

TORISEL
TEMSIROLIMUS
Solución para inyección
Caja , 1 Vial(es) , Solución para inyección , 1.2 Mililitros
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Principio Activo: Temsirolimus.
Nombre químico: (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R,34aS)-9,10,12,13,14,21, 22,23,24,25,26,27,32,33, 34,34a-Hexadecahidro-9,27-dihidroxi-3-[(1R)-2-[(1S,3R,4R)-4-hidroxi-3-metoxiciclohexil]-1- metiletil]-10,21-dimetoxi-6,8,12,14,20,26-hexametil-23,27-epoxi-3H-pirido[2,1-c][1,4] oxaazaciclohentriacontina-1,5,11,28,29(4H,6H,31H)-pentona 4'-[2,2-bis(hidroximetil)propionato]; o Rapamicina, 42-[3-hidroxi-2-(hidroximetil)-2-metilpropanoato]
Estructura
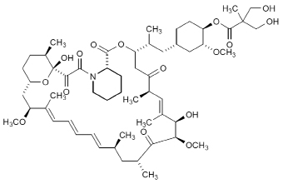
Fórmula molecular: C56H87NO16
Peso molecular: 1030,30
Características físicas: Temsirolimus es un polvo blanco a blancuzco. No es higroscópico. Temsirolimus es insoluble en agua y soluble en alcohol. No tiene grupos funcionales ionizables, y su solubilidad es independiente del pH.
Aplica únicamente a solución intravenosa para oncología: Temsirolimus para Inyección, 25 mg/mL.
Principio activo: Temsirolimus.
Diluente para el Concentrado de Temsirolimus para Inyección; 1,8 mL/vial
INDICACIONES
Carcinoma de células renales: Temsirolimus está indicado para el tratamiento del carcinoma de células renales avanzado o metastásico en pacientes con pobre pronóstico (es decir, que cumplen al menos 3 factores de pronóstico de riesgo).
FORMA FARMACÉUTICA
Aplica únicamente a soluciones intravenosas para oncología
— Concentrado Temsirolimus para Inyección, 25 mg/mL
— Diluente para el Concentrado de Temsirolimus para Inyección, 1.8 mL/Vial
PARTICULARIDADES FARMACÉUTICAS
Incompatibilidades: El Concentrado de Temsirolimus para Inyección no se debe adicionar directamente a soluciones de infusión acuosas. La adición directa de Temsirolimus para Inyección a soluciones acuosas produce precipitación del medicamento. Combine siempre el concentrado de Temsirolimus para inyección con su diluente antes de agregarlo a soluciones de infusión. Se recomienda que temsirolimus se administre en cloruro de sodio para inyección al 0,9% después de combinarlo con el diluente. No se ha evaluado la estabilidad del temsirolimus en otras soluciones para infusión. La adición de otros medicamentos o agentes nutricionales a las mezclas de temsirolimus en cloruro de sodio para inyección no se ha evaluado y se debe evitar. El temsirolimus se degrada tanto por los ácidos como por las bases, y por ello se deben evitar las combinaciones de temsirolimus con agentes capaces de modificar el pH de la solución.
Precauciones especiales de almacenamiento: El Concentrado de Temsirolimus para Inyección se debe almacenar refrigerado (2º- 8 °C) y protegido de la luz. Bajo estas condiciones de almacenamiento, el Concentrado de Temsirolimus para Inyección es estable durante al menos 36 meses.
El diluente para el Concentrado de Temsirolimus para Inyección se puede almacenar refrigerado y protegido de la luz. Por sí mismo, el diluente es química y físicamente estable durante 36 meses cuando se almacena entre 2° a 8 °C.
La mezcla de concentrado del medicamento y el diluente es estable durante hasta 24 horas a temperatura de 25 ºC.
La mezcla conteniendo temsirolimus con cloruro de sodio para inyección 0.9% debe usarse entre las siguientes 6 horas posterior a la preparación y debe almacenarse a temperatura ambiente y protegerse de luz excesiva.
Naturaleza del material de envase: El Concentrado de Temsirolimus para Inyección y su Diluente se envasan en viales de vidrio transparente con tapones de caucho de butilo. Se utilizan sellos codificados por colores en Temsirolimus para inyección y su diluente.
Manipulación: Durante la manipulación y preparación de las mezclas el Concentrado de Temsirolimus para Inyección se debe proteger de la luz ambiental y solar excesivas. El Concentrado Temsirolimus para Inyección se debe inspeccionar visualmente para determinar la presencia de material particulado y la decoloración luego de la reconstitución y antes de la administración. Las bolsas/contenedores que entran en contacto con el Concentrado Temsirolimus para Inyección deben estar fabricados de vidrio, poliolefina o polietileno.
Una vez combinado el concentrado con el diluente, inyecte la mezcla rápidamente dentro de cloruro de sodio para inyección al 0,9%. La administración de la solución final diluida para infusión se debe realizar dentro de las seis horas a partir del momento en que el concentrado se diluye por primera vez con el diluente.
PROPIEDADES FARMACOCINÉTICAS
Absorción: Después de la administración de una única dosis de 25 mg de temsirolimus intravenoso en pacientes con cáncer, la Cmax media en sangre entera fue 585 ng/ml (coeficiente de variación, CV=14%), y la media de la AUC en sangre fue 1627 ng·h/ml (CV=26%). Para pacientes que recibieron 175 mg semanales durante 3 semanas seguidos por 75 mg semanales, la Cmax estimada en sangre entera al final de la infusión fue 2457 ng/mL durante la semana 1 y 2574 ng/mL durante la semana 3.
Distribución: Temsirolimus exhibe una disminución poliexponencial en las concentraciones y en la distribución para sangre entera y se atribuye a la unión preferencial con el FKBP-12 en las células sanguíneas. La constate de disociación (Kd) media (desviación estándar, SD) de unión fue 5,1 (3,0) ng/ml, que indica la concentración a la que el 50% de los sitios de unión en las células sanguíneas se ocupaban. La distribución de temsirolimus es dependiente de la dosis con unión específica máxima media (10º, 90º percentiles) a las células sanguíneas de 1,4 mg (0,47 a 2,5 mg). Luego de una única dosis intravenosa de 25 mg, el volumen de distribución de estado estable medio en la sangre entera de los pacientes con cáncer fue 172 litros.
Metabolismo: El sirolimus, un metabolito igualmente potente para temsirolimus se observó como el metabolito principal en humanos luego de tratamiento intravenoso. Durante estudios in vitro del metabolismo del temsirolimus, se observaron sirolimus, seco-temsirolimus y seco-sirolimus; las vías metabólicas adicionales fueron la hidroxilación, la reducción y la desmetilación.Luego de una única dosis intravenosa de 25 mg en pacientes con cáncer, la AUC del sirolimus fue 2,7 veces la AUC del temsirolimus, debido principalmente a la vida media mayor del sirolimus.
Eliminación: Después de una única dosis de 25 mg de temsirolimus intravenoso en pacientes con cáncer, la depuración sistémica (CV) media del temsirolimus de la sangre entera fue 16,2 L/h (22%).Las medias de la vida media del temsirolimus y el sirolimus fueron 17,3 h y 54,6 h respectivamente. Después de la administración de temsirolimus etiquetado con [14C], la excreción fue predominantemente a través de las heces (78%), siendo la eliminación renal del medicamento y los metabolitos 4,6% de la dosis administrada.Los valores predichos por el modelo para depuración plasmática indican que la concentración mínima del metabolito del sirolimus es de 10,7 ng/mL.
• Farmacocinética poblacional
Ancianos: En los análisis de datos basados en la farmacocinética poblacional, la edad no tiene un efecto significativo en la disposición del temsirolimus o del metabolito sirolimus (ver sección Advertencias especiales y precauciones de uso).
Niños: En pacientes pediátricos con tumores sólidos recurrentes/resistentes al tratamiento, la depuración de temsirolimus fue baja y la exposición (AUC) fue mayor que en los adultos. En contraste, la exposición a temsirolimus fue proporcionalmente reducida en pacientes pediátricos, de modo que la exposición neta medida mediante la suma de las AUC (AUCsuma) de temsirolimus y sirolimus fue similar a la de los adultos (ver sección Advertencias especiales y precauciones de uso).
Género: En análisis de datos basados en farmacocinética poblacional, el género no tiene un efecto significativo sobre la disposición del temsirolimus o el metabolito sirolimus.
Peso corporal: En análisis de datos basados en farmacocinética poblacional, el cambio en la concentración mínima del metabolito principal del temsirolimus, el sirolimus, para pacientes con aumento del peso corporal (para pesos corporales entre 38,6 y 158,9 kg) fue limitado a un intervalo del doble (entre 8,28 y 16,7 ng/mL) en sangre entera.
Pacientes con deterioro renal: La eliminación de temsirolimus a través de los riñones es baja. Como las diferencias en la eliminación de creatinina no afectan la disposición del temsirolimus, no se requieren cambios en el régimen de tratamiento de temsirolimus intravenoso en pacientes con deterioro renal (ver sección Posología y método de administración).
Insuficiencia hepática o pacientes con deterioro hepático: El temsirolimus se elimina predominantemente por el hígado. El temsirolimus se debe utilizar con precaución en pacientes con deterioro hepático. El temsirolimus está contraindicado en pacientes con bilirrubina > 1,5 x ULN.
Efecto de los alimentos: No se ha examinado el efecto de los alimentos sobre la exposición luego de una dosis intravenosa de temsirolimus.
PROPIEDADES FARMACODINÁMICAS
Clase farmacológica: Temsirolimus es un inhibidor selectivo del mTOR (mammalian target of rapamycin).
Grupo farmacoterapéutico: Agente anti-neoplásico.
Mecanismo de acción: Temsirolimus es un inhibidor selectivo del mTOR (mammalian target of rapamycin). Temsirolimus se une a una proteína intracelular (FKBP-12), y el complejo proteína-medicamento se une e inhibe la actividad del mTOR que controla la división celular. In vitro, a altas concentraciones (10 - 20 µM) el temsirolimus se puede unir e inhibir mTOR en ausencia de FKBP-12. Se observó respuesta bifásica a la dosis de la inhibición del crecimiento celular. Las concentraciones altas produjeron inhibición completa del crecimiento celular in vitro, mientras que la inhibición medida por el complejo FKBP-12/temsirolimus sólo produjo aproximadamente una inhibición del 50% de la proliferación celular. La inhibición de la actividad del mTOR produce detención del crecimiento G1 en las células tumorales tratadas como resultado de la disrupción selectiva del transporte de las proteínas reguladoras del ciclo celular, como por ejemplo ciclinas tipo D, c-myc y ornitina decarboxilasa. Cuando se inhibe la actividad de mTOR, se bloquea la habilidad para la fosforilación y de esta forma se controla la actividad de los factores de transporte proteico (4E-BP1 y S6K, ambos encontrados después del mTOR en la ruta quinasa PI 3 /AKT) que controlan la división celular.
Además de regular las proteínas del ciclo celular, mTOR puede regular la traslación de los factores inducidos por hipoxia, alfa HIF-1 y HIF-2. Estos factores de transcripción regulan la actividad de los tumores para adaptarse a los microambientes hipóxicos y para producir el factor angiogénico, factor de crecimiento del endotelio vascular (VEGF). De este modo, el efecto antitumoral del temsirolimus, puede también en parte, derivarse de su habilidad para reducir los niveles de HIF y VEGF en el tumor o en el microambiente del tumor, deteriorando de esta forma el desarrollo del vaso sanguíneo.
Relación entre concentración y efecto: El efecto del tratamiento con temsirolimus intravenoso en la inhibición de la fosforilación de la proteína S6 ribosómica sobre los linfocitos circulantes se examinó en 30 sujetos sanos. Los datos indican que la inhibición de la fosforilación proteica fue rápida y dependiente de la dosis. Luego de una única dosis de 25 mg de temsirolimus, 20% y 50% de inhibición de la proteína S6 ribosómica se presentó durante al menos 8 y 3 días, respectivamente.
Efectos sobre la repolarización cardiaca (a través del Estudio de QT/QTc): En un estudio aleatorizado controlado con placebo y con moxifloxacina de dos periodos, transversales y un tercer periodo secuencial, 58 sujetos sanos recibieron una única dosis IV de 25 mg de temsirolimus. No se observó ningún efecto relacionado con la concentración en la duración del intervalo QT/QTc.
Eficacia clínica
Carcinoma de células renales: La seguridad y la eficacia de temsirolimus en el tratamiento de carcinoma avanzado de células renales (RCC) fue estudiada en los siguientes dos estudios clínicos aleatorios.
El Estudio 1 era un estudio de etiqueta abierta, aleatorio, de tres ramas, multicéntrico de fase 3, con pacientes sin tratamiento previo, con carcinoma avanzado de células renales y con 3 o más de 6 factores de pronóstico de riesgo (con menos de un año transcurrido desde diagnóstico inicial de carcinoma de células renales y la asignación aleatoria, estado de desempeño de Karnofsky de 60 o 70, hemoglobina menor del límite inferior de normalidad, calcio corregido mayor de 10 mg/dL, lactato deshidrogenasa > 1,5 veces el límite superior de normalidad, más de un sitio de órgano metastático). El punto final primario del estudio era la supervivencia total (OS). Los puntos finales secundarios incluyeron supervivencia sin progreso (PFS), tasa de respuesta objetiva (ORR), tasa de beneficio clínico, tiempo para el fracaso del tratamiento (TTF) y medición de la supervivencia con calidad ajustada. Los pacientes se estratificaron de acuerdo con el estado de nefrectomía previo dentro de las tres regiones geográficas y se asignaron de forma aleatoria (1:1:1) para recibir interferón alfa (IFN-a) solo (n=207), temsirolimus solo (25 mg semanalmente; n=209), o la combinación de IFN-a y temsirolimus (n=210).
La rama correspondiente a la combinación no exhibe un índice riesgo-beneficio clínico positivo comparado con IFN-a. El tratamiento con la combinación de temsirolimus 15 mg y IFN-a produjo un aumento estadísticamente significativo en la incidencia de ciertos eventos adversos grado 3-4 (pérdida de peso, anemia, neutropenia, trombocitopenia, e inflamación de las mucosas) cuando se compara con los eventos adversos observados en las ramas de IFN-a solo ó temsirolimus 25 mg solo. La combinación de temsirolimus 15 mg y IFN-a no produce un aumento significativo en la supervivencia total cuando se compara con IFN-a solo (mediana 8,4 vs. 7,3 meses, índice de peligro = 0,96, p = 0,6965).
En esta sección se describe la información relacionada con las ramas de temsirolimus 25 mg solo y IFN-a solo. Las características demográficas y de la enfermedad de la población del estudio se muestran en la Tabla 2. Las características demográficas y de la enfermedad al inicio del estudio se balancearon bien en las ramas de tratamiento.
|
Tabla 2. Características demográficas y otras características de los pacientes al Inicio del Estudio Clínico 1 |
||
|
Característica |
Temsirolimus Concentrado para Inyección |
IFN-a |
|
Total pacientes en la rama de tratamiento |
209 |
207 |
|
Edad < 65 años > 65 años |
145 (69,4) 64 (30,6) |
142 (68,6) 65 (31,4) |
|
Sexo Femenino Masculino |
70 (33,5) 139 (66,5) |
59 (28,5) 148 (71,5) |
|
Raza Blancos Asiáticos Negros Otros |
186 (89,0) 6 (2,9) 9 (4,3) 8 (3,8) |
191 (92,3) 4 (1,9) 8 (3,9) 4 (1,9) |
|
Nefrectomía previa No Si |
70 (33,5) 139 (66,5) |
68 (32,9) 139 (67,1) |
|
Etapa de la enfermedad al inicio Etapa IV Etapa II Recurrente Etapa III Recurrente |
200 (95,7) 1 (0,5) 8 (3,8) |
201 (97,1) 1 (0,5) 5 (2,4) |
|
Tipo de células primarias Claras Indeterminadas No claras Desconocidas |
169 (82,0) 24 (11,7) 13 (6,3) 3 |
170 (82,5) 23 (11,2) 13 (6,3) 1 |
En el Estudio 1, se asoció temsirolimus 25 mg con ventaja estadísticamente significativa sobre IFN-a en el punto final primario de OS (tiempo transcurrido desde la asignación aleatoria hasta la muerte). La rama de temsirolimus mostró 49% de aumento en la mediana del OS comparado con la rama IFN-a.
La Figura 1 es un gráfico de Kaplan-Meier del OS del Estudio 1. El temsirolimus se asoció también con ventajas estadísticamente significativas sobre IFN-a en los puntos finales secundarios de PFS (tiempo transcurrido desde el momento de asignación aleatoria hasta el progreso, de la enfermedad o muerte, censurado en la última fecha de evaluación del tumor), TTF (tiempo transcurrido desde el momento de asignación aleatoria hasta el progreso de la enfermedad, muerte, retiro del tratamiento debido a eventos adversos, retiro del consentimiento voluntario o pérdida del seguimiento) y tasa de beneficio clínico (respuesta completa, respuesta parcial o enfermedad estable durante > 24 semanas). Las evaluaciones de PFS, ORR, y la tasa de beneficios clínicos se basaron en evaluación radiológica independiente en estado ciego de la respuesta del tumor utilizando criterios basados en RECIST. Para el TTF se utilizó la evaluación del progreso realizada por los investigadores. Los resultados de eficacia se resumen en la Tabla 3.
Figura 1: Curvas de Kaplan-Meier para Supervivencia Total – Estudio 1
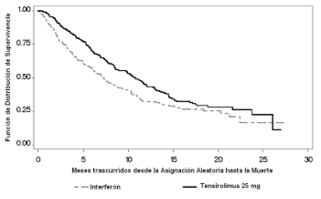
|
Tabla 3: Resumen de los Resultados de Eficacia para Temsirolimus Concentrado para Inyección en el Estudio Clínico 1 |
||||
|
Parámetro |
Temsirolimus Concentrado para Inyección n = 209 |
IFN-a |
Valor Pa |
Índice de Peligro |
|
Mediana de Supervivencia Total Meses (IC de 95%) |
10,9 (8,6; 12,7) |
7,3 (6,1; 8,8) |
0,0078* |
0,73 (0,58; 0,92) |
|
Mediana de Supervivencia sin Progreso Meses (IC de 95%) |
5,5 (3,9; 7,0) |
3,1 (2,2; 3,8) |
0,0001 |
0,66 (0,53; 0,81) |
|
Tasa Total de Respuesta % (IC de 95%) |
8,6 (4,8; 12,4) |
4,8 (1,9; 7,8) |
0,1232c |
NA |
|
Mediana del Tiempo para Fracaso del Tratamiento Meses (IC de 95%) |
3,8 (3,5; 3,9) |
1,9 (1,7; 1,9) |
<0,0001 |
0,61 (0,50; 0,74) |
|
Tasa de Beneficios Clínicos % (IC de 95%) |
32,1 (25,7; 38,4) |
15,5 (10,5; 20,4) |
<0,0001c |
NA |
|
IC = intervalo de confianza; NA = no aplica. * Una comparación se considera estadísticamente significativa si el valor de p es < 0,0159 (límite de O’Brien-a 446 muertes). a. Basado en la log-rank test estratificada por nefrectomía previa y región. b. Basado en el modelo de peligro proporcional de Cox estratificado por nefrología previa y región. c. Basado en la prueba de Cochran-Mantel-Haenszel estratificada por nefrología previa y región. |
||||
En el Estudio clínico 1, el 31% de los pacientes tratados con temsirolimus eran de 65 años o más. En pacientes menores de 65, la mediana de la supervivencia total era de 12 meses (IC de 95%: 9,9-14,5 con) con índice de peligro de 0,62 (IC de 95%: 0,47- 0,82), comparado con los pacientes tratados con IFN-a. En pacientes de 65 años o más, la mediana de la supervivencia total fue 8,6 meses (IC de 95%: 6,4-11,5 con) con índice de peligro de 1,08 (IC de 95%: 0,71-1,63) comparado con los pacientes tratados con IFN-a.
Supervivencia con calidad ajustada: La supervivencia con calidad ajustada se comparó para los grupos de tratamiento utilizando el enfoque Q-TWiST. La supervivencia se promedio por paciente con base en la presencia o ausencia de toxicidad o progreso mediante la aplicación de la escala EuroQoL 5D (EQ-5D) al inicio del estudio, en las semanas 12 y 32, cuando se reportaba un grado 3 o 4 de toxicidad, durante la recaída o el progreso, o durante el retiro del estudio. Temsirolimus 25 mg se asocia con un aumento estadísticamente significativo en el tiempo de supervivencia con calidad ajustada (Q-TWiST) de 1,3 meses (7,0 vs. 5,7 meses, 23%) estimado en comparación con IFN-a.
El Estudio 3 era un estudio aleatorio, doble ciego, multicéntrico con paciente externos que evaluaba la eficacia, la seguridad y la farmacocinética de tres niveles de dosis de temsirolimus cuando se administraba a pacientes tratados previamente con carcinoma de células renales avanzado. El punto de eficacia primario era la tasa de respuesta objetiva (ORR). Se evaluaron también la tasa de beneficio clínico, PFS, y OS. La PFS se definió como el tiempo transcurrido desde la primera dosis de temsirolimus hasta el progreso de la enfermedad o la muerte. Ciento once (111) pacientes fueron asignados de forma aleatoria a una razón de 1:1:1 para recibir semanalmente 25 mg, 75 mg, o 250 mg de temsirolimus intravenoso. En la rama de 25 mg, todos los pacientes tuvieron enfermedad metastática; 4 (11%) no se habían sometido previamente a quimioterapia o inmunoterapia; 17 (47%) se habían sometido a un tratamiento previo; y 15 (42%) se habían sometido a 2 o más tratamientos previos para RCC. Veintisiete (27,75%) se habían sometido a nefrectomía. Veinticuatro (24, 67%) se encontraban en un estado de desempeño (PS) = 1 de acuerdo con el Grupo de Oncología Cooperativa del Este (ECOG), y 12 (33%) tenían PS = 0 de ECOG.
Para pacientes tratados semanalmente con 25 mg de temsirolimus intravenoso, la mediana de supervivencia (OS) total fue 13,8 meses (IC de 95%: 9,0; 18,7 meses); la mediana de la supervivencia sin progreso (PFS) fue 6,3 meses (IC 95%: 3,6; 7,8 meses); la tasa de respuesta objetiva (ORR) fue 5,6% (IC de 95%: 0,7; 18,7%),y la tasa de beneficio clínico fue de 52,8% (IC 95%: 35,5; 69,6%).
CONTRAINDICACIONES: El temsirolimus intravenoso está contraindicado en personas con hipersensibilidad conocida al temsirolimus o cualquier otro componente de esta formulación.
Temsirolimus está contraindicado en pacientes con bilirrubina >1,5 x ULN (ver sección Advertencias especiales y precauciones de uso).
FERTILIDAD, EMBARAZO Y LACTANCIA
Fertilidad: Estudios en ratas han mostrado disminución de la fertilidad (ver sección Datos preclínicos de seguridad).
Embarazo: No existen datos disponibles del uso de temsirolimus en mujeres embarazadas.
No existen estudios adecuados y bien controlados con mujeres embarazadas que utilizan temsirolimus. Estudios en animales han demostrado toxicidad reproductiva (ver sección Datos preclínicos de seguridad). Se desconoce el posible riesgo para los humanos.
En estudios de toxicidad en animales con ratas y conejos, se aumentó la mortalidad del embrión/feto y disminuyó el desarrollo fetal (ver sección Datos preclínicos de seguridad).
Las mujeres en edad fértil deben utilizar durante y hasta 3 meses después del tratamiento un método de anticoncepción médicamente aceptado.
El temsirolimus intravenoso se debe utilizar durante el embarazo solamente si los beneficios potenciales justifican el riesgo potencial para el embrión/feto. Si una paciente queda embarazada durante el tratamiento con temsirolimus, ella y su médico deberán discutir en detalle el diagnóstico, las opciones alternas y los posibles riesgos del temsirolimus para el desarrollo del feto.
Además, los hombres deben tomar conciencia adecuada antes de iniciar el tratamiento con temsirolimus y deben comprender el posible peligro de tomar un medicamento con efectos desconocidos para el feto y el esperma. Los hombres con compañeras en edad fértil deben utilizar un método de anticoncepción médicamente aceptado durante el tratamiento y se recomienda continuarlo durante las 12 semanas después de recibir la última dosis de temsirolimus.
No existe información disponible sobre trabajo de parto y parto.
Lactancia: No se han realizado estudios de temsirolimus intravenoso relacionados con la lactancia.
Se desconoce si el temsirolimus se elimina en la leche materna. Debido a que muchos medicamentos se eliminan en la leche materna y que los efectos de la eliminación de temsirolimus en la leche materna no se han estudiado, se debe aconsejar a las mujeres no lactar mientras recibe temsirolimus.
EFECTOS INDESEABLES
La frecuencia esperada de las reacciones adversas se presenta en las categorías de frecuencia CIOMS:
Muy frecuente: >10%
Frecuente: >1 % y <10 %
Poco frecuente: >0,1% y <1%
Raro: >0,01% y <0,1%
Muy raro: <0,01%
Frecuencia desconocida: No se puede determinar con la información disponible.
Reaccciones adversas de carcinoma de células renales
|
Clasificación por órganos y sistemas |
Frecuencia |
Reacciones adversas |
Todos los grados |
Grado 3 y 4 |
|
Infecciones e infestaciones |
Muy frecuentes |
Infecciones virales y bacterianas (incluso infección, infección viral, celulitis, herpes zóster, herpes oral, gripe, herpes simple, infección bacteriana, bronquitis, absceso, infección de herida, infección de heridas postquirúrgicas) |
39 (18,8) |
3 (1,4) |
|
Infección urinaria (incluso cistitis) |
21 (10,1) |
5 (2,4) |
||
|
Frecuentes |
Neumoníaa |
16 (7,7) |
6 (2,9) |
|
|
Infección del tracto respiratorio superior |
13 (6,3) |
0 (0,0) |
||
|
Candidiasis (incluso candidiasis oral y anal) e infección fúngica/infecciones cutáneas fúngicas |
8 (3,9) |
0 (0,0) |
||
|
Faringitis |
4 (1,9) |
0 (0,0) |
||
|
Rinitis |
4 (1,9) |
0 (0,0) |
||
|
Sinusitis |
3 (1,4) |
0 (0,0) |
||
|
Foliculitis |
3 (1,4) |
0 (0,0) |
||
|
Poco frecuentes |
Laringitis |
1 (0,5) |
0 (0,0) |
|
|
Sepsis |
1 (0,5) |
1 (0,5) |
||
|
Trastornos de la sangre y el sistema linfático |
Muy frecuentes |
Anemia |
82 (39,4) |
34 (16,3) |
|
Trombocitopenia |
24 (11,5) |
3 (1,4) |
||
|
Frecuentes |
Neutropenia |
12 (5,8) |
6 (2,9) |
|
|
Leucopenia |
11 (5,3) |
1 (0,5) |
||
|
Linfopenia |
11 (5,3) |
9 (4,3) |
||
|
Trastornos del sistema inmunológico |
Frecuentes |
Reacciones de hipersensibilidad/hipersensibilidad a medicamentos |
19 (9,1) |
1 (0,5) |
|
Trastornos metabólicos y nutricionales |
Muy frecuentes |
Disminución del apetito |
66 (31,7) |
6 (2,9) |
|
Hiperglucemia |
48 (23,1) |
22 (10,6) |
||
|
Hipercolesterolemia |
48 (20,2) |
1 (0,5) |
||
|
Hipertrigliceridemia |
42 (20,2) |
7 (3,4) |
||
|
Frecuentes |
Hipocalemia |
19 (9,1) |
7 (3,4) |
|
|
Hipofosfatemia |
15 (7,2) |
9 (4,3) |
||
|
Deshidratación |
10 (4,8) |
5 (2,4) |
||
|
Hipocalcemia |
10 (4,8) |
2 (1,0) |
||
|
Diabetes mellitus |
9 (4,3) |
2 (1,0) |
||
|
Hiperlipidemia |
3 (1,4) |
0 (0,0) |
||
|
Trastornos psiquiátricos |
Muy frecuentes |
Insomnio |
26 (12,5) |
1 (0,5) |
|
Frecuentes |
Ansiedad |
16 (7,7) |
0 (0,0) |
|
|
Depresión |
8 (3,8) |
0 (0,0) |
||
|
Trastornos del sistema nervioso |
Muy frecuentes |
Disgeusia |
39 (18,8) |
0 (0,0) |
|
Cefalea |
32 (15,4) |
1 (0,5) |
||
|
Frecuentes |
Mareos |
20 (9,6) |
1 (0,5) |
|
|
Parestesia |
10 (4,8) |
1 (0,5) |
||
|
Somnolencia |
8 (3,8) |
1 (0,5) |
||
|
Ageusia |
2 (1,0) |
0 (0,0) |
||
|
Convulsión |
2 (1,0) |
0 (0,0) |
||
|
Poco frecuentes |
Hemorragia intracraneal |
1 (0,5) |
1 (0,5) |
|
|
Trastornos oculares |
Frecuentes |
Conjuntivitis (incluso conjuntivitis, trastorno del aparato lagrimal) |
10 (4,8) |
1 (0,5) |
|
Trastornos cardíacos |
Frecuentes |
Derrame pericárdico |
2 (1,0) |
1 (0,5) |
|
Trastornos vasculares |
Frecuentes |
Hipertensión |
13 (6,3) |
3 (1,4) |
|
Tromboembolia venosa (incluso trombosis venosa profunda) |
3 (1,4) |
3 (1,4) |
||
|
Tromboflebitis |
2 (1,0) |
0 (0,0) |
||
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy frecuentes |
Tos |
54 (26,0) |
1 (0,5) |
|
Disneaa |
53 (25,5) |
16 (7,7) |
||
|
Epistaxis |
27 (13,0) |
0 (0,0) |
||
|
Frecuentes |
Derrame pleural |
8 (3,8) |
6 (2,9) |
|
|
Embolia pulmonara |
2 (1,0) |
1 (0,5) |
||
|
Enfermedad pulmonar intersticial* |
6 (2,9) |
1 (0,5) |
||
|
Trastornos gastrointestinales |
Muy frecuentes |
Náuseas |
78 (37,5) |
5 (2,4) |
|
Diarrea |
60 (28,8) |
3 (1,4) |
||
|
Estomatitis |
44 (21,2) |
2 (1,0) |
||
|
Estreñimiento |
43 (20,7) |
0 (0,0) |
||
|
Vómitos |
42 (20,2) |
4 (1,9) |
||
|
Dolor abdominal |
31 (14,9) |
7 (3,4) |
||
|
Frecuentes |
Distensión abdominal |
13 (6,3) |
1 (0,5) |
|
|
Estomatitis aftosa |
8 (3,8) |
1 (0,5) |
||
|
Hemorragia gastrointestinal (incluso hemorragia rectal, hemorroidal, bucal y en los labios) |
7 (3,4) |
2 (1,0) |
||
|
Disfagia |
5 (2,4) |
0 (0,0) |
||
|
Gastritis |
2 (1,0) |
0 (0,0) |
||
|
Dolor en la cavidad oral |
3 (1,4) |
0 (0,0) |
||
|
Gingivitis |
3 (1,4) |
0 (0,0) |
||
|
Poco frecuentes |
Perforación intestinala |
1 (0,5) |
0 (0,0) |
|
|
Trastornos de la piel y del tejido subcutáneo |
Muy frecuentes |
Erupción (incluso erupción, erupción prurítica, erupción maculopapular, erupción generalizada, erupción macular, erupción papular) |
91 (43,8) |
10 (4,8) |
|
Prurito (incluso prurito generalizado) |
40 (19,2) |
1 (0,5) |
||
|
Sequedad de la piel |
21 (10,1) |
1 (0,5) |
||
|
Frecuentes |
Trastorno de las uñas |
16 (7,7) |
0 (0,0) |
|
|
Acné |
13 (6,3) |
0 (0,0) |
||
|
Erupción exfoliativa |
5 (2,4) |
0 (0,0) |
||
|
Dermatitis |
4 (1,9) |
0 (0,0) |
||
|
Trastornos musculoesqueléticos y del tejido conectivo |
Muy frecuentes |
Lumbalgia |
37 (17,8) |
5 (2,4) |
|
Artralgia |
35 (16,8) |
2 (1,0) |
||
|
Frecuentes |
Mialgia |
13 (6,3) |
0 (0,0) |
|
|
Trastornos renales y urinarios |
Frecuentes |
Insuficiencia renala |
2 (1,0) |
0 (0,0) |
|
Trastornos generales y afecciones en el lugar de la administración |
Muy frecuentes |
Edema (edema generalizado, edema facial y edema periférico) |
85 (40,9) |
7 (3,3) |
|
Fatiga |
82 (39,4) |
22 (10,6) |
||
|
Pirexia |
48 (23,1) |
1 (0,5) |
||
|
Asteniaa |
41 (19,7) |
5 (2,4) |
||
|
Inflamación de las mucosas |
38 (18,3) |
4 (1,9) |
||
|
Dolor de pecho |
24 (11,5) |
1 (0,5) |
||
|
Frecuentes |
Dolor |
20 (9,6) |
5 (2,4) |
|
|
Escalofríos |
13 (6,3) |
0 (0,0) |
||
|
Problemas de cicatrización |
2 (1,0) |
0 (0,0) |
||
|
Investigaciones |
Muy frecuentes |
Aumento de creatinina en sangre |
26 (12,5) |
4 (1,9) |
|
Frecuentes |
Aspartato aminotransferasa elevada |
19 (9,1) |
4 (1,9) |
|
|
Alanina aminotransferasa elevada |
13 (6,3) |
1 (0,5) |
||
|
a Se notificó un caso mortal * La enfermedad de pulmón intersticial enfermedad pulmonar intersticial es definida por un grupo de Términos Preferentes: enfermedad del pulmón intersticial (n=2), neumonitis (n=3), alveolitis (n=1). |
||||
Las reacciones adversas serias observadas en los estudios clínicos de temsirolimus para carcinoma de células renales incluyen: Anafilaxia, deterioro de la cicatrización de heridas, falla renal con resultados mortales, derrame pericárdico (incluye derrame pericárdico hemodinámicamente significativo que requiere intervención), convulsiones y embolia pulmonar.
Experiencia postcomercialización y otra experiencia clínica
|
Clasificación por órganos y sistemas |
Frecuencia |
Reacciones adversas |
|
Trastornos del sistema inmune |
No conocida* |
Reacciones de tipo edema angioneurótico |
|
Trastornos de la piel y del tejido subcutáneo |
No conocida |
Síndrome de Stevens-Johnson |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
No conocida |
Rabdomiólisis |
|
Infecciones e infestaciones |
Rara |
Neumonía por Pneumocystis jiroveci |
|
*no se puede calcular a partir de los datos disponibles |
||
Reacciones de tipo edema angioneurótico en algunos pacientes que recibieron temsirolimus e inhibidores de ACE simultáneamente.
Se han informado casos de neumonía por Pneumocystis jiroveci, algunos con resultados fatales (véase la sección Advertencias especiales y precauciones de uso).
EFECTO EN ACTIVIDADES QUE REQUIEREN CONCENTRACIÓN Y DESEMPEÑO: No se han desarrollado estudios que evalúen los efectos en la habilidad para manejar o uso de maquinas.
INTERACCIONES
Agentes inducidores del metabolismo de la CYP3A: La coadministración del Concentrado de Temsirolimus para Inyección con rifampicina, un potente inductor de la CYP3A4/5, no tiene efecto significativo sobre la Cmax (máxima concentración) y la AUC (área bajo la curva) del temsirolimus después de administración intravenosa; pero la Cmax del sirolimus disminuyó en 65% y la AUC del sirolimus en 56%, y la AUCsum (combinación de la AUC de temsirolimus más la AUC de sirolimus) en 41% comparado con el tratamiento con solo temsirolimus. Por ello, se debe evitar el tratamiento concomitante con agentes que tienen potencial de inducción de la CYP3A4/5. Si no se puede administrar un tratamiento alternativo, se debe considerar una dosis intravenosa semanal de temsirolimus de hasta 50 mg para pacientes con carcinoma de células renales (ver sección Advertencias especiales y precauciones de uso).
Agentes inhibidores del metabolismo de la CYP3A: La coadministración de temsirolimus con ketoconazol, un inhibidor potente de la CYP3A4, no tiene efectos significativos sobre la Cmax o la AUC del temsirolimus; sin embargo, se presentó aumento de la AUC del sirolimus en 3,1 veces y la AUCsum aumentó 2,3 veces en comparación con el temsirolimus solo. Las sustancias que son inhibidores potentes de la actividad de la CYP3A4 aumentan las concentraciones sanguíneas de sirolimus.
El tratamiento concomitante del Concentrado de Temsirolimus para Inyección con agentes que tienen fuerte potencial de inhibición de la CYP3A4 se debe evitar (ver sección Advertencias especiales y precauciones de uso). El tratamiento concomitante con inhibidores moderados de CYP3A4 debe administrarse únicamente con precaución en pacientes que reciben 25 mg y se debe evitar en pacientes que reciben dosis de temsirolimus mayores de 25 mg.
Interacciones con medicamentos metabolizados por la CYP2D6: En 23 sujetos sanos la concentración de desipramina, un sustrato de la CYP2D6, no se afectó cuando se coadministraban 25 mg de temsirolimus. No se anticipa efecto clínico significativo cuando el temsirolimus se coadministra con agentes que son metabolizados por la CYP2D6.
Interacciones con medicamentos que son sustratos de la glicoproteína P: En un estudio in vitro, el temsirolimus inhibió el transporte de digoxina, un sustrato de la glicoproteína P, con un valor CI50 de 2µM. Se desconocen las implicaciones clínicas relacionadas con la administración concomitante de los sustratos de la glicoproteína P.
DATOS PRECLÍNICOS DE SEGURIDAD
Embarazo: En estudios orales de toxicidad durante el desarrollo realizados en ratas, existió aumento en la mortalidad del embrión/feto y disminución del crecimiento fetal a dosis > 0,45 mg/kg/día (1,1 veces la dosis recomendada para humanos en mg/kg/día y 0,18 veces la dosis recomendada en mg/m2).
En estudios orales de toxicidad durante el desarrollo en conejos, existió aumento en la mortalidad del embrión/feto y disminución el crecimiento fetal a dosis > 0,6 mg/kg/día (1,4 veces la dosis recomendada para humanos en mg/kg/día y 0,47 la dosis recomendada en mg/m2). En conejos, existió un aumento en la incidencia de la protrusión intestinal en el abdomen a 0,9 mg/kg/día (2,1 veces la dosis recomendada para humanos en mg/kg/día y 0,7 veces la dosis recomendada en mg/m2).
Carcinogenicidad: No se han realizado estudios de carcinogenicidad con temsirolimus.
Mutagenicidad: Temsirolimus no fue genotóxico en una batería de ensayos in vitro (mutación bacteriana inversa en la Salmonella typhimurium y la Escherichia coli, mutación directa en células linfáticas de ratón, y aberraciones cromosómicas en células ováricas de hámster chino) y ensayos in vivo (micronúcleos del ratón).
Deterioro de la fertilidad: En ratas macho, la fertilidad se disminuyó a > 0,5 mg/kg/día (1,2 veces la dosis humana recomendada para humanos en mg/kg/día y 0,19 veces la dosis recomendada en mg/m2). Se presentó ausencia de fertilidad a 5 mg/kg/día (11,9 veces la dosis recomendada para humanos en mg/kg/día y 1,9 veces la dosis recomendada para humanos en mg/m2).
Estos efectos sobre la fertilidad en el macho estuvieron acompañados por degeneración tubular testicular, disminución en la concentración del esperma y motilidad, y pesos disminuidos en los órganos genitales a dosis > 0,5 mg/kg/día.
En ratas hembra, existieron aumentos en las incidencias de daños antes y después de la implantación a dosis > 0,7 mg/kg/día, que resultaron en disminución del número de fetos vivos (1,7 veces la dosis humana recomendada en mg/kg/día y 0,27 veces la dosis humana recomendada en mg/m2). Los pesos fetales se disminuyeron a dosis > 1 mg/kg/día (2,4 veces la dosis humana recomendada en mg/kg/día y 0,39 veces la dosis humana recomendada en mg/m2).
ADVERTENCIAS ESPECIALES Y PRECAUCIONES DE USO
Reacciones de hipersensibilidad o reacciones a la infusión: Las reacciones de hipersensibilidad o las reacciones a la infusión (incluidas reacciones que ponen en peligro la vida y reacciones mortales poco frecuentes), incluidas entre otras, rubefacción, dolor de pecho, disnea, hipotensión, apnea, pérdida de la conciencia, hipersensibilidad y anafilaxia, se han asociado con la administración del temsirolimus. Estas reacciones pueden ocurrir muy temprano en la primera infusión, pero pueden también presentarse en las infusiones posteriores. Los pacientes de deben monitorear al comienzo de la infusión y se debe tener disponible el apoyo apropiado. La infusión de temsirolimus se debe interrumpir en todos los pacientes con reacciones graves a la infusión y se deberá administrar terapia médica apropiada. En pacientes con reacciones graves o que ponen en peligro la vida se deberá realizar una evaluación beneficio-riesgo antes de continuar con la terapia de temsirolimus.
Sirolimus es el metabolito principal del temsirolimus; por ello, temsirolimus se debe administrar con precaución en pacientes con hipersensibilidad conocida al Sirolimus.
Como se recomienda que se administre una antihistamina H1 a los pacientes antes del inicio de la infusión intravenosa de temsirolimus, el temsirolimus se debe utilizar con precaución en pacientes con hipersensibilidad conocida a una antihistamina o que no pueden recibir una antihistamina por otras razones médicas.
Si un paciente desarrolla una reacción de hipersensibilidad durante la infusión de temsirolimus a pesar de la premedicación, se debe detener la infusión y el paciente se debe observar al menos durante 30 a 60 minutos (dependiendo de la severidad de la reacción). A discreción del médico, se puede reasumir el tratamiento con la administración de un antagonista del receptor H1 (como por ejemplo difenhidramina), si no se administró previamente, y/o un antagonista del receptor H2 (como por ejemplo famotidina 20 mg intravenosa o ranitidina 50 mg intravenosa) aproximadamente 30 minutos antes de reiniciar la infusión de temsirolimus. La infusión se puede reasumir a una menor tasa (durante hasta 60 minutos).
Deterioro hepático: El temsirolimus fue evaluado en un estudio de fase 1 de aumento de la dosis en 110 pacientes con diversos grados de deterioro hepático y normales, según se establecía mediante las concentraciones de AST y bilirrubina y pacientes con transplante hepático (Tabla 1). Durante el estudio, los pacientes con deterioro hepático moderado y grave presentaban aumentos en las tasas de eventos adversos y de las muertes, incluidas muertes debido al empeoramiento del cáncer subyacente (Tabla 1).
|
Tabla 1 – Eventos Adversos en Pacientes con Cánceres Avanzados y Función Hepática Normal o Deteriorada |
|||
|
Función Hepática* |
Intervalo de Dosis de Temsirolimus |
Eventos Adversos de Grado =3** |
Muerte General*** |
|
Normal (n = 25) |
25 – 175 |
20 (80,0) |
2 (8,0) |
|
Leve (n = 39) |
10 – 25 |
32 (82,1) |
5 (12,8) |
|
Moderada (n = 20) |
10 – 25 |
19 (95,0) |
8 (40,0) |
|
Grave (n = 24) |
7,5 – 15 |
23 (95,8) |
13 (54,2) |
|
Transplante Hepático (n = 2) |
10 |
1 (50,0) |
0 (0) |
|
* Grupos de Función Hepática: Normal = bilirrubina y AST £ ULN; Leve = bilirrubina > 1-1,5 x ULN o AST > ULN pero la bilirrubina £ ULN; Moderada = bilirrubina > 1,5-3 x ULN; Grave = bilirrubina > 3 x ULN, Transplante hepático = cualquier valor de bilirrubina y AST. ** Criterios de Terminología Común para Eventos Adversos, versión 3.0, incluidas todas las causalidades. *** Incluidas las muertes debido a empeoramiento del cáncer subyacente y reacciones adversas. |
|||
La seguridad y farmacocinética de temsirolimus fueron evaluadas en un estudio de fase 1 de aumento de la dosis en 110 pacientes con cáncer con deterioro hepático normal o de diversos grados. Los pacientes con bilirrubina al inicio del estudio > 1,5 x ULN experimentaron mayor toxicidad que los pacientes con bilirrubina al inicio del estudio = 1,5 x ULN cuando se trataban con temsirolimus. La frecuencia general de los evento adversos de grado = 3 y de las muertes, incluidas muertes debido a empeoramiento de la enfermedad fueron mayores en pacientes con bilirrubina al inicio del estudio > 1,5 x ULN. Temsirolimus está contraindicado en pacientes con bilirrubina > 1,5 x ULN debido al aumento del riesgo de muerte, incluidas muertes por empeoramiento del cáncer subyacente (ver sección Contraindicaciones).
Cuando se trate pacientes con deterioro hepático leve se debe tener precaución. Las concentraciones de temsirolimus y el metabolito sirolimus aumentaron en pacientes con concentraciones elevadas de AST o bilirrubina. Antes de iniciar el tratamiento con temsirolimus y después de iniciado se recomienda evaluar periódicamente las concentraciones de AST y bilirrubina.
Hiperglicemia/Intolerancia a la Glucosa: La utilización de Temsirolimus Concentrado para Inyección en pacientes con carcinoma de células renales se asoció con el aumento de la glucosa sérica. En el Estudio 1, un estudio clínico de fase 3 para el carcinoma de células renales (Estudio 3066K1-304), 26% de los pacientes informaron hiperglicemia como un evento adverso. Esto puede requerir del aumento en la dosis o el inicio de terapia con un agente hipoglicémico oral y/o insulina. Se debe instruir a los pacientes para que reporten si presentan sed excesiva o cualquier aumento en el volumen o frecuencia de la micción.
Infecciones: El paciente se puede inmunosuprimir y se debe observar cuidadosamente para determinar la ocurrencia de infecciones, incluyendo las infecciones oportunistas. Se han informado casos de neumonía por Pneumocystis jiroveci (NPJ), algunos con resultados fatales, en pacientes que recibieron temsirolimus; muchos de los cuales también recibieron corticosteroides u otros agentes inmunodepresivos. Para los pacientes que requieren el uso simultáneo de corticosteroides u otros agentes inmunodepresivos se puede considerar la profilaxis de NPJ.
Enfermedad pulmonar intersticial: Han existido casos de neumonitis intersticial no específica, incluyendo informes de casos fatales, que ocurrieron en pacientes que recibieron temsirolimus intravenoso semanalmente. Algunos pacientes fueron asintomáticos o presentaron síntomas mínimos con neumonitis detectada mediante escáner de tomografía computarizada o radiografía de tórax. Otros presentaron síntomas como disnea, tos y fiebre. Algunos pacientes necesitaron descontinuar el temsirolimus o el tratamiento con corticoesteroides y/o antibióticos, mientras que algunos pacientes continuaron el tratamiento sin intervención adicional.
Antes de iniciar el tratamiento con temsirolimus, se recomienda que los pacientes sean sometidos a evaluación radiográfica inicial mediante tomografía axial computarizada de los pulmones o radiografía de tórax. Se debe hacer seguimiento periódico a dichas evaluaciones, incluso en la ausencia se síntomas clínicos respiratorios.
Se recomienda que los pacientes sean controlados de cerca para detectar presencia de síntomas respiratorios clínicos.
Si se desarrollan síntomas respiratorios clínicos significativos, se debe considerar la interrupción de la administración del temsirolimus hasta que desaparezcan los síntomas y mejoren los resultados radiográficos relacionados con neumonitis. Las infecciones oportunistas, tales como la neumonía por Pneumocystis jiroveci (NPJ) se deberían considerar en el diagnóstico diferencial. Se puede considerar el tratamiento empírico con corticoesteroides y/o antibióticos. Para los pacientes que requieren el uso de corticosteroides se puede considerar la profilaxis de NPJ.
Hiperlipemia: La utilización de temsirolimus se asoció con el aumento en los triglicéridos y colesterol séricos. En el Estudio 1, se reportó hiperlipemia como un evento adverso en el 27% de los pacientes. En el Estudio 2, se reportó como evento adverso hiperlipemia en 9,3% de los pacientes. Esto puede requerir inicio o aumento en la dosis de agentes hipolipemiantes. El colesterol y los triglicéridos séricos se deben probar antes y durante el tratamiento con temsirolimus.
Perforación del intestino: Se han presentado casos de perforación del intestino (que incluyen resultados fatales) en pacientes que recibieron temsirolimus (ver sección Efectos indeseables).
Complicaciones en la cicatrización de las heridas: La utilización de temsirolimus se ha asociado con cicatrización anormal de las heridas. Por lo tanto, se debe tener precaución en la utilización de temsirolimus durante periodos periquirúrgicos.
Sangrado intracerebral: Los pacientes con tumores en el sistema nervioso central (tumores primarios o metástasis en el SNC) y/o que reciben terapia anticoagulante pueden tener mayor riesgo de desarrollar sangrado intracerebral (incluidos resultados fatales) mientras reciben terapia con temsirolimus.
Falla renal: Se ha observado falla renal (incluyendo resultados fatales) en pacientes que recibían temsirolimus por cáncer de células renales avanzado y/o con insuficiencia renal preexistente (ver sección Efectos indeseables).
Uso concomitante de Temsirolimus con Sunitinib: La combinación de temsirolimus y sunitinib produjo toxicidad limitadora de la dosis. Las toxicidades limitadoras de la dosis (de grado 3 sarpullido maculopapular eritematoso, gota/celulitis que requieren hospitalización) fueron observadas en dos de tres pacientes tratados en la primera cohorte de un estudio de fase I a dosis de 15 mg de temsirolimus intravenoso por semana y 25 mg de sunitinib oral por día (días 1-28 seguidos por un descanso de 2 semanas).
Uso concomitante de inhibidores de la enzima convertidora de la angiotensina (ECA): Se han observado reacciones tipo edema angioneurótico (que incluyen reacciones tardías que aparecieron dos meses después de iniciado el tratamiento) en algunos pacientes que recibieron de forma concomitante temsirolimus e inhibidores ECA.
Ancianos: No se observaron diferencias específicas en cuanto a seguridad entre pacientes menores a 65 años y pacientes mayores a 65 años.
Con base en los resultados del estudio de fase 3 para carcinoma de células renales, los pacientes ancianos tienen mayor probabilidad de experimentar ciertas reacciones adversas, incluidos edema, diarrea y neumonía.
La tasa de supervivencia en el subgrupo de pacientes de 65 años y mayores (n=64) tratados con temsirolimus en el estudio 1 fue más corta que los pacientes menores de 65 años (ver sección Propiedades farmacodinámicas). La relevancia clínica del análisis de este subgrupo no es clara.
Ningún ajuste específico de dosis es recomendado en pacientes ancianos.
Población pediátrica: No se recomienda la utilización de temsirolimus en pacientes pediátricos debido a que existen datos insuficientes sobre su eficacia.
Se encuentran disponibles datos limitados sobre la utilización de temsirolimus en pacientes pediátricos. La eficacia del temsirolimus en pacientes con tumores sólidos avanzados recurrentes/resistentes al tratamiento no se ha podido establecer.
Temsirolimus se ha estudiado en un total de 71 pacientes pediátricos (de un año a 21 años de edad) con tumores sólidos recurrentes/resistentes al tratamiento en un estudio de seguridad de fase I/II y farmacodinámica exploratorio.
En la parte 1, 19 pacientes pediátricos con tumores sólidos recurrentes/resistentes al tratamiento avanzados recibieron Temsirolimus a dosis que variaban entre 10 mg/m2 y 150 mg/m2 como infusión IV durante 60 minutos una vez a la semana y en ciclos de tres semanas. La dosis seleccionada para la parte 2 fue 75 mg/m2.
En la parte 2, 52 pacientes pediátricos con neuroblastoma, rabdomiosarcoma o glioma de alto grado recurrente/resistente al tratamiento recibieron Temsirolimus a una dosis semanal de 75 mg/m2. La tasa de respuesta general fue 1,9%. Se estableció que una dosis de 75 mg/m2 de Temsirolimus administrada semanalmente tenía suficiente eficacia en pacientes pediátricos con neuroblastoma, rabdomiosarcoma o glioma de alto grado recurrente/resistente al tratamiento.
Las reacciones adversas generales asociadas con Temsirolimus fueron similares a las observadas en adultos. Las reacciones adversas reportadas por el porcentaje más alto de pacientes fueron trastornos hematológicos (anemia, leucopenia, neutropenia y trombocitopenia), metabólicos (hipercolesterolemia, hiperlipemia, hiperglicemia, aumento del aspartato aminotransferasa sérico [AST] y en los niveles plasmáticos de alanina aminotransferasa [ALT]) y digestivos (mucositis, estomatitis, naúseas y vómito).
Cataratas: Se han observado cataratas en algunos pacientes que recibieron de forma combinada temsirolimus e interferón alfa (IFN-a).
Agentes inducidores del Metabolismo de la CYP3A: Los agentes como la carbamazepina, la fenitoina, los barbitúricos, la rifabutina, la rifampicina, y la Hierba de San Juan son inducidores fuertes de la CYP3A4/5 y pueden disminuir las exposiciones combinadas de los grupos activos, temsirolimus y su metabolito, sirolimus. Por ello, en pacientes con carcinoma de células renales, se debe evitar el tratamiento concomitante con agentes que tienen potencial de inducción de la CYP3A4/5. Si no se puede administrar tratamiento alterno, se debe considerar una dosis intravenosa semanal de hasta 50 mg (ver sección Interacciones).
Agentes inhibidores del Metabolismo de la CYP3A: Los agentes como los inhibidores de proteasa, los antifúngicos, los antibióticos macrólidos, la nefazodona, y los inhibidores selectivos de la serotonina son inhibidores fuertes de la CYP3A4 y pueden aumentar las concentraciones sanguíneas de los grupos activos, temsirolimus y su metabolito, sirolimus. Por ello, se debe evitar el tratamiento concomitante con agentes que tienen potencial de inhibición de la CYP3A4. El tratamiento concomitante con inhibidores moderados de CPY3A4 se deberá administrar únicamente con precaución en pacientes que reciben 25 mg y se deberá en evitar en pacientes que reciben dosis de temsirolimus mayores de 25 mg.
Se deben considerar los tratamientos alternativos con agentes que no tienen potencial de inhibición de la CYP3A4 (ver sección Interacciones).
Vacunaciones: La utilización de vacunas vivas se debe evitar durante el tratamiento con temsirolimus. Ejemplos de vacunas vivas son: vacunas contra el sarampión, las paperas, la rubéola, vacuna oral contra el polio, vacuna BCG, vacuna contra la fiebre amarilla, contra la varicela, y vacuna contra la tifoidea TY21a.
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN
Modo de administración: Intravenoso (IV).
Preparación y precauciones de administración: El tratamiento se debe continuar hasta que el paciente no se beneficie clínicamente más de la terapia o hasta que ocurra toxicidad inaceptable. No se requiere modificación especial de la dosis para ninguna de las poblaciones que se han estudiado (por ejemplo: según el género, ancianos).
Instrucciones para la administración intravenosa
— El Concentrado Temsirolimus para inyección y el diluyente se deben almacenar bajo refrigeración a 2º- 8 ºC y protegidos de la luz (ver sección Precauciones especiales de almacenamiento y Naturaleza del material de envase). Durante la manipulación y preparación de las mezclas, el Concentrado de Temsirolimus para Inyección se debe proteger de la luz ambiental y la luz solar excesivas.
— El concentrado de Temsirolimus para inyección y el diluyente se deben inspeccionar visualmente para determinar la presencia de material particulado y decoloración. Las bolsas/envases que entren en contacto con Concentrado de Temsirolimus para Inyección deben estar fabricados en vidrio, poliolefina o polietileno.
NO UTILIZAR SI HAY PRESENCIA DE PARTÍCULAS. USE UN NUEVO VIAL.
Premedicación: Los pacientes deben recibir medicación profiláctica de difenhidramina intravenosa 25 a 50 mg (o equivalente) aproximadamente 30 minutos antes del inicio de cada dosis de infusión de temsirolimus. Si una reacción de hipersensibilidad o una reacción a la infusión se desarrolla durante la infusión de temsirolimus, se debe detener la infusión. Una vez desaparezca la reacción, y a criterio del médico, se puede reiniciar el tratamiento con la administración de un antagonista del receptor H1 (o equivalente), si no se ha administrado previamente, y/o un antagonista del receptor H2 (como por ejemplo famotidina 20 mg intravenosa o ranitidina 50 mg intravenosa) aproximadamente 30 minutos antes de reiniciar la infusión de temsirolimus. La infusión se puede reiniciar a una tasa más lenta (hasta los 60 minutos) (ver sección Advertencias especiales y precauciones de uso).
Dilución: Cada vial de Temsirolimus se debe diluir con Diluyente de acuerdo con las instrucciones presentadas a continuación. El contenido diluido necesario de cada vial se debe combinar en una jeringa para inyección en 250 ml de cloruro de sodio para inyección 0,9%.
La solución diluida (Concentrado y diluyente) debe ser inspeccionada visualmente para detectar material particulado o decoloración.
Durante la preparación de la solución de administración de temsirolimus, siga el siguiente proceso de dilución de dos pasos de forma aséptica
— Paso 1: Inyecte 1,8 ml de diluente del Concentrado de Temsirolimus para Inyección dentro del vial del Concentrado de Temsirolimus para Inyección. Mezcle bien invirtiendo el vial. La concentración será 10 mg/ml. Espere el tiempo suficiente para que las burbujas de aire disminuyan. La solución será clara a levemente turbia, incolora entre amarillo claro y amarillo, esencialmente libre de partículas visibles. Un volumen de 1,2 ml del concentrado de Temsirolimus contiene un total de 30 mg del fármaco. Cuando 1,2 ml del concentrado de Temsirolimus se combina con 1,8 ml de diluente, se obtiene un volumen total de 3,0 ml. Treinta miligramos (30 mg) del fármaco por 3,0 ml es igual a 10 mg/ml del fármaco. La mezcla de la solución del concentrado de Temsirolimus y su diluente es estable por hasta 24 horas a temperatura controlada de 25 ºC.
— Paso 2: Extraiga la cantidad requerida de la mezcla de Diluente/Concentrado de Temsirolimus para Inyección del paso 1 (10 mg/ml) e inyéctela rápidamente dentro de 250 ml de cloruro de sodio al 0,9% para inyección asegurando la mezcla adecuada. Mezcle invirtiendo la bolsa o el frasco. Evite agitar de forma excesiva porque esto puede producir espuma.
La solución final diluida en bolsa o botella debe ser inspeccionada visualmente en búsqueda de material particulado.
Administración
— La administración de la solución final diluida para infusión se deberá finalizar dentro de las seis horas a partir del momento en que la mezcla de diluente y el Concentrado de Temsirolimus se adicionan al cloruro de sodio para inyección.
— El Concentrado de Temsirolimus para Inyección se administra mediante infusión durante un periodo de 30 - 60 minutos una vez a la semana. La utilización de una bomba de infusión es el método preferido de administración para asegurar la aplicación precisa del medicamento.
— Los materiales apropiados para la administración deben ser de vidrio, poliolefina o polietileno para evitar la pérdida excesiva del medicamento y disminuir la tasa de extracción de di-(2-etilhexil) ftalato (DEHP). Los materiales utilizados para la administración deben componerse de tuberías que no estén fabricadas de DEHP ni de cloruro de polivinilo (PVC) con el filtro apropiado. Para la administración se recomienda el uso de un filtro en línea de polietersulfona con tamaño de poro no mayor a 5 micrones, esto con el fin de evitar la infusión de partículas más grandes de 5 micrones. Si el set de administración disponible no tiene el filtro en línea incorporado un filtro debe ser adicionado al final del set (Por ejemplo un filtro terminal) antes de que la mezcla alcance la vena del paciente. Diferentes filtros terminales pueden ser usados con tamaños de poro entre 0.2 micrones y 5 micrones. No se recomienda el uso de ambos filtros en línea y terminales.
Es importante que se sigan estrictamente las recomendaciones dadas en esta sección.
El concentrado de Temsirolimus para inyección, cuando se constituye, contiene polisorbato 80, el cual se sabe, incrementa la tasa de extracción del di-(2-etilhexil)ftalato (DEHP) del PVC. Esto debe considerarse durante la preparación y administración del concentrado de Temsirolimus para inyección, incluyendo el tiempo de almacenamiento transcurrido en el contenedor de PVC seguida a la reconstitución. Es importante que se sigan muy de cerca las recomendaciones de esta sección.
Carcinoma de células renales: Las dosis recomendadas de Temsirolimus para carcinoma avanzado de células renales es de 25 mg, en infusión durante un periodo de 30 - 60 minutos, una vez a la semana.
El manejo de las reacciones que se sospecha son ocasionadas por el medicamento puede requerir interrupción temporal y/o la reducción de la dosis de la terapia con temsirolimus. Si una reacción sospechosa no se puede manejar con retrasos en las dosis, el Concentrado de Temsirolimus para Inyección se puede reducir mediante decrementos de 5 mg/semana.
Utilización en pacientes con deterioro renal: Después de una dosis intravenosa de 25 mg de temsirolimus etiquetado con [14C] en sujetos sanos, la eliminación renal de la radioactividad total fue 4,6% de la dosis administrada. La eliminación renal es una vía menor; por ello, no se espera que el deterioro renal influya de forma marcada en la exposición al medicamento, y no se requiere ajuste de la dosis del Concentrado Temsirolimus para Inyección en pacientes con deterioro renal. No se han realizado estudios en pacientes con diferentes grados de deterioro renal.
El Concentrado de Temsirolimus para Inyección no se ha estudiado en pacientes sometidos a hemodiálisis (ver sección Propiedades farmacocinéticas).
Utilización en pacientes con deterioro hepático: La seguridad y farmacocinética del temsirolimus fueron evaluadas en un estudio de fase 1 de escalamiento de dosis en 110 pacientes con cáncer con diversos grados de deterioro hepático o normal. Los pacientes con bilirrubina al inicio del estudio >1,5 x ULN experimentaron mayor toxicidad que los pacientes con bilirrubina <1,5 x ULN al inicio del estudio cuando se trataron con temsirolimus. La frecuencia general de reacciones adversas mayores a grado 3 y las muertes, incluidas muertes debidas a enfermedad progresiva, fueron mayores en pacientes con bilirrubina al inicio del estudio >1,5 x ULN. Temsirolimus está contraindicado en pacientes con bilirrubina >1,5 x ULN debido al mayor riesgo de muerte, incluidas muertes por empeoramiento del cáncer subyacente (ver sección Contraindicaciones).
Se debe tener precaución cuando se trata pacientes con deterioro hepático leve. Las concentraciones de temsirolimus y su metabolito sirolimus se aumentaron en pacientes con niveles elevados de AST o bilirrubina. Se recomienda antes del inicio del tratamiento y una vez iniciado el tratamiento, evaluar periódicamente las concentraciones de AST y bilirrubina.
Utilización en niños: Se encuentran disponibles datos limitados sobre la utilización de temsirolimus en pacientes pediátricos. La eficacia del temsirolimus en pacientes pediátricos con tumores sólidos avanzados recurrentes/resistentes al tratamiento no pudo establecerse (ver sección Advertencias especiales y precauciones de uso).
Utilización en pacientes ancianos: No se recomiendan ajustes específicos de la dosis en pacientes ancianos (ver sección Advertencias especiales y precauciones de uso).
SOBREDOSIS: No existe tratamiento específico para sobredosis de temsirolimus intravenoso; sin embargo, temsirolimus se ha administrado de forma segura en pacientes con cáncer en dosis intravenosas repetidas de hasta 220 mg/m2.
PRESENTACIÓN: TORISEL 30 mg concentrado para solución para Inyección (25 mg/mL) (Reg. San. INVIMA 2011M-0011801).
Es posible que la información de prescripción de este producto haya sido revisada y actualizada después de la fecha de impresión del PLM 2016. Para obtener información más actualizada comuníquese con la Dirección Médica de Pfizer S.A.S Teléfono: (1) 6002300 Ext. 2509 Bogotá – Colombia.
Título de documento: Temsirolimus
Fecha de CDS que reemplaza: 18 de Abril de 2013
Fecha Efectiva: 13 de Febrero de 2014
Version CDS: 20.0
LLD_Col_CDSv20.0_13Feb2014_v1.0_Aprobado por Resol. 2014038670_03Dic2014
PFIZER S.A.S.


