
MOZOBIL TM
PLERIXAFOR
Solución inyectable
Frasco(s) , Solución inyectable
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
FÓRMULA CUALICUANTITATIVA
Cada mL de SOLUCIÓN ESTÉRIL de MOZOBIL™ contiene 20 mg de plerixafor, 4,9 mg de cloruro de sodio en agua para inyección c.s ajustada a un pH de 6,0 a 7,5 con ácido clorhídrico e hidróxido de sodio. Cada frasco ampolla de uso único es llenado para suministrar 1,2 mL de solución estéril.
Industria Inglesa
Versión 1/2013
INDICACIONES: MOZOBIL™, en combinación con el factor estimulante de colonias de granulocitos (G-CSF), está indicado para movilizar células madre hematopoyéticas a sangre periférica, para su recolección y para su trasplante autólogo posterior en pacientes movilizadores pobres de células madre hematopoyéticas con linfoma no Hodgkin y mieloma múltiple.
USO EN POBLACIONES ESPECÍFICAS
Embarazo: Embarazo Categoría D
Plerixafor demostró ser teratogénico en animales. Cuando plerixafor es administrado en ratas preñadas induce toxicidad embriofetal incluyendo: Muerte fetal, incremento de resorciones y pérdida post-implantación, disminución del peso del feto, anoftalmía, acortamiento de los dígitos, defecto septal interventricular cardiaco, aorta anular, corazón globular, hidrocefalia, dilatación de los ventrículos olfatorios y retardo del desarrollo esquelético. La toxicidad embriofetal ocurre principalmente a la dosis de 90 mg/m2 (aproximadamente 10 veces la dosis recomendada en humanos de 0,24 mg/kg, cuando se compara sobre la base de mg/m2 o 10 veces el área bajo la curva AUC en sujetos con función renal normal que recibieron un dosis única de 0,24 mg/kg).
Madres en periodo de lactancia: Se desconoce si plerixafor es excretado en la leche humana. Dado que muchos fármacos se excretan en la leche humana, y debido al posible riesgo asociado a las reacciones adversas serias a MOZOBIL™ en infantes lactantes, se debe tomar la decisión de si es conveniente o no discontinuar la lactancia o discontinuar la droga, teniendo en cuenta la importancia de la droga para la madre.
Uso pediátrico: No se ha establecido la inocuidad y eficacia de MOZOBIL™ en pacientes pediátricos en estudios clínicos controlados.
Uso geriátrico: Del número total de sujetos que participaron en estudios clínicos controlados de MOZOBIL™, 24% de los pacientes tenían = 65 años de edad, mientras que 0,8% tenían = 75 años. En general, no se observaron diferencias en la seguridad y efectividad de la droga entre los pacientes ancianos y los pacientes jóvenes. En otros reportes clínicos, no se han identificado diferencias en las repuestas a plerixafor entre pacientes ancianos y jóvenes. Sin embargo, no se puede descartar una mayor sensibilidad a la droga en algunos ancianos o individuos geriátricos.
Dado que plerixafor es excretado principalmente por el riñón, no es necesario modificar la dosis en pacientes geriátricos con función renal normal. En general, se debe proceder con cuidado al seleccionar la dosis para pacientes geriátricos dado que con la edad avanzada se observa más frecuentemente una disminución de la función renal. Se recomienda el ajuste de la dosis en pacientes geriátricos cuyo CLCR sea = 50 mL/min [Véase Farmacología Clínica y Posología y Administración]
Insuficiencia renal: En pacientes con insuficiencia renal moderada y severa (CLCR = 50 mL/min) se debe reducir la dosis de plerixafor en un tercio a 0,16 mg/kg. (Véase Posología y Administración y Farmacología Clínica)
FORMA FARMACÉUTICA: Solución estéril inyectable para uso subcutáneo.
CONTRAINDICACIONES: Hipersensibilidad al principio activo o a alguno de sus excipientes.
INFORMACIÓN PARA EL PACIENTE: Informar a los pacientes sobre los signos y síntomas de posibles reacciones sistémicas como urticaria, hinchazón periorbital, disnea o hipoxia durante y después de la inyección de MOZOBIL™. (Véase Reacciones Adversas)
Los pacientes deben informar inmediatamente a su médico si presentan síntomas de reacciones vasovagales tales como hipotensión ortostática o síncope, durante o poco tiempo después de la inyección de MOZOBIL™. (Véase Reacciones Adversas)
Si los pacientes presentan prurito, exantema o una reacción en el sitio de la inyección, deben informar a su médico ya que estos síntomas se han tratado satisfactoriamente durante los estudios clínicos con medicamentos de venta libre. [Véase Reacciones Adversas] Informar a los pacientes que MOZOBIL™ puede causar trastornos gastrointestinales, incluyendo diarrea, náuseas, vómitos, flatulencia y dolor abdominal. Se debe indicar a los pacientes cómo manejar y/o prevenir trastornos gastrointestinales específicos e informar a su médico si se producen eventos graves después de la inyección de MOZOBIL™. (Véase Reacciones Adversas)
Recomendar a las pacientes con posibilidad de embarazarse que utilicen métodos anticonceptivos eficaces durante el tratamiento con MOZOBIL™. (Véase Uso en Poblaciones Específicas)
REACCIONES ADVERSAS: Las siguientes reacciones adversas graves son discutidas en la sección de advertencias y precauciones.
— Posible movilización de células tumorales en pacientes con leucemia.
— Aumento de leucocitos circulantes y disminución del recuento de plaquetas.
— Posible esplenomegalia.
Las reacciones adversas más comunes (= 10 %) informadas por pacientes que recibieron MOZOBIL™ conjuntamente con G-CSF sin tener en cuenta la causalidad y que fueron más frecuentes con MOZOBIL™ que en el grupo placebo durante la movilización de células madre hematopoyéticas y la aféresis fueron: Diarrea, náusea, fatiga, reacciones en el sitio de inyección, cefaleas, artralgia, mareos y vómitos.
Los datos de seguridad correspondientes a MOZOBIL™ administrado conjuntamente con G-CSF se obtuvieron de dos estudios controlados con placebo y de 10 estudios no controlados en 543 pacientes.
Los pacientes fueron tratados principalmente con dosis diarias de 0,24 mg/kg por vía subcutánea. La mediana de la exposición a MOZOBIL™ en estos estudios fue de 2 días (entre 1 a 7 días).
En dos estudios aleatorios en pacientes con linfoma no Hodgkin y mieloma múltiple, se trataron un total de 301 pacientes en el grupo de MOZOBIL™ y G-CSF y 292 pacientes en el grupo placebo y G-CSF. Los pacientes recibieron dosis matutinas diarias de G-CSF de 10 microgramos/kg durante los 4 días previos a la primera dosis de MOZOBIL™, o placebo cada mañana antes de la aféresis.
En la Tabla 7 se muestran las reacciones adversas que ocurrieron en = 5 % de los pacientes que recibieron MOZOBIL™, sin tomar en cuenta la causalidad, y que fueron más frecuentes con MOZOBIL™ que con placebo durante la movilización de células madre hematopoyéticas y la aféresis.
Dado que los estudios clínicos se llevan a cabo bajo condiciones variadas, los índices de reacciones adversas que se observan en los estudios clínicos de un fármaco no pueden compararse directamente con los índices observados en los estudios clínicos de otro fármaco, y pueden ser diferentes de los observados en la práctica.
|
Tabla 7: Reacciones adversas en = 5% de los pacientes con linfomas no Hodgkin y mieloma múltiple que recibieron MOZOBIL™y que fueron más frecuentes que en el grupo placebo durante la movilización de células madre hematopoyéticas y aféresis |
||||||
|
Porcentaje de pacientes (%) |
||||||
|
MOZOBIL™101 y G-CSF (n = 301) |
Placebo y G-CSF (n = 292) |
|||||
|
Todos los gradosa |
Grado 3 |
Grado 4 |
Todos los grados |
Grado 3 |
Grado 4 |
|
|
Trastornos gastrointestinales |
||||||
|
Diarrea |
37 |
< 1 |
o |
17 |
o |
o |
|
Náuseas |
34 |
1 |
o |
22 |
o |
o |
|
Vómitos |
10 |
< 1 |
o |
6 |
o |
o |
|
Flatulencia |
7 |
o |
o |
3 |
o |
o |
|
Trastornos generales y estado del área de administración |
||||||
|
Reacciones en el sitio de inyección |
34 |
o |
o |
10 |
o |
o |
|
Fatiga |
27 |
o |
o |
25 |
o |
o |
|
Trastornos osteomusculares y del tejido conjuntivo |
||||||
|
Artralgia |
13 |
o |
o |
12 |
o |
o |
|
Trastornos del sistema nervioso |
||||||
|
Cefalea |
22 |
<1 |
o |
21 |
1 |
o |
|
Mareos |
11 |
o |
o |
6 |
o |
o |
|
Trastornos psiquiátricos |
||||||
|
Insomnio |
7 |
o |
o |
5 |
o |
o |
|
a Los grados se basan en los criterios de la Organización Mundial de la Salud (OMS) |
||||||
En los estudios aleatorios, el 34% de los pacientes con linfoma no Hodgkin o mieloma múltiple tuvieron reacciones leves a moderadas en el sitio de inyección al administrar MOZOBIL™ en forma subcutánea. Estas incluyeron: Eritema, hematoma, hemorragia, induración, inflamación, irritación, dolor, parestesia, prurito, sarpullido, hinchazón y urticaria.
Aproximadamente 30 minutos después de la administración de MOZOBIL™ se observaron reacciones sistémicas leves a moderadas en menos del 1% de los pacientes. Los eventos incluyeron 1 o más de las siguientes reacciones: Urticaria (n = 2), hinchazón periorbital (n = 2), disnea (n = 1) o hipoxia (n = 1). Los síntomas generalmente respondieron al tratamiento (por ejemplo: Antihistamínicos, corticosteroides, hidratación o suplemento de oxígeno) o se resolvieron espontáneamente.
Después de la dosis subcutánea pueden ocurrir reacciones vasovagales, hipotensión ortostática y/o síncope. En estudios clínicos de MOZOBIL™ con sujetos normales y oncológicos, menos del 1% de los pacientes experimentaron reacciones vasovagales después de la administración subcutánea de MOZOBIL™ en dosis de =0,24 mg/kg. La mayoría de estos eventos ocurrieron durante 1 hora después de la administración de MOZOBIL™. Debido al potencial de estas reacciones se deben tomar medidas apropiadas.
Otras reacciones adversas observadas en < 5% de los pacientes pero que se informaron como relacionadas a MOZOBIL™ durante la movilización de células madre hematopoyéticas y la aféresis incluyeron: Dolor abdominal, hiperhidrosis, distensión abdominal, sequedad en la boca, eritema, molestias estomacales, malestar, hipoestesia oral, estreñimiento, dispepsia y dolor osteomuscular.
Infarto de miocardio: En los estudios clínicos, 7 de los 679 pacientes oncológicos sufrieron infartos de miocardio después de la movilización de células madre hematopoyéticas con plerixafor y G-CSF. Todos los acontecimientos se produjeron al menos 14 días después de la última administración de MOZOBIL™. Además, en el programa de uso compasivo, dos pacientes oncológicos del sexo femenino sufrieron infarto de miocardio tras la movilización de células madres hematopoyéticas con plerixafor y G-CSF. Una de estas reacciones se produjo 4 días después de la última administración de MOZOBIL™. La falta de relación temporal en 8 de los 9 pacientes junto con el perfil de riesgo de los pacientes con infarto de miocardio, no sugiere que MOZOBIL™ produzca un riesgo independiente de infarto de miocardio en pacientes que también recibieron G-CSF.
INTERACCIONES MEDICAMENTOSAS: En base a estudios in vitro, plerixafor no es sustrato, inhibidor o inductor de las isoenzimas humanas del citocromo P450. Es poco probable que plerixafor esté involucrado en interacciones farmacológicas in vivo (droga-droga) dependientes de las enzimas del grupo citocromo P450. (Véase Farmacología Clínica)
ESTUDIOS CLÍNICOS: La eficacia y seguridad de MOZOBIL™ administrado conjuntamente con G-CSF en pacientes con linfoma no Hodgkin y con mieloma múltiple fueron evaluadas en dos estudios controlados con placebo (Estudio 1 y 2). Los pacientes fueron asignados aleatoriamente para recibir ya fuera una dosis de 0,24 mg/kg de MOZOBIL™ o de placebo, durante cada noche antes de la aféresis. Los pacientes recibieron dosis matutinas diarias de G-CSF de 10 microgramos/kg durante los 4 días previos a la primera dosis de MOZOBIL™ o de placebo y cada mañana antes de la aféresis. En los análisis de eficacia primaria del Estudio 1 se incluyeron 298 pacientes con linfoma no Hodgkin. La edad media fue de 55,1 años (entre 29-75 años) y 57,5 años (entre 22-75) para los grupos de MOZOBIL™ y de placebo respectivamente, y 93 % de los sujetos eran caucásicos. En los análisis de eficacia primaria del Estudio 2 se incluyeron 302 pacientes con mieloma múltiple. La edad media fue de 58,2 años (entre 28-75 años) y 58,5 años (entre 28-75) para los grupos de MOZOBIL™ y de placebo respectivamente, y 81% de los sujetos eran caucásicos.
En el Estudio 1, 59% de los pacientes con linfoma no Hodgkin sometidos a movilización con MOZOBIL™ y con G-CSF recolectaron = 5 X 106 células CD34+/kg de sangre periférica a lo largo de 4 o menos sesiones de aféresis, en comparación con el 20% de los pacientes sometidos a movilización con placebo y G-CSF (p < 0,001). Otros resultados de movilización de células CD34+ mostraron hallazgos similares (Tabla 2).
|
Tabla 2: Resultados de eficacia del Estudio 1- Movilización de células CD34+ en pacientes con linfomas no Hodkin |
|||
|
Criterios de valoración de la eficacia |
MOZOBIL™ y G-CSF (n = 150) |
Placebo y G-CSF (n = 148) |
Valor de pa |
|
Pacientes que alcanzan = 5x106 células/kg en = 4 días de aféresis |
89 (59%) |
29 (20%) |
< 0,001 |
|
Pacientes que alcanzan = 2 x106 células/kg en = 4 días de aféresis |
130 (87%) |
70 (47%) |
< 0,001 |
|
a valor de p calculado con la prueba del Chi-cuadrado de Pearson. |
|||
La mediana de días para alcanzar la recolección de = 5 x 106 células CD34+/kg fue de 3 días para el grupo de MOZOBIL™ y no evaluable para el grupo de placebo. La Tabla 3 presenta la proporción de pacientes que alcanzaron a recolectar = 5 X 106 células CD34+/kg por días de aféresis.
|
Tabla 3: Resultados de eficacia delEstudio 1- Proporción de pacientes con linfomas no Hodgkin que alcanzaron a recolectar =5x106 células /kg CD34+ células/kg por días de aféresis. |
||
|
Días |
Proporcióna en el grupo MOZOBIL™ y G-CSF (n=147b) |
Proporcióna en el grupo placebo y G-CSF (n=142b) |
|
1 |
27,9% |
4,2% |
|
2 |
49,1% |
14,2% |
|
3 |
57,7% |
21,6% |
|
4 |
65,6% |
24,2% |
|
a Los porcentajes fueron calculados con el método Kaplan Meier b n incluye todos los pacientes que recibieron al menos un día de aféresis |
||
En el Estudio 2, 72 % de los pacientes con mieloma múltiple sometidos a movilización con MOZOBIL™ y con G-CSF recolectaron = 6 X 106 células CD34+/kg a partir de sangre periférica en 2 o menos sesiones de aféresis, en comparación con el 34% de los pacientes que movilizaron con placebo y G-CSF (p < 0,001). Otros resultados de movilización de células CD34+ mostraron hallazgos similares (Tabla 4).
|
Tabla 4: Resultados de eficacia del Estudio 2 - Movilización de células CD34+ en pacientes con mieloma múltiple |
|||
|
Criterios de valoración de la eficacia |
MOZOBIL™ y G-CSF (n = 148) |
Placebo y G-CSF (n = 154) |
Valor de pa |
|
Pacientes que alcanzan = 6 X 106 células/kg en = 2 días de aféresis |
106 (72%) |
53 (34%) |
< 0,001 |
|
Pacientes que alcanzan = 6 X 106 células/kg en = 4 días de aféresis |
112 (76%) |
79 (51%) |
< 0,001 |
|
Pacientes que alcanzan = 2 X 106 células/kg en = 4 días de aféresis |
141 (95%) |
136 (88%) |
0,028 |
|
a valor de p calculado con la prueba del Chi-cuadrado de Pearson. |
|||
La mediana de días para cumplir con la recolección de = 6 x 106 células CD34+/kg fue 1 día para el grupo de MOZOBIL™ y 4 para el grupo de placebo. La Tabla 5 presenta la proporción de pacientes que alcanzaron a recolectar = 6 X 106 células CD34+/kg por días de aféresis.
|
Tabla 5: Estudio 2- Proporción de pacientes con Mieloma Múltiple que alcanzaron a recolectar =6x106 células/kg CD34+ células/kg por días de aféresis. |
||
|
Días |
Proporcióna en el grupo MOZOBIL™ y G-CSF (n=144b) |
Proporcióna en el grupo placebo y G-CSF (n=150b) |
|
1 |
54,2% |
17,3% |
|
2 |
77,9% |
35,3% |
|
3 |
86,8% |
48,9% |
|
4 |
86,8% |
55,9% |
|
a Los porcentajes fueron calculados con el metodo Kaplan Meier b n incluye todos los pacientes que recibieron al menos un día de aféresis |
||
Factores múltiples pueden influenciar el tiempo de implantación del injerto y la duración del mismo, después del trasplante de células madres. En estudios de fase 3, para aquellos pacientes que recibieron trasplantes, el tiempo para la implantación del injerto de neutrófilos y de plaquetas y la duración del injerto fueron similares para todos los grupos de tratamiento.
FARMACOLOGÍA CLÍNICA:
Mecanismo de acción: Plerixafor es un inhibidor del receptor de quimiocina CXCR4 y bloquea la unión de su ligando análogo, el factor-1a derivado de células estromales (SDF-1a). SDF-1a y el CXCR4 juegan un papel importante en la circulación y alojamiento de las células madre hematopoyéticas humanas en el compartimento medular. Una vez en la médula, CXCR4 en las células madre puede ayudar a fijar estas células a la matriz medular, ya sea directamente mediante el factor SDF-1a o mediante la inducción de otras moléculas de adhesión. El tratamiento con plerixafor produce leucocitosis y aumento de células progenitoras hematopoyéticas circulantes en ratones, perros y humanos. En modelos de trasplante canino, las células CD34+ movilizadas por plerixafor fueron capaces de injertarse con capacidad de repoblar hasta 1 año posterior al tratamiento.
Farmacodinamia: En dos estudios clínicos controlados con placebo en pacientes con linfoma no Hodgkin y con mieloma múltiple (Estudio 1 y Estudio 2, respectivamente), se evaluó la magnitud del incremento en el recuento de células CD34+ en la sangre periférica (células/mcL) en el día de la aféresis. En la Tabla 1 se resume la magnitud del incremento en el recuento de células CD34+ (células/mcL) correspondiente a un periodo de 24 horas comenzando desde el día previo a la primera aféresis y culminando en la mañana siguiente, justo antes de la primera aféresis. Durante dicho periodo de 24 horas, se administró una única dosis de MOZOBIL™ o de placebo, 10-11 horas antes de la aféresis.
|
Tabla 1: Magnitud del incremento en el recuento de células CD34+ en la sangre periférica después del pretratamiento con G-CSF y la administración de Plerixafor. |
||||
|
Estudio |
MOZOBIL™ y G-CSF |
Placebo y G-CSF |
||
|
Mediana |
Media (DE) |
Mediana |
Media (DE) |
|
|
Estudio 1 |
5,0 |
6,2 (5,4) |
1,4 |
1,9 (1,5) |
|
Estudio 2 |
4,8 |
6,4 (6,8) |
1,7 |
2,4 (7,3) |
En estudios de farmacodinamia con MOZOBIL™ en voluntarios sanos, el pico de movilización de las células CD34+ se observó entre las 6 y 9 horas después de su administración. En estudios de farmacodinamia con MOZOBIL™ conjuntamente con G-CSF en voluntarios sanos, se observó un aumento sostenido en el recuento de células CD34+ en sangre periférica entre las 4 a 18 horas después de la administración de plerixafor, con un pico en el recuento de células CD34+ entre las 10 y 14 horas.
Farmacocinética: Las propiedades farmacocinéticas de plerixafor a dosis única, se evaluaron en pacientes con linfoma no Hodgkin y mieloma múltiple con una dosis de 0,24 mg/kg después del tratamiento previo con G-CSF (10 microgramos/kg una vez por día durante 4 días consecutivos). Plerixafor exhibe una cinética lineal entre las dosis de 0,04 mg/kg y 0,24 mg/kg. Los parámetros farmacocinéticos de plerixafor fueron similares en general en los estudios clínicos con sujetos sanos que recibieron plerixafor solo y pacientes con linfoma no Hodgkin y mieloma múltiple que recibieron plerixafor en combinación con G-CSF.
El análisis farmacocinético poblacional para plerixafor incluyó datos obtenidos de 63 sujetos (pacientes con linfoma no Hodgkin y mieloma múltiple, sujetos con diferente grado de insuficiencia renal y sujetos sanos) que recibieron una única dosis subcutánea de plerixafor (entre 0,04 mg/kg y 0,24 mg/kg). El modelo que describe adecuadamente el perfil concentración-tiempo de plerixafor fue un modelo bicompartimental con absorción y eliminación de primer orden. Se observaron relaciones significativas entre la depuración y el clearence de creatinina (CLCR), así como también entre el volumen central de distribución y el peso corporal. El tiempo de semivida plasmática (t1/2ß) se estimó en 0,3 horas y en pacientes con función renal normal el tiempo de semivida terminal poblacional (t1/2ß) fue 5,3 horas.
El análisis farmacocinético poblacional demostró que la posología ajustada en mg/kg, ocasiona un incremento en la exposición a plerixafor (área bajo la curva concentración plasmática vs. tiempo AUC0-24hs) a medida que aumenta el peso corporal. La experiencia de uso de la dosis 0,24 mg/kg en pacientes cuyo peso corporal es superior a 160 kg es limitada. Por lo tanto, la dosis no debería exceder aquella que se utiliza para un paciente de 160 kg (esto es 40 mg/día si CLCR > 50 mL/min y 27 mg/día si CLCR es > 50 mL/min) [Véase Posología y Administración]
Absorción: Las concentraciones plasmáticas máximas se presentaron 30-60 minutos después de la dosis subcutánea aproximadamente.
Distribución: Plerixafor se fija a las proteínas plasmáticas humanas hasta en un 58 %. El volumen de distribución aparente de plerixafor en el ser humano es de 0,3 L/kg, lo cual demuestra que plerixafor queda mayormente confinado, pero no exclusivamente limitado, al espacio del fluido extravascular.
Metabolismo: El metabolismo de plerixafor fue evaluado en ensayos in vitro. Los ensayos que utilizan microsomas hepáticos humanos o hepatocitos humanos primarios demuestran que plerixafor no es metabolizado y no exhibe actividad inhibitoria in vitro hacia de las enzimas metabolizantes principales del citocromo P450 (1A2, 2C9, 2C19, 2D6 y 3A4/5). Plerixafor no induce las enzimas CYP1A2, CYP2B6 o CYP3A4 en los estudios in vitro con hepatocitos humanos. Estos hallazgos sugieren que es improbable que plerixafor esté involucrado en las interacciones farmacológicas dependientes de citocromo P450.
Eliminación: La ruta de eliminación principal de plerixafor es la vía urinaria. Después de la administración de una dosis de 0,24 mg/kg a voluntarios sanos con función renal normal, aproximadamente 70 % de la dosis se excretó en la orina, como fármaco original, durante las primeras 24 horas después de la administración. En estudios con sujetos sanos y pacientes, la semivida terminal en plasma oscila entre 3 a 5 horas. La capacidad de plerixafor de actuar como sustrato o como un inhibidor de glucoproteína P no ha sido investigada.
Insuficiencia renal: Después de una dosis subcutánea única de 0,24 mg/kg, la depuración de plerixafor en sujetos con grados diferentes de insuficiencia renal se encontró reducida y se correlacionó positivamente con el clearance de creatinina (CLCR). El área bajo la curva de concentración plasmática vs. tiempo (AUC0-24hs) de plerixafor en sujetos con insuficiencia renal leve (CLCR 51-80 mL/min), moderada (CLCR 31-50 mL/min) y severa (CLCR< 31 mL/min) fue respectivamente 7 %, 32 % y 39 % mayor que la de los sujetos sanos con función renal normal. La insuficiencia renal no incidió en la Cmáx. El análisis farmacocinético poblacional indicó un incremento en la exposición (AUC0-24hs) en pacientes con insuficiencia renal moderada y severa comparada con pacientes cuyo CLCR > 50 mL/min. Estos resultados respaldan la disminución de la dosis en un tercio en aquellos pacientes con insuficiencia renal moderada a severa (CLCR £ 50 mL/min), con el objetivo de ajustar la exposición a plerixafor con la de los pacientes con función renal normal. El análisis farmacocinético poblacional demostró que la posología basada en mg/kg ocasiona un incremento en la exposición a plerixafor (AUC0-24hs) a medida que aumenta el peso corporal; por lo tanto si CLCR es £ 50 mL/min, la dosis no debería exceder los 27 mg/día [Véase Posología y Administración]
Plerixafor es fundamentalmente eliminado a través de los riñones. La administración concomitante de plerixafor con drogas que reducen la función renal o que compiten por la secreción tubular activa, puede incrementar la concentración sérica de plerixafor o de la droga coadministrada. No ha sido evaluado el efecto de la coadministración de plerixafor con otras drogas eliminadas a través del riñón o que se conoce que afectan la función renal.
Raza: Los datos clínicos muestran que los parámetros farmacocinéticos de plerixafor en la población caucásica y afroamericana son similares. No se ha estudiado el efecto en otras razas y/o grupos étnicos.
Género: Los datos clínicos no muestran diferencias en los parámetros farmacocinéticos de plerixafor respecto al género.
Edad: Los datos clínicos no muestran diferencias en los parámetros farmacocinéticas de plerixafor respecto a la edad.
ADVERTENCIAS Y PRECAUCIONES: Movilización de células tumorales en pacientes con leucemia
Con el propósito de movilizar células madre hematopoyéticas, MOZOBIL™ podría causar también la movilización de las células leucémicas y resultar en la contaminación subsiguiente del producto de la aféresis. Por lo tanto, MOZOBIL™ no ha sido concebido para la movilización y recolección de células madre hematopoyéticas en pacientes con leucemia.
Efectos hemáticos
Leucocitosis: La administración de MOZOBIL™ conjuntamente con G-CSF aumenta los leucocitos circulantes así como también las poblaciones de células madre hematopoyéticas. Se debe vigilar el recuento de glóbulos blancos en sangre durante el uso de MOZOBIL™. Se debe realizar una evaluación clínica al administrar MOZOBIL™ a pacientes cuyo recuento de neutrófilos en sangre periférica sea superior a 50.000 células/mcL.
Trombocitopenia: Se ha observado trombocitopenia en pacientes que reciben MOZOBIL™. Se debe supervisar el recuento de plaquetas de todos los pacientes que reciben MOZOBIL™ y que luego se someten a aféresis.
Posible movilización de células tumorales: Cuando MOZOBIL™ se administra conjuntamente con G-CSF para la movilización de células madre hematopoyéticas, se puede producir la liberación de células tumorales desde la médula ósea que serían luego recolectadas en el producto de la aféresis leucocitaria.
Todavía no se ha estudiado en detalle el efecto de la reinfusión potencial de células tumorales.
Esplenomegalia y posible ruptura esplénica: Se observó aumento del peso absoluto y relativo del bazo asociado a hematopoyesis extramedular después de la administración diaria prolongada (2 o 4 semanas) de plerixafor por vía subcutánea en ratas que recibieron dosis aproximadamente 4 veces superiores a la dosis recomendada en humanos, ajustada según el área de superficie corporal. En los estudios clínicos no fue evaluado específicamente el efecto de MOZOBIL™ sobre el tamaño del bazo de los pacientes. Se debe evaluar la integridad del bazo de aquellos pacientes que reciben MOZOBIL™ conjuntamente con G-CSF y que informan dolor en el cuadrante abdominal superior izquierdo y/o dolor escapular o en los hombros.
Embarazo: Embarazo Categoría D
MOZOBIL™ puede causar daño fetal cuando es administrado en mujeres embarazadas.
Plerixafor demostró ser teratogénico en animales. No existen estudios adecuados y controlados en mujeres embarazadas utilizando MOZOBIL™. A mujeres en edad reproductiva se les debe recomendar evitar el embarazo durante el tratamiento con MOZOBIL™. Si esta droga es utilizada durante el embarazo, o si la paciente queda embarazada durante el tratamiento, la paciente deberá ser informada acerca del riesgo potencial al feto.
POSOLOGÍA Y MODO DE ADMINISTRACIÓN: Dosis y administración recomendada.
Previo a su administración, los frascos ampolla debe ser inspeccionados visualmente para determinar la presencia de material particulado y decoloración. No deben ser utilizados si se observan partículas o si la solución se encuentra decolorada. Cada frasco ampolla de MOZOBIL™ está destinado exclusivamente para un uso único. Se debe desechar todo el resto del fármaco que no se haya usado en la inyección.
Iniciar el tratamiento con MOZOBIL™ una vez que el paciente haya recibido una dosis diaria de G-CSF por 4 días. Administrar MOZOBIL™ aproximadamente 11 horas antes de iniciar la aféresis por un periodo de hasta 4 días consecutivos.
La dosis recomendada de MOZOBIL™ es de 0,24 mg/kg de peso corporal, administrada por inyección subcutánea. Utilice el peso corporal actual del paciente para calcular el volumen de MOZOBIL™ que va ser administrado. Cada frasco ampolla suministra 1,2 mL de una solución de 20 mg/mL y el volumen a administrar a los pacientes debe ser calculado mediante la siguiente ecuación:
|
= |
0,012 X peso corporal actual del paciente (en kg) |
|
volumen a administrar (en mL) |
En estudios clínicos, la dosis de MOZOBIL™ se ha calculado en base al peso corporal actual en pacientes cuyo peso era de hasta 175 % del peso corporal ideal. No se ha investigado la dosis ni el tratamiento con MOZOBIL™ en pacientes cuyo peso fuera de más del 175 % del peso corporal ideal.
Debido a que la exposición a plerixafor incrementa con el aumento de peso corporal, la dosis de plerixafor no debe exceder los 40 mg/día. [Véase Farmacología Clínica]
Medicamentos concomitantes recomendados: Administrar dosis diarias matutinas de 10 microgramos/kg de factor estimulante de colonias de granulocitos (por sus siglas en inglés, G-CSF) durante los 4 días previos a la primera administración vespertina de MOZOBIL™ y cada día antes de la aféresis. (Véase Estudios Clínicos)
Posología en insuficiencia renal: En los pacientes con insuficiencia renal moderada y severa (clearance de creatinina estimado (CLCR) = 50 mL/min), reducir la dosis de MOZOBIL™ en un tercio a 0,16 mg/kg como se indica en la Tabla 6. Si CLCR es = 50 mL/min la dosis no debe exceder 27 mg/día, ya que la posología ajustada en mg/kg resulta en una exposición incrementada de plerixafor con el incremento del peso corporal. En pacientes con insuficiencia renal moderada y severa se puede predecir una exposición sistémica similar a la de los sujetos con función renal normal si la dosis es reducida en un tercio. [Véase Farmacología Clínica]
|
Tabla 6:Posología reromendada de plerixafor en pacientes con insuficiencia renal |
|
|
Clearence de creatinina estimado (mL/ min) |
Dosis |
|
> 50 |
0,24 mg/kg/ día (no exceder 40 mg/día diarios) |
|
= 50 |
0,16 mg/kg/ día (no exceder 27 mg/día diarios) |
La siguiente fórmula (Cockroft-Gault) se puede usar para calcular el clearance de creatinina :
|
Hombres: |
|
|
Clearance de creatinina (mL/min) |
= peso (ka) X ( 1 40 - edad en años) |
|
72 X creatinina en suero (mg/dL) |
|
|
Mujeres: |
||
|
= |
Clearance de creatinina (mL/min) |
|
|
0,85 X valor calculado para hombres |
No se cuenta con información suficiente para recomendar dosis para pacientes en hemodiálisis.
SOBREDOSIS: Según datos limitados, la frecuencia de desórdenes gastrointestinales, reacciones vasovagales, hipotensión ortostática y/o síncope puede ser mayor si se utilizan dosis subcutáneas por encima de la recomendada de 0,24 mg/kg.
CARCINOGÉNESIS, MUTAGÉNESIS Y EFECTOS SOBRE LA FERTILIDAD: No se han realizado estudios de carcinogenicidad con plerixafor.
Plerixafor no fue genotóxico en el estudio in vitro de mutación bacteriana (prueba Ames en Salmonella), en la prueba in vitro de aberración cromosómica usando células de ovario de hámster chino (V79), ni tampoco después de la administración subcutánea de hasta 25 mg/kg (150 mg/m2) en la prueba in vivo de micronúcleo de médula ósea en rata.
El efecto de plerixafor sobre la fertilidad humana es desconocido. No se han llevado a cabo estudios específicamente designados como estudios toxicológicos reproductivos, para evaluar los efectos potenciales de plerixafor sobre la fertilidad masculina o femenina. Las etapas de la espermatogénesis, medidas en un estudio de toxicidad de dosis repetida de 28 días en ratas, no revelaron ninguna anormalidad que se considerara relacionada a plerixafor. No se observaron signos histopatológicos de toxicidad en los órganos reproductores masculinos o femeninos durante los estudios de toxicidad de dosis repetida de 28 días.
DESCRIPCIÓN: Plerixafor es un movilizador de células madre hematopoyéticas cuyo nombre químico es 1,1’-[1,4-fenilenbis(metileno)]-bis-1,4,8,11-tetraazaciclotetradecano. Su fórmula molecular es C28H54N8. El peso molecular de plerixafor es 502,79 g/mol. En la Figura 1 se ilustra su fórmula estructural.
Figura 1: Fórmula estructural
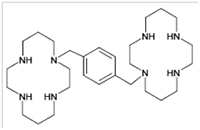
Plerixafor es un sólido higroscópico, cristalino blanco a blanco crema. Plerixafor posee un punto de fusión de 131,5°C. El coeficiente de partición 1-octanol/buffer acuoso a pH 7 es <0,1.
MOZOBIL™ (plerixafor inyectable) es una solución estéril, sin conservantes, transparente, incolora a amarillo pálido, isotónica para inyección subcutánea.
PRESENTACIÓN
Contenido y composición del envase: Cada frasco ampolla de uso único es llenado para suministrar 1,2 mL de una solución de 20 mg/mL, conteniendo 24 mg de plerixafor.
MOZOBIL™ se comercializa en un empaque de cartón conteniendo un frasco ampolla.
Manténgase fuera del alcance de los niños.
Condición de venta: Con formula médica, uso por especialistas.
MOZOBIL™ es una marca registrada de Genzyme Corporation
Revisado: Abril 2013
SANOFI-AVENTIS DE COLOMBIA S. A.
Transversal 23 No. 97-73, Pisos 8 y 9
Teléfono: 6214400, Fax: 7444237
Bogotá, D.C., Colombia

