
NEURONOX®. TOXINA BOTULÍNICA TIPO A
TOXINA BOTULÍNICA TIPO A
Vial
Caja , 1 Vial
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN: Cada VIAL contiene Toxina Botulínica Tipo A Purificada del Clostridium botulinum (Cepa Hall) 100 UI.
INDICACIONES:
Tratamiento de la hiperactividad muscular en las siguientes patologías: Oftalmología: Blefaroespasmo esencial benigno o asociado a distonía, estrabismo y distonía focal. Neurología: Coadyuvante o alternativo en parálisis cerebral, tremor esencial que no ha respondido a otros tratamientos orales, espasticidad, distonías, mioclonías que cursen con fenómenos distónicos, espasmo hemifacial, cefalea tensional, tortícolis espasmódica. Urología: Hiperactividad del músculo detrusor de la vejiga. Otorrinolaringología: Temblor palatal esencial, disfonía espasmódica. Dermatología: Hiperhidrosis refractaria a tratamientos convencionales. Traumatología/ortopedia: Coadyuvante en padecimientos espásticos, de cuello y espina dorsal asociado a contracturas patológicas que no han respondido a ninguna otra medida terapéutica. Bruxismo temporo-maxilar. Proctología: Fisura anal. Gastroenterología: Acalasia en casos de que no pueda hacerse dilatación neumática o cirugía. Tratamiento de líneas faciales hiperfuncionales. Alternativo en la profilaxis del dolor de cabeza en migraña crónica.
PROPIEDADES:
Mecanismo de acción: La toxina botulínica tipo A, bloquea la liberación de acetilcolina a nivel de las terminaciones nerviosas colinérgicas periféricas, por escindir SNAP-25, una proteína necesaria para que se produzca adecuadamente la fijación y liberación de acetilcolina de las vesículas situadas en las terminaciones nerviosas. Tras la inyección, la toxina inicialmente se une rápidamente y con elevada afinidad a receptores específicos de la superficie celular. A continuación, la toxina pasa a través de la membrana plasmática mediante endocitosis mediada por receptores, liberándose en el citosol. Este último paso va unido a una inhibición progresiva de la liberación de acetilcolina. Los signos clínicos se manifiestan a los 2-3 días, con un efecto máximo a las 5-6 semanas de la inyección. Normalmente la recuperación tras la inyección intramuscular se produce a las 12 semanas de la inyección a medida que las terminales nerviosas se ramifican y conectan nuevamente con las placas terminales. Después de la inyección intradérmica, donde el objetivo son las glándulas sudoríparas ecrinas, el efecto duró por término medio 7,5 meses tras la primera inyección en los pacientes tratados con 50 Unidades por axila. No obstante, en el 27,5% de los pacientes la duración del efecto fue de 1 año o superior. No se ha estudiado la recuperación de las terminaciones nerviosas simpáticas, que inervan las glándulas sudoríparas, tras la inyección intradérmica con toxina botulínica. Después de la administración de la inyección en el detrusor, la toxina botulínica afecta a las vías eferentes de la actividad de este músculo, inhibiendo la liberación de acetilcolina. Además, la toxina botulínica puede inhibir los neurotransmisores aferentes y las vías sensitivas.
FARMACOCINÉTICA:
Características generales de la sustancia activa: Estudios de distribución realizados en ratas muestran que tras su inyección, el complejo 125I-neurotoxina botulínica A, difunde lentamente por el músculo gemelo tibial, sufre un rápido metabolismo sistémico y se excreta vía urinaria. En el músculo, la cantidad de sustancia marcada se reduce hasta aproximadamente la mitad en aproximadamente 10 horas. En el punto de inyección, la radiactividad se une a grandes moléculas proteicas, mientras que en el plasma se une a moléculas pequeñas, lo que indica un rápido metabolismo sistémico del sustrato. En las 24 horas post inyección, el 60% de la radiactividad se excreta por orina. Probablemente, la toxina se metaboliza mediante proteasas y los componentes moleculares se reciclan a través de los circuitos metabólicos normales. Dada la naturaleza de este producto, no se han llevado a cabo los habituales estudios de absorción, distribución, biotransformación y eliminación. Características en pacientes: Se cree que la distribución sistémica de las dosis terapéuticas de toxina botulínica, es muy pequeña. Estudios clínicos, realizados empleando técnicas electromiográficas de fibra única, muestran una actividad muscular electrofisiológica aumentada en músculos alejados del punto de inyección, sin ir ésta, acompañada de ningún signo o síntoma clínico.
CONTRAINDICACIONES: Pacientes con hipersensibilidad conocida a cualquier componente de la fórmula. Pacientes con disturbios en la unión neuromuscular (ejemplo, miastenia grave, síndrome de Lambert-Eaton o esclerosis lateral amiotrófica. Las enfermedades pueden agravarse debido a la actividad del medicamento como relajante muscular). Cuando el medicamento se utiliza en el tratamiento de distonía cervical en los pacientes con disturbio respiratorio grave. Pacientes gestantes, mujeres con potencial para tener hijos o madres lactantes.
REACCIONES ADVERSAS: En general, las reacciones adversas se producen en la primera semana después de la inyección de NEURONOX®. Aunque generalmente son transitorias, pueden tener una duración de varios meses o más. El dolor relacionado con la aguja, trastornos del sistema nervioso, paresia facial, trastornos oculares, ptosis del párpado, ansiedad, pueden resultar en respuestas vasovagales (incluyendo, por ejemplo, síncope, hipotensión), que pueden requerir tratamiento médico adecuado. Infección, inflamación, sensibilidad, hinchazón, eritema, y/o sangrado/moretones pueden estar asociados con la inyección. Debilidad local del músculo(s) inyectado(s) representa la acción farmacológica esperada de la toxina botulínica. Sin embargo, la debilidad de los músculos cercanos también pueden ocurrir debido a la distribución de NEURONOX®. Se conocen notificaciones espontáneas raras de muerte, a veces asociada a disfagia, neumonía u otra debilidad significativa o anafilaxia, después del tratamiento con toxina botulínica. En un estudio de pacientes con blefarospasmo que recibieron una dosis promedio por ojo de 33 U (inyectado en 3 a 5 lugares) de otras inyecciones de toxina botulínica, las reacciones adversas más frecuentemente relatadas relacionadas al tratamiento fueron: Ptosis (20,8%), queratitis punteada superficial (6,3%) y sequedad ocular (6,3%). Todos esos eventos fueron de leves a moderados excepto en un caso de ptosis que fue considerado como grave. Otros eventos relatados en estudios clínicos anteriores con otras inyecciones de toxina botulínica, en orden decreciente de incidencia incluyen: irritación, lagrimeo, lagoftalmos, fotofobia, ectropión, queratitis, diplopía y entropión, erupción cutánea difusa e hinchazón local de la piel del párpado durante varios días después de la inyección en el párpado. En dos casos de alteraciones del nervio VIl (un caso de ojo afáquico), el número reducido de pestañeo a partir de otras inyecciones de toxina botulínica en el músculo orbicular llevó a exposición corneana grave, defecto epitelial persistente y úlceras corneanas. Ocurrió perforación en el ojo afáquico y requirió un injerto de córnea. Hubo un relato de glaucoma agudo de ángulo cerrado un día después de recibir una inyección de toxina botulínica por blefaroespasmo con recuperación luego de cuatro meses después de iridotomía con láser y trabeculectomía. Parálisis facial focal, síncope y empeoramiento de la Miastenia gravis fueron relatadas también después del tratamiento de blefaroespasmo. Fueron relatadas con frecuencia, anopia o conjuntivitis, que exigieron medidas apropiadas. En 660 pacientes con otras inyecciones de toxina botulínica (durante 6 años en Corea), un total de 41 pacientes (6,2%) presentaron reacciones adversas. Las reacciones adversas incluyeron ptosis en 17 pacientes (2,6%), hinchazón local en 5 (0,8%), disturbios lagrimales en 3 (0,5%), irritación bulbar en 3 (0,5%), lagoftalmo en 3 (0,5%), debilidad muscular en 3 (0,5%). sequedad ocular en 3. Reacciones adversas con causalidad incierta incluyeron contracción en el lugar de la inyección en 2 pacientes (0,3%), hipertonía en 2 (0,3%), congestión conjuntival en 2 (0,3%) y dolor ocular en 1 (0,2%). Los eventos adversos relatados con mayor frecuencia con otras inyecciones de toxina botulínica en el tratamiento de tortícolis espasmódica incluyeron: dolor y sensibilidad en los lugares de la inyección, debilidad local, debilidad general sintomática y fatiga. Mientras tanto, la fatiga también fue relatada en pacientes tratados con placebo. La disfagia y debilidad local pueden ser atribuidas a una extensión de la farmacología de la toxina botulínica resultante de la distribución de la toxina en los músculos que recibieron la inyección. Considerando que las reacciones adversas asociadas a la posología son observadas con mayor frecuencia en pacientes del sexo femenino, la masa muscular debe ser tenida en cuenta al seleccionar la dosis adecuada. Otros eventos adversos incluyen: Náuseas, vértigo, dolor de cabeza entumecimiento, rigidez y heridas. Fueron realizadas pruebas de seguridad de uso de la toxina botulínica en el tratamiento de la deformidad dinámica en pie equino debido a espasticidad en pacientes con parálisis cerebral pediátrica. De acuerdo a lo esperado en cualquier procedimiento de inyección intramuscular, el dolor localizado fue asociado a la inyección aplicada a los pacientes. En un ensayo clínico llevado a cabo en Corea, 60 pacientes quienes recibieron NEURONOX® mostraron reacciones adversas frecuentes (> 1%) como: nasofaringitis (5%), infección respiratoria superior (1.67%), pirexia (3.3%), alteración de la marcha (1.67%), constipación (1.67%), dolor en la extremidad (1.67%), alteración del tejido musculoesquelético o conectivo (1.67%), convulsión febril (1.67%), constipación (1.67%), fractura de extremidad inferior (1.67%). Adicionalmente, las reacciones adversas frecuentes (> 1%) también aparecieron en 59 pacientes a quienes se administró el medicamento referencia, así: nasofaringitis (5.08%), infección por Haemophilus (1.69%), neumonía (1.69%), pirexia (5.08%), astenia (1.69%), contractura articular (1.69%), debilidad muscular (1.69%), cefalea (1.69%), conjuntivitis (1.69%), anemia (1.69%), discrepancia en la longitud de la extremidad (1.69%). Estas reacciones pueden presentarse dependiendo de las características de los pacientes. En la literatura sobre otros productos de toxina botulínica, se reportan reacciones adversas similares.
INTERACCIONES:
El efecto de la toxina botulínica puede ser potencializado por antibióticos aminoglicósidos u otros medicamentos que interfieren con la transmisión neuromuscular por ejemplo los relajantes musculares del tipo tubocurarina. El uso concomitante de NEURONOX® con aminoglicósidos o espectinomicina está contraindicado. Polimixinas, tetraciclinas y lincomicinas deben ser utilizadas con cuidado en pacientes tratados con NEURONOX®. El efecto de la administración de diferentes serotipos de neurotoxina botulínica simultáneamente o en el plazo de varios meses cada una, es desconocida. La debilidad neuromuscular excesiva puede ser agravada por la administración de otras toxinas botulínicas antes de la disminución de los efectos de una toxina botulínica previamente administrada.
PRECAUCIONES: Pacientes en tratamiento con otros relajantes musculares (ejemplo, cloruro de Tubocurarina, Dantrolene sódico, etc) (el relajamiento muscular puede ser potenciado o los riesgos de disfagia pueden ser aumentados). Pacientes en tratamientos con medicamentos de actividad relajante muscular, ejemplo, Espectinomicina HCl, antibióticos aminoglicósidos (Sulfato de Gentamicina, Sulfato de Neomicina, etc.), antibióticos polipeptídicos (Sulfato de Polimixina B, etc.), antibióticos a base de tetraciclina, antibióticos a base de Lincomicina (Lincosamidas), relajantes musculares (Baclofen etc.), agentes anticolinérgicos (Butilbromuro de Escopolamina, Trihexilfenidil HCl, etc.), Benzodiacepina y drogas similares (Diazepam, Etizolam, etc.), medicamentos a base de benzamida (Tiaprida HCl, sulpirida, etc.). (El relajamiento muscular puede ser potenciado o los riesgos de disfagia pueden ser aumentados). Dado que el principio activo de este medicamento es la toxina botulínica del tipo A de Clostridium botulinum, una neurotoxina derivada del Clostridium botulinum, las dosis y las frecuencias de administración recomendadas deben ser observadas con una total comprensión de las precauciones en su utilización. Los médicos que administran el medicamento deben conocer la anatomía relevante neuromuscular y/u orbital del área involucrada y cualesquiera alteración en la anatomía debido a procedimientos quirúrgicos anteriores. También es necesaria una comprensión de las técnicas electromiográficas estándares para la administración del medicamento. La dosis y la frecuencia de la administración recomendadas no deben ser excedidas. Este producto contiene albúmina humana. El riesgo de transmisión de una infección viral no puede excluirse con absoluta certeza después del uso de sangre humana o hemoderivados. No debe sustituir ni la marca ni el tipo de toxina botulinica que le sea administrada. Reacciones de hipersensibilidad: Reacciones serias y/o inmediatas de hipersensibilidad fueron poco relatadas con otras inyecciones de toxina botulínica. Esas reacciones incluyen anafilaxia, urticaria, edema de tejido blando y disnea. Fue relatado un caso fetal de anafilaxia en que la lidocaína fue usada como diluyente, pero el agente causal no pudo ser determinado con seguridad. Si ocurre tal reacción, deberán suspenderse otras inyecciones del medicamento y la terapia médica adecuada debe ser inmediatamente instituida.
Disturbios neuromusculares preexistentes: Individuos con enfermedades neuropáticas del sistema motor periférico (ejemplo Esclerosis lateral amiotrófica, o neuropatía motora) o disturbios de las uniones neuromuscular (ejemplo Miastenia grave o síndrome de Lambert-Eaton) pueden tener un riesgo mayor de efectos sistémicos clínicamente significativos, incluyendo disfagia grave y compromiso respiratorio a partir de dosis típicas de inyección de toxina botulínica. La literatura médica publicada sobre la inyección de otra toxina botulínica tiene escasos relatos de casos de administración de una toxina botulínica en pacientes con disturbios neuromusculares conocidos o desconocidos, donde los pacientes mostraron extrema sensibilidad a los efectos sistémicos de las dosis clínicas típicas. En algunos de esos casos, la disfagia duró varios meses y exigió la colocación de un tubo de alimentación gástrica.
Disfagia: La disfagia es un evento adverso relatado con frecuencia después del tratamiento de pacientes con distonía cervical con todas las toxinas botulínicas. En esos pacientes, hay escasos relatos de casos de disfagia grave, lo suficiente para justificar la inserción de un tubo de alimentación gástrica. Hay también escasos relatos de casos donde luego de descubierta la disfagia un paciente desarrolló pneumonía por aspiración y falleció. Hay también escasos relatos de eventos adversos con inyección de otra toxina botulínica involucrando el sistema cardiovascular, incluyendo arritmia e infarto del miocardio, algunos con consecuencias fatales. Algunos de esos pacientes presentaban factores de riesgo, incluyendo enfermedad cardiovascular.
Inyección en o cerca de estructuras anatómicas vulnerables: Se deben tomar cuidados cuando se inyectan en o cerca de estructuras anatómicas vulnerables. Serios eventos adversos incluyendo resultados fatales han sido reportados en pacientes quienes han recibido otros productos de toxina botulínica inyectado directamente dentro de las glándulas salivales, la región buco-lingual-faringea, esófago y estómago. Algunos pacientes tienen disfagia pre-existente o debilidad significativa. (Seguridad y efectividad no han sido establecidas para indicaciones pertenecientes a este sitio de inyección). Se ha reportado pneumotórax asociado a un procedimiento de inyección para administración de otro producto de toxina botulínica cerca del tórax. Se recomienda precaución cuando es inyectado en proximidades del pulmón, particularmente el ápice. Efectos pulmonares por productos de toxina botulínica en pacientes con estado de compromiso respiratorio tratado para espasticidad o para hiperactividad del detrusor asociado con condición neurológica. En pacientes con estado respiratorio comprometido tratados con otra toxina botulínica para espasticidad de las extremidades superiores, reportes de reducción de la función del pulmón e infecciones del tracto respiratorio superior, y en pacientes con hiperactividad del detrusor asociada con una condición neurológica tratada con NEURONOX®, se reportó una reducción de la función del pulmón.
Bronquitis e infecciones del tracto respiratorio superior en pacientes tratados por espasticidad: Reportes de bronquitis más frecuentemente como una reacción adversa en pacientes tratados para espasticidad de las extremidades superiores con toxina botulínica, comparado al placebo. En pacientes con función del pulmón reducida tratados para espasticidad de extremidades superiores, infecciones del tracto respiratorio superior también fueron reportadas más frecuentemente como reacción adversa en pacientes tratados con toxina botulínica comparada con placebo. El uso seguro y efectivo de NEURONOX® depende del almacenamiento adecuado del producto, de la selección de la dosis correcta y de las técnicas adecuadas de reconstitución y administración. Los médicos que administran NEURONOX® deben comprender la anatomía neuromuscular del área comprometida y las alteraciones de la anatomía debido a procedimientos quirúrgicos anteriores.
También se requiere del conocimiento de técnicas electromiográficas estándar para la administración de NEURONOX®. No se debe exceder la dosis recomendada ni la frecuencia de la administración de NEURONOX®. En algunos casos, el efecto de NEURONOX® puede observarse más allá del sitio de inyección. Los síntomas pueden incluir astenia, debilidad muscular generalizada, diplopía, ptosis, disfagia, disartria, incontinencia urinaria y dificultad respiratoria. Estas reacciones pueden llegar a ser fatales. Debe advertirse a los pacientes o a quienes los cuidan, buscar atención médica inmediata en caso de surgir alteraciones de la deglución, del lenguaje o respiratorias. Se debe tener cuidado cuando se inyecta en o cerca de estructuras anatómicas vulnerables. Los eventos adversos graves, incluyendo resultados fatales, fueron reportados en pacientes que utilizaron otras toxinas botulínicas, cuando se inyectan directamente en las glándulas salivales, la región buco-lingual-faringe, el esófago y el estómago.
La seguridad y eficacia no se han establecido para las indicaciones relativas a estos sitios de inyección. Algunos pacientes tenían disfagia preexistente o debilidad significativa. El neumotórax asociado con el proceso de inyección se ha reportado después de la administración de NEURONOX® cerca del tórax. Se debe tener precaución cuando se inyecta en la proximidad al pulmón, en particular los ápices. Las reacciones de hipersensibilidad seria y/o inmediata han sido raramente reportadas, incluyendo anafilaxia, urticaria, edema de tejido blando y disnea. Si se producen dichas reacciones, debe interrumpirse el tratamiento y debe instaurarse un tratamiento médico de inmediato (como epinefrina).
Ha habido escasos reportes de eventos adversos que involucran el sistema cardiovascular, incluyendo arritmia e infarto de miocardio, algunos con consecuencias fatales. Algunos de estos pacientes tienen un riesgo cardíaco pre-existentes o enfermedad cardiovascular. La relación exacta de estos eventos con la inyección de toxina botulínica no fue establecida. Se debe tener precaución cuando se administra a pacientes con enfermedad cardiovascular preexistente. La disminución del pestañeo, luego de la inyección de toxina botulínica en el músculo orbicular, puede llevar a exposición corneana, defecto epitelial persistente y úlceras corneanas, principalmente en pacientes con trastornos del nervio craneal VII. Debe utilizarse una prueba minuciosa de la sensación corneana en los ojos previamente operados, se debe evitar la inyección en el área del párpado inferior para evitar ectropión y debe aplicarse un tratamiento efectivo para cualquier defecto epitelial, que puede incluir colirios de protección, ungüentos, lentes de contacto, geles o cubrimiento del ojo con vendas u otros medios. Se debe tener precaución cuando NEURONOX® se utiliza en presencia de inflamación en el sitio de la inyección propuesto(s), o cuando la debilidad o atrofia excesiva está presente en el músculo objetivo(s).
DOSIFICACIÓN:
1. Blefaroespasmo: Para blefaroespasmo, NEURONOX® reconstituido (ver tabla de dilución) se inyecta utilizando una aguja estéril calibre 27-30 sin guía electromiográfica. La dosis inicial recomendada es de 1.25-2.5 U (0.05 mL a 0.1 mL de volumen en cada sitio) inyectada en el orbicularis oculi pretarsal medio y lateral del párpado superior y en el orbicularis oculi pretarsal lateral del párpado inferior. En general el efecto inicial de las inyecciones se observa dentro de los tres días y alcanza un máximo en una o dos semanas después del tratamiento. Cada tratamiento dura aproximadamente tres meses, luego de dicho periodo se puede repetir el tratamiento. En las sesiones de repetición de tratamiento, la dosis se puede aumentar hasta el doble si la respuesta al tratamiento inicial se considera insuficiente - normalmente definida como un efecto que no dura más de dos meses. Sin embargo, parece existir poco beneficio de inyectar más de 5U por sitio. Se puede encontrar alguna tolerancia cuando se usa el medicamento en el tratamiento del blefaroespasmo si los tratamientos se aplican con más frecuencia que cada tres meses y es raro conseguir que el efecto sea permanente. La dosis acumulada del tratamiento con NEURONOX® en un periodo de 30 días no debe exceder 200 U.
2. Parálisis cerebral infantil - Deformidad en pie equino: En parálisis cerebral infantil, NEURONOX® reconstituido (ver tabla de dilución) se inyecta utilizando una aguja estéril calibre 26-30. Se recomienda inyectar cada una de las cabezas (medial y lateral) del músculo gastrocnemio. Se recomienda una dosis total de 4 U/kg de peso corporal en el músculo gastrocnemio afectado, en pacientes con hemiplejía y, en pacientes con diplejía, la dosis recomendada es 6 U/kg de peso corporal dividida en ambas piernas. La dosis máxima administrada no debe exceder 200 U/paciente a la vez. Después de la inyección, el paciente debe monitorearse durante al menos 30 minutos en cuanto a la presencia de algún evento adverso agudo. Se puede esperar mejoría clínica dentro de las 4 semanas después de la inyección. Las inyecciones se pueden repetir cuando el efecto de la inyección anterior haya disminuido pero generalmente no antes de 12 semanas.
3. Espasticidad muscular: La dosis exacta y el número de inyecciones de NEURONOX® deben ser adaptadas al individuo, basado en el tamaño, número y ubicación de los músculos involucrados, la severidad de la espasticidad, presencia de debilidad muscular local y a la respuesta del paciente a tratamientos previos. El mejoramiento clínico en el tono del músculo se ve entre cuatro a seis semanas después del tratamiento. En ensayos clínicos controlados, las siguientes dosis son administradas, sin sobrepasar 360 U:
|
Dosis |
Dosis total: Número de sitios |
|
Bíceps braquial |
100-200 U: hasta 4 sitios |
|
Flexor profundo |
15-50 U: 1-2 sitios |
|
Flexor sublimis |
15-50 U: 1-2 sitios |
|
Flexor radial del carpo |
15-50 U: 1-2 sitios |
|
Flexor cubital del carpo |
15-50 U: 1-2 sitios |
NEURONOX® reconstituido es inyectado usando una aguja estéril de calibre 24-30 para músculo superficial y una aguja más larga puede ser usada para musculatura más profunda. Se recomienda la localización de los músculos involucrados con la orientación electromiográfica o técnicas de estimulación del nervio.
4. Espasmo hemifacial: La dosis de NEURONOX® recomendada es de 12 a 15 U inyectadas en dosis divididas de 2.5 a 5 U en las áreas afectadas.
5. Cefalea tensional: La dosis de NEURONOX® requerida es de 50-100 U, distribuidas en varios puntos: Área Dosis (U)
|
Área |
Dosis (U) |
|
Músculo frontal |
2 x 2* |
|
Músculo temporal |
4 |
|
Músculo esternocleidomastoideo |
10 |
|
Músculo trapecio |
12 |
|
Músculo esplenio de la cabeza |
10 |
|
Músculo semiespinoso de la cabeza |
10 |
|
* Dos sitios de inyección por cada lado |
|
6. Hiperactividad del músculo detrusor de la vejiga: La dosis de NEURONOX® recomendada es de 300 U (100-400 U) aplicadas en aproximadamente 30 sitios en el músculo detrusor, a través de un cistoscopio flexible o rígido, como se indica en la figura 1:
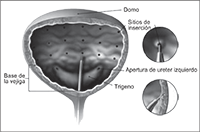
Figura 1. Puntos de aplicación de toxina botulínica en la vejiga
7. Temblor palatal esencial: Se recomienda administrar 10 U de NEURONOX® por vía intramuscular (5 U en el músculo tensor del velo del paladar y 5 U en el elevador del velo del paladar, de forma bilateral).
8. Disfonía espasmódica: En pacientes con disfonía espasmódica aductora, la dosis inicial de NEURONOX® es habitualmente de 1.25 U en cada músculo tiroaritenoideo. En pacientes con disfonía espasmódica abductora, la dosis es de 5 U en el cricoaritenoideo posterior unilateral. Puede aplicarse una segunda inyección en el músculo contralateral, si los síntomas no son suficientemente controlados con la dosis inicial. Antes de esta inyección, debe realizarse una laringoscopia flexible, para confirmar que la abducción de la cuerda vocal está debilitada pero es adecuada. Si la abducción es mínima, existe riesgo significativo de compromiso de la vía aérea debido a la inyección contralateral, por lo cual, la aplicación de NEURONOX® debe posponerse. Alternativamente, 1.25 U pueden administrarse en un lado, mientras 5-7 U pueden administrarse en el otro, durante el mismo procedimiento. La aplicación de toxina botulínica para el tratamiento de la disfonía espasmódica abductora es más difícil que para la disfonía espasmódica aductora y se asocia con mayor riesgo, incluyendo estridor leve a severo, causado por parálisis del cricoaritenoideo posterior.
9. Hiperhidrosis refractaria: La dosis recomendada de NEURONOX® en el tratamiento de la hiperhidrosis axilar es de 50 U por cada axila, inyectando por vía intradérmica, 2.5-4 U de NEURONOX® cada 2 cm en el área afectada. Debe identificarse el área hiperhidrótica, utilizando una técnica de tinción estándar. Puede repetirse la aplicación, cuando el efecto clínico disminuya. En el caso de la hiperhidrosis palmar, se requieren 100-150 U para tratar una sola palma, divididas en 50 a 60 inyecciones intradérmicas de 2-3 U. Se debe realizar bloqueo anestésico regional de los nervios mediano, cubital y radial.
10. Bruxismo temporo-maxilar: Se requiere una dosis de toxina botulínica entre 25-60 U por músculo. En un ensayo clínico que evaluó el efecto de NEURONOX® en el tratamiento del bruxismo en pacientes que no respondieron al manejo con férula, se aplicaron 25 U en cada músculo, usando una aguja calibre 29, como se muestra en la figura 2.

Figura 2. Puntos de aplicación de toxina botulínica en los músculos temporal (A) y masetero (B)
11. Fisura anal: Se recomiendan 5 a 15 U de NEURONOX® inyectadas en la cara interna y externa del esfínter anal.
12. Acalasia: La dosis recomendada de NEURONOX® es de 80 a 100 U inyectadas en el esfínter esofágico inferior.
13. Líneas faciales hiperfuncionales (arrugas glabelares, líneas del cantus lateral): Para el tratamiento de las arrugas glabelares, se recomienda aplicar 5 U por vía intramuscular, en cada uno de los 5 sitios señalados en la figura 3, para una dosis total de 25 U. Normalmente la dosis inicial de NEURONOX® reconstituido induce una denervación química de los músculos inyectados uno o dos días después de la inyección, incrementando la intensidad durante la primera semana. La duración del efecto de NEURONOX® para las líneas glabelares es de aproximadamente 3-4 meses.

Figura 3. Puntos de aplicación de toxina botulínica en el tratamiento de las arrugas glabelares
La seguridad y la efectividad de la dosis mayor a 3 meses no han sido evaluadas clínicamente. En el caso de las líneas del cantus lateral (patas de gallina), administrar 0.1 mL (5 U) en cada uno de los 3 sitios señalados en la figura, por cada ojo (6 puntos de inyección en total) en el músculo orbicular lateral del ojo, para una dosis total de 30 U/0.6 mL (15 U por lado). La primera inyección debe ser aproximadamente 1.5 a 2 cm en las líneas temporales del cantus lateral y para el reborde orbitario. Si las líneas de expresión están por encima del ángulo lateral y por debajo del cantus lateral, se inyectan según la figura 2. Alternativamente, si las líneas de expresión se encuentran en la región del ángulo lateral por debajo del cantus lateral, se inyectan según la figura 4.
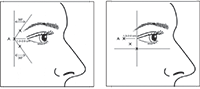
Figura 4. Puntos de aplicación de toxina botulínica en el tratamiento de las líneas del cantus lateral
14. Profilaxis de migraña: Las dosis recomendadas dependen del sitio de aplicación NEURONOX®, así:
|
Área cabeza/cuello |
Dosis recomendada |
|
Frontal** |
20 U divididas en 4 sitios |
|
Corrugador** |
10 U divididas en 2 sitios |
|
Procerus |
5 U divididas en 1 sitios |
|
Occipital** |
30 U divididas en 6 sitios |
|
Temporal** |
40 U divididas en 8 sitios |
|
Trapecio** |
30 U divididas en 6 sitios |
|
Cervical paraespinal** |
20 U divididas en 4 sitios |
|
Dosis total |
155 U divididas en 31 sitios |
|
* Cada sitio de inyección IM = 5 U/0.1 mL de NEURONOX® **Todas las dosis deben dividirse y administrarse bilateralmente. |
|
Técnica de reconstitución: Antes de la inyección, reconstituir el NEURONOX® con solución salina estéril sin conservantes. El Cloruro de Sodio (NaCl) al 0.9% es el diluyente recomendado. Retire la cantidad adecuada de diluyente en la jeringa de tamaño apropiado. El diluyente se debe inyectar suavemente dentro del vial. Descarte el vial si un vacío no empuja el diluyente al interior del vial. Mezcle NEURONOX® suavemente con la solución salina, girando el vial. Registre la fecha y el horario de la reconstitución en el espacio en blanco de la etiqueta. NEURONOX® debe ser administrado dentro de las veinticuatro (24) horas de su reconstitución. Durante este periodo de tiempo, NEURONOX® reconstituido deberá mantenerse en la nevera (2~8°C). NEURONOX® reconstituido debe ser claro, incoloro y libre de material particulado. Los medicamentos parenterales deben ser inspeccionados visualmente para detectar partículas y decoloración antes de su administración. Debido a que NEURONOX® y el diluyente no contienen ningún conservante, el vial de NEURONOX® debe ser utilizado en un único paciente.
|
Tabla de dilución de NEURONOX® 100 UI |
|
|
Diluyente adicionado (inyección de 0.9% de Cloruro de Sodio) |
Dosis resultante |
|
1.0 mL |
10.0 U |
|
2.0 mL |
5.0 U |
|
4.0 mL |
2.5 U |
|
8.0 mL |
1.25 U |
Nota: Estas diluciones son calculadas para un volumen de inyección de 0.1 mL. También es posible reducir o aumentar la dosis administrando un volumen mayor o menor de inyección de 0.05 mL (reducción del 50% en la dosis) a 0.15 mL (aumento de 50% en la dosis).
SOBREDOSIFICACIÓN:
Los signos de sobredosificación no son aparentes inmediatamente después de la inyección. En caso de inyección o ingestión accidental se debe someter al paciente a supervisión médica durante varios días, para detectar posibles signos o síntomas de debilidad sistémica o parálisis muscular. Aquellos pacientes que muestren síntomas de intoxicación por toxina botulínica tipo A (debilidad generalizada, ptosis, diplopía, alteraciones del habla y de la deglución, o paresia de los músculos respiratorios) deben ser hospitalizados. Al aumentar la dosis, se produce parálisis muscular profunda y generalizada. En caso de que se vea afectada la musculatura de la orofaringe y el esófago, se puede producir aspiración que puede llevar al paciente a sufrir una neumonía por aspiración. En caso de que se paralicen los músculos respiratorios, será necesario proceder a entubar y aplicar respiración asistida hasta la recuperación.
PRESENTACIÓN: Caja por 1 vial vidrio tipo I con tapón clorobutilo, tapa FLIP-OFF en polipropileno/ aluminio, conteniendo el polvo liofilizado. Reg. San. INVIMA 2013M-0014471.
HUMAX PHARMACEUTICAL, S. A.
CONSERVACIÓN:
Almacenar entre 2 °C y 8 °C en su envase y empaque original. El producto una vez reconstituido en solucion salina normal (NaCl 0,9%) es estable durante 24 horas almacenado en refrigeración (2°C-8°C).

