
JEVTANA
CABAZIXATEL
Solución concentrada para infusión
Caja , 1 Vial(es) , Solución concentrada para diluir
Solución concentrada para infusión
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN
Cada frasco ampolla con 1,5 ml de SOLUCIÓN concentrada contiene cabazitaxel (como solvato acetónico) 60 mg.
Excipientes: Polisorbato 80 DF RPR-2: 1,56 g; etanol anhidro (<1,5% P/V)
Cada frasco ampolla con diluyente para la primera dilución contiene etanol 573,3 mg y agua para inyectable c.s.p. 4,5 ml.
No utilizar si los frascos no están intactos.
Tanto el frasco ampolla para inyección de JEVTANA® como el frasco ampolla de diluyente contienen un sobrellenado para compensar la pérdida de líquido durante la preparación.
Contenido del envase, un envase de JEVTANA® contiene 2 frascos ampolla
Cada frasco ampolla con JEVTANA® contiene 60 mg de Cabazitaxel en 1,5 ml de volumen nominal (volumen de llenado: 1,83 ml con 73,2 mg de Cabazitaxel). Este volumen de llenado ha sido establecido para compensar la pérdida de líquido durante la preparación de premezcla.
Este sobrellenado garantiza que después de la dilución con la totalidad del diluyente, contenido en el frasco ampolla acompañante, hay un volumen de premezcla mínimo extraíble de 6 ml, conteniendo 10 mg/ml de JEVTANA®, que corresponde al valor declarado de 60 mg/frasco ampolla.
Cada frasco ampolla con diluyente contiene 4,5 ml de volumen nominal (volumen de llenado: 5,67 ml).
Este volumen de llenado garantiza que luego del agregado del total del contenido de éste frasco ampolla con diluyente, al contenido de frasco ampolla con JEVTANA® 60 mg solución concentrada, la concentración de la solución premezcla es de 10 mg/ml.
ACCIÓN TERAPÉUTICA: Agente antineoplásico (Taxano).
Código ATC: L01CD04
INDICACIONES Y USO: JEVTANA® es un inhibidor de los microtúbulos indicado en combinación con prednisona o prednisolona para el tratamiento de pacientes con cáncer de próstata metastásico refractario a hormonas, que hayan recibido previamente un régimen de tratamiento que contenga docetaxel.
CARACTERÍSTICAS FARMACOLÓGICAS/PROPIEDADES
Mecanismo de acción: Cabazitaxel es un inhibidor de los microtúbulos. Cabazitaxel se une a la tubulina y promueve su acoplamiento a los microtúbulos y simultáneamente inhibe su desacoplamiento. Esto conduce a la estabilización de los microtúbulos, la cual da como resultado la inhibición de las funciones celulares de la mitosis y la interfase.
FARMACOCINÉTICA: Se realizó un análisis farmacocinético poblacional en 170 pacientes con tumores sólidos con dosis que oscilaron entre 10 y 30 mg/m2 semanalmente o cada tres semanas.
Absorción: En un análisis farmacocinético poblacional, después de una dosis endovenosa de cabazitaxel 25 mg/m2 cada tres semanas, la Cmáx promedio en pacientes con cáncer de próstata metastásico fue de 226 ng/ml (Coeficiente de Variación [CV] 107%) y se alcanzó al finalizar la infusión de una hora (Tmax). El Área Bajo la Curva (ABC) promedio en los pacientes con cáncer de próstata metastásico fue de 991 ng.h/ml (CV 34%).
No se observó una desviación importante en la proporcionalidad de la dosis de 10 a 30 mg/m2 en pacientes con tumores sólidos avanzados.
Distribución: El volumen de distribución (Vss) fue de 4870 L (2640 L/m2 para un paciente con un Área de Superficie Corporal (ASC) promedio de 1,84 m2) en estado estable.
In vitro, la unión de cabazitaxel a las proteínas séricas humanas fue de 89 a 92% y no fue saturable hasta 50000 ng/ml, que cubren la concentración máxima observada en ensayos clínicos. Cabazitaxel se une principalmente a la albúmina sérica humana (82%) y a las lipoproteínas (88% para HDL, 70% para LDL y 56% para VLDL). La relación entre la concentración sanguínea y la plasmática in vitro en sangre humana osciló entre 0,90 y 0,99, indicando que cabazitaxel se distribuye por igual en sangre y plasma.
Metabolismo: Cabazitaxel se metaboliza extensamente a nivel del hígado (=95%), principalmente por la isoenzima CYP3A (80% a 90%). Cabazitaxel es la principal fracción circulante en plasma humano. Fueron detectados siete metabolitos en plasma (incluyendo a los 3 metabolitos activos derivados de la O-desmetilación), donde el principal de ellos representa el 5% de la exposición a cabazitaxel.
Alrededor de 20 metabolitos de cabazitaxel se excretan por la orina y heces humanas.
Sobre la base de estudios in vitro, el riesgo potencial de inhibición por cabazitaxel, a concentraciones clínicamente relevantes, es posible respecto a los fármacos que son principalmente sustrato de CYP3A. Cabazitaxel no inhibe otras enzimas CYP. Además, cabazitaxel no indujo isoenzimas CYP (CYP1A, CYP2C, Y CYP3A) in vitro.
Estudios de interacción en humanos han demostrado que cabazitaxel (25 mg/m2, administrado como una sola infusión de 1 hora) no modificó los niveles plasmáticos de midazolam, un sustrato de prueba de CYP3A. Por lo tanto, cabazitaxel no es un inhibidor in vivo de CYP3A.
Eliminación: Después de una infusión endovenosa de una hora de [14C]-cabazitaxel 25 mg/m2, aproximadamente el 80% de la dosis administrada fue eliminada dentro de las dos semanas posteriores a la administración. Cabazitaxel se excreta principalmente a través de las heces en forma de numerosos metabolitos (76% de la dosis); mientras que la excreción renal de cabazitaxel y sus metabolitos representa el 3,7% de la dosis (2,3% como cabazitaxel no modificado en la orina).
En un análisis farmacocinético poblacional, cabazitaxel tuvo una depuración plasmática de 48,5 L/h (CV 39%; 26,4 L/h/m2 para un paciente con una mediana de ASC de 1,84 m2) en pacientes con cáncer de próstata metastásico. Luego de una infusión endovenosa de una hora, las concentraciones plasmáticas de cabazitaxel pueden describirse mediante un modelo farmacocinético tricompartimental con vidas medias a, ß y ? de 4 minutos, 2 horas y 95 horas, respectivamente.
Insuficiencia renal: Cabazitaxel se excreta mínimamente a través del riñón (2,3% de la dosis). El análisis farmacocinético poblacional realizado en 170 pacientes, que incluyó 14 pacientes con insuficiencia renal moderada (30 ml/min <CLcr <50 ml/min) y 59 pacientes con insuficiencia renal leve (50 ml/min <CLcr <80 ml/min) mostró que la insuficiencia renal leve a moderada no tuvo efectos significativos sobre la farmacocinética de cabazitaxel. Esto fue confirmado por un estudio farmacocinético comparativo especial en pacientes con cáncer sólido con función renal normal (8 pacientes), moderada (8) e insuficiencia renal grave (9), que recibieron varios ciclos de cabazitaxel en una sola infusión intravenosa de hasta 25 mg/m2.
Insuficiencia hepática: Cabazitaxel se elimina principalmente mediante el metabolismo hepático.
Un estudio realizado en 43 pacientes con cáncer e insuficiencia hepática no mostró influencia de la insuficiencia hepática leve (niveles plasmáticos de bilirrubina total > 1 a = 1,5 veces el LSN o AST > 1,5 x LSN) o moderada (niveles plasmáticos de bilirrubina total > 1,5 a = 3,0 veces el LSN) en la farmacocinética de cabazitaxel. La dosis máxima tolerada de cabazitaxel (DMT) fue de 20 y 15 mg/m2, respectivamente.
En 3 pacientes con insuficiencia hepática severa (niveles plasmáticos de bilirrubina total > 3 veces el LSN), se observó una reducción del 39% en el clearance en comparación con los pacientes con insuficiencia hepática leve, lo que indica algún efecto de la insuficiencia hepática severa sobre la farmacocinética de cabazitaxel. No se estableció la DMT de cabazitaxel en pacientes con insuficiencia hepática severa.
De acuerdo a los datos de seguridad y tolerancia, la dosis de cabazitaxel debe reducirse en pacientes con insuficiencia hepática leve (ver Uso en Poblaciones Específicas). Cabazitaxel está contraindicado en pacientes con insuficiencia hepática severa (ver Contraindicaciones).
FARMACODINAMIA: Cabazitaxel demostró actividad antitumoral contra tumores humanos avanzados implantados en ratones.
Cabazitaxel es activo en tumores sensibles a docetaxel. Además, cabazitaxel demostró actividad en los modelos de tumores insensibles a quimioterapia incluyendo a docetaxel.
CONTRAINDICACIONES
JEVTANA® no debe utilizarse en pacientes con
— Recuento de neutrófilos =1500/mm3
— Antecedentes de hipersensibilidad severa a cabazitaxel, polisorbato 80 o a cualquiera de los componentes de la fórmula.
— Compromiso hepático severo (bilirrubina total = 3 x Límite Superior Normal (LSN).
REACCIONES ADVERSAS
Las siguientes reacciones adversas serias se discuten en mayor detalle en otra sección del prospecto
— Neutropenia (ver Advertencias y precauciones).
— Reacciones de hipersensibilidad (ver Advertencias y Precauciones).
— Síntomas gastrointestinales (ver Precauciones).
— Insuficiencia renal (ver Precauciones).
Experiencia en estudios clínicos: Dado que los estudios clínicos se realizan bajo condiciones ampliamente variables, los índices de reacciones adversas observados no se pueden comparar directamente con los índices de otros estudios y pueden no reflejar los índices observados en la práctica clínica.
La seguridad de JEVTANA® en combinación con prednisona o prednisolona se evaluó en 371 pacientes con cáncer de próstata metastásico refractario a hormonas en un estudio único aleatorizado, comparado con mitoxantrona más prednisona o prednisolona.
Se informaron muertes debidas a causas diferentes a progresión de la enfermedad dentro de los 30 días posteriores a la última dosis del medicamento en estudio en 18 pacientes (5%) tratados con JEVTANA® y en 3 pacientes (<1%) tratados con mitoxantrona. Las reacciones adversas fatales más frecuentes en pacientes tratados con JEVTANA® fueron infecciones (n=5) e insuficiencia renal (n=4). La mayoría (4 ó 5 pacientes) de las reacciones adversas fatales relacionadas con infecciones ocurrieron luego de una dosis única de JEVTANA®. Otras reacciones adversas fatales en los pacientes tratados con JEVTANA® incluyeron fibrilación ventricular, hemorragia cerebral y disnea.
Las reacciones adversas más comunes (=10%) de grado 1-4 fueron anemia, leucopenia, neutropenia, trombocitopenia, diarrea, cansancio, náuseas, vómitos, estreñimiento, astenia, dolor abdominal, hematuria, dolor de espalda, anorexia, neuropatía periférica, fiebre, disnea, disgeusia, tos, artralgia y alopecía.
Las reacciones adversas más comunes (=5%) de grado 3-4 en pacientes que recibieron JEVTANA® fueron neutropenia, leucopenia, anemia, neutropenia febril, diarrea, cansancio y astenia.
Las discontinuaciones del tratamiento debidas a reacciones adversas al medicamento ocurrieron en el 18% de los pacientes que recibieron JEVTANA® y en el 8% de los pacientes que recibieron mitoxantrona. Las reacciones adversas más comunes que provocaron la discontinuación del tratamiento en el grupo de JEVTANA® fueron neutropenia e insuficiencia renal. Se informaron reducciones de dosis en el 12% de los pacientes tratados con JEVTANA® y en el 4% de los pacientes tratados con mitoxantrona. Se informaron retrasos en las dosis en el 28% de los pacientes tratados con JEVTANA® y en el 15% de los pacientes tratados con mitoxantrona.
|
Tabla 2 – Incidencia de reacciones adversas informadas* y valores hematológicos anormales en =5% de los pacientes que recibieron JEVTANA® en combinación con prednisona, y pacientes recibiendo mitoxantrona en combinación con prednisona (tasa de incidencia de al menos un 2% más alta en el grupo JEVTANA®, comparado con mitoxantrona). |
||||
|
JEVTANA® 25 mg/m2 cada 3 semanas con prednisona 10 mg diarios n = 371 |
Mitoxantrona 12 mg/m2 cada 3 semanas con prednisona 10 mg diarios n = 371 |
|||
|
Grado 1-4 |
Grado 3-4 |
Grado 1-4 |
Grado 3-4 |
|
|
Cualquier reacción adversa |
||||
|
Trastornos sanguíneos y del sistema linfático |
||||
|
Neutropenia† |
347 (93,5%) |
303 (81,7%) |
325 (87,6%) |
215 (58%) |
|
Neutropenia febril |
--- |
28 (7,5%) |
--- |
5 (1,3%) |
|
Anemia† |
361 (97,3%) |
39 (10,5%) |
302 (81,4%) |
18 (4,9%) |
|
Leucopenia† |
355 (95,7%) |
253 (68,2%) |
343 (92,5%) |
157 (42,3%) |
|
Trompocitopenia † |
176 (47,4%) |
15 (4%) |
160 (43,1%) |
6 (1,6%) |
|
Trastornos cardíacos |
||||
|
Arritmia‡ |
18 (5%) |
4 (1%) |
6 (2%) |
1 (<1%) |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
173 (46,6%) |
23 (6,2%) |
39 (10,5%) |
1 (0.3%) |
|
Náuseas |
127 (34,2%) |
7 (1,9%) |
85 (22,9%) |
1 (0,3%) |
|
Vómitos |
84 (22,6%) |
7 (1,9%) |
38 (10,2%) |
0 |
|
Estreñimiento |
76 (20,5%) |
4 (1,1%) |
57 (15,4%) |
2 (0,5%) |
|
Dolor abdominal§ |
43 (11,6%) |
7 (1,9%) |
13 (3,5%) |
0 |
|
Dispepsia¶ |
25 (6,7%) |
0 |
6 (1,6%) |
0 |
|
Dolor abdominal superior |
20 (5,4%) |
0 |
5 (1,3%) |
0 |
|
Hemorroides |
14 (3,8%) |
0 |
3 (0,8%) |
0 |
|
Reflujo gastroesofágico |
12 (3,2%) |
0 |
3 (0,8%) |
0 |
|
Trastornos generales y reacciones en el sitio de la administración |
||||
|
Cansancio |
136 (36,7%) |
18 (4,9%) |
102 (27,5%) |
11 (3,0%) |
|
Astenia |
76 (20,5%) |
17 (4,6%) |
46 (12,4%) |
9 (2,4%) |
|
Fiebre |
45 (12,1%) |
4 (1,1%) |
23 (6,2%) |
1 (0,3%) |
|
Edema periférico |
34 (9%) |
2 (<1%) |
34 (9%) |
2 (<1%) |
|
Inflamación de mucosas |
22 (5,9%) |
1 (0,3%) |
10 (2,7%) |
1 (0,3%) |
|
Dolor |
20 (5%) |
4 (1%) |
18 (5%) |
7 (2%) |
|
Infecciones e infestaciones |
||||
|
Infección del tracto urinario# |
27 (7,3%) |
4 (1,1%) |
11 (3%) |
3 (0,8%) |
|
Investigaciones |
||||
|
Disminución de peso |
32 (9%) |
0 |
28 (8%) |
1 (<1%) |
|
Trastornos del metabolismo y trastornos nutricionales |
||||
|
Anorexia |
59 (15,2%) |
3 (0,8%) |
39 (10,5%) |
3 (0,8%) |
|
Deshidratación |
18 (4,9%) |
8 (2,2%) |
10 (2,7%) |
3 (0,8%) |
|
Trastornos músculoesqueléticos y del tejido conectivo |
||||
|
Dolor de espalda |
60 (16,2%) |
14 (3,8%) |
45 (12,1%) |
11 (3,0%) |
|
Artralgias |
39 (10,5%) |
4 (1,1%) |
31 (8,4%) |
4 (1,1%) |
|
Espasmos musculares |
27 (7,3%) |
0 |
10 (2,7%) |
0 |
|
Trastornos del sistema nervioso |
||||
|
Neuropatía periférica ? |
30 (8,1%) |
2 (0,5%) |
4 (1,1%) |
1 (0,3%) |
|
Disgeusia |
41 (11,1%) |
0 |
15 (4%) |
0 |
|
Mareos |
30 (8,1%) |
0 |
21 (5,7%) |
2 (0,5%) |
|
Cefalea |
28 (7,5%) |
0 |
19 (5,1%) |
0 |
|
Neuropatía sensorial periférica |
20 (5,4%) |
1 (0,3%) |
5 (1,3%) |
0 |
|
Trastornos renales y del tracto urinario |
||||
|
Hematuria |
62 (16,7%) |
7 (1,9%) |
14 (3,8%) |
2 (0,5%) |
|
Disuria |
25 (6,7%) |
0 |
5 (1,3%) |
0 |
|
Incontinencia urinaria |
9 (2,4%) |
0 |
1 (0,3%) |
0 |
|
Insuficiencia renal aguda |
8 (2,2%) |
6 (1,6%) |
0 |
0 |
|
Trastornos respiratorios, torácicos y mediastinales |
||||
|
Disnea |
44 (11,9%) |
5 (1,3%) |
17 (4,6%) |
3 (0,8%) |
|
Tos |
40 (10,8%) |
0 |
22 (5,9%) |
0 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Alopecía |
37 (10%) |
0 |
18 (4,9%) |
0 |
|
Trastornos vasculares |
||||
|
Hipotensión |
20 (5,4%) |
2 (0,5%) |
9 (2,4%) |
1 (0,3%) |
|
Duración media del tratamiento |
6 ciclos |
4 ciclos |
||
|
*Clasificado utilizando NCI CTCAE versión 3 † Basado en valores de laboratorio, cabazitaxel: n=369, mitoxantrona: n=370. ‡Incluye fibrilación auricular, flúter auricular, taquicardia auricular, bloqueo auriculoventricular completo, bradicardia, palpitaciones, taquicardia supraventricular, taquiarritmia y taquicardia. § Incluye malestar abdominal, dolor abdominal inferior, dolor abdominal superior, sensibilidad abdominal y dolor gastrointestinal ¶ Incluye enfermedad por reflujo gastroesofágico y gastritis con reflujo # Incluye infección enterocóccica del tracto urinario e infección fúngica del tracto urinario ? Incluye neuropatía motora periférica y neuropatía sensorial periférica |
||||
Neutropenia y eventos clínicos asociados: La incidencia de neutropenia de grado =3 sobre la base de datos de laboratorio fue del 81,7%. La incidencia de grado =3 de neutropenia clínica y neutropenia febril, de reacciones adversas fue, respectivamente, 21,3% y 7,5%. La reacción adversa más común que provocó la discontinuación del tratamiento fue neutropenia (2,4%). Complicaciones neutropénicas incluyendo infecciones neutropénicas (0,5%), sepsis neutropénica (0,8%), y el shock séptico (1,1%), que en algunos casos resultó en un desenlace fatal.
Se ha demostrado que el uso de G-CSF limita la incidencia y gravedad de neutropenia (ver Preacuciones).
Trastornos cardiacos y arritmias: Todos los grados de eventos entre los trastornos cardiacos fueron más frecuentes en el grupo JEVTANA®, de los cuales 6 pacientes (1,6%) tenían grado =3 de arritmias cardiacas. La incidencia de taquicardia en el grupo JEVTANA® fue de 1,6%, ninguno de los cuales fueron de grado =3. La incidencia de fibrilación auricular fue de 1,1% en el grupo JEVTANA®.
Trastornos renales y del tracto urinario: Se obrservó insuficiencia renal en el 2,2% en todos los grados y 1,6% en los grados =3 en el grupo JEVTANA®.
Trastornos gastrointestinales: Han sido observados: Colitis, enterocolitis, gastritis, enterocolitis neutropénica. También han sido reportados: Hemorragia gastrointestinal y perforación, ileo y obstrucción intestinal.
Hematuria: Los eventos adversos de hematuria, incluyendo aquéllos que requirieron intervención médica, fueron más comunes en los pacientes tratados con JEVTANA®. La incidencia de hematuria de grado =2 fue del 6% en los pacientes tratados con JEVTANA® y del 2% en los pacientes tratados con mitoxantrona. Otros factores asociados a hematuria estuvieron bien equilibrados entre los brazos del estudio y no explican la tasa incrementada de hematuria en el brazo con JEVTANA®.
Investigaciones: La incidencia de AST/GOT, ALT/GPT y bilirrubina incrementada de grado 3-4 basados en anormalidades de laboratorio fueron, 0,9%, 1,1% y 0,6%, respectivamente.
La incidencia de anemia de grado =3 fue del 10,6%.
Se observó disminución de peso en el 8,6% y 7,5% en todos los grados, y el 0% y el 0,3% en los grados =3 en los grupos JEVTANA® y mitoxantrona, respectivamente.
Población de pacientes de edad avanzada: De los 371 pacientes tratados con JEVTANA® en el estudio del cáncer de próstata, 240 pacientes tenían 65 años o más, incluyendo 70 pacientes mayores de 75 años.
Las siguientes reacciones adversas de grado 1 - 4 se reportaron con tasas más altas del =5% en pacientes de 65 o más años de edad en comparación con pacientes más jóvenes: Cansancio (40,4% vs. 29,8%), neutropenia (24,2% vs. 17,6%), astenia (23,8% vs. 14,5%), fiebre (14,6% vs. 7,6%), mareo (10,0% vs. 4,6%), infección del tracto urinario (9,6% vs. 3,1%) y deshidratación (6,7% vs. 1,5%), respectivamente.
La incidencia de las siguientes reacciones adversas de grado 3 - 4 fue más elevada en pacientes >65 años de edad en comparación con pacientes más jóvenes: Neutropenia (86,3% vs. 73,3%), neutropenia clínica (23,8% vs. 16,8%) y neutropenia febril (8,3% vs. 6,1%) (ver Uso en Poblaciones Específicas).
INTERACCIONES MEDICAMENTOSAS: Cabazitaxel se metaboliza principalmente por el CYP3A.
La administración repetida de ketoconazol (400 mg/día), un potente inhibidor de CYP3A, resultó en un 20% de disminución en el aclaramiento de cabazitaxel, que correspondió a un incremento del 25% de la AUC (área bajo la curva). La administración concomitante de aprepitant, un inhibidor moderado de CYP3A, no tuvo efecto sobre el aclaramiento ni la exposición a cabazitaxel.
La administración repetida de rifampicina (600 mg/día), un potente inductor de CYP3A, resultó en un incremento del 21% del aclaramiento del cabazitaxel, correspondiendo a una disminución del 17% en la AUC.
Prednisona o prednisolona administradas a 10 mg diarios no afectó la farmacocinética de cabazitaxel.
In-vitro, cabazitaxel no inhibió las proteínas 1 o 2 de resistencia a múltiples fármacos (MRP1 y MRP2) ni el Transportador de Catión Orgánico (OCT1). In-vitro, cabazitaxel inhibió el transporte de la glicoproteína P (gp-P), la Proteína de Resistencia del Cáncer de Mama (BCRP, por su sigla en inglés) y los Polipéptidos de Transporte de Anión Orgánico (OATP1B3) (CCK8) a concentraciones de por lo menos 17 veces la que se observó en condiciones clínicas mientras que inhibe el transporte de OATP1B1 (Estradiol-17ß-glucuronide) en concentraciones de tan sólo 5 veces lo que se observó en la práctica clínica. Por lo tanto, el riesgo in vivo de cabazitaxel de inhibir a los sustratos de MRPs, OCT1, P-gp y OATP1B3 es poco probable a una dosis de 25 mg/m2.
El riesgo de interacción con el trasportador de OATP1B1 es, posiblemente, notable durante la duración de la infusión (1 hora) y hasta 20 minutos después del final de la infusión.
In vitro, cabazitaxel es un sustrato de la gp-P pero no un sustrato de las MRP1 y MRP2, o la BCRP.
Efecto en el electrocardiograma: En un ensayo multicéntrico, abierto, de un solo grupo, 94 pacientes evaluables con tumores sólidos recibieron cabazitaxel a una dosis de 25 mg/m2 cada 3 semanas. Evaluaciones durante el ciclo 1 el día 1 hasta las 24 horas no mostraron cambios >10 ms en el intervalo medio QTc desde el inicio. Sin embargo, un pequeño aumento en el intervalo medio QTc (p. ej., <10 ms), debido a cabazitaxel no puede ser excluido debido a limitaciones en el diseño del estudio.
ADVERTENCIAS
— Se han informado muertes por neutropenia. Se deben realizar recuentos sanguíneos frecuentes para monitorear la existencia de neutropenia en todos los pacientes que reciban JEVTANA®. No administrar JEVTANA® si los recuentos de neutrófilos son =1500 células/mm3.
— Puede ocurrir hipersensibilidad severa que puede incluir erupciones cutáneas/eritema generalizado, hipotensión y broncoespasmo. Descontinuar JEVTANA® inmediatamente si ocurren reacciones severas y administrar el tratamiento apropiado.
PRECAUCIONES
Neutropenia: Cinco pacientes experimentaron eventos adversos infecciosos fatales (sepsis o shock séptico). Todos tuvieron neutropenia grado 4 y uno tuvo neutropenia febril. La muerte de pacientes adicionales se atribuyó a neutropenia sin infección documentada.
Se puede administrar G-CSF para reducir el riesgo de complicaciones por neutropenia asociadas con el uso de JEVTANA®. La profilaxis primaria con G-CSF debe considerarse en pacientes con características clínicas de alto riesgo (edad >65 años, pobre estado funcional, episodios previos de neutropenia febril, extensas zonas de radiación previa, estado nutricional pobre u otras comorbilidades serias) que predispongan a complicaciones mayores debidas a una neutropenia prolongada. El uso terapéutico de G-CSF y profilaxis secundaria deben contemplarse en todos los pacientes considerados de alto riesgo de complicaciones por neutropenia.
La neutropenia es el evento adverso más común de JEVTANA®. El monitoreo semanal de recuento sanguíneo completo es esencial durante el ciclo 1 y antes de cada ciclo de tratamiento de allí en adelante para que la dosis pueda ajustarse, en caso de ser necesario (ver Posología/Dosificación - Modo de administración).
No se debe usar JEVTANA® en pacientes con recuento de neutrófilos =1500/mm3 (ver Contraindicaciones).
Si un paciente experimenta neutropenia febril o neutropenia prolongada (más de una semana) a pesar de la medicación apropiada (p. ej., G-CSF), la dosis de JEVTANA® debe reducirse (ver Posología/Dosificación - Modo de Administración). Los pacientes pueden reiniciar el tratamiento con JEVTANA® solamente cuando los recuentos de neutrófilos se recuperen a un nivel >1500/mm3 (ver Contraindicaciones).
Reacciones de hipersensibilidad: Todos los pacientes deben ser premedicados antes del inicio de la infusión de JEVTANA® (ver Posología/Dosificación - Modo de Administración). Los pacientes deben ser observados atentamente para detectar si presentan reacciones de hipersensibilidad, especialmente durante la primera y segunda infusión. Las reacciones de hipersensibilidad pueden ocurrir unos pocos minutos después del inicio de la infusión de JEVTANA®, por lo tanto, deben estar disponibles instalaciones y equipos para el manejo de la hipotensión y el broncoespasmo. Pueden ocurrir reacciones severas de hipersensibilidad que incluyen erupciones cutáneas/eritema generalizado, hipotensión y broncoespasmo. Las reacciones severas de hipersensibilidad requieren la discontinuación inmediata de la infusión de JEVTANA® y terapia apropiada. Los pacientes con antecedentes de reacciones severas de hipersensibilidad no deben ser medicados nuevamente con JEVTANA® (ver Contraindicaciones).
Síntomas gastrointestinales: En ocasiones, pueden ocurrir náuseas, vómitos y diarrea severa. En el estudio clínico aleatorizado ocurrió muerte relacionada con diarrea y desequilibrio electrolítico. Ante diarrea severa y desequilibrio electrolítico, se requieren medidas intensivas. Los pacientes deben ser tratados con rehidratación, medicamentos antidiarreicos o antieméticos, según sea necesario. Puede ser necesario retrasar el tratamiento o reducir la dosis si los pacientes experimentan diarrea Grado =3 (ver Posología/Dosificación - Modo de Administración). Si los pacientes presentan náuseas o vómitos, pueden ser tratados con los antieméticos comúnmente utilizados.
En pacientes tratados con cabazitaxel han sido reportados: Hemorragia gastrointestinal (GI) y perforación, ileo, colitis, inclusive con resultado fatal. Se recomienda precaución en el tratamiento de pacientes con mayor riesgo de desarrollar complicaciones gastrointestinales: Aquellos con neutropenia, los ancianos, uso concomitante de AINEs, terapia antiplaquetaria o anticoagulantes, y pacientes con historia previa de radioterapia pélvica, trastorno gastrointestinal, tales como ulceración y sangrado gastrointestinal.
Síntomas tales como dolor y sensibilidad abdominal, fiebre, constipación persistente, diarrea, con o sin neutropenia, pueden ser claras manifestaciones de toxicidad gastrointestinal seria, y deben ser evaluados y tratados rápidamente. Puede ser necesario retrasar o discontinuar el tratamiento con cabazitaxel.
Anemia: Se ha observado presencia de anemia en los pacientes que reciben cabazitaxel (ver Reacciones adversas). La hemoglobina y el hematocrito se deben revisar antes del tratamiento con cabazitaxel y si los pacientes presentan signos o síntomas de anemia o pérdida de sangre. Se recomienda precaución en pacientes con valores de hemoglobina <10 g/dl y se deben tomar las medidas adecuadas según lo indicado clínicamente.
Insuficiencia renal: En el estudio clínico aleatorizado se reportó insuficiencia renal, incluyendo cuatro casos con desenlace fatal. La mayoría de los casos ocurrieron en asociación con sepsis, deshidratación severa debido a diarrea, vómitos o uropatía obstructiva (ver Reacciones Adversas). Algunas de las muertes debidas a insuficiencia renal no tuvieron una etiología clara. Deben adoptarse medidas apropiadas para identificar las causas de insuficiencia renal y tratarlas agresivamente. La función renal debe ser monitoreada.
Arritmias cardiacas: Se han reportado arritmias cardiacas, con mayor frecuencia taquicardia y fibrilación auricular (ver Reacciones adversas).
Pacientes de edad avanzada: En el estudio clínico aleatorizado, 3 de 131 pacientes <65 años de edad (2%) y 15 de 240 =65 años de edad (6%) murieron por causas diferentes a la progresión de la enfermedad dentro de los 30 días de la última dosis de cabazitaxel. Es más probable que los pacientes =65 años de edad experimenten ciertas reacciones adversas, incluyendo neutropenia y neutropenia febril (ver Reacciones Adversas y Uso en Poblaciones Específicas).
Insuficiencia hepática: Cabazitaxel se metaboliza extensamente en el hígado.
JEVTANA® está contraindicado en pacientes con afección hepática severa (niveles plasmáticos de bilirrubina > 3 veces por encima del límite superior normal) (ver Contraindicaciones).Para pacientes con insuficiencia hepática leve (niveles plasmáticos de bilirrubina > 1 a = 1,5 veces por encima del límite superior normal o AST > 1,5 veces por encima del límite superior normal) la dosis debe ser reducida (ver Poblaciones especiales e Interacciones medicamentosas).
Interacciones medicamentosas: El metabolismo del cabazitaxel es modificado por la administración concomitante de compuestos que se sabe que son potentes inhibidores (p. ej., ketoconazol, itraconazol, claritromicina, atazanavir, indinavir, nefazodona, nelfinavir, ritonavir, saquinavir, telitromicina, vorconazol, etc.); o potentes inductores (por ejemplo, rifampicina, carbamazepina o fenitoína, etc.) de CYP3A.
La coadministración con potentes inhibidores de CYP3A debería ser evitada. Si la coadministración con inhibidores potentes de la CYP3A no puede ser evitada, debería ser considerado un monitoreo estricto de toxicidad y considerarse una eventual reducción de la dosis de cabazitaxel (ver Poblaciones especiales e Interacciones medicamentosas).
La coadministración de cabazitaxel con potentes inductores de CYP3A debería ser evitada ya que pueden disminuir la exposición al mismo (ver Poblaciones especiales e Interacciones medicamentosas).
In vitro, se ha demostrado que cabazitaxel también inhibe las proteínas de transporte del anión orgánico transportador de polipéptidos OATP1B1. El riesgo de interacción con sustratos de OATP1B1 (p. ej., estatinas, valsartán, repaglinida) es posible sobretodo durante la duración de la infusión (1 hora) y hasta 20 minutos después del final de la infusión, y puede conducir a un aumento de la exposición de sustratos de OATP1B1.
Prednisona o prednisolona administradas a 10 mg diarios no afectaron la farmacocinética del cabazitaxel.
La administración de vacunas vivas o vivas atenuadas, en pacientes inmunocomprometidos por agentes quimioterapéuticos pueden dar lugar a infecciones graves o mortales. La vacunación con una vacuna viva atenuada se debe evitar en los pacientes tratados con cabazitaxel. Las vacunas muertas o inactivadas se pueden administrar; sin embargo, la respuesta a tales vacunas puede verse disminuida.
• Uso en poblaciones específicas
Embarazo: Debido a la potencial exposición vía líquido seminal, los hombres con parejas en edad fértil deben utilizar un método anticonceptivo confiable durante todo el tratamiento y se recomienda continuar con éste hasta 6 meses después de la última dosis de JEVTANA®.
JEVTANA® puede provocar daño fetal cuando se administra a mujeres embarazadas. No existen estudios adecuados y bien controlados de JEVTANA® en mujeres embarazadas.
Estudios no-clínicos en ratas y conejos han mostrado que cabazitaxel es embriotóxico, fetotóxico y abortivo con exposiciones significativamente menores que aquellas recomendadas como dosis en humanos. El cabazitaxel cruza la barrera placentaria. JEVTANA® no está recomendado durante el embarazo. En ratas embarazadas, en el día 17 de la gestación, cabazitaxel mostró atravesar la barrera placentaria dentro de las 24 horas de la administración endovenosa de una dosis única de 0,08 mg/kg (aproximadamente 0,02 veces la dosis máxima recomendada en humanos – DMRH).
Cabazitaxel, administrado una vez por día a ratas hembra durante la organogénesis a una dosis de 0,16 mg/kg/día (aproximadamente 0,02-0,06 veces la Cmáx de la dosis recomendada en humanos en pacientes con cáncer) ocasionó toxicidad materna y embriofetal consistente en aumento de la pérdida postimplantación, embrioletalidad y muertes fetales. Se observó una disminución de la media del peso de los fetos al nacer asociada con retrasos en la osificación esquelética a dosis >0,08 mg/kg (aproximadamente 0,02 veces la Cmáx a la DMRH).
La exposición in utero a cabazitaxel no dio como resultado anormalidades fetales en ratas o conejos a niveles de exposición significativamente inferiores a las exposiciones esperadas en humanos.
Si se utiliza este medicamento durante el embarazo o si la paciente queda embarazada mientras estuviera recibiendo el mismo, se debe informar a la paciente sobre los potenciales riesgos para el feto. Se debe aconsejar a las mujeres con potencial reproductivo que eviten quedar embarazadas mientras estén recibiendo JEVTANA®.
Embarazo: Categoría D.
Madres en periodo de lactancia: Cabazitaxel o los metabolitos de cabazitaxel se excretan a través de la leche materna de animales lactantes. No se sabe si el fármaco se excreta en la lecha humana. Dentro de las 2 horas siguientes a la administración endovenosa única de cabazitaxel a ratas lactantes a una dosis de 0,08 mg/kg (aproximadamente 0,02 veces la dosis máxima recomendada en humanos), se detectó radioactividad relacionada con cabazitaxel en los estómagos de las crías lactantes. Esto fue detectable hasta 24 horas después de la dosis. Se calculó que aproximadamente el 1,5% de la dosis administrada a la madre estaba presente en la leche materna. Dado que muchos fármacos son excretados en la lecha humana y debido a las potenciales reacciones adversas serias en bebés lactantes, por JEVTANA®, debe adoptarse una decisión con respecto a discontinuar la lactancia o discontinuar el medicamento, teniendo en cuenta la importancia de éste para la madre. JEVTANA® no debe ser usado durante la lactancia.
Uso pediátrico: No se han establecido la seguridad y la eficacia de JEVTANA® en pacientes pediátricos.
Uso geriátrico: Basado en análisis farmacocinéticos poblacionales, no se observó diferencia significativa en la farmacocinética de cabazitaxel entre pacientes <65 años (n=100) y mayores (n=70).
De los 371 pacientes con cáncer de próstata tratados con JEVTANA® cada tres semanas más prednisona o prednisolona, 240 pacientes (64,7%) tenían 65 y más años de edad, mientras que 70 pacientes (18,9%) tenían 75 años de edad y más. No se observaron diferencias globales en la efectividad entre los pacientes >65 años de edad y los pacientes más jóvenes. Los pacientes de edad avanzada (>65 de edad) pueden tener más probabilidades de experimentar ciertas reacciones adversas. La incidencia de neutropenia, cansancio, astenia, fiebre, mareos, infección del tracto urinario y deshidratación se presentó en porcentajes =5% más elevadas en pacientes que tenían 65 años de edad o más, comparados con los pacientes más jóvenes (ver Reacciones Adversas).
Insuficiencia renal: No se realizó ningún estudio con JEVTANA® en insuficiencia renal. En un análisis farmacocinético poblacional, no se observó diferencia significativa en la depuración en pacientes con compromiso leve de la depuración de creatinina (50 ml/min =CLcr <80 ml/min). Existe limitada información en pacientes con insuficiencia renal moderada (30 ml/min =CLcr <50 ml/min) y en pacientes con insuficiencia renal severa (CLcr <30 ml/min) o enfermedad renal en estadío terminal (ver Características farmacológicas/Propiedades). En consecuencia se debe actuar con precaución en estos pacientes y los mismos deben ser cuidadosamente monitoreados durante el tratamiento. (Ver Posología/dosificación - Modo de administración / Poblaciones especiales).
Insuficiencia hepática: Dado que cabazitaxel se metaboliza extensamente a nivel del hígado, es probable que la insuficiencia hepática aumente las concentraciones de cabazitaxel. (Ver Precauciones). Los pacientes con compromiso de la función hepática severa no deben recibir JEVTANA® (ver Contraindicaciones).
Uso concomitante de fármacos: Debe evitarse el uso concomitante con fármacos que son potentes inductores de CYP3A o potentes inhibidores de CYP3A (ver Interacciones e Interacciones medicamentosas). Sin embargo, si los pacientes requieren la coadministración de un potente inhibidor de CYP3A, una reducción del 25% de la dosis de cabazitaxel debería considerarse.
POBLACIONES ESPECIALES
Uso pediátrico: No se han establecido la seguridad y la eficacia de JEVTANA® en pacientes pediátricos.
Uso geriátrico: No se recomienda modificar la dosis de JEVTANA® en pacientes geriátricos (ver Precauciones y Reacciones adversas).
Pacientes con insuficiencia hepática
Cabazitaxel se metaboliza extensamente en el hígado.
— Los pacientes con afección hepática leve (niveles plasmáticos de bilirrubina total > 1 a = 1,5 veces el Límite Superior Normal (LSN) o AST > 1,5 veces el LSN), deben reducir la dosis de cabazitaxel a 20 mg/m2. La administración de cabazitaxel a pacientes con afección hepática leve debe realizarse con precaución y mediante un estrecho monitoreo de la seguridad.
— Están disponibles datos limitados de eficacia para cabazitaxel a 15 mg/m2, la dosis máxima tolerada en pacientes con insuficiencia hepática moderada (niveles plasmáticos de bilirrubina total > 1,5 a = 3,0 veces el LSN), para recomendar esta dosis a esta población (ver Uso en Poblaciones Específicas).
— JEVTANA® no debe administrarse a pacientes con insuficiencia hepática severa (niveles plasmáticos de bilirrubina total > 3 veces el LSN) (ver Uso en Poblaciones Específicas).
Pacientes con insuficiencia renal
JEVTANA® es mínimamente excretado a través del riñon. No se requiere un ajuste en la dosis en pacientes con insuficiencia renal que no requieren hemodiálisis. Los pacientes que presentan insuficiencia renal en etapa terminal (CLCR < 15 mL/min/1,73 m2), por su condición y por la escasa cantidad de información disponible, deben ser tratados con precaución y monitoreados cuidadosamente durante el tratamiento (ver Uso en Poblaciones Específicas).
Uso concomitante de fármacos
Debe evitarse el uso concomitante con fármacos que son potentes inductores de CYP3A o potentes inhibidores de CYP3A (ver Interacciones e Interacciones medicamentosas). Sin embargo, si los pacientes requieren la coadministración de un potente inhibidor de CYP3A, una reducción del 25% de la dosis de cabazitaxel debería considerarse.
Premedicación
Se recomienda la premedicación previa al tratamiento (ver Posología/Dosificación – Modo de administración).
Premedicar por lo menos 30 minutos antes de cada dosis de JEVTANA® con los siguientes medicamentos por vía endovenosa para reducir el riesgo y/o la severidad de la hipersensibilidad
— Antihistamínico (dexclorfeniramina 5 mg o difenhidramina 25 mg o antihistamínico equivalente).
— Corticosteroide (dexametasona 8 mg o esteroide equivalente).
— Antagonista de H2 (ranitidina 50 mg o antagonista de H2 equivalente) (ver Precauciones).
Se recomienda profilaxis antiemética y puede administrarse por vía oral o endovenosa según sea necesario.
• Precauciones de administración
— Requiere 2 diluciones.
— Administrar por infusión IV sólo después de la segunda dilución.
JEVTANA® es un medicamento citotóxico anticancerígeno, por lo tanto deben tomarse todas las precauciones correspondientes a este grupo de fármacos, al manipular y preparar soluciones de JEVTANA®, teniendo en cuenta el uso de dispositivos para contención, equipos de protección personal (por ejemplo guantes) y procedimientos de preparación. Por favor remitirse a Manipulación y Descarte.
En caso de que la inyección de JEVTANA® con la primera dilución o con la segunda dilución (final) entrara en contacto con la piel, lavar inmediatamente y por completo con abundante agua y jabón. En caso de que la inyección de JEVTANA® con la primera dilución o con la segunda dilución (final) para infusión entrara en contacto con las mucosas, lavar inmediatamente y por completo con agua.
JEVTANA® debe ser preparado y administrado por personal entrenado debidamente calificado en el manejo de agentes citotóxicos. Las mujeres embarazadas, no deben manipular este medicamento.
• Instrucciones para la preparación
No utilizar envases de infusión de PVC ni equipos de infusión de poliuretano para la preparación y administración de la solución para infusión de JEVTANA®.
Como la solución para infusión está sobresaturada, puede cristalizar con el tiempo. En ese caso, la solución no debe ser utilizada y debe ser descartada.
— Pasos para la preparación
Leer esta sección COMPLETA y atentamente antes de mezclar y diluir. JEVTANA® requiere dos diluciones antes de la administración. Por favor seguir las instrucciones de preparación que se suministran más abajo.
Nota: Tanto el frasco ampolla de inyección de JEVTANA® (volumen de llenado: 73,2 mg de cabazitaxel/1,83 ml) como el de diluyente (Volumen de llenado: 5,67 ml), contienen un sobrellenado para compensar la pérdida de líquido durante la preparación. Este sobrellenado asegura que luego de la dilución con el contenido COMPLETO del diluyente acompañante, haya una solución inicial diluida que contenga 10 mg/ml de JEVTANA®.
Debe realizarse el siguiente proceso de dilución en dos pasos bajo condiciones asépticas para preparar la solución para infusión.
Paso 1: PRIMERA DILUCIÓN
Paso 1.1
Examinar el frasco ampolla de solución concentrada de JEVTANA® 60 mg/1,5 ml y el del diluyente provisto. La solución concentrada debe ser clara.
Paso 1.2
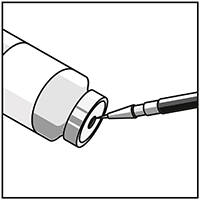
Utilizando una jeringa equipada con aguja, retirar asépticamente el contenido COMPLETO de diluyente provisto, invirtiendo parcialmente el vial.
Paso 1.3
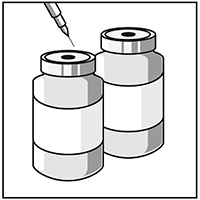
Inyectar el contenido completo en el frasco ampolla de JEVTANA® correspondiente.
Para limitar, al máximo posible, la aparición de espuma cuando se inyecta el diluyente, dirigir la aguja hacia la pared interior del frasco ampolla con solución concentrada e inyectar lentamente.
Una vez reconstituída, la solución resultante contiene 10 mg/ml de JEVTANA®
Paso 1.4
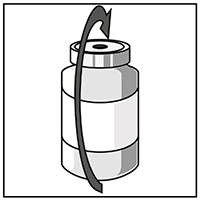
Retirar la jeringa y la aguja y mezclar manualmente y suavemente con inversiones repetidas, hasta obtener una solución clara y homogénea. Esto podría tardar 45 segundos aproximadamente.
Paso 1.5
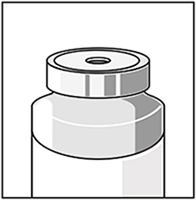
Dejar la solución en reposo durante 5 minutos aproximadamente y verificar que la solución sea homogénea y clara.
Es normal que la espuma persista luego de este periodo de tiempo.
Esta mezcla concentrado-diluyente resultante contiene 10 mg/ml de JEVTANA® (al menos 6 ml de volumen extraíble). La segunda dilución debe realizarse inmediatamente (dentro de 1 hora) como se detalla en el Paso 2.
Puede ser necesario más de un vial de la solución diluída inicial para administrar la dosis prescripta.
Por ejemplo, una dosis de 45 mg de JEVTANA®, requeriría 4,5 ml de la mezcla concentrado-diluyente preparada siguiendo el Paso 1.
Paso 2: Segunda dilución (final) para infusión
Paso 2.1
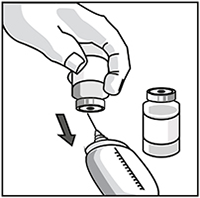
Retirar asépticamente la cantidad requerida de la solución inicial diluída de JEVTANA® (10 mg/ml de JEVTANA®), con una jeringa graduada y equipada con aguja.
Paso 2.2
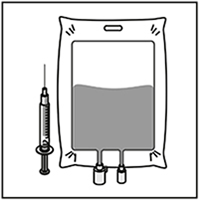
Inyectar en un recipiente estéril libre de PVC y/o poliuretanos, ya sea con solución de glucosa al 5% o solución de cloruro de sodio al 0,9% para infusión. La concentración de la solución para infusión debe estar entre 0,10 mg/ml y 0,26 mg/ml.
Dado que la espuma puede persistir en la pared del frasco ampolla de esta solución, siguiendo la preparación descrita en el Paso 1, es preferible colocar la aguja de la jeringa en el medio al extraer.
Paso 2.3
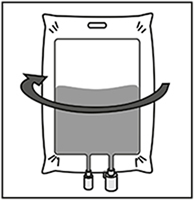
Retirar la jeringa y mezclar el contenido de la bolsa de infusión o el frasco manualmente con movimientos oscilantes.
Paso 2.4
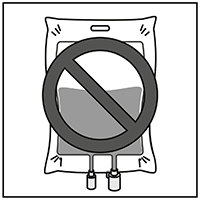
Como con todos los productos para uso parenteral, la solución para infusión resultante debe inspeccionarse visualmente antes de su uso. Si la solución contiene precipitado, debe ser descartada.
Administración: La solución final de JEVTANA® para infusión debe administrarse vía endovenosa como una infusión de 1 hora a temperatura ambiente.
Usar un filtro en línea de 0,22 micrómetros de tamaño nominal de poro durante la administración.
La solución final de JEVTANA® para infusión debe utilizarse inmediatamente. Sin embargo, el tiempo de almacenamiento durante el uso puede ser mayor bajo condiciones específicas, es decir 8 horas bajo condición ambiente (incluyendo la infusión de una hora) o por un total de 24 si se refrigera (incluyendo la infusión de una hora) (ver Posología/Dosificación – Modo de administración).
JEVTANA® no debe mezclarse con ningún otro medicamento.
POSOLOGÍA/DOSIFICACIÓN - MODO DE ADMINISTRACIÓN
Información general sobre la dosis
— La dosificación individual de JEVTANA® debe basarse en el cálculo del ASC y es de 25 mg/m2, administrados como una infusión endovenosa de una hora, cada tres semanas en combinación con prednisona o prednisolona oral 10 mg administrados diariamente durante el tratamiento con JEVTANA®.
— Se recomienda premedicación antes del tratamiento (ver Posología/Dosificación - Modo de Administración).
— JEVTANA® debe administrarse bajo supervisión de un médico experimentado en el uso de medicamentos antineoplásicos. El manejo apropiado de las complicaciones, es posible sólo cuando estén disponibles instalaciones de diagnóstico y tratamiento adecuadas.
— El frasco ampolla de uso único para la inyección de JEVTANA® requiere dos diluciones antes de ser administrada (ver Posología/Dosificación - Modo de Administración).
— No utilizar envases de infusión de PVC ni equipos de infusión de poliuretano para la preparación y administración de la solución de infusión de JEVTANA® (ver Posología/Dosificación - Modo de Administración).
— Tanto el frasco ampolla para inyección de JEVTANA® como el frasco ampolla de diluyente contienen un sobrellenado para compensar la pérdida de líquido durante la preparación.
Modificaciones de las dosis
La dosis de JEVTANA® debe reducirse a 20 mg/m2 si el paciente experimenta las siguientes reacciones adversas.
|
Tabla 1: Modificaciones de dosis recomendadas por reacciones adversas en pacientes tratados con JEVTANA® |
|
|
reacciones adversas |
Modificación de las dosis |
|
Neutropenia de grado =3 prolongada (más de una semana) a pesar de una medicación apropiada incluyendo G-CSF |
Retrasar el tratamiento hasta que el recuento de neutrófilos sea >1500 células/mm3, luego reducir la dosis de JEVTANA® a 20 mg/m2. Usar G-CSF para profilaxis secundaria. |
|
Neutropenia febril o infección neutropénica |
Retrasar el tratamiento hasta mejoría o resolución y hasta que el recuento de neutrófilos sea >1500 células/mm3, luego reducir la dosis de JEVTANA® a 20 mg/m2. Usar G-CSF para profilaxis secundaria. |
|
Diarrea de grado =3 o diarrea persistente a pesar de la medicación apropiada, reemplazo de líquidos y electrolitos. |
Retrasar el tratamiento hasta mejoría o resolución, luego reducir la dosis de JEVTANA® a 20 mg/m2. |
|
Neuropatía periférica de grado >2 |
Retrasar el tratamiento hasta mejoría, y luego considerar una reducción de dosis |
Discontinuar el tratamiento con JEVTANA® si un paciente continúa experimentando cualquiera de éstas cuatro reacciones con 20 mg/m2.
SOBREDOSIFICACIÓN: No se conoce ningún antídoto para la sobredosis de JEVTANA®. Las complicaciones anticipadas de la sobredosis incluyen la exacerbación de las reacciones adversas tales como supresión de la médula ósea y trastornos gastrointestinales. En caso de sobredosis, el paciente debe permanecer en una unidad especializada donde puedan controlarse muy de cerca los signos vitales, la bioquímica y las funciones particulares. Los pacientes deben recibir tratamiento con G-CSF tan pronto como sea posible luego de descubrirse la sobredosis. Deben adoptarse otras medidas sintomáticas adecuadas, según sea necesario.
ANTE LA EVENTUALIDAD DE UNA SOBREDOSIFICACIÓN CONCURRIR AL HOSPITAL MÁS CERCANO O COMUNICARSE CON LOS CENTROS DE TOXICOLCOGÍA (VER AL FINAL DEL PROSPECTO).
• Información para asesoramiento del paciente
— Educar a los pacientes sobre el riesgo de una potencial hipersensibilidad asociada con JEVTANA®. Confirmar que los pacientes no tengan antecedentes de reacciones de hipersensibilidad severa a cabazitaxel o a otros medicamentos con polisorbato 80 en su formulación. Instruir a los pacientes para que informen de manera inmediata los signos de una reacción de hipersensibilidad.
— Explicar la importancia de los recuentos de rutina de células sanguíneas. Instruir a los pacientes para que controlen su temperatura en forma frecuente y que informen de inmediato cualquier episodio de fiebre al oncólogo a cargo del tratamiento.
— Explicar que es importante tomar la prednisona o prednisolona oral tal como está prescrita. Instruir a los pacientes que informen si no cumplieron con el régimen oral de corticosteroides.
— Explicar a los pacientes que se han asociado con la exposición a cabazitaxel infecciones fatales, deshidratación e insuficiencia renal. Los pacientes deben informar de inmediato fiebre, vómitos o diarrea significativos, disminución de la producción de orina y hematuria al oncólogo a cargo del tratamiento.
— Informar a los pacientes sobre el riesgo de interacciones medicamentosas y sobre la importancia de suministrar una lista de medicamentos de venta bajo receta y de venta libre al oncólogo a cargo del tratamiento (ver Interacciones Medicamentosas).
— Informar a los pacientes de edad avanzada que ciertos efectos colaterales pueden ser más frecuentes o severos.
Última Revisión: CCDS V5_JEVTANA _sav005/Ene15
SANOFI-AVENTIS DE COLOMBIA S. A.
Transversal 23 No. 97-73, Pisos 8 y 9
Teléfono: 6214400, Fax: 7444237
Bogotá, D.C., Colombia

