
ONGLYZA
SAXAGLIPTINA
Comprimidos recubiertos
Caja , 14 Comprimidos recubiertos , 5 Miligramos
Caja , 28 Comprimidos , 5 Miligramos
Caja , 28 Comprimidos , 2.5 Miligramos
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
INDICACIONES Y USO: Monoterapia y terapia combinada: ONGLYZA® (saxagliptina) está indicado como complemento de la dieta y el ejercicio para mejorar el control glucémico en adultos con diabetes mellitus tipo 2 en múltiples entornos clínicos [ver Estudios Clínicos].
CARCINOGÉNESIS, MUTAGÉNESIS, DETERIORO DE LA FERTILIDAD: La saxagliptina no indujo tumores en ratones (50, 250 y 600 mg/kg) ni en ratas (25, 75, 150 y 300 mg/kg) con las dosis más altas evaluadas. Las dosis más altas evaluadas en ratones fueron equivalentes a alrededor de 870 (en machos) y 1165 (en hembras) veces la exposición humana con la máxima dosis humana recomendada (MDHR) de 5 mg/día. En ratas, las exposiciones fueron de aproximadamente 355 (machos) y 2217 (hembras) veces la MDHR.
La saxagliptina no fue mutagénica ni clastogénica, con o sin activación metabólica, en un ensayo bacteriano de Ames in vitro, un ensayo in vitro de citogenética en linfocitos humanos primarios, un ensayo oral in vivo de micronúcleos en ratas, un estudio oral in vivo de reparación del ADN en ratas, y un estudio oral de citogenética in vivo/in vitro en linfocitos de la sangre periférica de ratas. El metabolito activo no fue mutagénico en un ensayo bacteriano in vitro de Ames.
En un estudio de fertilidad de ratas, los machos fueron tratados con dosis orales administradas por sonda esofágica durante 2 semanas antes del apareamiento, durante el apareamiento, y hasta la fecha de sacrificio prevista (aproximadamente 4 semanas en total) y las hembras fueron tratadas con dosis orales administradas por sonda esofágica durante 2 semanas antes del apareamiento y hasta el séptimo día de gestación. No se observaron efectos adversos sobre la fertilidad con exposiciones de alrededor de 603 (machos) y 776 (hembras) veces la MDHR. Con dosis mayores, tóxicas para la madre, se observó aumento de resorción fetal (aproximadamente 2069 y 6138 veces la MDHR). Se observaron efectos adicionales sobre el ciclo de celo, la fertilidad, la ovulación y la implantación con aproximadamente 6138 veces la MDHR.
Toxicología animal: La saxagliptina produjo cambios adversos en la piel (costras y/o úlceras en la cola, los dedos, el escroto, y/o la nariz) en las extremidades de macacos de Java. Las lesiones cutáneas fueron reversibles con dosis equivalentes a >20 veces la MDHR pero en algunos casos fueron irreversibles y necrosantes cuando la exposición fue mayor. No se observaron cambios cutáneos adversos con exposiciones similares (1 a 3 veces) a la MDHR de 5 mg. No se han observado correlaciones clínicas de las lesiones cutáneas en los monos en los ensayos clínicos humanos de saxagliptina.
Estudios clínicos:
Mejoría en el control glicémico: Se ha estudiado ONGLYZA® como monoterapia y en combinación con metformina, gliburida, y tiazolidinedionas (pioglitazona y rosiglitazona).
Se aleatorizó a un total de 4148 pacientes con diabetes mellitus tipo 2 en seis estudios clínicos doble ciego controlados para evaluar la seguridad y la eficacia de ONGLYZA® en el control de la glucemia. En estos estudios se trató a un total de 3021 pacientes con ONGLYZA®. La edad promedio de los pacientes fue de 54 años, y el 71% de los pacientes eran blancos, 16% eran asiáticos, 4% eran negros, y el 9% de otros grupos raciales. Un grupo adicional de 423 pacientes, incluidos 315 que recibieron ONGLYZA®, participó en un estudio controlado con placebo de rangos de dosis de 6 a 12 semanas de duración.
En estos seis estudios doble ciego, se evaluó ONGLYZA® con dosis de 2,5 mg y 5 mg una vez al día. Tres de estos estudios también evaluaron una dosis diaria de 10 mg de saxagliptina. La dosis de 10 mg diarios de saxagliptina no brindó una mayor eficacia que la dosis diaria de 5 mg. La dosis de 10 mg no está aprobada., El tratamiento con dosis de 5 mg y 2,5 mg de ONGLYZA® produjo mejoras clínicamente importantes y estadísticamente significativas en la A1C, glucosa plasmática en ayunas (GPA), y glucosa postprandial a las 2 horas (GPP), después de la prueba estándar oral de tolerancia a la glucosa (POTG), en comparación con el control. Se observaron reducciones en la A1C en todos los subgrupos basados en el género, la edad, la raza y el IMC de referencia.
ONGLYZA® no estuvo asociado con cambios significativos respecto del nivel de referencia del peso corporal o los lípidos séricos en ayunas, en comparación con el placebo.
ONGLYZA® también ha sido evaluado en cuatro ensayos adicionales realizados en pacientes con diabetes tipo 2: Un ensayo con control activo que comparó ONGLYZA® con la glipizida en 858 pacientes inadecuadamente controlados con metformina sola, un ensayo controlado con placebo en 455 pacientes que no habían logrado un control adecuado con la insulina sola o con una asociación de insulina + metformina, un estudio que comparó ONGLYZA® con un placebo en 257 pacientes que no habían logrado un control adecuado con la combinación de metformina y una sulfonilurea y un ensayo con control de placebo que comparó ONGLYZA® con placebo en 170 pacientes con diabetes tipo 2 e insuficiencia renal moderada o grave, o ESRD.
Monoterapia: Un total de 766 pacientes con diabetes tipo 2 inadecuadamente controlada con dieta y ejercicio (A1C >7% a <10%) participaron en dos estudios a doble ciego controlados con placebo de 24 semanas de duración para evaluar la eficacia y seguridad de la monoterapia con ONGLYZA®.
En el primer estudio, tras un periodo de 2 semanas de inducción con dieta, ejercicio y placebo simple ciego, se aleatorizó a 401 pacientes para recibir 2,5 mg, 5 mg o 10 mg de ONGLYZA® o placebo. Los pacientes que no lograron responder a determinados objetivos glucémicos durante el estudio recibieron tratamiento con una terapia de rescate con metformina, añadida al placebo o a ONGLYZA®. Se evaluó la eficacia en la última medición previa a la terapia de rescate para los pacientes que requerían rescate. En este estudio no se permitió hacer ajustes de la dosis de ONGLYZA®.
El tratamiento con 2,5 mg y 5 mg de ONGLYZA® por día proporcionó mejorías significativas en la A1C, GPA y GPP en comparación con el placebo (Tabla 5). El porcentaje de pacientes que descontinuaron a causa de falta de control glucémico o que fueron rescatados debido a que cumplían con criterios glucémicos preespecificados fue del 16% en el grupo de tratamiento con 2,5 mg de ONGLYZA®, del 20% en el grupo de tratamiento con 5 mg de ONGLYZA® y del 26% en el grupo de placebo.
|
Tabla 5. Parámetros glucémicos en la semana 24 en un estudio controlado con placebo de monoterapia con ONGLYZA® en pacientes con diabetes tipo 2* |
|||
|
Parámetro de eficacia |
ONGLYZA® 2,5 mg N = 102 |
ONGLYZA® 5 mg N = 106 |
Placebo N = 95 |
|
Hemoglobina A1C (%) |
N = 100 |
N = 103 |
N = 92 |
|
Valor de referencia (media) |
7,9 |
8,0 |
7,9 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-0,4 |
-0,5 |
+0,2 |
|
Diferencia con el placebo (media ajustada†) |
-0,6‡ |
-0,6 ‡ |
|
|
Intervalo de confianza del 95% |
(-0,9, -0,3) |
(-0,9, -0,4) |
|
|
Porcentaje de pacientes que alcanzaron A1C <7% |
35% (35/100) |
38%§ (39/103) |
24% (22/92) |
|
Glucosa plasmática en ayunas (mg/dL) |
N = 101 |
N = 105 |
N = 92 |
|
Valor de referencia (media) |
178 |
171 |
172 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-15 |
-9 |
+6 |
|
Diferencia con el placebo (media ajustada†) |
-21§ |
-15§ |
|
|
Intervalo de confianza del 95% |
(-31, -10) |
(-25, -4) |
|
|
Glucosa postprandial a las 2 horas (mg/dL) |
N = 78 |
N = 84 |
N = 71 |
|
Valor de referencia (media) |
279 |
278 |
283 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-45 |
-43 |
-6 |
|
Diferencia con respecto al placebo (media ajustada†) |
-39¶ |
-37§ |
|
|
Intervalo de confianza del 95% |
(-61, -16) |
(-59, -15) |
|
|
* Población por intención de tratar usando la última observación durante el estudio o la última observación antes de la terapia de rescate con metformina para pacientes que requirieron rescate. † Media de mínimos cuadrados ajustada en función del valor de referencia. ‡ Valor de p <0,0001 en comparación con el placebo. § Valor de p < 0,05 en comparación con el placebo. ¶ No se evaluó la significancia para la GPP a las 2 horas para la dosis de 2,5 mg de ONGLYZA®. |
|||
Se realizó un segundo estudio de monoterapia de 24 semanas de duración para evaluar una gama de regímenes de dosificación para ONGLYZA®. Los pacientes sin tratamiento previo con diabetes inadecuadamente controlada (A1C =7% a =10%) fueron sometidos a un periodo de inducción de 2 semanas de duración de dieta, ejercicio y placebo simple ciego. Un total de 365 pacientes fueron aleatorizados para recibir ONGLYZA® 2,5 mg cada mañana, 5 mg cada mañana, 2,5 mg con posible ajuste de la dosis a 5 mg cada mañana o 5 mg cada noche, o placebo. Los pacientes que no lograron responder a determinados objetivos glucémicos durante el estudio fueron tratados con una terapia de rescate con metformina, añadida a placebo o a ONGLYZA®; el número de pacientes aleatorizados por grupo de tratamiento varió de 71 a 74.
El tratamiento con ONGLYZA® 5 mg cada mañana o de 5 mg cada noche brindó mejoras significativas en la A1C frente al placebo (reducciones medias corregidas por el efecto placebo de -0,4% y -0,3%, respectivamente). El tratamiento con ONGLYZA® 2,5 mg cada mañana también brindó mejoras significativas en la A1C frente al placebo (reducción media corregida por el efecto placebo de -0,4%).
Terapia de combinación
Adición de ONGLYZA® al tratamiento con la metformina: Un total de 743 pacientes con diabetes tipo 2 participaron en este estudio aleatorizado, doble ciego, controlado con placebo de 24 semanas de duración para evaluar la eficacia y seguridad de ONGLYZA® en combinación con metformina en pacientes con control glucémico inadecuado (A1C =7% y =10%) con metformina sola. A los pacientes se les solicitó mantener una dosis estable de metformina (1500 mg a 2550 mg al día) durante al menos 8 semanas para poder inscribirse en este estudio.
Los pacientes que cumplieron los criterios de elegibilidad se inscribieron en un periodo de inducción de dieta, ejercicio y placebo simple ciego de 2 semanas de duración, durante el cual recibieron metformina en la dosis que recibían antes del estudio, de hasta 2500 mg al día. Luego del periodo de inducción, los pacientes elegibles fueron aleatorizados para recibir 2,5 mg, 5 mg o 10 mg de ONGLYZA® o placebo, además de su actual dosis de metformina abierta. Los pacientes que no lograron responder a determinados objetivos glucémicos durante el estudio fueron tratados con una terapia de rescate con pioglitazona, añadida a los medicamentos del estudio existente. En este estudio no se permitió hacer ajustes a la dosis de ONGLYZA® y metformina.
En combinación con metformina, ONGLYZA® 2,5 mg y 5 mg proporcionó mejorías significativas en la A1C, GPA y GPP en comparación con el grupo de placebo más metformina (Tabla 6). En la Figura 1 se muestran los cambios medios respecto del valor de referencia para la A1C a lo largo del tiempo y en el criterio de valoración. La proporción de pacientes que suspendió por falta de control de la glucemia o que fueron rescatados por cumplir con criterios previamente especificados de glucemia fue del 15% en el grupo de ONGLYZA® 2,5 mg más metformina, del 13% en el grupo de ONGLYZA® 5 mg más metformina y del 27% en el grupo de placebo más metformina.
|
Tabla 6. Parámetros glucémicos en la semana 24 en un estudio controlado con placebo sobre la adición de ONGLYZA® al tratamiento con la metformina* |
|||
|
Parámetro de eficacia |
ONGLYZA® 2,5 mg + metformina |
ONGLYZA® 5 mg + metformina |
Placebo |
|
Hemoglobina A1C (%) |
N = 186 |
N = 186 |
N = 175 |
|
Valor de referencia (media) |
8,1 |
8,1 |
8,1 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-0,6 |
-0,7 |
+0,1 |
|
Diferencia con el placebo (media ajustada†) |
-0,7‡ |
-0,8‡ |
|
|
Intervalo de confianza del 95% |
(-0,9; -0,5) |
(-1,0; -0,6) |
|
|
Porcentaje de pacientes que alcanzaron A1C <7% |
37%§ (69/186) |
44%§ (81/186) |
17% (29/175) |
|
Glucosa plasmática en ayunas (mg/dL) |
N = 188 |
N = 187 |
N = 176 |
|
Valor de referencia (media) |
174 |
179 |
175 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-14 |
-22 |
+1 |
|
Diferencia con el placebo (media ajustada†) |
-16§ |
-23§ |
|
|
Intervalo de confianza del 95% |
(-23, -9) |
(-30, -16) |
|
|
Glucosa postprandial a las 2 horas (mg/dL) |
N = 155 |
N = 155 |
N = 135 |
|
Valor de referencia (media) |
294 |
296 |
295 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-62 |
-58 |
-18 |
|
Diferencia con respecto al placebo (media ajustada†) |
-44§ |
-40§ |
|
|
Intervalo de confianza del 95% |
(-60, -27) |
(-56, -24) |
|
|
* Población por intención de tratar usando la última observación durante el estudio o la última observación antes de la terapia de rescate con pioglitazona para pacientes que requirieron rescate. † Media de mínimos cuadrados ajustada en función del valor de referencia. ‡ Valor de p <0,0001 en comparación con el placebo + metformina. § Valor de p < 0,05 en comparación con el placebo + metformina. |
|||
FIGURA 1. Cambio medio con respecto a la referencia en la A1C en un estudio controlado con placebo sobre la adición de ONGLYZA® al tratamiento con metformina*
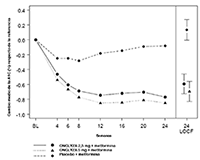
* Incluye los pacientes con un valor de referencia y un valor de la semana 24.
La semana 24 (LOCF) incluye la población por intención de tratar usando la última observación del estudio antes de la terapia de rescate con pioglitazona para los pacientes que requirieron rescate. El cambio medio respecto de la referencia se ajustó según el valor de referencia.
Adición de ONGLYZA® al tratamiento con una tiazolidinediona: Un total de 565 pacientes con diabetes tipo 2 participaron en este estudio aleatorizado, doble ciego, controlado con placebo de 24 semanas de duración para evaluar la eficacia y seguridad de ONGLYZA® en combinación con una tiazolidinediona (TZD) en pacientes con control glucémico inadecuado (A1C =7% a =10,5%) con tiazolidinediona sola. A los pacientes se les solicitó mantener una dosis estable de pioglitazona (30 mg a 45 mg una vez al día) o rosiglitazona (4 mg una vez al día o bien 8 mg una vez al día o en dos dosis de 4 mg) durante un mínimo de 12 semanas para poder inscribirse en este estudio.
Los pacientes que cumplieron con los criterios de elegibilidad se inscribieron en un periodo de inducción de dieta, ejercicio y placebo simple ciego de 2 semanas, durante el cual los pacientes recibieron TZD en la dosis que recibían previo al estudio. Luego del periodo de inducción, los pacientes elegibles fueron asignados al azar a 2,5 mg o 5 mg de ONGLYZA® o placebo, además de su dosis actual de TZD. Los pacientes que no lograron responder a determinados objetivos glucémicos durante el estudio fueron tratados con una terapia de rescate con metformina, añadida a los medicamentos del estudio existente. Durante el estudio no se permitieron ajustes a la dosis de ONGLYZA® o TZD. Se permitió un cambio en el régimen de TZD de rosiglitazona a pioglitazona en dosis terapéuticas determinadas, equivalentes, a discreción del investigador si lo consideraba médicamente apropiado.
En combinación con TZD, ONGLYZA® 2,5 mg y 5 mg proporcionó mejorías significativas en la A1C, GPA y GPP en comparación con el grupo de tratamiento de placebo más TZD (Tabla 7). La proporción de pacientes que suspendió por falta de control de la glucemia o que fueron rescatados por cumplir con criterios glucémicos previamente especificados fue del 10% en el grupo de ONGLYZA® 2,5 mg más TZD, del 6% en el grupo de ONGLYZA® 5 mg más TZD y del 10% en el grupo de placebo más TZD.
|
Tabla 7. Parámetros glucémicos en la semana 24 en un estudio controlado con placebo sobre la adición de ONGLYZA® al tratamiento con una tiazolidinediona* |
|||
|
Parámetro de eficacia |
ONGLYZA® 2,5 mg + TZD |
ONGLYZA® 5 mg + TZD |
Placebo + TZD |
|
Hemoglobina A1C (%) |
N = 192 |
N = 183 |
N = 180 |
|
Valor de referencia (media) |
8,3 |
8,4 |
8,2 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-0,7 |
-0,9 |
-0,3 |
|
Diferencia con el placebo (media ajustada†) |
-0,4‡ |
-0,6 ‡ |
|
|
Intervalo de confianza del 95% |
(-0,6; -0,2) |
(-0,8; -0,4) |
|
|
Porcentaje de pacientes que alcanzaron A1C <7% |
42%§ (81/192) |
42%§ (77/184) |
26% (46/180) |
|
Glucosa plasmática en ayunas (mg/dL) |
N = 193 |
N = 185 |
N = 181 |
|
Valor de referencia (media) |
163 |
160 |
162 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-14 |
-17 |
-3 |
|
Diferencia con el placebo (media ajustada†) |
-12§ |
-15§ |
|
|
Intervalo de confianza del 95% |
(-20, -3) |
(-23, -6) |
|
|
Glucosa postprandial a las 2 horas (mg/dL) |
N = 156 |
N = 134 |
N = 127 |
|
Valor de referencia (media) |
296 |
303 |
291 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-55 |
-65 |
-15 |
|
Diferencia con respecto al placebo (media ajustada†) |
-40§ |
-50§ |
|
|
Intervalo de confianza del 95% |
(-56, -24) |
(-66, -34) |
|
|
* Población con intención de tratar usando la última observación durante el estudio o la última observación antes de la terapia de rescate con metformina para pacientes que requieren rescate. † Media de mínimos cuadrados ajustada en función del valor de referencia. ‡ Valor de p <0,0001 en comparación con el placebo + TZD. § Valor de p < 0,05 en comparación con el placebo + TZD. |
|||
Adición de ONGLYZA® al tratamiento con gliburida: Un total de 768 pacientes con diabetes tipo 2 participaron en este estudio aleatorizado, doble ciego, controlado con placebo de 24 semanas de duración para evaluar la eficacia y seguridad de ONGLYZA® en combinación con sulfonilurea (SU) en pacientes con control glucémico inadecuado al momento de la inscripción (A1C =7,5% a =10%) con una dosis submáxima de SU sola. A los pacientes se les solicitó mantener una dosis submáxima de la SU durante 2 meses o más para poder inscribirse en este estudio. En este estudio, ONGLYZA® en combinación con una dosis intermedia fija de SU, se comparó con el ajuste a una dosis mayor de SU.
Los pacientes que cumplieron los criterios de elegibilidad se inscribieron en un periodo de inducción de dieta, ejercicio y placebo simple ciego de 4 semanas, y se les administró gliburida 7,5 mg una vez al día. Luego del periodo de inducción, los pacientes elegibles con A1C =7% a =10% fueron asignados al azar para recibir 2,5 mg o 5 mg de ONGLYZA® más gliburida 7,5 mg o placebo además de una dosis diaria total de gliburida de 10 mg. Los pacientes que recibieron placebo fueron elegibles para recibir gliburida, ajustando la dosis hasta una dosis total diaria de 15 mg. No se permitió aumentar la dosis de gliburida en pacientes que recibieron ONGLYZA® 2,5 mg o 5 mg. La gliburida podría ajustarse hacia abajo en cualquier grupo de tratamiento una vez durante el periodo de 24 semanas de duración del estudio debido a la hipoglucemia, cuando el investigador lo considerara necesario. Aproximadamente el 92% de los pacientes del grupo de placebo más gliburida recibieron un incremento en la dosis a un total diario de 15 mg durante las primeras 4 semanas del periodo de estudio. Los pacientes que no respondieron a determinados objetivos glucémicos durante el estudio fueron tratados con terapia de rescate de metformina, añadida a la medicación existente del estudio. Durante el estudio no se permitieron ajustes a la dosis de ONGLYZA®.
En combinación con gliburida, ONGLYZA® 2,5 mg o 5 mg proporcionó mejorías significativas en la A1C, GPA y GPP, en comparación con el grupo de placebo más gliburida con ajuste de la dosis (Tabla 8). La proporción de pacientes que suspendió por falta de control de la glucemia o que fueron rescatados por cumplir con criterios previamente especificados de glucemia fue del 18% en el grupo de ONGLYZA® 2,5 mg más gliburida, del 17% en el grupo de ONGLYZA® 5 mg más gliburida y del 30% en el grupo de placebo más gliburida con ajuste de la dosis.
|
Tabla 8. Parámetros glucémicos en la semana 24 en un estudio controlado con placebo sobre la adición de ONGLYZA® al tratamiento con gliburida* |
|||
|
Parámetro de eficacia |
ONGLYZA® 2,5 mg + gliburida 7,5 mg |
ONGLYZA® 5 mg + gliburida 7,5 mg |
Placebo + gliburida con ajuste de la dosis |
|
Hemoglobina A1C (%) |
N = 246 |
N = 250 |
N = 264 |
|
Valor de referencia (media) |
8,4 |
8,5 |
8,4 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-0,5 |
-0,6 |
+0,1 |
|
Diferencia con la gliburida con ajuste de la dosis (media ajustada†) |
-0,6‡ |
-0,7‡ |
|
|
Intervalo de confianza del 95% |
(-0,8; -0,5) |
(-0,9; -0,6) |
|
|
Porcentaje de pacientes que alcanzaron A1C <7% |
22%§ (55/246) |
23%§ (57/250) |
9% (24/264) |
|
Glucosa plasmática en ayunas (mg/dl) |
N = 247 |
N = 252 |
N = 265 |
|
Valor de referencia (media) |
170 |
175 |
174 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-7 |
-10 |
+1 |
|
Diferencia con la gliburida con ajuste de la dosis (media ajustada†) |
-8§ |
-10§ |
|
|
Intervalo de confianza del 95% |
(-14, -1) |
(-17, -4) |
|
|
Glucosa postprandial a las 2 horas (mg/dl) |
N = 195 |
N = 202 |
N = 206 |
|
Valor de referencia (media) |
309 |
315 |
323 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-31 |
-34 |
+8 |
|
Diferencia con respecto a gliburida con ajuste de la dosis (media ajustada†) |
-38§ |
-42§ |
|
|
Intervalo de confianza del 95% |
(-50, -27) |
(-53, -31) |
|
|
* Población por intención de tratar usando la última observación durante el estudio o la última observación antes de la terapia de rescate con metformina para pacientes que requirieron rescate. † Media de mínimos cuadrados ajustada en función del valor de referencia. ‡ Valor de p <0,0001 en comparación con el placebo + gliburida con ajuste de la dosis. § Valor de p < 0,05 en comparación con el placebo + gliburida con ajuste de la dosis. |
|||
Coadministración con metformina a pacientes sin tratamiento previo: Un total de 1306 pacientes sin tratamiento previo con diabetes mellitus tipo 2 participaron en este estudio aleatorizado, doble ciego, controlado con un fármaco de referencia, de 24 semanas de duración para evaluar la eficacia y seguridad de ONGLYZA® coadministrado con metformina a pacientes con control glucémico inadecuado (A1C =8% a =12%) solo con dieta y ejercicio. Uno de los requisitos para poder inscribirse en este estudio consistió en que los pacientes no hubieran recibido tratamiento previo.
Los pacientes que cumplieron los criterios de elegibilidad se inscribieron en un periodo de inducción de dieta, ejercicio y placebo simple ciego de 1 semana. Los pacientes fueron asignados al azar a uno de los cuatro grupos de tratamiento: ONGLYZA® 5 mg + 500 mg de metformina, saxagliptina 10 mg + 500 mg de metformina, saxagliptina 10 mg + placebo, o 500 mg de metformina + placebo. ONGLYZA® se administró una vez al día. En los 3 grupos de tratamiento con metformina, se aumentó la dosis de metformina en forma semanal por incrementos de 500 mg por día, según tolerancia, hasta un máximo de 2000 mg por día en base a la GPA. Los pacientes que no cumplieron con los objetivos específicos de glucemia durante los estudios recibieron tratamiento de rescate con pioglitazona añadida.
La coadministración de ONGLYZA® 5 mg más metformina proporcionó mejorías significativas en la A1C, GPA y GPP en comparación con el placebo más metformina (Tabla 9).
|
Tabla 9. Parámetros glucémicos en la semana 24 en un estudio controlado con placebo de coadministración de ONGLYZA® con metformina a pacientes sin tratamiento previo* |
||
|
Parámetro de eficacia |
ONGLYZA® 5 mg + metformina |
Placebo + metformina |
|
Hemoglobina A1C (%) |
N = 306 |
N = 313 |
|
Valor de referencia (media) |
9,4 |
9,4 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-2,5 |
-2,0 |
|
Diferencia con el placebo + metformina (media ajustada†) |
-0,5‡ |
|
|
Intervalo de confianza del 95% |
(-0,7; -0,4) |
|
|
Porcentaje de pacientes que alcanzaron A1C <7% |
60%§ (185/307) |
41% (129/314) |
|
Glucosa plasmática en ayunas (mg/dl) |
N = 315 |
N = 320 |
|
Valor de referencia (media) |
199 |
199 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-60 |
-47 |
|
Diferencia con el placebo + metformina (media ajustada†) |
-13§ |
|
|
Intervalo de confianza del 95% |
(-19, -6) |
|
|
Glucosa postprandial a las 2 horas (mg/dl) |
N = 146 |
N = 141 |
|
Valor de referencia (media) |
340 |
355 |
|
Cambio con respecto al valor de referencia (media ajustada†) |
-138 |
-97 |
|
Diferencia con el placebo + metformina (media ajustada†) |
-41§ |
|
|
Intervalo de confianza del 95% |
(-57, -25) |
|
|
* Población por intención de tratar usando la última observación durante el estudio o la última observación antes de la terapia de rescate con pioglitazona para pacientes que requirieron rescate. † Media de mínimos cuadrados ajustada en función del valor de referencia. ‡ Valor de p <0,0001 en comparación con el placebo + metformina. § Valor de p < 0,05 en comparación con el placebo + metformina. |
||
Comparación de la adición de ONGLYZA® o de glipizida al tratamiento con metformina: En este ensayo con control activo de 52 semanas de duración, un total de 858 pacientes con diabetes tipo 2 y control glucémico inadecuado (A1C >6,5% y =10%) con metformina solamente fueron aleatorizados a la terapia adyuvante a doble ciego con ONGLYZA® o glipizida. Los pacientes debían estar tomando una dosis estable de metformina (de al menos 1500 mg por día) durante al menos 8 semanas antes del enrolamiento.
Los pacientes que cumplían con los criterios de elegibilidad fueron enrolados en un periodo de inducción de dieta, ejercicio y placebo a simple ciego, de 2 semanas de duración, durante el cual recibieron metformina (1500-3000 mg según su dosis previa al estudio). Luego del periodo de inducción, los pacientes elegibles fueron aleatorizados para recibir 5 mg de ONGLYZA® o 5 mg de glipizida además de su dosis de metformina sin enmascaramiento. Los pacientes del grupo de glipizida más metformina tuvieron un ajuste ciego de la dosis de glipizida durante las primeras 18 semanas del ensayo hasta una dosis máxima de glipizida de 20 mg por día. El ajuste de la dosis se basó en un nivel objetivo de glucemia en ayunas = 110 mg/dl o la dosis tolerable más alta de glipizida. El 50% de los pacientes tratados con glipizida tuvieron un ajuste hasta la dosis de 20 mg por día; el 21% de los pacientes tratados con glipizida tuvieron una dosis diaria final de glipizida de 5 mg o menos. La dosis diaria final media de glipizida fue de 15 mg.
Luego de 52 semanas de tratamiento, ONGLYZA® y glipizida dieron como resultado reducciones medias similares en A1C desde el nivel basal cuando se agregaron a la terapia con metformina (Tabla 10). Esta conclusión se puede limitar a pacientes con un nivel basal de A1C comparable con aquellos del ensayo (el 91% de los pacientes tenía un nivel basal de A1C <9%).
A partir de un peso corporal medio basal de 89 kg, hubo una reducción media estadísticamente significativa de 1,1 kg en pacientes tratados con ONGLYZA® en comparación con un aumento de peso medio de 1,1 kg en pacientes tratados con glipizida (p<0,0001).
|
Tabla 10. Parámetros glucémicos en la semana 52 en un estudio controlado con agente activo de ONGLYZA® versus glipizida en combinación con metformina* |
||
|
Parámetro de eficacia |
ONGLYZA® 5 mg + metformina |
Glipizida titulada + metformina |
|
Hemoglobina A1C (%) |
N = 423 |
N = 423 |
|
Valor basal (media) |
7,7 |
7,6 |
|
Cambio respecto del valor basal (media ajustada†) |
-0,6 |
-0,7 |
|
Diferencia respecto de glipizida + metformina (media ajustada†) |
0,1 |
|
|
Intervalo de confianza del 95% |
(-0,02; 0,2)‡ |
|
|
Glucosa plasmática en ayunas (mg/dl) |
N = 420 |
N = 420 |
|
Valor basal (media) |
162 |
161 |
|
Cambio respecto del valor basal (media ajustada†) |
-9 |
-16 |
|
Diferencia respecto de glipizida + metformina (media ajustada†) |
6 |
|
|
Intervalo de confianza del 95% |
(2, 11)§ |
|
|
* Población por intención de tratar usando la última observación durante el estudio. † Media de mínimos cuadrados ajustada en función del valor basal. ‡ Saxagliptina + metformina se considera no inferior a glipizida + metformina porque el límite superior de este intervalo de confianza es menor que el margen de no inferioridad preespecificado del 0,35%. § Significación no evaluada. |
||
Adición de ONGLYZA® al tratamiento con insulina (con o sin metformina): Un total de 455 pacientes con diabetes tipo 2 participaron en este ensayo de 24 semanas, aleatorizado, doble ciego, controlado con placebo, que evaluó la eficacia y la seguridad de ONGLYZA® coadministrado con insulina en pacientes que no habían conseguido un control glucémico adecuado (A1C entre = 7.5% y = 11%) con insulina solamente (N=141) o con insulina + una dosis estable de metformina (N=314). Los pacientes debían haber estado recibiendo una dosis estable de insulina (entre =30 y =150 unidades al día) con una variación = 20% de la dosis diaria total durante = 8 semanas antes de la selección para el ensayo, con o sin metformina. Los pacientes recibían insulina de acción intermedia o prolongada, o insulina premezclada. Se excluyeron los pacientes tratados con insulina de acción corta, a menos que ésta formara parte de la insulina premezclada.
Los pacientes que cumplieron los criterios de elegibilidad fueron admitidos en un periodo de preinclusión de 4 semanas, con un diseño ciego simple, con dieta, ejercicio y un placebo. Durante este periodo, los pacientes recibieron la dosis de insulina asignada antes del estudio (y metformina, si procedía). Después del periodo de preinclusión, los pacientes elegibles fueron distribuidos al azar para recibir 5 mg de ONGLYZA® o un placebo además de la dosis habitual de insulina (y de metformina si procedía). Cuando fue posible, se mantuvo una dosis estable de insulina. Los pacientes que no cumplieron determinados objetivos de glucemia o que necesitaron un aumento de la dosis de insulina >20% recibieron un tratamiento de rescate y luego cambiaron a un régimen de administración flexible de insulina. No se permitió ajustar la dosis de ONGLYZA® ni de metformina (si procedía) durante el estudio.
La adición de 5 mg de ONGLYZA® al tratamiento con insulina (con o sin metformina) dio lugar a mejorías significativas de la A1C y la GPP frente a la adición de un placebo al tratamiento con insulina (con o sin metformina) (Tabla 11). Se consiguieron reducciones similares de la A1C frente al placebo (del -0.4% y -0.4%, respectivamente) al añadir 5 mg de ONGLYZA® a la monoterapia con insulina o al añadir 5 mg de ONGLYZA® al tratamiento con insulina y metformina La proporción de pacientes que suspendieron el tratamiento por un control glucémico insuficiente o que recibieron un tratamiento de rescate fue del 23% en grupo tratado con 5 mg de ONGLYZA® + insulina y del 32% en el grupo que recibió el placebo + insulina.
|
Tabla 11. Parámetros glucémicos en la semana 24 de un ensayo controlado con placebo sobre la adición de ONGLYZA® al tratamiento con insulina* |
||
|
Parámetro de eficacia |
ONGLYZA® 5 mg + insulina |
Placebo + insulina |
|
Hemoglobina A1C (%) |
N=300 |
N=149 |
|
Valor inicial (media) |
8.7 |
8.7 |
|
Variación respecto del valor inicial (media ajustada†) |
-0.7 |
-0.3 |
|
Diferencia con respecto al placebo (media ajustada†) |
-0.4‡ |
|
|
Intervalo de confianza del 95% |
(-0.6, -0.2) |
|
|
Porcentaje de pacientes que alcanzaron una A1C <7% |
17%§ (52/300) |
7% (10/149) |
|
Glucemia posprandial de 2 horas (mg/dl) |
N=262 |
N=129 |
|
Valor inicial (media) |
251 |
255 |
|
Variación respecto del valor inicial (media ajustada†) |
-27 |
-4 |
|
Diferencia con respecto al placebo (media ajustada†) |
-23¶ |
|
|
Intervalo de confianza del 95% |
(-37, -9) |
|
|
Glucemia en ayunas (mg/dl) |
N=300 |
N=149 |
|
Valor inicial (media) |
173 |
173 |
|
Variación respecto del valor inicial (media ajustada†) |
-10 |
-6 |
|
Diferencia con respecto al placebo (media ajustada†) |
-4# |
|
|
Intervalo de confianza del 95% |
(-13, 5) |
|
|
Dosis media diaria total de insulina (unidades) |
N=299 |
N=151 |
|
Valor inicial (media) |
53 |
55 |
|
Variación respecto del valor inicial (media ajustada†) |
2 |
5 |
|
Diferencia con respecto al placebo (media ajustada†) |
-3§ |
|
|
Intervalo de confianza del 95% |
(-6, -1) |
|
|
* Población por intención de tratar usando la última observación realizada durante el estudio o la última observación previa al tratamiento de rescate con insulina en los pacientes que lo necesitaron. Dosis media diaria total de insulina: Población por intención de tratar usando la última observación realizada durante el estudio. † Media de mínimos cuadrados ajustada en función del valor inicial y del uso inicial de metformina. ‡ Valor de p < 0.0001 en comparación con el placebo + insulina § No se evaluó el nivel de significación ¶ Valor de p < 0.05 en comparación con el placebo + insulina # Diferencia no estadísticamente significativa |
||
Adición de ONGLYZA® al tratamiento con la combinación de metformina y una sulfonilurea: Un total de 257 pacientes con diabetes tipo 2 participaron en este estudio de 24 semanas, aleatorizado, doble ciego, controlado con placebo, que evaluó la eficacia y la seguridad de ONGLYZA® combinado con metformina y una sulfonilurea en pacientes que no habían conseguido un control glucémico adecuado (A1C = 7% y = 10%). Los pacientes debían haber estado recibiendo durante un periodo = 8 semanas previo a su admisión en el estudio, dosis estables de la combinación de metformina de liberación prolongada o de liberación inmediata (dosis máxima tolerada, con una dosis mínima de 1500 mg en el momento de la admisión) y una sulfonilurea (dosis máxima tolerada, con una dosis mínima en el momento de la admisión = 50% de la dosis máxima recomendada).
Los pacientes que cumplieron los criterios de elegibilidad fueron admitidos en un periodo de preinclusión de 2 semanas durante el cual se evaluaron los criterios de inclusión y de exclusión. Al cabo de este periodo de 2 semanas, los pacientes elegibles fueron distribuidos al azar para recibir ONGLYZA® (5 mg una vez al día) o un placebo equiparable durante un periodo de 24 semanas según un diseño doble ciego. Durante dicho periodo, los pacientes debían recibir las mismas dosis contantes de metformina y sulfonilurea definidas durante el periodo de preinclusión. La dosis de sulfonilurea podía disminuirse una vez en caso de producirse un episodio grave de hipoglucemia o episodios hipoglucémicos sin gravedad, pero recurrentes. En ausencia de hipoglucemia se prohibió cualquier ajuste (aumento o reducción) de las dosis de los medicamentos en investigación durante el periodo de tratamiento.
La adición de ONGLYZA® a la combinación de metformina y una sulfonilurea produjo mejorías significativas de A1C y GA frente a un placebo más la combinación de metformina y una sulfonilurea (Tabla 12).
|
Tabla 12. Parámetros glucémicos en la semana 24 de un estudio controlado con placebo sobre la adición de ONGLYZA® al tratamiento con la combinación de metformina y una sulfonilurea* |
||
|
Parámetro de eficacia |
ONGLYZA® 5 mg + metformina y sulfonilurea |
Placebo + metformina y sulfonilurea |
|
Hemoglobina A1C (%) |
N=127 |
N=127 |
|
Valor inicial (media) |
8.4 |
8.2 |
|
Variación respecto del valor inicial (media ajustada†) |
-0.7 |
-0.1 |
|
Diferencia con respecto al placebo (media ajustada†) |
-0.7‡ |
|
|
Intervalo de confianza del 95% |
(-0.9, -0.5) |
|
|
Porcentaje de pacientes que alcanzaron una A1C <7% |
31%§ (39/127) |
9% (12/127) |
|
Glucemia en ayunas (mg/dl) |
N=121 |
N=123 |
|
Valor inicial (media) |
162 |
155 |
|
Variación respecto del valor inicial (media ajustada†) |
-5 |
3 |
|
Diferencia con respecto al placebo (media ajustada†) |
-8¶ |
|
|
Intervalo de confianza del 95% |
(-17, 1) |
|
|
Glucemia posprandial de 2 horas (mg/dl) |
N=115 |
N=113 |
|
Valor inicial (media) |
268 |
262 |
|
Variación respecto del valor inicial (media ajustada†) |
-12 |
5 |
|
Diferencia con respecto al placebo (media ajustada†) |
-17# |
|
|
Intervalo de confianza del 95% |
(-32, -2) |
|
|
* Población por intención de tratar usando la última observación previa a la retirada del estudio. † Media de mínimos cuadrados ajustada en función del valor inicial. ‡ Valor de p < 0.0001 en comparación con la combinación de placebo + metformina y sulfonilurea § No se probó la significación estadística ¶ Diferencia no estadísticamente significativa # Valor de p < 0.05 en comparación con la combinación de placebo + metformina y sulfonilurea |
||
Insuficiencia renal: Un total de 170 pacientes participaron en un ensayo aleatorizado, a doble ciego, con control de placebo, de 12 semanas de duración, realizado para evaluar la eficacia y la seguridad de ONGLYZA® 2,5 mg una vez por día en comparación con el placebo en pacientes con diabetes tipo 2 e insuficiencia renal moderada (n=90) o grave (n=41), o ESRD (n=39). En este ensayo, el 98% de los pacientes estaban tomando medicaciones antidiabéticas de base (el 75% estaba usando insulina y el 31% estaba usando medicaciones antidiabéticas orales, principalmente sulfonilureas).
Luego de 12 semanas de tratamiento, ONGLYZA® 2,5 mg proporcionó una significativa mejora en A1C en comparación con el placebo (Tabla 13). En el subgrupo de pacientes con ESRD, ONGLYZA® y el placebo dieron como resultado reducciones comparables de A1C respecto del nivel basal hasta la Semana 12. Este hallazgo no es concluyente, ya que el ensayo no tenía la potencia adecuada para demostrar la eficacia dentro de subgrupos específicos de insuficiencia renal.
Luego de 12 semanas de tratamiento, el cambio medio en la glucemia en ayunas fue -12 mg/dl con ONGLYZA® 2,5 mg y -13 mg/dl con el placebo. En comparación con el placebo, el cambio medio en la glucemia en ayunas con ONGLYZA® fue -12 mg/dl en el subgrupo de pacientes con insuficiencia renal moderada, -4 mg/dl en el subgrupo de pacientes con insuficiencia renal grave, y +44 mg/dl en el subgrupo de pacientes con ESRD. Estos hallazgos no son concluyentes, ya que el ensayo no tenía la potencia adecuada para demostrar la eficacia dentro de subgrupos específicos de insuficiencia renal.
|
Tabla 13. A1C en la Semana 52 en un estudio controlado con placebo de ONGLYZA® en pacientes con insuficiencia renal* |
||
|
Parámetro de eficacia |
ONGLYZA® 2,5 mg |
Placebo |
|
Hemoglobina A1C (%) |
N = 81 |
N = 83 |
|
Valor basal (media) |
8,4 |
8,1 |
|
Cambio respecto del valor basal (media ajustada†) |
-0,9 |
-0,4 |
|
Diferencia respecto del placebo (media ajustada†) |
-0,4‡ |
|
|
Intervalo de confianza del 95% |
(-0,7; -0,1) |
|
|
* Población por intención de tratar usando la última observación durante el estudio. † Media de mínimos cuadrados ajustada en función del valor basal. ‡ Valor p <0,01 en comparación con el placebo. |
||
Seguridad cardiovascular: En el Estudio de Evaluación de Saxagliptina sobre Resultados Vasculares Registrados en Pacientes con Diabetes mellitus-Trombolisis en Infarto de Miocardio (SAVOR), se evaluó el efecto de ONGLYZA® sobre la ocurrencia de eventos mayores de enfermedad cardiovascular (ECV) en 16.492 pacientes adultos con diabetes tipo 2, quienes tenían ECV establecida o múltiples factores de riesgo para enfermedad vascular, incluyendo pacientes con daño renal moderado o severo. Se enrolaron pacientes con edad =40 años, diagnosticados con diabetes tipo 2, que tenían A1C =6.5%, y con ECV establecida o múltiples factores de riesgo CV.
Los pacientes se asignaron aleatoriamente a placebo (n=8212) o saxagliptina (5 mg o 2.5 mg para pacientes con insuficiencia renal moderada o severa) una vez al día (n=8280). La aleatorización a los grupos saxagliptina y placebo se estratificó según el riesgo CV, con 3533 pacientes (21.4%) que tenían solo factores de riesgo CV y 12.959 pacientes (78.6%) con ECV establecida, y según el daño renal, incluyendo 13.916 sujetos (84.4%) con función renal normal a daño leve, 2240 sujetos (13.6%) con daño moderado, y 336 sujetos (2.0%) con daño renal severo. Los pacientes con ECV establecida se definieron por una historia de enfermedad cardiaca isquémica, enfermedad vascular periférica, o accidente cerebrovascular isquémico. Los pacientes con factores de riesgo CV tenían solo la edad como factor de riesgo CV (hombres =55 años y mujeres =60 años) más por lo menos un factor adicional de dislipidemia, hipertensión, o hábito de fumar actual.
Se equilibraron las características demográficas y basales de los sujetos entre los grupos saxagliptina y placebo. La población del estudio fue de 67% hombres y 33% mujeres, con una edad promedio en la aleatorización de 65 años. De los 16.492 pacientes aleatorizados, 8561 (52%) tenían 65 años de edad y más y 2330 (14%) tenían 75 años y más.
Todos los sujetos del estudio tenían diabetes mellitus tipo 2 de una duración promedio de 12 años (mediana = 10.3) y un nivel promedio de A1C de 8.0% (mediana = 7.6%). En general, el 25% de los sujetos tenía niveles basales de A1C <7%. Los pacientes fueron seguidos durante un tiempo promedio de 2 años (mediana = 2.0).
El uso de medicación concomitante fue similar para los dos grupos de tratamiento. En general, el uso de medicamentos para diabetes fue consistente con la práctica de tratamiento local y el programa clínico de saxagliptina (metformina 69%, insulina 41%, sulfonilúreas 40%, y TZDs 6%). El uso de medicamentos para ECV también fue coherente con las guías locales actuales (inhibidores de ECA o BRAs 79%, estatinas 78%, aspirina 75%, beta-bloqueadores 62%, y medicamentos antiplaquetarios diferentes a aspirina 24%). Aproximadamente el 6% de los sujetos fue tratado únicamente con dieta y ejercicio en el nivel basal. Los medicamentos concomitantes se manejaron a todo lo largo del estudio de acuerdo con las metas de la guía local para control glicémico y reducción del riesgo CV con el fin de minimizar las diferencias entre los dos grupos de tratamiento, particularmente en cuanto al control glicémico.
El punto final primario de seguridad y eficacia fue un punto final compuesto, constituido por el tiempo hasta primera ocurrencia de cualquiera de los siguientes eventos adversos CV mayores (MACE, por su sigla en inglés): Muerte CV, IM no fatal, o accidente cerebrovascular isquémico no fatal.
El objetivo primario de seguridad de este estudio fue establecer que el límite superior del IC 95% bilateral para el índice de riesgo estimado comparando la incidencia del punto final compuesto de muerte CV, IM no fatal, o accidente cerebrovascular isquémico no fatal observado con saxagliptina, con el observado en el grupo placebo, era <1.3.
El objetivo primario de eficacia fue determinar, como una evaluación de superioridad, si el tratamiento con saxagliptina, comparado con placebo al adicionarse al tratamiento actual, producía una reducción significativa en el punto final primario de MACE.
El primer punto final secundario de eficacia fue un punto final compuesto, constituido por el tiempo hasta primera ocurrencia de MACE más hospitalización por falla cardiaca, hospitalización por angina pectoris inestable, u hospitalización por revascularización coronaria (MACE plus). El siguiente punto final secundario de eficacia fue determinar si el tratamiento con saxagliptina, comparado con placebo, al ser adicionado al tratamiento de fondo actual en sujetos con diabetes mellitus tipo 2, produciría una reducción en la mortalidad por todas las causas.
En el estudio SAVOR se evaluó la seguridad cardiovascular de saxagliptina y se estableció que la saxagliptina no incrementaba el riesgo cardiovascular (muerte CV, IM no fatal, o accidente cerebrovascular isquémico no fatal) en pacientes con diabetes mellitus tipo 2 comparada con placebo, al adicionarse al tratamiento actual (HR: 1.00; IC 95%: 0.89, 1.12; P<0.001 para no inferioridad).
El punto final primario de eficacia no demostró una diferencia estadísticamente significativa en los eventos adversos coronarios mayores para saxagliptina en pacientes con diabetes mellitus tipo 2 comparada con placebo al adicionarse al tratamiento actual.
|
Tabla 14. Punto final clínico primario y secundario por grupo de tratamiento en el Estudio SAVOR* |
|||||
|
Punto final |
ONGLYZA® |
Placebo |
Índice de riesgo |
||
|
Sujetos con eventos |
Índice de eventos por 100 pacientes-años |
Sujetos con eventos |
Índice de eventos por 100 pacientes-años |
||
|
Punto final primario compuesto: MACE |
613 |
3.76 |
609 |
3.77 |
1.00 |
|
Punto final secundario compuesto: MACE plus |
1059 |
6.72 |
1034 |
6.60 |
1.02 |
|
Mortalidad por todas las causas |
420 |
2.50 |
378 |
2.26 |
1.11 |
|
* Población por intención de tratar † Índice de riesgo ajustado por categoría de función renal basal y categoría de riesgo de ECV basal. ‡ Valor P <0.001 para no inferioridad (basado en un HR <1.3) comparado con placebo. § Valor P= 0.99 para superioridad (basado en HR <1.0) comparado con placebo. ¶ No se examinó la significancia. |
|||||
FIGURA 2. Porcentaje acumulativo de tiempo hasta primer evento CV para el Punto Final Primario Compuesto*
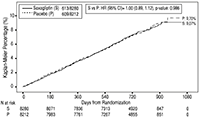
* Población por intención de tratar
Un componente del punto final secundario compuesto, la hospitalización por falla cardiaca, ocurrió a una tasa más alta en el grupo saxagliptina (3.5%) comparado con el grupo placebo (2.8%), con una significancia estadística nominal (i.e., sin ajuste por examen de múltiples puntos finales) que favorece al placebo (HR=1.27; [95% CI: 1.07, 1.51]; P=0.007). No se pudieron identificar en forma definitiva los factores clínicamente relevantes, predictivos de riesgo relativo incrementado con el tratamiento con saxagliptina. Se pudieron identificar los sujetos en riesgo más alto de hospitalización por falla cardiaca, independientemente de la asignación a tratamiento, mediante factores de riesgo conocidos para falla cardiaca, tales como historia inicial de falla cardiaca o alteración de la función renal. Sin embargo, los sujetos en tratamiento con saxagliptina, con una historia de falla cardiaca o función renal alterada en el nivel basal, no estuvieron en riesgo incrementado en relación con el placebo para el punto final primario o secundario compuesto o para mortalidad por todas las causas.
No se observó un riesgo incrementado para el punto final primario entre saxagliptina y placebo en ninguno de los siguientes subgrupos: ECV, múltiples factores de riesgo para ECV, daño renal leve, moderado o severo, edad, sexo, raza, región, duración de la diabetes tipo 2, historia de falla cardiaca, A1C basal, índice albúmina/creatinina, medicamento antidiabético basal, o uso inicial de estatinas, aspirina, inhibidores de ECA, BRAs, beta-bloqueadores, o medicamentos antiplaquetarios.
A pesar del manejo activo de tratamiento antidiabético concomitante en ambos brazos del estudio, los niveles promedio de A1C fueron más bajos en el grupo saxagliptina comparado con el grupo placebo en el año 1 (7.6% versus 7.9%, diferencia de -0.35% [IC 95%: -0.38, -0.31]) y en el año 2 (7.6% versus 7.9%, diferencia de -0.30% [95% CI: -0.34, -0.26]). Las proporciones de sujetos con A1C <7% en el grupo saxagliptina comparado con el grupo placebo fueron de 38% versus 27% en el año 1 y de 38% versus 29% en el año 2.
Comparada con placebo, la saxagliptina dio como resultado una menor necesidad de iniciar medicamentos nuevos o de incrementar los antidiabéticos orales actuales o la insulina. Se observaron mejorías en la A1C y en la proporción de sujetos que alcanzó las metas de la A1C entre los sujetos tratados con saxagliptina, a pesar de los índices más bajos de ajustes ascendentes en los medicamentos para diabetes o del inicio de nuevos medicamentos antidiabéticos o insulina comparados con placebo.
FARMACOCINÉTICA: La farmacocinética de la saxagliptina y de su metabolito activo, 5-hidroxisaxagliptina, fue similar en los sujetos sanos y en los pacientes con diabetes mellitus tipo 2. La Cmáx y el ABC de la saxagliptina y de su metabolito activo aumentaron proporcionalmente dentro del rango de dosis de 2,5 a 400 mg. Después de una sola dosis oral de 5 mg de saxagliptina administrada a sujetos sanos, los valores medios del ABC en plasma de saxagliptina y su metabolito activo fueron 78 ng•h/mL y 214 ng•h/mL, respectivamente. Los valores de Cmáx en plasma correspondientes fueron 24 ng/mL y 47 ng/mL, respectivamente. La variabilidad media (%CV) del ABC y la Cmáx de saxagliptina y su metabolito activo fue inferior al 25%.
No se observó una acumulación apreciable de saxagliptina o de su metabolito activo con la administración repetida de una dosis diaria, independientemente del nivel de dosis. No se observaron dependencia de la dosis ni del tiempo en el clearance de la saxagliptina y de su metabolito activo a lo largo de 14 días de administración de una dosis diaria de saxagliptina, dentro del rango de dosis de 2,5 a 400 mg.
Absorción: La mediana de tiempo hasta la concentración máxima (tmáx) tras la administración de una dosis diaria de 5 mg fue de 2 horas para la saxagliptina y 4 horas para su metabolito activo La administración con una comida rica en grasas provocó un incremento del tmáx de saxagliptina de aproximadamente 20 minutos en comparación con la administración en ayunas. Hubo un incremento del 27% del ABC de saxagliptina cuando se la administró con la comida respecto de la administración en ayunas. ONGLYZA® puede administrarse con o sin alimentos.
Distribución: La fijación a las proteínas in vitro de saxagliptina y su metabolito activo en el suero humano es insignificante. De modo que no son de esperar cambios en los niveles de proteína en la sangre en el caso de diversas enfermedades (p. ej., insuficiencia renal o hepática) que alteren la disposición de la saxagliptina.
Metabolismo: El metabolismo de saxagliptina está mediado principalmente por el citocromo P450 3A4/5 (CYP3A4/5). El principal metabolito de la saxagliptina es también un inhibidor de la DPP4 con la mitad de potencia de la saxagliptina. Por lo tanto, los inhibidores e inductores potentes del CYP3A4/5 alteran la farmacocinética de la saxagliptina y de su metabolito activo [ver Interacciones medicamentosas].
Excreción: La saxagliptina se elimina tanto por vía renal como por vía hepática. Tras una única dosis de 50 mg de 14C-saxagliptina, el 24%, 36% y el 75% de la dosis se excretó en la orina como saxagliptina, su metabolito activo, y como radiactividad total, respectivamente. La media de clearance renal de saxagliptina (~230 mL/min) fue mayor que la tasa de filtración glomerular promedio estimada (~120 mL/min), lo que sugiere cierta excreción renal activa. Un total del 22% de la radiactividad administrada se recuperó en las heces que representa la fracción de la dosis de saxagliptina excretada en la bilis y/o del fármaco no absorbido del tracto gastrointestinal. Tras la administración de una única dosis oral de ONGLYZA® 5 mg a sujetos sanos, las vidas medias terminales (t1/2) plasmáticas de saxagliptina y de su metabolito activo fueron de 2,5 y 3,1 horas, respectivamente.
Poblaciones especiales
Insuficiencia renal: Se realizó un estudio sin enmascaramiento, de dosis única, para evaluar la farmacocinética de saxagliptina (dosis de 10 mg) en sujetos con diversos grados de insuficiencia renal crónica (N=8 por grupo), comparados con sujetos con función renal normal. El estudio incluyó pacientes con insuficiencia renal clasificados sobre la base de clearance de creatinina como leve (>50 a =80 mL/min), moderada (30 a =50 mL/min), y grave (<30 mL/min), así como pacientes con enfermedad renal en estadio terminal (ESRD) en hemodiálisis. Se calculó el clearance de creatinina a partir de la creatinina sérica sobre la base de la fórmula de Cockcroft-Gault:
|
CrCl = [140 -edad (años)] x peso (kg) {x 0,85 para pacientes mujeres}
[72 x creatinina sérica (mg/dL)] |
El grado de insuficiencia renal no afectó la Cmáx de saxagliptina o de su metabolito activo. En sujetos con insuficiencia renal leve, los valores ABC de saxagliptina y su metabolito activo fueron 20% y 70% mayores, respectivamente, que los valores del ABC de los sujetos con función renal normal. Debido a que los aumentos de esta magnitud no son considerados clínicamente relevantes, no es recomendable un ajuste de la dosis en pacientes con insuficiencia renal leve. En sujetos con insuficiencia renal moderada o grave, los valores del ABC de saxagliptina y su metabolito activo fueron hasta 2,1 y 4,5 veces mayores, respectivamente, que los valores del ABC en sujetos con función renal normal. Para lograr exposiciones en plasma de saxagliptina y su metabolito activo similares a las de pacientes con función renal normal, la dosis recomendada es de 2,5 mg una vez al día en pacientes con insuficiencia renal moderada y grave, así como en pacientes con enfermedad renal en estadio terminal que requieren hemodiálisis. La saxagliptina se elimina por hemodiálisis.
Insuficiencia hepática: En sujetos con insuficiencia hepática (clases Child-Pugh A, B y C), los valores medios del ABC y la Cmáx de saxagliptina fueron hasta 8% y 77% mayores, respectivamente, en comparación con controles sanos con las mismas características, tras la administración de una sola dosis de 10 mg de saxagliptina. Los valores correspondientes de la Cmáx y el ABC del metabolito activo fueron hasta 59% y 33% inferiores, respectivamente, en comparación con controles sanos de las mismas características. Estas diferencias no se consideran clínicamente significativas. No se recomienda ajustar la dosis en los pacientes con insuficiencia hepática.
Índice de masa corporal: No se recomienda ajustar la dosis sobre la base del índice de masa corporal (IMC), que no se identificó como una covariable significativa en el clearance aparente de saxagliptina o de su metabolito activo en un análisis farmacocinético poblacional.
Género: No se recomienda ajustar la dosis sobre la base del género. No se observaron diferencias en cuanto a la farmacocinética de la saxagliptina entre hombres y mujeres. En comparación con los hombres, las mujeres tuvieron valores de exposición aproximadamente 25% más altos para el metabolito activo que los hombres, pero es poco probable que esta diferencia sea de relevancia clínica. El género no se identificó como una covariable significativa en el clearance aparente de saxagliptina y su metabolito activo en un análisis farmacocinético poblacional.
Pacientes geriátricos: No se recomienda ajustar la dosis en base a la edad solamente. Los sujetos de edad avanzada (65-80 años) tuvieron una media geométrica de 23% y 59% más elevada en los valores de la Cmáx y el ABC, respectivamente, para la saxagliptina que los sujetos jóvenes (18-40 años). Las diferencias en la farmacocinética del metabolito activo entre los sujetos de edad avanzada y los jóvenes, en general, reflejaron las diferencias observadas en la farmacocinética de saxagliptina. La diferencia entre la farmacocinética de la saxagliptina y el metabolito activo en sujetos jóvenes y de edad avanzada es probable que se deba a múltiples factores, entre ellos la disminución de la función renal y de la capacidad metabólica con el aumento de la edad. La edad no se identificó como una covariable significativa en el clearance aparente de saxagliptina y su metabolito activo en un análisis farmacocinético poblacional.
Pacientes pediátricos: No se han realizado estudios que caractericen la farmacocinética de saxagliptina en pacientes pediátricos.
Raza y etnia: No se recomienda ajustar la dosis en base a la raza. En un análisis farmacocinético poblacional se comparó la farmacocinética de saxagliptina y su metabolito activo en 309 sujetos blancos con 105 sujetos no blancos (que comprendían seis grupos raciales). No se detectaron diferencias significativas en la farmacocinética de saxagliptina y su metabolito activo entre estas dos poblaciones.
Interacciones medicamentosas
Evaluación in vitro de las interacciones medicamentosas: El metabolismo de la saxagliptina es mediado principalmente por CYP3A4/5.
En los estudios in vitro, la saxagliptina y su metabolito activo no inhibieron las enzimas CYP1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1 o 3A4, ni indujeron las enzimas CYP1A2, 2B6, 2C9 o 3A4. Por lo tanto, no es de esperar que la saxagliptina altere el clearance metabólico de los medicamentos coadministrados que son metabolizados por estas enzimas. La saxagliptina es un sustrato de la glucoproteína P (P-gp), pero no es un inhibidor o un inductor importante de la Pgp.
Evaluación in vivo de las interacciones medicamentosas
|
Tabla 3. Efecto de los fármacos coadministrados sobre la exposición sistémica de saxagliptina y su metabolito activo, 5-hidroxi-saxagliptina |
|||||
|
Fármaco coadministrado |
Dosis del fármaco coadministrado* |
Dosis de saxagliptina* |
Relación de medias geométricas |
||
|
ABC† |
Cmáx |
||||
|
No se requiere ajuste de dosis para los siguientes: |
|||||
|
Metformina |
1000 mg |
100 mg |
saxagliptina 5-hidroxi-saxagliptina |
0,98 0,99 |
0,79 0,88 |
|
Gliburida |
5 mg |
10 mg |
saxagliptina 5-hidroxi-saxagliptina |
0,98 ND |
1,08 ND |
|
Pioglitazona‡ |
45 mg una vez al día durante 10 días |
10 mg una vez al día durante 5 días |
saxagliptina 5-hidroxi-saxagliptina |
1,11 ND |
1,11 ND |
|
Digoxina |
0,25 mg cada 6 h el primer día, luego cada 12 h el segundo día, y después cada 24 h durante 5 días |
10 mg una vez al día durante 7 días |
saxagliptina 5-hidroxi-saxagliptina |
1,05 1,06 |
0,99 1,02 |
|
Simvastatina |
40 mg una vez al día durante 8 días |
10 mg una vez al día durante 4 días |
saxagliptina 5-hidroxi-saxagliptina |
1,12 1,02 |
1,21 1,08 |
|
Diltiazem |
360 mg LA una vez al día durante 9 días |
10 mg |
saxagliptina 5-hidroxi-saxagliptina |
2,09 0,66 |
1,63 0,57 |
|
Rifampicina§ |
600 mg una vez al día durante 6 días |
5 mg |
saxagliptina 5-hidroxi-saxagliptina |
0,24 1,03 |
0,47 1,39 |
|
Omeprazol |
40 mg una vez al día durante 5 días |
10 mg |
saxagliptina 5-hidroxi-saxagliptina |
1,13 ND |
0,98 ND |
|
Hidróxido de aluminio + hidróxido de magnesio + simeticona |
Hidróxido de aluminio: 2400 mg Hidróxido de magnesio: 2400 mg Simeticona: 240 mg |
10 mg |
saxagliptina 5-hidroxi-saxagliptina |
0,97 ND |
0,74 ND |
|
Famotidina |
40 mg |
10 mg |
saxagliptina 5-hidroxi-saxagliptina |
1,03 ND |
1,14 ND |
|
Limitar la dosis de ONGLYZA® a 2,5 mg una vez al día cuando se coadministra con inhibidores potentes de CYP3A4/5 |
|||||
|
Ketoconazol |
200 mg dos veces al día durante 9 días |
100 mg |
saxagliptina 5-hidroxi-saxagliptina |
2,45 0,12 |
1,62 0,05 |
|
Ketoconazol |
200 mg dos veces al día durante 7 días |
20 mg |
saxagliptina 5-hidroxi-saxagliptina |
3,67 ND |
2,44 ND |
|
* Dosis única, a menos que se indique lo contrario. † ABC = ABC(0-8) para los fármacos administrados en dosis única y ABC = ABC(0-t) para los fármacos administrados en dosis múltiples. ‡ Los resultados excluyen a un paciente. § La inhibición de la actividad de dipeptidil peptidasa-4 (DPP4) en plasma durante un intervalo de administración de 24 horas no resultó afectada por la rifampicina. ND = no determinado; LA = acción prolongada |
|||||
|
Tabla 4. Efecto de la saxagliptina sobre la exposición sistémica a los fármacos coadministrados |
|||||
|
Fármaco coadministrado |
Dosis del fármaco coadministrado* |
Dosis de saxagliptina* |
Relación de medias geométricas |
||
|
ABC† |
Cmáx |
||||
|
No se requiere ajuste de dosis para los siguientes: |
|||||
|
Metformina |
1000 mg |
100 mg |
metformina |
1,20 |
1,09 |
|
Gliburida |
5 mg |
10 mg |
gliburida |
1,06 |
1,16 |
|
Pioglitazona‡ |
45 mg una vez al día durante 10 días |
10 mg una vez al día durante 5 días |
pioglitazona hidroxi-pioglitazona |
1,08 ND |
1,14 ND |
|
Digoxina |
0,25 mg cada 6 h el primer día, luego cada 12 h el segundo día, y después cada 24 h durante 5 días |
10 mg una vez al día durante 7 días |
digoxina |
1,06 |
1,09 |
|
Simvastatina |
40 mg una vez al día durante 8 días |
10 mg una vez al día durante 4 días |
simvastatina ácido de simvastatina |
1,04 1,16 |
0,88 1,00 |
|
Diltiazem |
360 mg LA una vez al día durante 9 días |
10 mg |
diltiazem |
1,10 |
1,16 |
|
Ketoconazol |
200 mg dos veces por día durante 9 días |
100 mg |
ketoconazol |
0,87 |
0,84 |
|
Etinilestradiol y norgestimato |
Etinilestradiol 0,035 mg y norgestimato 0,250 mg durante 21 días |
5 mg una vez al día durante 21 días |
etinilestradiol norelgestromina norgestrel |
1,07 1,10 1,13 |
0,98 1,09 1,17 |
|
* Dosis única, a menos que se indique lo contrario. † ABC = ABC(0-8) para los fármacos administrados en dosis única y ABC = ABC(0-t) para los fármacos administrados en dosis múltiples. ‡ Los resultados incluyen a todos los pacientes. ND = no determinado; LA = acción prolongada |
|||||
FARMACODINAMIA: En pacientes con diabetes mellitus tipo 2, la administración de ONGLYZA® inhibe la actividad de la enzima DPP4 por un lapso de 24 horas. Tras una carga de glucosa por vía oral o de una comida, esta inhibición de la DPP4 dio lugar a un incremento de entre 2 y 3 veces de los niveles circulantes de GLP-1 y GIP activos, a una disminución de las concentraciones de glucagón y a un aumento de la secreción de insulina dependiente de la glucosa por parte de las células beta pancreáticas. La elevación de los niveles de insulina y la reducción de los niveles de glucagón se asociaron a la disminución de las concentraciones de glucosa en ayunas y a una menor fluctuación de los niveles de glucosa tras una carga de glucosa por vía oral o una comida.
Electrofisiología cardiaca: En un estudio aleatorizado, doble ciego, con comparador activo, controlado con placebo, de diseño cruzado de 4 ramas, en el cual se administró moxifloxacina a 40 sujetos sanos, la administración de ONGLYZA® no se asoció a una prolongación significativa desde el punto de vista clínico del intervalo QTc o la frecuencia cardiaca con dosis diarias de hasta 40 mg (8 veces la MDHR).
CONTRAINDICACIONES: Antecedentes de reacciones graves de hipersensibilidad a ONGLYZA® (p.ej. anafilaxia, edema angioneurótico, afecciones cutáneas exfoliativas) [ver Advertencias y precauciones y Reacciones adversas].
LIMITACIONES DE USO: ONGLYZA® no debe usarse para el tratamiento de la diabetes mellitus tipo 1 ni de la cetoacidosis diabética, ya que no sería eficaz en esos casos.
Hay limitada experiencia con ONGLYZA® en pacientes con antecedentes de pancreatitis. No se sabe si en tales pacientes es mayor el riesgo de presentar pancreatitis durante el tratamiento con ONGLYZA® [ver Advertencias y precauciones].
REACCIONES ADVERSAS
Experiencia en ensayos clínicos: Dado que los ensayos clínicos se llevan a cabo en condiciones muy variadas, las tasas de reacciones adversas observadas en los ensayos clínicos de un fármaco no pueden compararse directamente con las observadas en los ensayos clínicos de otro fármaco y es posible que no reflejen las observadas en la práctica.
En estudios clínicos aleatorizados, controlados, doble-ciegos, más de 17.000 pacientes con diabetes tipo 2 han sido tratados con ONGLYZA®.
Reacciones adversas asociadas con ONGLYZA® en el Estudio SAVOR: El estudio SAVOR incluyó 8240 pacientes tratados con ONGLYZA® 5 mg o 2.5 mg una vez al día y 8173 pacientes con placebo. La duración media de exposición a ONGLYZA®, independientemente de las interrupciones, fue de 1.8 años. Un total de 3698 sujetos (45%) fueron tratados con ONGLYZA® durante un periodo de 2 a 3 años.
La incidencia total de eventos adversos en pacientes tratados con ONGLYZA® en este estudio fue similar a la observada con placebo (72.5% versus 72.2%, respectivamente). La descontinuación del tratamiento debido a eventos adversos fue similar entre los dos grupos de tratamiento (4.9% en el grupo ONGLYZA® y 5.0% en el grupo placebo). Los eventos adversos serios fueron similares entre los dos grupos de tratamiento (24.2% en el grupo ONGLYZA® y 23.7% en el grupo placebo).
En el estudio SAVOR se evaluó la seguridad cardiovascular de saxagliptina, y se estableció que saxagliptina no incrementó el riesgo cardiovascular (CV) (muerte CV, infarto de miocardio [IM] no fatal, o accidente cerebrovascular isquémico no fatal) en pacientes con diabetes mellitus tipo 2 comparada con placebo al adicionarse al tratamiento actual (Índice de riesgo [HR]: 1.00; intervalo de confianza [IC] 95%: 0.89, 1.12; P<0.001 para no inferioridad) [véase Estudios clínicos].
En el estudio SAVOR, la incidencia de eventos adjudicados a pancreatitis fue de 0.3% tanto en pacientes tratados con ONGLYZA® como en los que recibieron placebo en la población por intención de tratar.
La incidencia de reacciones de hipersensibilidad fue de 1.1% en pacientes tratados con ONGLYZA® y en los que recibieron placebo.
La incidencia de fracturas óseas fue de 2.6% tanto en pacientes tratados con ONGLYZA® como en los que recibieron placebo.
Hipoglucemia: En el estudio SAVOR, la incidencia total de hipoglucemia reportada fue de 17.1% en pacientes tratados con ONGLYZA® y de 14.8% en los que recibieron placebo.
El porcentaje de sujetos con eventos de hipoglucemia mayor (definidos como eventos que requirieron ayuda de otra persona), reportados bajo tratamiento, fue mayor en el grupo saxagliptina que en el grupo placebo (2.1% y 1.6%, respectivamente).
El riesgo incrementado de hipoglucemia total e hipoglucemia mayor observada en el grupo tratado con saxagliptina se produjo principalmente en sujetos tratados con una sulfonilúrea inicialmente y no en sujetos bajo tratamiento con insulina o monoterapia con metformina en el nivel basal.
El riesgo incrementado de hipoglucemia total y mayor se observó principalmente en sujetos con hemoglobina A1c (A1C) <7% en el nivel basal.
Reacciones adversas con monoterapia y con terapia combinada adicionada en estudios de control glucémico: En dos estudios de monoterapia controlados con placebo cuya duración fue de 24 semanas, se trató a pacientes con 2,5 mg de ONGLYZA® por día, 5 mg de ONGLYZA® por día y placebo. También se realizaron tres estudios de 24 semanas de duración sobre la adición de ONGLYZA® o un placebo al tratamiento con metformina, con una tiazolidinediona (pioglitazona o rosiglitazona) o con gliburida. En estos tres estudios, se aleatorizó a los pacientes a recibir tratamiento adicional con ONGLYZA® 2,5 mg por día, ONGLYZA® 5 mg por día o placebo. En uno de los estudios de monoterapia y en el estudio sobre la adición de ONGLYZA® al tratamiento con metformina se incluyó un grupo de tratamiento con saxagliptina 10 mg por día.
En un análisis conjunto predefinido de los resultados a la semana 24 (independientemente del rescate glucémico) de los dos estudios de monoterapia, del estudio de adición a metformina, del estudio de adición a la tiazolidinediona (TZD) y del estudio de adición a gliburida, la incidencia global de eventos adversos en pacientes tratados con ONGLYZA® 2,5 mg y ONGLYZA® 5 mg fue similar a la observada en los que recibieron placebo (72,0% y 72,2% frente a 70,6% respectivamente). La tasa de descontinuación del tratamiento a causa de eventos adversos fue de 2,2%, 3,3% y 1,8% de los pacientes tratados con ONGLYZA® 2,5 mg, ONGLYZA® 5 mg y placebo, respectivamente. Los eventos adversos más comunes (notificados al menos en 2 pacientes tratados con ONGLYZA® 2,5 mg o al menos en 2 pacientes tratados con ONGLYZA® 5 mg) asociados con la descontinuación prematura del tratamiento fueron linfopenia (0,1% y 0,5% frente a 0%, respectivamente), erupción cutánea (0,2% y 0,3% frente a 0,3%), aumento de la creatinina en sangre (0,3% y 0% frente a 0%) y aumento de la creatinina fosfocinasa en sangre (0,1% y 0,2% frente a 0%). La Tabla 1 muestra las reacciones adversas observadas en este análisis conjunto, informadas (independientemente de la evaluación de causalidad por parte del investigador) en =5% de los pacientes tratados con ONGLYZA® 5 mg y más comúnmente que en los pacientes tratados con placebo.
|
Tabla 1. Reacciones adversas (independientemente de la evaluación de causalidad por parte del investigador) en estudios controlados con placebo*, informadas en =5% de los pacientes tratados con ONGLYZA® 5 mg y más comúnmente que en los pacientes tratados con placebo |
||
|
Número (%) de pacientes |
||
|
ONGLYZA® 5 mg |
Placebo |
|
|
Infección de las vías respiratorias superiores |
68 (7,7) |
61 (7,6) |
|
Infección del tracto urinario |
60 (6,8) |
49 (6,1) |
|
Cefalea |
57 (6,5) |
47 (5,9) |
|
* Los cinco estudios controlados con placebo incluyen dos estudios de monoterapia y un estudio de terapia combinada adicional con cada uno de los siguientes fármacos: Metformina, tiazolidinediona o gliburida. La tabla muestra los datos a 24 semanas, independientemente del rescate glucémico. |
||
En los pacientes tratados con ONGLYZA® 2,5 mg, la cefalea (6,5%) fue la única reacción adversa notificada con una tasa >5% y más comúnmente que en los pacientes tratados con placebo.
En este análisis conjunto, las reacciones adversas que se informaron en =2% de los pacientes tratados con ONGLYZA® 2,5 mg u ONGLYZA® 5 mg y con una frecuencia =1% mayor que con el placebo incluyeron las siguientes: Sinusitis (2,9% y 2,6% frente a 1,6% respectivamente), dolor abdominal (2,4% y 1,7% frente a 0,5%), gastroenteritis (1,9% y 2,3% frente a 0,9%) y vómito (2,2% y 2,3% frente a 1,3%).
En el estudio de tratamiento adicional a la TZD, la incidencia de edema periférico fue mayor con ONGLYZA® 5 mg que con el placebo (8,1% y 4,3%, respectivamente). La incidencia de edema periférico con ONGLYZA® 2,5 mg fue de 3,1%. Ninguna de las reacciones adversas de edema periférico notificadas dio origen a la descontinuación del fármaco. Las tasas de edema periférico con ONGLYZA® 2,5 mg y ONGLYZA® 5 mg frente al placebo fueron de 3,6% y 2% frente a 3% cuando se administró como monoterapia, 2,1% y 2,1% frente a 2,2% cuando se administró como terapia adicional a la metformina y 2,4% y 1,2% frente a 2,2% cuando se administró como terapia adicional a la gliburida.
La incidencia de fracturas fue de 1,0 y 0,6 por cada 100 pacientes-años con ONGLYZA® (análisis combinado de 2,5 mg, 5 mg y 10 mg) y placebo respectivamente. La tasa de incidencia de eventos de fractura en los pacientes tratados con ONGLYZA® no aumentó a lo largo del tiempo. No se ha establecido la relación causal y no hay estudios preclínicos que hayan demostrado efectos adversos de saxagliptina sobre el hueso.
En el programa clínico se observó un evento de trombocitopenia, consistente con un diagnóstico de púrpura trombocitopénica idiopática. Se desconoce la relación de este evento con ONGLYZA®.
Uso en pacientes con insuficiencia renal: ONGLYZA® 2,5 mg se comparó con placebo en un ensayo de 12 semanas realizado en 170 pacientes con diabetes tipo 2 e insuficiencia renal moderada o grave, o enfermedad renal en estadio terminal (ESRD). La incidencia de eventos adversos, incluidos eventos adversos serios y discontinuaciones debido a eventos adversos, fue similar entre ONGLYZA® y el placebo.
Reacciones adversas asociadas con ONGLYZA® coadministrado con metformina a pacientes con diabetes tipo 2 no tratados previamente
La Tabla 2 muestra las reacciones adversas informadas (independientemente de la evaluación de causalidad por parte del investigador) en =5% de los pacientes participantes en un estudio adicional de tratamiento inicial con ONGLYZA® en combinación con metformina, con control activo, de 24 semanas de duración en pacientes no tratados previamente.
|
Tabla 2. Tratamiento inicial con una combinación de ONGLYZA® y metformina en pacientes no tratados previamente: Reacciones adversas informadas (independientemente de la evaluación de causalidad por parte del investigador) en =5% de los pacientes que recibieron tratamiento combinado con ONGLYZA® 5 mg más metformina (y más comúnmente que en los pacientes tratados con metformina sola) |
||
|
Número (%) de pacientes |
||
|
ONGLYZA® 5 mg más metformina* |
Metformina* |
|
|
Cefalea |
24 (7,5) |
17 (5,2) |
|
Rinofaringitis |
22 (6,9) |
13 (4,0) |
|
* La metformina comenzó a administrarse en una dosis inicial de 500 mg por día y luego se fue aumentando gradualmente hasta llegar a un máximo de 2000 mg por día. |
||
Hipoglucemia: Las reacciones adversas de hipoglucemia se basaron en todos los casos de hipoglucemia notificados; no se exigió una medición concurrente de la glucosa. En el estudio de tratamiento adicional a la gliburida, la incidencia general de hipoglucemia notificada fue mayor con el tratamiento con ONGLYZA® 2,5 mg y ONGLYZA® 5 mg (13,3% y 14,6%) que con el placebo (10,1%). La incidencia de hipoglucemia confirmada en este estudio, definida como síntomas de hipoglucemia acompañados de un valor de glucosa por punción digital de <50 mg/dL, fue de 2,4% y 0,8% con ONGLYZA® 2,5 mg y ONGLYZA® 5 mg y de 0,7% con el placebo. La incidencia de hipoglucemia notificada con ONGLYZA® 2,5 mg y ONGLYZA® 5 mg en comparación con el placebo administrados como monoterapia fue de 4,0% y 5,6% contra 4,1%, respectivamente; 7,8% y 5,8% contra 5% cuando se administró como tratamiento adicional a la metformina y de 4,1% y 2,7% contra 3,8% cuando se administró como tratamiento adicional de la TZD. La incidencia de hipoglucemia notificada fue de 3,4% en los pacientes no tratados previamente a los cuales se les administró ONGLYZA® 5 mg más metformina y de 4,0% en los pacientes a los cuales solo se les administró metformina.
En el estudio con control activo que comparó la terapia adyuvante con ONGLYZA® 5 mg respecto de glipizida en pacientes que presentaban un control inadecuado con metformina solamente, la incidencia de hipoglucemia reportada fue del 3% (19 eventos en 13 pacientes) con ONGLYZA® 5 mg versus 36,3% (750 eventos en 156 pacientes) con glipizida. No se informó ningún caso de hipoglucemia sintomática confirmada (nivel asociado de glucosa en sangre por punción capilar =50 mg/dl) en ninguno de los pacientes tratados con ONGLYZA®, pero sí en 35 pacientes tratados con glipizida (8,1%) (p<0,0001).
Durante 12 semanas de tratamiento en pacientes con insuficiencia renal moderada o grave, o ESRD, la incidencia global de hipoglucemia reportada fue del 20% entre los pacientes tratados con ONGLYZA® 2,5 mg y del 22% entre los pacientes tratados con placebo. Cuatro pacientes tratados con ONGLYZA® (4,7%) y tres pacientes tratados con placebo (3,5%) informaron al menos un episodio de hipoglucemia sintomática confirmada (nivel asociado de glucosa por punción capilar =50 mg/dl).
En el estudio sobre la adición de ONGLYZA® a la insulina, la incidencia total de hipoglucemia fue del 18.4% con 5 mg de ONGLYZA® y del 19.9% con el placebo.
En el estudio sobre la adición de ONGLYZA® a la combinación de metformina + sulfonilurea, la incidencia total de hipoglucemia fue del 10.1% con 5 mg de ONGLYZA® y del 6.3% con el placebo. La incidencia de hipoglucemia confirmada fue del 1.6% en los pacientes tratados con ONGLYZA®, mientras que no se notificó ningún caso en el grupo placebo [ver Advertencias y precauciones].
Reacciones de hipersensibilidad: Se notificaron eventos relacionados con la hipersensibilidad, tales como urticaria y edema facial, en el análisis combinado de los 5 estudios hasta la semana 24 en 1,5%, 1,5% y 0,4% de los pacientes tratados con ONGLYZA® 2,5 mg, ONGLYZA® 5 mg y placebo, respectivamente. En los pacientes tratados con ONGLYZA®, ninguno de estos eventos requirió hospitalización y los investigadores no los notificaron como eventos con riesgo de vida. Un paciente tratado con saxagliptina en este análisis combinado descontinuó el tratamiento a causa de urticaria generalizada y edema facial.
Infecciones: Hasta la fecha, en la base de datos de ensayos clínicos controlados sin enmascaramiento sobre la saxagliptina, se han informado 6 casos (0,12%) de tuberculosis entre los 4959 pacientes tratados con saxagliptina (1,1 por cada 1000 años-paciente) en comparación con ningún caso de tuberculosis entre los 2868 pacientes tratados con el fármaco comparador. Dos de estos seis casos fueron confirmados con análisis de laboratorio. Los casos restantes tuvieron información limitada o un diagnóstico presunto de tuberculosis. Ninguno de los seis casos se produjo en Estados Unidos ni en Europa occidental. Un caso se registró en Canadá, en un paciente proveniente originalmente de Indonesia, que había visitado recientemente dicho país. La duración del tratamiento con saxagliptina hasta que se informó tuberculosis osciló entre 144 y 929 días. Los recuentos de linfocitos post-tratamiento estuvieron consistentemente dentro del rango de referencia en cuatro casos. Un paciente tuvo linfopenia antes de iniciar terapia con saxagliptina, que permaneció estable durante todo el tratamiento con dicho fármaco. El último paciente tuvo un recuento aislado de linfocitos por debajo del rango normal aproximadamente cuatro meses antes del informe de tuberculosis. No se han producido informes espontáneos de tuberculosis asociada con el uso de saxagliptina. No se ha estimado la causalidad, y hasta la fecha ha habido muy pocos casos para determinar si la tuberculosis está relacionada con el uso de saxagliptina.
Hasta la fecha se ha registrado un caso de potencial infección oportunista en la base de datos de ensayos clínicos controlados sin enmascaramiento en un paciente tratado con saxagliptina, que desarrolló presunta sepsis mortal por Salmonella tras una intoxicación por alimentos luego de aproximadamente 600 días de tratamiento con saxagliptina. No ha habido informes espontáneos de infecciones oportunistas asociadas con el uso de saxagliptina.
En el estudio SAVOR, la incidencia de infecciones oportunistas (definidas como eventos que aparecen en la Lista del CDC 1993 de Diagnósticos que Definen el SIDA y que son específicos para inmunosupresión) fue similar en pacientes tratados con ONGLYZA® (<0.1%) versus pacientes tratados con placebo (0.1%).
Signos vitales: No se observaron cambios clínicamente significativos en los signos vitales de los pacientes tratados con ONGLYZA®.
Análisis de laboratorio
Recuento absoluto de linfocitos: Se observó una reducción media relacionada con la dosis del recuento absoluto de linfocitos con ONGLYZA®. A partir de un recuento absoluto de linfocitos basal de aproximadamente 2200 células/microlitro, en un análisis combinado de cinco estudios clínicos controlados con placebo se observaron a las 24 semanas reducciones medias de aproximadamente 100 y 120 células/microlitro con ONGLYZA® 5 mg y 10 mg, respectivamente, en comparación con el placebo. Se observaron efectos similares cuando se administró la dosis de ONGLYZA® 5 mg en una combinación inicial con metformina en comparación con la metformina en monoterapia. No se observó ninguna diferencia con ONGLYZA® 2,5 mg en relación con el placebo. La proporción de pacientes en los cuales se notificó un recuento de linfocitos <750 células/microlitro fue de 0,5%, 1,5%, 1,4% y 0,4% en los grupos tratados con saxagliptina 2,5 mg, 5 mg, 10 mg y placebo, respectivamente. En la mayoría de los pacientes, no se observó recurrencia con la exposición repetida a ONGLYZA®, aunque algunos pacientes tuvieron una reducción recurrente cuando se les readministró el fármaco, provocando la suspensión del tratamiento con ONGLYZA®. Las reducciones del recuento de linfocitos no estuvieron asociadas con reacciones adversas clínicamente relevantes.
Se desconoce la importancia clínica de esta reducción del recuento de linfocitos respecto del placebo. Cuando estuviera clínicamente indicado, por ejemplo en cuadros de infección inusual o prolongada, se debe determinar el recuento de linfocitos. Se desconoce el efecto de ONGLYZA® sobre el recuento de linfocitos de los pacientes con anormalidades linfocíticas (p.ej., virus de inmunodeficiencia humana).
En el estudio SAVOR, se reportaron recuentos de linfoncitos disminuidos en el 0.5% de pacientes tratados con ONGLYZA® y en el 0.4% de pacientes que recibieron placebo. Experiencia posterior a la comercialización: Desde la aprobación de ONGLYZA® se han identificado otras reacciones adversas. Dado que tales reacciones se han notificado de manera espontánea en una población de tamaño indeterminado, es imposible obtener una estimación fiable de su frecuencia o establecer una relación causal con la exposición al medicamento.
• Reacciones de hipersensibilidad que incluyen anafilaxia, edema angioneurótico y afecciones cutáneas exfoliativas [ver Contraindicaciones y Advertencias y precauciones].
• Pancreatitis aguda [ver Indicaciones y uso y Advertencias y precauciones].
• Artralgia (ver Advertencias y precauciones].
INTERACCIONES MEDICAMENTOSAS
Inhibidores potentes de las enzimas CYP3A4/5: El ketoconazol aumentó significativamente la exposición a la saxagliptina. Son de esperar aumentos significativos similares de las concentraciones plasmáticas de saxagliptina con otros inhibidores potentes del CYP3A4/5 (p.ej., atazanavir, claritromicina, indinavir, itraconazol, nefazodona, nelfinavir, ritonavir, saquinavir y telitromicina). La dosis de ONGLYZA® debe limitarse a 2,5 mg cuando se coadministra con un inhibidor potente del CYP3A4/5 [ver Dosis y administración y Farmacología clínica].
Uso en poblaciones específicas
Embarazo: Categoría B de riesgo durante el embarazo.
No se han realizado estudios adecuados y bien controlados en mujeres embarazadas. Dado que los estudios de reproducción animal no siempre tienen validez predictiva en cuanto a la respuesta en seres humanos, ONGLYZA®, al igual que otros medicamentos antidiabéticos, solo debe usarse durante el embarazo en caso de necesidad absoluta.
La saxagliptina no fue teratogénica en ninguna de las dosis evaluadas administradas en ratas y conejas preñadas durante el periodo de organogénesis. Se observó una osificación incompleta de la pelvis, una forma de retraso del desarrollo, en las ratas tratadas con una dosis de 240 mg/kg, o aproximadamente 1503 y 66 veces la exposición humana a la saxagliptina y al metabolito activo, respectivamente, con la máxima dosis humana recomendada (MDHR) de 5 mg. Se observaron toxicidad materna y peso corporal fetal reducido con una exposición 7986 y 328 veces superior a la exposición que se alcanza con la MDHR a saxagliptina y al metabolito activo, respectivamente. Se observaron variaciones esqueléticas menores en conejos con una dosis tóxica para la progenitora de 200 mg/kg, o aproximadamente 1432 y 992 veces la MDHR.
La coadministración de saxagliptina y metformina a ratas y conejas gestantes durante el periodo de organogénesis no resultó embrioletal ni teratogénica en ninguna de las especies cuando se evaluó en dosis que arrojaban exposiciones sistémicas (ABC) hasta 100 y 10 veces la MRHD (saxagliptina 5 mg y metformina 2000 mg), respectivamente, en ratas; y 249 y 1,1 veces la MRHD en conejas. En las ratas, la leve toxicidad en el desarrollo se limitó a una mayor incidencia de costillas onduladas; la toxicidad materna asociada se limitó a descensos de peso del 11% al 17% durante el transcurso del estudio, y reducciones relacionadas del consumo materno de alimento. En las conejas, la coadministración fue poco tolerada en un subconjunto de hembras (12 de 30), lo cual dio como resultado casos de muerte, estado moribundo o aborto. Sin embargo, entre las hembras sobrevivientes con crías evaluables, la toxicidad materna se limitó a reducciones marginales del peso corporal durante los días de gestación 21 a 29; y la toxicidad en el desarrollo asociada en estas crías se limitó a descensos del peso corporal fetal del 7%, y una baja incidencia de osificación retardada del hioides fetal.
La administración de saxagliptina a ratas hembra entre el día 6 de gestación y el día 20 de lactancia provocó disminuciones en el peso corporal de las crías macho y hembra solo a dosis tóxicas para las progenitoras (exposiciones =1629 y 53 veces superiores a la de la saxagliptina y su metabolito activo con la MDHR). No se observó ninguna toxicidad funcional ni de conducta en las crías de las ratas a las que se administró saxagliptina, cualquiera fuese la dosis.
Se determinó que la saxagliptina atraviesa la placenta y llega al feto tras su administración a ratas preñadas.
Madres en periodo de lactancia: La saxagliptina se excreta en la leche de las ratas lactantes en una relación de 1:1 con las concentraciones plasmáticas del fármaco. Se ignora si la saxagliptina se excreta en la leche humana. Dado que muchos fármacos se excretan en la leche humana, debe tenerse precaución al administrar ONGLYZA® a mujeres en periodo de lactancia.
Uso pediátrico: No se ha determinado la seguridad ni la eficacia de ONGLYZA® en pacientes pediátricos.
Uso geriátrico: De los 16.492 pacientes aleatorizados en el estudio SAVOR, 8561 (51.9%) pacientes tenían 65 años de edad o más y 2330 (14.1%) tenían 75 años o más. El número de sujetos tratados con ONGLYZA® en el estudio SAVOR, que tenían 65 años y más fue de 4290 y el de sujetos con 75 años o más fue de 1169.
Del número total de 4148 pacientes aleatorizados en seis estudios clínicos controlados, doble ciego, sobre la seguridad y eficacia de ONGLYZA®, 634 (15,3%) pacientes tenían 65 años de edad o más, y 59 pacientes (1,4%) tenían 75 años de edad o más.
No se observaron diferencias globales en la seguridad o efectividad entre pacientes con edad =65 años, =75 años y los pacientes más jóvenes.
La saxagliptina y su metabolito activo se eliminan parcialmente por el riñón. Dado que es más probable que los pacientes de edad avanzada tengan una función renal reducida, debe tenerse cuidado al seleccionar la dosis en adultos mayores, teniendo en cuenta su función renal [ver Dosis y administración y Farmacología clínica].
FARMACOLOGÍA CLÍNICA
Mecanismo de acción: En respuesta a las comidas, el intestino delgado libera en el torrente sanguíneo concentraciones más elevadas de hormonas incretinas, tales como el péptido similar al glucagón tipo 1 (GLP-1) y el polipéptido insulinotrópico dependiente de la glucosa (GIP). Estas hormonas dan origen a la liberación de insulina por parte de las células beta pancreáticas de manera dependiente de la glucosa, pero son inactivadas por la enzima dipeptidil peptidasa 4 (DPP4) en cuestión de minutos. El GLP-1 también reduce la secreción de glucagón por parte de las células alfa pancreáticas, lo cual reduce la producción hepática de glucosa. En los pacientes con diabetes tipo 2, las concentraciones de GLP-1 están reducidas, pero la respuesta de la insulina al GLP-1 está conservada. La saxagliptina es un inhibidor competitivo de la DPP4 que retarda la inactivación de las hormonas incretinas y así incrementa sus concentraciones en el torrente sanguíneo y reduce las concentraciones en ayunas y postprandiales de glucosa de manera dependiente de la glucosa en pacientes con diabetes mellitus tipo 2.
ADVERTENCIAS Y PRECAUCIONES
Pancreatitis: Desde la comercialización del producto se han notificado casos de pancreatitis aguda en pacientes tratados con ONGLYZA®. Al empezar el tratamiento con ONGLYZA® se requiere una supervisión cuidadosa de los signos y síntomas de pancreatitis. Si se sospecha de pancreatitis, ONGLYZA® debe suspenderse inmediatamente e iniciar un tratamiento adecuado. No se sabe si en pacientes con antecedentes de pancreatitis es mayor el riesgo de presentar pancreatitis durante el tratamiento con ONGLYZA® [ver Reacciones adversas]. Los pacientes deben ser informados sobre los síntomas característicos de pancreatitis.
En el Estudio de Evaluación de Saxagliptina de Resultados Vasculares Registrados en Pacientes con Diabetes mellitus-Trombolisis en Infarto de Miocardio (SAVOR), la incidencia de eventos adjudicados a pancreatitis fue de 0.3% en pacientes tratados con ONGLYZA® y en los que recibieron placebo en la población por intención de tratar [ver Reacciones adversas].
Uso con medicamentos que son causa conocida de hipoglucemia: Los secretagogos de insulina como las sulfonilureas, causan hipoglucemia. Por lo tanto, es posible que se requiera una dosis menor del secretagogo de insulina, para reducir el riesgo de hipoglucemia cuando se lo usa en combinación con ONGLYZA® [ver Reacciones adversas].
Reacciones de hipersensibilidad: Desde la comercialización del producto se han notificado reacciones graves de hipersensibilidad en pacientes tratados con ONGLYZA®, que han incluido anafilaxia, edema angioneurótico y afecciones cutáneas exfoliativas. Estas reacciones se presentaron en los 3 primeros meses de tratamiento con ONGLYZA®, y algunas después de la primera dosis. Si se sospecha de una reacción de hipersensibilidad grave, suspender ONGLYZA®, investigar otras causas posibles e iniciar un tratamiento antidiabético alternativo [ver Reacciones adversas].
Se requiere precaución en los pacientes con antecedentes de edema angioneurótico con otro inhibidor de la dipeptidilpeptidasa-4 (DPP4) porque no se sabe si presentarán una predisposición al edema angioneurótico con ONGLYZA®.
Artralgia: En reportes post-comercialización se ha informado dolor articular producido por inhibidores de DPP4. Los reportes de casos sugieren que el dolor articular puede ser severo e incapacitante. El tiempo transcurrido hasta la aparición de los síntomas luego del inicio de tratamiento con el medicamento varió desde un día hasta años. Los pacientes experimentaron alivio de los síntomas al descontinuar el medicamento; un subgrupo de pacientes experimentó recurrencia de los síntomas cuando volvió a ser sometido a prueba de provocación con el mismo fármaco u otro inhibidor de DPP4. Los inhibidores de DPP4 se deben considerar como una posible causa de que cualquier paciente presente dolor articular severo, y se debe evaluar en forma individual la continuación del tratamiento farmacológico
Falla cardíaca: En el estudio SAVOR, se observó un incremento en la tasa de hospitalización por falla cardiaca en los pacientes tratados con saxagliptina, comparada con placebo, aunque no se ha establecido una relación causal. Se justifica tener precaución al utilizar ONGLYZA® en pacientes que tengan factores de riesgo conocidos para hospitalización por falla cardiaca, tales como una historia de falla cardiaca o daño renal moderado a severo. Se debe brindar asesoría a los pacientes sobre los síntomas característicos de la falla cardiaca, y recomendarles que reporten inmediatamente dichos síntomas [véase Reacciones adversas y Estudios clínicos].
Resultados macrovasculares: No se han realizado estudios clínicos que hayan presentado evidencias concluyentes de reducción del riesgo macrovascular con ONGLYZA® o algún otro agente antidiabético. En el estudio clínico placebo-controlado, aleatorizado SAVOR, el uso de ONGLYZA® no se asoció a un riesgo incrementado de eventos adversos cardiovasculares importantes [ver Reacciones adversas y Estudios clínicos].
DOSIS Y ADMINISTRACIÓN
Dosis recomendada: La dosis recomendada de ONGLYZA® es de 2,5 o 5 mg una vez al día solo o con las comidas. Los comprimidos de ONGLYZA® no deben ser divididos o partidos.
Pacientes con insuficiencia renal: No se recomienda ningún ajuste de la dosis de ONGLYZA® en pacientes con insuficiencia renal leve (depuración de creatinina [CrCl]) >50 mL/min).
Se recomienda un ajuste de dosis en pacientes con insuficiencia renal moderada o grave, y en pacientes con enfermedad renal en estado terminal que requieran hemodiálisis.
La dosis de ONGLYZA® es de 2,5 mg una vez por día (solo o con las comidas) en pacientes con insuficiencia renal moderada o grave, o con enfermedad renal en estadio terminal (ESRD) que requieran hemodiálisis (depuración de creatinina [CrCl]) =50 mL/min) [ver Farmacología clínica y Estudios clínicos]. ONGLYZA® debe administrarse después de la hemodiálisis. ONGLYZA® no ha sido estudiado en pacientes sometidos a diálisis peritoneal.
Dado que la dosis de ONGLYZA® debe limitarse a 2,5 mg en base a la función renal, se recomienda evaluar la función renal antes de iniciar la administración de ONGLYZA® y posteriormente en forma periódica. La función renal puede estimarse a partir de los niveles de creatinina sérica empleando la fórmula de Cockcroft-Gault o la fórmula de Modificación de la Dieta en la Enfermedad Renal [ver Farmacología clínica].
Inhibidores potentes del sistema CYP3A4/5: Se recomienda un ajuste de dosis en pacientes que estén consumiendo inhibidores potentes del CYP3A4/5
La dosis de ONGLYZA® es de 2,5 mg una vez al día cuando se la coadministra con inhibidores potentes del sistema P450 3A4/5 (CYP3A4/5) (tales como ketoconazol, atazanavir, claritromicina, indinavir, itraconazol, nefazodona, nelfinavir, ritonavir, saquinavir y telitromicina) [ver Interacciones medicamentosas y Farmacología clínica].
FORMAS DE DOSIFICACIÓN Y CONCENTRACIONES
• Los comprimidos de 5 mg de ONGLYZA® (saxagliptina) son comprimidos recubiertos, de color rosa, biconvexos, redondos, con el número “5” impreso en uno de sus lados y el número “4215” impreso en el reverso, en tinta de color azul.
• Los comprimidos de 2,5 mg de ONGLYZA® (saxagliptina) son comprimidos recubiertos, de color entre amarillo pálido y amarillo claro, biconvexos, redondos, con el número “2.5” impreso en uno de sus lados y el número “4214” impreso en el reverso, en tinta de color azul.
SOBREDOSIS: En un estudio clínico controlado la administración oral de ONGLYZA® una vez por día a sujetos sanos en dosis de hasta 400 mg diarios durante 2 semanas (80 veces la MDHR) no causó reacciones clínicas adversas ni efectos clínicos significativos relacionados con la dosis sobre el intervalo QTc o la frecuencia cardiaca.
En caso de sobredosis, debe iniciarse un tratamiento de apoyo apropiado de acuerdo con el estado clínico del paciente. La saxagliptina y su metabolito activo se eliminan por hemodiálisis (23% de la dosis en 4 horas).
INFORMACIÓN DE ASESORAMIENTO PARA EL PACIENTE
Guía de medicación: Los médicos deben indicar a sus pacientes que lean la Guía de medicación antes de comenzar a tomar ONGLYZA® y que la vuelvan a leer cada vez que se les surta una nueva receta, y que informen a su médico si desarrollan algún síntoma inusual o si persiste o empeora algún síntoma existente.
Se debe informar a los pacientes de los riesgos y beneficios potenciales de ONGLYZA® y de modos alternativos de terapia. Los pacientes también deben ser informados acerca de la importancia de cumplir con las instrucciones de dieta, la actividad física regular, el monitoreo periódico de la glucosa y las pruebas de A1C, el reconocimiento y el manejo de la hipoglucemia y la hiperglucemia, y la evaluación de las complicaciones de la diabetes. Durante los periodos de estrés como fiebre, trauma, infección, o cirugía, pueden cambiar los requisitos de medicación y se debe advertir a los pacientes que acudan al médico sin demora.
Pancreatitis: Es necesario informar a los pacientes que desde la comercialización de ONGLYZA® se han notificado casos de pancreatitis aguda. Antes de iniciar un tratamiento con ONGLYZA®, debe interrogarse a los pacientes sobre otros factores de riesgo de pancreatitis como antecedentes de pancreatitis, alcoholismo, cálculos biliares o hipertrigliceridemia. Los pacientes también deben saber que un dolor abdominal intenso y persistente, acompañado o no de vómito y que a veces se difunde a la espalda, constituye el síntoma característico de la pancreatitis aguda. Debe informarse a los pacientes que si presentan un dolor abdominal intenso y persistente deben suspender ONGLYZA® y consultar a su médico [ver Advertencias y precauciones].
Reacciones de hipersensibilidad: Es necesario informar a los pacientes que desde la comercialización de ONGLYZA® se han notificado reacciones alérgicas (hipersensibilidad) graves tales como edema angioneurótico, anafilaxia y afecciones cutáneas exfoliativas. Los pacientes deben suspender el tratamiento con ONGLYZA® y buscar asistencia médica rápidamente si presentan síntomas de tales reacciones alérgicas (p.ej. exantema, descamación o exfoliación de la piel, urticaria, inflamación de la piel, la cara, los labios, la lengua y la garganta que puede dificultar la respiración o la deglución).
Artralgia: Informe a los pacientes que se han recibido reportes post-comercialización de dolor articular, algunas veces severo, con esta clase de medicamentos. Instruya a los pacientes para que busquen asesoría médica si se presenta dolor articular severo [véase Advertencias y precauciones].
Dosis olvidada: Debe informarse a los pacientes que si olvidan una dosis de ONGLYZA®, deben tomar la siguiente de la manera prescrita, a menos que su médico indique otra cosa. Los pacientes también deben saber que no deben tomar una dosis adicional de ONGLYZA® el siguiente día.
Instrucciones de administración: Se debe informar a los pacientes que el comprimido de ONGLYZA® debe tomarse entero sin triturarlo ni masticarlo
Pruebas de laboratorio: Se debe informar a los pacientes que la respuesta a todos los tratamientos para diabéticos debe ser supervisada por mediciones periódicas de la glucosa en la sangre y de los niveles de A1C, con el objetivo de disminuir estos niveles hacia el rango normal. El nivel de A1C es especialmente útil para evaluar el control de la glucemia a largo plazo. Los pacientes deben ser informados de la posible necesidad de ajustar sus dosis basándose en los cambios en las pruebas de función renal realizadas con el tiempo.
Acción terapéutica: Hipoglucemiante oral.
Código ATC: A10BH.
DESCRIPCIÓN: La saxagliptina es un inhibidor oralmente activo de la enzima DPP4.
La descripción química del monohidrato de saxagliptina es (1S,3S,5S)-2-[(2S)-2-amino-2-(3-hidroxitriciclo[3.3.1.13,7]dec-1-il)acetil]-2-azabiciclo[3.1.0]hexano-3-carbonitrilo, monohidrato o (1S,3S,5S)-2-[(2S)-2-amino-2-(3-hidroxiadamantano-1-il)acetil]-2-azabiciclo[3.1.0]hexano-3-carbonitrilo hidrato. Su fórmula empírica es C18H25N3O2•H2O y su peso molecular, 333,43. Su fórmula estructural es:
El monohidrato de saxagliptina es un polvo cristalino no higroscópico de color entre blanco y amarillo claro o marrón claro. Es bastante soluble en agua a 24 °C ± 3 °C, poco soluble en acetato de etilo, y soluble en metanol, etanol, alcohol isopropílico, acetonitrilo, acetona y polietilenglicol 400 (PEG 400) [ver Fórmula cualicuantitativa].
PRESENTACIÓN: Los comprimidos de ONGLYZA® (saxagliptina) tienen marcas en ambos lados y están disponibles en las concentraciones y envases que figuran en la Tabla 15.
|
Tabla 15. Presentación de los comprimidos de ONGLYZA® |
|||
|
Concentración del comprimido |
Comprimidos recubiertos Color/Forma |
Marcas en los |
Tamaño del envase: Número de comprimidos recubiertos por blíster |
|
5 mg |
rosados |
“5” en una cara y “4215” en el reverso, en tinta de color azul |
Caja con 14 Comprimidos Recubiertos Caja con 28 Comprimidos Recubiertos |
|
2,5 mg |
amarillo pálido a amarillo claro biconvexos, redondos |
“2.5” en una cara y “4214” en el reverso, en tinta de color azul |
Caja con 28 Comprimidos Recubiertos |
Almacenamiento y manipulación: Consérvese a temperatura ambiente a no más de 30 °C.
ONGLYZA® 2.5 mg. Caja con 28 comprimidos recubiertos (Reg. San. INVIMA 2016M-0011547-R1).
ONGLYZA® 5 mg. Caja con 14 comprimidos recubiertos. Caja con 28 comprimidos recubiertos (Reg. San. INVIMA 2016M-0011400-R1).
IPP Clave: 1-2015.
Fuente: Doc ID-002951175. Versión 1.0.
Texto basado en USPI/Junio 2015.
Fecha de preparación de la versión: Julio de 2015.
Mayor información Departamento Médico de
AstraZeneca Colombia S.A.S.
Bogotá, D.C., Colombia


