
HECTOROL
DOXERCALCIFEROL
Cápsula blanda
Frasco , 50 Cápsulas , 0.5 mcg
Frasco , 50 Cápsulas , 2.5 mcg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN - FÓRMULA CUALICUANTITATIVA
HECTOROL® cápsulas blandas 2,5 mcg
Cada CÁPSULA BLANDA contiene: Doxercalciferol 2,5 mcg, triglicérido de aceite de coco fraccionado, gelatina, glicerina, dióxido de titanio, etanol, butilhidroxianisol, óxido de hierro amarillo.
HECTOROL® cápsulas blandas 0,5 mcg
Cada CÁPSULA BLANDA contiene: Doxercalciferol 0,5 mcg, triglicérido de aceite de coco fraccionado, gelatina, glicerina, dióxido de titanio, etanol, óxido de hierro amarillo, butilhidroxianisol, Rojo FD&C Nº 40.
INDICACIONES Y USO
Pacientes en diálisis: HECTOROL® está indicado para el tratamiento del hiperparatiroidismo secundario en pacientes con insuficiencia renal crónica en diálisis.
Pacientes en prediálisis: HECTOROL® está indicado para el tratamiento del hiperparatiroidismo secundario en pacientes con insuficiencia renal crónica de estadío 3 o estadío 4.
FORMA FARMACÉUTICA: Cápsulas blandas.
FARMACOCINÉTICA Y METABOLISMO
El doxercalciferol se absorbe del tracto gastrointestinal y la CYP 27 hepática lo activa para formar la 1a,25-(OH)2D2 (metabolito principal) y la 1a,24-dihidroxivitamina D2 (metabolito minoritario). La activación del doxercalciferol no requiere la participación de los riñones.
En voluntarios sanos los niveles sanguíneos máximos de 1a,25-(OH)2D2, el metabolito principal del doxercalciferol, se alcanzan a las 11-12 horas de la administración de dosis orales repetidas de 5 a 15 mcg de HECTOROL® , y el promedio de vida media de eliminación de la 1a,25-(OH)2D2 es de aproximadamente 32 a 37 horas, con un rango de hasta 96 horas.
El promedio de vida media de eliminación en los pacientes con insuficiencia renal terminal (IRT) en diálisis parece ser similar. La hemodiálisis causa un aumento temporal de las concentraciones medias de 1a,25-(OH)2D2, presumiblemente debido a la contracción del volumen. La 1a,25-(OH)2D2 no se elimina de la sangre durante la hemodiálisis.
Estudios clínicos
Diálisis: Se evaluaron la seguridad y la eficacia de HECTOROL® en dos estudios clínicos doble ciego, controlados con placebo, multicéntricos (Estudio A y Estudio B) en un total de 138 pacientes con insuficiencia renal crónica en hemodiálisis (IRC de estadío 5). En el Estudio A los pacientes tenían un promedio de edad de 52 años (rango: 22-75); el 55% eran hombres; el 58% eran de raza negra, el 31% caucásicos y el 11% hispanos; y habían estado en hemodiálisis durante un promedio de 53 meses. En el Estudio B los pacientes tenían un promedio de edad de 52 años (rango: 27-75); el 45% eran hombres; el 99% eran de raza negra y el 1% caucásicos; y habían estado en hemodiálisis durante un promedio de 56 meses. Luego de la distribución aleatoria en dos grupos, los pacientes que cumplieron los requisitos de inclusión fueron sometidos a un periodo de lavado de 8 semanas, durante el cual ninguno de los grupos recibió derivados de la vitamina D. A continuación, todos los pacientes recibieron HECTOROL® en la modalidad a rótulo abierto durante 16 semanas seguidas de un periodo doble ciego de 8 semanas durante el cual los pacientes recibieron HECTOROL® o placebo. La dosis inicial de HECTOROL® durante la fase a rótulo abierto fue de 10 mcg después de cada sesión de diálisis (3 veces a la semana) para un total de 30 mcg/semana. El investigador ajustó la dosis de HECTOROL® según fuera necesario con la intención de conseguir que los niveles de hormona paratiroidea intacta (PTHi) estuvieran dentro del rango buscado de 150 a 300 pg/mL. Se puso un límite máximo de dosis de 20 mcg después de cada sesión de diálisis (60 mcg/semana). Si en algún momento durante el ensayo la concentración de PTHi descendía por debajo de 150 pg/mL se suspendía HECTOROL® inmediatamente y a la semana siguiente se retomaba la administración con una dosis menor.
Resultados: De los 138 pacientes tratados con HECTOROL® durante la fase a rótulo abierto de 16 semanas, 106 alcanzaron niveles de PTHi = 300 pg/mL. De estos pacientes, 94 presentaron niveles plasmáticos de PTHi = 300 pg/mL en al menos tres ocasiones. Durante la fase a rótulo abierto de su participación en el estudio, 87 pacientes presentaron niveles plasmáticos de PTHi <150 pg/mL en al menos una ocasión.
En el Estudio A, las dosis semanales medias durante el periodo a rótulo abierto de 16 semanas variaron de 14,8 mcg a 28,7 mcg. En el Estudio B, las dosis semanales medias durante el periodo a rótulo abierto de 16 semanas variaron de 19,2 mcg a 28,0 mcg.
Las disminuciones de los valores plasmáticos de PTHi en relación a los valores basales se calcularon utilizando como valor basal el promedio de los últimos tres valores obtenidos durante la fase de lavado de 8 semanas y se muestra en la tabla siguiente:
|
PTHi (pg/mL) medias ± d.e. (n*) Valor de p respecto del valor basal Valor de p respecto del placebo |
|||
|
HECTOROL® |
Placebo |
||
|
Estudio A |
Valor basal |
797,2 ± 443,8 (30) no corresponde, 0,97 |
847,1 ± 765,5 (32) |
|
Semana 16 (a rótulo abierto) |
384,3 ± 397,8 (24) < 0,001 0,72 |
526,5 ± 872,2 (29) < 0,001 |
|
|
Semana 24 (doble ciego) |
404,4 ± 262,9 (21) < 0,001 0,008 |
672,6 ± 356,9 (24) 0,70 |
|
|
Estudio B |
Valor basal |
973,9 ± 567,0 (41) no corresponde, 0,81 |
990,4 ± 488,3 (35) |
|
Semana 16 (a rótulo abierto) |
476,1 ± 444,5 (37) < 0,001 0,91 |
485,9 ± 443,4 (32) < 0,001 |
|
|
Semana 24 (doble ciego) |
459,8 ± 443,0 (35) < 0,001 < 0,001 |
871,9 ± 623,6 (30) < 0,065 |
|
|
* todos los sujetos; se trasladó el último valor al momento del retiro del estudio |
|||
En ambos estudios, los niveles de PTHi aumentaron progresiva y significativamente en el 65,9% de los pacientes durante el periodo de lavado de 8 semanas (control) durante el cual no se administraron derivados de la vitamina D. Por el contrario, en más del 93,5% de los 138 pacientes tratados, el tratamiento con HECTOROL® resultó en una reducción estadísticamente significativa de los valores medios de los niveles de la PTHi (respecto del valor basal) durante el periodo de tratamiento a rótulo abierto de 16 semanas. Durante el periodo doble ciego (semanas 17 a 24), se mantuvo la reducción de los niveles medios de PTHi en el grupo HECTOROL®, en comparación con un retorno a niveles cercanos a los basales en el grupo placebo.
En los ensayos clínicos, los valores de la PTHi variaron ampliamente de paciente a paciente y de semana en semana en pacientes individuales. La tabla siguiente muestra la cantidad de pacientes de cada grupo que alcanzaron y mantuvieron niveles de PTHi por debajo de 300 pg/mL durante las fases a rótulo abierto y doble ciego. De 138 pacientes, 74 (53,6%) presentaron durante las semanas 14-16 niveles plasmáticos de PTHi dentro del rango buscado (150-300 pg/mL).
|
Número de veces que tuvieron TPI = 300 pg/mL |
|||||||
|
1 |
2 |
= 3 |
|||||
|
HECTOROL® |
Placebo |
HECTOROL® |
Placebo |
HECTOROL® |
Placebo |
||
|
Estudio A |
Semanas 1-16 (a rótulo abierto) |
2/30 |
2/32 |
0/30 |
0/32 |
22/30 |
23/32 |
|
Semanas 17-24 (doble ciego) |
0/24 |
9/29 |
3/24 |
1/29 |
17/24 |
5/29 |
|
|
Estudio B |
Semanas 1-16 (a rótulo abierto) |
2/41 |
4/35 |
1/41 |
0/35 |
29/41 |
21/35 |
|
Semanas 17-24 (doble ciego) |
2/37 |
6/32 |
1/37 |
4/32 |
26/37 |
4/32 |
|
Durante la fase doble ciego de 8 semanas, la cantidad de pacientes tratados con HECTOROL® que lograron alcanzar el rango deseado de valores de PTHi y mantenerse en dicho rango fue mayor que con placebo.
Prediálisis: La seguridad y la eficacia de HECTOROL® se evaluaron en dos estudios clínicos en 55 pacientes con insuficiencia renal crónica de estadío 3 o estadío 4. El 82% de los pacientes eran hombres; el promedio de edad fue de 64,6 años; el 51% eran caucásicos, el 40% de raza negra; y el valor sérico medio de PTHi al inicio fue de 194,6 pg/mL. Aunque no se evaluaron los valores basales de 25-(OH) vitamina D, las evaluaciones retrospectivas sobre suero almacenado revelaron que la media ± DE de la 25-(OH) vitamina D sérica era de 18,5 ± 8,1 ng/mL (rango: <5 a 54 ng/mL) en la población en estudio.
Luego de la distribución aleatoria en dos grupos, los pacientes que cumplieron los requisitos de inclusión fueron sometidos a un periodo de lavado de 8 semanas durante el cual ninguno de los grupos recibió derivados de la vitamina D. A continuación, un grupo recibió HECTOROL® y el otro placebo durante un periodo doble ciego de 24 semanas. La dosis inicial de HECTOROL® fue de 1 mcg por día. El investigador ajustó la dosis según fuera necesario con la intención de conseguir que los niveles de hormona paratiroidea intacta (PTHi) se redujeran hasta el objetivo buscado de ³ 30% por debajo de los valores basales posteriores al periodo de lavado.
Se puso un límite máximo de dosis de 3,5 mcg/día. Si en algún momento durante el estudio la PTHi descendía por debajo de 15 pg/mL, se suspendía HECTOROL® inmediatamente y a la semana siguiente se retomaba la administración a una dosis menor.
Resultados: Las disminuciones en el promedio de la PTHi plasmática en relación a los valores basales se calcularon utilizando como valor basal el promedio de los 2 últimos valores obtenidos durante la fase de lavado de 8 semanas. En los análisis de los datos combinados de los dos estudios, los niveles de PTHi disminuyeron en comparación con los basales en un promedio de 101,4 pg/mL en el grupo HECTOROL® y de 4,4 pg/mL en el grupo placebo (p< 0,001). En cada estudio, las reducciones de la PTHi que se observaron fueron mayores con HECTOROL® que con el placebo. De los 27 sujetos del grupo HECTOROL®, 20 (74%) alcanzaron una supresión media de la PTHi plasmática de ³ 30% con respecto al nivel basal, para las últimas cuatro semanas de tratamiento, en tanto que 2 de los 28 sujetos (7%) tratados con placebo alcanzaron este grado de supresión de la PTHi.
En los pacientes tratados con HECTOROL® las reducciones en la PTHi plasmática se asociaron con una reducción en suero de la fosfatasa alcalina específica del hueso.
CONTRAINDICACIONES: HECTOROL® no debe administrarse a pacientes con tendencia a la hipercalcemia o evidencia actual de toxicidad por vitamina D.
REACCIONES ADVERSAS: Diálisis: Se evaluó HECTOROL® en lo que respecta a la seguridad en estudios clínicos con 165 pacientes con insuficiencia renal crónica en hemodiálisis. En dos estudios multicéntricos, doble ciego, controlados por placebo, se interrumpió la terapia como consecuencia de algún evento adverso en el 2,9% de los 138 pacientes tratados con HECTOROL® durante cuatro a seis meses (dosis ajustada para alcanzar los niveles deseados de PTHi, véase Farmacología clínica/Estudios clínicos) y en el 3,3% de 61 pacientes tratados con placebo durante dos meses. Los eventos adversos que tuvieron lugar en el grupo HECTOROL® con una frecuencia de 2% o mayor y más frecuentemente que en el grupo placebo se presentan en la siguiente tabla:
|
Eventos adversos informados por = 2% de los pacientes tratados con HECTOROL® y más frecuentemente que en el grupo placebo durante la fase doble ciego de dos estudios clínicos |
||
|
Evento adverso |
HECTOROL® (n=61) % |
Placebo (n=61) % |
|
Cuerpo en general |
||
|
Absceso |
3,3 |
0,0 |
|
Cefalea |
27,9 |
18,0 |
|
Malestar |
27,9 |
19,7 |
|
Sistema cardiovascular |
||
|
Bradicardia |
6,6 |
4,9 |
|
Sistema digestivo |
||
|
Anorexia |
4,9 |
3,3 |
|
Estreñimiento |
3,3 |
3,3 |
|
Dispepsia |
4,9 |
1,6 |
|
Náuseas/Vómitos |
21,3 |
19,7 |
|
Sistema músculoesquelético |
||
|
Artralgia |
4,9 |
0,0 |
|
Metabólico y nutricional |
||
|
Edema |
34,4 |
21,3 |
|
Aumento de peso |
4,9 |
0,0 |
|
Sistema nervioso |
||
|
Mareos |
11,5 |
9,8 |
|
Trastornos del sueño |
3,3 |
0,0 |
|
Sistema respiratorio |
||
|
Disnea |
11,5 |
6,6 |
|
Piel |
||
|
Prurito |
8,2 |
6,6 |
|
Un paciente que informase sobre un único término médico más de una vez se contó solamente una vez para ese término médico. |
||
Prediálisis: La seguridad del tratamiento con HECTOROL® fue evaluada en estudios clínicos en 55 pacientes (27 activo y 28 placebo) con insuficiencia renal crónica de estadío 3 o estadío 4. En dos estudios multicéntricos, doble ciego, controlados por placebo, la interrupción de la terapia debida a algún evento adverso tuvo lugar en uno de los 27 pacientes (3,7%) tratados con HECTOROL® durante 24 semanas (dosis ajustada para alcanzar los niveles deseados de la PTHi, véase Farmacología clínica/Estudios clínicos) y en tres de los 28 pacientes (10,7%) tratados con placebo durante 24 semanas. Los eventos adversos que tuvieron lugar en el grupo HECTOROL® con una frecuencia de 5% o más y más frecuentemente que en el grupo placebo fueron los siguientes: Cuerpo en General: Infección, dolor torácico; Sistema Digestivo: Estreñimiento, dispepsia; Sistema Hematológico y Linfático: Anemia; Metabólico y Nutricional: Deshidratación; Sistema Nervioso: Depresión, hipertonia, insomnia, parestesia; Sistema Respiratorio: Aumento de la Tos, disnea, rinitis.
Los eventos adversos potenciales de HECTOROL® son, en general, similares a aquellos observados en la ingesta excesiva de vitamina D. Los signos y síntomas tempranos y tardíos de la intoxicación con vitamina D asociados con la hipercalcemia incluyen:
Tempranos: Debilidad, cefalea, somnolencia, náuseas, vómitos, sequedad de boca, estreñimiento, dolor muscular, dolor óseo, sabor metálico y anorexia.
Tardíos: Poliuria, polidipsia, anorexia, pérdida de peso, nocturia, conjuntivitis (calcific), pancreatitis, fotofobia, rinorrea, prurito, hipertermia, libido deprimida, nitrógeno úrico sanguíneo (BUN) elevado, albuminuria, hipercolesterolemia, aspartato transaminasa (AST) y alanina transaminasa (ALT) séricas elevadas, calcificación ectópica, hipertensión, arritmias cardiacas, trastornos sensoriales, deshidratación, apatía, detención del crecimiento, infecciones del tracto urinario y, raramente, psicosis manifiesta.
Un aumento de la creatinina sérica puede ser observado en pacientes en pre-diálisis. Esto puede reflejar la inhibición reversible de la secreción tubular de creatinina por análogos de la vitamina D.
INTERACCIONES MEDICAMENTOSAS: No se han realizado estudios específicos de interacción medicamentosa. Se ha informado que la colestiramina reduce la absorción intestinal de vitaminas liposolubles; por consiguiente, ella podría disminuir la absorción intestinal del doxercalciferol. Los antiácidos que contienen magnesio y HECTOROL® no deberán usarse concomitantemente, porque tal uso podría conducir al desarrollo de hipermagnesemia (ver Advertencias). El uso de aceite mineral u otras sustancias que pudieran afectar la absorción de grasas puede influenciar la absorción y disponibilidad de HECTOROL®. Aunque no se ha examinado específicamente, los inductores enzimáticos (como glutetimida o fenobarbital) pueden afectar la 25-hidroxilación de HECTOROL® y podría ser necesario realizar ajustes de la dosis. Los inhibidores del citocromo P450 (tales como el ketoconazol y la eritromicina) pueden inhibir la 25-hidroxilación de HECTOROL®. Por lo tanto, se puede interferir la formación de la molécula activa de HECTOROL®.
Carcinogenia, mutagenia y efecto sobre la fertilidad: En un estudio de carcinogenicidad de 104 semanas en ratas, se produjo un aumento en la incidencia de feocromocitomas suprarrenales benignos y malignos en machos y hembras a dosis orales de 0,04, 0,13 y 0,39 mcg / kg / día (= 1 exposición humana de pacientes en pre-diálisis con una dosis máxima recomendada de 3,5 mcg / día o 24.5 mcg / semana). Este aumento en la incidencia de feocromocitomas en ratas puede ser debido a la homeostasis del calcio alterada por doxercalciferol.
En un ensayo in vitro de la capacidad mutagénica bacteriana (ensayo de Ames) o en el ensayo de la mutación del gen de linfoma murino no se observó evidencia de toxicidad genética. El doxercalciferol causó aberraciones estructurales de la cromátida y del cromosoma en un ensayo in vitro de clastogenia linfocítica humana con activación metabólica. Sin embargo, el doxercalciferol fue negativo en un ensayo de clastogenia in vivo en micronúcleos de ratón. El doxercalciferol no tuvo efecto sobre la fertilidad masculina o femenina en ratas a dosis orales de hasta 2,5 mcg/kg/día (aproximadamente 3 veces la dosis máxima recomendada para el hombre —60 mcg/semana— en términos de mcg por m2 de área corporal).
Uso durante el embarazo:
Embarazo categoría B: En estudios sobre reproducción en ratas y conejos a dosis de hasta 20 mcg/kg/día y 0,1 mcg/kg/día, dosis que —en términos de mcg por m2 de área corporal— representan unas 25 veces más que la dosis máxima recomendada para el hombre (60 mcg/semana) y menos que dicha dosis máxima recomendada, respectivamente, no se ha revelado ningún efecto teratógeno o fetotóxico del doxercalciferol. Sin embargo, no hay estudios adecuados y bien controlados en mujeres embarazadas. Debido a que los estudios sobre la reproducción en animales no son siempre predictivos de la respuesta en el hombre, este fármaco debe usarse durante el embarazo sólo si existe una necesidad clara.
Madres en lactancia: No se conoce si el doxercalciferol se excreta en la leche humana. Debido a que otros derivados de la vitamina D se excretan en la leche humana y debido al potencial de reacciones adversas serias al doxercalciferol en infantes lactantes, debe tomarse una decisión entre interrumpir el amamantamiento o interrumpir el medicamento, teniendo en cuenta la importancia de este último para la madre.
Uso pediátrico: No se ha establecido la seguridad y eficacia de HECTOROL® en pacientes pediátricos.
Uso geriátrico: De los 138 pacientes tratados con cápsulas de HECTOROL® en dos estudios de fase 3, 30 pacientes tenían 65 años o más. En estos estudios no se observaron diferencias generales en la eficacia y seguridad entre estos pacientes de 65 años o más y los más jóvenes.
Insuficiencia hepática: Debido a que los pacientes con insuficiencia hepática pueden no metabolizar el HECTOROL® correctamente, el medicamento se debe usar con precaución en pacientes con función hepática deteriorada. En estos individuos debe efectuarse un monitoreo más frecuente de los niveles de PTHi, calcio y fósforo.
FARMACOLOGÍA CLÍNICA
Los niveles de vitamina D en los seres humanos dependen de dos fuentes: (1) exposición a los rayos ultravioletas del sol para la conversión en la piel del 7-dehidrocolesterol en vitamina D3 (colecalciferol) y (2) la ingesta alimentaria de la vitamina D2 (ergocalciferol) o de la vitamina D3. La vitamina D2 y la vitamina D3 deben activarse metabólicamente en el hígado y en el riñón antes de tornarse completamente activas en los órganos destinatarios. El paso inicial del proceso de activación es la introducción de un grupo hidroxilo en el carbono 25 de la cadena lateral mediante la enzima hepática CYP 27 (una hidroxilasa del carbono 25 de la vitamina D). Los productos de esta reacción son 25-(OH)D2 y 25-(OH)D3, respectivamente.
Una hidroxilación adicional de estos metabolitos tiene lugar en la mitocondria del tejido renal, catalizada por la 25-hidroxivitamina D-1-a-hidroxilasa renal, para producir 1a,25-(OH)2D2, la forma biológicamente activa primaria de la vitamina D2, y 1a,25-(OH)2D3 (calcitriol), la forma biológicamente activa de la vitamina D3.
Mecanismo de acción: El calcitriol (1a,25-(OH)2D3) y la 1a,25-(OH)2D2 regulan el calcio sanguíneo a los niveles que se requieren para las funciones corporales esenciales. En concreto, los metabolitos biológicamente activos de la vitamina D controlan la absorción intestinal del calcio de la dieta, la reabsorción tubular de calcio por los riñones y, conjuntamente con la hormona paratiroidea (PTH), la movilización ósea del calcio. Actúan directamente sobre las células óseas (osteoblastos) para estimular el crecimiento esquelético, y sobre las glándulas paratiroideas para suprimir la síntesis y secreción de la PTH.
Estas funciones están mediadas por la interacción de estos metabolitos biológicamente activos con proteínas receptoras específicas en los diferentes tejidos destinatarios. En pacientes con insuficiencia renal crónica (IRC), la producción deficiente de metabolitos biológicamente activos de la vitamina D (debido a la falta de la 25-hidroxivitamina D-1-a-hidroxilasa o a la actividad deficiente de la misma) conduce a un hiperparatiroidismo secundario, que contribuye al desarrollo de la enfermedad metabólica ósea.
ADVERTENCIAS: La sobredosis de cualquier forma de vitamina D, incluyendo HECTOROL®, es peligrosa (ver sobredosis). La hipercalcemia progresiva debida a sobredosis con vitamina D y sus metabolitos puede ser tan severa como para requerir atención de emergencia. La hipercalcemia aguda puede exacerbar tendencias de arritmias y convulsiones y puede potenciar la acción de drogas digitálicas. La hipercalcemia crónica puede conducir a calcificación vascular generalizada y otras calcificaciones de tejido blando. El producto calcio-fósforo sérico (Ca x P) debe mantenerse en < 55 mg2/dL2 en pacientes con insuficiencia renal crónica. La evaluación radiográfica de las regiones anatómicas comprometidas puede ser útil en la evaluación temprana de esta condición.
Dado que el doxercalciferol es un precursor de la 1a,25-(OH)2D2, un potente metabolito de la vitamina D2, debe suspenderse la administración de dosis farmacológicas de la vitamina D y sus derivados durante el tratamiento con HECTOROL® para evitar posibles efectos aditivos e hipercalcemia.
Deben utilizarse quelantes de fosfato orales a base de calcio u otros no alumínicos y una dieta baja en fosfato, para controlar los niveles de fósforo sérico en pacientes con insuficiencia renal crónica. El fósforo sérico no controlado exacerba el hiperparatiroidismo secundario y puede disminuir la eficacia de HECTOROL® para reducir los niveles sanguíneos de la PTH. Si ocurre hipercalcemia luego de comenzar la terapia con HECTOROL®, debe reducirse la dosis de HECTOROL® y/o de quelantes de fosfato que contienen calcio. Si ocurre hiperfosfatemia luego de iniciada la terapia con HECTOROL®, debe reducirse la dosis de HECTOROL® y/o debe aumentarse la dosis de quelantes de fosfato (Ver las recomendaciones sobre posología para HECTOROL® en la sección Posología y modo de empleo).
Los antiácidos que contienen magnesio y HECTOROL® no deben usarse concomitantemente en pacientes en diálisis renal crónica debido a que su utilización podría conducir al desarrollo de hipermagnesemia.
Un aumento de la creatinina sérica puede ser observado en pacientes en pre-diálisis. Esto puede reflejar la inhibición reversible de la secreción tubular de creatinina por análogos de la vitamina D.
PRECAUCIONES
General: Los esteroles activos de vitamina D no deben utilizarse como tratamiento inicial de la deficiencia nutricional de vitamina D (definida como 25-hidroxi Vitamina D baja). Antes de iniciar el tratamiento con HECTOROL®, se deben controlar y tratar a los pacientes por su deficiencia nutricional de vitamina D.
Los principales efectos adversos del tratamiento con HECTOROL® son hipercalcemia, hiperfosfatemia, hipercalciuria y supresión excesiva de la PTHi. La hipercalcemia prolongada puede conducir a la calcificación de los tejidos blandos, incluyendo el corazón y las arterias, y la hiperfosfatemia puede exacerbar el hiperparatiroidismo. La hipercalciuria puede acelerar el inicio de la falla renal a través de la nefrocalcinosis. La supresión excesiva de la PTHi puede conducir al síndrome óseo adinámico. Todos estos efectos adversos potenciales se deben manejar mediante un control regular del paciente y ajustes adecuados de la posología. Durante el tratamiento con HECTOROL®, los pacientes usualmente requieren un ajuste de la dosis así como también un ajuste de la terapia conjunta (es decir, quelantes del fosfato alimentario) con el fin de lograr y sostener la supresión de la PTH y a la vez mantener el calcio y fósforo séricos dentro de los rangos prescritos.
Diálisis: En cuatro estudios adecuados y bien controlados, la incidencia de hipercalcemia e hiperfosfatemia aumentaron durante la terapia con HECTOROL®. Los aumentos observados durante el tratamiento con HECTOROL®, aunque en un porcentaje bajo, enfatizan la importancia de un control regular de los niveles séricos de calcio y fósforo a lo largo del tratamiento. Los pacientes que previo al tratamiento tenían niveles séricos más elevados de calcio (> 10,5 mg/dL) o de fósforo (> 6,9 mg/dL) fueron más propensos a experimentar hipercalcemia o hiperfosfatemia. Por lo tanto, HECTOROL® no debe administrarse a pacientes con antecedentes recientes de hipercalcemia o hiperfosfatemia, o evidencia de toxicidad por vitamina D.
Prediálisis: En dos estudios clínicos, las incidencias de hipercalcemia y de hiperfosfatemia durante la terapia con HECTOROL® fueron similares a las de la terapia con placebo y no se observaron episodios de hipercalciuria. El nivel basal medio de 25-(OH) vitamina D de los pacientes enrolados en estos estudios fue de 17,2 ng/mL. El 93% de los pacientes tenían niveles de 25-(OH) vitamina D inferiores a 30 ng/mL; el 26% tenía niveles de 25-(OH) vitamina D ³ 20 a < 30 ng/mL; el 58% tenía niveles >10 a < 20 ng/mL, el 7% tenía niveles > 5 a < 10 ng/mL, y el 2% tenía niveles <5 ng/mL. Las incidencias de hipercalcemia, hiperfosfatemia e hipercalciuria en pacientes en prediálisis tratados con HECTOROL®, debido a hiperparatiroidismo relacionado con la insuficiencia renal, no se han estudiado completamente cuando los niveles de 25-(OH) vitamina D son superiores o iguales a 30 ng/mL.
Información para el paciente: Se debe informar al paciente, cónyuge o guardián acerca del cumplimiento de las instrucciones sobre posología, el seguimiento de las instrucciones referentes a la dieta, suplementación con calcio y abstinencia del uso de medicamentos de venta sin receta, sin aprobación previa de su médico. Asimismo, se debe informar bien a los pacientes sobre los síntomas de la hipercalcemia (véase sección Reacciones adversas).
Los pacientes deben tener una ingesta combinada de calcio elemental (quelantes de fosfato y alimenticio) que no exceda los 2 g diarios.
Análisis de laboratorio: Se deben determinar periódicamente la PTHi sérica o plasmática y el calcio, el fósforo y la fosfatasa alcalina en suero. En pacientes en fase temprana de tratamiento de diálisis, se deben determinar la PTHi, el calcio sérico y el fósforo sérico antes de iniciar el tratamiento con HECTOROL® y luego una vez por semana. En los pacientes en prediálisis, los niveles séricos de calcio y fósforo y los niveles plasmáticos de PTHi deben monitorearse al menos cada dos semanas durante los 3 meses posteriores al inicio del tratamiento con HECTOROL® o luego de los ajustes de dosis de la terapia con HECTOROL®, luego mensualmente durante 3 meses y posteriormente cada 3 meses.
POSOLOGÍA Y MODO DE EMPLEO
Administración en adultos: La dosis óptima de HECTOROL® debe determinarse cuidadosamente para cada paciente. La tabla siguiente provee los niveles terapéuticos de la PTHi buscados actualmente en pacientes con insuficiencia renal crónica que se recomiendan:
|
Rango buscado de la PTH intacta plasmática según el estadío de insuficiencia renal crónica (IRC) |
||
|
Estadío de IRC |
TFG (tasa de filtración glomerular) (mL/min/1,73 m2) |
PTH “intacta” buscada (pg/mL) |
|
3 |
30-59 |
35-70 |
|
4 |
15-29 |
70-110 |
|
5 |
< 15 (o diálisis) |
150-300 |
|
Obtenida de la Tabla 15 de la National Kidney Foundation. K/DOQI Clinical Practice Guidelines for Bone Metabolism and Disease in Chronic Kidney Disease. Am J Kidney Dis 42:S1-S202, 2003 (suppl 3) |
||
Diálisis: La dosis inicial recomendada de HECTOROL® es de 10 mcg administrada tres veces a la semana en la diálisis (aproximadamente día por medio). La dosis inicial debe ajustarse, según fuera necesario, con el fin de disminuir la PTHi sanguínea hasta el rango de 150-300 pg/mL. Si la PTHi no disminuyera en un 50% y se fallara en lograr el rango buscado, la dosis puede aumentarse a intervalos de 8 semanas en 2,5 mcg. La dosis máxima recomendada de HECTOROL® es de 20 mcg administrada 3 veces a la semana en la diálisis, totalizando 60 mcg/semana. Si la PTHi cae por debajo de 100 pg/mL la administración del fármaco debe suspenderse y una semana después retomarse a una dosis de al menos 2,5 mcg menor que la última dosis administrada. Durante el ajuste de dosis deben obtenerse semanalmente los niveles de PTHi, de calcio sérico y de fósforo sérico. Si se advierten hipercalcemia, hiperfosfatemia o un producto calcio-fósforo sérico mayor de 55 mg2/dL2, debe disminuirse o suspenderse la dosis de HECTOROL® y/o ajustarse adecuadamente la dosis de quelantes de fosfato. De suspenderse, debe retomarse la administración del fármaco a una dosis de por lo menos 2,5 mcg menor.
La dosis debe individualizarse y basarse en los niveles de PTHi con monitoreo de los niveles séricos de calcio y fósforo. A continuación se presenta una aproximación sugerida para el ajuste de la dosis:
|
Posología inicial |
|
|
Nivel de PTHi |
Dosis de HECTOROL® |
|
> 400 pg/mL |
10 mcg tres veces a la semana en la diálisis |
|
Ajuste de la dosis |
|
|
Nivel de PTHi |
Dosis de HECTOROL® |
|
> 300 pg/ml |
Aumento de 2,5 mcg cada 8 semanas según fuera necesario. |
|
150-300 pg/mL |
Mantener |
|
< 100 pg/mL |
Suspender por una semana, luego reanudar a una dosis que sea de al menos 2,5 mcg menor. |
Prediálisis: La dosis inicial recomendada de HECTOROL® es de 1 mcg administrada una vez al día. La dosis inicial debe ajustarse, según fuera necesario, con el fin de disminuir la PTHi sanguínea hasta el rango deseado (véase tabla siguiente). La dosis puede incrementarse a intervalos de 2 semanas en 0,5 mcg hasta alcanzar el rango buscado de PTHi. La dosis máxima recomendada de HECTOROL® es de 3,5 mcg administrada una vez al día.
Los niveles séricos de calcio y fósforo y los niveles plasmáticos de PTHi deben monitorearse al menos cada dos semanas durante los 3 meses posteriores al inicio del tratamiento con HECTOROL® o luego de los ajustes de dosis de la terapia con HECTOROL®, luego mensualmente durante 3 meses y posteriormente cada 3 meses.
Si se advirtiera hipercalcemia, hiperfosfatemia o un producto calcio-fósforo séricos mayor de 55 mg2/dL2, debe reducirse o suspenderse la dosis de HECTOROL® y/o ajustarse adecuadamente la dosis de quelantes de fosfato. De suspenderse, debe retomarse la administración del fármaco a una dosis de por lo menos 0,5 mcg menor.
La dosis debe individualizarse y basarse en los niveles de PTHi con monitoreo de los niveles de calcio y fósforo séricos. A continuación se presenta una aproximación sugerida para el ajuste de la dosis:
|
Posología inicial |
|
|
Nivel de PTHi |
Dosis de HECTOROL® |
|
> 70 pg/mL (Estadío 3) > 110 pg/mL (Estadío 4) |
1 mcg una vez al día |
|
Ajuste de la dosis |
|
|
Nivel de PTHi |
Dosis de HECTOROL® |
|
> 70 pg/mL (Estadío 3) 110 pg/mL (Estadío 4) |
Aumento de 0,5 mcg cada 2 semanas según fuera necesario |
|
35-70 pg/mL (Estadío 3) 70-110 pg/mL (Estadío 4) |
Mantener |
|
< 35 pg/mL (Estadío 3) < 70 pg/mL (Estadío 4) |
Suspender por una semana, luego reanudar a una dosis que sea de al menos 0,5 mcg menor |
SOBREDOSIS: La administración de HECTOROL® en exceso a pacientes, puede causar hipercalcemia, hipercalciuria, hiperfosfatemia y supresión excesiva de la secreción de la PTH, resultando, en algunos casos, a enfermedad ósea adinámica. La ingesta elevada de calcio y fosfato concomitantemente con HECTOROL® puede conducir a anormalidades semejantes. Altos niveles de calcio en la solución de diálisis pueden contribuir a la hipercalcemia.
Tratamiento de la hipercalcemia y la sobredosis: El tratamiento general de la hipercalcemia (> 1 mg/dL por encima del límite superior del rango normal en pacientes en diálisis; > 10,7 mg/dL en pacientes en prediálisis) consiste en la suspensión inmediata de la terapia con HECTOROL®, la institución de una dieta baja en calcio y la eliminación de los suplementos de calcio. Los niveles de calcio sérico deben determinarse al menos semanalmente, hasta que la normocalcemia se restituya. La hipercalcemia normalmente se resuelve en 2 a 7 días. Cuando los niveles de calcio sérico han retornado a los límites normales, puede reinstituirse la terapia con HECTOROL® a una dosis menor (al menos 2,5 mcg en pacientes en diálisis y 0,5 mcg en pacientes en prediálisis) que la terapia previa. En los pacientes en diálisis, los niveles de calcio sérico deben obtenerse semanalmente luego de cada cambio de posología y durante el subsiguiente ajuste de la dosis. Se pueden corregir los niveles de calcio sérico persistente o marcadamente elevados mediante la diálisis contra una solución de diálisis reducida en calcio o libre de calcio.
Tratamiento de la sobredosis accidental de doxercalciferol: El tratamiento de la sobredosis aguda accidental de HECTOROL® debe consistir en medidas generales de asistencia. Si la ingestión del fármaco se descubre en un tiempo relativamente corto (10 minutos), la inducción de emesis o el lavado gástrico pueden ser beneficiosos para prevenir una absorción mayor. Si la ingestión del fármaco se descubre en un tiempo superior a 10 minutos, la administración de aceite mineral puede promover su eliminación fecal. Se deben obtener determinaciones seriales de electrolitos séricos (especialmente calcio), tasa de excreción urinaria de calcio y evaluación de anormalidades electrocardiográficas debidas a la hipercalcemia. Este monitoreo es crítico en los pacientes que reciben digitálicos. En los accidentes por sobredosis también están indicadas la interrupción de suplementos cálcicos y la institución de una dieta baja en calcio.
Si aparecen niveles de calcio sérico persistente y marcadamente elevados, pueden considerarse una variedad de alternativas terapéuticas. Estas incluyen el uso de fármacos como fosfatos y corticosteroides, así como medidas para inducir la diuresis. También puede considerarse la diálisis contra una solución de diálisis libre de calcio.
Ante la eventualidad de una sobredosis, acudir al hospital más cercano.
DESCRIPCIÓN
El principio activo de HECTOROL®, doxercalciferol, es un análogo sintético de la vitamina D2 que sufre activación metabólica in vivo para dar lugar a la 1a, 25-dihidroxivitamina D2 (1a, 25-(OH)2 D2), una forma biológicamente activa natural de la vitamina D2. HECTOROL® se encuentra disponible bajo la forma de cápsulas blandas de gelatina que contienen 0,5 mcg o 2,5 mcg de doxercalciferol. Cada cápsula también contiene triglicérido de aceite de coco fraccionado, etanol, y butilhidroxianisol (BHA). La cubierta de las cápsulas contiene gelatina, glicerina y dióxido de titanio. Además, la cápsula de 0,5mcg contiene óxido de hierro amarillo,y Rojo FD&C Nº 40, y la cápsula de 2,5 mcg contiene óxido de hierro amarillo.
El doxercalciferol es un compuesto cristalino incoloro con un peso molecular calculado de 412,66 y una fórmula molecular de C28H44 O2. Es soluble en grasas y solventes orgánicos, aunque relativamente insoluble en agua. Químicamente, el doxercalciferol es (1a,3ß,5Z,7E,22E)-9,10-secoergosta-5,7,10(19),22-tetraeno-1,3-diol y tiene la siguiente fórmula estructural:
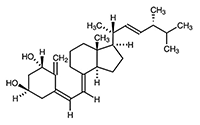
Otros nombres que se suelen usar para designar el doxercalciferol son 1a-hidroxivitamina D2,1a-OH-D2 y 1a-hidroxiergocalciferol.
PRESENTACIÓN: Contenido y composición del envase
HECTOROL® cápsulas blandas 2,5 mcg: Frascos plásticos con tapa a prueba de niños. Contiene 50 cápsulas blandas (Reg. San. INVIMA2009M-0010183).
HECTOROL® cápsulas blandas 0,5 mcg: Frascos plásticos con tapa a prueba de niños. Contiene 50 cápsulas blandas (Reg. San. INVIMA2009M-0010186).
HECTOROL® y Genzyme son marcas registradas de Genzyme Corporation.
Elaborado por:
Catalent Pharma Solutions
Para Genzyme Corporation
Cambridge, MA, EE. UU.
Importado por:
Genzyme de Colombia S.A.
Ref:US PI- 6800 (08/08)
Last revision date: 02/2012
Importado y distribuido por:
SANOFI-AVENTIS DE COLOMBIA S. A.
Transversal 23 No. 97-73, Pisos 8 y 9
Teléfono: 6214400, Fax: 7444237
Bogotá, D.C., Colombia
CONDICIONES ESPECIALES DE CONSERVACIÓN: No conservar a mas de 30°C: Se permiten variaciones entre 15º y 30ºC.
Mantener el envase bien cerrado.
Manténgase fuera del alcance de los niños.

