

SUTENT
SUNITINIB
Cápsulas
Caja , Frasco(s) , 28 Cápsulas , 12.5 Miligramos
Caja , Frasco(s) , 28 Cápsulas , 25 Miligramos
Caja , Frasco(s) , 28 Cápsulas , 50 Miligramos
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Cada CÁPSULA contiene 12,5; 25; 50 mg de sunitinib (como malato).
INDICACIONES TERAPÉUTICAS: Tratamiento de los tumores malignos del estroma gastrointestinal (TEGI) luego del fracaso del tratamiento con imatinib, debido a resistencia o intolerancia. (Ver la sección Propiedades Farmacodinámicas). Tratamiento del carcinoma de células renales avanzado y/o metastásico (CCRM) (ver la sección Propiedades Farmacodinámicas) y para el tratamiento de sujetos con tumores neuroendocrinos bien diferenciados del páncreas (TNEp).
PROPIEDADES FARMACOCINÉTICAS: La farmacocinética de sunitinib y del malato de sunitinib, fue evaluada en 135 voluntarios saludables y en 266 sujetos con tumores sólidos.
Absorción: Las concentraciones plasmáticas máximas (Cmáx) se observaron generalmente entre 6 - 12 horas (Tmáx) después de la administración oral. Los alimentos no afectaron la biodisponibilidad de sunitinib.
Distribución: La unión in vitro de sunitinib y su metabolito primario activo a las proteínas plasmáticas humanas, fue 95% y 90%, respectivamente, sin dependencia aparente de la concentración en el rango de 100 - 4000 ng/ml. El volumen de distribución aparente (Vd/F) para sunitinib fue grande 2,230 l, indicando su distribución hacia los tejidos. En el rango de dosis de 25 - 100 mg, el área bajo la curva de concentración plasmática-tiempo (ABC) y la Cmáx aumentaron proporcionalmente con la dosis.
Metabolismo: Los valores de Ki calculados para todas las isoformas CYP evaluadas in vitro (CYP1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP3A4/5 y CYP4A9/11), indicaron que es improbable que el sunitinib y su metabolito activo principal tengan interacciones fármaco-fármaco clínicamente relevantes con fármacos que puedan ser metabolizados por estas enzimas.
Estudios in vitro indicaron que el sunitinib no induce, ni inhibe, enzimas CYP principales, incluida la CYP3A4 (ver la Sección Interacción con otros medicamentos y otras formas de interacción).
Sunitinib es metabolizado principalmente por la enzima del citocromo P450, CYP3A4, para producir su metabolito activo principal, que posteriormente es metabolizado por la CYP3A4.El metabolito activo principal comprende de 23 a 37% de la exposición total.
Eliminación: La excreción ocurre principalmente vía heces (61%), con eliminación renal del fármaco y sus metabolitos que representa el 16% de la dosis administrada. El sunitinib y su metabolito activo principal, fueron los principales compuestos relacionados con el fármaco identificados en plasma, orina y heces, representando 91,5%, 86,4% y 73,8% de la radioactividad en muestras combinadas, respectivamente. Se identificaron metabolitos menores en orina y heces, pero generalmente no se detectaron en el plasma. La depuración oral total (CL/F) varió entre 34 - 62 l/h, con una variabilidad inter-paciente de 40%. Después de la administración de una dosis oral individual en voluntarios sanos, los tiempos de vida media terminal de sunitinib y su metabolito activo primario desetilo fueron de aproximadamente 40 - 60 horas y 80 - 110 horas, respectivamente.
• Farmacocinética en grupos especiales de pacientes
Insuficiencia hepática: El sunitinib y su metabolito primario son metabolizados principalmente por el hígado. Las exposiciones sistémicas después de una sola dosis de sunitinib, fueron similares en sujetos con insuficiencia hepática leve (Clase A Child-Pugh) o moderada (Clase B Child-Pugh), comparadas con las de sujetos con función hepática normal. El sunitinib no ha sido estudiado en sujetos con insuficiencia hepática severa (Clase C Child-Pugh).
Insuficiencia renal: Los análisis farmacocinéticos poblacionales, demostraron que la farmacocinética de sunitinib se mantuvo inalterada en los sujetos con depuraciones de creatinina calculadas en el rango de 42 - 347 ml/min. Las exposiciones sistémicas después de una única dosis de sunitinib fueron similares en personas con deterioro renal severo (CLcr <30 ml/min) comparadas con personas con función renal normal (CLcr >80 ml/min). Aunque sunitinib y su metabolito primario no se eliminaron a través de hemodiálisis en pacientes con IRT, las exposiciones sistémicas totales fueron menores en un 47% para sunitinib y en 31% para su metabolito principal comparadas con personas con función renal normal.
Electrofisiología cardíaca: La prolongación del intervalo QT se investigó en un estudio de Fase I con 24 sujetos evaluables, de 20 - 87 años de edad con malignidades avanzadas. En las concentraciones plasmáticas terapéuticas, el cambio promedio máximo desde el período inicial en el QTcF, fue de 9,6 mseg (IC 90% límite superior de 15,1 mseg). A concentraciones que eran aproximadamente el doble de las concentraciones terapéuticas, el cambio promedio máximo desde el período inicial en el QTcF, fue de 15,4 mseg (IC 90% límite superior de 22,4 mseg). La moxifloxacina (400 mg), usada como un control positivo, mostró un cambio promedio máximo desde el período inicial en el QTcF de 5,6 mseg. Ningún sujeto experimentó un efecto sobre el intervalo QTc, mayor que Grado 2 (CTCAE, versión 3.0). Ningún paciente presentó arritmia cardíaca (ver la Sección Advertencias y precauciones especiales para el uso).
Farmacocinética plasmática: Después de la administración de una sola dosis oral a voluntarios sanos, las vidas medias de eliminación de sunitinib y su metabolito activo principal fueron aproximadamente, 40 - 60 horas y 80 - 110 horas, respectivamente. Con la administración diaria repetida, el sunitinib se acumuló de 3- a 4 veces, mientras que el metabolito activo principal se acumuló de 7- a 10 veces. Las concentraciones en estado estable de sunitinib y su metabolito activo principal, se alcanzan en el lapso de 10 a 14 días. Para el día 14, las concentraciones plasmáticas combinadas de sunitinib y su metabolito activo son de 62,9-101 ng/ml, que son las concentraciones objetivo, predichas con base en los datos preclínicos para inhibir in vitro la fosforilación del receptor y que resultan en el estancamiento/reducción del crecimiento tumoral in vivo. No se observaron cambios significativos en la farmacocinética de sunitinib o del metabolito activo principal, con la administración diaria repetida, ni con la repetición de los ciclos, en los regímenes de dosificación evaluados.
Las farmacocinéticas fueron similares en todas las poblaciones de tumores sólidos evaluados y en los voluntarios sanos.
Farmacocinética poblacional: Los análisis farmacocinéticos poblacionales de los datos demográficos, indicaron que no hubo efectos clínicamente relevantes de la edad, peso corporal, depuración de creatinina, género, raza o puntaje ECOG, sobre la farmacocinética de sunitinib o del metabolito activo primario.
Estado funcional, peso: Los análisis farmacocinéticos de la población de los datos demográficos indican que no son necesarios ajustes de la dosis inicial por peso o el estado funcional ECOG.
Género: Los datos disponibles indican que las mujeres pueden tener una depuración aparente aproximadamente 30% menor (CL/F) de sunitinib que los hombres: Esta diferencia, sin embargo, no necesita ajustes en dosis inicial.
PROPIEDADES FARMACODINÁMICAS: Sunitinib inhibe múltiples receptores de tirosina-quinasas (RTK), que están implicados en el crecimiento tumoral, la angiogénesis patológica y la progresión metastásica del cáncer. Sunitinib fue identificado como un inhibidor de los receptores del factor de crecimiento derivado de las plaquetas (PDGFRa y PDGFRß), de los receptores del factor de crecimiento vascular endotelial (VEGFR1, VEGFR2 y VEGFR3), del receptor del factor de células madres (KIT), del receptor de la tirosina quinasa-3 Fms-similar (FLT3), del receptor del factor estimulador de colonias Tipo 1 (CSF-1R) y del receptor del factor neurotrófico derivado de la línea celular glial (RET). La inhibición ejercida por sunitinib sobre la actividad de esos RTK, ha sido demostrada en ensayos bioquímicos y celulares, y la inhibición de la función ha sido demostrada en ensayos de proliferación celular. El metabolito principal exhibió una potencia similar, cuando se comparó con sunitinib en ensayos bioquímicos y celulares.
El sunitinib inhibió la fosforilación de múltiples RTK (PDGFRb, VEGFR2, KIT), en xenoinjertos tumorales con expresión de blancos RTK in vivo y demostró capacidad de inhibición del crecimiento tumoral o regresión tumoral, o inhibió las metástasis en algunos modelos experimentales de cáncer. Sunitinib demostró capacidad para inhibir in vitro el crecimiento de células tumorales expresando blancos RTK desregulados (PDGFR, RET o KIT) y para inhibir in vivo la angiogénesis tumoral dependiente del PDGFR b- y el VEGFR2.
Estudios clínicos: La seguridad y eficacia clínica de sunitinib, han sido estudiadas en sujetos con TEGI maligno quienes eran resistentes a imatinib (es decir, quienes experimentaron progresión de la enfermedad durante o después del tratamiento con imatinib) o en sujetos que no toleraron el imatinib (p. ej., quienes experimentaron una toxicidad significativa durante el tratamiento con imatinib, lo cual imposibilitó la continuación del tratamiento), en sujetos con carcinoma de células renales metastásico (CCRM) y en sujetos con TNEp no removible con cirugía.
La eficacia se estableció con base en el tiempo transcurrido hasta la progresión del tumor (Time to Tumor Progression-TTP) y en un aumento de la supervivencia, para el TEGI.
La eficacia se estableció con base en la supervivencia libre de progresión (SLP) y en las tasas de respuestas objetivas (TRO), para el CCRM sin tratamiento previo (naïve) y para el CCRM resistente a la citocina, respectivamente y con base en SSA para TNEp.
Tumores estromales gastrointestinales (TEGI): Se realizó un estudio inicial, abierto de aumento de dosis, en sujetos con TEGI, después del fracaso de imatinib (mediana de la dosis diaria máxima 800 mg) debido a resistencia o intolerancia. Se reclutaron noventa y siete sujetos en varias dosis y regímenes de dosificación; 55 sujetos recibieron 50 mg en el régimen de dosificación recomendado de 4 semanas en tratamiento ON / 2 semanas sin tratamiento OFF (Esquema 4/2). En este estudio, la mediana del tiempo hasta la progresión del tumor (TTP) y de supervivencia libre de progresión (SLP), fue de 34,0 semanas (IC 95%: 22,0 – 46,0 semanas).
Se realizó un estudio de Fase 3, aleatorizado, doble-ciego, controlado con placebo de sunitinib, en sujetos con TEGI quienes eran intolerantes, o habían experimentado progresión de la enfermedad durante o después del tratamiento con imatinib (mediana de la dosis diaria máxima 800 mg). En este estudio, 312 sujetos fueron aleatorizados (2:1) para recibir 50 mg de sunitinib o placebo, por vía oral una vez al día en el Esquema 4/2, hasta la progresión de la enfermedad o su retiro del estudio por otra razón (207 sujetos recibieron sunitinib y 105 sujetos recibieron placebo). El criterio de valoración primario de eficacia del estudio fue el TTP (como fue evaluado por la Revisión Independiente), definido como el tiempo transcurrido desde la aleatorización, hasta la primera documentación de progresión objetiva del tumor. Los objetivos secundarios, incluyeron la SSA, TRO y supervivencia global (SG).
En el momento de análisis preliminar preespecificado, la mediana del TTP para sunitinib, fue 28,9 semanas (IC 95%: 21,3-34,1 semanas) evaluado por el Investigador y 27,3 semanas (95% IC: 16,0-32,1 semanas) evaluado por la Revisión Independiente y fue desde el punto de vista estadístico significativamente más prolongado que el TTP de 5,1 semanas (IC 95%: 4,4-10,1 semanas) que el evaluado por el Investigador y 6,4 semanas (95% IC: 4,4-10,0 semanas) que el evaluado por la Revisión Independiente. La diferencia en la SG fue estadísticamente en favor para el sunitinib (hazard ratio [HR]: 0,491 [IC 95% 0,290- 0,831]); el riesgo de muerte fue 2 veces mayor en los sujetos del brazo placebo, en comparación con los del brazo sunitinib. Información adicional de eficacia se presenta a continuación en la Tabla 5.
Después del análisis preliminar positivo de eficacia y seguridad, por la recomendación de la Junta de Monitoreo de Seguridad y Datos (DSMB) independiente, el estudio fue descubierto (se retiró el ciego del estudio) y se ofreció a los sujetos en el grupo de placebo, un tratamiento de sunitinib abierto.
Un total de 255 sujetos recibieron sunitinib en la fase de tratamiento abierto del estudio, Incluyendo 99 sujetos quienes se trataron inicialmente con placebo. En este análisis final, el grupo de placebo incluyó aquellos sujetos aleatorizados al placebo quienes posteriormente recibieron el tratamiento de sunitinib abierto.
Los análisis finales de criterios de valoración primarios y secundarios del estudio reafirmaron los resultados obtenidos en el tiempo del análisis preliminar, como se muestra en la Tabla 5 a continuación:
|
Tabla 5: Resumen de criterios de valoración de eficacia (población ITT) |
|||||
|
Tratamiento doble ciego a |
|||||
|
Mediana (95% IC) |
Razón de riesgo (HR) |
Tratamiento de grupo cruzado de placebo b |
|||
|
Criterio de valoración |
Sunitinib |
Placebo |
(95% IC) |
p |
|
|
primario TTP (semanas) |
|||||
|
Preliminar |
27,3 (16,0 a 32,1) |
6,4 (4,4 a 10,0) |
0,329 (0,233 a 0,466) |
<0,001 |
- |
|
Final |
26,6 (16,0 a 32,1) |
6,4 (4,4 a 10,0) |
0,339 (0,244 a 0,472) |
<0,001 |
10,4 (4,3 a 22,0) |
|
Secundario |
|||||
|
Preliminar SSA (semanas) c |
24,1 (11,1 a 28,3) |
6,0 (4,4 a 9,9) |
0,333 (0,238 a 0,467) |
<0,001 |
- |
|
TRO (%) d |
6,8 (3,7 a 11,1) |
(0)- |
NA |
0,006 |
- |
|
SG (semanas) e |
- |
- |
0,491 (0,290 a 0,831) |
0,007 |
- |
|
Final |
|||||
|
SSA (semanas) |
22,9 (10,9 a 28,0) |
6,0 (4,4 a 9,7) |
0,347 (0,253 a 0,475) |
<0,001 |
- |
|
TRO (%) d |
6,6 (3,8 a 10,5) |
0 (-) |
NA |
0,004 |
10,1 (5,0 a 17,8) |
|
SG (semanas) |
72,7 (61,3 a 83,0) |
64,9 (45,7 a 96,0) |
0,876 (0,679 a 1,129) |
0,306 |
- |
|
a. Los resultados del tratamiento doble ciego pertenecen a la población ITT y se utiliza la medida de radiólogo central, como sea apropiado. b. Los resultados de eficacia para los 99 sujetos quienes pasaron del tratamiento con placebo a sunitinib después del descubrimiento. La línea basal se reajustó al momento del cambio y los análisis de eficacia se basaron en la evaluación de los investigadores. c. Los números SSA preliminares se han actualizado con base en el recálculo de los datos originales. d. Los resultados de TRO se presentan como el porcentaje de sujetos con respuesta confirmada con el 95% IC. e. No se alcanzó la mediana porque los datos no eran maduros todavía. |
|||||
De aquellos sujetos aleatorizados al grupo sunitinib, 62.7% sobrevivieron más de un año, 35,5% sobrevivieron más de 2 años y 22,3% sobrevivieron más de 3 años.
En general, el estudio demostró una mejora estadística y clínicamente significativa en TTP, el criterio de valoración primario, para sunitinib más la mejor asistencia sintomática comparada con la mejor asistencia sintomática más placebo.
Tumores Neuroendocrinos del páncreas (TNEp): Un estudio multicéntrico, abierto de Fase 2 evaluó la eficacia y seguridad de sunitinib 50 mg diarios como monoterapia bajo la pauta posológica 4/2 en sujetos con TNEp avanzado no removible con cirugía. En una cohorte con insulinoma de páncreas de 66 sujetos, se observó una TRO del 17%.
Se realizó un estudio fundamental de Fase 3, multicéntrico, internacional, aleatorizado, doble ciego, controlado con placebo con sunitinib como monoterapia en sujetos con TNEp no removible con cirugía.
Se pidió a los sujetos documentar la evolución de la enfermedad, con base en Criterios de Evaluación de Respuestas en Tumores Sólidos (RECIST), dentro de los 12 meses previos y se asignaron aleatoriamente (1:1) para recibir 37,5 mg de sunitinib una vez al día, sin un periodo sin tratamiento programado (n=86) o para recibir placebo (n=85).
El objetivo principal fue comparar la SLP en sujetos que estaban recibiendo sunitinib con respecto a los sujetos que estaban recibiendo placebo. Otros criterios de valoración incluyeron la SG, TRO, resultados informados por el paciente (RRP) y seguridad. Las características demográficas fueron similares entre los grupos de sunitinib y placebo. Adicionalmente, 49% de los sujetos con sunitinib presentaron tumores no funcionales con respecto a 52% de los sujetos con placebo y 92% de los sujetos de ambos grupos presentaron metástasis hepática. La utilización de análogos de la somatostatina se permitió en el estudio. Un total de 66% de los sujetos con sunitinib recibieron tratamiento sistémico previo comparado con 72% de los sujetos con placebo. Además, 24% de los sujetos con sunitinib habían recibido análogos de la somatostatina comparados con 22% de los sujetos con placebo.
Se observó una ventaja clínicamente significativa de la SLP evaluada por el investigador para sunitinib sobre placebo. La mediana de la SLP fue 11,4 meses para el grupo con sunitinib comparado con 5,5 meses para el grupo con placebo [HR: 0,418 (IC 95%: 0,263; 0,662), valor p=0,0001]. Se observaron resultados similares cuando las evaluaciones de las respuestas obtenidas del tumor basadas en la aplicación de RECIST a las mediciones del tumor realizadas por el investigador se utilizaban para determinar el avance de la enfermedad, como se presenta en la Tabla 6. Se observó una HR que favorecía a sunitinib en todos los subgrupos de características iniciales evaluadas, incluido un análisis por número de tratamientos sistémicos previos. Un total de 29 sujetos en el grupo de sunitinib y 24 en el grupo de placebo no habían recibido tratamiento sistémico previo, de estos sujetos, la razón de riesgo para SSA fue 0,365 (IC 95%: 0,156; 0,857), p=0,0156. De manera similar, entre 57 sujetos del grupo con sunitinib (incluidos 28 con 1 tratamiento sistémico previo y 29 con 2 o más tratamientos sistémicos previos) y 61 sujetos en el grupo de placebo (incluidos 25 con 1 tratamiento sistémico previo y 36 con 2 o más tratamientos sistémicos previos) , la razón de riesgo para SSA fue 0,456 (IC 95%: 0,264; 0,787), p=0,0036.
Se realizó un análisis de sensibilidad para SLP en el que la evolución de la enfermedad se basaba en las mediciones del tumor informadas por el investigador y en el que todos los pacientes censurados por razones diferentes a la finalización del estudio fueron tratados como si presentaran eventos SLP. Este análisis proporcionó una estimación conservadora del efecto del tratamiento con sunitinib y respaldó el análisis primario, demostrando una razón de riesgo de 0,507 (IC 95%: 0,350; 0,733, p=0,000193). El estudio fundamental de TNEp se finalizó anticipadamente por recomendación de un Comité de Farmacovigilancia Independiente y el criterio de valoración primario se basó en la evaluación del investigador, lo que pudo haber afectado las estimaciones del efecto del tratamiento.
Para descartar sesgos de la evaluación de SLP del investigador, se realizó una revisión central independiente en condición enmascarada de las gammagrafías y, como se observa en la Tabla 6 respaldaron la evaluación del investigador. La curva de Kaplan-Meier se muestra en la Figura 1.
|
Tabla 6 –Resultados de Eficacia para TNEp obtenidos en el estudio de Fase 3 |
||||
|
Parámetro de Eficacia |
Sunitinib |
Placebo |
HR (IC 95%) |
Valor p |
|
SLP [Mediana, Meses (IC 95%)] Evaluada por el Investigador |
11,4 (7,4; 19,8) |
5,5 |
0,418 |
0,0001a |
|
SLP [Mediana, Meses (IC 95%)] por evaluación de la respuesta derivada basada en la aplicación de RECIST a las evaluaciones de los tumores del investigador |
12,6 (7,4; 16,9) |
5,4 (3,5; 6,0) |
0,401 (0,252; 0,640) |
0,000066a |
|
SLP [Mediana, Meses (IC 95%)] por Revisión Central Independiente en Condición Enmascarada de las Evaluaciones de los Tumores |
12,6 (11,1; 20,6) |
5,8 (3,8; 7,2) |
0,315 (0,181; 0,546) |
0,000015 a |
|
SG [mediana, meses (IC 95%)] |
20,6 (20,6; NR) |
NR (15,5; NR) |
0,409 (0,187; 0,894) |
0,0204a |
|
TRO [%, (IC 95%)] |
9,3 (3,2; 15,4) |
0 |
NA |
0,0066b |
|
IC= Intervalo de Confianza, HR=Razón de riesgo, NA=No aplica, NR=No alcanzada a2- Prueba de orden logarítmico bilateral no estratificado b Prueba exacta de Fisher |
||||
Figura 1. Curva de Kaplan-Meier de SSA en el estudio de Fase 3 de TNEp
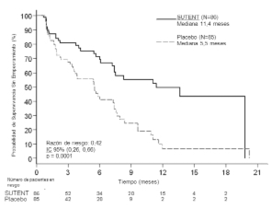
Al momento del análisis los datos SG no eran los definitivos. Se presentaron 9 muertes en el grupo con sunitinib y 21 muertes en el grupo con placebo. Se observó una diferencia estadísticamente significativa en TRO que favorecía a sunitinib sobre placebo.
Debido al agravamiento de la enfermedad, a los sujetos que perdieron la condición enmascarada y los sujetos con placebo se les ofrecía ingresar a un estudio de extensión separado abierto con sunitinib. Debido al cierre anticipado del estudio, se desenmascaró a los demás sujetos y se les ofreció participar en un estudio de extensión abierto con sunitinib. Un total de 59 sujetos del grupo de placebo recibió sunitinib en el estudio de extensión.
Los resultados del Cuestionario de Calidad de Vida de la Organización Europea para la Investigación y Tratamiento del Cáncer (EORTC QLQ-C30) mostraron que la calidad de vida relacionada con la salud global general y los cinco dominios de desempeño (físico, roles, cognitivo, emocional y social) se mantuvieron para los sujetos bajo tratamiento con sunitinib en comparación con placebo con efectos sintomáticos adversos limitados.
• Carcinoma de células renales
CCRM sin tratamiento previo (Naïve): Se realizó un estudio de Fase 3 aleatorizado, comparativo, de sunitinib con el interferón-a, como agentes únicos, en sujetos con CCRM sin tratamiento previo (naïve). El objetivo primario fue comparar la SLP en sujetos que recibían sunitinib versus sujetos que recibían IFN-a. Los objetivos secundarios incluyeron TTP, TRO, seguridad SG y resultados informados por los pacientes (RRP). Se aleatorizaron setecientos cincuenta (750) sujetos (1:1) para recibir ya fuera 50 mg de sunitinib, una vez al día con el Esquema 4/2 o interferón-a administrado subcutáneamente 9 MUI, tres veces a la semana. Los sujetos se trataron hasta la progresión de la enfermedad o el retiro del estudio por otra razón.
La población ITT, incluyó 750 sujetos, 375 aleatorizados a sunitinib y 375 aleatorizados a IFN-a. La edad, género, raza y estado funcional del Grupo Oncológico Cooperativo del Este (ECOG) iniciales fueron comparables y estaban balanceados entre los grupos sunitinib e IFN-a. En la Tabla 7 se muestran las características demográficas y de los pacientes. Los sitios de metástasis más comunes presentes en el tamizaje fueron el pulmón (78% versus 80%, respectivamente), seguido por los nódulos linfáticos (58% versus 53%, respectivamente) y el hueso (30% en cada brazo). La mayoría de los sujetos tuvieron múltiples (2 o más) sitios metastásicos en el período inicial (80% versus 77%, respectivamente).
|
Tabla 7. Datos demográficos iniciales del estudio CCRM sin tratamiento previo (Naïve) |
||
|
CCRM sin tratamiento previo |
||
|
sunitinib (n=375) |
IFN-a (n=375) |
|
|
Género [n (%)] |
||
|
Masculino |
267 (71) |
269 (72) |
|
Femenino |
108 (29) |
106 (28) |
|
Raza Auto-Declarada [n (%)] |
||
|
Blanca |
354 (94) |
340 (91) |
|
Asiática |
7 (2) |
12 (3) |
|
Negra |
4 (1) |
9 (2) |
|
No informada |
10 (3) |
14 (4) |
|
Grupo de Edad [n (%)] |
||
|
< 65 años |
223 (59) |
252 (67) |
|
= 65 años |
152 (41) |
123 (33) |
|
Estado funcional [n (%)] |
||
|
0 |
231 (62) |
229 (61) |
|
1 |
144 (38) |
142 (38) |
|
2 |
0 (0) |
4 (1)a |
|
Tratamiento Anterior [n (%)] |
||
|
Nefrectomía |
340 (91) |
335 (89) |
|
Radioterapia |
53 (14) |
54 (14) |
|
a Los sujetos tuvieron un estado funcional ECOG de 1 en el tamizaje el cual cambió a 2 en el período inicial |
||
La duración media del tratamiento fue de 11,1 meses (rango: 0,4-46,1) para el tratamiento con sunitinib y 4,1 meses (rango: 0,1-45,6) para tratamiento con IFN-a. Las interrupciones de dosis ocurrieron en 202 sujetos (54%) en sunitinib y 141 sujetos (39%) en IFN-a. Las reducciones de dosis ocurrieron en 194 sujetos (52%) en sunitinib y 98 sujetos (27%) en IFN-a. Las tasas de interrupción debido a reacciones adversas fueron 20% para sunitinib y 23% para IFN-a. Los sujetos fueron tratados hasta la progresión de la enfermedad o retiro del estudio. El criterio de valoración de eficacia primario fue la SLP. Un análisis de interino planeado, mostró una ventaja estadísticamente significativa para sunitinib, sobre el IFN-a, en el criterio de valoración primario de SLP, siendo la SLP para sunitinib más del doble que la del IFN-a (47,3 semanas y 22,0 semanas, respectivamente). El criterio de valoración secundario de TRO de sunitinib fue más de cuatro veces mayor que el del IFN-a (27,5% y 5,3%, respectivamente). Sin embargo, la información no estaba lo suficientemente madura para determinar el beneficio en supervivencia (sobrevida) total; para el momento del análisis interino, 374 de 750 sujetos reclutados continuaron en el estudio, 248 de 375 (66%) en el brazo sunitinib y 126 de 375 (34%) en el brazo IFN-a.
En el momento del análisis final hubo una ventaja estadísticamente significativa para el sunitinib sobre el IFN-a en el criterio de valoración de SLP (ver Tabla 8 y Figura 2). En los factores preespecificados de estratificación de lactato dehidrogenasa (LDH) (>1,5 ULN contra =1,5 ULN), el estado funcional ECOG (0 contra 1), y nefrectomía previa (sí vs no), el HR favoreció a sunitinib sobre IFN-a. La evaluación principal de radiología se descontinuó después de que se alcanzó el criterio de valoración primario. La TRO determinado por la evaluación del investigador fue de 46% (95% IC: 41,51) para el grupo de sunitinib y 12% (95% IC: 9,16) para el grupo IFN-a [p<0,001] (ver Tabla 8).
Los resultados fueron similares en los análisis complementarios y se mostraron robustos cuando se controlaron los factores demográficos (edad, género, raza y estado funcional) y los factores de riesgo conocidos. Para 262 de los 750 sujetos (35%) sin factores de riesgo conocidos, la mediana de la SLP fue 64,1 semanas en el brazo sunitinib y 34,1 semanas en el brazo IFN-a (HR 0,447; IC 95% 0,313–0,640); para los 424 (56%) sujetos con 1 o 2 factores de riesgo, la mediana de la SLP fue 46,6 semanas en el brazo sunitinib y de 16,1 semanas en el brazo IFN-a (HR 0,547; IC 95% 0,423 - 0.707); y para los 47 sujetos (6%) con =3 factores de riesgo, la mediana de la SLP fue 12,1 semanas en el brazo sunitinib y de 5,7 semanas en el brazo IFN-a (HR 0,679; IC 95% 0,330–1,398).
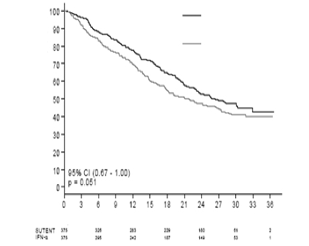
Como se muestra en la Figura 3, el tratamiento de sunitinib fue asociado con supervivencia mayor en comparación con IFN-a. La mediana SG fue 114,6 semanas para el grupo de sunitinib (95% IC: 100,1-142.9) y 94.9 semanas para el grupo IFN-a (95% IC: 77.7-117,0) [HR = 0,821; 95% IC: 0,673-1,001; p=0,0510 por prueba de rango logarítmico (Log-rank test), p=0,013 por prueba Wilcoxon] En el análisis estratificado (LDH> contra =1,5 x ULN, estado de desempeño ECOG 0 contra =1, en ausencia o presencia de nefrectomía previa), el HR fue 0,818 (95% IC: 0,699-0.999; p=0,049 por prueba de rango logarítmico) La mediana SG para el grupo IFN-a incluye 25 sujetos que interrumpieron el tratamiento con IFN-a debido a progresión de enfermedad y cruzaron a tratamiento con sunitinib. Después de la interrupción del estudio, 213 sujetos en el grupo IFN-a recibieron tratamiento de cáncer posterior al estudio, incluyendo 32% que recibieron sunitinib; 182 sujetos en el grupo de sunitinib que recibieron tratamiento para el cáncer posterior al estudio, incluyendo 11% que recibieron sunitinib. En análisis post-hoc los sujetos censurados que pasaron del tratamiento con IFN-a al tratamiento con sunitinib, mediana de SG al momento del cambio fue 114,6 contra 86.7 semanas (rango de peligro no estratificado: 0,808; p=0,0361 por prueba de rango logarítmico; p=0,0081 por prueba Wilcoxon). Cuando se excluyeron los sujetos que recibieron terapias anticáncer pos-estudio, la mediana SG fue 121.9 contra 61,3 semanas en sunitinib contra IFN-a (HR 0,647; 95% IC: 0,482-0,867; p= 0,0033 por prueba de rango logarítmico (log-rank test); p=0,0013 por prueba Wilcoxon).
|
Tabla 8. Resultados de eficacia en CCRM |
|||||
|
Parámetro de eficacia |
CCRM sin tratamiento previo |
||||
|
Sunitinib |
IFN-a |
Valor-P |
HR (IC 95%) |
||
|
SLPa [mediana, semanas (IC 95%)] |
48,3 (46,4, 58,3) |
22,1 (17,1, 24,0) |
<0,00001 1 |
0,516 (0,419, (0,635) |
|
|
TPTa [mediana, semanas (IC 95%)] |
49,1 (46,6, 59,1) |
22,4 (21.9, 33,1) |
<0,00001 |
0,516 (0,419, (0,635) |
|
|
PROa [%, (IC 95%)] |
38,7 (33,7, 43,8) |
7,7 (5,2,10.9) |
<0,0001 |
NA |
|
|
Parámetro de Eficacia |
CCRM Resistente a Citocina |
||||
|
Estudio 1 (n=106) |
Estudio 2 (n=63) |
||||
|
Porcentaje de respuesta objetiva [%, (IC 95%)] |
34,0a |
36,5b |
|||
|
Duración de la respuesta |
* |
54b |
|||
|
IC= Intervalo de confianza, NA = No aplicable * La mediana de DR no se había alcanzado todavía ** La información aún no estaba suficientemente madura para determinar el límite de confianza superior a Evaluado por un laboratorio de radiología central ciego: Para el momento del análisis no habían sido leídas las tomografías (escaneos) de 90 sujetos b Evaluado por los investigadores |
|||||
Figura 2. Curva de Kaplan-Meier de SLP en el estudio de tratamiento de CCRM -Naïve (población con intención de tratar)
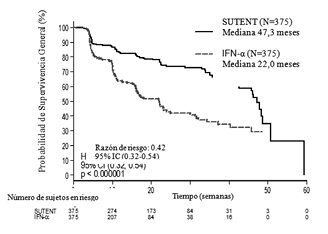
Figura 3 Curva Kaplan-Meier para SG en Estudio de Tratamiento Inicial RCC (población con intención de tratar)
Los resultados informados por los pacientes se midieron utilizando el Índice Funcional de Evaluación de la Terapia del Cáncer – Síntomas del Cáncer Renal Avanzado (FKSI) y la Evaluación Funcional de la Terapia General del Cáncer (FACT-G). Los objetivos criterios de valoración RRP incluyen el puntaje del índice FKSI, es la calificación en la subescala de Síntomas Relacionados con la Enfermedad (FKSI – DRS), el puntaje total FACT-G y de sus cuatro subescalas (Bienestar Físico [PWB], Bienestar Social/Familiar [SWB], Bienestar Emocional [EWB] y Bienestar Funcional [FWB]. El FKSI-DRS fue preespecificado como el criterio de valoración primario RRP y utilizado para evaluar los síntomas relacionados con cáncer renal informados por los pacientes (falta de energía/fatiga, dolor/dolor óseo, pérdida de peso, disnea, tos, fiebre y hematuria) en 719 sujetos. Los sujetos tratados con sunitinib informaron puntajes significativamente mejores del índice FKSI-DRS (p=0,0071), puntajes FKSI (p=0,0133), puntajes totales FACT-G (p=0,0244), puntajes PWB (p=0,0208) y FWB (p=0,0044), que los de los sujetos tratados con IFN-a, en todos los puntos de tiempo de evaluación posterior al período inicial hasta por 20 ciclos de tratamiento. Para PWB, SWB y EWB, el nivel de significación estadística se incrementó por arriba del nivel 0,05 después del ciclo 13, ciclo 15 día 1 y el ciclo 10, respectivamente. Comparadas con las diferencias mínimas clínicamente importantes preestablecidas para esos criterios de valoración, las diferencias entre los tratamientos para los síntomas relacionados con cáncer renal (FKSI en todos los puntos de tiempo de post-línea basal y FKSI-DRS después del ciclo 3, día 1) y la calidad de vida total (FACT-G) en todos los puntos de tiempo de post-línea basal se consideraron clínicamente significativas.
CCRM Resistente a Citocina: Se realizó un estudio de Fase 2 de sunitinib, en sujetos que fueron resistentes a una terapia previa con citocina, con interleuquina-2 o IFN-a. Sesenta y tres (63) sujetos recibieron una dosis inicial de 50 mg de sunitinib oralmente, una vez al día, con el Esquema 4/2. El criterio de valoración primario de eficacia fue la TRO, basada en RECIST. Los criterios de valoración secundarios, incluyeron la evaluación del TTP, SLP, duración de la respuesta (DR) y SG.5,53,54
En este estudio, la FRO fue de 36,5% (IC 95%: 24,7%-49,6%), la mediana del THP/SSP fue 37,7 semanas (IC 95%: 24,0-46,4 semanas).
Un estudio de confirmación, abierto, de un solo brazo, multicéntrico, de evaluación de la eficacia y seguridad de sunitinib, se realizó en sujetos con CCRM que fueron resistentes a un tratamiento previo con citosina. Ciento seis (106) sujetos recibieron por lo menos una dosis de 50 mg de sunitinib, con el régimen 4/2. El criterio de valoración primario de este estudio, fue la FRO. Los criterios de valoración secundarios, incluyeron el TTP, SLP, DR y SG.
En este estudio, la TRO fue de 34,0% (IC 95%: 25,0%–43,8%). Todavía no se han alcanzado las medianas de TTP, SLP, DR y SG.
CONTRAINDICACIONES: Sujetos con hipersensibilidad al sunitinib malato o a cualquier otro constituyente de las cápsulas. Embarazo (Categoría D).
FERTILIDAD, EMBARAZO Y LACTANCIA
Embarazo: No se han realizado estudios en mujeres embarazadas usando sunitinib.
Los estudios en animales han evidenciado toxicidad reproductiva, incluyendo malformaciones fetales (ver la sección Información Preclínica de Seguridad). El sunitinib no se debe usar en el embarazo, ni en ninguna mujer que no esté empleando contracepción adecuada, a menos que el posible beneficio justifique el riesgo potencial para el feto. Si se usa sunitinib durante el embarazo, o si la paciente queda embarazada mientras lo está recibiendo, la paciente debe ser informada de los posibles riesgos para el feto. A las mujeres en capacidad de procrear (en edad fértil), se les debe aconsejar que eviten quedar embarazadas, mientras estén recibiendo tratamiento con sunitinib.
Sunitinib (0,3; 1,0; 3,0 mg/kg/día) fue evaluado en un estudio de desarrollo pre y postnatal en ratas embarazadas. Los aumentos de peso corporal de la madre se redujeron durante la gestación y la lactancia en >1 mg/kg/día pero no se observó ninguna toxicidad para la reproducción en la madre hasta los 3 mg/kg/día (exposición estimada >2,3 veces el ABC en sujetos que recibieron la dosis diaria recomendada [DDR]). A 3 mg/kg/día se observaron reducciones en los pesos corporales de las crías durante los periodos antes y después del destete. A 1 mg/kg/día no se observó toxicidad para el desarrollo (exposición aproximada de >0,9 veces el ABC en sujetos que recibieron la DDR).
Fertilidad: De acuerdo con hallazgos no-clínicos, la fertilidad masculina y femenina se podría ver comprometida por el tratamiento con sunitinib (ver la sección Información Preclínica de Seguridad).
Lactancia: Sunitinib o sus metabolitos se excretan en la leche de ratas. No se sabe si sunitinib o su metabolito activo principal se excretan en la leche humana. Como los fármacos se excretan comúnmente en la leche humana y considerando el potencial de reacciones adversas serias en los infantes lactantes, las mujeres no deben amamantar mientras estén tomando sunitinib.
EFECTOS SOBRE LA CAPACIDAD PARA MANEJAR Y USAR MAQUINARIAS: No se han realizado estudios sobre la capacidad de manejar u operar maquinaria. Los sujetos deben ser advertidos de que podrían experimentar mareos durante el tratamiento con sunitinib.
REACCIONES ADVERSAS: Las frecuencias de ADR (reacciones adversas) que se presentan en esta sección representan las frecuencias de los eventos que se produjeron en sujetos tratados con sunitinib, sin tomar en cuenta la evaluación de causalidad.
Las reacciones adversos serias más importantes, asociadas con el tratamiento de sujetos con tumores sólidos con sunitinib fueron, embolismo pulmonar , trombocitopenia , hemorragia tumoral, neutropenia febril e hipertensión (ver también la sección Advertencias y precauciones especiales para el uso).
Las reacciones adversas al medicamento más comunes de cualquier grado, incluyeron: Fatiga; trastornos gastrointestinales, tales como diarrea, náuseas, estomatitis, dispepsia y vómitos; cambios en la coloración de la piel; erupción cutánea; eritrodisestesia palmoplantar; piel seca; cambios en el color del cabello; inflamación mucosa, astenia, disgeusia, anorexia e hipertensión. Fatiga, hipertensión y neutropenia, fueron las reacciones adversas al medicamento más comunes, con una severidad máxima de Grado 3, y lipasa aumentada fue la reacción adversa al medicamento con una severidad máxima de Grado 4, que ocurrió más frecuentemente en sujetos con tumores sólidos.
Epistaxis fue la reacción adversa hemorrágica más frecuente al medicamento, siendo informada en aproximadamente la mitad de los sujetos con tumores sólidos que experimentaron eventos hemorrágicos (ver también la sección Advertencias y precauciones especiales para el uso).
En los estudios clínicos de sunitinib, se observaron convulsiones en sujetos con evidencia radiológica de metástasis cerebrales. Adicionalmente, hubo informes (<1%), algunos fatales, de sujetos que presentaron convulsiones y evidencia radiológica de SLPR (ver también la sección Advertencias y precauciones especiales para el uso).
Eventos Tromboembólicos Venosos (ETV): En la fase de tratamiento doble ciego en un estudio TEGI, 7 sujetos (3%) bajo sunitinib y ninguno bajo placebo experimentaron ETV; 5 de los 7 fueron trombosis venosas profundas (TVP) de Grado 3 y 2 fueron de Grado 1 o 2. A cuatro de esos 7 sujetos con TEGI, se les discontinuó el tratamiento después de la primera observación de TVP. Trece sujetos (3%) que recibían sunitinib para el tratamiento de CCRM sin tratamiento previo (naïve) y 4 (2%) sujetos en los 2 estudios de CCRM5,6 resistentes a citocina, tuvieron informes de ETV. Nueve de esos sujetos tuvieron embolismo pulmonar, 1 fue de Grado 2 y 8 fueron de Grado 4, y ocho sujetos tuvieron TVP, 1 Grado 1, 2 Grado 2, 4 de Grado 3 y 1 de grado 4. Un sujeto con embolismo pulmonar en el estudio de CCRM resistente a citocina requirió la interrupción de la dosis. Entre los sujetos que recibían IFN-a para el tratamiento de CCRM naïve, ocurrieron 6 (2%) ETV; 1 paciente (<1%) experimentó una TVP de Grado 3 y 5 (1%) sujetos tuvieron embolismo pulmonar, todos Grado 4.
El embolismo pulmonar se informó en aproximadamente el 2,2% de los sujetos con tumores sólidos, que recibían sunitinib. Ninguno de esos eventos resultó en una interrupción del tratamiento con sunitinib; sin embargo, en unos pocos casos hubo una reducción temporal de la dosis o un retraso del tratamiento. Después de reasumir el tratamiento, no se presentaron eventos de embolismo pulmonar en estos sujetos.
Las reacciones adversas al medicamento de frecuencia de toda causalidad, surgidas del tratamiento, informadas sujetos que recibían sunitinib en estudios de agente único en CCR avanzado, TEGI y TNEp y de la experiencia posterior a la comercialización están listadas a continuación por clasificación por órganos y sistemas (SOC), frecuencia y grado de severidad.
Las frecuencias están definidas como: Muy común: (=1/10), común: (=1/100 a <1/10), poco común: (=1/1000 a <1/100), raro: (=1/10.000 a <1/1000), muy raro: (<1/10.000).
|
Tabla 1. Reacciones adversas relacionadas con el tratamiento, Las frecuencias de toda causalidad, en la fase de tratamiento doble ciego del estudio de GIST |
|||||
|
Clase de sistema orgánico |
Frecuencia |
Evento |
Todos los grados n (%) |
Grado 3 n (%) |
Grado 4 n (%) |
|
Infecciones e infestaciones |
Muy Común |
Infecciones* |
2956 (41.5) |
528 (7.4) |
83 (1.2) |
|
Trastornos de la sangre y sistema linfático |
Muy común |
Neutropenia |
1224 (17.2) |
484 (6.8) |
46 (0.6) |
|
Muy común |
Leucopenia |
725 (10.2) |
141 (2.0) |
9 (0.1) |
|
|
Muy común |
Trombocitopenia |
1563 (22.0) |
460 (6.5) |
115 (1.6) |
|
|
Muy común |
Anemia |
1697 (23.9) |
462 (6.5) |
103 (1.4) |
|
|
Muy común |
Linfopenia |
155 (2.2) |
49 (0.7) |
2 (0.028) |
|
|
Rara |
Microangiopatía trombótica |
1 (0.014) |
1 (0.014) |
0 (0.0) |
|
|
Trastornos del sistema inmune |
Poco común |
Hipersensibilidad |
45 (0.6) |
7 (0.098) |
0 (0.0) |
|
Rara |
Angioedema |
7 (0.098) |
3 (0.042) |
0 (0.0) |
|
|
Trastornos endocrinos |
Muy Común |
Hipotiroidismo |
890 (12.5) |
52 (0.7) |
6 (0.084) |
|
Poco Común |
Hipertiroidismo |
52 (0.7) |
5 (0.07) |
0 (0.0) |
|
|
Poco Común |
Tiroides |
6 (0.084) |
0 (0.0) |
0 (0.0) |
|
|
Trastornos del metabolismo y desorden nutricional |
Muy común |
Disminución del apetito* |
2644 (37.2) |
218 (3.1) |
3 (0.0042) |
|
Común |
Deshidratación** |
501 (7.0) |
192 (2.7) |
15 (0.2) |
|
|
Común |
Hipoglucemia |
106 (1.5) |
28 (0.4) |
16 (0.2) |
|
|
Rara |
Síndrome lisis tumoral |
4 (0.056) |
3 (0.042) |
0 (0.0) |
|
|
Trastornos pediátricos |
Muy Común |
Insomnio |
759 (10.7) |
12 (0.2) |
0 (0.0) |
|
Común |
Depresión |
379 (5.3) |
18 (0.3) |
3 (0.042) |
|
|
Trastornos del sistema nervioso |
Muy común |
Disgeusia |
2048 (28.8) |
32 (0.4) |
0 (0.0) |
|
Muy común |
Cefalea |
1406 (19.8) |
85 (1.2) |
5 (0.070) |
|
|
Común |
Mareo |
684 (9.6) |
34 (0.5) |
3 (0.042) |
|
|
Común |
Parestesia |
382 (5.4) |
13 (0.2) |
1 (0.014) |
|
|
Poco común |
Hemorragia cerebral** |
23 (0.3) |
2 (0.028) |
4 (0.056) |
|
|
Poco común |
Accidente cerebrovascular** |
32 (0.4) |
8 (0.1) |
11 (0.2) |
|
|
Poco común |
Ataque isquémico transitorio |
21 (0.3) |
8 (0.1) |
3 (0.042) |
|
|
Raro |
Infarto cerebral |
6 (0.084) |
2 (0.028) |
2 (0.028) |
|
|
Raro |
Síndrome de encefalopatía reversible posterior |
5 (0.070) |
3 (0.042) |
1 (0.014) |
|
|
Raro |
Ageusia |
3 (0.042) |
- |
- |
|
|
Trastornos de ojo |
Común |
Edema periorbitario |
333 (4.7) |
3 (0.042) |
0 (0.0) |
|
Común |
Edema en párpados |
276 (3.9) |
9 (0.1) |
0 (0.0) |
|
|
Común |
Aumento de lagrimeo |
394 (5.5) |
1 (0.01) |
0 (0.0) |
|
|
Trastornos cardíacos |
Común |
Isquemia del miocardio** |
87 (1.2) |
27 (0.4) |
3 (0.0) |
|
Común |
Fracción de eyección disminuidab |
152 (2.1) |
27 (0.4) |
0 (0.0) |
|
|
Poco común |
Infarto del miocardio** |
62 (0.9) |
10 (0.1) |
33 (0.5) |
|
|
Poco común |
Falla cardíaca** |
51 (0.7) |
22 (0.3) |
8 (0.1) |
|
|
Poco común |
Falla cardíaca congestiva |
32 (0.4) |
22 (0.3) |
4 (0.056) |
|
|
Poco común |
Electrocardiograma QT prolongado |
23 (0.3) |
4 (0.056) |
2 (0.028) |
|
|
Poco común |
Cardiomiopatía** |
15 (0.2) |
5 (0.070) |
1 (0.014) |
|
|
Poco común |
Falla ventricular izquierda** |
7 (0.098) |
5 (0.070) |
0 (0.0) |
|
|
Rara |
Torsade de Pointes |
1 (0.014) |
0 (0.0) |
1 (0.014) |
|
|
Trastornos vasculares |
Muy común |
Hipertensión |
1991 (28.0) |
505 (7.1) |
15 (0.2) |
|
Poco común |
Tumor hemorrágico** |
49 (0.7) |
26 (0.4) |
3 (0.042) |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy común |
Dispnea |
1443 (20.3) |
322 (4.5) |
75 (1.1) |
|
Muy común |
Epistaxis |
1080 (15.2) |
43 (0.6) |
4 (0.056) |
|
|
Común |
Dolor orofaríngeod |
455 (6.4) |
6 (0.1) |
0 (0.0) |
|
|
Común |
Hemoptsise** |
360 (5.1) |
25 (0.4) |
5 (0.070) |
|
|
Común |
Embolismo pulmonar** |
119 (1.7) |
33 (0.5) |
52 (0.7) |
|
|
Trastornos gastrointestinales |
Muy común |
Diarrea |
3729 (52.4) |
430 (6.0) |
13 (0.2) |
|
Muy común |
Náusea |
3035 (42.7) |
246 (3.5) |
4 (0.056) |
|
|
Muy común |
Vómito |
2416 (34.0) |
287 (4.0) |
17 (0.2) |
|
|
Muy común |
Dolor abdominalf |
2162 (30.4) |
406 (5.7) |
38 (0.5) |
|
|
Muy común |
Estomatitisg |
2011 (28.3) |
189 (2.7) |
2 (0.028) |
|
|
Muy común |
Constipación |
1653 (23.2) |
67 (0.9) |
3 (0.042) |
|
|
Muy común |
Dispepsia |
1564 (22.0) |
36 (0.5) |
1 (0.014) |
|
|
Común |
Hemorragia gastrointestinal** |
29 (11,3) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Esofagitis |
22 (8,6) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Reflujo gastro- esofágico |
465 (6.5) |
13 (0.2) |
0 (0.0) |
|
|
Común |
Dolor oral |
582 (8.2) |
23 (0.3) |
0 (0.0) |
|
|
Común |
Glosodinia |
430 (6.0) |
13 (0.2) |
0 (0.0) |
|
|
Común |
Distensión abdominal |
451 (6.3) |
32 ( 0.4) |
2 ( 0.028) |
|
|
Común |
Sangrado gingival |
147 (2.1) |
6 (0.1) |
0 (0.0) |
|
|
Común |
Boca seca |
483 (6.8) |
2 (0.028) |
0 (0.0) |
|
|
Común |
Flatulencia |
501 (7.0) |
2 (0.028) |
0 (0.0) |
|
|
Poco común |
Pancreatitis |
17 (0.2) |
6 (0.084) |
1 (0.014) |
|
|
Poco común |
Perforación gastrointestinal** |
15 (0.2) |
7 (0.098) |
4 (0.056) |
|
|
Trastornos hepatobiliares |
Poco común |
Colecistitis |
33 (0.5) |
16 (0.2) |
4 (0.056) |
|
Poco común |
Falla hepática** |
23 (0.3) |
4 (0.056) |
8 (0.1) |
|
|
Trastornos de la piel y tejidos subcutáneos |
Muy común |
Síndrome de eritrodisestesia palmar-plantar |
1984 (27.9) |
551 (7.7) |
3 (0.042) |
|
Muy común |
Decoloración de la pieli |
1761 (24.8) |
13 (0.2) |
0 (0.0) |
|
|
Muy común |
Erupciónj |
1595 (22.4) |
73 (1.0) |
2 (0.028) |
|
|
Muy común |
Cambios en el color del cabello |
858 (12.1) |
10 (0.1) |
0 (0.0) |
|
|
Muy común |
Piel seca |
805 (11.3) |
5 (0.070) |
0 (0.0) |
|
|
Común |
Alopecia |
564 (7.9) |
1 (0.014) |
0 (0.0) |
|
|
Común |
Eritema |
488 (6.9) |
15 (0.2) |
0 (0.0) |
|
|
Común |
Prurito |
460 (6.5) |
3 (0.042) |
0 (0.0) |
|
|
Común |
Exfoliación cutánea |
373 (5.2) |
15 (0.2) |
0 (0.0) |
|
|
Común |
Ampollas |
257 (3.6) |
27 (0.4) |
1 (0.014) |
|
|
Común |
Lesión cutánea |
190 (2.7) |
14 (0.2) |
0 (0.0) |
|
|
Común |
Reacción cutánea |
180 (2.5) |
11 (0.2) |
0 (0.0) |
|
|
Común |
Trastorno de las uñas |
176 (2.5) |
3 (0.042) |
0 (0.0) |
|
|
Poco común |
Dermatitis exfoliativa |
21 (0.3) |
2 (0.028) |
0 (0.0) |
|
|
Rara |
Eritema multiforme** |
5 (0.070) |
0 (0.0) |
0 (0.0) |
|
|
Rara |
Síndrome de Stevens-Johnson** |
2 (0.028) |
1 (0.014) |
1 (0.014) |
|
|
Rara |
Pioderma gangrenoso |
1 (0.014) |
0 (0.0) |
0 (0.0) |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Muy común |
Dolor en extremidades |
1237 (17.4) |
125 (1.8) |
13 (0.2) |
|
Muy común |
Artralgia |
1023 (14.4) |
97 (1.4) |
5 (0.070) |
|
|
Común |
Mialgia |
650 (9.1) |
34 (0.5) |
0 (0.0) |
|
|
Poco común |
Osteonecrosis mandibular |
31 (0.4) |
12 (0.2) |
0 (0.0) |
|
|
Poco común |
Formación de fístula** |
13 (0.2) |
3 (0.042) |
2 (0.028) |
|
|
Rara |
Rabdomiólisis** |
7 (0.098) |
2 (0.028) |
1 (0.014) |
|
|
Rara |
Miopatía |
7 (0.098) |
0 (0.0) |
0 (0.0) |
|
|
Trastornos renales y urinarios |
Común |
Falla renal |
153 (2.2) |
66 (0.9) |
18 (0.3) |
|
Común |
Cromaturia |
197 (2.8) |
0 (0.0) |
0 (0.0) |
|
|
Común |
Proteinuria |
105 (1.5) |
39 (0.5) |
4 (0.056) |
|
|
Poco común |
Deterioro renal |
29 (0.4) |
9 (0.1) |
1 (0.0) |
|
|
Poco común |
Hemorragia en el tracto urinario |
8 (0.1) |
2 (0.028) |
0 (0.0) |
|
|
Rara |
Síndrome nefrótico |
7 (0.098) |
1 (0.014) |
4 (0.056) |
|
|
Trastornos generales y condiciones de sitio de administración |
Muy común |
Fatigak |
4746 (66.7) |
1211 (17.0) |
87 (1.2) |
|
Muy común |
Inflamación de las mucosas |
1928 (27.1) |
180 (2.5) |
10 (0.1) |
|
|
Muy común |
Edemal |
1723 (24.2) |
87 (1.2) |
2 (0.028) |
|
|
Pirexia |
1252 (17.6) |
72 (1.0) |
8 (0.1) |
||
|
Escalofríos |
430 (6.0) |
11 (0.2) |
1 (0.014) |
||
|
Enfermedad semejante a influenza |
155 (2.2) |
4 (0.056) |
0 (0.0) |
||
|
Investigaciones |
Común |
Aumento de lipasa |
105 (1.5) |
46 (0.6) |
26 (0.4) |
|
Común |
Aumento de amilasam |
76 (1.1) |
31 (0.4) |
4 (0.056) |
|
|
Común |
Aumento de ácido úrico en sangre |
98 (1.4) |
4 (0.056) |
22 (0.3) |
|
|
Común |
Disminución del recuento de glóbulos blancos |
274 (3.9) |
95 (1.3) |
7 (0.098) |
|
|
Común |
Disminución del recuento de plaquetas |
307 (4.3) |
94 (1.3) |
15 (0.2) |
|
|
Común |
Disminución de hemoglobina |
269 (3.8) |
62 (0.9) |
12 (0.2) |
|
|
Común |
Disminución de peso |
701 (9.9) |
29 (0.4) |
1 (0.014) |
|
|
Rara |
Aumento de creatina fosfoquinasa en sangre |
60 (0.8) |
12 (0.2) |
5 (0.07) |
|
|
Rara |
Aumento de hormona estimulante de la tiroides en sangre |
45 (0.6) |
7 (0.098) |
0 (0.0) |
|
|
a. La Isquemia miocárdica: Los siguientes términos se han combinados: Síndrome coronario agudo, Angina de pecho, Angina inestable, Oclusión arterial coronaria, Isquemia miocárdica. b. Disminución de fracción de eyección: se han combinado los siguientes términos: disminución de fracción de eyección y fracción de eyección anormal. c. Infarto de miocardio: Los siguientes términos se han combinado: Infarto agudo de miocardio, Infarto de miocardio, Infarto de miocardio silencioso d. Dolor orofaríngeo: se han combinado los siguientes términos: dolor faringeolaríngeo y dolor orofaríngeo. e. Hemóptisis: se han combinado los siguientes términos: hemóptisis y hemorragia pulmonar. f. Dolor abdominal: se han combinado los siguientes términos: dolor abdominal, dolor abdominal inferior, dolor abdominal superior. g. Estomatitis: se han combinado los siguientes términos: estomatitis y estomatitis aftosa. h. Colecistitis: se han combinado los siguientes términos: colecistitis y colecistitis acalculosa. i. Decoloración de la piel: se han combinado los siguientes términos: decoloración de la piel, piel amarilla, trastorno de pigmentación j. Erupción: se han combinado los siguientes términos: dermatitis psoriasiforme, erupción exfoliativa, erupción, erupción eritomatosa, erupción folicular, erupción generalizada, erupción macular, erupción maculopapular, erupción popular, erupción prurítica. k. Fatiga: se han combinado los siguientes términos: fatiga y astenia. l. Edema: se han combinado los siguientes términos: edema facial, edema periférico. m. Aumento de la amilasa: se han combinado los siguientes términos: amilasa, aumento de la amilasa. * Las infecciones e infestaciones se describen en el subapartado Descripción de reacciones adversas seleccionadas. ** El evento puede ser mortal. EMA= Agencia Europea de Medicamentos; SOC= Clase de sistema orgánico.. |
|||||
La Tabla 2 presenta las ADR, incluidas en la sección Reacciones adversas, reportadas en pacientes con CCRM resistente a citosina.
|
Tabla 2. Reacciones adversas relacionadas con el tratamiento. Las frecuencias de toda causalidad, en pacientes con CCR resistente a citocina |
|||||
|
Clase de sistema orgánico |
Frecuencia |
Evento |
Todos los grados n (%) |
Grado 3 n (%) |
Grado 4 n (%) |
|
Trastornos de la sangre y sistema linfático |
Muy común |
Neutropenia |
18 (10,7) |
8 (4,7) |
2 (1,2) |
|
Común |
Leucopenia |
14 (8,3) |
7 (4,1) |
0 (0,0) |
|
|
Común |
Trombocitopenia |
16 (9,5) |
5 (3,0) |
2 (1,2) |
|
|
Muy común |
Anemia |
22 (13,0) |
8 (4,7) |
1 (0,6) |
|
|
Trastornos de los ojos |
Común |
Edema periorbital |
14 (8,3) |
0 (0,0) |
0 (0,0) |
|
Común |
Lacrimosa aumentada |
10 (5,9) |
0 (0,0) |
0 (0,0) |
|
|
Trastornos del metabolismo y desorden nutricional |
Muy común |
Disminución del apetito * |
68 (40,3) |
1 (0,6) |
0 (0,0) |
|
Muy común |
Deshidratación |
19 (11,2) |
5 (3,0) |
0 (0,0) |
|
|
Trastornos del sistema nervioso |
Muy común |
Disgeusia |
72 (42,6) |
0 (0,0) |
0 (0,0) |
|
Muy común |
Cefalea |
43 (25,4) |
2 (1,2) |
0 (0,0) |
|
|
Muy común |
Mareo |
27 (16,0) |
3 (1,8) |
0 (0,0) |
|
|
Muy común |
Paraestesia |
17 (10,1) |
0 (0,0) |
0 (0,0) |
|
|
Trastornos vasculares |
Muy común |
Hipertensión |
47 (27,8) |
10 (5,9) |
0 (0,0) |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy común |
Disnea |
38 (22,5) |
9 (5,3) |
0 (0,0) |
|
Muy común |
Epistaxis |
29 (17,2) |
0 (0,0) |
0 (0,0) |
|
|
Trastornos gastrointestinales |
Muy común |
Diarrea |
93 (55,0) |
8 (4,7) |
0 (0,0) |
|
Muy común |
Náusea |
92 (54,4) |
4 (2,4) |
0 (0,0) |
|
|
Muy común |
Dispepsia |
78 (46,2) |
1 (0,6) |
0 (0,0) |
|
|
Muy común |
Estomatitis |
70 (41,4) |
6 (3,6) |
0 (0,0) |
|
|
Muy común |
Vómito |
63 (37,3) |
7 (4,1) |
0 (0,0) |
|
|
Muy común |
Constipación |
57 (33,7) |
1 (0,6) |
0 (0,0) |
|
|
Muy común |
Dolor abdominal** |
34 (20,1) |
5 (3,0) |
0 (0,0) |
|
|
Muy común |
Glosodinia |
25 (14,8) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Flatulencia |
24 (14,2) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Distensión abdominal |
10 (5,9) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Boca seca |
10 (5,9) |
0 (0,0) |
0 (0,0) |
|
|
Trastornos de la piel y tejidos subcutáneos |
Muy común |
Exantema*** |
60 (35,6) |
1 (0,6) |
0 (0,0) |
|
Muy común |
Decoloración de la piel |
55 (32,5) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Piel seca |
29 (17,2) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Cambios en el color de cabello |
26 (15,4) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Eritema |
26 (15,4) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Síndrome de eritrodisestesia palmo-plantar |
21 (12,4) |
6 (3,6) |
0 (0,0) |
|
|
Muy común |
Alopecia |
17 (10,1) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Ampollas |
15 (8,9) |
4 (2,4) |
0 (0,0) |
|
|
Común |
|||||
|
Común |
Dermatitis exfoliativa |
10 (5,9) |
2 (1,2) |
0 (0,0) |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Muy común |
Dolor en extremidades |
39 (23,1) |
1 (0,6) |
0 (0,0) |
|
Muy común |
Mialgia |
29 (17,2) |
1 (0,6) |
0 (0,0) |
|
|
Trastornos generales y condiciones de sitio de administración |
Muy común |
Fatiga**** |
140 (82,9) |
23 (13,6) |
0 (0,0) |
|
Muy común |
Inflamación de mucosas |
30 (17,8) |
1 (0,6) |
0 (0,0) |
|
|
Investigaciones |
Muy común |
Incremento de lipasa |
20 (11,8) |
15 (8,9) |
3 (1,8) |
|
Muy común |
Disminución de peso |
19 (11,2) |
1 (0,6) |
0 (0,0) |
|
|
Común |
Aumento de amilasa en sangre**** |
9 (5,3) |
6 (3,6) |
0 (0,0) |
|
|
Común |
Disminución de WBC |
10 (5,9) |
3 (1,8) |
0 (0,0) |
|
|
Común |
Fracción de eyección anormal |
16 (9,5) |
1 (0,6) |
0 (0,0) |
|
|
Común |
Reducción de conteo de plaquetas |
13 (7,7) |
3 (1,8) |
2 (1,2) |
|
|
Cualquier evento adverso |
169 (100) |
89 (52,7) |
26 (15,4) |
||
|
Fuente: Tabla A-2.1.1, pág. 4311, Resumen de Seguridad Clínica (SCS) 2005 Malato de Sunitinib (SU011248 L-Malate) *Disminución del apetito: los siguientes términos se han combinado: disminución del apetito y anorexia. **Dolor abdominal: los siguientes términos se han combinado: dolor abdominal, dolor abdominal superior y dolor abdominal inferior ***Exantema: los siguientes términos se han combinado: exantema, exantema eritematoso, exantema macular, exantema escamoso, exantema papular, exantema prurítico y exantema maculopapular. ****Fatiga: los siguientes términos se han combinado: fatiga y astenia. *****Aumento de amilasa en sangre: los siguientes términos se han combinado: amilasa en sangre y aumento de amilasa en sangre. MedDRA= diccionario médico para actividades regulatorias; CCRm: carcinoma de células renales matastásico. |
|||||
La Tabla 3 presenta las ADR, incluidas en la sección Reacciones adversas, informadas en pacientes con TNEp que recibieron sunitinib.
|
Tabla 3. Reacciones adversas relacionadas con el tratamiento, Las frecuencias de toda causalidad, en pacientes con pNET que recibieron sunitinib |
|||||
|
Clase de sistema orgánico |
Frecuencia |
Reacciones adversas |
Todos los grados n (%) |
Grado 3 n (%) |
Grado 4 n (%) |
|
Trastornos de la sangre y sistema linfático |
Muy común |
Neutropenia |
24 (28,9) |
6 (7,2) |
4 (4,8) |
|
Muy común |
Leucopenia |
9 (10,8) |
4 (4,8) |
1 (1,2) |
|
|
Muy común |
Trombocitopenia |
14 (16,9) |
2 (2,4) |
1 (1,2) |
|
|
Trastornos endocrinos |
Común |
Hipotiroidismo |
6 (7,2) |
0 (0,0) |
0 (0,0) |
|
Trastornos del metabolismo y desorden nutricional |
Muy común |
Disminución del apetito * |
23 (27,7) |
2 (2,4) |
0 (0,0) |
|
Trastornos psiquiátricos |
Muy común |
Insomnio |
15 (18,1) |
0 (0,0) |
0 (0,0) |
|
Trastornos del sistema nervioso |
Muy común |
Disgeusia |
17 (20,5) |
0 (0,0) |
0 (0,0) |
|
Muy común |
Cefalea |
15 (18,1) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Mareo |
5 (6,0) |
1 (1,2) |
0 (0,0) |
|
|
Trastornos de los ojos |
Común |
Edema palpebral |
6 (7,2) |
1 (1,2) |
0 (0,0) |
|
Trastornos vasculares |
Muy común |
Hipertensión |
22 (26,5) |
8 (9,6) |
0 (0,0) |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy común |
Epistaxis |
17 (20,5) |
1 (1,2) |
0 (0,0) |
|
Muy común |
Disnea |
10 (12,0) |
1 (1,2) |
0 (0,0) |
|
|
Trastornos gastrointestinales |
Muy común |
Diarrea |
49 (59,0) |
4 (4,8) |
0 (0,0) |
|
Muy común |
Náusea |
37 (44,6) |
1 (1,2) |
0 (0,0) |
|
|
Muy común |
Dolor abdominal** |
34 (41,0) |
5 (6,0) |
0 (0,0) |
|
|
Muy común |
Vómito |
28 (33,7) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Estomatitis |
18 (21,7) |
3 (3,6) |
0 (0,0) |
|
|
Muy común |
Dispepsia |
12 (14,5) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Constipación |
12 (14,5) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Boca seca |
7 (8,4) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Sangrado gingival |
7 (8,4) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Flatulencia |
5 (6,0) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Estomatitis aftosa |
5 (6,0) |
0 (0,0) |
0 (0,0) |
|
|
Trastornos de la piel y tejidos subcutáneos |
Muy común |
Cambios en el color de cabello |
24 (28,9) |
1 (1,2) |
0 (0,0) |
|
Muy común |
Síndrome de eritrodisestesia palmo-plantar |
19 (22,9) |
5 (6,0) |
0 (0,0) |
|
|
Muy común |
Exantema*** |
19 (22,9) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Piel seca |
12 (14,5) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Trastorno de las uñas |
8 (9,6) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Eritema |
8 (9,6) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Piel amarilla |
6 (7,2) |
0 (0,0) |
0 (0,0) |
|
|
Común |
Alopecia |
5 (6,0) |
0 (0,0) |
0 (0,0) |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Muy común |
Artralgia |
12 (14,5) |
0 (0,0) |
0 (0,0) |
|
Común |
Dolor en extremidades |
8 (9,6) |
0 (0,0) |
0 (0,0) |
|
|
Trastornos generales y condiciones de sitio de administración |
Muy común |
Fatiga**** |
55 (66,2) |
7 (8,4) |
1 (1,2) |
|
Muy común |
Inflamación de mucosas |
13 (15,7) |
1 (1,2) |
0 (0,0) |
|
|
Investigaciones |
Muy común |
Disminución de peso |
13 (15,7) |
1 (1,2) |
0 (0,0) |
|
Cualquier evento adverso |
82 (98,8) |
30 (36,1) |
11 (13,3) |
||
|
Fuente: Estudio A6181111 CSR: A6181111 Tablas de resumen: Tabla 13.6.2.4.1. *Disminución del apetito: los siguientes términos se han combinado: disminución del apetito y anorexia. **Dolor abdominal: los siguientes términos se han combinado: dolor abdominal, dolor abdominal superior y dolor abdominal inferior ***Exantema: los siguientes términos se han combinado: exantema, exantema macular, exantema exfoliativo y exantema papular. ****Fatiga: los siguientes términos se han combinado: fatiga y astenia. MedDRA= diccionario médico para actividades regulatorias; pNET: tumor neuroendocrino pancreático. |
|||||
La Tabla 4 presenta los ADR, incluidos en la sección Reacciones adversas, que se informaron en pacientes con CCRm sin tratamiento previo que recibieron sunitinib o interferón-a (IFN-a).
|
Tabla 4. Reacciones adversas al medicamento informadas en pacientes con CCR sin tratamiento previo que recibieron sunitinib o IFN-a |
||||||
|
Clase de sistema orgánico |
Frecuencia |
Evento |
Todos los grados Sunitinib n (%) |
Grado 3/4 Sunitinib n (%) |
Todos los grados IFN-a n (%) |
Grado 3/4 IFN-a n (%) |
|
Trastornos de la sangre y sistema linfático |
Muy común |
Neutropenia |
70 (18,7) |
41 (10,9) |
33 (9,2) |
14 (3,9) |
|
Muy común |
Leucopenia |
40 (10,7) |
13 (3,5) |
16 (4,4) |
4 (1,1) |
|
|
Muy común |
Trombocitopenia |
72 (19,2) |
34 (9,1) |
15 (4,2) |
2 (0,6) |
|
|
Muy común |
Anemia |
81 (21,6) |
32 (8,5) |
58 (16,1) |
24 (6,7) |
|
|
Común |
Linfopenia |
19 (5,1) |
11 (2,9) |
21 (5,8) |
15 (4,2) |
|
|
Trastornos endocrinos |
Muy común |
Hipotiroidismo |
61 (16,3) |
6 (1,6) |
3 (0,8) |
0 (0,0) |
|
Trastornos de los ojos |
Común |
Lacrimosa aumentada |
31 (8,3) |
0 (0,0) |
0 (0,0) |
0 (0,0) |
|
Trastornos del metabolismo y desorden nutricional |
Muy común |
Disminución del apetito* |
199 (53,1) |
11 (2,9) |
156 (43,3) |
7 (1,9) |
|
Común |
Deshidratación |
37 (9,9) |
10 (2,7) |
19 (5,3) |
3 (0,8) |
|
|
Trastornos psiquiátricos |
Muy común |
Insomnio |
57 (15,2) |
3 (0,8) |
37 (10,3) |
0 (0,0) |
|
Muy común |
Depresión |
40 (10,7) |
0 (0,0) |
51 (14,2) |
5 (1,4) |
|
|
Trastornos del sistema nervioso |
Muy común |
Disgeusia |
174 (46,4) |
1 (0,3) |
53 (14,7) |
0 (0,0) |
|
Muy común |
Cefalea |
86 (22,9) |
4 (1,1) |
69 (19,2) |
0 (0,0) |
|
|
Muy común |
Mareo |
43 (11,5) |
2 (0,5) |
50 (13,9) |
2 (0,6) |
|
|
Común |
Paraestesia |
35 (9,3) |
0 (0,0) |
7 (1,9) |
1 (0,3) |
|
|
Trastornos vasculares |
Muy común |
Hipertensión |
127 (33,9) |
50 (13,3) |
13 (3,6) |
1 (0,3) |
|
Trastornos respiratorios, torácicos y mediastínicos |
Muy común |
Disnea |
99 (26,4) |
24 (6,4) |
71 (19,7) |
15 (4,2) |
|
Muy común |
Epistaxis |
80 (21,3) |
5 (1,3) |
9 (2,5) |
1 (0,3) |
|
|
Muy común |
Dolor faríngeo-laríngeo** |
51 (13,6) |
2 (0,5) |
9 (2,5) |
0 (0,0) |
|
|
Trastornos gastrointestinales |
Muy común |
Diarrea |
246 (65,6) |
37 (9,9) |
76 (21,1) |
1 (0,3) |
|
Muy común |
Náusea |
216 (57,6) |
21 (5,6) |
147 (40,8) |
6 (1,7) |
|
|
Muy común |
Vómito |
148 (39,5) |
19 (5,1) |
62 (17,2) |
4 (1,1) |
|
|
Muy común |
Dispepsia |
128 (34,1) |
8 (2,1) |
16 (4,4) |
0 (0,0) |
|
|
Muy común |
Dolor abdominal*** |
127 (33,9) |
20 (15,3) |
44 (12,2) |
5 (1,4) |
|
|
Muy común |
Estomatitis |
114 (30,4) |
5 (1,3) |
12 (3,3) |
1 (0,3) |
|
|
Muy común |
Constipación |
85 (22,7) |
4 (1,1) |
49 (13,6) |
1 (0,3) |
|
|
Muy común |
Dolor oral |
54 (14,4) |
2 (0,5) |
2 (0,6) |
0 (0,0) |
|
|
Muy común |
Flatulencia |
52 (13,9) |
0 (0,0) |
8 (2,2) |
0 (0,0) |
|
|
Muy común |
Boca seca |
50 (13,3) |
0 (0,0) |
27 (7,5) |
1 (0,3) |
|
|
Muy común |
Enfermedad por reflujo gastroesofágico |
47 (12,5) |
1 (0,3) |
3 (0,8) |
0 (0,0) |
|
|
Muy común |
Glosodinia |
40 (10,7) |
0 (0,0) |
2 (0,6) |
0 (0,0) |
|
|
Común |
Distensión abdominal |
28 (7,5) |
1 (0,3) |
6 (1,7) |
0 (0,0) |
|
|
Trastornos de la piel y tejidos subcutáneos |
Muy común |
Exantema**** |
140 (37,5) |
6 (1,6) |
51 (11,4) |
3 (0,9) |
|
Muy común |
Síndrome de eritrodisestesia palmo-plantar |
108 (28,8) |
32 (8,5) |
3 (0,8) |
0 (0,0) |
|
|
Muy común |
Decoloración de la piel |
94 (25,1) |
1 (0,3) |
0 (0,0) |
0 (0,0) |
|
|
Muy común |
Piel seca |
85 (22,7) |
1 (0,3) |
26 (7,2) |
0 (0,0) |
|
|
Muy común |
Cambios en el color de cabello |
75 (20,0) |
0 (0,0) |
1 (0,3) |
0 (0,0) |
|
|
Muy común |
Alopecia |
51 (13,6) |
0 (0,0) |
34 (9,4) |
0 (0,0) |
|
|
Muy común |
Eritema |
46 (12,3) |
2 (0,5) |
5 (1,4) |
0 (0,0) |
|
|
Muy común |
Prurito |
44 (11,7) |
1 (0,3) |
24 (6,7) |
1 (0,3) |
|
|
Común |
Exfoliación de la piel |
37 (9,9) |
3 (0,8) |
5 (1,4) |
0 (0,0) |
|
|
Común |
Lesión en la piel |
26 (6,9) |
1 (0,3) |
2 (0,6) |
0 (0,0) |
|
|
Común |
Reacción en la piel |
22 (5,9) |
3 (0,8) |
1 (0,3) |
0 (0,0) |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Muy común |
Artralgia |
111 (29,6) |
10 (2,7) |
69 (19,2) |
4 (1,1) |
|
Muy común |
Dolor en extremidades |
101 (26,9) |
10 (2,7) |
31 (8,6) |
4 (1,1) |
|
|
Muy común |
Mialgia |
46 (12,3) |
4 (1,1) |
68 (18,9) |
3 (0,8) |
|
|
Trastornos generales y condiciones de sitio de administración |
Muy común |
Fatiga |
233 (62,1) |
55 (14,7) |
202 (56,1) |
54 (15,0) |
|
Muy común |
Edema***** |
114 (30,4) |
7 (1,9) |
21 (5,8) |
2 (0,6) |
|
|
Muy común |
Inflamación de mucosas |
100 (26,7) |
8 (2,1) |
7 (1,9) |
1 (0,3) |
|
|
Muy común |
Astenia |
96 (25,6) |
42 (11,2) |
81 (22,5) |
21 (5,8) |
|
|
Muy común |
Pirexia |
84 (22,4) |
3 (0,8) |
134 (37,2) |
1 (0,3) |
|
|
Muy común |
Escalofríos |
53 (14,1) |
3 (0,8) |
111 (30,8) |
0 (0,0) |
|
|
Común |
Enfermedad parecida a la influenza |
18 (4,8) |
0 (0,0) |
54 (15,0) |
1 (0,3) |
|
|
Investigaciones |
Muy común |
Fracción de eyección reducida |
61 (16,3) |
10 (2,7) |
19 (5,3) |
6 (1,7) |
|
Muy común |
Disminución de peso |
60 (16,0) |
1 (0,3) |
60 (16,7) |
3 (0,8) |
|
|
Común |
Conteo de plaquetas reducido |
32 (8,5) |
12 (3,2) |
3 (0,8) |
0 (0,0) |
|
|
Cualquier evento adverso |
372 (99,2) |
290 (77,3) |
355 (98,6) |
197 (54,7) |
||
|
Fuente: Estudio A6181034-a CSR Tabla 13.6.2.4. *Disminución del apetito: los siguientes términos se han combinado: disminución del apetito y anorexia. **Dolor faríngeo-laríngeo: los siguientes términos se han combinado: dolor faríngeo-laríngeo y dolor orofaríngeo. ***Dolor abdominal: los siguientes términos se han combinado: dolor abdominal y dolor abdominal superior. ****Exantema: los siguientes términos se han combinado: exantema, exantema eritematoso, exantema exfoliativo, exantema folicular, exantema papular, exantema prurítico, exantema maculopapular, exantema macular, exantema generalizado y dermatitis psoriaforme. *****Edema: los siguientes términos se han combinado: edema y edema periférico. MedDRA= diccionario médico para actividades regulatorias; CCRm: carcinoma de células renales metastásico; IFN-a: interferón-a. |
||||||
Experiencia postcomercialización: Las reacciones adversas que han sido identificadas durante el uso después de la comercialización de sunitinib desde cualquier fuente (ensayos clínicos, informes espontáneos y otras fuentes) se listan a continuación (ver también sección Advertencias y precauciones especiales para el uso). Debido a que estas reacciones son informadas voluntariamente por una población de un tamaño incierto, no siempre es posible estimar la frecuencia o establecer una relación causal con la exposición al medicamento.
Trastornos de la sangre y sistema linfático: Se han informado casos de microangiopatía trombótica. Se recomienda la suspensión temporal de sunitinib; después de su solución, se puede retomar el tratamiento a discreción del médico.
Trastornos cardíacos: Insuficiencia cardíaca, insuficiencia cardíaca congestiva, insuficiencia ventricular izquierda, intervalo QT prolongado y torsade de pointes. Se ha informado cardiomiopatía isquemia de miocardio e infarto de miocardio, en algunos casos mortal.
Trastornos endocrinos: Se han informado casos de hipertiroidismo, algunos seguidos de hipotiroidismo, en ensayos clínicos y a lo largo de la experiencia postcomercialización (ver sección Advertencias y precauciones especiales para el uso). Se han informado casos de tiroiditis.
Trastornos gastrointestinales: Pancreatitis, perforación gastrointestinal y esofagitis.
Eventos hemorrágicos: Se han informado casos de hemorragia pulmonar, gastrointestinal, tumoral, cerebral y del tracto urinario, algunas mortales, en pacientes tratados con sunitinib.
Trastornos hepatobiliares: Insuficiencia hepática y colecistitis, en particular colecistitis acalculosa.
Trastornos del sistema inmune: Han sido informadas reacciones de hipersensibilidad, incluyendo angioedema.
Infecciones e infestaciones: Casos de infecciones serias (con o sin neutropenia), en algunos casos con desenlace fatal, han sido informados. Las infecciones observadas más frecuentemente con el tratamiento con sunitinib son infecciones típicamente encontradas en los pacientes con cáncer, por ejemplo infecciones respiratorias, (tales como neumonía, bronquitis, etc.) - común, infecciones de las vías urinarias - común, infecciones cutáneas (por ejemplo celulitis) - común, sepsis/choque séptico - poco común y abscesos (por ejemplo orales, genitales, anorrectales en la piel, en las extremidades, viscerales) - común. Las infecciones pueden ser bacterianas (por ejemplo osteomielitis intrabdominal) - común, virales (por ejemplo nasofaringitis, herpes oral) - común o micóticas (por ejemplo candidiasis: oral, esofágica) – común. Se han informado casos raros de fascitis necrotizante, incluida la de perineo, algunas veces fatal (ver sección Advertencias y precauciones especiales para el uso).
Investigaciones: Se han informado TSH e incremento de ácido úrico en sangre.
Trastornos del metabolismo y la nutrición: Se han informado casos de TLS, algunos mortales, en pacientes tratados con sunitinib.
Se informaron disminuciones del azúcar en la sangre, en ciertos casos con síntomas clínicos, con el tratamiento con sunitinib.
Desórdenes de tejido musculoesquelético y del tejido conectivo: Casos de miopatía y/o rabdomiólisis, con o sin insuficiencia renal aguda, en algunos casos con desenlace mortal, han sido informados. La mayoría de estos pacientes tenían factores de riesgo preexistentes y/o estaban recibiendo medicación concomitante conocida de estar asociada con estas reacciones adversas. Pacientes con signos o síntomas de toxicidad muscular deben ser manejados de acuerdo a las prácticas médicas estándares.
Se han informado casos de formación de fístulas, algunas veces asociados con necrosis o regresión de tumor, en algunos casos con desenlaces mortales.
Los casos de osteonecrosis del maxilar inferior (ONJ) han sido informados en pacientes tratados con sunitinib, de los cuales la mayoría ocurrieron en pacientes que han identificado factores de riesgo por ONJ, en particular la exposición a bifosfonatos IV o antecedentes de enfermedades dentales que requieren procedimientos dentales invasivos (ver sección Advertencias y precauciones especiales para el uso).
Trastornos del sistema nervioso: Han sido informadas alteraciones del gusto, incluyendo ageusia.
Trastornos renales y urinarios: Se han informado casos de daño y/o insuficiencia renal, en algunos casos con desenlaces mortales. Se han informado casos de proteinuria y síndrome nefrótico (ver sección Advertencias y precauciones especiales para el uso).
Trastornos respiratorios, torácicos y mediastínicos: Se ha informado embolismo pulmonar, en algunos casos con desenlace mortal.
Trastornos de la piel y el tejido subcutáneo: Se han informado casos de piodermia gangrenosa, eritema multiforme y síndrome de Stevens-Johnson.
Trastornos vasculares: En sujetos tratados con sunitinib se han informado casos de eventos tromboembólicos arteriales (ETA), algunas veces mortales. Los eventos más frecuentes incluyeron accidente cerebrovascular, ataque isquémico transitorio e infarto cerebral. Los factores de riesgo asociados con ETA, además de la enfermedad maligna subyacente y edad ³65 años, incluyeron la hipertensión, la diabetes mellitus y enfermedad tromboembólica previa.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN
Fármacos que pueden aumentar las concentraciones plasmáticas de sunitinib: La administración concomitante de sunitinib con el potente inhibidor de la CYP3A4 ketoconazol, resultó en un incremento de 49% y de 51% de los valores de Cmáx y ABC0-¥ del complejo [sunitinib + metabolito activo principal], respectivamente, después de la administración de una dosis única de sunitinib a voluntarios saludables.
La administración de sunitinib con inhibidores potentes de la familia CYP3A4 (p.ej., ritonavir, itraconazol, eritromicina, claritromicina, jugo de toronja/pomelo), puede resultar en concentraciones de sunitinib aumentadas. Se debe evitar la administración concomitante con inhibidores o se debe considerar la selección de una medicación concomitante alterna que no inhiba, o tenga un potencial mínimo para inhibir, a la CYP3A4. Si esto no es posible, puede ser necesaria la reducción de la dosis de sunitinib (ver la sección Posología y método de administración).
Drogas que pueden disminuir las concentraciones plasmáticas de sunitinib: La administración concomitante de sunitinib con el inductor de la CYP3A4 rifampicina, resultó en una disminución de 23% y 46% de los valores de Cmáx y ABC0-¥ del complejo [sunitinib + metabolito activo principal], respectivamente, después de la administración de una dosis única de sunitinib a voluntarios sanos.
La administración de sunitinib con inductores potentes de la familia CYP3A4 (p.ej., dexametasona, fenitoína, carbamazepina, rifampicina, fenobarbital o Hypericum perforatum, también conocido como hierba de San Juan), puede disminuir las concentraciones de sunitinib. Por consiguiente, se debe evitar la administración concomitante con inductores, o se debe considerar una medicación concomitante alterna que no induzca, o tenga un potencial mínimo para inducir a la CYP3A4. Si esto no es posible, puede ser necesario un aumento de la dosis de sunitinib (ver la sección Posología y método de administración).
INFORMACIÓN PRECLÍNICA DE SEGURIDAD: En los estudios de toxicidad de dosis repetidas en ratas y monos, de hasta 9 meses de duración, los efectos en los órganos blancos principales se identificaron en el tracto gastrointestinal (emesis y diarrea en monos); glándula suprarrenal (congestión cortical o hemorragia en ratas y monos, con necrosis seguida por fibrosis en las ratas); sistema hemato-linfopoyético (hipocelularidad de la médula ósea y depleción linfoide del timo, bazo y ganglios linfáticos); páncreas exocrino (desgranulación de las células acinares con necrosis celular simple); glándulas salivares (hipertrofia acinar); articulación ósea (engrosamiento de la placa de crecimiento); útero (atrofia) y ovarios (desarrollo folicular disminuido). Todos los hallazgos ocurrieron en niveles de exposición a concentraciones plasmáticas de sunitinib clínicamente relevantes. Otros efectos adicionales, observados en otros estudios, incluyeron prolongación del intervalo QTc, disminución de la FEVI, y atrofia testicular tubular, matriz mesangial aumentada en el riñón, hemorragia en el tracto GI y la mucosa oral e hipertrofia de las células pituitarias anteriores. Se piensa que los cambios en el útero (atrofia endometrial) y en la placa de crecimiento del hueso (engrosamiento de la fisis o displasia del cartílago), estarían relacionados con la acción farmacológica de sunitinib. La mayoría de los hallazgos fueron reversibles, después de 2 a 6 semanas sin tratamiento.
Genotoxicidad: El potencial genotóxico de sunitinib fue evaluado in vitro e in vivo. El sunitinib no fue mutagénico en bacterias, usando la activación metabólica brindada por el hígado de rata. El sunitinib no indujo aberraciones cromosómicas estructurales en células linfocíticas de sangre periférica humana in vitro. En linfocitos de sangre periférica humana in vitro se observó poliploidia (aberraciones cromosómicas numéricas), tanto en presencia como en ausencia de activación metabólica. El sunitinib no fue clastogénico en la médula ósea de la rata in vivo. El metabolito activo principal no fue evaluado para determinar su potencial de toxicidad genética.
Carcinogenicidad: En un estudio de determinación de la dosis oral administrada con sonda, de 1 mes (0, 10, 25, 75 o 200 mg/kg/día) con administración diaria continua en ratones transgénicos rasH2 se observaron carcinoma e hiperplasia de las glándulas de Brunner del duodeno a la dosis más alta evaluada (200 mg/kg/día).
En un estudio de carcinogenicidad con administración de la dosis por sonda vía oral de 6 meses de duración (0, 8, 25 o 75 [reducida a 50] mg/kg/día), administrada diariamente en ratones transgénicos rasH2 se observaron carcinomas gastroduodenales, aumento en la incidencia de hemangiosarcomas espontáneos o hiperplasia de las mucosas a dosis =25 mg/kg/día después de 1 o 6 meses de duración (=7,3 veces el AUC en sujetos que recibieron la RDD).
En un estudio de carcinogenicidad en ratas de 2 años de duración (0; 0,33; 1 o 3 mg/kg/día), la administración de sunitinib en ciclos de 28 días seguidos por periodos sin dosis de 7 días produjo aumento en la incidencia de feocromocitomas e hiperplasia en la médula suprarrenal de las ratas macho que recibieron 3 mg/kg/día después de >1 año de administración (=7,8 veces la AUC en sujetos que recibieron la RDD). El carcinoma de glándulas de Brunner ocurrió en el duodeno a =1 mg/kg/día en las hembras y a 3 mg/kg/día en los machos y la hiperplasia de células mucosas fue evidente en el estómago glandular a 3 mg/kg/día en los machos, lo cual ocurrió a =0,9; 7,8 y 7,8 veces la AUC en sujetos que recibieron la RDD, respectivamente. La relevancia para los humanos de los hallazgos neoplásicos observados en el ratón (transgénicos rasH2) y en los estudios de carcinogenicidad en la rata con el tratamiento con sunitinib no queda clara.
Toxicidad reproductiva y del desarrollo: No se observaron efectos sobre la fertilidad, en ratas machos dosificadas por 58 días, antes de aparearlas con hembras no-tratadas. No se observaron efectos sobre la reproducción, en ratas hembras tratadas por 14 días antes de aparearlas con machos no-tratados, con dosis que resultaron en una exposición sistémica de aproximadamente 5 veces la exposición sistémica en humanos. Sin embargo, en los estudios de toxicidad de dosis repetidas realizados en ratas y monos, se observaron efectos sobre la fertilidad femenina bajo la forma de atresia folicular, degeneración del cuerpo lúteo, cambios endometriales en el útero y disminución del peso del útero y ovarios, con niveles de exposición sistémica clínicamente relevantes. Además, en los estudios de toxicidad de dosis repetidas realizados en ratas, los efectos sobre la fertilidad masculina se vieron bajo la forma de atrofia tubular en los testículos, reducción de los espermatozoides en los epidídimos y depleción del coloide en la próstata y vesículas seminales, con niveles de exposición plasmática 18 veces la exposición sistémica en humanos. No todos los efectos observados en las ratas machos, se habían revertido al finalizar el período de recuperación (6 semanas).
En las ratas, la mortalidad embriofetal relacionada con el tratamiento fue evidente en términos de reducciones significativas en el número de fetos vivos, el mayor número de resorciones (tempranas y totales), el incremento correspondiente en la pérdida post-implantación y la pérdida de la camada completa en 8 de 28 hembras preñadas, con niveles de exposición plasmática 5,5 veces la exposición sistémica en humanos. En los conejos, las disminuciones de los pesos de los úteros grávidos y del número de fetos vivos, se debieron al aumento del número de las resorciones (tempranas y totales), al incremento en la pérdida post-implantación y a la pérdida de la camada completa en 4 de 6 hembras preñadas, con niveles de exposición plasmática 3 la exposición sistémica en humanos.
El tratamiento con sunitinib en ratas durante la organogénesis, resultó en efectos sobre el desarrollo a ³5 mg/kg/día, consistente en incidencia aumentada de malformaciones esqueléticas fetales, caracterizadas predominantemente como osificación retardada de las vértebras torácicas/lumbares. Los defectos del desarrollo en las ratas, ocurrieron con niveles de exposición plasmática 6 la exposición sistémica en humanos. En los conejos, los efectos sobre el desarrollo consistieron en una mayor incidencia de labio hendido, con niveles de exposición plasmática aproximadamente iguales a los observados en la clínica y labio hendido y paladar hendido, con niveles de exposición plasmática 2,7 veces la exposición sistémica en humanos.
No se efectuó un estudio definitivo de toxicidad en el desarrollo embriofetal en conejos, ya que los efectos embriofetales quedaron demostrados claramente en la rata y se informaron en el estudio preliminar efectuado en conejos.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES PARA EL USO: Hepatotoxicidad, disfunción ventricular izquierda y trastornos hemorrágicos.
Piel y tejidos: La coloración de la piel, posiblemente debido al color (amarillo) de la sustancia activa, fue una reacción adversa muy común en ensayos clínicos de pacientes/sujetos. Los sujetos también deben ser advertidos de que podría ocurrir despigmentación del cabello o de la piel, durante el tratamiento con sunitinib. Otros efectos dermatológicos posibles, incluyen resequedad, engrosamiento o agrietamiento de la piel, ampollas y ocasionalmente erupción en las palmas de las manos y plantas de los pies.
Los eventos antes mencionados no fueron acumulativos, típicamente fueron reversibles y generalmente no resultaron en la interrupción del tratamiento.
Se informaron reacciones cutáneas graves, entre ellas casos de eritema multiforme (EM) y casos indicativos de síndrome de Stevens-Johnson (SSJ), algunas de las cuales fueron mortales. Si hay signos o síntomas de SSJ o EM (p.ej., erupción cutánea progresiva habitualmente con ampollas o lesiones de la mucosa), se debe interrumpir el tratamiento con sunitinib. Si se confirma el diagnóstico de SSJ, no se deber reiniciar. En ciertos casos en que se sospecha de EM, los sujetos toleraron la reintroducción del tratamiento con una dosis menor de sunitinib después de la resolución de la reacción; algunos de estos sujetos también recibieron tratamiento concomitante con corticosteroides o antihistamínicos.
Eventos hemorrágicos: Eventos hemorrágicos han sido informados a través de la experiencia postcomercialización, algunos de los cuales fueron fatales, entre los que están incluidos hemorragias gastrointestinales (GI), respiratorias, tumorales, del tracto urinario y del cerebro. En estudios clínicos, en aproximadamente el 2% de los sujetos con TEGI, ocurrió hemorragia del tumor. Estos eventos pueden ocurrir súbitamente, y en el caso de los tumores de pulmón, se pueden presentar como hemoptisis o hemorragia pulmonar, con riesgo para la vida. Casos de hemorragia pulmonar, algunos con resultados mortales se han observado en ensayos clínicos y se han informado en la experiencia postcomercialización en sujetos tratados con sunitinib para CCRM, TEGI y Carcinoma de pulmón de célula no pequeña metastásico (CPCNP). El sunitinib no está aprobado para uso en sujetos con CPCNP.
Eventos de sangrado emergente durante el tratamiento ocurrieron en 18% de los sujetos que recibieron sunitinib en la fase de tratamiento doble ciego de un estudio de TEGI, comparado con 17% de los sujetos que recibieron placebo. En los sujetos que recibían sunitinib para el tratamiento de CCRM (Carcinoma de Células Renales Metastásico) no tratado previamente (naïve), el 39% de los sujetos tuvieron eventos de sangrado, comparado con 11% de los sujetos que recibían interferón-a (IFN-a). Diecisiete (4,5%) de los sujetos en sunitinib versus 5 (1,7%) de los sujetos en IFN-a experimentaron eventos hemorrágicos Grado 3 o mayores. De los sujetos que recibían sunitinib para CCRM resistente a citocina, 26% experimentó sangrado. Los eventos hemorrágicos, excluida epistaxis, ocurrieron en 21,7% de los sujetos que estaban recibiendo sunitinib en el estudio de Fase 3 de TNEp comparados con el 9,85% de los sujetos que estaban recibiendo placebo. Los exámenes habituales para evaluar estos eventos, incluyen recuento sanguíneo completo y examen físico.
Tracto gastrointestinal: Complicaciones GI serias, algunas veces fatales, incluyendo perforación GI, han ocurrido raramente en sujetos con malignidades intrabdominales tratados con sunitinib.
Eventos gastrointestinales: Náuseas, diarrea, estomatitis, dispepsia y vómitos han sido los eventos más frecuentes asociados con el tratamiento. La asistencia complementaria en estos casos puede incluir anti-eméticos y anti- diarreicos.
Pancreatitis: Se ha informado pancreatitis en ensayos clínicos de sunitinib. Se han observado en sujetos con varios tumores sólidos que recibieron sunitinib, un aumento sérico de los niveles de lipasa y amilasa. Los aumentos de los niveles de lipasa fueron transitorios y generalmente no asociados a signos y síntomas de pancreatitis en pacientes con varios tumores sólidos. Si están presentes síntomas de pancreatitis, los sujetos deben suspender sunitinib y se les deberá proporcionar cuidado sintomático apropiado.
Hepatotoxicidad: En sujetos tratados con sunitinib se ha observado hepatotoxicidad. En <1% de los sujetos con tumores sólidos tratados con sunitinib, se observó insuficiencia hepática algunos casos con desenlaces fatales. Antes de iniciar el tratamiento, durante cada ciclo de tratamiento y como clínicamente sea indicado, se deberán controlar las pruebas de la función hepática (alanina transaminasa [ALT], aspartato transaminasa [AST], concentraciones de bilirrubina). Sunitinib se debe interrumpir durante eventos adversos hepáticos Grado 3 o 4 y deberá suspenderse si no se observa resolución.
Hematológicos: Recuentos absolutos de neutrófilos disminuidos y recuentos de plaquetas disminuidos fueron informados en ensayos clínicos. Tales eventos no fueron acumulativos, típicamente fueron reversibles y generalmente no resultaron en la interrupción del tratamiento. En adición, algunos casos de hemorragia fatal asociada con trombocitopenia fueron informados a través de la experiencia post-mercadeo.
Al inicio de cada ciclo de tratamiento, se les deben realizar recuentos sanguíneos completos a los sujetos que estén recibiendo tratamiento con sunitinib.
Cardiovasculares: Eventos cardiovasculares, como insuficiencia cardíaca, cardiomiopatía , isquemia del miocardio einfarto miocárdicos, algunos de los cuales fueron fatales, han sido informados a través de la experiencia post-mercadeo. Utilice sunitinib con precaución en pacientes que corran riesgo de sufrir, o que tengan antecedentes de haber sufrido, estas patologías.En los estudios clínicos ocurrieron disminuciones ³20% y valores por debajo del límite inferior de la normalidad (LIN) de la fracción de eyección ventricular izquierda (FEVI), en aproximadamente el 2% de los sujetos con TEGI tratados con sunitinib, en el 4% de los sujetos con CCRM resistente a citocina y en el 2% de los sujetos tratados con placebo. Estos descensos en la FEVI no fueron progresivos y frecuentemente mejoraron a medida que continuaba el tratamiento.
En el estudio de CCRM sin tratamiento previo (naïve), el 27% y 15% de los sujetos en sunitinib e IFN-a, respectivamente, tuvieron un valor de FEVI por debajo del límite inferior de la normalidad (Lower Limit of Normal-LLN). Dos sujetos (<1%), que recibieron sunitinib, fueron diagnosticados con insuficiencia cardíaca congestiva (ICC).
Insuficiencia cardíaca, insuficiencia cardíaca congestiva o insuficiencia ventricular izquierda se reportaron en 0,8% de los sujetos con tumores sólidos y en 1% de los sujetos tratados con placebo.
En el estudio de Fase 3 de TNEp, 1 (1,2%) sujeto que estaba recibiendo sunitinib presentó insuficiencia cardíaca mortal relacionada con el tratamiento.
Los sujetos que habían presentado eventos cardíacos, tales como infarto del miocardio (incluyendo angina severa/inestable), injerto de derivación de arteria coronaria/periférica, insuficiencia cardíaca congestiva (ICC) sintomática, accidente cerebrovascular o ataque isquémico transitorio, o embolismo pulmonar dentro de los 12 meses previos a la administración de sunitinib, fueron excluidos de los estudios clínicos de sunitinib. Se desconoce si los pacientes con esas afecciones concomitantes puedan estar en mayor riesgo de desarrollar disfunción ventricular izquierda relacionada con el fármaco. A los médicos se les aconseja sopesar este riesgo, contra los posibles beneficios del medicamento. Los sujetos deben ser monitoreados cuidadosamente, para identificar signos y síntomas clínicos de ICC, mientras estén recibiendo sunitinib. También se debe considerar la realización de evaluaciones periódicas de la FEVI, mientras el paciente esté recibiendo sunitinib. En los sujetos sin factores de riesgo cardíaco, se debe considerar la realización de una evaluación basal de la fracción de eyección.
Si se presentan manifestaciones clínicas de ICC, se recomienda la interrupción de sunitinib. La dosificación de sunitinib debe ser interrumpida o reducida en los sujetos sin evidencia clínica de ICC, pero con una fracción de eyección <50% y >20% por debajo de la basal.
Prolongación del intervalo QT: Se ha evidenciado que en concentraciones aproximadamente el doble de las terapéuticas, sunitinib prolonga (corrección de Fridericia) el intervalo QTcF (ver la sección Propiedades farmacocinéticas). Ningún sujeto tuvo una prolongación de intervalo QT/QTc, mayor que un Grado 2 de los Criterios de Terminologías Comunes para Eventos Adversos (CTCAE) v3.0. La prolongación del intervalo QT puede llevar a un mayor riesgo de arritmias ventriculares, incluyendo torsade de pointes. El torsade de pointes ha sido observado en <0,1% de los sujetos expuestos a sunitinib. Sunitinib se debe usar con precaución en los sujetos con antecedentes conocidos de prolongación del intervalo QT, en los sujetos que estén tomado antiarrítmicos o en los sujetos con enfermedad cardíaca relevante pre existente, bradicardia o trastornos electrolíticos. El tratamiento concomitante con inhibidores potentes de la CYP3A4, que pueden aumentar las concentraciones plasmáticas de sunitinib, se debe implementar con precaución y reducir la dosis de sunitinib.
Hipertensión: La hipertensión fue una reacción adversa muy común informada en ensayos clínicos de sujetos con tumores sólidos, incluidos principalmente GIST y CCR resistente a citocina. La dosis de sunitinib se redujo o retrasó temporalmente en aproximadamente el 2,7% de esta población de pacientes. A ninguno de ellos se le discontinuó el tratamiento con sunitinib. Se observó hipertensión severa (sistólica >200 mmHg o diastólica >110 mmHg) en el 4,7% de esta población de pacientes. Hipertensión se informó en aproximadamente el 33,9% de los sujetos que recibían sunitinib para el tratamiento de CCRM sin tratamiento previo (naïve), comparado con el 3,6% de los sujetos que recibían IFN-a. La hipertensión relacionada con el tratamiento fue informada en 26,5% de los sujetos que estaba recibiendo sunitinib en el estudio de Fase 3 de TNEp, comparados con el 4,9% de los pacientes que estaban recibiendo placebo. Se presentó hipertensión severa en el 10% de los sujetos con TNEp bajo tratamiento con sunitinib y en 3% de los sujetos con placebo. Los sujetos deben ser sometidos a un tamizaje para hipertensión y controlados en la forma más apropiada. Se recomienda la suspensión temporal en los sujetos con hipertensión severa, que no estén siendo controlados con manejo médico. El tratamiento se puede reasumir cuando la hipertensión esté apropiadamente controlada.
Disfunción tiroidea: Se recomienda la medición basal de la función tiroidea por el laboratorio y en el caso de los sujetos con hipotiroidismo o hipertiroidismo, estos deben ser tratados antes de iniciar el manejo con SUTENT, según la práctica médica estándar. Durante el tratamiento con SUTENT, todos los sujetos deben ser cuidadosamente observados para la aparición de signos o síntomas de disfunción tiroidea. Los sujetos con signos o síntomas sugestivos de disfunción tiroidea, deben ser monitoreados con exámenes de laboratorio de función tiroidea y se deben tratar de acuerdo con la práctica médica estándar.
Hipotiroidismo adquirido emergente durante el tratamiento, se observó en el 6,2% de los sujetos con TEGI que recibían sunitinib versus 1% con placebo. Hipotiroidismo fue informado como un evento adverso en el 16% de los sujetos en tratamiento con sunitinib del estudio de sujetos con CCRM sin tratamiento previo (naïve) y en 3 sujetos (<1%) en el brazo del IFN-a, y en el 4% de los sujetos durante los 2 estudios de CCRM resistente a citocina. Adicionalmente, en el 2% de los sujetos con CCRM resistente a citocina se informaron aumentos de la hormona estimuladora de la tiroides (TSH). En total, el 7% de la población con CCRM resistente a citocina tuvo evidencia clínica o de laboratorio de hipotiroidismo emergente del tratamiento. En el estudio de Fase 3 de TNEp, se informó hipotiroidismo relacion en 6 (7,2%) sujetos que estaban recibiendo sunitinib y en 1 (1,2) sujeto que estaba recibiendo placebo.
Casos de hipertiroidismo, algunos seguidos por hipotiroidismo, se han informado en ensayos clínicos y durante la experiencia post-mercadeo.
Convulsiones: En los estudios clínicos de sunitinib, se observaron convulsiones en sujetos con evidencia radiológica de metástasis cerebrales. Adicionalmente, hubo informes raros (<1%), algunos fatales, de sujetos que presentaron convulsiones y evidencia radiológica del síndrome de leucoencefalopatía posterior reversible (SLPR). Los sujetos con convulsiones y signos/síntomas compatibles con SLPR, tales como hipertensión, cefalea, estado de alerta disminuido, funcionamiento mental alterado y pérdida visual, incluyendo ceguera cortical, se deben controlar con atención médica, incluyendo control de hipertensión. Se recomienda la suspensión temporal de sunitinib; después de la resolución, el tratamiento se puede reasumir a discreción del médico tratante.
Procedimientos quirúrgicos: Durante el tratamiento con sunitinib se han informado casos de afectación del sanado de la herida quirúrgica. En sujetos sometidos a procedimientos quirúrgicos mayores se recomienda por precaución la interrupción temporal del tratamiento con sunitinib. La experiencia clínica con relación al momento de reiniciación del tratamiento después de intervención quirúrgica mayor es limitada. Por lo tanto, la decisión de reiniciar el tratamiento con sunitinib después de una intervención quirúrgica mayor se debe basar en el criterio clínico de recuperación de la cirugía.
Osteonecrosis de maxilar inferior (ONJ): ONJ ha sido observada con poca frecuencia en ensayos clínicos y ha sido informada en la experiencia post-comercialización en los sujetos tratados con sunitinib. La mayoría de los casos ocurrieron en sujetos que habían recibido tratamiento previo o concomitante con bifosfonatos intravenosos (IV), para lo cual ONJ ha sido identificada como un riesgo. Por tanto, se debe tener atención y cuidado cuando se utilizan sunitinib y bifosfonatos IV de forma simultánea o secuencial.
Los procedimientos dentales invasivos son también un factor de riesgo identificado para la ONJ. Antes del tratamiento con sunitinib, se debe considerar un examen dental y una apropiada odontología preventiva. En sujetos tratados con sunitinib, que previamente han recibido o están recibiendo bifosfonatos IV, en caso de ser posible se deben evitar procedimientos dentales invasivos.
Síndrome de lisis tumoral (SLT): Casos de SLT, algunos de ellos mortales, se han observado raramente en ensayos clínicos y se han informado en la experiencia postcomercialización en sujetos tratados con sunitinib. Generalmente los sujetos en riesgo de SLT son los que tienen elevada masa tumoral antes del tratamiento. Estos sujetos se deben controlar de cerca y tratar de la forma clínicamente indicada.
Fascitis necrotizante: Se han informado casos raros de fascitis necrotizante, incluida la del perineo, algunas veces fatal. Debe interrumpirse el tratamiento con Sunitinib en sujetos que desarrollan fascitis necrotizante y debe iniciarse con prontitud el tratamiento apropiado.
Proteinuria: Se han informado casos de proteinuria y síndrome nefrótico. Se recomienda análisis de orina basal y debe monitorearse a los sujetos por si desarrollan proteinuria o si esta empeora. La seguridad del tratamiento continuado con sunitinib en sujetos con proteinuria moderada a severa no ha sido evaluada sistemáticamente. Suspenda el sunitinib en sujetos con síndrome nefrótico.
Hipoglucemia: Se informaron disminuciones del azúcar en la sangre, en ciertos casos con síntomas clínicos, con el tratamiento con sunitinib. Los niveles de glucemia en los pacientes diabéticos se deben controlar con regularidad para evaluar si es necesario ajustar la dosis de los hipoglucemiantes para minimizar el riesgo de hipoglucemia.
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN: Para TEGI y CCRM, la dosis recomendada de sunitinib es 50 mg tomada oralmente todos los días, durante 4 semanas consecutivas, seguidas por un período de descanso de 2 semanas (Régimen 4/2), para abarcar un ciclo completo de 6 semanas.
Para TNEp, la dosis recomendada de sunitinib es 37,5 mg una vez al día vía oral sin periodo de reposo programado.
Sunitinib se puede ingerir con o sin alimentos.
Si se olvida una dosis, el paciente no debe tomarse una dosis adicional. El paciente debe tomarse la dosis usual prescrita el día siguiente.
Modificación de la dosis
Seguridad y tolerabilidad: Para TEGI y CCRM, las dosis se pueden modificar, según la seguridad y tolerabilidad individuales, en incrementos o decrementos de 12,5 mg hasta subir a 75 mg o disminuir hasta 25 mg.
Para TNEp, se pueden aplicar modificaciones de la dosis en incrementos o decrementos de 12,5 mg con base en la seguridad y tolerabilidad individual. La dosis máxima administrada en el estudio de Fase 3 de TNEp fue 50 mg diarios.
De acuerdo con la seguridad y tolerabilidad individual podrían requerirse interrupciones de la dosis.
CYP3A4 Inducción/Inhibición: La coadministración de sunitinib con inductores potentes de la CYP3A4, tales como la rifampicina, se debe evitar (ver la sección Interacción con otros medicamentos y otras formas de interacción). Si lo anterior no es posible, podría ser necesario aumentar progresivamente la dosis de sunitinib, en incrementos de 12,5 mg, hasta máximo 87,5 mg (TEGI y RCC) o 62,5 mg (TNEp) diarios, con base en un monitoreo cuidadoso de la tolerabilidad.
La coadministración de sunitinib con inhibidores potentes de la CYP3A4, tales como ketoconazol, debe ser evitada (ver la sección Interacción con otros medicamentos y otras formas de interacción). Si esto no es posible, podría ser necesario disminuir la dosis de sunitinib en 12,5 mg hasta llegar a un mínimo de 37,5 mg (TEGI y RCC) o 25 mg (TNEp) diarios.
Se recomienda seleccionar una medicación concomitante alternativa, con mínimo o ningún potencial para inducir o inhibir la CYP3A4.
Uso en pediatría: No se ha establecido la seguridad y la eficacia de sunitinib en sujetos pediátricos.
Uso en pacientes de edad avanzada: No se requieren ajustes de dosis en los sujetos de edad avanzada. Aproximadamente el 34% de los sujetos participantes en los estudios clínicos de sunitinib, tenían 65 o más años de edad. No se observaron diferencias significativas en seguridad o eficacia entre los sujetos jóvenes y mayores.
Insuficiencia hepática: No es necesario ajustar la dosis cuando se administra sunitinib a sujetos con insuficiencia hepática leve (Clase A Child-Pugh) o moderada (Clase B Child-Pugh). Sunitinib no ha sido estudiado en sujetos con insuficiencia hepática severa (Clase C Child-Pugh) (ver la sección Propiedades farmacocinéticas).
Insuficiencia renal: Cuando se administra sunitinib a sujetos con deterioro renal (leve a severo) o con enfermedad renal en estado terminal (ESRD) a los que se les realiza hemodiálisis no se requieren ajustes de la dosis inicial. Los ajustes de las dosis subsiguientes se deben basar en la seguridad y tolerabilidad individuales.
SOBREDOSIS: No existe ningún antídoto específico para la sobredosificación con sunitinib y el tratamiento de las sobredosis deberá consistir de las medidas de asistencia generales. Si es indicado, se puede intentar la eliminación de la droga no-absorbida por emesis o lavado gástrico. Se han reportado casos de sobredosis; estos casos fueron asociados con reacciones adversas consistentes con el perfil de seguridad conocido de sunitinib.
PRESENTACIÓN:
SUTENT 12,5 mg cápsulas (Reg. San. INVIMA 2006M-0006444).
SUTENT 25 mg cápsulas (Reg. San. INVIMA 2006M-0006443).
SUTENT 50 mg cápsulas (Reg. San. INVIMA 2006M-0006442).
Es posible que la información de prescripción de este producto haya sido revisada y actualizada después de la fecha de impresión del PLM 2016. Para obtener información más actualizada comuníquese con la Dirección Médica de Pfizer S.A.S Teléfono: (1) 6002300 Ext. 2509 Bogotá – Colombia.
Título del Documento de Producto: Malato de Sunitinib
Fecha de reemplazo: 6 de Marzo de 2014
Fecha de vigencia: 14 de agosto de de 2014
Versión CDS: 37.0
LLD_Col_CDSv37.0_14Aug2014
PFIZER S.A.S.


