

REMINYL / REMINYL ER
GALANTAMINA
Solución oral
Solución, 100 Mililitros
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN: REMINYL® SOLUCIÓN Oral contiene hidrobromuro de galantamina, equivalente a 4 mg/mL de galantamina base. REMINYL® CÁPSULAS de liberación prolongada contiene hidrobromuro de galantamina, equivalente a 8 y 16 mg de galantamina base.
INDICACIONES TERAPÉUTICAS: REMINYL® está indicado para el tratamiento de la demencia de tipo Alzheimer leve a moderada.
FORMA FARMACÉUTICA:
Solución oral 4 mg/mL: Solución clara e incolora.
Cápsulas de liberación prolongada para uso oral. 8 mg de galantamina, cápsulas de gelatina dura, de color blanco opaco, con la inscripción “GAL8”, con gránulos blancos a blanco crema. 16 mg de galantamina, cápsulas de gelatina dura, de color rosado opaco, con la inscripción “GAL16”, con gránulos blancos a blanco crema.
PROPIEDADES FARMACOCINÉTICAS:
Absorción: Después de una ingesta oral de una dosis única de 8 mg de galantamina como tabletas, la absorción es rápida, con una concentración plasmática máxima de 43 ± 13 ng/ml, el cual es alcanzado después de 1.2 horas y un ABC¥ promedio de 427 ± 102 ng.h/ml. La biodisponibilidad oral absoluta de galantamina es de 88.5%. La ingesta oral de galantamina tabletas con la comida disminuye la velocidad de absorción (Cmax reducida alrededor de 25%), pero no afecta el grado a la cual es absorbida (ABC).
Después de repetir la dosis oral de 12 mg de galantamina dos veces al día como tabletas, las concentraciones plasmáticas máximas y mínimas promedio fluctúan entre 30 y 90 mg/ml. La farmacocinética de galantamina es lineal en el rango de dosis de 4 – 16 mg dos veces al día.
Biodisponibilidad de las formulaciones de liberación inmediata frente a la de liberación prolongada: En un estudio de biodisponibilidad en el estado estacionario, REMINYL® ER Cápsulas de liberación prolongada, 24 mg una vez al día, mostró ser bioequivalente a las tabletas de liberación inmediata, 12 mg dos veces al día, con respecto al ABC(24hr) y la Cmin. El valor de la Cmax de la cápsula de liberación prolongada de 24 mg una vez al día, el cual se alcanzó después de 4.4 horas, fue alrededor de 24 % menos que la de las tabletas de liberación inmediata de 12 mg dos veces al día. La comida no tuvo ningún efecto en la bioequivalencia en el estado estacionario de las cápsulas de liberación prolongada de 24 mg. En un estudio de proporcionalidad de dosis de REMINYL® ER Cápsulas de liberación prolongada en ancianos sanos y pacientes adultos jóvenes, la concentración plasmática en el estado estacionario se alcanzó dentro de los 6 días con todas las dosis (8 mg, 16 mg y 24 mg) en los dos grupos. La farmacocinética en el estado estacionario fue proporcional a las dosis dentro del rango de la dosis estudiada de 8 mg a 24 mg en ambos grupos.
Figura 3: Grafica lineal comparativa de la media de la concentración de galantamina en plasma vs perfil de tiempo
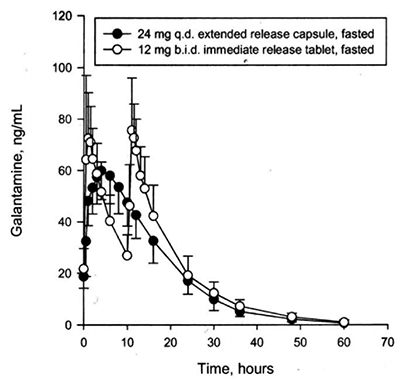
Distribución: La galantamina tiene un volumen de distribución moderado (Vdss promedio, 175l).
La unión a proteínas plasmáticas de la galantamina es bajo: 17.7 ±0.8%. En la sangre entera, la galantamina es principalmente distribuida a la células sanguíneas (52.7%) y al agua plasmática (39.0%), mientras que la fracción de galantamina unida a proteínas plasmáticas es solo del 8.4%. La proporción de concentración sangre/plasma de galantamina es de 1.17.
Metabolismo: Las principales vías metabólicas fueron la N-oxidación, N-demetilación, O-demetilación, glucuronidación y epimerización. La O-demetilación fue mucho importante en los metabolizadores rápidos del CYP2D6. Los niveles de excreción de la radioactividad total en la orina y heces no fueron diferentes entre los metabolizadores lentos y rápidos. Estudios in vitro confirmaron que el 2D6 y 3A4 del citocromo P450 fueron las principales isoenzimas del citocromos P450 involucradas en el metabolismo de la galantamina.
En el plasma de los metabolizadores lentos y rápidos, la galantamina intacta y su glucuronato presentaron la mayor parte de la radiactividad de las muestra. El glucuronato de la O-desmetilgalantamina también es importante en el plasma de los metabolizadores rápidos.
Ninguno de los metabolitos activos de galantamina (norgalantamina, O-desmetilgalantamina y O-desmetil-norgalantamina) deberían ser detectados en su forma no conjugada después de una dosis única en el plasma de los metabolizadores lentos o rápidos. Después de múltiples dosis, norgalantamina fue detectada en el plasma de pacientes, pero no representó más que 10% de los niveles de galantamina.
Eliminación: La galantamina es un medicamento de baja depuración (depuración plasmática de aproximadamente 300 ml/min). La eliminación de galantamina es bi-exponencial, con una vida media terminal en el orden de 7 – 8 h.
Siete días después de una dosis oral de 4 mg de 3H-galantamina, se recuperó en la orina el 90-97% de la radioactividad y 2.2 – 6.3% en las heces. Después de la administración oral e intravenosa, 18 – 22 % de la dosis fue excretada como galantamina inalterada en la orina en 24 horas, con una depuración renal de alrededor de 65 ml/min., lo cual representa 20 – 25% de la depuración plasmática total.
• Poblaciones especiales:
Insuficiencia renal: La disposición de la galantamina se estudió en sujetos jóvenes con grados variables de función renal. La eliminación de la galantamina disminuyó con la disminución de la depuración de la creatinina. Las concentraciones plasmáticas de la galantamina aumentaron en los sujetos con deterioro de la función renal: 38% en los sujetos con insuficiencia renal moderada (depuración de creatinina = 52-104 ml/min) o 67% en sujetos con insuficiencia renal severa (depuración de creatinina = 9-51 ml/min), en comparación con los sujetos sanos emparejados por edad y peso (depuración de creatinina ≥ 121 ml/min). El análisis y las simulaciones de la farmacocinética poblacional indican que no es necesario ajustar la dosis en pacientes con Alzheimer con insuficiencia renal siempre que la depuración de creatinina sea por lo menos 9 ml/min (Ver sección Posología y Método de administración - Poblaciones Especiales) ya que la depuración de galantamina es más baja en la población con Alzheimer.
Insuficiencia hepática: La farmacocinética de la galantamina en sujetos con insuficiencia hepática leve (puntuación de Child-Pugh de 5-6) fue comparable con la observada en sujetos sanos. En pacientes con deterioro hepático moderado (puntuación de Child-Pugh de 7-9), el ABC y la vida media de la galantamina aumentaron alrededor del 30% (ver sección Posología y Método de Administración - Poblaciones Especiales).
Características en pacientes con enfermedad de Alzheimer: Información de ensayos clínicos en pacientes indican que las concentraciones plasmáticas de galantamina en pacientes con enfermedad de Alzheimer es de 30 – 40% mayor en sujetos jóvenes sanos.
PROPIEDADES FARMACODINÁMICAS:
Grupo farmacoterapéutico: Fármacos antidemencia: Código ATC: N06DA04
Mecanismo de acción: La galantamina, un alcaloide terciario, es un inhibidor selectivo, competitivo y reversible de la acetilcolinesterasa. Adicionalmente, la galantamina aumenta las acciones intrínsecas de la acetilcolina sobre el receptor nicotínico, probablemente a través de la unión a un sitio alostérico del receptor. En consecuencia, un aumento de la actividad en el sistema colinérgico asociado con una mejoría de la función cognitiva pueden ser alcanzadas en los pacientes con demencia de tipo Alzheimer.
CONTRAINDICACIONES: REMINYL® no debe ser administrado a pacientes con hipersensibilidad conocida al hidrobromuro de galantamina o a cualquiera de los excipientes usados en las formulaciones.
EMBARAZO Y LACTANCIA:
Embarazo: Estudios de reproducción realizados en ratas preñadas con dosis hasta de 16 mg/kg (o alrededor de 25 veces la dosis terapéutica humana) y en conejas preñadas con dosis hasta de 40 mg/kg (o alrededor de 63 veces la dosis terapéutica humana) no mostraron ninguna evidencia de potencial teratogénico. Un incremento no significativo en la incidencia de anormalidades mínimas en el esqueleto fue notado con dosis de 16 mg/kg en las ratas.
No hay estudios disponibles del uso de REMINYL® en mujeres embarazadas. REMINYL® debe ser usado durante el embarazo solamente si el beneficio potencial justifica el potencial riesgo al feto.
Lactancia: Se desconoce si REMINYL® se excreta en la leche materna humana y no hay estudios en mujeres en periodo de lactancia. Por lo tanto, las mujeres que toman REMINYL® no deben dar de lactar.
EFECTOS EN LA CAPACIDAD PARA MANEJAR O UTILIZAR MÁQUINAS: La enfermedad Alzheimer puede causar deterioro gradual en la capacidad para manejar o comprometer la habilidad de usar maquinaria. Además, como otros colinomiméticos, REMINYL® puede causar reacciones adversas (como mareos y somnolencia), las cuales podrían afectar la habilidad de manejar o usar maquinaria, especialmente durante las primeras semanas después de iniciado el tratamiento. (Ver sección Reacciones adversas).
REACCIONES ADVERSAS: En toda esta sección se presentan las reacciones adversas. Las reacciones adversas son eventos adversos que fueron considerados estar razonablemente asociados con el uso de bromhidrato de galantamina, según la evaluación detallada de la información disponible sobre el evento adverso. No es posible establecer de manera confiable una relación causal con el bromhidrato de galantamina en los casos individuales. Además, debido a que los estudios clínicos se realizan en condiciones muy variadas, las tasas de las reacciones adversas observadas en los estudios clínicos de un medicamento no pueden compararse directamente con las tasas de estudios clínicos de otro medicamento y podrían no reflejar las tasas observadas en la práctica clínica.
INCOMPATIBILIDADES: No aplicable.
INTERACCIONES FARMACOCINÉTICAS: Varias vías metabólicas y la excreción renal están involucradas en la eliminación de galantamina. Basados en estudios in vitro, la CYP2D6 y CYP3A4 fueron las principales enzimas involucradas en el metabolismo de la galantamina.
La inhibición de la secreción del ácido gástrico no impide la absorción de la galantamina.
Otros medicamentos que influyen en el metabolismo de la galantamina: Los medicamentos que son potentes inhibidores del CYP2D6 o CYP3A4 pueden incrementar el área bajo la curva (ABC) de la galantamina. Estudios farmacocinéticos de dosis múltiple demostraron que el ABC de la galantamina se incrementó en 30% y 40%, respectivamente, durante la coadministración de ketoconazol y paroxetina. Así como cuando se coadministró con eritromicina, otro inhibidor del CYP3A4, el ABC de la galantamina sólo se incrementó alrededor del 10%. El análisis farmacocinético de la población para pacientes con enfermedad de Alzheimer, mostró que la depuración de galantamina descendió entre 25 - 33% por la administración concomitante de amitriptilina, fluoxetina, fluvoxamina, paroxetina y quinidina, conocidos inhibidores del CYP2D6.
Por consiguiente, durante la iniciación del tratamiento con inhibidores potentes del CYP2D6 o CYP3A4, los pacientes podrían experimentar una incidencia incrementada de efectos secundarios colinérgicos, predominantemente náuseas y vómitos. En estas circunstancias, basado en la tolerabilidad, se puede considerar una disminución de la dosis de mantenimiento de la galantamina. (Ver sección Posología y método de administración – Poblaciones Especiales).
Memantina, un antagonista del receptor N-metil-D-aspartato (NMDA), a una dosis de 10 mg/día por dos días seguido de 10mg dos veces al día por 12 días no tuvo efecto sobre la farmacocinética de galantamina a dosis de 16 mg/día en el estado estacionario.
Efectos de la galantamina en el metabolismo de otros medicamentos: Las dosis terapéuticas de galantamina (12 mg dos veces al día) no tuvieron efecto en la cinética de la digoxina y de la warfarina. La galantamina no afectó el incremento en el tiempo de protombina inducido por la warfarina.
Estudios in vitro indicaron que el potencial de inhibición de la galantamina con respecto a las formas principales del citocromo P450 humano es muy bajo.
INTERACCIONES FARMACODINÁMICAS: Debido a su mecanismo de acción, la galantamina no debe ser administrada concomitantemente con otros colinomiméticos. La galantamina antagoniza el efecto de los medicamentos anticolinérgicos. Como es de esperar con los colinomimeticos, una interacción farmacodinámica es posible con los medicamentos que significativamente reducen la frecuencia cardiaca (p. ej., digoxina y beta-bloqueadores).
La galantamina, como colinomimético, es probable que aumente la relajación muscular tipo succinilcolina durante la anestesia.
ESTUDIOS CLÍNICOS: Las dosis de REMINYL® que demostraron ser eficaces en los estudios clínicos controlados en la enfermedad de Alzheimer fueron 16, 24 y 32 mg/día. De estas dosis, se determinó que 16 y 24 mg/día tienen la mejor relación riesgo/beneficio y son las dosis recomendadas. La eficacia de la galantamina ha sido estudiada usando la medición de cuatro criterios de evaluación específicos: El ADAS-cog (una medición de la cognición basada en el desempeño), el CIBIC-plus (una evaluación global realizada por un médico independiente basado en una entrevista clínica con el paciente y el cuidador), varias mediciones de las actividades cotidianas diarias y el Cuestionario Neuropsiquiátrico (NPI, una escala que mide los trastornos de la conducta).
En los estudios clínicos, el desempeño de los pacientes tratados con galantamina en el ADAS-cog (ver Figura) y en el CIBIC-plus fue estadísticamente consistente y significativamente mejor que el de los pacientes tratados con el placebo. Los pacientes que fueron tratados durante 6 meses con galantamina tuvieron puntuaciones ADAS-cog que fueron significativamente mejores en comparación con sus puntuaciones basales. En comparación con los pacientes no tratados hubo un beneficio sustancial y sostenido en el funcionamiento cognitivo. El tratamiento con galantamina también preservó significativamente las actividades cotidianas diarias como vestirse, higiene y preparación de las comidas. Estos fueron valorados usando la Evaluación de la Discapacidad en la Demencia (DAD) y por la evaluación realizada por el cuidador con el Inventario del Estudio Cooperativo de la Enfermedad de Alzheimer (ADCS)-ADL. Las dosis de galantamina de 16 y 24 mg al día mantuvieron la puntuación NPI a lo largo del periodo de observación, mientras que la puntuación de los pacientes tratados con placebo mostró un claro deterioro como resultado del surgimiento de trastornos del comportamiento.
Figura 1: Cambio promedio (± SE) a partir del valor basal en la puntuación ADAS-cog/11 en el tiempo (datos observados) (datos agrupados GAL-USA-1 y GAL-INT-1)
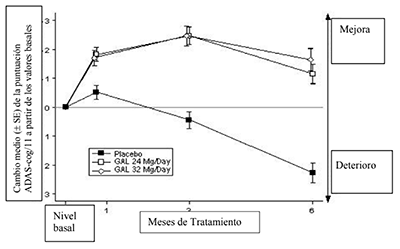
Figura 2: Cambio promedio (± SE) a partir del valor basal en la puntuación ADAS-cog/11 en el tiempo (todos los pacientes, datos observados) (GAL-USA-10)
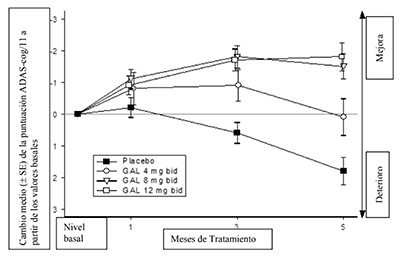
El tratamiento a largo plazo (combinación de 6 meses de tratamiento doble ciego seguidos de 6 meses de tratamiento abierto) sugirió que el desempeño cognitivo y funcional de los pacientes se mantuvo durante un año completo.
La eficacia de REMINYL® ER Cápsulas de liberación prolongada fue estudiado en un estudio aleatorizado, doble ciego, controlado con placebo en enfermedad de Alzheimer. Los pacientes recibieron galantamina 8 mg/día por 4 semanas, seguido de galantamina 16 mg/día por 4 semanas. En la semana 8, la dosis pudo ser incrementada a 24 mg/día basado en la seguridad y tolerabilidad, y pudo ser reducido a 16 mg/día en la semana 12. La dosis seleccionada en la semana 12 se fijó por el resto de los 6 meses. En el análisis de la eficacia primaria especificada por el protocolo para los dos criterios de evaluación (ADAS-cog/11 y CIBIC-plus) en el mes 6 simultáneamente, REMINYL® liberación prolongada mostró una mejoría estadísticamente significativa frente al placebo solamente para ADAS-cog/11. Además, REMINYL® liberación prolongada fue estadísticamente y significativamente mejor que el placebo en el mejoramiento de las actividades cotidianas diarias (ADCS-ADL), una medida secundaria clave de la eficacia. Los resultados de la eficacia fueron similares para REMINYL® ER Cápsulas de liberación prolongada y galantamina tabletas, las cuales sirvieron como un control activo en este estudio.
Eficacia y seguridad a largo plazo (2 años) en enfermedad de Alzheimer leve a moderadamente severa: Un estudio multicéntrico, aleatorizado, doble ciego, controlado con placebo, con grupo paralelo evaluó la eficacia y seguridad a largo plazo (2 años) de las cápsulas de liberación prolongada de galantamina en el tratamiento de pacientes con enfermedad de Alzheimer leve a moderadamente severa. Aleatoriamente se distribuyeron 1023 pacientes al grupo con placebo y 1028 al grupo con galantamina. Las características demográficas basales fueron similares entre los grupos. La mayoría de los pacientes fueron mujeres (65%) y de raza blanca (99.9%). La mediana de la edad fue 74 años de edad y la puntuación basal del examen del estado mini mental (MMSE) fue19.
En el criterio de evaluación principal de la eficacia (definido como el cambio en la puntuación MMSE desde la base hasta el mes 24), hubo un daño cognitivo significativamente menor en el grupo con galantamina en comparación con el grupo con placebo en lo referente al cambio en el MMSE desde la base hasta el mes 24 (-1.41 vs -2.14; p<0.001). En los criterios claves de evaluación secundaria de la eficacia (definidos como el cambio en el MMSE en el mes 6 y cambio en la calificación DAD en el mes 24), hubo una mejoría significativamente mayor en el MMSE desde la base hasta el mes 6 en el grupo con galantamina en comparación con el grupo con placebo (cambio medio 0.15 vs -0.28; p <0.001) y un daño significativamente menor en la calificación DAD en el mes 24 en el grupo con galantamina en comparación con el grupo con placebo (-8.2 vs -10.8; p = 0.002).
En el criterio principal de evaluación de la seguridad (mortalidad), hubo un total de 89 muertes; 56 (5.5%) muertes en el grupo con placebo y 33 (3.2%) muertes en el grupo con galantamina. Esto representa una tasa de mortalidad significativamente mayor en el grupo con placebo en comparación con el grupo con galantamina [índice de riesgo e intervalos de confianza al 95% de 0.58 (0.37-0.89) (p = 0.011)].
Deterioro cognitivo leve (DCL): Dos estudios controlados, de 2 años de duración, en sujetos con DCL, no cumplieron los dos criterios principales de evaluación. Aunque la mortalidad fue baja (0.7%), se registraron inicialmente más muertes en los sujetos asignados aleatoriamente al grupo de galantamina (13/1026) que en los sujetos del grupo con placebo (1/1022); sin embargo, la incidencia de eventos adversos graves fue idéntica (19%) en los dos grupos de tratamiento.
El análisis con intención de tratamiento a 24 meses registró 20 muertes entre los sujetos asignados aleatoriamente para recibir placebo, en comparación a las 34 muertes registradas entre los sujetos asignados aleatoriamente para recibir galantamina (riesgo relativo [IC del 95%] = 1.70 [1.00, 2.90]; p = 0.051). De los sujetos que murieron, en el periodo especificado en el protocolo, 30 días después de la descontinuación del medicamento en el estudio doble ciego, hubo 14 pacientes en el grupo con galantamina y 3 en el grupo con placebo (riesgo relativo [IC del 95%] = 4.08 [1.57, 10.57]; p = 0.004). Se determinó que 13 muertes en el grupo con placebo y 20 muertes en el grupo con galantamina estaban directamente relacionadas con los eventos adversos desarrollados mientras los pacientes eran expuestos al medicamento del estudio doble ciego (riesgo relativo [CI del 95%] = 1.54 (0.78, 3.04); p = 0.218).
Más sujetos tratados con placebo que con galantamina descontinuaron el tratamiento antes de la muerte, lo que podría haber influenciado la diferencia en la mortalidad inicial registrada. Cuando se incluyeron los datos procedentes de la amplia proporción de pacientes de ambos grupos que descontinuaron el tratamiento antes de la finalización del periodo doble ciego (GAL-COG-3002), se identificaron un total de 102 muertes, 56 en el grupo de galantamina y 46 en el grupo con placebo (riesgo relativo [IC del 95%] = 1.24 [0.84, 1.83]; p = 0.274).
Las muertes se debieron a varias causas que no fueron inesperadas entre la población de edad avanzada, aproximadamente la mitad de las muertes, en ambos grupos, se debieron a causas vasculares.
DATOS DE ESTUDIOS CLÍNICOS:
Datos de estudios clínicos doble-ciego – Reacciones adversas reportadas con una frecuencia ≥ 1%: La seguridad de REMINYL® se evaluó en 6502 sujetos con demencia de tipo Alzheimer leve a moderadamente severa que participaron en 8 estudios clínicos doble-ciego controlados con placebo. La información descrita en esta sección se derivó de los datos agrupados.
Las reacciones adversas reportadas por ≥ 1% de sujetos tratados con REMINYL® en estos estudios se muestran en la Tabla 1.
|
Tabla 1. Reacciones adversas reportadas por ≥ 1% de los sujetos tratados con REMINYL® en 8 estudios clínicos doble-ciego controlados con placebo |
||
|
Sistema de clasificación de órganos Reacción adversa |
REMINYL® (n=3956) |
Placebo |
|
Trastornos del metabolismo y la nutrición |
||
|
Disminución del apetito |
7.4 |
2.1 |
|
Trastornos psiquiátricos |
||
|
Depresión |
3.6 |
2.3 |
|
Trastornos del sistema nervioso |
||
|
Mareo |
6.8 |
2.9 |
|
Cefalea |
7.1 |
5.5 |
|
Temblor |
1.6 |
0.7 |
|
Síncope |
1.4 |
0.6 |
|
Letargo |
1.3 |
0.4 |
|
Somnolencia |
1.5 |
0.8 |
|
Trastornos cardiacos |
||
|
Bradicardia |
1.0 |
0.3 |
|
Trastornos gastrointestinales |
||
|
Náuseas |
20.7 |
5.5 |
|
Vómito |
10.5 |
2.3 |
|
Diarrea |
7.4 |
4.9 |
|
Dolor abdominal |
2.0 |
0.6 |
|
Dolor de la parte alta del abdomen |
1.9 |
1.4 |
|
Dispepsia |
1.5 |
1.0 |
|
Malestar abdominal |
2.1 |
0.7 |
|
Trastornos musculoesqueléticos y del tejido conectivo |
||
|
Espasmos musculares |
1.2 |
0.5 |
|
Trastornos generales y condiciones en el lugar de la administración |
||
|
Fatiga |
3.5 |
1.8 |
|
Astenia |
2.0 |
1.5 |
|
Malestar general |
1.1 |
0.5 |
|
Investigaciones |
||
|
Disminución de peso |
4.7 |
1.5 |
|
Lesión, intoxicación y complicaciones de procedimientos |
||
|
Caída |
3.9 |
3.0 |
|
Laceración |
1.1 |
0.5 |
En un estudio clínico aleatorizado, doble-ciego, controlado con placebo, el perfil de seguridad del tratamiento con REMINYL® ER cápsulas de liberación prolongada una vez al día fue similar en frecuencia y naturaleza al observado con las tabletas.
Las náuseas y los vómitos, las reacciones adversas más frecuentes, producidos principalmente durante los periodos de titulación, duraron menos de una semana en la mayoría de los casos y casi todos los pacientes tuvieron un episodio. La prescripción de antieméticos y asegurar un aporte adecuado de líquidos pueden ser útiles en estos casos.
Datos de estudios clínicos doble ciego y abierto – Reacciones adversas reportadas con una frecuencia <1%: Además de los estudios clínicos doble ciego, la seguridad de REMINYL® se evaluó en 1454 sujetos con demencia de tipo Alzheimer leve a moderada severa que participaron en 5 estudios clínicos abiertos.
Las reacciones adversas adicionales no reportadas en la Tabla 1 que ocurrieron en <1% de los sujetos tratados con REMINYL® (n = 5410) en los grupos de datos de los 8 estudios doble ciego y 5 estudios abiertos se describen en la Tabla 2.
|
Tabla 2. Reacciones adversas reportadas por <1% de los sujetos tratados con REMINYL® en los estudios clínicos doble-ciego o abierto |
|
|
Sistema de clasificación de órganos Reacción adversa |
REMINYL® |
|
Trastornos del metabolismo y la nutrición |
|
|
Deshidratación |
0.96 |
|
Trastornos del sistema nervioso |
|
|
Disgeusia |
0.31 |
|
Hipersomnia |
0.55 |
|
Parestesia |
0.33 |
|
Trastornos oculares |
|
|
Visión borrosa |
0.31 |
|
Trastornos cardiacos |
|
|
Bloqueo auriculoventricular de primer grado |
0.30 |
|
Palpitaciones |
0.41 |
|
Bradicardia sinusal |
0.55 |
|
Extrasístoles supraventriculares |
0.46 |
|
Trastornos vasculares |
|
|
Enrojecimiento |
0.24 |
|
Hipotensión |
0.52 |
|
Trastornos gastrointestinales |
|
|
Arcadas |
0.22 |
|
Trastornos de la piel y el tejido subcutáneo |
|
|
Hiperhidrosis |
0.85 |
|
Trastornos musculoesqueléticos y del sistema conectivo |
|
|
Debilidad muscular |
0.61 |
Datos posteriores a la comercialización: Además de las reacciones adversas reportadas durante los estudios clínicos y descritas anteriormente, se han reportado las siguientes reacciones adversas durante la experiencia posterior a la comercialización.
La tabla 3 proporciona las frecuencias de las reacciones adversas de acuerdo a la siguiente la convención:
• Muy frecuente: ≥ 1/10 (≥ 10%)
• Frecuente: ≥ 1/100 y <1/10 (≥ 1% y < 10%)
• Poco frecuente: ≥ 1/1000 y < 1/100 (≥ 0.1% y < 1%)
• Raro: ≥ 1/10 000 y < 1/1000 (≥ 0.01% y < 0.1%)
• Muy raro: < 1/10 000 (< 0.01%), incluidos reportes aislados.
• Desconocido: No puede estimarse a partir de los datos disponibles.
|
Tabla 3. Reacciones adversas identificadas durante la experiencia posterior a la comercialización de REMINYL® |
|
|
Sistema de clasificación de órganos Reacción adversa |
Categoría de frecuencia estimada según las tasas de reportes espontáneos |
|
Trastornos del Sistema Inmunológico |
|
|
Hipersensibilidad |
Muy raro |
|
Trastornos Psiquiátricos |
|
|
Alucinaciones |
Muy raro |
|
Alucinaciones visuales |
Muy raro |
|
Alucinaciones auditivas |
Muy raro |
|
Trastornos del sistema nervioso |
|
|
Convulsiones |
Muy raro |
|
Trastornos del oído y del laberinto |
|
|
Tinnitus |
Muy raro |
|
Trastornos cardiacos |
|
|
Bloqueo auriculoventricular completo |
Muy raro |
|
Trastornos vasculares |
|
|
Hipertensión |
Muy raro |
|
Trastornos hepatobiliares |
|
|
Hepatitis |
Muy raro |
|
Trastornos de la piel y del tejido subcutáneo |
|
|
Síndrome de Stevens-Johnson |
Muy raro |
|
Pustulosis exantemática generalizada aguda |
Muy raro |
|
Eritema multiforme |
Muy raro |
|
Investigaciones |
|
|
Aumento de la enzima hepática |
Muy raro |
DATOS DE SEGURIDAD PRECLÍNICA: El resto de los datos de seguridad preclínica relevantes para el prescriptor han sido incluidos en las secciones apropiadas.
ADVERTENCIAS Y PRECAUCIONES:
Tipos de demencia distintos a la demencia de Alzheimer: REMINYL® está indicado en pacientes con demencia de tipo Alzheimer leve a moderada. No se ha demostrado el beneficio de REMINYL® en pacientes con otros tipos de demencia u otros tipos de deterioro de la memoria.
Reacciones cutáneas graves: Se han reportado reacciones cutáneas graves (síndrome de Stevens-Johnson y pustulosis exantématica generalizada aguda) en pacientes que reciben REMINYL® (ver sección Reacciones Adversas). Se recomienda informar a los pacientes sobre los signos de las reacciones cutáneas graves y que el uso de REMINYL® se descontinúe ante la primera aparición de un exantema cutáneo.
Control del peso: Los pacientes con enfermedad de Alzheimer pierden peso. El tratamiento con inhibidores de la colinesterasa, incluyendo galantamina, se ha asociado con la pérdida de peso en estos pacientes. El peso de los pacientes debe ser monitoreado durante la terapia.
Condiciones que requieren precaución:
Como con otros colinomiméticos, REMINYL® debe ser administrado con precaución en las siguientes condiciones:
Condiciones cardiovasculares: Debido a su acción farmacológica, los colinomiméticos pueden tener efectos vagotónicos en la frecuencia cardiaca, incluyendo bradicardia y todos los tipos de bloqueo del nódulo auriculoventricular (ver sección Reacciones Adversas). El potencial para esta acción puede ser particularmente importante para pacientes con “síndrome del seno enfermo” u otras alteraciones de la conducción cardiaca supraventricular o para quienes usan concomitantemente medicamentos que reducen significativamente la frecuencia cardiaca, tal como digoxina y beta-bloqueadores. En estudios clínicos, el uso de REMINYL® ha sido asociado con sincope y raramente con bradicardia severa.
Condiciones gastrointestinales: Los pacientes con alto riesgo de desarrollar úlceras pépticas, como por ejemplo, pacientes con antecedente de enfermedad ulcerosa o pacientes con predisposición a estos trastornos, incluyendo aquellos que están recibiendo concomitantemente medicamentos antiinflamatorios no esteroides (AINES), deben ser vigilados ante la aparición de síntomas. Sin embargo, los estudios clínicos con REMINYL® no mostraron ningún incremento, con relación al placebo, en la incidencia de enfermedad ulcerosa péptica o de hemorragia gastrointestinal. El uso de REMINYL® no está recomendado en pacientes con obstrucción gastrointestinal o en pacientes en recuperación de una cirugía gastrointestinal.
Condiciones Neurológicos: Se han reportado convulsiones con REMINYL® (ver sección Reacciones adversas – Datos posteriores a la comercialización). La actividad convulsiva puede ser también una manifestación de la enfermedad de Alzheimer.
Condiciones pulmonares: Debido a las acciones colinomiméticas, los colinomiméticos deben ser prescritos con precaución en pacientes con antecedente de asma severo o enfermedad pulmonar obstructiva.
Genitourinario: El uso de REMINYL® no está recomendado en pacientes con obstrucción del flujo urinario o en pacientes en recuperación de una cirugía de vejiga.
Seguridad en sujetos con deterioro cognitivo leve (DCL): REMINYL® no está indicado en aquellas personas con deterioro cognitivo leve (DCL); es decir, aquellos que demuestran deterioro aislado de la memoria superior al esperado por su edad y educación, pero que no cumplen los criterios de la enfermedad de Alzheimer.
Dos estudios controlados, de 2 años de duración, en pacientes con DCL no cumplieron los dos principales criterios de valoración de la eficacia. Aunque la mortalidad fue baja en ambos grupos de tratamiento, se registraron inicialmente más muertes en los pacientes asignados aleatoriamente a galantamina que al placebo; pero la incidencia de eventos adversos graves fue idéntica entre los grupos de tratamiento. Las muertes se debieron a varias causas que no son inesperadas en una población de edad avanzada. Cuando se incluyeron los datos procedentes de la amplia proporción de pacientes que descontinuaron el tratamiento antes de la finalización del periodo doble ciego, no hubo evidencia de un incremento del riesgo de muerte en los sujetos tratados con REMINYL® a través del tiempo. Más sujetos del grupo del placebo que del grupo de galantamina descontinuaron el tratamiento antes de la muerte, lo cual podría explicar las diferencias de la mortalidad registradas inicialmente.
Los resultados del estudio para el DCL discrepan de los observados en estudios para la enfermedad de Alzheimer. En los estudios agrupados para la enfermedad de Alzheimer (n = 4614), la tasa de mortalidad fue numéricamente mayor en el grupo del placebo que en el grupo de REMINYL®.
ADMINISTRACIÓN: REMINYL® se administra por vía oral.
REMINYL® Solución oral debe ser administrado dos veces al día, de preferencia con los alimentos de la mañana y de la noche.
REMINYL® ER Cápsulas de liberación prolongada debe ser administrado una vez al día en la mañana, de preferencia con la comida.
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN:
Posología - Adultos: Asegurar una adecuada ingesta de líquidos durante el tratamiento.
Dosis inicial: La dosis inicial recomendada de REMINYL® Solución oral es 4 mg dos veces al día por 4 semanas.
La dosis inicial recomendada de REMINYL® ER Cápsulas de liberación prolongada es 8 mg por día durante 4 semanas.
Conversión de formulaciones de liberación inmediata a cápsulas de liberación prolongada.
Los pacientes actualmente tratados con REMINYL® Solución oral pueden cambiar a REMINYL® ER Cápsulas de liberación prolongada, tomando su última dosis de REMINYL® ER Solución oral en la noche e iniciar tratamiento con REMINYL® ER Cápsulas de liberación prolongada una vez al día al siguiente mañana. Cuando se cambia de un régimen de dosificación de dos veces al día con REMINYL® Solución oral a un régimen de dosificación de una vez al día con REMINYL® ER Cápsulas de liberación prolongada, se debe administrar la misma dosis total diaria.
Mantenimiento de la dosis: La dosis inicial recomendada de mantenimiento es 16 mg/día (8 mg dos veces al día con solución oral o 16 mg una vez al día con cápsulas) y los pacientes deben mantenerse con 16 mg/día por lo menos por 4 semanas.
Un incremento a la dosis máxima de mantenimiento recomendada de 24 mg/día (12 mg dos veces al día con la solución oral o 24 mg una vez al día con las cápsulas) debe considerarse después de una evaluación apropiada incluyendo la evaluación del beneficio clínico y la tolerabilidad.
Retiro del tratamiento: Cuando se descontinua abruptamente el tratamiento (p. ej., preparación para procedimientos quirúrgicos) no se produce efecto rebote.
• Poblaciones especiales:
Población pediátrica: El uso de REMINYL® en niños no está recomendado. No está disponible información sobre el uso de REMINYL® en pacientes pediátricos.
Insuficiencia renal: Las concentraciones plasmáticas de galantamina pueden incrementarse en los pacientes con insuficiencia renal moderada (depuración de creatinina = 52-104 mL/min) a severa (depuración de creatinina = 9-51 ml/min).
En pacientes con depuración de creatinina ≥ 9 ml/min no se requiere ajustar la dosis. (Ver sección Propiedades farmacocinéticas – Poblaciones especiales).
El uso de REMINYL® en pacientes con depuración de creatinina menor a 9 ml/min no está recomendado debido a que no existen datos disponibles.
Insuficiencia hepática: Las concentraciones plasmáticas de galantamina pueden incrementarse en pacientes con insuficiencia hepática moderada a severa.
En pacientes con insuficiencia moderada de la función hepática (puntuación de Child-Pugh 7-9), basado en el modelo farmacocinético, la dosificación debe iniciarse con 4 mg una vez al día con formulaciones de liberación inmediata, preferentemente tomadas en la mañanas por lo menos por una semana. Basado en el modelo farmacocinético, con cápsulas de liberación prolongada, la dosificación debe iniciarse con 8 mg cada dos días por lo menos por una semana, preferentemente tomadas en las mañanas. Después de ello, los pacientes deben continuar con 4 mg dos veces al día con formulaciones de liberación inmediata u 8 mg una vez al día con cápsulas de liberación prolongada por lo menos por cuatro semanas. En esos pacientes la dosis diaria total no debe exceder 16 mg.
En pacientes con insuficiencia hepática severa (puntuación de Child-Pugh > 9), el uso de REMINYL® no está recomendado.
Tratamiento concomitante: En pacientes tratados con potentes inhibidores del CYP2D6 o CYP3A4, la reducción de la dosis puede ser considerada. (Ver sección Interacciones - Otros medicamentos que influyen en el metabolismo de la galantamina).
SOBREDOSIS:
Síntomas y signos: Es previsible que los signos y síntomas de una sobredosis significativa de galantamina sean similares a aquellas sobredosis de otro colinomimético. Estos efectos generalmente involucran el sistema nervioso central, el sistema nervioso parasimpático y la unión neuromuscular. Además de la debilidad muscular o fasciculaciones, algunos o todos los signos de una crisis colinérgica pueden desarrollarse: Nauseas severas, vómitos, calambres gastrointestinales, salivación, lagrimeo, micción, defecación, sudoración, bradicardia, hipotensión, colapsos y convulsiones. El incremento de la debilidad muscular junto con la hipersecreción traqueal y los broncoespasmos, podrían producir compromiso vital de la vía aérea.
Existen reportes posteriores a la comercialización en los cuales se han notificado Torsade de Pointes, prolongación del intervalo QT, bradicardia, taquicardia ventricular y pérdida momentánea de la consciencia asociada a sobredosis accidentales de galantamina. En un caso en el que la dosis era conocida, se ingirió 8 comprimidos de 4 mg (en total, 32 mg) en un solo día.
Dos casos adicionales de ingestión accidental de 32 mg (náuseas, vómitos y sequedad de boca; náuseas, vómitos y dolor de pecho subesternal) y uno de 40 mg (vómitos) causaron periodos breves de hospitalización para observación con recuperación completa. Un paciente, con antecedente de alucinaciones en los dos últimos años, a quien se había prescrito una dosis de 24 mg/día, recibió por error 24 mg dos veces al día, durante 34 días y desarrolló alucinaciones requiriendo hospitalización. Otro paciente, a quien se prescribió una dosis de 16 mg/día de la solución oral, ingirió por error 160 mg (40 mL) y experimentó sudoración, vómitos, bradicardia y presíncope una hora después, el cual requirió tratamiento hospitalario. Sus síntomas se resolvieron dentro de 24 horas.
Tratamiento: Como en cualquier caso de sobredosis, medidas generales de soporte deben ser usadas. En casos severos, los anticolinérgicos como la atropina pueden ser usados como un antídoto general para los colinomiméticos. Una dosis inicial de 0.5 a 1.0 mg por vía intravenosa es recomendada, con dosis subsecuentes basadas en la respuesta clínica.
Debido a que las estrategias para el manejo de sobredosis están evolucionando constantemente, se recomienda contactar a un centro de control de intoxicaciones, para determinar las últimas recomendaciones y para el manejo de la sobredosis.
VIDA UTIL:
REMINYL® Solución oral y REMINYL® ER cápsulas de liberación prolongada: 24 meses para todas las zonas climáticas.
Después del primer uso, úsese REMINYL® solución oral dentro de los 3 meses siguientes.
PRESENTACIONES: REMINYL® ER 8 mg por 7 y 14 cápsulas de liberación prolongada (Reg. San. INVIMA 2016M-0004734-R1); 16 mg por 7 y 14 cápsulas de liberación prolongada (Reg. San. INVIMA 2016M-0004731-R1). REMINYL® Solución 4 mg/mL por 100 mL (Reg. San. INVIMA 2013M-0001946-R1).
JANSSEN-CILAG, S. A.
Av. Calle 26 No. 69-76 - Edificio Elemento
Torre 2 - Piso 11 - PBX: (+57) 1 9271200 - Bogotá, D. C.
ALMACENAMIENTO: REMINYL® Cápsulas de liberación prolongada, almacénese a temperatura de inferior a 30 °C.
REMINYL® solución oral, almacénese a temperatura de inferior a 30 °C, protéjase de la congelación, úsese dentro de los tres meses siguientes al primer uso.
Manténgase fuera del alcance de los niños.


