadas en pacientes con insuficiencia renal grave (depuración de creatinina: CrCL < 30 ml/min).
Insuficiencia hepática: Nintedanib se elimina primordialmente a través de la excreción por vía biliar/fecal (> 90 %). La exposición aumentó en pacientes con insuficiencia hepática (Child Pugh A, Child Pugh B; véase la sección Farmacocinética).
No se requiere ningún ajuste de la dosis inicial para los pacientes con insuficiencia hepática leve sobre la base de los datos clínicos disponibles (Child Pugh A; véase la sección Advertencias y precauciones especiales).
La seguridad y la eficacia de nintedanib no han sido investigadas en pacientes con insuficiencia hepática clasificada como Child Pugh B o C. No se recomienda el tratamiento con VARGATEF® en los pacientes con insuficiencia hepática moderada (Child Pugh B) o grave (Child Pugh C); véase la sección Farmacocinética.
• Modo de administración:
Las cápsulas de VARGATEF® deben tomarse por vía oral, preferentemente con alimentos; deben tragarse enteras con agua y no deben masticarse ni triturarse. En el caso de omitirse una dosis, la administración debe reanudarse en el siguiente momento programado según la dosis recomendada. En el caso de omitirse una dosis, no debe administrarse una dosis adicional.
• Modo de empleo / manipulación:
No corresponde.
SOBREDOSIS: No existe ningún antídoto ni tratamiento específico para la sobredosis de VARGATEF®. La dosis única más alta de nintedanib administrada en los estudios de fase I fue 450 mg una vez al día. Asimismo, 2 pacientes del programa de oncología tuvieron una sobredosis de un máximo de 600 mg dos veces al día (b.i.d.) durante un total de hasta ocho días. Los eventos adversos observados fueron coherentes con el perfil de seguridad conocido de nintedanib, es decir, elevación de las enzimas hepáticas y síntomas gastrointestinales. Ambos pacientes se recuperaron de dichas reacciones adversas.
En el caso de una sobredosis, debe interrumpirse el tratamiento y deben iniciarse medidas de soporte generales según corresponda.
TOXICOLOGÍA: Toxicología general: Los estudios de toxicidad de dosis únicas en ratas y en ratones indicaron un bajo potencial de toxicidad aguda para nintedanib. En los estudios de toxicología de dosis repetidas en ratas, los efectos adversos (p. ej., engrosamiento de las placas epifisarias, lesiones de los incisivos) estuvieron mayormente relacionados con el mecanismo de acción (es decir, inhibición del VEGFR-2) de nintedanib. Estos cambios son efectos conocidos de otros inhibidores del VEGFR-2 y pueden considerarse efectos de la clase farmacológica.
Se observó un cuadro de diarrea y vómitos acompañado de una disminución del consumo de alimentos y descenso del peso corporal en los estudios de toxicidad efectuados en no roedores.
No hubo evidencia de elevación de las enzimas hepáticas en ratas, perros ni monos Cynomolgus. Las elevaciones leves de las enzimas hepáticas que no se debieron a efectos adversos serios, como la diarrea, se observaron únicamente en los monos Rhesus.
Toxicidad para la reproducción: Un estudio de fertilidad masculina y desarrollo embrionario temprano en ratas que abarcó hasta la fase de implantación no reveló ningún efecto sobre la fertilidad masculina ni el aparato reproductor de los machos.
En las ratas se observó letalidad embriofetal y efectos teratogénicos con una exposición inferior a la exposición humana, con la dosis recomendada máxima en los seres humanos (MRHD) de 200 mg dos veces al día. También se observaron efectos sobre el desarrollo del esqueleto axial y sobre el desarrollo de las grandes arterias en niveles de exposición subterapéuticos.
En los conejos, se observó letalidad embriofetal y efectos teratogénicos comparables a los detectados en las ratas con una exposición ligeramente superior a la de las ratas.
En las ratas, se observó la eliminación de pequeñas cantidades de nintedanib radiomarcado y/o sus metabolitos en la leche (≤ 0,5 % de la dosis administrada).
A partir de los estudios de carcinogenia de 2 años de duración realizados en ratones y ratas, no surgió evidencia alguna de un potencial carcinogénico de nintedanib.
Los estudios de genotoxicidad no indicaron ningún potencial mutagénico para nintedanib.
• Anexo de frecuencias de reacciones adversas:
VARGATEF®
Categorías de frecuencias:
— Muy frecuentes: ≥1/10
— Frecuentes: ≥1/100 <1/10
— Poco frecuentes: ≥1/1000 <1/100
— Raros: ≥1/10000 <1/1000
— Muy raros: <1/10000
— Desconocidos: No pueden estimarse en base a los datos disponibles.
Nota: Las categorías de frecuencias mencionadas anteriormente se basan en los lineamientos de SmPC de la UE (septiembre de 2009). Por consiguiente, en países fuera de la Unión Europea, es posible que sean apropiadas otras definiciones.
|
Tabla 14 VARGATEF®: Categorización de frecuencias de reacciones adversas al medicamento sobre la base de eventos adversos en pacientes que padecen NSCLC con histología tumoral de adenocarcinoma. |
||
|
Terminología de la clasificación por sistema y órgano del MedDRA |
Reacciones adversas de nintedanib según el término literal de la CCDS TP del MedDRA (versión 15.1) |
Categoría de frecuencias según los lineamientos de SmPC de la UE |
|
Infecciones e infestaciones |
Septicemia |
Frecuentes |
|
Abscesos |
Frecuentes |
|
|
Neutropenia febril |
Frecuentes |
|
|
Trastornos de la sangre y del sistema linfático |
Neutropenia** |
Muy frecuentes |
|
Trombocitopenia |
Frecuentes |
|
|
Trastornos del metabolismo y de la nutrición |
Deshidratación |
Frecuentes |
|
Desequilibrio electrolítico |
Muy frecuentes |
|
|
Disminución del apetito |
Muy frecuentes |
|
|
Descenso de peso |
Frecuentes |
|
|
Trastornos vasculares |
Hipertensión |
Frecuentes |
|
Tromboembolia venosa |
Frecuentes |
|
|
Sangrado |
Muy frecuentes |
|
|
Trastornos gastrointestinales |
Diarrea |
Muy frecuentes |
|
Vómitos |
Muy frecuentes |
|
|
Dolor abdominal |
Muy frecuentes |
|
|
Perforación |
Poco frecuentes |
|
|
Náuseas |
Muy frecuentes |
|
|
Estomatitis |
Muy frecuentes |
|
|
Pancreatitis |
Poco frecuentes |
|
|
Trastornos hepatobiliares |
Lesión hepática producida por medicamentos |
Poco frecuentes |
|
Elevación de la alanina aminotransferasa |
Muy frecuentes |
|
|
Elevación de la aspartato aminotransferasa |
Muy frecuentes |
|
|
Elevación de la fosfatasa alcalina en sangre |
Muy frecuentes |
|
|
Elevación de la gamma glutamiltransferasa |
Frecuentes |
|
|
Hiperbilirrubinemia |
Frecuentes |
|
|
Trastornos de la piel y del tejido subcutáneo |
Mucositis*** |
Muy frecuentes |
|
Clasificación por sistema y órgano -terminología MedDRA- |
Término literal de la CCDS TP del MedDRA (versión 15.1) |
Categoría de frecuencias según los lineamientos de SmPC de la UE |
|
Exantema |
Muy frecuentes |
|
|
Prurito |
Frecuentes |
|
|
Trastornos del sistema nervioso |
Neuropatía periférica |
Muy frecuentes |
|
** Incluye neutropenia febril *** Incluye estomatitis |
||
INDICACIONES/USO: VARGATEF® Tratamiento de segunda línea de pacientes con CPNM localmente avanzado, metástasico o localmente recidivante con histología de adenocarcinoma tras una primera línea de quimioterapia, siempre y cuando la primera línea no haya incluido docetaxel o inhibidores del VEGF, con excepción de Bevacizumab.
PRESENTACIÓN: VARGATEF® 100 mg: Caja por 120 cápsulas en blíster (Reg. San. No. INVIMA 2019M-0018841). VARGATEF® 150 mg: Caja por 60 cápsulas blandas en blíster (Reg. San. No. INVIMA 2019M-0018796)
“¡Almacenar en un lugar seguro; fuera del alcance de los niños!
La información de seguridad del producto puede cambiar, consulte la información vigente en la Dirección Médica.
Teléfono: (601) 319 91 00 - e-mail: medfora.co@boehringer-ingelheim.com
Carrera 11 No. 84A-09 Piso 5, Bogotá D.C. Colombia.
BOEHRINGER INGELHEIM S.A.
Versión-14 del 15 de febrero de 2019

