
GIOTRIF
AFATINIB
Tabletas recubiertas
Caja, 28 Tabletas recubiertas, 20 mg
Caja, 28 Tabletas recubiertas, 30 mg
Caja, 28 Tabletas recubiertas, 40 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN: 1 COMPRIMIDO Recubierto contiene 50 mg, 40 mg, 30 mg o 20 mg de afatinib (= base libre) que corresponden a:
73,9 mg, 59,12 mg, 44,34 mg o 29,56 mg de 2-butenamida, N-[4-[3(-cloro-4-fluorofenil)amino]-7-[[(3S)-tetrahidro-3-furanil]oxi]-6- quinazolinil]-4-(dimetilamino)-,(2E)-, (2Z)-2-butenodioato (1:2) (= afatinib dimaleato)
MECANISMO DE ACCIÓN: Afatinib es un bloqueador irreversible potente y selectivo de la familia ErbB. Afatinib se une mediante enlace covalente y bloquea en forma irreversible la señalización de todos los homo y heterodímeros formados por los siguientes integrantes de la familia ErbB: EGFR (ErbB1), HER2 (ErbB2), ErbB3 y ErbB4.
Efectos farmacodinámicos: La señalización aberrante de ErbB desencadenada, por ejemplo, por las mutaciones y/o la amplificación del EGFR, por la amplificación o la mutación del HER2 [45] y/o por la sobreexpresión del ligando o receptor ErbB, contribuye al fenotipo maligno en subconjuntos de pacientes en diversos tipos de cáncer.
En modelos no clínicos de esta patología con desregulación de la vía ErbB, afatinib administrado como agente único bloquea efectivamente la señalización del receptor ErbB, lo cual conduce a la inhibición del crecimiento tumoral o bien a la regresión del tumor. Los modelos de NSCLC con mutaciones L858R o Del 19 del EGFR son particularmente sensibles al tratamiento con afatinib.
En NSCLC, la adquisición de una mutación T790M secundaria es el principal mecanismo de resistencia adquirida a afatinib y la dosis génica del alelo que contiene la mutación T790M se correlaciona con el grado de resistencia in vitro. La mutación T790M se encuentra aproximadamente en el 50% de los tumores de los pacientes tras la progresión de la enfermedad con afatinib, para los cuales los EGFR TKI dirigidos a la mutación T790M se pueden considerar como una opción de línea de tratamiento posterior.
Estudios clínicos:
GIOTRIF® en cáncer de pulmón de células no pequeñas (NSCLC): La eficacia y la seguridad de la monoterapia de GIOTRIF® en el tratamiento de pacientes con NSCLC con mutaciones del EGFR se demostró en 4 estudios aleatorizados y controlados (LUX- Lung 3; 1200.32, LUX-Lung 6; 1200.34; LUX-Lung 1; 1200.23 y LUX-Lung 7; 1200.123), en un
estudio a gran escala de Fase III (LUX-Lung 5; 1200.42) y en un estudio a gran escala de rama única de Fase II (LUX-Lung 2, 1200.22). En los estudios LUX-Lung 3, LUX-Lung 6, LUX-Lung 7 y LUX-Lung 2 se reclutaron pacientes con mutación positiva del EGFR no tratados previamente con un EGFR TKI. En los estudios LUX-Lung 1 y LUX-Lung 5 se reclutaron pacientes clínicamente enriquecidos en términos de mutaciones del EGFR que habían recibido tratamiento previo con un EGFR TKI (gefitinib o erlotinib) y tuvieron progresión de la enfermedad con dicho tratamiento. Se esperaba que las poblaciones de los estudios LUX-Lung 1 y 5 contuvieran una gran proporción de pacientes con mutación de resistencia T790M, la cual es detectable en aproximadamente el 50% de los pacientes con NSCLC previamente responsivos que presentan resistencia a erlotinib y/o gefitinib.
La eficacia y la seguridad de GIOTRIF® como tratamiento de segunda línea para pacientes con NSCLC de histología escamosa se investigó en un estudio de diseño abierto controlado con tratamiento activo LUX-Lung 8.
GIOTRIF® en pacientes con mutación positiva del EGFR no tratados previamente con un EGFR TKI:
LUX-Lung 3 (1200.32): En el contexto del tratamiento de primera línea, la eficacia y seguridad de GIOTRIF® en pacientes con NSCLC localmente avanzado o metastásico (estadio IIIB o IV) positivo para mutación del EGFR se evaluaron en un estudio internacional, aleatorizado, multicéntrico, de diseño abierto (LUX-Lung 3). Los pacientes que no habían recibido tratamiento sistémico previo para su enfermedad avanzada o metastásica fueron sometidos a pruebas destinadas a detectar la presencia de 29 mutaciones del EGFR diferentes, usando un método basado en una reacción de la cadena polimerasa (PCR) (TheraScreen®: EGFR29 Mutation Kit, Qiagen Manchester Ltd.). Los pacientes (N = 345) fueron aleatorizados (en una proporción de 2:1) a recibir 40 mg de GIOTRIF® por vía oral una vez al día (N = 230) o bien un total de hasta 6 ciclos de pemetrexed/cisplatino (N = 115). La aleatorización se estratificó en función del estado de mutación del EGFR (L858R; Del 19;otra) y de la raza (asiática, no asiática). Se permitió el ajuste creciente de la dosis de GIOTRIF® hasta 50 mg después del primer ciclo (21 días) de tratamiento, si los pacientes no presentaran ningún evento adverso relacionado con el medicamento o de que, en el caso de presentarse, el evento adverso fuera limitado (es decir, ausencia de diarrea, exantema, estomatitis y/u otros eventos relacionados con el fármaco > Grado 1 de los CTCAE), y de que cumplieran y no hubieran tenido ninguna reducción de la dosis en el pasado.
El criterio de valoración primario de sobrevida libre de progresión (PFS) (revisión independiente, 221 eventos) indicó una mejoría estadísticamente significativa en la PFS para los pacientes tratados con GIOTRIF® comparados con aquellos tratados con quimioterapia (mediana de PFS 11,1 vs. 6,9 meses). Cuando se compararon los subgrupos preespecificados de mutaciones frecuentes del EGFR (L858R o Del 19), las diferencias en términos de PFS fueron más pronunciadas (mediana de PFS: 13,6 vs. 6,9 meses). Los porcentajes de pacientes que estaban vivos y libres de progresión (tasa de PFS) a los 12 meses fueron del 46,5% en los pacientes tratados con GIOTRIF® y del 22% en los pacientes tratados con quimioterapia para la población total del estudio, y del 51,1% versus un 21,4% en el caso del subgrupo de mutaciones frecuentes.
Las curvas de Kaplan-Meier del análisis primario de PFS se presentan en la Figura 1, y los resultados de eficacia se resumen en la Tabla 5. Al momento de la realización del análisis primario de PFS, un total de 45 (20%) pacientes tratados con GIOTRIF® y 3 (3%) pacientes tratados con quimioterapia estaban vivos y libres de progresión; de esta manera, sus datos están censurados en la Figura 1.
Figura 1. Curva de Kaplan-Meier para la PFS, por revisión independiente, por grupo de tratamiento, en el Estudio LUX-Lung 3 (población total)
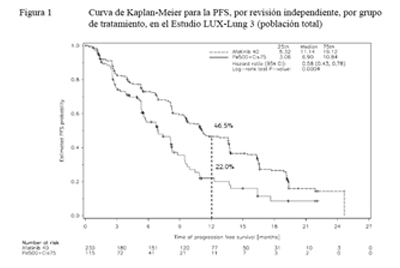
Referencias de la figura:
Estimated PFS Probability: Probabilidad de PFS estimada
Time of progression free survival (months): Tiempo de sobrevida libre de progresión (meses)
Median: Mediana
Hazard ratio: Razón de riesgos
Log-rank test: Prueba de rangos logarítmicos
P-value: Valor p
Number at risk: Cantidad de pacientes en riesgo
|
Tabla 4 Resultados de eficacia de GIOTRIF® vs. pemetrexed/cisplatino (Estudio LUX- Lung 3) obtenidos sobre la base del análisis primario de la PFS al 9 de febrero de 2012 (revisión independiente) |
|||
|
GIOTRIF® (N = 230) |
Pemetrexed/ Cisplatino (N = 115) |
Razón de riesgos (HR) / Cociente de probabilidades (OR) (IC del 95%) Valor p4 |
|
|
PFS, Población general del estudio Meses (mediana) |
11,1 |
6,9 |
HR 0,58 (0,43-0,78) |
|
Tasa de PFS a 1 año Tasa de PFS a 18 meses |
46,5% 26,4% |
22% 8,6% |
0,0004 |
|
PFS, Pacientes con mutaciones L858R o Del 191 |
|||
|
Meses (mediana) |
13,6 |
6,9 |
HR 0,47 |
|
Tasa de PFS a 1 año |
51,1% |
21,4% |
(0,34-0,65) |
|
Tasa de PFS a 18 meses |
28,6% |
7,4% |
< 0,0001 |
|
Tasa de respuesta objetiva (CR+PR) 2 |
56,1% |
22,6% |
OR 4,66 (2,77-7,83) < 0,0001 |
|
Tasa de control de la enfermedad (CR+PR+SD) 2 |
90,0% |
80,9% |
OR 2,14 (1,13-4,04) 0,0189 |
|
Duración de la respuesta Meses (mediana) |
11,1 |
5,5 |
— |
|
Sobrevida general (OS), Población del estudio total Meses (mediana) 3 |
28,2 |
28,2 |
HR 0,88 (0,66, 1,17) 0,39 |
|
1 N = 308 (GIOTRIF®: 204, pemetrexed/cisplatino: 104) 2 CR = respuesta completa; PR = respuesta parcial; SD = enfermedad estable 3 Análisis de OS actualizado a diciembre de 2013 4 Valor p para la PFS/OS basado en la prueba de rangos logarítmicos estratificada; Valor p para la Tasa de Respuesta Objetiva y Tasa de Control de la Enfermedad basado en regresión logística. |
|||
El análisis de la PFS basado en la revisión del investigador arrojó resultados similares (mediana de PFS 11,1 vs. 6,7 meses, HR = 0,49, p < 0,0001) según el análisis basado en la revisión independiente. El efecto sobre la PFS fue concordante en todos los principales subgrupos, lo que incluyó el sexo, la edad, la raza, el estado ECOG y el tipo de mutación (L858R, Del 19) tanto en la revisión independiente como en la revisión del investigador. Sobre la base de la revisión del investigador, la ORR fue 69,1% vs. 44,3% y la DCR fue 90,0% vs. 82,6% en los pacientes tratados con GIOTRIF® en comparación con aquellos tratados con quimioterapia. En el subgrupo predefinido de mutaciones frecuentes de EGFR (Del 19, L858R) para GIOTRIF (N = 203) y quimioterapia (N = 104), la mediana de OS fue 31,6 meses versus 28,2 meses (HR = 0,78, IC del 95% (0,58, 1,06), p = 0,1090). En los subgrupos predefinidos de mutaciones de EGFR, la mediana de OS con GIOTRIF como tratamiento de primera línea vs. quimioterapia fue 33,3 meses vs. 21,1 meses (HR = 0,54 (IC del 95% 0,36-0,79), p = 0,0015) en pacientes con Del 19 (n = 169) y 27,6 meses vs. 40,3 meses (HR=1,30 (IC del 95% 0,80-2,11), p = 0,2919) en pacientes con L858R (n = 138).
El beneficio en términos de PFS de GIOTRIF® estuvo acompañado por una mejoría en los síntomas relacionados con la enfermedad, según lo determinado mediante los Cuestionarios de Calidad de Vida (QLQ-C30 y QLQ-LC13) de la Organización Europea para la Investigación y el Tratamiento del Cáncer (EORTC). GIOTRIF® prolongó significativamente el tiempo hasta el deterioro de los síntomas preespecificados de tos (HR 0,60; p = 0,0072) y disnea (HR 0,68; p = 0,0145) a razón de más de 7 meses al comparar con la quimioterapia. El tiempo hasta el deterioro del dolor también se prolongó con GIOTRIF®, pero no alcanzó un grado de significación estadística (HR 0,83; p = 0,1913). Se observó un porcentaje significativamente mayor de mejoría de la disnea entre los pacientes tratados con GIOTRIF® en comparación con la quimioterapia (64% vs. 50%; p = 0,0103). Se observó una tendencia a favor de GIOTRIF® en lo que respecta al dolor (59% vs. 48%; p = 0,0513), con ítems de dolor individuales que alcanzaron relevancia (“Con dolor”: 56,0% vs. 40,0%; p = 0,0095; “Dolor en el pecho”: 51,0% vs. 37,0%; p = 0,0184; “Dolor en el brazo o en el hombro”: 41,0% vs. 26,0%; p = 0,0103). En lo que a la tos se refiere, un número mayor de pacientes mejoró con GIOTRIF® (67% vs. 60%; p = 0,2444).
Los puntajes medios a través del tiempo para la calidad de vida relacionada con la salud (HRQoL) se midieron utilizando la herramienta QLQ-C30 de la EORTC. Los puntajes medios a lo largo del tiempo para la calidad de vida general y el estado de salud general fueron significativamente mejores con GIOTRIF® que con la quimioterapia. Los puntajes medios fueron significativamente mejores en 3 de los 5 dominios funcionales (físico, rol, cognitivo) y no indicaron ninguna diferencia en los dominios de desempeño social y emocional.
LUX-Lung 6 (1200.34): La eficacia y la seguridad de GIOTRIF® en pacientes asiáticos con adenocarcinoma de pulmón localmente avanzado o metastásico (estadio IIIB/IV) positivo para mutación del EGFR se evaluaron en un estudio aleatorizado, multicéntrico, de diseño abierto (LUX-Lung 6). Al igual que en el LUX-Lung 3, los pacientes que no habían recibido tratamiento sistémico previo para su enfermedad avanzada o metastásica fueron sometidos a pruebas destinadas a detectar la presencia de 29 mutaciones del EGFR diferentes, usando un método basado en una reacción de la cadena polimerasa (PCR) (TheraScreen®: EGFR29 Mutation Kit, Qiagen Manchester Ltd.). Los pacientes (N = 364) fueron aleatorizados (en una proporción de 2:1) a recibir 40 mg de GIOTRIF® por vía oral una vez al día (N = 242) o bien un total de hasta 6 ciclos de gemcitabina/cisplatino (N = 122). La aleatorización se estratificó en función del estado de mutación del EGFR (L858R; Del 19; otra). Se permitió el ajuste creciente de la dosis de GIOTRIF® hasta 50 mg después del primer ciclo (21 días) de tratamiento en el caso de que los pacientes no presentaran ningún evento adverso relacionado con el medicamento o de que, en el caso de presentarse, el evento adverso fuera limitado (es decir, ausencia de diarrea, exantema cutáneo, estomatitis y/u otros eventos relacionados con el fármaco > Grado 1 de los CTCAE), de que cumplieran y no hubieran tenido ninguna reducción de la dosis en el pasado. Entre los pacientes aleatorizados, el 65% era de sexo femenino, la mediana de edad fue de 58 años y todos los pacientes fueron asiáticos. Los pacientes con mutación del EGFR frecuente (L858R o Del 19) representaron el 89% de la población del estudio.
El criterio de valoración primario de sobrevida libre de progresión (PFS) (revisión independiente central, 221 eventos) indicó una mejoría estadísticamente significativa en la PFS para los pacientes tratados con GIOTRIF® comparados con aquellos tratados con quimioterapia (mediana de PFS 11,0 vs. 5,6 meses). Cuando se compararon los subgrupos preespecificados de mutaciones frecuentes del EGFR (L858R o Del 19), las diferencias en términos de la mediana de PFS fueron constantes (11,0 vs. 5,6 meses). Los porcentajes de pacientes vivos y libres de progresión (tasa de PFS) a los 12 meses fueron del 46,7% en los pacientes tratados con GIOTRIF® y del 2,1% en los pacientes tratados con quimioterapia para la población total del estudio, y del 56,4% versus 4,4% en el caso del subgrupo de mutaciones frecuentes.
Las curvas de Kaplan-Meier del análisis primario de PFS se presentan en la Figura 2 y los resultados de eficacia se resumen en la Tabla 6. Al momento de la realización del análisis primario de PFS, un total de 57 (15,7%) pacientes tratados con GIOTRIF® estaba vivo y libre de progresión; de esta manera, sus datos están censurados en la Figura 2.
Figura 2. Curvas de Kaplan-Meier para la PFS, por revisión independiente, por grupo de tratamiento, en el Estudio LUX-Lung 6 (análisis primario, población total)
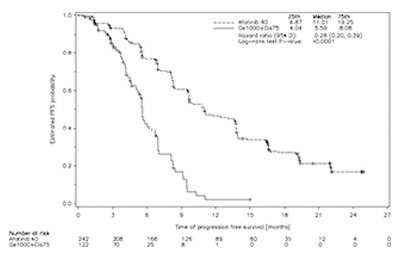
Referencias de la figura:
Estimated PFS Probability: Probabilidad de PFS estimada
Time of progression free survival (months): Tiempo de sobrevida libre de progresión (meses)
Median: Mediana
Hazard ratio: Razón de riesgos
Log-rank test: Prueba de rangos logarítmicos
P-value: Valor p
Number at risk: Cantidad de pacientes en riesgo
|
Tabla 5. Resultados de eficacia de GIOTRIF® vs. gemcitabina/cisplatino (Estudio LUX-Lung 6) obtenidos sobre la base del análisis primario de la PFS al 29 de octubre de 2012 (revisión independiente) GI |
|||
|
GIOTRIF® (N = 242) |
Gemcitabina/ Cisplatino (N = 122) |
Razón de riesgos (HR) / Cociente de probabilidades (OR) (IC del 95%) Valor p4 |
|
|
PFS, Población general del estudio Meses (mediana) Tasa de PFS a 1 año Tasa de PFS a 18 meses |
11,0 46,7% 26,8% |
5,6 2,1% 0,0% |
HR 0,28 (0,20-0,39) <0,0001 |
|
PFS, Pacientes con mutaciones L858R o Del 191 Meses (mediana) Tasa de PFS a 1 año Tasa de PFS a 18 meses |
11,0 56,4% 26,8% |
5,6 4,4% 0,0% |
HR 0,25 (0,18-0,35) < 0,0001 |
|
Tasa de respuesta objetiva (CR+PR)2 |
66,9% |
23,0% |
OR 7,28 (4,36-12,18) < 0,0001 |
|
Tasa de control de la enfermedad (CR+PR+SD)2 |
92,6% |
76,2% |
OR 3,84 (2,04-7,24) <0,0001 |
|
Duración de la respuesta Meses (mediana) |
9,7 |
4,3 |
— |
|
Sobrevida general (OS), Población del estudio total Meses (mediana)3 |
23,1 |
23,5 |
HR 0,93 (0,72, 1,22) 0,6137 |
|
1 N = 324 (GIOTRIF®: 216, gemcitabina/cisplatino: 108) 2 CR = respuesta completa; PR = respuesta parcial; SD = enfermedad estable 3 Análisis de OS actualizado al 27 de diciembre de 2013 (cuando fallecieron 246 pacientes) 4 Valor p para la PFS/OS basado en la prueba de rangos logarítmicos estratificada; Valor p para la Tasa de Respuesta Objetiva y Tasa de Control de la Enfermedad basado en regresión logística. |
|||
El análisis de la PFS basado en la revisión del investigador arrojó resultados similares (HR = 0,26, IC=95%, 0,19-0,36; p <0,0001; mediana de PFS 13,7 vs. 5,6 meses) según el análisis basado en la revisión independiente. El efecto sobre la PFS fue concordante en todos los principales subgrupos, lo que incluyó el sexo, la edad, la raza, el estado ECOG y el tipo de mutación (L858R, Del 19) tanto en la revisión independiente como en la revisión del investigador. Sobre la base de la revisión del investigador, la ORR fue 74,4% vs. 31,1% y la DCR fue 93,0% vs. 75,4% en los pacientes tratados con GIOTRIF® en comparación con aquellos tratados con quimioterapia. En el subgrupo predefinido de mutaciones frecuentes de EGFR (Del 19, L858R) para GIOTRIF (N =216) y quimioterapia (N = 108), la mediana de OS fue 23,6 meses versus 23,5 meses (HR = 0,83, IC del 95% (0,62, 1,09), p = 0,1756). En los subgrupos predefinidos de mutaciones de EGFR, la mediana de OS con GIOTRIF como tratamiento de primera línea vs. quimioterapia fue 31,4 meses vs. 18,4 meses (HR = 0,64 (IC del 95% 0,44-0,94), p = 0,0229) en pacientes con Del 19 (n= 186) y 19,6 meses vs. 24,3 meses (HR=1,22 (IC del 95% 0,81-1,83), p = 0,3432) en pacientes con L858R (n = 138).
El beneficio en términos de PFS de GIOTRIF® estuvo acompañado por una mejoría en los síntomas relacionados con la enfermedad, según lo determinado mediante los Cuestionarios de Calidad de Vida (QLQ-C30 y QLQ-LC13) de la Organización Europea para la Investigación y el Tratamiento del Cáncer (EORTC). GIOTRIF® prolongó significativamente el tiempo hasta el deterioro de los síntomas preespecificados de tos (HR 0,453; IC del 95% 0,299, 0,685: p =0,0001), disnea (HR 0,536; IC del 95% 0,395, 0,727: p = < 0,0001) y dolor (HR 0,703; IC del95% 0,514, 0,961: p = 0,0265) al comparar con la quimioterapia. Se observó un porcentaje significativamente mayor de mejoría entre los pacientes tratados con GIOTRIF® en comparación con la quimioterapia en lo que respecta a tos (75,9% de los pacientes vs. 55,4%; p=0,0003), disnea (70,9% vs. 47,5%; p < 0,0001) y dolor (64,3% vs. 46,5%; p=0,0029)Los puntajes medios a través del tiempo para la calidad de vida relacionada con la salud (HRQoL) se midieron utilizando la herramienta QLQ-C30 de la EORTC. Los puntajes medios a lo largo del tiempo para la calidad de vida general, el estado de salud general y los dominios funcionales (físico, rol, cognitivo, social y emocional) fueron significativamente mejores con GIOTRIF® que con la quimioterapia.
LUX-Lung 2 (1200.22): El estudio LUX-Lung 2 fue un ensayo de diseño abierto, de rama única, de Fase II, en el cual se investigó la eficacia y seguridad de GIOTRIF® en 129 pacientes con adenocarcinoma pulmonar localmente avanzado o metastásico (estadio IIIB o IV) con mutaciones del EGFR no tratados previamente con un EGFR TKI. Los pacientes fueron reclutados en un régimen de primera línea (N = 61) o de segunda línea (N = 68) (es decir, después del fracaso de 1 régimen de quimioterapia previo). Los pacientes fueron seleccionados centralmente en función de las mutaciones del EGFR. Los pacientes recibieron 40 mg (N = 30) o 50 mg (N = 99) de GIOTRIF® una vez al día.
El criterio de valoración primario fue la ORR. Los criterios de valoración secundarios fueron PFS, DCR y OS.
En 61 pacientes tratados con el régimen de primera línea, la ORR confirmada fue 65,6% y la DCR fue 86,9% de acuerdo con la revisión independiente. La mediana de PFS fue de 12,0 meses según la revisión independiente y de 15,6 meses de acuerdo a la evaluación del investigador. La mediana de OS no se alcanzó en la población que recibió el régimen de primera línea. La eficacia fue similarmente alta en el grupo de pacientes que había recibido quimioterapia previa (N = 68; ORR 57,4%; PFS de 8 meses según la revisión independiente y de 10,5 meses según la evaluación del investigador; DCR 77,9%). La mediana de la OS en los pacientes que recibieron el régimen de segunda línea fue 23,3 meses (IC del 95% 18,5-38).
LUX-Lung 7 (1200.123): El estudio LUX-Lung 7 es un ensayo internacional, aleatorizado, de diseño abierto, de Fase IIb, en el cual se investigó la eficacia y seguridad de GIOTRIF® en pacientes con adenocarcinoma pulmonar localmente avanzado o metastásico (estadio IIIB o IV) con mutaciones del EGFR en un régimen de primera línea. Los pacientes fueron seleccionados en función de la presencia de mutaciones activadoras del EGFR (Del 19 y/o L858R) usando el método TheraScreen® EGFR RGQ PCR Kit, Qiagen Manchester Ltd. Los pacientes (N = 319) fueron aleatorizados (1:1) a recibir 40 mg de GIOTRIF® por vía oral una vez al día (N = 160) o 250 mg de gefitinib por vía oral una vez al día (N = 159). La aleatorización se estratificó en función del estado de mutación del EGFR (Del 19; L858R) y de la presencia de metástasis cerebral (sí; no).
Entre los pacientes aleatorizados, el 62% era de sexo femenino, la mediana de edad fue de 63 años, el 16% de los pacientes tenía metástasis cerebral, el estado funcional ECOG inicial fue 0 (31%) o 1 (69%), el 57% eran asiáticos y el 43% eran no-asiáticos. Los pacientes tenían una muestra tumoral con una mutación del EGFR categorizada como deleción del exón 19 (58%) o sustitución del exón 21 L858R (42%).
Los criterios de valoración co-primarios son PFS determinada por la revisión independiente, tiempo hasta el fracaso del tratamiento (TTF) y OS. Los criterios de valoración secundarios incluyen ORR y DCR. El riesgo de progresión se redujo significativamente en el caso de afatinib versus gefitinib (ver Tabla 6) y la ORR fue 70% para afatinib y 56% para gefitinib. El análisis primario de la OS se llevará a cabo luego de que hayan ocurrido la cantidad de eventos requeridos según protocolo.
|
Tabla 6. Resultados de eficacia de GIOTRIF® vs. gefitinib (Estudio LUX-Lung 7) obtenidos sobre la base del análisis primario a agosto de 2015 |
|||
|
GIOTRIF® (N = 160) |
Gefitinib (N = 159) |
Razón de riesgos / Cociente de probabilidades (IC del 95%) Valor p2 |
|
|
Mediana de PFS (meses), Población general del estudio Tasa de PFS a 18 meses Tasa de PFS a 24 meses |
11,0 27% 18% |
10,9 15% 8% |
HR 0,73 (0,57-0,95) 0,0165 |
|
Tiempo hasta fracaso del tratamiento (meses) Tasa de TTF a 18 meses Tasa de TTF a 24 meses |
13,7 35% 25% |
11,5 27% 13% |
HR 0,73 (0,58-0,92) 0,0073 |
|
Mediana de OS (meses)1, Población general del estudio |
27,9% |
25% |
HR 0,87 (0,65, 1,15) 0,33 |
|
1 Análisis de OS en desarrollo a agosto de 2015 2 Valor p para la PFS/TTF/OS basado en la prueba de rangos logarítmicos estratificada |
|||
La razón de riesgos de la PFS para pacientes con mutaciones Del 19 y mutaciones L858R fue 0,76 (IC del 95% [0,55, 1,06]; p = 0,1071), y 0,71 (IC del 95% [0,47, 1,06]; p = 0,0856) respectivamente para afatinib vs. gefitinib.
Análisis de la eficacia de GIOTRIF en pacientes sin tratamiento previo con EGFR TKI que presentan tumores que albergan mutaciones poco comunes del EGFR (LUX-Lung 2, 3 y 6: En tres estudios clínicos de GIOTRIF® con genotipificación tumoral prospectiva (estudios de Fase 3 LUX-Lung 3 y - 6, y estudio de rama única de Fase 2 LUX-Lung 2), se llevó a cabo un estudio de datos de un total de 75 pacientes sin tratamiento previo con TKI que padecían adenocarcinomas pulmonares avanzados (estadio IIIb-IV) que albergaban mutaciones poco comunes del EGFR, definidas como todas las mutaciones excepto las mutaciones Del 19 y L858R. Los pacientes fueron tratados con GIOTRIF® 40 mg (en los tres estudios) o 50 mg (LUX- Lung 2) por vía oral, una vez al día.
En los pacientes con tumores que albergaban mutaciones de tipo sustitución ya sea G719X (N=18), L861Q (N=16) o S768I (N=8), la ORR confirmada fue del 72,2%, 56,3%, 75,0%, respectivamente, y la mediana de duración de la respuesta fue de 13,2 meses, 12,9 meses y 26,3 meses, respectivamente.
En pacientes con tumores que albergan inserciones en el exón 20 (N=23) la ORR confirmada fue del 8,7% y la mediana de duración de la respuesta fue de 7,1 meses. En pacientes con tumores que albergan mutaciones T790M de-novo (N=14) la ORR confirmada fue del 14,3% y la mediana de duración de la respuesta fue de 8,3 meses.
GIOTRIF® en pacientes con tratamiento previo con un EGFR TKI:
LUX-Lung 1 (1200.23): En un estudio internacional, doble ciego, comparativo con placebo, de Fase IIb/III (LUX-Lung 1), se evaluó la eficacia y seguridad de GIOTRIF® en pacientes con NSCLC localmente avanzado o metastásico (estadio IIIB o IV) que habían recibido previamente 1 o 2 líneas de quimioterapia y que tuvieron progresión después del tratamiento con un EGFR TKI (gefitinib o erlotinib). En este estudio se reclutaron 585 pacientes que fueron aleatorizados (2:1) a recibir 50 mg de GIOTRIF® una vez al día más el mejor cuidado de soporte (BSC) (N = 390) o placebo más BSC (N = 195).
La población del estudio estuvo clínicamente enriquecida por mutaciones del EGFR, al requerir que los pacientes hubieran tenido tratamiento previo con un EGFR TKI durante al menos 12 semanas.
El criterio de valoración primario del estudio fue la OS. Los criterios de valoración secundarios incluyeron PFS, ORR, DCR y calidad de vida relacionada con la salud (HRQoL), evaluada mediante los cuestionarios QLQ-C30, QLQ-LC13 y EQ-5D de la EORTC. La PFS fue evaluada por el investigador y por un comité de revisión independiente.
Entre los 585 pacientes aleatorizados, el 60% había recibido 1 línea y el 39% había recibido 2 líneas de quimioterapia previa para la enfermedad metastásica. El 55% de los pacientes había recibido tratamiento previo con erlotinib como EGFR TKI, el 40% había recibido gefitinib y el 5% había recibido ambos. Un total de 214 (36,5%) pacientes cumplieron los criterios de resistencia adquirida a erlotinib/gefitinib (es decir, CR/PR a erlotinib/gefitinib o SD ≥ 6 meses con el régimen previo de erlotinib/gefitinib, progresión dentro de las últimas 4 semanas con erlotinib/gefitinib y ningún tratamiento sistémico de intervención previo al tratamiento con GIOTRIF®).
Se realizaron pruebas opcionales de mutaciones del EGFR en 141 pacientes, de los cuales 96 (68%) tuvieron un resultado positivo, lo que indica una elevada tasa de positividad para mutaciones en la población total del estudio. Una duración prolongada del tratamiento previo con un EGFR TKI (≥ 48 semanas) y/o una respuesta del tumor (CR/PR) al EGFR TKI previo estuvieron asociadas con una mayor probabilidad de positividad para mutaciones del EGFR (subpoblación altamente enriquecida en términos de mutaciones del EGFR; ver Tabla 8).
|
Tabla 7. Enriquecimiento clínico en términos de mutaciones del EGFR dentro de la población total del estudio LUX-Lung 1 (N = 585) |
||||
|
Subpoblación altamente enriquecida en mutaciones del EGFR (CR/PR a EGFR TKI previo y/o ≥ 48 semanas de EGFR TKI)1 |
Cantidad de pacientes de la subpoblación |
Cantidad de pacientes con resultados interpretables en la prueba de mutaciones del EGFR2 |
Cantidad de pacientes con resultados positivos en la prueba de mutaciones del EGFR |
Proporción estimada de pacientes con mutaciones del EGFR |
|
Sí |
391 |
103 |
86 |
83% |
|
No |
194 |
38 |
10 |
26% |
|
1 CR = respuesta completa; PR = respuesta parcial 2 Sobre la base de las pruebas del tejido archivado (analizado en forma central o local) |
||||
Los resultados de eficacia se resumen en la Tabla 7. El criterio de valoración primario de OS no indicó ninguna diferencia estadística entre los pacientes tratados con GIOTRIF® y aquellos que recibieron placebo (10,8 meses vs. 12,0 meses). Lo más probable es que este resultado sea atribuible al tiempo inesperadamente prolongado de sobrevida observado tras la progresión en ambos grupos de tratamiento y a la mayor duración y la elevada frecuencia de tratamientos oncológicos posteriores, que evidenciaron un desequilibrio a favor del grupo de placebo. El criterio de valoración secundario de PFS evidenció una mejoría estadísticamente significativa en la mediana de la PFS entre los pacientes tratados con GIOTRIF® y aquellos que recibieron placebo (3,3 meses vs. 1,1 meses). El porcentaje de pacientes vivos y sin progresión a los 6 meses fue del 26,1% en el caso de GIOTRIF® y del 6,0% en el caso del placebo. Esta diferencia fue más pronunciada cuando se compararon las subpoblaciones con un elevado grado de enriquecimiento clínico por mutaciones del EGFR (N = 391) (PFS 4,4 meses vs. 1,0 mes; tasa de PFS a 6 meses 29,9% vs. 3,7%). En la subpoblación complementaria que no cumplió con los criterios de alto enriquecimiento clínico (N = 194), las diferencias entre GIOTRIF® y el placebo fueron menores (PFS 2,76 vs. 1,84 meses, tasa de PFS a 6 meses 18,7% vs. 11,5%).
|
Tabla 8. Resultados de eficacia de GIOTRIF® más BSC vs. placebo más BSC en el estudio LUX-Lung 1 obtenidos a partir del análisis primario al 8 de julio de 2010 |
||||||
|
Población total del estudio (N = 585; est. 68% positivo para mutaciones del EGFR) |
Subpoblación altamente enriquecida (N = 391; est. 83% positivo para mutaciones del EGFR) |
|||||
|
GIOTRIF® + BSC |
Placebo + BSC |
Razón de riesgos / Cociente de probabilidades (IC del 95%) Valor p4 |
GIOTRIF® + BSC |
Placebo + BSC |
Razón de riesgos / Cociente de probabilidades (IC del 95%) Valor p4 |
|
|
Cantidad de pacientes |
390 |
195 |
- |
257 |
134 |
- |
|
OS Meses (mediana) 358 eventos |
10,8 |
12,0 |
HR 1,08 (0,86-1,35) 0,7428 |
11,8 |
11,2 |
HR 0,90 (0,69-1,18) 0,433 |
|
OS actualizada1 Meses (mediana) 501 eventos |
10,9 |
11,7 |
HR 1,01 (0,84-1,22) 0,5445 |
12,0 |
11,2 |
HR 0,91 (0,73-1,14) 0,419 |
|
PFS Meses (mediana)2 Tasa de PFS a 6 meses |
3,3 26,1% |
1,1 6,0% |
HR 0,38 (0,31-0,48) < 0,0001 |
4,4 29,9% |
1,0 3,7% |
HR 0,28 (0,21-0,36) < 0,0001 |
|
Tasa de respuesta objetiva (CR+PR)2,3 |
7,4% |
0,5% |
OR 15,61 (2,1-115) 0,0071 |
8,9% |
0,7% |
OR 13,07 (1,7-97,9) 0,0123 |
|
Tasa de control de la enfermedad (CR+PR+SD)2,3 |
58,2% |
18,5 % |
OR 6,28 (4,1-9,5) < 0,0001 |
63,8% |
15,7% |
OR 9,49 (5,6-16,1) < 0,0001 |
|
1 Análisis de sobrevida actualizado al 9 de febrero de 2012 2 Sobre la base de la revisión independiente 3 CR = respuesta completa; PR = respuesta parcial; SD = enfermedad estable 4 Valor p para la PFS/OS basado en la prueba de rangos logarítmicos estratificada; valor p para la Tasa de Respuesta Objetiva y Tasa de Control de la Enfermedad basado en regresión logística. |
||||||
En un análisis de los pacientes que cumplieron con los criterios de resistencia adquirida (133 [34%] pacientes tratados con GIOTRIF®, 81 [42%] de los pacientes que recibieron placebo), la mediana de PFS determinada por la revisión independiente fue de 4,5 meses (IC del 95% 2,73- 4,73) para los pacientes tratados con GIOTRIF® en comparación con 1,0 mes (IC del 95% 0,95- 1,71) para los pacientes que recibieron placebo.
La actividad de GIOTRIF® en este subgrupo (en el cual el intervalo entre el final del tratamiento previo con un EGFR TKI y el inicio del tratamiento del estudio fue ≤ 4 semanas) sugiere que el efecto de GIOTRIF® no es un mero resultado de la reexposición a un tratamiento orientado al EGFR.
El beneficio en términos de la PFS estuvo acompañado de una mejoría en los síntomas relacionados con la enfermedad, según lo determinado por las herramientas QLQ-C30 y QLQ- LC13 de la EORTC. GIOTRIF® prolongó significativamente el tiempo hasta el deterioro de los síntomas preespecificados de tos a razón de más de 3 meses (HR 0,60; p < 0,001) en comparación con el placebo. El tiempo hasta el deterioro de la disnea (HR 0,84; p = 0,1701) y el dolor (HR 0,88; p = 0,2876) también fue más prolongado con GIOTRIF®, pero no alcanzó significación estadística. Una cantidad significativamente mayor de pacientes tratados con GIOTRIF® en comparación con aquellos tratados con placebo tuvo mejorías en la tos (46% vs. 25%, p < 0,0001), la disnea (51% vs. 36%, p = 0,0006) y el dolor (50% vs. 32%, p < 0,0001).
Los puntajes medios a través del tiempo para la HRQoL se midieron utilizando la herramienta QLQ-C30 de la EORTC. Los puntajes medios a lo largo del tiempo para la calidad de vida general y el estado de salud general fueron significativamente mejores para GIOTRIF® que para el placebo. Los puntajes medios fueron significativamente mejores en 1 de los 5 dominios funcionales (físico) y favorecieron a GIOTRIF® en el dominio del rol, y no indicaron ninguna diferencia en 3 de los 5 dominios (desempeño social, cognitivo y emocional).
LUX-Lung 5 (1200.42): LUX-Lung 5 es un estudio internacional, de diseño abierto, aleatorizado, de Fase III, en una población de pacientes enriquecida con mutaciones del EGFR similar a la del estudio LUX-Lung 1. En la Parte A, los pacientes con NSCLC metastásico, de estadio IIIB/IV, confirmado patológicamente, luego de ≥ 1 línea de quimioterapia y progresión tras el tratamiento con gefitinib o erlotinib durante generalmente 12 semanas o más, recibieron 50 mg de GIOTRIF® una vez al día hasta la progresión de la enfermedad. Después de la progresión, en la Parte B, los pacientes con beneficio clínico (≥ 12 semanas) fueron elegibles para continuar con 40 mg de GIOTRIF® más paclitaxel o para recibir la quimioterapia elegida por el investigador.
El criterio de valoración primario para la Parte A fue la evaluación de la PFS efectuada por el investigador. Los criterios de valoración secundarios fueron ORR, DCR y OS.
Se llevó a cabo un análisis intermedio de la Parte A, el cual confirmó los resultados del estudio LUX-Lung 1. La mediana de PFS en la población total del estudio (N = 1154) fue 3,3 meses, la tasa de PFS a 6 meses fue 24,5%, y la ORR y la DCR fueron del 7,6% y del 63,6%, respectivamente. En forma similar a lo observado en el estudio LUX-Lung 1, la eficacia en el subgrupo altamente enriquecido clínicamente (N = 598) fue más pronunciada, con una mediana de PFS de 4,2 meses y una ORR y una DCR de 9,5% y 72,2%, respectivamente.
La cantidad de pacientes con un evento de OS fue 301 (26,1%). La mediana de la OS fue 13,70 meses.
GIOTRIF en pacientes con NSCLC de histología escamosa
LUX-Lung 8 (1200.125): La eficacia y seguridad de GIOTRIF® como tratamiento de segunda línea para pacientes con NSCLC avanzado de histología escamosa se investigó en un estudio internacional de diseño abierto aleatorizado de Fase III LUX-Lung 8. Los pacientes que recibieron como mínimo 4 ciclos de terapia a base de platino en el contexto del tratamiento de primera línea posteriormente fueron aleatorizados 1:1 a una dosis diaria de 40 mg de GIOTRIF® o 150 mg de erlotinib hasta la progresión. Se permitió el aumento escalonado de la dosis de GIOTRIF® hasta 50 mg después del primer ciclo (28 días) de tratamiento en el caso de que el paciente no presentara ningún evento adverso relacionado con el medicamento o de que, en el caso de presentarse, el evento adverso fuera limitado (es decir, ausencia de diarrea, exantema, estomatitis y/u otros eventos relacionados con el fármaco > Grado 1 de los CTCAE), y de que cumpliera con el régimen posológico y no hubiera tenido ninguna reducción de la dosis en el pasado. La aleatorización se estratificó en función de la raza (asiática oriental vs. asiática no oriental). El criterio de valoración primario fue la PFS (analizado cuando fueron informados al menos 372 eventos a partir de una revisión independiente); la OS fue el criterio de valoración secundario clave (analizado al producirse las primeras 632 muertes). Otros criterios de valoración secundarios incluyeron ORR, DCR, cambio en el tamaño del tumor y HRQOL.
Entre los 795 pacientes aleatorizados, la mayoría fueron de sexo masculino (83,8%), de raza blanca (72,8%), fumadores o ex-fumadores (91,6%) con estado funcional ECOG 1 inicial (66,8%).
GIOTRIF® como tratamiento de segunda línea mejoró significativamente la PFS y la OS de pacientes con NSCLC escamoso en comparación con erlotinib. En el análisis primario de PFS la mediana de PFS fue de 2,43 meses en el grupo de GIOTRIF® y de 1,94 meses en el de erlotinib (HR=0,82, IC del 95% (0,676, 0,998), p=0,0427). El análisis final de PFS que incluyó a todos los pacientes aleatorizados confirmó los resultados anteriores (Tabla 10). El análisis primario de OS demostró una reducción significativa del riesgo de muerte para pacientes tratados con GIOTRIF® en comparación con erlotinib (HR=0,81, IC del 95% (0,69, 0,95), p=0,0077) con proporciones significativamente más altas de pacientes tratados con GIOTRIF® que se hallaban con vida en los parámetros de referencia a lo largo del período de observación, tales como 12 y 18 meses posteriores a la aleatorización.
Las tasas de respuesta tumoral objetiva y estabilización de la enfermedad fueron mayores con GIOTRIF®. La mediana de duración de la respuesta fue de 7,29 meses en el caso de GIOTRIF® y de 3,71 en el caso de erlotinib.
Tabla 9. Resultados de eficacia de GIOTRIF® vs. erlotinib en el estudio LUX-Lung 8, obtenidos a partir del análisis primario de OS, incluidos todos los pacientes aleatorizados
|
Tabla 9. Resultados de eficacia de GIOTRIF® vs. erlotinib en el estudio LUX-Lung 8, obtenidos a partir del análisis primario de OS, incluidos todos los pacientes aleatorizados |
|||
|
GIOTRIF® (N = 398) |
Erlotinib (n =397) |
Razón de riesgos/ Cociente de probabilidades (IC del 95%) valor p1 |
|
|
PFS Meses (mediana) |
2,63 |
1,94 |
HR 0,81 (0,69, 0,96) 0,0103 |
|
OS Meses (mediana) Vivo a los 12 meses Vivo a los 18 meses |
7,92 36,4% 22,0% |
6,77 28,2% 14,4% |
HR 0,81 (0,69, 0,95) 0,0077 |
|
Tasa de respuesta objetiva (CR+PR)* |
5,5% |
2,8% |
OR 2,06 (0,98, 4,32) 0,0551 |
|
Tasa de control de la enfermedad (CR+PR+SD)* |
50,5% |
39,5% |
OR 1,56 (1,18, 2,06) 0,0020 |
|
*CR = respuesta completa; PR = respuesta parcial; SD = enfermedad estable 1Valor p para la PFS/OS basado en la prueba de rangos logarítmicos estratificada; valor p para la Tasa de Respuesta Objetiva y Tasa de Control de la Enfermedad basado en regresión logística. |
|||
Figura 3. Curvas de Kaplan-Meier para la OS por grupo de tratamiento, en el Estudio LUX-Lung 8
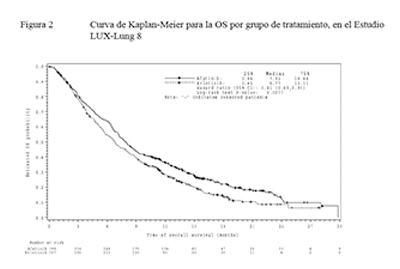
Referencias de la figura: Estimated OS Probability: Probabilidad de OS estimada.
Time of overall survival (months): Tiempo de sobrevida general (meses).
Median: Mediana.
Hazard ratio: Razón de riesgos.
Los análisis de los resultados informados por pacientes, según los cuestionarios QLQ-C30 y QLQ-LC13 favorecieron a GIOTRIF®. Una cantidad significativamente mayor de pacientes en el grupo tratado con GIOTRIF® informó una mejoría del estado de salud general/calidad de vida en comparación con erlotinib (35,7% vs. 28,3%, p=0,0406). Una mayor proporción de pacientes tratados con GIOTRIF® presentó una mejoría de la tos (43,4% vs. 35,2%, p=0,0294) y la disnea (51,3% vs. 44,1%, p=0,0605), mientras que no se observaron diferencias en relación al dolor (40,2% vs. 39,2%, p=0,7752). GIOTRIF® retrasó significativamente el tiempo hasta el deterioro de la disnea (HR 0,79, p=0,0078). Los puntajes medios a lo largo del tiempo para la tos, la disnea, y el dolor, así como para los dominios funcionales físico, rol, cognitivo, y de desempeño emocional fueron significativamente mejores con GIOTRIF® que con erlotinib.
Farmacocinética:
Absorción y distribución: Tras la administración oral de GIOTRIF®, las concentraciones máximas (Cmáx) de afatinib se observan aproximadamente de 2 a 5 horas después de la administración de la dosis. La media de los valores de Cmáx y AUC0-∞ aumentó en forma ligeramente más que proporcional en el rango de dosis de 20 mg a 50 mg de GIOTRIF®. La exposición sistémica a afatinib disminuyó en un 50% (Cmáx) y un 39% (AUC0-∞) cuando se administró con una comida de alto contenido graso en comparación con la administración en ayunas. Sobre la base de los datos de farmacocinética poblacional obtenidos de estudios clínicos en diversos tipos de tumores, se observó una disminución promedio del 26% en el AUCτ,ss cuando se consumieron alimentos dentro del lapso de 3 horas antes o 1 hora después de la toma de GIOTRIF®. Por lo tanto, no se deben consumir alimentos 3 horas antes y 1 hora después, como mínimo, de la toma de GIOTRIF® (ver las secciones Posología y Administración e Interacciones). Después de la administración de GIOTRIF®, la biodisponibilidad relativa media fue del 92% (cociente de la media geométrica ajustada del AUC0-∞) cuando se la comparó con una solución oral. La unión in vitro de afatinib a las proteínas plasmáticas humanas es de aproximadamente un 95%.
Metabolismo y excreción: Las reacciones metabólicas catalizadas por enzimas juegan un papel insignificante para afatinib in vivo. Los aductos covalentes a proteínas son los principales metabolitos circulantes de afatinib.
Tras la administración de una solución oral de 15 mg de afatinib, el 85,4% de la dosis se recuperó en las heces y el 4,3% en la orina. El compuesto original afatinib representó el 88% de la dosis recuperada. La vida media terminal aparente es de 37 horas. Las concentraciones plasmáticas de afatinib en estado de equilibrio dinámico se alcanzan dentro de los 8 días de la administración de dosis múltiples de afatinib, lo cual conduce a una acumulación de 2,77 veces (AUC) y 2,11 veces (Cmáx).
Insuficiencia renal: Menos del 5% de una dosis única de afatinib se excreta a través de los riñones. [78] Se comparó la exposición a afatinib de sujetos con insuficiencia renal y la de voluntarios sanos tras la administración de una dosis única de 40 mg de GIOTRIF® [16]. La exposición de los sujetos con insuficiencia renal moderada (índice de filtración glomerular estimado [eGFR] de 30 a 59 ml/min según la fórmula MDRD) fue 101% (Cmax) y 122% (AUC0-tz) en comparación con los voluntarios sanos. La exposición de los sujetos con insuficiencia renal grave (eGFR de 15 a 29 ml/min de acuerdo con la fórmula MDRD) fue 122% (Cmax) y 150% (AUC0-tz) en comparación con los voluntarios sanos. Según este estudio y el análisis farmacocinético poblacional de los datos obtenidos de ensayos clínicos efectuados en diversos tipos de tumor, se concluye que no es necesario ajustar la dosis inicial de los pacientes con insuficiencia renal leve (eGFR 60-89 ml/min), moderada (eGFR 30-59 ml/min) o grave (eGFR 15-29 ml/min), pero que se debe realizar un monitoreo de los pacientes que sufren insuficiencia grave (ver las secciones Análisis farmacocinético poblacional en poblaciones especiales a continuación y Posología y Administración). No se ha estudiado GIOTRIF® en pacientes con un eGFR < 15 ml/min o sometidos a diálisis.
Insuficiencia hepática: Afatinib es eliminado principalmente por vía de excreción biliar/fecal. Los sujetos con insuficiencia hepática leve (Child Pugh A) o moderada (Child Pugh B) tuvieron una exposición similar en comparación con los voluntarios sanos después de una dosis única de 50 mg de GIOTRIF®. Esto coincide con los datos de farmacocinética poblacional obtenidos de estudios clínicos en diversos tipos de tumor (ver la sección Análisis farmacocinético poblacional en poblaciones especiales). No se considera necesario ningún ajuste de dosis inicial en los pacientes con insuficiencia hepática leve o moderada (ver la sección Posología y Administración). No se ha estudiado la farmacocinética de afatinib en sujetos con disfunción hepática grave (Child Pugh C) (ver la sección Advertencias y Precauciones especiales).
Análisis farmacocinético en poblaciones objetivo: Se realizó un análisis de farmacocinética poblacional en 927 pacientes con cáncer que recibieron monoterapia de GIOTRIF®. No se considera necesario ningún ajuste de dosis inicial para ninguna de las siguientes covariables estudiadas.
Edad: No se pudo observar ninguna repercusión significativa de la edad (rango: 28 a 87 años) sobre la base de la farmacocinética de afatinib.
Peso corporal: La exposición plasmática (AUCτ,ss) se incrementó un 26% para un paciente de 42 kg (percentil 2,5) y se redujo un 22% para un paciente de 95 kg (percentil 97,5) en relación con un paciente con un peso corporal de 62 kg (mediana del peso corporal de los pacientes de la población general del estudio).
Sexo: Las mujeres tuvieron una exposición plasmática (AUCτ,ss, con corrección para peso corporal) un 15% más alta que los hombres.
Raza: No existe ninguna diferencia estadísticamente significativa en la farmacocinética de afatinib entre los pacientes asiáticos y los caucásicos. Tampoco se detectaron diferencias evidentes en la farmacocinética para los pacientes indoamericanos, nativos de Alaska o negros, sobre la base de los limitados datos disponibles de estas poblaciones (6 y 9 de 927 pacientes incluidos en el análisis, respectivamente).
Insuficiencia renal: La exposición a GIOTRIF® se incrementó moderadamente conforme menor fue el nivel de depuración de creatinina (CrCL), es decir, para un paciente con una CrCL de 60 o 30 ml/min, la exposición (AUCτ,ss) a afatinib aumentó un 13% y un 42%, respectivamente, y disminuyó un 6% y un 20% para un paciente con una CrCL de 90 o 120 ml/min, respectivamente, en comparación con un paciente con una CrCL de 79 ml/min (mediana de CrCL de la población de pacientes total analizada).
Insuficiencia hepática: La insuficiencia hepática leve y moderada, identificada mediante resultados anómalos en las pruebas de función hepática, no se correlacionó con ningún cambio significativo en la exposición de afatinib.
Otras características/factores intrínsecos de los pacientes: Otras características/factores intrínsecos de los pacientes que tuvieron una repercusión significativa sobre la exposición a afatinib fueron: estado funcional ECOG, niveles de lactato deshidrogenasa, niveles de fosfatasa alcalina y proteínas totales. Las magnitudes del efecto individual de estas covariables no se consideraron clínicamente relevantes.
Los antecedentes de tabaquismo, el consumo de alcohol o la presencia de metástasis hepáticas no tuvieron ninguna repercusión significativa en la farmacocinética de afatinib.
USO EN POBLACIONES ESPECIALES: Embarazo, lactancia y fertilidad: Embarazo: Los estudios no clínicos con afatinib no han revelado ningún signo de teratogenia hasta los niveles de dosis letales para las madres, inclusive. Los cambios adversos estuvieron limitados a los niveles de dosis francamente tóxicos (ver la sección Toxicología).
No existe ningún estudio en embarazadas en los que se haya utilizado GIOTRIF®. Por lo tanto, el potencial de riesgo para los seres humanos es desconocido. Se debe advertir a las mujeres con capacidad reproductiva que eviten quedar embarazadas mientras reciben tratamiento con GIOTRIF®. Se deben utilizar métodos anticonceptivos adecuados durante el tratamiento y durante por lo menos 2 semanas después de la última dosis. Si GIOTRIF® se usa durante el embarazo o si la paciente queda embarazada mientras recibe GIOTRIF®, se le debe informar de los posibles riesgos para el feto.
Lactancia: Sobre la base de los datos no clínicos (ver la sección Toxicología), es probable que afatinib sea excretado en la leche humana. No se puede excluir un riesgo para el lactante. Se debe informar a las madres que no deben amamantar mientras reciban GIOTRIF®.
Fertilidad: No se han realizado estudios de fertilidad en seres humanos con GIOTRIF®. Los datos de toxicología no clínicos disponibles han indicado efectos sobre los órganos reproductores con dosis elevadas (ver la sección Toxicología). Por lo tanto, no se puede descartar la posibilidad de que el tratamiento con GIOTRIF® tenga efectos adversos sobre la fertilidad en los seres humanos.
PROPIEDADES FARMACOLÓGICAS: Grupo farmacoterapéutico: Otros agentes antineoplásicos - inhibidores de la proteína cinasa.
Código ATC: L01XE13.
CONTRAINDICACIONES: GIOTRIF® está contraindicado en pacientes con hipersensibilidad conocida a afatinib o a cualquiera de los excipientes.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR VEHÍCULOS Y OPERAR MAQUINARIA: No se ha realizado ningún estudio de los efectos sobre la capacidad para conducir vehículos u operar maquinarias.
REACCIONES ADVERSAS: La evaluación de seguridad de GIOTRIF® se basa en datos obtenidos de estudios clínicos y de la experiencia posterior a la comercialización.
NSCLC
Estudios controlados: En el estudio pivote LUX-Lung 3 (1200.32), un total de 229 pacientes no tratados previamente con un EGFR TKI fueron tratados con GIOTRIF® con una dosis inicial de 40 mg administrada una vez al día. Un total de 111 pacientes fueron tratados con pemetrexed/cisplatino. La incidencia general de reacciones adversas al medicamento (RAM) en los pacientes tratados con GIOTRIF® fue similar a la observada con pemetrexed/cisplatino (100% vs. 96%). La incidencia de las RAM diarrea (95% vs. 15%) y exantema/acné (89% vs. 6%) fue más alta en los pacientes tratados con GIOTRIF® que en aquellos tratados con pemetrexed/cisplatino, respectivamente. En el 57% de los pacientes tratados con GIOTRIF® se efectuaron reducciones de dosis a raíz de los eventos adversos. Las reducciones de la dosis observadas en el conjunto de pacientes condujeron a una menor frecuencia de eventos adversos comunes (p. ej., después de la primera reducción de la dosis, la frecuencia de la diarrea, independientemente de la causalidad, se redujo del 96% al 52%).
La interrupción del tratamiento a raíz de RAM fue menor en los pacientes que recibieron 40 mg de GIOTRIF® una vez al día en comparación con aquellos que recibieron pemetrexed/cisplatino (8% vs. 12%). En los pacientes tratados con GIOTRIF®, las tasas de interrupción debido a las RAM diarrea y exantema/acné fueron del 1,3% y del 0%, respectivamente.
En el estudio LUX-Lung 6 (1200.34), un total de 239 pacientes no tratados previamente con un EGFR TKI fueron tratados con GIOTRIF® con una dosis inicial de 40 mg, administrada una vez al día. Un total de 113 pacientes fueron tratados con gemcitabina/cisplatino. La incidencia general de RAM en pacientes tratados con GIOTRIF® fue similar a la observada con gemcitabina/ cisplatino (98,7% vs. 99,1%). La incidencia de las RAM diarrea (88,7% vs. 10,6%) y exantema/acné (81,2% vs. 8,8%) fue más alta en los pacientes tratados con GIOTRIF® que en aquellos tratados con gemcitabina/cisplatino. Hubo reducciones de dosis debido a los eventos adversos en el 33,1% de los pacientes tratados con GIOTRIF® y en el 26,5% de los pacientes tratados con gemcitabina/cisplatino. Las interrupciones de la medicación del estudio debido a las RAM fueron menos frecuentes en los pacientes que recibieron GIOTRIF® en comparación con gemcitabina/cisplatino (6,3% vs. 39,8%). En los pacientes tratados con GIOTRIF®, las tasas de interrupción debido a las RAM diarrea y exantema/acné fueron 0% y 2,5%, respectivamente.
En el estudio de aval controlado con placebo LUX-Lung 1 (1200.23), un total de 390 pacientes tratados previamente con un EGFR TKI fueron aleatorizados a GIOTRIF® y recibieron una dosis inicial de 50 mg una vez al día. Un total de 195 pacientes recibieron placebo. La incidencia general de RAM en los pacientes tratados con GIOTRIF® fue superior que en aquellos que recibieron placebo (95% vs. 38%). La incidencia de las RAM diarrea (85% vs. 6%) y exantema/acné (77% vs. 13%) fue más alta en los pacientes tratados con GIOTRIF®. En el 38% de los pacientes tratados con GIOTRIF® hubo reducciones de dosis a raíz de los eventos adversos. Las reducciones de la dosis observadas en el conjunto de pacientes condujeron a una menor frecuencia de eventos adversos comunes (p. ej., después de la primera reducción de la dosis, la frecuencia de la diarrea, independientemente de la causalidad, se redujo del 87% al 49%).
La tasa de interrupción del tratamiento a raíz de RAM fue más alta en los pacientes que recibieron 50 mg de GIOTRIF® una vez al día en comparación con aquellos que recibieron placebo (8% vs.< 1%). En los pacientes tratados con GIOTRIF®, las tasas de interrupción como consecuencia de eventos de diarrea y exantema/acné fueron del 3,6% y del 1,8%, respectivamente.
En el estudio pivote LUX-Lung 8 (1200.125), un total de 392 pacientes con NSCLC escamoso fueron tratados con GIOTRIF® con una dosis inicial de 40 mg administrada una vez al día y un total de 395 pacientes fueron tratados con 150 mg de erlotinib una vez al día. Después del primer ciclo de tratamiento (28 días) se aumentó gradualmente la dosis de GIOTRIF® a 50 mg en 39 (10%) pacientes. La incidencia general de RAM en los pacientes tratados con GIOTRIF® o erlotinib fue del 93% vs. 81% respectivamente. La incidencia de la RAM diarrea fue más alta en los pacientes tratados con GIOTRIF® en comparación con aquellos que recibieron erlotinib (70% vs. 33%), mientras que la incidencia de exantema/acné fue similar en ambos grupos (67% vs.67%). En el 27% de los pacientes tratados con GIOTRIF® hubo reducciones de dosis debido a los eventos adversos. El tratamiento se interrumpió a raíz de RAM en el 11% de los pacientes tratados con GIOTRIF®, y en el 5% de los pacientes tratados con erlotinib.
Todos los estudios de NSCLC con dosis diarias de 40 mg o 50 mg de GIOTRIF®
La seguridad de la monoterapia de GIOTRIF® con dosis iniciales de 40 mg o 50 mg administradas una vez al día se evaluó en análisis combinados de estudios de NSCLC en pacientes con mutaciones del EGFR o enriquecidos por ellas. El tipo de histología predominante en esta población de pacientes fue el adenocarcinoma de pulmón. Los tipos de RAM estuvieron generalmente asociados con el modo de acción inhibitorio del EGFR que posee afatinib y el perfil de las RAM fue concordante entre los estudios LUX-Lung 3 y LUX-Lung 1, respectivamente.
Las RAM de Grado 1 o 2 de los CTCAE se produjeron en el 58,8% y en el 53,1% de los pacientes tratados con 40 mg y 50 mg de GIOTRIF®, respectivamente. Las RAM de Grado 3 o 4 de los CTCAE se produjeron en el 38% y en el 41% de los pacientes tratados con 40 mg y 50 mg de GIOTRIF®, respectivamente. Para ambas dosis, en estas 2 poblaciones de pacientes diferentes, la mayoría de las RAM fueron Grado 1 o 2 de los CTCAE. Las RAM fueron tratables, según se describe en las secciones “Posología y administración” y “Advertencias y Precauciones especiales”. Esto se vio reflejado en las bajas tasas de interrupción del tratamiento debido a las RAM observadas para ambas dosis iniciales (7% y 11,7%).
En la Tabla 2 se proporciona un resumen de las RAM frecuentes diarrea y exantema/acné en una población con NSCLC positivo para mutación del EGFR o enriquecida por ella, tratada con la monoterapia de GIOTRIF®.
|
Tabla 2. Análisis combinados de eventos de diarrea y exantema/acné relacionados con el fármaco tomados de una población con NSCLC positivo para mutación del EGFR o enriquecida por ella que recibió monoterapia de GIOTRIF® en estudios clínicos |
||
|
Pacientes no tratados previamente con un EGFR TKI (dosis inicial 40 mg/día) N = 497 |
Pacientes tratados previamente con un EGFR TKI (dosis inicial 50 mg/día) N = 1638 |
|
|
Exantema/acné de Grado 3 según CTCAE |
14,3% |
11,8% |
|
Diarrea de Grado 3 según CTCAE |
9,9% |
17,6% |
|
Interrupción debido a exantema/acné (todos los grados) |
1,2% |
1,9% |
|
Interrupción debido a diarrea (todos los grados) |
0,6% |
4,5% |
De los pacientes que recibieron una dosis inicial de 40 mg, 1 paciente (0,2%) tuvo en evento de exantema/acné de Grado 4. Entre los pacientes que recibieron una dosis inicial de 50 mg, 1 paciente (0,1%) tuvo en evento de exantema/acné de Grado 4, y 3 pacientes (0,2%) tuvieron diarrea de Grado 4.
La seguridad de la monoterapia de GIOTRIF® se evaluó en el estudio LUX-Lung 8 en pacientes con carcinoma de pulmón de células escamosas que recibieron una dosis inicial de 40 mg. Las RAM más frecuentes estuvieron asociadas con el modo de acción inhibitorio del EGFR que posee GIOTRIF® y fueron concordantes con las observadas en los estudios LUX-Lung 3 y LUX-Lung 1 en pacientes con adenocarcinoma de pulmón. La mayoría de los pacientes con RAM (65%) tuvo eventos de Grado 1 o 2. La RAM de Grado 3 / 4 de los CTCAE diarrea se produjo en el 9,9% / 0,5% de los pacientes. La tasa de exantema de grado 3 de los CTCAE relacionada con el fármaco fue del 5,9%. Las RAM condujeron a la interrupción del tratamiento en el 11% de los pacientes. La interrupción del tratamiento debido a las RAM diarrea y exantema/acné, independientemente del grado de gravedad, se produjo en el 3,8% y el 2,0% de los pacientes.
Resumen tabulado de las reacciones adversas: En la Tabla 3 se presentan las RAM clasificadas por SOC y términos preferentes del MedDRA reportadas a partir de cualquiera de los grupos de dosis de GIOTRIF®, ordenados por población, obtenidos a partir del total de estudios de NSCLC, con dosis iniciales de 40 mg y 50 mg de GIOTRIF® .
Tabla 3. Reacciones adversas identificadas para NSCLC (40 mg y 50 mg) y de GIOTRIF® como monoterapia:
Infecciones e infestaciones:
— Paroniquia
— Cistitis
Trastornos del metabolismo y de la nutrición:
— Disminución del apetito
— Deshidratación
— Hipopotasemia
Trastornos del sistema nervioso:
— Disgeusia
Trastornos oculares:
— Conjuntivitis
— Sequedad ocular
— Queratitis
Trastornos respiratorios, torácicos y mediastínicos:
— Epistaxis
— Rinorrea
— Enfermedad pulmonar intersticial
Trastornos gastrointestinales:
— Diarrea
— Estomatitis
— Queilitis
— Dispepsia
— Náuseas
— Vómitos
— Pancreatitis
— Perforación gastrointestinal
Trastornos hepatobiliares:
— Elevación de la alanina aminotransferasa
— Elevación de la aspartato aminotransferasa
Trastornos de la piel y del tejido subcutáneo:
— Exantema
— Dermatitis acneiforme
— Prurito
— Piel seca
— Alteraciones en las uñas
— Síndrome de eritrodisestesia palmoplantar
— Síndrome de Stevens-Johnson*
— Necrólisis epidérmica tóxica*
Trastornos musculoesqueléticos y del tejido conectivo:
— Espasmos musculares
Trastornos renales y urinarios:
— Insuficiencia renal / falla renal
Trastornos generales y afecciones del sitio de administración:
— Pirexia
Investigaciones:
— Descenso de peso
*derivados de la experiencia posterior a la comercialización
INTERACCIONES: Interacciones con BCRP: Estudios in vitro han mostrado que afatinib es un sustrato y un inhibidor del transportador BCRP. Afatinib puede aumentar la biodisponibilidad de sustratos de la BCRP administrados por via oral (entre ellos rosuvastatina y sulfazalasina).
Interacciones con la glucoproteína P (P-gp): Sobre la base de los datos in vitro, se ha determinado que afatinib es un sustrato de la P-gp. Los datos clínicos indican que la administración concomitante de inhibidores o inductores potentes de la P-gp pueden alterar la exposición a afatinib. Los resultados de un estudio de interacción farmacológica demostraron que GIOTRIF® puede combinarse de manera segura con inhibidores de la P-gp (como ritonavir) siempre que el inhibidor se administre de forma simultánea al GIOTRIF® o después de éste. Si se administran antes de GIOTRIF®, los inhibidores potentes de la P-gp (incluidos, entre otros, ritonavir, ciclosporina A, ketoconazol, itraconazol, eritromicina, verapamilo, quinidina, tacrolimus, nelfinavir, saquinavir y amiodarona) pueden aumentar la exposición a afatinib y deben utilizarse con precaución (ver las secciones Posología y Administración, Advertencias y Precauciones especiales y Farmacocinética).
Los inductores potentes de P-gp (incluidos, entre otros, rifampicina, carbamazepina, fenitoína, fenobarbital o hierba de San Juan) pueden reducir la exposición a afatinib (ver las secciones Advertencias y Precauciones especiales y Farmacocinética).
Efecto de los alimentos sobre afatinib: La coadministración de una comida de alto contenido graso con GIOTRIF® dio como resultado una disminución significativa de la exposición a afatinib de aproximadamente un 50% en lo que respecta a la Cmáx y de un 39% en lo que respecta al AUC0-∞. GIOTRIF® se debe administrar lejos de las comidas (ver las secciones Posología y Administración y Farmacocinética).
INTERACCIONES MEDICAMENTOSAS FARMACOCINÉTICAS: Transportadores del fármaco: Glucoproteína P (P-gp): Efecto de los inhibidores e inductores de la P-gp sobre afatinib: Se realizaron dos estudios para evaluar el efecto de ritonavir, un potente inhibidor de la P-gp, sobre la farmacocinética de afatinib. En un estudio, se investigó la biodisponibilidad relativa de afatinib cuando se administró ritonavir (200 mg dos veces al día durante tres días), ya sea en forma simultánea o 6 horas después de una dosis única de 40 mg de GIOTRIF®. La biodisponibilidad relativa de afatinib fue del 119% (AUC0-∞) y el 104% (Cmáx) cuando se administró de manera simultánea con ritonavir y de 111% (AUC0-∞) y el 105% (Cmáx) cuando ritonavir se administró 6 horas después de GIOTRIF®. En un segundo estudio, cuando ritonavir (200 mg dos veces al día durante tres días) se administró 1 hora antes de una dosis única de 20 mg de GIOTRIF®, la exposición a afatinib aumentó un 48% (AUC0-∞) y un 39% (Cmáx) (ver las secciones Posología y Administración, “Advertencias y Precauciones especiales e Interacciones).
El pretratamiento con rifampicina (600 mg por día durante 7 días), un potente inductor de la P-gp, disminuyó la exposición plasmática a afatinib un 34% (AUC0-∞) y un 22% (Cmáx) después de la administración de una dosis única de 40 mg de GIOTRIF® (ver las secciones Advertencias y Precauciones Especiales e Interacciones).
Efecto de afatinib sobre los sustratos de la P-gp: Sobre la base de los datos in vitro, se ha determinado que afatinib es un inhibidor moderado de la P-gp. Se considera improbable que el tratamiento con GIOTRIF® se traduzca en cambios en las concentraciones plasmáticas de otros sustratos de la P-gp.
Proteína de resistencia del cáncer de mama (BCRP)
Los estudios in vitro indicaron que afatinib es un sustrato y un inhibidor del transportador BCRP.
Sistemas de transporte de captación del fármaco: Los datos in vitro indicaron que se considera improbable que se produzcan interacciones medicamentosas con afatinib como consecuencia de la inhibición de los transportadores OATB1B1, OATP1B3, OATP2B1, OAT1, OAT3, OCT1, OCT2 y OCT3.
Enzimas metabolizadoras del fármaco:
Enzimas del citocromo P450 (CYP): Efectos de las enzimas del CYP inductoras e inhibidoras de afatinib: Los datos in vitro indicaron que es improbable que se produzcan interacciones medicamentosas con afatinib como consecuencia de una inhibición o inducción de las enzimas del CYP por la medicación concomitante. En los seres humanos, se ha determinado que las reacciones metabólicas catalizadas por enzimas desempeñan un papel insignificante en el metabolismo de afatinib. Aproximadamente el 2% de la dosis de afatinib fue metabolizada por FMO3 y la N- desmetilación dependiente del CYP3A4 fue demasiado baja para ser detectada en forma cuantitativa.
Efecto de afatinib sobre las enzimas del CYP: Afatinib no es un inhibidor ni un inductor de las enzimas del CYP. Por lo tanto, es improbable que GIOTRIF® afecte el metabolismo de otros fármacos que dependan de las enzimas del CYP.
UDP-glucuronosiltransferasa 1A1 (UGT1A1): Sobre la base de datos in vitro, se considera improbable que se produzcan interacciones medicamentosas con afatinib como consecuencia de la inhibición de UGT1A1.
Farmacodinamia:
Electrofisiología cardíaca: GIOTRIF® en dosis diarias de 50 mg no provocó ninguna prolongación significativa del intervalo QTcF tras la administración de dosis únicas y múltiples en pacientes con tumores sólidos recidivantes o refractarios. No hubo ningún hallazgo cardíaco de seguridad que fuera un motivo de inquietud desde el punto de vista clínico. Esto sugiere que GIOTRIF® no tiene un efecto relevante sobre el intervalo QTcF.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES: Evaluación del estado de mutación del EGFR: Cuando se evalúe el estado de mutación del EGFR de un paciente, es importante elegir una metodología bien validada y robusta para evitar resultados falsos negativos o falsos positivos.
Diarrea: Se ha informado diarrea, incluso diarrea grave, durante el tratamiento con GIOTRIF® (ver la sección Reacciones Adversas). La diarrea puede causar deshidratación con o sin insuficiencia renal, cuadro éste que, en casos raros, ha tenido un desenlace fatal. La diarrea usualmente se presentó dentro de las 2 primeras semanas de tratamiento. La diarrea de Grado 3 se presentó con mayor frecuencia dentro de las primeras 6 semanas de tratamiento. El tratamiento proactivo de la diarrea, que incluye la hidratación adecuada combinada con agentes antidiarreicos, en especial dentro de las primeras 6 semanas de tratamiento, es importante y debe iniciarse cuando aparecen los primeros signos de diarrea. Deben utilizarse antidiarreicos (p. ej., loperamida) y, de ser necesario, debe aumentarse gradualmente la dosis hasta llegar a la dosis aprobada recomendada máxima.
Los antidiarreicos deben estar fácilmente accequibles para los pacientes, de manera que el tratamiento pueda iniciarse ante los primeros signos de diarrea y continuarse hasta que las deposiciones blandas hayan cesado durante 12 horas. Puede ser necesario que los pacientes con diarrea grave tengan que suspender o reducir la dosis o interrumpir el tratamiento con GIOTRIF® (ver la sección Posología y administración). En caso de deshidratación, puede ser necesario que los pacientes reciban electrolitos y líquidos por vía intravenosa.
Eventos adversos cutáneos relacionados: En pacientes tratados con GIOTRIF® se ha informado exantema/acné (ver la sección Reacciones adversas). En general, el exantema se manifiesta como una erupción eritematosa y acneiforme leve o moderada, que puede producirse o empeorar en las zonas expuestas al sol. Se aconseja el uso de ropa de protección y/o el uso de pantalla solar en los pacientes que se expongan al sol. La intervención temprana (p. ej., emolientes, antibióticos) de las reacciones dermatológicas puede facilitar la continuación del tratamiento con GIOTRIF®.
Los pacientes con reacciones cutáneas graves o prolongadas también pueden requerir la suspensión temporaria del tratamiento, una reducción de la dosis (ver la sección Posología y administración), intervenciones terapéuticas adicionales o derivación a un especialista con experiencia en el tratamiento de estos efectos dermatológicos. Se han notificado cuadros cutáneos bullosos, vesiculares y exfoliativos, que incluyen casos raros indicativos de síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. Se debe suspender o interrumpir el tratamiento con GIOTRIF® si el paciente presenta cuadros bullosos, vesiculares o exfoliativos graves.
Sexo femenino, bajo peso corporal e insuficiencia renal subyacente: Se ha observado una mayor exposición a afatinib en pacientes de sexo femenino, en pacientes con bajo peso corporal y en pacientes con insuficiencia renal subyacente (ver la sección Farmacocinética). Esto podría dar como resultado un mayor riesgo de padecer eventos adversos mediados por el EGFR, como diarrea, exantema/acné y estomatitis. Se recomienda un monitoreo más estrecho en los pacientes con dichos factores de riesgo.
Enfermedad pulmonar intersticial (ILD): Se han informado casos de ILD o eventos similares a la ILD (como infiltración pulmonar, neumonitis, síndrome de distrés respiratorio agudo, alveolitis alérgica), que incluyen casos con desenlace fatal, en pacientes que recibieron GIOTRIF® para el tratamiento del NSCLC. Los eventos similares a la ILD relacionados con el fármaco de todos los grados se informaron en el 0,7% de pacientes tratados con GIOTRIF® en todos los estudios clínicos (0,5% de pacientes con NSCLC (ver la sección Reacciones adversas). No se han estudiado pacientes con antecedentes de ILD. Se debe realizar una evaluación cuidadosa de todos los pacientes con un comienzo agudo y/o un empeoramiento inexplicable de los síntomas pulmonares (disnea, tos, fiebre) para excluir la posibilidad de un cuadro de ILD. Se debe suspender la administración de GIOTRIF® mientras se investigan dichos síntomas. Si se diagnostica ILD, GIOTRIF® se interrumpirá de forma permanente y se instaurará el tratamiento adecuado según sea necesario (ver la sección Posología y administración).
Insuficiencia hepática grave: Se ha notificado insuficiencia hepática, que incluye casos con desenlace fatal, durante el tratamiento con GIOTRIF® en menos del 1% de los pacientes. En dichos pacientes, existían variables de distorsión tales como hepatopatía preexistente y/o comorbilidades asociadas con la progresión de la enfermedad maligna subyacente. Se recomienda la realización de pruebas de función hepática periódicas en los pacientes con afecciones hepáticas preexistentes. La suspensión de la dosis de GIOTRIF® puede llegar a resultar necesaria en pacientes que presenten empeoramiento de la función hepática (ver la sección Posología y administración). Se debe interrumpir el tratamiento con GIOTRIF® en los pacientes que desarrollen una insuficiencia hepática grave mientras reciban dicho fármaco.
Perforaciones gastrointestinales: Se han informado perforaciones gastrointestinales, inclusive casos fatales, durante el tratamiento con GIOTRIF® en el 0,2% de los pacientes en todos los estudios clínicos controlados aleatorizados. En la mayoría de los casos, se asoció la perforación gastrointestinal con otros factores de riesgo conocidos, inclusive las medicaciones concomitantes como los corticoides, AINE o agentes antiangiogénicos, antecedente subyacente de úlcera gastrointestinal, enfermedad diverticular subyacente, edad o metástasis en intestinos, en los sitios de perforación. En los pacientes que presentan perforación gastrointestinal mientras están recibiendo GIOTRIF®, se debe interrumpir el tratamiento de manera definitiva
Queratitis: Los pacientes que presenten síntomas como inflamación ocular aguda o que empeora, lagrimeo, sensibilidad a la luz, visión borrosa, dolor ocular y/u ojos rojos deben ser derivados sin demora a un especialista en oftalmología. Si se confirma un diagnóstico de queratitis ulcerativa, se debe suspender o interrumpir el tratamiento con GIOTRIF®. Si se diagnostica queratitis, los beneficios y riesgos de continuar el tratamiento deben ser cuidadosamente sopesados. GIOTRIF® debe utilizarse con precaución en pacientes con antecedentes de queratitis, queratitis ulcerativa o sequedad ocular grave. El uso de lentes de contacto constituye también un factor de riesgo para la queratitis y la ulceración (ver la sección Reacciones adversas).
Función ventricular izquierda: La disfunción ventricular izquierda se ha asociado con la inhibición del HER2. Sobre la base de los datos de estudios clínicos disponibles, no existen indicios de que GIOTRIF® provoque efectos adversos sobre la contractilidad cardíaca. Sin embargo, GIOTRIF® no ha sido estudiado en pacientes con anomalías de la fracción de eyección ventricular izquierda (FEVI) o antecedentes de afecciones cardíacas importantes. En los pacientes que tengan factores de riesgo cardíacos y en los pacientes con trastornos que puedan afectar la FEVI, debe considerarse un control cardíaco, incluida una evaluación de la FEVI al inicio del tratamiento con GIOTRIF® y durante el mismo. En los pacientes que desarrollan signos/síntomas cardíacos relevantes durante el tratamiento, debe considerarse un control cardíaco, que incluya la evaluación de la FEVI.
En los pacientes cuya fracción de eyección sea menor que el límite normal inferior de la institución, debe considerarse la realización de una consulta cardiológica y la suspensión o interrupción del tratamiento con GIOTRIF®.
Interacciones con la glucoproteína P (P-gp): Los inhibidores potentes de la P-gp pueden conducir a una mayor exposición a afatinib si se administran antes de la toma de GIOTRIF®, y es por ello que se deben utilizar con precaución. Si fuera necesario administrar inhibidores de la P-gp, deben administrarse en forma simultánea con la toma de GIOTRIF® o posteriormente. El tratamiento concomitante con inductores potentes de la P-gp puede reducir la exposición a afatinib (ver las secciones Posología y administración, Interacciones y Farmacocinética).
Lactosa: Este medicamento contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, deficiencia total de lactasa o problemas de absorción de glucosa o galactosa, no deben tomar este medicamento.
POSOLOGÍA Y ADMINISTRACIÓN: La dosis recomendada de GIOTRIF®:
• para los pacientes con NSCLC positivo para mutación del EGFR, no tratados previamente con un inhibidor de la cinasa de tirosina del EGFR (pacientes no tratados previamente con un EGFR TKI) es 40 mg por vía oral una vez al día.
• para los pacientes con NSCLC positivo para mutación del EGFR tratados previamente con un EGFR TKI es 50 mg por vía oral una vez al día.
• para los pacientes con NSCLC escamoso que han recibido previamente régimen de primera línea con platino es 40 mg por vía oral una vez al día.
GIOTRIF® se debe administrar sin alimentos. No se deben consumir alimentos al menos 3 horas antes y 1 hora después, como mínimo, de haber tomado GIOTRIF® (ver las secciones Interacciones y Farmacocinética). Los comprimidos deben tragarse enteros, con agua.
El tratamiento con GIOTRIF® se debe continuar hasta la progresión de la enfermedad o hasta que el paciente ya no lo tolere (ver Tabla 1 a continuación).
Método alternativo de administración: Si no es posible administrar los comprimidos enteros, los comprimidos de GIOTRIF® se pueden disolver en aproximadamente 100 ml de agua mineral sin gas. No se deben usar otros tipos de bebidas. El comprimido debe colocarse en el vaso con agua, sin aplastarlo, y se debe revolver de tanto en tanto el líquido durante 15 minutos hasta que el comprimido se desintegre en partículas muy pequeñas. La dispersión se debe ingerir de inmediato. El vaso se debe enjuagar con aproximadamente 100 ml de agua, que también se debe beber. La dispersión también se puede administrar mediante una sonda nasogástrica.
Posología:
Aumento escalonado de dosis: Se puede considerar un aumento escalonado de dosis hasta un máximo de 50 mg/día en los pacientes que toleren una dosis inicial de 40 mg/día (es decir, ausencia de diarrea, exantema, estomatitis y otros eventos relacionados con el fármaco de Grado > 1 según los CTCAE) en el primer ciclo de tratamiento (para la definición de ciclo de tratamiento ver Propiedades farmacológicas). La dosis no se debe aumentar en los pacientes con una reducción de dosis anterior. No está recomendado el aumento escalonado de dosis en pacientes tratados previamente con un EGFR TKI.
La dosis diaria máxima en todos los casos es 50 mg.
Ajuste de dosis por reacciones adversas: Las reacciones adversas al medicamento sintomáticas (p. ej., diarrea grave/persistente o reacciones adversas cutáneas) pueden tratarse exitosamente mediante la interrupción del tratamiento y la implementación de reducciones de la dosis de GIOTRIF® según se detalla en la Tabla 1 (ver la sección Reacciones adversas; para más detalles sobre el tratamiento de los eventos adversos (EA) específicos relacionados con el medicamento, ver la sección Advertencias y Precauciones especiales).
|
Tabla 1. Información sobre el ajuste de dosis por reacciones adversas |
||
|
Evento adverso relacionado con el medicamento según los CTCAEa |
Posología recomendada de GIOTRIF® |
|
|
Grado 1 o Grado 2 |
No interrumpirb |
Ningún ajuste de dosis |
|
Grado 2 (prolongadoc o intolerable) o Grado > 3 |
Interrumpir hasta Grado 0/1b |
Reanudar con reducción de dosis en disminuciones de 10 mgd |
|
a Criterios Terminológicos Comunes del NCI para Eventos Adversos v3.0 [12] b En el caso de diarrea, se deben administrar medicamentos antidiarreicos (p. ej., loperamida) de inmediato y continuarlos en caso de diarrea persistente hasta que cesen las deposiciones líquidas. c >48 horas de diarrea y/o >7 días de exantema d Si un paciente no puede tolerar 20 mg/día, se debe considerar la interrupción permanente de GIOTRIF® |
||
Si un paciente presenta un cuadro de síntomas respiratorios agudo o un empeoramiento de tales síntomas, se debe considerar la posibilidad de enfermedad pulmonar intersticial (ILD), en cuyo caso GIOTRIF® debe interrumpirse hasta tanto esté disponible el resultado de la evaluación. Si se diagnostica ILD, GIOTRIF® se debe interrumpir y se debe instaurar el tratamiento adecuado, según sea necesario [ver la sección Advertencias y Precauciones especiales].
Dosis omitidas: Si se omite una dosis de GIOTRIF®, se debe tomar durante el mismo día tan pronto como el paciente lo recuerde. No obstante, si la próxima dosis programada se debe tomar dentro de las 8 horas, entonces la dosis omitida se debe saltear.
POBLACIONES ESPECIALES: Pacientes con insuficiencia renal: Se observó un aumento de la exposición a afatinib en los pacientes con insuficiencia renal moderada o grave (ver la sección Farmacocinética). No es necesario realizar ajustes en la dosis inicial de los pacientes con insuficiencia renal leve, moderada o grave (eGFR 15- 29 ml/min). Debe realizarse un monitoreo de los pacientes con insuficiencia renal grave y ajustar la dosis de GIOTRIF® en caso de intolerancia. No se recomienda el tratamiento con GIOTRIF® en pacientes con eGFR < 15 ml/min o sometidos a diálisis.
Pacientes con insuficiencia hepática: La exposición a afatinib no cambia significativamente en pacientes con insuficiencia hepática leve (Child Pugh A) o moderada (Child Pugh B) (ver la sección Farmacocinética). No es necesario realizar ajustes en la dosis inicial en los pacientes con insuficiencia hepática leve o moderada. GIOTRIF® no ha sido estudiado en pacientes con insuficiencia hepática grave (Child Pugh C). No está recomendado el tratamiento con GIOTRIF® en esta población.
Edad, raza, sexo: No es necesario ningún ajuste de dosis en función de la edad, la raza o el sexo del paciente (ver la sección Farmacocinética).
Población pediátrica: La seguridad y la eficacia de GIOTRIF® no han sido estudiadas en pacientes pediátricos. Por lo tanto, no se recomienda el tratamiento con GIOTRIF® en niños o adolescentes.
Uso de inhibidores de la glucoproteína P (P-gp): En el caso de que sea necesario el tratamiento con inhibidores de la P-gp, los mismos deben administrarse en forma simultánea con GIOTRIF® o después de la toma de este fármaco (ver las secciones Advertencias y Precauciones especiales, Interacciones y Farmacocinética).
SOBREDOSIS: Síntomas: La dosis más alta de GIOTRIF® estudiada en una cantidad limitada de pacientes en estudios clínicos de Fase I es de 160 mg una vez al día durante 3 días y de 100 mg una vez al día durante 2 semanas. Las reacciones adversas observadas con esta dosis fueron principalmente de tipo dermatológico (exantema/acné) y gastrointestinal (especialmente diarrea). Los casos de sobredosis en 2 adolescentes sanos, que involucraron la ingesta de 360 mg de GIOTRIF® cada uno (como parte de una ingesta farmacológica mixta), estuvieron asociados con reacciones adversas al medicamento que consistieron en náuseas, vómitos, astenia, mareos, cefalea, dolor abdominal y elevación de la amilasa (< 1,5 veces el límite normal superior). Ambos sujetos se recuperaron de dichos eventos adversos.
Tratamiento: No existe un antídoto específico para la sobredosis de GIOTRIF®. Cuando se sospeche de una sobredosis, se debe suspender GIOTRIF® e instituir cuidado de soporte.
Si está indicado, se puede lograr la eliminación del afatinib no absorbido mediante vómitos o lavado gástrico.
TOXICOLOGÍA: La administración oral de dosis únicas a ratones y a ratas indicó un bajo potencial tóxico agudo de afatinib. En estudios de dosis orales repetidas de hasta 26 semanas de duración en ratas, o de 52 semanas en cerdos miniatura, los principales efectos se identificaron en la piel (cambios dérmicos, atrofia epitelial y foliculitis en ratas), en el aparato gastrointestinal (diarrea, erosiones estomacales, atrofia epitelial en ratas y cerdos miniatura) y en los riñones (necrosis papilar en ratas). Dependiendo de los hallazgos, dichos cambios se produjeron ante niveles de exposición ubicados por encima, por debajo o dentro del rango de valores clínicamente relevantes. Además, en varios órganos se observó atrofia del epitelio mediada farmacodinámicamente en ambas especies.
Toxicidad reproductiva: Debido a su mecanismo de acción, GIOTRIF® tiene el potencial de causar daño fetal. Los estudios de desarrollo embriofetal realizados con afatinib no revelaron ningún indicio de teratogenia en niveles que alcanzan las concentraciones letales para las madres. Los cambios identificados estuvieron limitados a alternaciones esqueléticas que consistieron en osificaciones incompletas/elementos no osificados (ratas) y abortos en niveles de dosis tóxicos para las madres, reducción del peso fetal, como también variaciones principalmente viscerales y dérmicas (conejos). Los respectivos valores de exposición sistémica total (AUC) de los animales fueron ligeramente más altos (2,2 veces en las ratas) o más bajos (0,3 veces en los conejos) que los niveles observados en los pacientes.
Afatinib radiomarcado administrado por vía oral a ratas en el Día 11 del período de lactancia se excretó en la leche de las hembras. Las concentraciones promedio en la leche en los momentos de medición de 1 h y 6 h después de la administración de la dosis fueron aproximadamente 80 y 150 veces superiores a la concentración respectiva en plasma.
Un estudio de fertilidad en ratas macho y hembra de administración por vía oral en niveles de hasta la máxima dosis tolerada no reveló ninguna repercusión significativa sobre la fertilidad. La exposición sistémica total (AUC0-24) que se pudo alcanzar en las ratas macho y hembra se ubicó dentro o por debajo del rango de los valores observados en los pacientes (1,3 veces y 0,51 veces, respectivamente).
Un estudio en ratas de administración por vía oral hasta la máxima dosis tolerada no reveló ninguna repercusión significativa en el desarrollo pre/post natal. Los efectos se vieron limitados a pesos inferiores al nacer y un menor aumento del peso corporal de las crías, pero sin que ello afectara significativamente el logro de los parámetros de desarrollo, la maduración o el desempeño sexual en las evaluaciones conductuales. La exposición sistémica total más elevada (AUC0-24) que se pudo alcanzar en las ratas hembra fue inferior a la observada en los pacientes (0,23 veces).
Fototoxicidad: Se realizó una prueba de fototoxicidad 3T3 in vitro con afatinib. Se concluyó que GIOTRIF® puede tener potencial de fototoxicidad.
Carcinogenia: No se han realizado estudios de carcinogenia con afatinib.
Se observó una respuesta marginal a afatinib en una prueba de mutagenia en células bacterianas (Ames) con cepa única. Sin embargo, no se pudo identificar potencial mutagénico ni genotóxico en una prueba de aberración cromosómica in vitro efectuada en concentraciones no citotóxicas , y tampoco en el ensayo de micronúcleo de médula ósea in vivo , en el ensayo Comet in vivo ni en el estudio in vivo de mutación oral de 4 semanas en Muta™ Mouse.
ANEXO
Frecuencia de reacciones adversas GIOTRIF®
Categorías de frecuencia:
— Muy frecuente: ≥ 1/10
— Frecuente: ≥ 1/100 - < 1/10
— Poco frecuente: ≥ 1/1000 - < 1/100
— Rara: ≥ 1/10000 - < 1/1000
— Muy rara: < 1/10000
— Frecuencia desconocida: no se puede estimar a partir de los datos disponibles
Nota: Las categorías de frecuencia mencionadas anteriormente están basadas en la Guía de SmPC de la UE (Septiembre 2009); por consiguiente, en los países no pertenecientes a la Unión Europea pueden resultar aplicables otras definiciones.
La evaluación de seguridad de GIOTRIF® se basa en los datos de estudios clínicos y de la experiencia posterior a la comercialización provenientes de más de 7300 pacientes, incluidos más de 1638 pacientes con NSCLC tratados con una dosis diaria de 50 mg de GIOTRIF® como monoterapia, más de 497 pacientes con NSCLC que recibieron 40 mg de GIOTRIF® como monoterapia una vez al día, más de 392 pacientes con NSCLC escamoso que recibieron una dosis inicial de 40 mg de GIOTRIF® como monoterapia.
|
Tabla 1. Reacciones adversas mencionadas en la CCDS y las correspondientes frecuencias de acuerdo con la Guía de SmPC de la UE para GIOTRIF® en NSCLC con mutaciones del EGFR o enriquecidos por ellas (afatinib 40 mg y 50 mg), NSCLC escamoso (afatinib 40 mg y 50 mg). |
|||
|
Terminología del MedDRA Clasificación por sistema y órgano |
Reacciones adversas de afatinib con término textual de la CCDS TP del MedDRA (versión 21.0) |
Frecuencias según la Guía de SmPC de la UE |
|
|
NSCLC con mutaciones del EGFR o enriquecidas por ellas (afatinib 40 mg y 50 mg) |
NSCLC escamoso (afatinib 40 mg y 50 mg) |
||
|
Infecciones e infestaciones |
Paroniquia Cistitis |
Muy frecuente Frecuente |
Muy frecuente Poco frecuente |
|
Trastornos del metabolismo y de la nutrición |
Disminución del apetito Deshidratación Hipopotasemia |
Muy frecuente Frecuente Frecuente |
Muy frecuente Frecuente Frecuente |
|
Trastornos del sistema nervioso |
Disgeusia |
Frecuente |
Frecuente |
|
Trastornos oculares |
Conjuntivitis Sequedad ocular Queratitis |
Frecuente Frecuente Poco frecuente |
Frecuente Frecuente Poco frecuente |
|
Trastornos respiratorios, torácicos y mediastínicos |
Epistaxis Rinorrea Enfermedad pulmonar intersticial |
Muy frecuente Frecuente Poco frecuente |
Frecuente Frecuente Poco frecuente |
|
Trastornos gastrointestinales |
Diarrea Estomatitis Náuseas Vómitos Queilitis Dispepsia Pancreatitis Perforación gastrointestinal |
Muy frecuente Muy frecuente Muy frecuente Muy frecuente Frecuente Frecuente Poco frecuente Poco frecuente |
Muy frecuente Muy frecuente Muy frecuente Frecuente Poco frecuente Frecuente Poco frecuente Poco frecuente |
|
Trastornos hepatobiliares |
Elevación de la alaninaaminotransferasa Elevación de la aspartatoaminotransferasa |
Frecuente Frecuente |
Frecuente Poco frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
Exantema Dermatitis acneiforme Prurito Piel seca Alteraciones en las uñas Síndrome de eritrodisestesia palmoplantar Síndrome de Stevens-Johnson2 Necrólisis epidérmica tóxica2 |
Muy frecuente Muy frecuente Muy frecuente Muy frecuente Frecuente Frecuente Rara* Rara $ |
Muy frecuente Muy frecuente Frecuente Frecuente Frecuente Frecuente Rara* Rara $ |
|
Trastornos musculoesqueléticos y del tejido conectivo |
Espasmos musculares |
Frecuente |
Frecuente |
|
Trastornos renales y urinarios |
Insuficiencia renal / falla renal |
Frecuente |
Frecuente |
|
Trastornos generales y afecciones del sitio de administración |
Pirexia |
Frecuente |
Poco frecuente |
|
Investigaciones |
Descenso de peso |
Frecuente |
Frecuente |
|
¹ Para los eventos de ILD y pancreatitis con frecuencia baja, se utilizó un conjunto de datos combinados (SAF-5) que consistió en todos los pacientes tratados con afatinib en los estudios clínicos patrocinados por BI, para calcular la frecuencia. 2 Sobre la base de la experiencia posterior a la comercialización *Se incluye el SJS sobre la base de la experiencia posterior a la comercialización. Tal como se establece en la Guía europea de SmPC 2009 “En caso de detectarse una reacción adversa inesperada a partir de un informe espontáneo, cada estudio adecuadamente diseñado donde esta reacción adversa pudo haberse detectado debe ser revisado a fin de elegir una categoría de frecuencia”. Se revisaron los datos SAF-5 y se identificó 1 evento adverso relacionado con el fármaco cuya frecuencia se calculó como ‘rara’. $ Se incluye la NET sobre la base de la experiencia posterior a la comercialización, sin que se hayan informado EA relacionados con el fármaco en el conjunto de datos de referencia (SAF-5). Después de utilizar la norma 3/X (Guía europea de SmPC 2009), se calculó la frecuencia como ‘rara’. |
|||
INDICACIONES/USO: GIOTRIF® está indicado para el tratamiento de pacientes con:
Cáncer de pulmón de células no pequeñas (NSCLC) localmente avanzado o metastásico con mutación(es) del receptor del factor de crecimiento epidérmico (EGFR).
Cáncer de pulmón de células no pequeñas (NSCLC) localmente avanzado o metastásico de histología escamosa, con progresión al recibir quimioterapia basada en platino.
PRESENTACIÓN: GIOTRIF 20 mg: Caja de cartón con blíster por 28 tabletas recubiertas (Reg. San No. INVIMA 2021M-0016753-R1)
GIOTRIF 30 mg: Caja de cartón con blíster por 28 tabletas recubiertas (Reg. San No. INVIMA 2021M-0016747-R1)
GIOTRIF 40 mg: Caja de cartón con blíster por 28 tabletas recubiertas (Reg. San No. INVIMA 2021M-0016748-R1)
“¡Almacenar en un lugar seguro; fuera del alcance de los niños!
La información de seguridad del producto puede cambiar, consulte la información vigente en la Dirección Médica.
Teléfono: (601) 319 91 00, e-mail: medfora.co@boehringer-ingelheim.com
Carrera 11 No. 84A-09 Piso 5, Bogotá D.C. Colombia.
BOEHRINGER INGELHEIM S.A.
Versión - 09 del 28 de Febrero de 2019

