

FORXIGA
DAPAGLIFLOZINA
Comprimidos recubiertos
Caja , Blíster , 14 y 28 Comprimidos recubiertos , 10 Miligramos
Caja , Blíster , 14 y 28 Comprimidos recubiertos , 10 Miligramos
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
INDICACIONES TERAPÉUTICAS
Monoterapia: FORXIGA® está indicado para pacientes con diabetes mellitus tipo 2 en monoterapia en quienes no pueden utilizar metformina.
Adición al tratamiento con otros fármacos: FORXIGA® está indicado para pacientes con diabetes mellitus tipo 2 en terapia combinada con otros medicamentos hipoglicemiantes, incluyendo insulina, cuando estos junto con la dieta y el ejercicio no proveen adecuado control glicémico.
PROPIEDADES FARMACÉUTICAS
Lista de excipientes: Cada comprimido recubierto de FORXIGA® contiene 5 mg o 10 mg de dapagliflozina y los siguientes componentes inactivos: Celulosa microcristalina, lactosa anhidra, crospovidona, dióxido de silicio y estearato de magnesio. Además, la película de recubrimiento contiene los siguientes componentes inactivos: Alcohol polivinílico, dióxido de titanio, polietilenglicol, talco y óxido de hierro amarillo.
Incompatibilidades: No procede.
Plazo de caducidad: Véase la caja externa.
Conservación: Véase la caja externa.
Naturaleza y contenido del envase: Los comprimidos recubiertos de 5 mg de FORXIGA® (dapagliflozina propanodiol) son amarillos, biconvexos, redondos y llevan la FORXIGA® “5” grabada en una cara y “1427” en la otra.
Los comprimidos recubiertos de 10 mg de FORXIGA® (dapagliflozina propanodiol) son amarillos, biconvexos, romboidales y llevan la FORXIGA® “10” grabada en una cara y “1428” en la otra.
Instrucciones especiales de uso, manipulación y desecho: Ninguna instrucción especial. Todo el producto no utilizado y el material sobrante deben desecharse de conformidad con la reglamentación local.
PROPIEDADES FARMACOLÓGICAS
Farmacodinamia
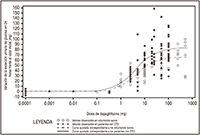
Aspectos generales: Después de la administración de la dapagliflozina se observaron aumentos de la cantidad de glucosa excretada en la orina tanto en voluntarios sanos como en pacientes con diabetes tipo 2 (véase la Figura 1). En pacientes con diabetes tipo 2, se excretaron aproximadamente 70 gramos al día de glucosa en la orina (lo que corresponde a 280 kcal/día) con una dosis de dapagliflozina de 10 mg al día administrada durante 12 semanas. Esta velocidad de eliminación de la glucosa se acercó a la excreción máxima observada con 20 mg de dapagliflozina al día. Se comprobó la excreción sostenida de glucosa en pacientes con diabetes tipo 2 tratados con 10 mg al día de dapagliflozina durante un periodo de hasta 2 años.
Esta excreción urinaria de glucosa inducida por la dapagliflozina también provoca diuresis osmótica y aumentos del volumen de orina. En pacientes con diabetes tipo 2 tratados con 10 mg de FORXIGA®, tales aumentos del volumen de orina se mantuvieron después de 12 semanas y ascendieron a aproximadamente 375 ml al día.135 El aumento del volumen de orina se acompañó de un aumento pequeño y transitorio de la natriuresis, sin modificaciones de las concentraciones séricas de sodio.
La excreción urinaria de ácido úrico también aumentó de manera transitoria (entre 3 y 7 días) y se acompañó de una reducción de la concentración sérica de ácido úrico. Después de 24 semanas, las disminuciones de las concentraciones séricas de ácido úrico fluctuaron entre -0.33 mg/dl y -0.87 mg/dl.
FIGURA 1. Diagrama de dispersión y curva ajustada de la variación de la cantidad de glucosa excretada en la orina en 24 horas frente al valor inicial, en función de la dosis de dapagliflozina en voluntarios sanos y en pacientes con diabetes tipo 2 (DT2) (escala semilogarítmica)
Electrofisiología cardiaca: En un estudio realizado en voluntarios sanos, la dapagliflozina no provocó una prolongación de importancia clínica del intervalo QTc con dosis diarias de hasta 150 mg (15 veces la dosis recomendada). Por otro lado, después de la administración a voluntarios sanos de dosis únicas de hasta 500 mg de dapagliflozina (50 veces la dosis recomendada), tampoco se observaron efectos de importancia clínica en el intervalo QTc.
Mecanismo de acción: La dapagliflozina es un inhibidor extremadamente potente, selectivo y reversible del cotransportador 2 de sodio-glucosa (SGLT2), que mejora el control glucémico en pacientes con diabetes tipo 2 reduciendo la reabsorción renal de glucosa y provocando su excreción en la orina (glucuresis). FORXIGA® se administra por vía oral una vez al día.
El SGLT2 se expresa selectivamente en los riñones y no se detecta expresión alguna en más de 70 tejidos diferentes como el hígado, el músculo-esquelético, el tejido adiposo, la mama, la vejiga y el cerebro. El SGLT2 es el principal transportador responsable de la reabsorción de glucosa desde el filtrado glomerular hacia la circulación. En la diabetes tipo 2, pese a la presencia de hiperglucemia, continúa la reabsorción de la glucosa filtrada. Dapagliflozina reduce un 55% el transporte tubular máximo de glucosa y disminuye la reabsorción renal de glucosa de tal modo que la glucosa aparece en la orina a niveles plasmáticos normales de glucosa. De este modo, la dapagliflozina mejora tanto la glucemia en ayunas como la glucemia posprandial reduciendo la reabsorción renal de glucosa y provocando su excreción en la orina. Este efecto glucurético se observa desde la administración de la primera dosis, continúa durante el intervalo de administración de 24 horas y se mantiene durante todo el periodo de tratamiento. La cantidad de glucosa eliminada por los riñones a través de este mecanismo depende de la concentración de glucosa en la sangre (glucemia) y de la TFG. Así pues, en voluntarios sanos normoglucémicos, la dapagliflozina muestra una baja propensión a causar hipoglucemia. La dapagliflozina no altera la producción endógena normal de glucosa en respuesta a la hipoglucemia. El efecto de la dapagliflozina es independiente de la secreción y acción de la insulina. En los estudios clínicos de la dapagliflozina se ha observado que la función de las células beta (HOMA-2) mejora con el tiempo.
La excreción urinaria de glucosa (glucuresis) inducida por la dapagliflozina se acompaña de pérdida calórica y disminución de peso. La mayor parte de la reducción de peso se explica por la pérdida de masa adiposa (incluida la masa adiposa visceral), más que por la pérdida de tejidos magros o de líquidos, según los análisis realizados por absorciometría de rayos X de doble energía (DXA) y por resonancia magnética nuclear (RMN). La inhibición del cotransporte de glucosa y de sodio por parte de la dapagliflozina también se acompaña de diuresis leve y natriuresis transitoria.
La dapagliflozina no inhibe otras sustancias importantes para el transporte de la glucosa a los tejidos periféricos, y su efecto en el SGLT2 es muy selectivo, siendo 1400 veces mayor que su efecto en el SGLT1, que es el principal transportador responsable de la absorción de glucosa en el intestino.152
FARMACOCINÉTICA
Absorción: Después de la administración oral, la dapagliflozina es absorbida bien y rápidamente, por lo que puede administrarse con o sin alimentos. En general, las concentraciones plasmáticas máximas de dapagliflozina (Cmáx) se alcanzan en un plazo de 2 horas después de la administración en ayunas. Los valores de Cmáx y ABC (área bajo la curva de concentraciones plasmáticas en función del tiempo) aumentan proporcionalmente al incremento de la dosis de dapagliflozina. La biodisponibilidad oral absoluta de la dapagliflozina es del 78% tras la administración de una dosis de 10 mg.156 El consumo de alimentos tiene un efecto relativamente limitado en la farmacocinética de la dapagliflozina en voluntarios sanos. La administración de la dapagliflozina junto con una comida con un alto contenido de lípidos reduce la Cmáx hasta un 50% y prolonga el Tmáx aproximadamente 1 hora, sin modificar el ABC con respecto a la administración en ayunas. Se considera que estos cambios carecen de importancia clínica.
Distribución: La dapagliflozina se une aproximadamente un 91% a las proteínas. La unión a las proteínas no se ve afectada por distintos estados patológicos (por ejemplo, insuficiencia renal o hepática).
Metabolismo: La dapagliflozina es un glucósido con enlace C, lo cual significa que la aglicona se une a la glucosa por un enlace carbono-carbono que le confiere una gran estabilidad en presencia de enzimas glucosidasas. La vida media terminal plasmática (t½) de la dapagliflozina es, en promedio, de 12.9 horas tras una dosis oral única de FORXIGA® de 10 mg en voluntarios sanos. La dapagliflozina es objeto de un extenso metabolismo que da lugar básicamente a la formación del 3-O-glucurónido de dapagliflozina, que es un metabolito inactivo. El 3-O-glucurónido de dapagliflozina representa el 61% de una dosis de 50 mg de [14C]dapagliflozina y es el principal componente relacionado con el fármaco en el plasma humano, representando el 42% (basándose en el ABC[0-12 horas]) de la radiactividad total recuperada en el plasma, es decir, un porcentaje similar a la contribución del fármaco original (39%). Basándose en el ABC, ningún otro metabolito representa más del 5% de la radiactividad total en el plasma en ninguna de las mediciones realizadas. Ni el 3-O-glucorónido de dapagliflozina ni otros metabolitos contribuyen a los efectos hipoglucemiantes del medicamento. La formación del 3O-glucurónido de dapagliflozina es mediada por la enzima UGT1A9 presente en el hígado y los riñones, y el metabolismo mediado por el citocromo P 450 (CYP) es una vía de eliminación poco importante en el ser humano.
Eliminación: La dapagliflozina y sus metabolitos relacionados se eliminan básicamente por excreción urinaria, mientras que menos del 2% se elimina en forma de dapagliflozina intacta. Después de la administración de 50 mg de [14C]-dapagliflozina, se recupera el 96% de la dosis: El 75% en la orina y el 21% en las heces. En las heces, alrededor del 15% de la dosis se excreta en forma del fármaco original.
Poblaciones específicas: No se recomienda ajustar la dosis en función de los resultados de los análisis farmacocinéticos en caso de insuficiencia renal leve a moderada, insuficiencia hepática leve, moderada y grave, ni en función de la edad, el sexo, la raza o el peso corporal del paciente.
Insuficiencia renal: FORXIGA® no debe usarse en pacientes con insuficiencia renal moderada o grave (TFGe < 45 ml/min/1.73 m2 persistente o DEPCr <60 ml/min persistente) (véanse los apartados Insuficiencia renal y Uso en pacientes con insuficiencia renal). En el estado de equilibrio (20 mg de dapagliflozina una vez al día durante 7 días), los pacientes con diabetes tipo 2 y insuficiencia renal leve, moderada o grave (determinada por la depuración de iohexol) presentaron exposiciones sistémicas medias a la dapagliflozina un 32%, un 60% y un 87% mayores, respectivamente, que los pacientes con diabetes tipo 2 y una función renal normal. Con la dosis de 20 mg de dapagliflozina una vez al día, la mayor exposición sistémica a la dapagliflozina en pacientes con diabetes tipo 2 e insuficiencia renal no condujo a un aumento correspondiente de la depuración renal de glucosa ni de la excreción de glucosa en 24 horas. La depuración renal de glucosa y la excreción de glucosa en 24 horas fueron menores en pacientes con insuficiencia renal moderada o grave con respecto a los pacientes con una función renal normal o con insuficiencia renal leve. La excreción urinaria de glucosa en 24 horas en el estado de equilibrio dependió en gran medida de la función renal, y los pacientes con diabetes tipo 2 y una función renal normal o con insuficiencia renal leve, moderada o grave excretaron 85, 52, 18 y 11 gramos de glucosa al día, respectivamente. No se encontraron diferencias en cuanto a la unión de la dapagliflozina a proteínas entre los pacientes con insuficiencia renal y los voluntarios sanos. Se desconocen los efectos de la hemodiálisis en la exposición a la dapagliflozina.
Insuficiencia hepática: Véanse en el apartado Insuficiencia hepática las dosis recomendadas en los pacientes con insuficiencia hepática moderada o grave. Se llevó a cabo un estudio de farmacología clínica con una dosis única de 10 mg de dapagliflozina en pacientes con insuficiencia hepática leve, moderada o grave (clases A, B y C de la escala de Child-Pugh, respectivamente) y en controles sanos equiparados, con el fin de comparar las características farmacocinéticas de la dapagliflozina entre estas poblaciones. No se encontraron diferencias en cuanto a la unión de la dapagliflozina a proteínas entre los pacientes con insuficiencia hepática y los sujetos sanos. En pacientes con insuficiencia hepática leve o moderada, los valores medios de Cmáx y ABC de la dapagliflozina aumentaron un 12% y un 36%, respectivamente, frente a los de controles sanos equiparados. Se considera que estas diferencias carecen de importancia clínica por lo que no se propone ningún ajuste específico de la dosis en estas poblaciones con respecto a la dosis usual propuesta de 10 mg de dapagliflozina una vez al día. En pacientes con insuficiencia hepática grave (clase C de la escala de Child-Pugh), los valores medios de Cmáx y ABC de la dapagliflozina aumentaron un 40% y un 67%, respectivamente, con respecto a los de controles sanos equiparados. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática grave. No obstante, es preciso evaluar individualmente la relación beneficio-riesgo de la dapagliflozina en pacientes con insuficiencia hepática grave dado que no se han investigado específicamente su seguridad y su eficacia en esta población.
Edad: No se recomienda ningún ajuste de la dosis de dapagliflozina en función de la edad con respecto a la dosis propuesta de 10 mg una vez al día. El efecto de la edad (jóvenes: =18-<40 años [n=105], ancianos: =65 años [n=224]) se evaluó como una covariable en un modelo farmacocinético poblacional mediante una comparación con pacientes de =40-<65 años, utilizando los datos procedentes de estudios realizados en voluntarios sanos y en pacientes. Se estimó que la exposición sistémica media a la dapagliflozina (ABC) en pacientes jóvenes era un 10.4% menor que en el grupo de referencia (IC del 90%: [87.9%; 92.2%]) y un 25% mayor en los pacientes de edad avanzada que en el grupo de referencia (IC del 90%: [123%; 129%]). Se considera que estas diferencias de exposición sistémica carecen de importancia clínica.
Niños y adolescentes: No se ha investigado la farmacocinética en la población pediátrica y adolescente.
Sexo: No se recomienda ningún ajuste de la dosis de dapagliflozina con respecto a la dosis propuesta de 10 mg una vez al día en función del sexo del paciente. Se evaluó el sexo como una covariable en un modelo farmacocinético poblacional que utilizó los datos procedentes de estudios realizados en voluntarios sanos y en pacientes. Se estima que el ABC media de la dapagliflozina en mujeres (n=619) fue un 22% mayor que en los varones (n=634) (IC del 90%:[117,124]).
Raza: No se recomienda ningún ajuste de la dosis de dapagliflozina con respecto a la dosis propuesta de 10 mg una vez al día en función de la raza del paciente. La raza (blanca, negra o asiática) se evaluó como una covariable en un modelo farmacocinético poblacional que utilizó los datos procedentes de estudios realizados en voluntarios sanos y en pacientes.168 Las diferencias de exposición sistémica fueron muy reducidas entre las distintas razas. Con respecto a los sujetos de raza blanca (n=1147), los sujetos asiáticos (n=47) no mostraron ninguna diferencia en cuanto a la exposición sistémica media estimada a la dapagliflozina (IC del 90%: [3.7% menor, 1% mayor]). Con respecto a los sujetos de raza blanca, los de raza negra (n=43) presentaron una exposición sistémica media estimada un 4.9% menor (IC del 90%: [7.7% menor, 3.7% menor]).
Peso corporal: No se recomienda ningún ajuste de la dosis de dapagliflozina con respecto a la dosis propuesta de 10 mg una vez al día en función del peso corporal del paciente.
En un análisis farmacocinético poblacional que utilizó los datos procedentes de estudios realizados en voluntarios sanos y en pacientes, se estimó que la exposición sistémica en sujetos de peso elevado (= 120 kg; n=91) representaba el 78.3% (IC del 90%: [78.2%; 83.2%]) de los valores obtenidos en sujetos de referencia con un peso de 75 a 100 kg. Se considera que esta diferencia es pequeña; en consecuencia, no se recomienda ningún ajuste de la dosis de dapagliflozina con respecto a la dosis propuesta de 10 mg una vez al día en los pacientes con diabetes tipo 2 y un peso corporal =120 kg.
Los sujetos de bajo peso (<50 kg) no estaban bien representados en los estudios realizados en voluntarios sanos y en el análisis farmacocinético poblacional. En consecuencia, se simularon las exposiciones sistémicas a la dapagliflozina usando una gran cantidad de sujetos. La exposición sistémica media simulada a la dapagliflozina en sujetos de bajo peso fue un 29% mayor que en el grupo de referencia. Se considera que esta diferencia es pequeña por lo que, en vista de estos resultados, no se recomienda ningún ajuste de la dosis de dapagliflozina con respecto a la dosis propuesta de 10 mg una vez al día en los pacientes con diabetes tipo 2 y un peso <50 kg.
CONTRAINDICACIONES: FORXIGA® está contraindicado en pacientes con antecedentes de reacciones graves de hipersensibilidad al principio activo o a alguno de los excipientes.
Insuficiencia renal severa (TFGe <30 mL/min/1.73 m2 calculada mediante la fórmula MDRD o con una DepCr =30 mL/min calculada mediante la fórmula de Cockcroft-Gault),
Menores de 18 años.
EMBARAZO Y LACTANCIA
Embarazo: FORXIGA® no debe usarse durante el segundo y tercer trimestres del embarazo. Durante el periodo correspondiente al segundo y tercer trimestres de gestación con respecto a la maduración renal humana, la exposición de ratas gestantes a la dapagliflozina se asoció con un aumento de la incidencia y/o intensidad de dilataciones pélvicas y tubulares renales en las crías (véase el apartado Potencial cancerígeno, potencial mutágeno y alteración de la fecundidad).
En estudios convencionales sobre el desarrollo embrionario y fetal en ratas y conejos, la dapagliflozina se administró durante los intervalos correspondientes al primer trimestre de la organogénesis no renal en el ser humano. En los conejos no se observaron efectos tóxicos en el desarrollo con ninguna de las dosis examinadas (1191 veces la dosis máxima recomendada en el ser humano [DMRH]. En las ratas, la dapagliflozina no fue embrioletal ni teratógena (1441 x DMRH) en ausencia de toxicidad materna.
No se han realizado estudios adecuados y bien controlados con FORXIGA® en mujeres embarazadas. Si se detecta un embarazo, debe suspenderse el tratamiento con FORXIGA®
Lactancia: Las mujeres que amamantan no deben usar FORXIGA®. Los estudios realizados en ratas mostraron que FORXIGA® se excreta en la leche. La administración directa e indirecta de FORXIGA® a ratas jóvenes recién destetadas y la exposición durante la última fase de la gestación se asocian, cada una, con un aumento de la incidencia y/o intensidad de dilataciones pélvicas y tubulares renales en las crías, aunque se desconocen las consecuencias funcionales de estos efectos a largo plazo. Estos periodos de exposición coinciden con un intervalo de tiempo crítico para la maduración renal de las ratas. Dado que, en el ser humano, la maduración funcional de los riñones continúa durante los 2 primeros años de vida, las dilataciones pélvicas y tubulares renales que se observaron en ratas jóvenes con FORXIGA® podrían representar un riesgo potencial para la maduración renal humana durante los 2 primeros años de vida. Por otro lado, los efectos negativos en el aumento de peso asociados con la exposición durante la lactancia en ratas jóvenes recién destetadas indican que el tratamiento con FORXIGA® debe evitarse durante los 2 primeros años de vida (véase el apartado Potencial cancerígeno, potencial mutágeno y alteración de la fecundidad).
No se sabe si FORXIGA® y/o su metabolito se excretan en la leche materna humana.
Parto
Uso pediátrico: No se han establecido la seguridad y la eficacia de FORXIGA® en pacientes pediátricos.
Uso geriátrico: No se recomienda el inicio de terapia con dapaglifozina en pacientes mayores de 75 años debido a la limitada experiencia terapéutica.
No se recomienda ningún ajuste de la dosis en función de la edad. Un total de 2403 (26%) de los 9339 pacientes tratados tenía 65 años de edad y más, y 327 (3.5%) pacientes tenían 75 años y más en el grupo de 21 estudios doble-ciegos, controlados, de seguridad clínica y eficacia de FORXIGA®. Tras efectuar la corrección correspondiente al grado de insuficiencia renal (TFGe), no se encontraron pruebas convincentes de que la edad fuera un factor independiente en la eficacia del tratamiento. En total, las proporciones de pacientes que presentaron reacciones adversas fueron equilibradas entre las poblaciones mayores y menores de 65 años de edad. En los pacientes mayores de 65 años, la proporción que presentó reacciones relacionadas con disfunción o insuficiencia renal fue mayor con FORXIGA® que con el placebo. Los eventos adversos más comúnmente reportados, relacionados con daño y falla renal en los pacientes mayores de 65 años de todos los grupos consistieron en disminuciones de la depuración renal de creatinina, insuficiencia renal y elevaciones de la creatininemia.
La probabilidad de insuficiencia renal es mayor en los pacientes de edad avanzada. Las recomendaciones relativas a la función renal que se aplican a todos los pacientes también son válidas para los ancianos (véanse los apartados Uso en pacientes con insuficiencia renal, Insuficiencia renal y Experiencia adquirida durante los estudios clínicos).
Hallazgos macrovasculares: Ningún ensayo clínico ha demostrado de manera convincente la reducción del riesgo macrovascular con FORXIGA® o con algún otro fármaco antidiabético. En un meta-análisis de 21 estudios clínicos, el uso de FORXIGA® no se asoció con un mayor riesgo de reacciones adversas cardiovasculares (véase el apartado Experiencia adquirida durante los estudios clínicos).33
EFECTOS EN LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: No se han realizado estudios sobre los efectos de FORXIGA® en la capacidad para conducir y utilizar máquinas.
Insuficiencia renal
Pacientes con insuficiencia renal leve (TFGe = 60-< 90 ml/min/1.73 m2): El grupo de 21 estudios doble-ciegos, activo- y placebo-controlados, de seguridad clínica y eficacia incluyó 53% (4906/9339) de pacientes con insuficiencia renal leve. La eficacia se evaluó en un análisis combinado de 9 estudios clínicos en los que participaron 2226 pacientes con insuficiencia renal leve. La variación media de la hemoglobina A1c (HbA1c) después de 24 semanas frente al valor inicial y la variación media de la HbA1c corregida tomando en cuenta el efecto placebo fueron de -1.03% y -0.54%, respectivamente, con la dosis de FORXIGA® de 10 mg (n = 562). El perfil de seguridad en pacientes con insuficiencia renal leve es similar al de la población general.
Pacientes con insuficiencia renal moderada (TFGe =30-<60 ml/min/1.73 m2): El grupo de 21 estudios clínicos activo- y placebo-controlados incluyó 11% (1055/9339) de pacientes con insuficiencia renal moderada. La eficacia en estos pacientes se evaluó en un análisis combinado de 9 estudios clínicos (366 pacientes, 87% con una TFGe =45-<60 ml/min/1.73 m2). La variación media de la HbA1c después de 24 semanas frente al valor inicial y la variación media de la HbA1c corregida tomando en cuenta el efecto placebo fueron de -0.87% y -0.39%, respectivamente, con la dosis de 10 mg de FORXIGA® (n=85).
La eficacia de FORXIGA® también se evaluó por separado en un estudio realizado en pacientes diabéticos con insuficiencia renal moderada (252 pacientes con una TFGe media de 45 mL/min/1.73 m2). La variación media de la HbA1c después de 24 semanas frente al valor inicial y la variación media de la HbA1c corregida tomando en cuenta el efecto placebo fueron de -0.44% y -0.11%, respectivamente, con la dosis de 10 mg de FORXIGA® (n=82).
En este estudio, después de 24 semanas se llevó a cabo un análisis adicional de subgrupos basados en el valor de TFGe (=45 o <45 mL/min/1.73 m2). En los pacientes con una TFGe inicial =45-<60 mL/min/1.73 m2, la variación media de la HbA1c después de 24 semanas frente al valor inicial y la variación media de la HbA1c corregida tomando en cuenta el efecto placebo fueron de -0.44% y -0.33%, respectivamente, con la dosis de 10 mg de FORXIGA® (n=32). En los pacientes de este estudio que tenían una TFGe =30-<45 ml/min/1.73 m2, la variación media de la HbA1c después de 24 semanas frente al valor inicial y la reducción media de la HbA1c corregida tomando en cuenta el efecto placebo fueron de -0.45% y 0.07%, respectivamente, con la dosis de 10 mg de FORXIGA® (n=45). Estos resultados son compatibles con el modo de acción de FORXIGA®, que depende de la función renal (véase el apartado Mecanismo de acción).
La seguridad en pacientes con insuficiencia renal moderada se evaluó en un análisis combinado de 12 estudios clínicos (384 pacientes, 88% con una TFGe =45-<60 ml/min/1.73 m2), que no incluyó el estudio realizado específicamente en pacientes diabéticos con insuficiencia renal moderada. Después de 24 semanas, la seguridad fue similar a la observada en el programa general de ensayos clínicos, salvo que una mayor proporción de pacientes presentaron al menos una reacción adversa relacionada con disfunción o insuficiencia renal (7.9% con la dosis de 10 mg de FORXIGA® frente al 5.6% con el placebo). La más frecuente de estas reacciones adversas fue el aumento de la concentración sérica de creatinina (FORXIGA® 10 mg: 6.7%; placebo: 2.8%). Los aumentos de las concentraciones medias de parathormona (PTH) y fósforo sérico, que se registraron con FORXIGA® durante el programa general de ensayos clínicos, también se observaron en el análisis combinado. En este análisis no se detectó ningún desequilibrio en la incidencia de fracturas. Según los datos de seguridad combinados de los estudios a corto plazo y largo plazo de hasta 102 semanas, se mantuvo un perfil de seguridad similar.
La seguridad también fue evaluada en el estudio realizado en pacientes con insuficiencia renal moderada, después de 52 semanas, FORXIGA® produjo variaciones de las medias de TFGe y DEPCre frente a los valores iniciales (TFGe con FORXIGA® 10 mg: -4.46 ml/min/1.73 m2 y placebo: 2.58 ml/min/ 1.73 m2; DEPCre con FORXIGA® 10 mg: -7.27 ml/min y placebo: 2.56 ml/min). A la semana 104, estos cambios persistieron (TFGe: FORXIGA® 10 mg -3.50 mL/min/1.73 m2 y placebo -2.38 mL/min/1.73 m2) (DEPCre: FORXIGA® 10 mg -6.32 mL/min y placebo -2.35 mL/min). Con la administración de 10 mg de FORXIGA®, estas TFGe y DEPCre reducciones se manifestaron desde la primera semana y permanecieron estables hasta la semana 104, mientras que los pacientes que recibieron un placebo mostraron una disminución lenta y continua hasta la semana 52 que fue estabilizada hasta la semana 104.
En la semana 52 y persistiendo hasta la semana 104, los aumentos de las concentraciones medias de PTH y fósforo sérico fueron más importantes en este estudio con FORXIGA® 10 mg comparada con placebo, en el que los valores iniciales de estos parámetros eran más elevados. Las elevaciones de potasio =6 mEq/L fueron más frecuentes en pacientes tratados con placebo (12.0%) que en quienes recibieron FORXIGA® 5 mg y 10 mg (4.8% para ambos grupos) durante el periodo de tratamiento acumulativo de 104 semanas. La proporción de pacientes que descontinuaron el tratamiento por potasio elevado, ajustado en función del potasio inicial, fue mayor para el grupo placebo (14.3%) que para los grupos FORXIGA® (6.9% y 6.7% para los grupos que recibieron 5 mg y 10 mg, respectivamente).
En general, hubo 13 pacientes con un evento adverso de fractura ósea reportada en este estudio hasta la semana 104, de los cuales, 8 ocurrieron en el grupo tratado con FORXIGA® 10 mg, 5 en el grupo FORXIGA® 5 mg, y ninguno en el grupo placebo. Ocho (8) de estas 13 fracturas se produjeron en pacientes que tenían TFGe de 30 a 45 mL/min/1.73 m2 y 11 de las 13 fracturas fueron reportadas dentro de las primeras 52 semanas. No hubo un patrón aparente con respecto al sitio de fractura.
Pacientes con insuficiencia renal grave (TFGe <30 mL/min/1.73 m2): Los estudios clínicos no incluyeron pacientes con insuficiencia renal grave (TFGe <30 ml/min/1.73 m2) o ERET, dado su modo de acción, no se esperaba que FORXIGA® fuese eficaz en estas poblaciones.
Uso en pacientes con diabetes y afecciones cardiovasculares: En el marco de dos estudios controlados con placebo de 24 semanas de duración y periodos de extensión de 80 semanas, un total de 1887 pacientes con diabetes tipo 2 y enfermedad cardiovascular (ECV) recibieron un tratamiento con 10 mg de FORXIGA® o un placebo. En estos estudios participaron pacientes con enfermedad CV establecida y un control glucémico inadecuado (HbA1c =7.0% y =10.0%), pese a recibir un tratamiento estable con un antidiabético oral (ADO) o insulina (sola o combinada) antes de su admisión en el estudio. Los pacientes fueron estratificados en función de su edad (<65 o =65 años), del uso o no de insulina y del tiempo transcurrido desde el evento cardiovascular cualificador más reciente (>1 año o <1 año antes de su admisión en el estudio). Sumando los dos estudios, 942 pacientes recibieron un tratamiento con 10 mg de FORXIGA® y 945 con el placebo. El 96% de aquellos tratados con FORXIGA® en los dos estudios tenían hipertensión en el momento de su admisión, la mayoría desde hace más de 10 años. El evento cardiovascular cualificador más frecuente fue la enfermedad coronaria (75%) o el accidente cerebrovascular (22%). Aproximadamente el 19% de los pacientes tomaban diuréticos de asa en el momento de la admisión y el 15% padecían insuficiencia cardiaca congestiva (el 2% de clase III según la NYHA). Aproximadamente el 37% de los pacientes tratados con 10 mg de FORXIGA® también recibían una combinación de metformina + un antidiabético oral en el momento de su admisión (sulfonilurea, inhibidor de la DPP4 u otro antidiabético asociado o no con insulina), el 39% recibían insulina + al menos un antibiabético oral y el 18% solamente insulina.
En esta población, la adición de 10 mg de FORXIGA® al tratamiento antidiabético existente produjo una mejora significativa durante el periodo de 24 semanas de las covariables principales de la HbA1c y del beneficio clínico compuesto frente al placebo. También se observaron reducciones significativas en el peso corporal total y presión arterial sistólica en posición sentada (véase sección Estudios complementarios). Estos beneficios se extendieron hasta las 104 semanas de tratamiento. En estos estudios, el perfil de seguridad de FORXIGA® concordó con el observado en la población general del estudio a todo lo largo de las 104 semanas de tratamiento (véase el apartado Experiencia adquirida durante los estudios clínicos).
Infecciones urinarias: Teniendo en cuenta la potencial ocurrencia de infecciones urinarias se debe advertir a los pacientes de consultar con su médico si se presentan síntomas de infecciones del tracto urinario.
Falla cardiaca: La experiencia en NYHA clase I-II es limitada y no hay experiencia en estudios clínicos con dapagliflozina en NYHA clase III-IV.
Falla hepática: La seguridad y eficacia de dapaglifozina no ha sido estudiada específicamente en pacientes con insuficiencia hepática severa o falla hepática.
Uso en pacientes tratados con pioglitazona: Aunque es poco probable una relación causal entre la dapaglifozina y el cáncer de vejiga, como medida preventiva, no se recomienda el uso de dapaglifozina en pacientes con terapia concomitante con pioglitazona. Los datos epidemiológicos disponibles para pioglitazona sugieren un incremento menor en el riesgo de cáncer de vejiga en pacientes diabéticos tratados con pioglitazona.
REACCIONES ADVERSAS
Experiencia adquirida durante los estudios clínicos
Se utilizaron dos grupos importantes de pacientes para evaluar las reacciones con FORXIGA® 10 mg versus control, un grupo de estudios placebo-controlados y un grupo más grande de estudios activo- y placebo-controlados.
Estudios placebo-controlados: El primero es un grupo pre-especificado de pacientes de 13 estudios a corto plazo, placebo-controlados, usados para evaluar y presentar todos los datos de seguridad diferentes a enfermedades malignas, pruebas hepáticas e hipoglucemia (evaluadas por estudio individual). Este grupo incluyó los estudios de monoterapia, varios estudios adicionales (metformina, sulfonilurea, pioglitazona, inhibidor de DPP4, insulina, y dos estudios con una combinación de terapias adicionales), y una combinación inicial con el estudio de metformina. A través de estos 13 estudios, 2360 pacientes recibieron tratamiento una vez al día con FORXIGA® 10 mg y 2295 fueron tratados con placebo (como monoterapia o en combinación con otros tratamientos antidiabéticos).
Estos 13 estudios proveen una duración media de exposición de 22 semanas. La edad promedio de la población era de 59 años y 4% era mayor de 75 años. Cincuenta y ocho por ciento (58%) de la población era de sexo masculino; 84% era de raza blanca, 9% asiática y 3% de raza negra o afroamericana. En el nivel basal, la población había tenido diabetes por 9 años, la HbA1c media era de 8.2%, y la función renal era normal o estaba ligeramente alterada en 88% de los pacientes, y moderadamente alterada en el 11%.
Estudios activo- y placebo-controlados: El segundo es un grupo de pacientes de 21 estudios activo- y placebo-controlados, usados para evaluar y presentar datos de enfermedades malignas y pruebas hepáticas. En este grupo, 5936 pacientes fueron tratados con FORXIGA® y 3403 recibieron control (como monoterapia o en combinación con otros tratamientos antidiabéticos).
Estos 21 estudios proveen una duración media de exposición a FORXIGA® 10 mg de 55 semanas (6247 pacientes-años). A través de ambos grupos de tratamiento, la edad promedio de la población era de 58 años y 3.5% era mayor de 75 años. Cincuenta y seis por ciento (56%) de la población era de sexo masculino; 77% era de raza blanca, 16% asiática, y 4% de raza negra o afroamericana. En el nivel basal, la población había tenido diabetes durante un promedio de 7 años, 34% de los pacientes tenía historia de enfermedad cardiovascular, la HbA1c media era de 8.2%, y la función renal basal era normal o ligeramente alterada en 89% de los pacientes, y moderadamente alterada en 11% de los pacientes.
Adicionalmente, FORXIGA® 5 mg fue evaluado en un grupo de 12 estudios a corto plazo, placebo-controlados, que incluyeron 1145 pacientes tratados con FORXIGA® 5 mg como monoterapia o en combinación con otro tratamiento antidiabético (exposición media = 22 semanas) y 1393 pacientes tratados con placebo como monoterapia o en combinación con otro tratamiento antidiabético (exposición media = 21 semanas). Todos los datos de seguridad presentados para FORXIGA® 5 mg provienen de este grupo.
La incidencia global de eventos adversos para el grupo de 13 estudios a corto plazo, placebo-controlados (tratamiento a corto plazo) en pacientes tratados con FORXIGA® 10 mg fue de 60.0% comparada con 55.7% para el grupo placebo. La descontinuación del tratamiento debido a eventos adversos en pacientes que recibieron FORXIGA® 10 mg fue de 4.3% comparada con 3.6% para el grupo placebo. Los eventos más comúnmente reportados que condujeron a descontinuación y que fueron reportados por lo menos en 3 pacientes tratados con FORXIGA® 10 mg fueron insuficiencia renal (0.8%), disminución en la depuración de creatinina (0.6%), aumento en la creatinina sérica (0.3%), infecciones del tracto urinario (0.2%), e infección micótica vulvovaginal (0.1%).
En la Tabla 1 se muestran las reacciones adversas en este análisis agrupado de 13 estudios placebo-controlados (independientemente de la evaluación de causalidad por el investigador) reportadas en =2% de los pacientes tratados con FORXIGA® 10 mg, y con una frecuencia =1% que en los pacientes tratados con placebo.
|
Tabla 1. Reacciones adversas (independientemente de la evaluación de causalidad por el investigador) en estudios placebo-controlados, reportadas en =2% de los pacientes tratados con FORXIGA® 10 mg, y con una frecuencia =1% que en los pacientes tratados con placebo (excluyendo hipoglucemia)*,† |
|
|
Clase de sistema orgánico Término preferido |
FORXIGA® 10 mg N=2360 |
|
Infecciones e infestaciones Infección genital‡ |
Frecuente |
|
Infecciones e infestaciones Infección del tracto urinario§ |
Frecuente |
|
Trastornos musculoesqueléticos y del tejido conectivo Dolor de espalda |
Frecuente |
|
Trastornos del metabolismo y la nutrición Poliuria¶ |
Frecuente |
|
* Los 13 estudios placebo-controlados incluyeron 3 sobre la monoterapia, 1 sobre el tratamiento combinado inicial con metformina, 2 sobre la adición al tratamiento con metformina, 2 sobre la adición al tratamiento con insulina, 1 sobre la adición al tratamiento con pioglitazona, 1 sobre la adición al tratamiento con sitagliptina, 1 sobre la adición al tratamiento con glimepirida, y 2 estudios con terapia combinada adicional. La Tabla muestra datos hasta de 24 semanas (corto plazo) independientemente del tratamiento de rescate glucémico. † Véase información sobre hipoglucemia en la sub-sección Hipoglucemia. ‡ La infección genital incluye los siguientes términos preferidos, enumerados en orden de frecuencia reportada: Infección micótica vulvovaginal, balanitis, infección vaginal, infección fungosa genital, infección genital, candidiasis vulvovaginal, balanitis causada por cándida, vulvovaginitis, candidiasis genital, vulvitis, balanopostitis, infección genital masculina, infección del tracto genitourinario, absceso peneano, infección peneana, postitis, absceso vulvar, y vaginitis bacteriana. § La infección del tracto urinario incluye los siguientes términos preferidos, enumerados en orden de frecuencia reportada: Infección del tracto urinario, cistitis, infección del tracto urinario por Escherichia, infección del tracto genitourinario, pielonefritis, trigonitis, uretritis, infección renal, y prostatitis. ¶ Poliuria incluye los términos preferidos, enumerados en orden de frecuencia reportada: Polaquiuria, poliuria, aumento de la producción de orina. |
|
A continuación se describen, por cada régimen terapéutico, las demás reacciones adversas notificadas en =5% de los pacientes tratados con 10 mg de FORXIGA®, con una incidencia = 1% mayor que en los pacientes tratados con un placebo o con el fármaco de referencia, y que se observaron en al menos 3 pacientes o más del grupo tratado con 10 mg de FORXIGA®, independientemente de la relación causal evaluada por el investigador.
Estudios sobre la adición de la dapagliflozina al tratamiento con metformina: Cefalea (FORXIGA® 10 mg: 5.3%; placebo: 3.1%).
Hipovolemia: Se reportaron eventos relacionados con depleción de volumen (incluyendo reportes de deshidratación, hipovolemia o hipotensión) en 1.1% y 0.7% de los pacientes que recibieron FORXIGA® 10 mg y placebo, respectivamente, en el grupo de 13 estudios a corto plazo, placebo-controlados. Ocurrieron eventos graves en =0.2% de los pacientes a través de los 21 estudios activo- y placebo-controlados, y fueron equilibrados entre FORXIGA® 10 mg y el comparador. En el análisis de subgrupo de pacientes que estaban tomando diuréticos de ASA, o con edad =65 años en el grupo de 13 estudios placebo-controlados, la proporción de pacientes con eventos relacionados con depleción de volumen fue ligeramente más alta en quellos tratados con FORXIGA® 10 mg que en quienes recibieron placebo (eventos en pacientes que estaban tomando diuréticos de ASAs: 2.5% vs. 1.5%; eventos en pacientes =65 años de edad: 1.7% vs. 0.8%, respectivamente).
Se reportaron eventos relacionados con depleción de volumen en 0.6% de los pacientes que recibieron FORXIGA® 5 mg comparados con 0.4% de quienes recibieron placebo en el grupo de 12 estudios a corto plazo, placebo-controlados. Ningún paciente que estaba tomando diurético de ASA y solo 1 paciente =65 años de edad (0.5%) tuvo un evento relacionado con depleción de volumen durante el tratamiento con FORXIGA® 5 mg comparado con 1 paciente que estaba recibiendo diurético de asa (1.8%) y 1 paciente =65 años de edad (0.4%) tratado con placebo (véanse secciones Pacientes expuestos a un riesgo de hipovolemia y Uso en pacientes con riesgo de hipovolemia).
Adicionalmente, en el subgrupo de pacientes con insuficiencia renal moderada, con TFGe =45 a <60 mL/min/1.73m2, la proporción de pacientes con eventos relacionados con depleción de volumen fue más alta en pacientes con FORXIGA® 10 mg (4.7%) y FORXIGA® 5 mg (2.3%) que en aquellos tratados con placebo (1.4%).
Infecciones genitales: En el grupo de 13 estudios controlados con placebo a corto plazo se notificaron infecciones genitales en el 5.5% y el 0.6% de los pacientes tratados con 10 mg de FORXIGA® y con un placebo, respectivamente. Todas las infecciones genitales notificadas en pacientes tratados con 10 mg de FORXIGA® fueron de intensidad leve a moderada. La mayoría de las infecciones genitales respondieron a un ciclo inicial de tratamiento convencional y solo en raras ocasiones provocaron el retiro del paciente del estudio (FORXIGA® 10 mg: 0.2%; placebo: 0%). Las infecciones fueron más frecuentes en las mujeres (FORXIGA® 10 mg: 8.4%; placebo: 1.2%) que en los varones (FORXIGA® 10 mg: 3.4%; placebo: 0.2%). Las infecciones genitales notificadas con mayor frecuencia consistieron en micosis vulvovaginales en mujeres, y en balanitis en los varones.
En 9 de los 13 estudios del grupo placebo-controlado, con tratamiento a largo plazo se tuvieron datos disponibles. Para este grupo a corto plazo más el grupo a largo plazo placebo-controlado (duración media del tratamiento: 439.5 días con 10 mg de FORXIGA® y 419.0 días con el placebo), las proporciones de pacientes con infecciones genitales fueron del 7.7% (156/2026) con el grupo de FORXIGA® 10 mg y del 1.0% (19/1956) con el grupo placebo. De los 156 pacientes tratados con 10 mg de FORXIGA® que desarrollaron una infección, 106 (67.9%) solo tuvieron un episodio infeccioso y 17 (10.9%) tres o más. De los 19 pacientes tratados con un placebo que presentaron una infección, 17 (89.5%) tuvieron un solo episodio infeccioso y ninguno tres o más.
En el grupo de 13 estudios a corto plazo, placebo-controlados, los pacientes con antecedentes de infecciones genitales recurrentes tuvieron una mayor probabilidad de contraer una infección genital durante el estudio (33.3% de los pacientes con antecedentes infecciosos tratados con 10 mg de FORXIGA® y 9.5% con el placebo) que los pacientes que no tenían una historia de infección (FORXIGA® 10 mg: 5.2%; placebo: 0.5%).
Globalmente, los tratamientos con 5 mg y 10 mg de FORXIGA® fueron similares.
Infecciones urinarias: Se reportaron eventos de infecciones urinarias en 4.7% y 3.5% de los pacientes que recibieron FORXIGA® 10 mg y placebo, respectivamente, en el grupo de 13 estudios a corto plazo, placebo-controlados.La mayoría de las infecciones urinarias de los pacientes tratados con 10 mg de FORXIGA® fueron de intensidad leve a moderada. La mayoría de los pacientes respondieron a un ciclo inicial de tratamiento convencional y las infecciones urinarias solo provocaron la retirada del paciente del estudio en raras ocasiones (FORXIGA® 10 mg: 0.2%; placebo: 0.1%). Las infecciones fueron más frecuentes en las mujeres (FORXIGA® 10 mg: 8.5%; placebo: 6.7%) que en los varones (FORXIGA® 10 mg: 1.8%; placebo: 1.3%).
En 9 de los 13 estudios en el grupo placebo-controlado, estuvieron disponibles datos de tratamiento a largo plazo. Para este análisis combinado de los estudios controlados con placebo a corto plazo y a largo plazo (duración media del tratamiento: 439.5 días con FORXIGA® 10 mg y 419.0 días con el placebo), las proporciones de pacientes que contrajeron infecciones urinarias fueron del 8.6% (174/2026) con el grupo de FORXIGA® 10 mg y del 6.2% (121/1956) con el grupo placebo. De los 174 pacientes tratados con 10 mg de FORXIGA® que contrajeron una infección, 135 (77.6%) tuvieron un solo episodio infeccioso y 11 (6.3%) tuvieron tres o más. De los 121 pacientes tratados con un placebo que contrajeron una infección, 94 (77.7%) tuvieron un solo episodio infeccioso y 12 (9.9%) tuvieron tres o más.
En el grupo de 13 estudios a corto plazo, placebo-controlados, los pacientes con antecedentes de infecciones urinarias recurrentes tuvieron una mayor probabilidad de contraer una infección urinaria durante el estudio (6.0% de los pacientes con antecedentes infecciosos tratados con 10 mg de FORXIGA® y 5.9% con el placebo) que los pacientes sin una historia de infección (FORXIGA® 10 mg: 4.4%; placebo: 3.0%).97
Globalmente, los tratamientos con 5 mg y 10 mg de FORXIGA® fueron similares.
Hipoglucemia: La frecuencia de hipoglucemia dependió del tipo de terapia de fondo usada en cada estudio. Los estudios de FORXIGA® adicionado a sulfonilúrea o como una adición a insulinoterapia tuvo índices de hipoglucemia con el tratamiento con FORXIGA® más altos que con el tratamiento con placebo (veáse sección Uso con medicamentos con efectos hipoglucemiantes conocidos).
En estudios con FORXIGA® usado como monoterapia, adicionado a metformina, y combinación inicial con metformina hasta por 102 semanas, no se produjeron reportes de episodios de hipoglucemia mayor. En un estudio de FORXIGA® 10 mg adicionado a sitagliptina (con o sin metformina) hasta por 48 semanas, se reportó un episodio mayor de hipoglucemia en un paciente tratado con FORXIGA® 10 mg más sitagliptina (sin metformina). En estos estudios, la frecuencia de episodios de hipoglucemia menor fue similar (<5%) a través de los grupos de tratamiento, incluyendo el grupo placebo.
En un estudio con FORXIGA® 10 mg adicionado a glimepirida hasta por 48 semanas, que también incluyó otras dosis de FORXIGA®, se produjo un episodio de hipoglucemia mayor en un paciente tratado con dapagliflozina 2.5 mg más glimepirida. Se reportaron episodios menores de hipoglucemia en 7.9% de los pacientes tratados con FORXIGA® 10 mg más glimepirida y 2.1% de los pacientes tratados con placebo más glimepirida.
En un estudio de adición a metformina, que comparó FORXIGA® con glipizide hasta por 104 semanas, hubo 3 episodios (0.7%) de hipoglucemia mayor en pacientes tratados con glipizide más metformina, y ninguno en pacientes tratados con FORXIGA® más metformina. Se reportaron episodios menores de hipoglucemia en 2.5% de los pacientes tratados con FORXIGA® más metformina y 42.4% de los pacientes tratados con glipizide más metformina.
En un estudio de adición a insulina (con o sin 2 agentes antidiabéticos orales adicionales, incluyendo metformina) que comparó FORXIGA® 10 mg más insulina con placebo más insulina hasta por 24 semanas, se produjo un episodio (0.5%) de hipoglucemia mayor en un paciente tratado con FORXIGA® 10 mg más insulina y 1 (0.5%) episodio en un paciente tratado con placebo más insulina. A la semana 104, se reportaron episodios de hipoglucemia mayor en 1.0% y 0.5% de los pacientes tratados con FORXIGA® 10 mg o placebo adicionado a insulina, respectivamente. Se reportaron episodios menores en 40.3% de los pacientes tratados con FORXIGA® 10 mg más insulina y en 34% de los pacientes tratados con placebo más insulina hasta por 24 semanas. A la semana 104, se reportaron episodios menores en 53.1% y 41.6% de los pacientes tratados con FORXIGA® 10 mg o placebo adicionado a insulina, respectivamente. En dos estudios adicionales que también incluyeron una gran proporción de pacientes que recibieron insulina como terapia de fondo (sola o con uno o más tratamientos antidiabéticos orales) (véase sección Estudios complementarios), el índice de episodios menores de hipoglucemia también se incrementó en pacientes tratados con FORXIGA® 10 mg comparados con aquellos tratados con placebo.
Cáncer: En el grupo de 21 estudios activo- y placebo-controlados, la proporción global de pacientes con tumores malignos o no especificados fue similar entre aquellos tratados con FORXIGA® (1.50%) y placebo/comparador (1.50%), y no hubo señales de carcinogenicidad o mutagenicidad en datos de animales (véase sección Potencial cancerígeno, potencial mutágeno y alteración de la fecundidad). Al considerar los casos de tumores que ocurren en los diferentes sistemas orgánicos, el riesgo relativo asociado con FORXIGA® fue superior a 1 para algunos tumores (por ej., vejiga y seno) e inferior a 1 para otros (por ej., sangre y tejido linfático, ovarios, tracto renal). Ni los incrementos ni las reducciones en el riesgo fueron estadísticamente significativos en ninguno de los sistemas orgánicos. Teniendo en cuenta la ausencia de hallazgos de tumores en estudios no clínicos, como también la corta latencia entre la primera exposición al medicamento y el diagnóstico de tumor, se considera improbable una relación causal con ningún tipo de tumor.
Pruebas de la función hepática: En el grupo de 21 estudios activo- y placebo-controlados, no hubo desequilibrio a través de los grupos de tratamiento en la incidencia de elevaciones de la alanina aminotransferasa sérica (ALT) o aspartato aminotransferasa (AST). Se reportó alanina aminotransferasa (ALT) >3 x ULN en 1.2% de los pacientes tratados con FORXIGA® 10 mg y 1.6% tratados con comparador. Se reportó alanina aminotransferasa (ALT) o AST >3 x ULN y bilirrubina >2 x ULN en 7 pacientes (0.1%) que estaban recibiendo cualquiera de las dosis de FORXIGA®, 5 pacientes (0.2%) recibiendo FORXIGA® 10 mg, y 4 pacientes (0.1%) tratados con comparador.
Seguridad cardiovascular: Se llevó a cabo un meta-análisis de las reacciones cardiovasculares notificadas en los 21 estudios activo- y placebo-controlados y confirmadas por un comité de evaluación independiente. La variable principal fue el tiempo transcurrido hasta el primero de los siguientes acontecimientos: Muerte de origen cardiovascular, accidente vascular cerebral, infarto de miocardio y hospitalización debida a angina inestable. La incidencia de acontecimientos incluidos en la variable principal fue del 1.46 por 100 pacientes-años en pacientes tratados con FORXIGA® y del 2.14 en pacientes tratados con el comparador, por 100 pacientes-años. La razón de riesgos entre FORXIGA® y el fármaco de referencia fue de 0.79 (intervalo de confianza [IC] del 95%: 0.58, 1.10). El tratamiento con FORXIGA® no se asocia con un aumento del riesgo cardiovascular en pacientes con diabetes tipo 2.
Experiencia posterior a la comercialización del producto: No procede.
Constantes vitales: En el grupo de 13 estudios placebo-controlados, se observó una reducción en la presión arterial en pacientes tratados con FORXIGA® 10 mg(variación media entre el valor inicial y la semana 24 de -3.7 mmHg en la presión arterial sistólica en posición sentada y de -1.8 mmHg en la presión arterial diastólica en posición sentada con FORXIGA® 10 mg, frente a variaciones respectivas de -0.5 mmHg y -0.5 mmHg en el grupo placebo). La medición de la presión arterial postural durante el periodo de tratamiento de 24 semanas reveló hipotensión ortostática en el 13.1% de los pacientes tratados con 10 mg de FORXIGA®, frente al 11.3% de los pacientes del grupo placebo.
Adicionalmente, en 2 estudios en pacientes con diabetes tipo 2 e hipertensión, la medición de la presión postural reveló hipotensión ortostática en 3.2% de los pacientes tratados con FORXIGA® 10 mg versus 1.7% de los pacientes tratados con placebo a través de los 2 estudios durante el periodo de tratamiento de 12 semanas (véase sección Estudios complementarios).
No se han observado otras variaciones de importancia clínica de las constantes vitales en los pacientes tratados con FORXIGA®.
Hallazgos de laboratorio
Hematocrito: En el grupo de 13 estudios placebo-controlados, los pacientes tratados con FORXIGA® presentaron aumentos del hematocrito frente al valor inicial que comenzaron en la semana 1 y continuaron hasta la semana 16, momento en el que se registró la diferencia máxima con respecto al valor inicial. Al cabo de 24 semanas, las variaciones medias del hematocrito frente a los valores iniciales fueron del 2.30% en el grupo tratado con 10 mg de FORXIGA® frente a -0.33% en el grupo placebo. Al cabo de 102 semanas, las variaciones medias fueron del 2.68% frente a -0.46%, respectivamente. En la semana 24 se registraron valores del hematocrito >55% en el 1.3% de los pacientes tratados con 10 mg de FORXIGA® frente al 0.4% de aquellos que recibieron un placebo. Los resultados fueron similares combinando los periodos a corto plazo y a largo plazo de los estudios (la mayoría de los pacientes recibieron el medicamento más de un año).
Concentración sérica de fósforo inorgánico: En el grupo de 13 estudios placebo-controlados, en la semana 24 se notificaron elevaciones de las concentraciones séricas medias de fósforo frente a los valores iniciales en los pacientes tratados con 10 mg de FORXIGA® con respecto al grupo placebo (aumentos medios de 0.13 mg/dL frente a -0.04 mg/dL, respectivamente). Se observaron resultados similares después de 102 semanas.121 Las proporciones de pacientes con hiperfosfatemia importante (=5.6 mg/dl en pacientes de 17 a 65 años o =5.1 mg/dl en pacientes mayores de 66 años) fueron más elevadas en el grupo tratado con 10 mg de FORXIGA® que en el grupo placebo después de 24 semanas (1.7% frente al 0.9%, respectivamente) y durante los periodos a corto plazo y largo plazo combinados (3.0% frente al 1.6%, respectivamente). Se desconoce la importancia clínica de estas observaciones.
Lípidos: En el grupo de 13 estudios placebo-controlados, se registraron pequeñas variaciones de las cifras medias de lípidos después de 24 semanas frente a las cifras iniciales en los pacientes tratados con 10 mg de FORXIGA® comparados con los del grupo placebo. Las medias de los porcentajes de variación entre el valor inicial y la semana 24 con 10 mg de FORXIGA® y con el placebo, respectivamente, fueron las siguientes: Colesterol total: 2.5% frente a 0.0%; colesterol de HDL: 6.0% frente al 2.7%; colesterol de LDL: 2.9% frente a -1.0%; triglicéridos: -2.7% frente al -0.7%. Las medias de los porcentajes de variación entre el valor inicial y la semana 102 con 10 mg de FORXIGA® y con el placebo, respectivamente, fueron las siguientes: Colesterol total: 2.1% frente a -1.5%; colesterol de HDL: 6.6% frente al 2.1%; colesterol de LDL: 2.9% frente a -2.2%; triglicéridos: -1.8% frente al -1.8%.126,127,128,129 Después de 24 semanas, la razón entre el colesterol de LDL y el colesterol de HDL disminuyó en ambos grupos de tratamiento.130
INTERACCIONES CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: El metabolismo de la dapagliflozina consiste básicamente en glucuronidación dependiente de la UGT1A9. Su principal metabolito, el 3-O-glucurónido de dapagliflozina, no es un inhibidor del SGLT2.
En estudios in vitro, la dapagliflozina y el 3-O-glucurónido de dapagliflozina no inhibieron las enzimas CYP 1A2, 2C9, 2C19, 2D6 o 3A4 y no indujeron las enzimas CYP 1A2, 2B6 o 3A4. En consecuencia, se prevé que la dapagliflozina no alterará la depuración metabólica de los fármacos coadministrados cuyo metabolismo depende de dichas enzimas y que los fármacos coadministrados que inhiben o inducen dichas enzimas tampoco alterarán la depuración metabólica de la dapagliflozina. La dapagliflozina es un sustrato débil del transportador activo de glucoproteína P (P-gp), mientras que el 3-O-glucurónido de dapagliflozina es un sustrato del transportador activo OAT3.51 Ni la dapagliflozina ni el 3-O-glucurónido de dapagliflozina inhibieron de manera significativa los transportadores activos P-gp, OCT2, OAT1 u OAT3. De manera general, es improbable que la dapagliflozina afecte la farmacocinética de fármacos coadministrados que sean sustratos de los transportadores P-gp, OCT2, OAT1 u OAT3.
Efectos de otros fármacos en la dapagliflozina: En estudios realizados en voluntarios sanos, los siguientes fármacos no alteraron la farmacocinética de la dapagliflozina: Metformina, pioglitazona (sustrato importante de CYP2C8 y sustrato menor de CYP3A4), sitagliptina (sustrato de hOAT-3 y de P-gp), glimepirida, voglibosa, hidroclorotiazida, bumetanida, valsartán y simvastatina. Tras la coadministración de dapagliflozina con rifampicina (un inductor de distintos transportadores activos y de enzimas responsables del metabolismo de los medicamentos) se observó una disminución del 22% de la exposición sistémica a la dapagliflozina, mientras que tras la coadministración de dapagliflozina con ácido mefenámico (un inhibidor de UGT1A9) se observó un aumento del 51% de la exposición sistémica a la dapagliflozina, aunque en ninguno de estos casos se observó un efecto de importancia clínica en la excreción urinaria de glucosa de 24 horas.
Metformina: La coadministración de una dosis única de dapagliflozina (20 mg) y de metformina (1000 mg), un sustrato de hOCT-1 y hOCT-2, no alteró la farmacocinética de la dapagliflozina. En consecuencia, no se prevén interacciones importantes entre FORXIGA® y otros sustratos de hOCT-1 y hOCT-2.
Pioglitazona: La coadministración de una dosis única de dapagliflozina (50 mg) y de pioglitazona (45 mg), un sustrato importante de CYP2C8 y un sustrato menor de CYP3A4, no alteró la farmacocinética de la dapagliflozina. En consecuencia, no se prevén interacciones importantes entre FORXIGA® y otros sustratos de CYP2C8.
Sitagliptina: La coadministración de una dosis única de dapagliflozina (20 mg) y de sitagliptina (100 mg), un sustrato de hOCT-3, no alteró la farmacocinética de la dapagliflozina. En consecuencia, no se prevén interacciones importantes entre FORXIGA® y otros sustratos de hOCT-3.
Glimepirida: La coadministración de una dosis única de dapagliflozina (20 mg) y de glimepirida (4 mg), un sustrato de CYP2C9, no alteró la farmacocinética de la dapagliflozina. En consecuencia, no se prevén interacciones importantes entre FORXIGA® y otros sustratos de CYP2C9.
Voglibosa (inhibidor de la a -glucosidasa): La coadministración de una dosis única de dapagliflozina (10 mg) y de voglibosa (0.2 mg tres veces al día) no alteró la farmacocinética de la dapagliflozina.
Hidroclorotiazida: La coadministración de una dosis única de dapagliflozina (50 mg) y de hidroclorotiazida (25 mg) no alteró la farmacocinética de la dapagliflozina.
Bumetanida: La coadministración de dosis repetidas de dapagliflozina (10 mg) y de bumetanida (1 mg) una vez al día no alteró la farmacocinética de la dapagliflozina. La coadministración de dapagliflozina y bumetanida no modificó de manera importante el efecto farmacodinámico de la dapagliflozina, que consiste en aumentar la excreción urinaria de glucosa en sujetos sanos.
Valsartán: La coadministración de una dosis única de dapagliflozina (20 mg) y de valsartán (320 mg) no alteró la farmacocinética de la dapagliflozina.
Simvastatina: La coadministración de una dosis única de dapagliflozina (20 mg) y de simvastatina (40 mg), un sustrato de CYP3A4, no alteró la farmacocinética de la dapagliflozina. En consecuencia, no se prevén interacciones importantes entre FORXIGA® y otros sustratos de CYP3A4.
Rifampicina: La coadministración de una dosis única de dapagliflozina (10 mg) y de dosis de rifampicina (un inductor de distintos transportadores activos y de enzimas responsables del metabolismo de los medicamentos) que permitieron alcanzar el estado de equilibrio (600 mg al día), produjo disminuciones del 7% de la Cmáx de la dapagliflozina y del 22% de su ABC. La cantidad media de glucosa excretada en la orina en las 24 horas siguientes a la administración de la dapagliflozina sola (51 g) no varió de manera pronunciada con la coadministración de rifampicina (45 g). No se recomienda ningún ajuste de la dosis de dapagliflozina en caso de coadministración con la rifampicina.
Ácido mefenámico: La coadministración de una dosis única de dapagliflozina (10 mg) y de dosis de ácido mefenámico (un inhibidor de UGT1A9) que permitieron alcanzar el estado de equilibrio (250 mg cada 6 horas), produjo aumentos del 13% de la Cmáx de la dapagliflozina y del 51% de su ABC. La cantidad media de glucosa excretada en la orina en las 24 horas siguientes a la administración de la dapagliflozina sola no varió de manera pronunciada con la coadministración de ácido mefenámico. No se recomienda ningún ajuste de la dosis de dapagliflozina en caso de coadministración con el ácido mefenámico.
Efecto de la dapagliflozina en otros fármacos: En los estudios realizados en voluntarios sanos que se describen a continuación, la dapagliflozina no alteró la farmacocinética de los siguientes fármacos: Metformina, pioglitazona, sitagliptina, glimepirida, hidroclorotiazida, bumetanida, valsartán, simvastatina, digoxina y warfarina.
Metformina: La coadministración de una dosis única de dapagliflozina (20 mg) y de metformina (1000 mg), un sustrato de hOCT-1 y hOCT-2, no alteró la farmacocinética de la metformina. En consecuencia, FORXIGA® no es un inhibidor del transporte mediado por hOCT-1 y hOCT-2.
Pioglitazona: La coadministración de una dosis única de dapagliflozina (50 mg) y de pioglitazona (45 mg), un sustrato importante de CYP2C8 y un sustrato menor de CYP3A4, no alteró la farmacocinética de la pioglitazona. En consecuencia, FORXIGA® no inhibe de manera significativa el metabolismo mediado por la CYP2C8.
Sitagliptina: La coadministración de una dosis única de dapagliflozina (20 mg) y de sitagliptina (100 mg), un sustrato de hOAT-3, no alteró la farmacocinética de la sitagliptina. En consecuencia, FORXIGA® no es un inhibidor del transportador hOAT-3.
Glimepirida: La coadministración de una dosis única de dapagliflozina (20 mg) y de glimepirida (4 mg), un sustrato de CYP2C9, no alteró la farmacocinética de la glimepirida. En consecuencia, FORXIGA® no es un inhibidor del metabolismo mediado por la CYP2C9.
Hidroclorotiazida: La coadministración de una dosis única de dapagliflozina (50 mg) y de hidroclorotiazida (25 mg) no alteró la farmacocinética de la hidroclorotiazida.
Bumetanida: La coadministración de dosis repetidas de dapagliflozina (10 mg) y de bumetanida (1 mg) administradas una vez al día elevó un 13% tanto la Cmáx como el ABC de la bumetanida. La coadministración de la dapagliflozina no alteró de manera importante las respuestas farmacodinámicas en el estado de equilibrio (excreción urinaria de sodio, volumen de orina) a la bumetanida en sujetos sanos.
Valsartán: La coadministración de una dosis única de dapagliflozina (20 mg) y de valsartán (320 mg) no alteró la farmacocinética del valsartán.
Simvastatina: La coadministración de una dosis única de dapagliflozina (20 mg) y de simvastatina (40 mg), un sustrato de CYP3A4, no alteró la Cmáx de la simvastatina pero sí elevó un 20% su ABC; este efecto no se consideró de importancia clínica. En consecuencia, FORXIGA® no inhibió de manera significativa el metabolismo mediado por la CYP3A4.
Digoxina: La coadministración de dapagliflozina (10 mg una vez al día después de una dosis de carga de 20 mg) y una dosis única de digoxina (0.25 mg), un sustrato de la glucoproteína P, no alteró la farmacocinética de la digoxina. En consecuencia, la dapagliflozina no inhibe ni induce de manera significativa el transporte mediado por la glucoproteína P.
Warfarina: La coadministración de dapagliflozina (10 mg una vez al día después de una dosis de carga de 20 mg) y una dosis única de warfarina (25 mg) no alteró la farmacocinética de la S-warfarina, un sustrato de la CYP2C19. En consecuencia, la dapagliflozina no inhibe ni induce de manera significativa el metabolismo mediado por la CYP2C19. La dapagliflozina no alteró la farmacocinética de la R-warfarina. Por otro lado, la dapagliflozina no alteró el efecto anticoagulante de la warfarina, medido por el tiempo de protrombina (índice normalizado internacional [INR]).
Otras interacciones: No se han estudiado específicamente los efectos del tabaquismo, de la dieta o del consumo de hierbas medicinales y de alcohol en la farmacocinética de la dapagliflozina.
INFORMACIÓN DE ESTUDIOS CLÍNICOS: FORXIGA® se ha investigado en monoterapia y en asociación con los siguientes fármacos: Metformina, glimepirida, sitagliptina o insulina. FORXIGA® también se ha estudiado en pacientes con diabetes tipo 2 y enfermedad cardiovascular y en aquellos con diabetes tipo 2 e hipertensión. Un total de 9412 pacientes con diabetes tipo 2 participaron en 16 ensayos clínicos controlados con un diseño doble ciego que evaluaron la seguridad y la eficacia de FORXIGA®; en estos estudios, 5952 pacientes recibieron el tratamiento con FORXIGA®. Trece estudios incluyeron un periodo terapéutico de 24 semanas, dos de 12 semanas y un estudio duró 52 semanas. De los 16 estudios, 11 tuvieron extensiones a largo plazo de 24 a 80 semanas (duración total de hasta 104 semanas). Combinando los 16 estudios clínicos, la media de la edad de los pacientes fue de 57 años (18 a 92 años) y la duración media de la diabetes fue de 8 años (entre <1 a 54 años). El 55% de los pacientes eran varones, el 82% de raza blanca, el 10% asiáticos, el 4% de raza negra. El 81% de los pacientes tenían un índice de masa corporal (IMC) = 27 kg/m2. FORXIGA® también se ha investigado en pacientes con insuficiencia renal leve (53% de la población examinada) o moderada (12% de la población examinada).
El tratamiento con FORXIGA®, en monoterapia o en asociación con metformina, glimepirida, sitagliptina o insulina, produjo mejorías de importancia clínica y estadísticamente significativas de la variación media entre los valores iniciales y los valores medidos después de 24 semanas de la HbA1c, la glucemia en ayunas (GPA) y la glucemia posprandial determinada 2 horas después de comer (GPP de 2 horas), con respecto al grupo de control. Estos efectos de importancia clínica en la glucemia se mantuvieron durante las extensiones a largo plazo de hasta 104 semanas de duración. Se observaron reducciones de la HbA1c en todos los subgrupos basados en el sexo, la edad, la raza, la duración de la enfermedad y el IMC inicial. Además, después de 24 semanas se observaron reducciones de importancia clínica y estadísticamente significativas de las variaciones medias del peso corporal frente al valor inicial con las asociaciones de FORXIGA® y otro fármaco frente al grupo de control. Las reducciones del peso corporal se mantuvieron durante las extensiones a largo plazo de hasta 104 semanas. En un estudio clínico específico, la disminución del peso corporal se atribuyó principalmente a una reducción de la masa adiposa medida por DXA.
En dos estudios de FORXIGA® 10 mg en pacientes con diabetes tipo 2 y enfermedad cardiovascular, se observaron mejorías estadísticamente representativas en la HbA1c y reducciones significativas en el peso corporal y presión arterial sistólica en posición sentada a la semana 24 en pacientes tratados con FORXIGA® 10 mg comparados con los que recibieron placebo, y se mantuvieron hasta la semana 104. En dos estudios de FORXIGA® 10 mg en pacientes con diabetes tipo 2 e hipertensión, también se observaron reducciones estadísticamente significativas en la presión arterial sistólica media en posición sentada en pacientes tratados con FORXIGA® 10 mg combinado con otras terapias antidiabéticas orales y tratamientos antihipertensivos (un IECA o BRA en un estudio y un IECA o BRA más un tratamiento antihipertensivo adicional en otro estudio) comparados con aquellos tratados con placebo a la semana 12.
En 14 de los 16 estudios con diseño doble ciego se evaluó la dosis de FORXIGA® de 10 mg una vez al día. Se examinaron igualmente dosis de 2.5 mg de dapagliflozina y 5 mg de FORXIGA® en algunos de estos estudios; la dosis de 2.5 mg no mostró una eficacia constante para el control glucémico y la dosis de 10 mg de FORXIGA® mostró una eficacia numéricamente superior y una seguridad comparable a la de la dosis de 5 mg.
Monoterapia: Un total de 840 pacientes no tratados anteriormente, con diabetes tipo 2 insuficientemente controlada, participaron en dos estudios controlados con placebo cuyo objetivo consistió en evaluar la eficacia y la seguridad de la monoterapia con FORXIGA®.
En un estudio sobre la monoterapia, un total de 558 pacientes no tratados anteriormente, con diabetes insuficientemente controlada, participaron en un estudio de 24 semanas con un periodo de extensión de 78 semanas controlado y con diseño ciego. Tras un periodo de introducción de 2 semanas con dieta, ejercicio y tratamiento con un placebo, 485 pacientes con una HbA1c =7% y =10% fueron aleatorizados entre los siguientes grupos: 2.5 mg de dapagliflozina o 5 mg o 10 mg de FORXIGA® una vez al día por la mañana (1 v/d por la mañana, cohorte principal) o por la tarde (1v/día por la tarde), o bien, un placebo por la mañana solamente.
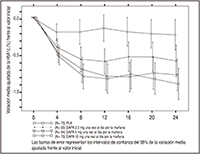
Después de 24 semanas, el tratamiento con 10 mg de FORXIGA® 1 v/d por la mañana produjo mejorías significativas de la HbA1c y la GA frente al placebo (Tabla 2, Figura 2). Globalmente, la administración de FORXIGA® por la tarde mostró un perfil de eficacia y seguridad comparable al de la administración de FORXIGA® por la mañana. Las variaciones medias ajustadas de HbA1c y GA fueron de -0.61% y -27.0 mg/dl, respectivamente, después de 102 semanas en el grupo tratado con 10 mg de FORXIGA® 1 v/d por la mañana, y de -0.17% y -6.9 mg/dL, respectivamente, en los pacientes tratados con un placebo, basándose en un análisis longitudinal de mediciones repetidas y excluyendo los datos obtenidos después del tratamiento de rescate.
La proporción de pacientes de la cohorte principal que necesitaron un tratamiento de rescate o que abandonaron el estudio por la falta de control glucémico después de 24 semanas (tras un ajuste en función del valor inicial de HbA1c) fue mayor en el grupo placebo (12.0%) que en el tratado con 10 mg de FORXIGA® (0.0%). Después de 102 semanas (ajustado en función de la HbA1c inicial), la proporción de pacientes que necesitaron un tratamiento de rescate fue mayor en pacientes tratados con el placebo (44.0%) que en pacientes tratados con FORXIGA® 10 mg (35.0%).
|
Tabla 2. Resultados obtenidos después de 24 semanas (método LOCF*) en un estudio controlado con placebo sobre la monoterapia con FORXIGA® en pacientes con diabetes tipo 2 (cohorte principal tratada por la mañana) |
||
|
Parámetro de eficacia |
FORXIGA® 10 mg |
Placebo |
|
HbA1c (%) |
||
|
Valor inicial (media) |
8.01 |
7.79 |
|
Variación frente al valor inicial (media ajustada‡) |
-0.89 |
-0.23 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-0.66§ |
|
|
Porcentaje de pacientes que alcanzaron un valor de HbA1c <7% (tras un ajuste en función del valor inicial) |
50.8%¶ |
31.6% |
|
Variación de HbA1c frente al valor inicial en pacientes con una HbA1c inicial = 9% (media ajustada‡) |
-2.04¶ |
0.19 |
|
Glucemia en ayunas (mg/dl) |
||
|
Valor inicial (media) |
166.6 |
159.9 |
|
Variación frente al valor inicial (media ajustada‡) |
-28.8 |
-4.1 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-24.7§ |
|
|
Peso corporal (kg) |
||
|
Valor inicial (media) |
94.13 |
88.77 |
|
Variación frente al valor inicial (media ajustada‡) |
-3.16 |
-2.19 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-0.97 |
|
|
* LOCF: Método de reutilización del resultado de la última observación (antes del tratamiento de rescate, en su caso). † Todos los pacientes aleatorizados que tomaron al menos una dosis del medicamento en investigación durante el periodo con un diseño doble ciego a corto plazo. ‡ Media de mínimos cuadrados ajustada en función del valor inicial. § p < 0.0001 frente al placebo. ¶ No se determinó si la diferencia fue estadísticamente significativa debido al procedimiento secuencial empleado para analizar los variables secundarias. |
||
FIGURA 2. Variación media ajustada de la HbA1c (%) frente al valor inicial en función del tiempo (método LOCF) en un estudio de 24 semanas controlado con placebo sobre la monoterapia con FORXIGA® en pacientes con diabetes tipo 2 (grupo 1 tratado por la mañana)
Otro estudio de 24 semanas sobre la monoterapia que comparó dosis de 1 mg y 2.5 mg de dapagliflozina y 5 mg de FORXIGA® con un placebo, también reveló mejorías de importancia clínica y estadísticamente significativas de los parámetros glucémicos y del peso.
Tratamiento combinado: Se investigó la adición de FORXIGA® al tratamiento con metformina, con una sulfonilurea (glimepirida), con un inhibidor de la DPP4 (sitagliptina) y con insulina (con o sin otros antidiabéticos).
Adición de FORXIGA® al tratamiento con metformina: Se llevaron a cabo 4 estudios sobre la asociación de la dapagliflozina y la metformina. Dos de ellos evaluaron el tratamiento inicial con la asociación de FORXIGA® y metformina, uno evaluó el efecto de la adición de FORXIGA® en pacientes que ya estaban tomando metformina, y uno evaluó el efecto de la adición de FORXIGA® al tratamiento con la metformina comparada con una sulfonilurea adicionada a metformina.
Tratamiento inicial con la asociación de FORXIGA® y metformina: Un total de 1241 pacientes no tratados anteriormente, con diabetes tipo 2 insuficientemente controlada (HbA1c =7.5% y =12%), participaron en dos estudios controlados con un medicamento de referencia de 24 semanas de duración que evaluaron la eficacia y la seguridad del tratamiento inicial con la asociación de 5 mg o 10 mg de FORXIGA® y una formulación de metformina de liberación prolongada (metformina XR).
En un estudio, después de un periodo de introducción de 1 semana, 638 pacientes se distribuyeron al azar entre tres grupos de tratamiento: FORXIGA® 10 mg + metformina XR (hasta 2000 mg al día), FORXIGA® 10 mg + placebo, o metformina XR (hasta 2000 mg al día) + placebo. Se aumentó progresivamente la dosis de metformina XR por incrementos semanales de 500 mg en función de la tolerabilidad, alcanzando una mediana de 2000 mg.
La asociación de FORXIGA® 10 mg + metformina XR produjo mejorías significativas de la HbA1c y la GPA con respecto a cualquiera de las monoterapias, y reducciones significativas del peso corporal frente al tratamiento con la metformina XR sola (Tabla 4, Figuras 3 y 4). La monoterapia con 10 mg de FORXIGA® también produjo mejorías significativas de la GPA y reducciones significativas del peso corporal frente a la metformina XR sola, y la reducción de la HbA1c no fue inferior a la obtenida con la metformina XR en monoterapia. La proporción de pacientes que necesitaron un tratamiento de rescate o que abandonaron el estudio por la falta de control glucémico durante el periodo terapéutico con un diseño doble ciego de 24 semanas (tras un ajuste en función del valor inicial de HbA1c) fue mayor en el grupo tratado con metformina XR + placebo (13.5%) que en los grupos tratados con FORXIGA® 10 mg + placebo (7.8%) y con FORXIGA® 10 mg + metformina XR (1.4%).
|
Tabla 3. Resultados obtenidos después de 24 semanas (método LOCF*) en un estudio controlado con un medicamento de referencia sobre el tratamiento inicial con una asociación de FORXIGA® y metformina XR |
|||
|
Parámetro de eficacia |
FORXIGA® 10 mg + metformina XR |
FORXIGA® 10 mg |
Metformina XR |
|
HbA1c (%) |
|||
|
Valor inicial (media) |
9.10 |
9.03 |
9.03 |
|
Variación frente al valor inicial (media ajustada‡) |
-1.98 |
-1.45 |
-1.44 |
|
Diferencia con respecto a FORXIGA® (media ajustada‡) (IC del 95%) |
-0.53§ (-0.74, -0.32) |
||
|
Diferencia con respecto a metformina XR (media ajustada‡) (IC del 95%) |
-0.54§ (-0.75, -0.33) |
-0.01¶ |
|
|
Porcentaje de pacientes que alcanzaron un valor de HbA1c <7% (tras un ajuste en función del valor inicial) |
46.6%# |
31.7% |
35.2% |
|
Variación de HbA1c frente al valor inicial en pacientes con una HbA1c inicial = 9% (media ajustada‡) |
-2.59# |
-2.14 |
-2.05 |
|
Glucemia en ayunas (mg/dl) |
|||
|
Valor inicial (media) |
189.6 |
197.5 |
189.9 |
|
Variación frente al valor inicial (media ajustada‡) |
-60.4 |
-46.4 |
-34.8 |
|
Diferencia con respecto a FORXIGA® (media ajustada‡) (IC del 95%) |
-13.9§ |
||
|
Diferencia con respecto a metformina XR (media ajustada‡) (IC del 95%) |
-25.5§ |
-11.6¶ |
|
|
Peso corporal (kg) |
|||
|
Valor inicial (media) |
88.56 |
88.53 |
87.24 |
|
Variación frente al valor inicial (media ajustada‡) |
-3.33 |
-2.73 |
-1.36 |
|
Diferencia con respecto a metformina XR (media ajustada‡) (IC del 95%) |
-1.97§ |
-1.37§ |
|
|
* LOCF: Método de reutilización del resultado de la última observación (antes del tratamiento de rescate, en su caso). † Todos los pacientes aleatorizados que tomaron al menos una dosis del medicamento en investigación durante el periodo con un diseño doble ciego a corto plazo. ‡ Media de mínimos cuadrados ajustada en función del valor inicial. § p < 0.0001. ¶ No inferioridad frente a metformina XR. # p <0.05. |
|||
FIGURA 3. Variación media ajustada de la HbA1c (%) frente al valor inicial en función del tiempo (método LOCF) en un estudio de 24 semanas controlado con un medicamento de referencia sobre el tratamiento inicial con la asociación de FORXIGA® y metformina XR
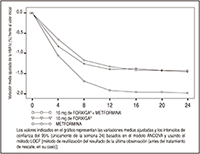
Los valores indicados en el gráfico representan las variaciones medias ajustadas y los intervalos de confianza del 95% (únicamente de la semana 24) basados en el modelo ANCOVA y usando el método LOCF [método de reutilización del resultado de la última observación (antes del tratamiento de rescate, en su caso)].
FIGURA 4. Variación media ajustada del peso corporal total (kg) frente al valor inicial en función del tiempo (método LOCFa) en un estudio de 24 semanas controlado con un medicamento de referencia sobre el tratamiento inicial con la asociación de FORXIGA® y metformina XR
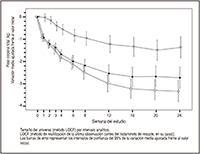
Otro estudio de 24 semanas que evaluó la asociación de FORXIGA® 5 mg + metformina XR demostró mejorías de importancia clínica y estadísticamente significativas de los parámetros glucémicos frente a la monoterapia con 5 mg de FORXIGA® y a la monoterapia con metformina XR.
Adición de FORXIGA® al tratamiento con metformina: Un total de 546 pacientes con diabetes tipo 2 y un control glucémico insuficiente (HbA1c =7% y =10%) participaron en un estudio controlado con placebo de 24 semanas con un periodo de extensión de 78 semanas, controlado y con diseño ciego, que evaluó la asociación de FORXIGA® y metformina. En este estudio participaron pacientes tratados con una dosis de metformina de al menos 1500 mg al día que, al cabo de un periodo de introducción de 2 semanas con un diseño simple ciego durante el cual recibieron un placebo, fueron distribuidos al azar entre los siguientes grupos: 2.5 mg de dapagliflozina, 5 mg o 10 mg de FORXIGA® o un placebo, además de la dosis de metformina que ya estaban tomando.
Al añadirse al tratamiento con metformina, FORXIGA® 10 mg produjo mejorías significativas de la HbA1c y la GPA y una reducción significativa en el peso corporal frente al placebo después de 24 semanas (Tabla 4). Después de 102 semanas, las variaciones medias ajustadas frente a los valores iniciales de la HbA1c (véase la Figura 5), la GPA y el peso corporal fueron de -0.78%, -24.5 mg/dl y -2.81 kg, respectivamente, en los pacientes tratados con la asociación de FORXIGA® 10 mg + metformina, y de 0.02%, -10.4 mg/dl y -0.67 kg en los pacientes tratados con un placebo + metformina, basándose en el análisis longitudinal de mediciones repetidas y excluyendo los datos obtenidos después del tratamiento de rescate (Figura 5). La proporción de pacientes que necesitaron un tratamiento de rescate o que abandonaron el estudio por la falta de control glucémico durante el periodo terapéutico de 24 semanas con un diseño doble ciego (tras un ajuste en función del valor inicial de HbA1c) fue mayor en el grupo placebo + metformina (15.0%) que en el grupo tratado con FORXIGA® 10 mg + metformina (4.4%). Después de 102 semanas (ajustado en función de la HbA1c inicial), la proporción de pacientes que necesitaron un tratamiento de rescate fue mayor en el grupo placebo + metformina (60.1%) que en el grupo tratado con FORXIGA® 10 mg + metformina (44.0%).
|
Tabla 4. Resultados obtenidos después de 24 semanas (método LOCF*) en un estudio controlado con placebo sobre la adición de FORXIGA® al tratamiento con metformina |
||
|
Parámetro de eficacia |
FORXIGA® 10 mg + metformina N=135† |
Placebo + metformina |
|
HbA1c (%) |
||
|
Valor inicial (media) |
7.92 |
8.11 |
|
Variación frente al valor inicial (media ajustada‡) |
-0.84 |
-0.30 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-0.54§ |
|
|
Porcentaje de pacientes que alcanzaron un valor de HbA1c <7% (tras un ajuste en función del valor inicial) |
40.6%¶ |
25.9% |
|
Variación de HbA1c frente al valor inicial en pacientes con una HbA1c inicial = 9% (media ajustada‡) |
-1.32¶ |
-0.53 (N=22) |
|
Glucemia en ayunas (mg/dl) |
||
|
Valor inicial (media) |
156.0 |
165.6 |
|
Variación después de 24 semanas frente al valor inicial (media ajustada‡) |
-23.5 |
-6.0 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-17.5§ |
|
|
Variación entre el valor inicial y el valor medido en la semana 1 (media ajustada‡) |
-16.5§ |
1.2 |
|
Peso corporal (kg) |
||
|
Valor inicial (media) |
86.28 |
87.74 |
|
Variación frente al valor inicial (media ajustada‡) |
-2.86 |
-0.89 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-1.97§ |
|
|
* LOCF: Método de reutilización del resultado de la última observación (antes del tratamiento de rescate, en su caso). † Todos los pacientes aleatorizados que tomaron al menos una dosis del medicamento en investigación durante el periodo con un diseño doble ciego a corto plazo. ‡ Media de mínimos cuadrados ajustada en función del valor inicial. § p < 0.00001 frente a la asociación de placebo + metformina. ¶ p <0.05 frente a la asociación de placebo + metformina. |
||
FIGURA 5. Variación media ajustada de la HbA1c (%) frente al valor inicial en función del tiempo en un estudio de 102 semanas controlado con placebo sobre la asociación de FORXIGA® y metformina (análisis longitudinal de mediciones repetidas, excluyendo los datos obtenidos después del tratamiento de rescate)
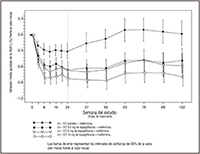
Estudio controlado con la glipizida sobre la adición de FORXIGA® al tratamiento con metformina: Un total de 816 pacientes con diabetes tipo 2 y un control glucémico insuficiente (HbA1c >6.5% y =10%) fueron aleatorizados en un estudio de no inferioridad de 52 semanas, controlado con la glipizida y con un periodo de extensión de 156 semanas, que evaluó la adición de FORXIGA® al tratamiento con metformina. Los pacientes, que estaban recibiendo una dosis de metformina de al menos 1500 mg al día fueron distribuidos al azar, al cabo de un periodo de introducción de 2 semanas con placebo, entre el tratamiento con la glipizida o con 5 mg o 2.5 mg de dapagliflozina, aumentando progresivamente la dosis durante 18 semanas hasta que se alcanzara un efecto óptimo en la glucemia (GPA < 110 mg/dl, < 6.1 mmol/l)) o bien la dosis máxima (20 mg de glipizida y 10 mg de FORXIGA®), en función de la tolerabilidad. Posteriormente se mantuvieron dosis constantes, salvo en los casos de ajuste a la baja para la prevención de la hipoglucemia.
Al final del periodo de ajuste de la dosis, el 87% de los pacientes tratados con FORXIGA® habían alcanzado la dosis máxima (10 mg) frente al 73% de aquellos tratados con la glipizida (20 mg). FORXIGA® produjo una reducción media de la HbA1c entre el valor inicial y la semana 52 similar a la que se consiguió con la glipizida, lo cual demostró la no inferioridad (Tabla 5). El tratamiento con FORXIGA® produjo una reducción media significativa del peso corporal entre el valor inicial y la semana 52, mientras que en el grupo de la glipizida se registró un aumento medio del peso corporal.
Según el análisis longitudinal de mediciones repetidas, la variación media ajustada de la HbA1c y del peso corporal fue de -0.32% y de -3.70 kg, respectivamente, entre el inicio y la semana 104 en los pacientes tratados con FORXIGA®, y de -0.14% y 1.36 kg en los que recibieron la glipizida (Figuras 6 y 7). El 23.8% de los pacientes tratados con FORXIGA® tuvieron una pérdida de peso =5% (valor ajustado), frente al 2.8% de los que recibieron la glipizida.
En las semanas 52 y 104, la proporción de pacientes que habían abandonado el estudio por la falta de control glucémico (tras un ajuste en función del valor inicial de HbA1c) fue mayor en el grupo de la glipizida + metformina (3.6% y 21.6%, respectivamente) que en el grupo de FORXIGA® + metformina (0.2% y 14.5%, respectivamente). En este estudio no se previó un tratamiento de rescate en caso de descontrol glucémico. En las semanas 52 y 104, respectivamente, la proporción de pacientes que habían sufrido al menos un episodio de hipoglucemia fue significativamente menor en pacientes tratados con FORXIGA® (3.5% y 4.3%) que con la glipizida (40.8% y 47.0%).
|
Tabla 5. Resultados obtenidos después de 52 semanas (método LOCF*) en un estudio controlado con un medicamento de referencia que comparó la adición de FORXIGA® o de glipizida al tratamiento con metformina |
||
|
Parámetro de eficacia |
FORXIGA® + metformina |
Glipizida + metformina |
|
HbA1c (%) |
||
|
Valor inicial (media) |
7.69 |
7.74 |
|
Variación frente al valor inicial (media ajustada‡) |
-0.52 |
-0.52 |
|
Diferencia con respecto a la asociación de glipizida + metformina (media ajustada‡) (IC del 95%) |
0.00§ (-0.11, 0.11) |
|
|
Peso corporal (kg) |
||
|
Valor inicial (media) |
88.44 |
87.60 |
|
Variación frente al valor inicial (media ajustada‡) |
-3.22 |
1.44 |
|
Diferencia con respecto a la asociación de glipizida + metformina (media ajustada‡) (IC del 95%) |
-4.65¶ (-5.14, -4.17) |
|
|
Porcentaje de pacientes con una pérdida de peso >5% (valor ajustado) (IC del 95%) |
33.3%¶ |
2.5% |
|
* LOCF: Método de reutilización del resultado de la última observación. † Pacientes aleatorizados y tratados que fueron objeto de una evaluación inicial de la eficacia y al menos una evaluación posterior. ‡ Media de mínimos cuadrados ajustada en función del valor inicial. § No inferioridad frente a la asociación de glipizida + metformina. ¶ p <0.0001 |
||
FIGURA 6. Variación media ajustada de la HbA1c (%) frente al valor inicial en función del tiempo en un estudio de 104 semanas controlado con un fármaco de referencia que comparó la adición de FORXIGA® o glipizida al tratamiento con metformina (análisis longitudinal de mediciones repetidas, excluyendo los datos obtenidos después del tratamiento de rescate)
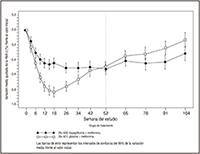
FIGURA 7. Variación media ajustada del peso corporal (kg) frente al valor inicial en función del tiempo en un estudio de 104 semanas controlado con un fármaco de referencia que comparó la adición de FORXIGA® o glipizida al tratamiento con metformina (análisis longitudinal de mediciones repetidas, excluyendo los datos obtenidos después del tratamiento de rescate)
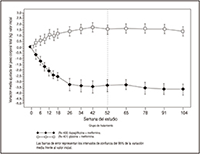
Adición al tratamiento con otros antidiabéticos orales
Adición de FORXIGA® al tratamiento con una sulfonilurea: Un total de 597 pacientes con diabetes tipo 2 y un control glucémico insuficiente (HbA1c =7% y =10%) fueron aleatorizados en este estudio controlado con placebo de 24 semanas con una extensión de 24 semanas, que evaluó la asociación de FORXIGA® y glimepirida (una sulfonilurea).
Los pacientes recibieron al menos la mitad de la dosis máxima recomendada de glimepirida en monoterapia (4 mg) durante un periodo de introducción de al menos 8 semanas antes de ser distribuidos al azar entre los siguientes grupos: 2.5 mg de dapagliflozina, 5 mg o 10 mg de FORXIGA® o un placebo, además de 4 mg al día de glimepirida. Se permitió reducir la dosis de glimepirida a 2 mg o incluso a 0 mg en caso de hipoglucemia durante el periodo terapéutico; en cambio, no se permitió aumentar la dosis de glimepirida.
La asociación de glimepirida y 10 mg de FORXIGA® produjo una mejoría significativa de la HbA1c, la GPA, la GPP de 2 horas y una reducción significativa en el peso corporal frente a la asociación de placebo + glimepirida después de 24 semanas (Tabla 6, Figura 8). Después de 48 semanas, las variaciones medias ajustadas de la HbA1c, la GPA y el peso corporal frente a los valores iniciales fueron de -0.73%, -28.8 mg/dl y -2.41 kg, respectivamente, en los pacientes tratados con FORXIGA® 10 mg + glimepirida, y de -0.04%, 2.6 mg/dl y -0.77 kg en los pacientes tratados con placebo + glimepirida, basándose en el análisis longitudinal de mediciones repetidas y excluyendo los datos obtenidos después del tratamiento de rescate.
Después de las 24 semanas, la proporción de pacientes que necesitaron un tratamiento de rescate o que abandonaron el estudio por la falta de control glucémico (tras un ajuste en función del valor inicial de HbA1c) fue mayor en el grupo del placebo + glimepirida (16.2%) que en el grupo tratado con FORXIGA® 10 mg + glimepirida (2.0%). Después de 48 semanas (ajustado en función de la HbA1c inicial), la proporción de pacientes que necesitaron un tratamiento de rescate fue mayor en el grupo tratado con placebo + glimepirida (52.1%) que en el grupo tratado con FORXIGA® 10 mg + glimepirida (18.4%).
Adición de FORXIGA® al tratamiento con insulina: Un total de 808 pacientes con diabetes tipo 2 y un control glucémico insuficiente (HbA1c =7.5% y =10.5%) fueron aleatorizados en un estudio controlado con placebo de 24 semanas, con un periodo de extensión de 80 semanas, que evaluó la adición de FORXIGA® al tratamiento con insulina. Los pacientes que habían recibido un régimen estable con una dosis media de al menos 30 UI de insulina inyectable al día durante un periodo de al menos 8 semanas antes de su admisión en el estudio, y un máximo de dos antidiabéticos orales (ADO), incluida la metformina, fueron distribuidos al azar, al cabo de un periodo de introducción de 2 semanas, entre los siguientes grupos: 2.5 mg de dapagliflozina, 5 mg o 10 mg de FORXIGA® o un placebo, además de la dosis de insulina y, en su caso, de otros ADO que estaban tomando anteriormente. Se efectuó una estratificación basada en la presencia o ausencia de un tratamiento de fondo con ADO. El aumento o la reducción de la dosis de insulina durante el periodo terapéutico solo se autorizó en los pacientes que no alcanzaron objetivos específicos en materia de control glucémico. No se permitieron modificaciones de la dosis del medicamento administrado con un diseño ciego ni de los ADO durante el periodo terapéutico, salvo una disminución de la dosis del ADO en caso de que surgieran problemas de hipoglucemia tras la interrupción del tratamiento con insulina.
En este estudio, el 50% de los pacientes recibían inicialmente una monoterapia con insulina, mientras que el 50% tomaban uno o dos ADO además de la insulina. Después de 24 semanas, la dosis de 10 mg de FORXIGA® produjo una mejoría significativa de la HbA1c, y de la dosis media de insulina, y una significativa reducción del peso corporal frente al grupo tratado con placebo + insulina, al usar hasta dos ADO o sin ellos (Tabla 6). El efecto de FORXIGA® en la HbA1c fue similar en los pacientes tratados con insulina sola o con insulina + ADO. Después de 48 semanas, las variaciones medias ajustadas frente a los valores iniciales de la HbA1c, la GA y el peso corporal fueron de -0.93%, -21.5 mg/dl y -1.79 kg, respectivamente, en los pacientes tratados con FORXIGA® 10 mg + insulina, y de -0.43%, -4.4 mg/dl y -0.18 kg, respectivamente, en los pacientes del grupo placebo + insulina, basándose en el análisis longitudinal de mediciones repetidas y excluyendo los datos obtenidos después del tratamiento de rescate. En la semana 104, las variaciones medias ajustadas frente a los valores iniciales de la HbA1c, la GPA y el peso corporal fueron de -0.71%, -18.2 mg/dl y 1.97 kg, respectivamente, en los pacientes tratados con FORXIGA® 10 mg + insulina, y de 0.06%, -11.2 mg/dl y -0.91 kg, respectivamente, en los pacientes del grupo placebo + insulina, basándose en el análisis longitudinal de mediciones repetidas y excluyendo los datos obtenidos después del tratamiento de rescate (véase la Figura 10).
Después de 24 semanas, la proporción de pacientes que lograron reducir al menos un 10% su dosis de insulina fue mayor con 10 mg de FORXIGA® que con el placebo. La proporción de pacientes que necesitaron un aumento de la dosis de insulina o que abandonaron el estudio por la falta de control glucémico (tras un ajuste en función del valor inicial de HbA1c) fue mayor en el grupo tratado con placebo + insulina (29.2%) que en el grupo tratado con FORXIGA® 10 mg + insulina (9.7%). En las semanas 48 y 104, la dosis de insulina permaneció estable en los pacientes tratados con 10 mg de FORXIGA® con una dosis media de 76 UI al día, mientras que siguió aumentando (aumento medio de 10.5 UI y 18.3 IU, respectivamente frente al valor inicial) en los pacientes tratados con un placebo. Después de 48 y 104 semanas (ajustado en función de la HbA1c inicial), la proporción de pacientes que necesitaron un aumento de la dosis de insulina para mantener el control glucémico, o que suspendieron el tratamiento debido a la falta de control glucémico, fue mayor en los pacientes tratados con el placebo (42.8% y 50.4%, respectivamente) en comparación con los pacientes tratados con FORXIGA® 10 mg (15.3% y 25.5%, respectivamente).
|
Tabla 6. Resultados obtenidos después de 24 semanas (método LOCF*) en los estudios controlados con placebo sobre la asociación de FORXIGA® y antidiabéticos |
||
|
Parámetro de eficacia |
FORXIGA® 10 mg |
Placebo |
|
Asociación con una sulfonilurea (glimepirida) |
||
|
Población por intención de tratar |
N=151† |
N=145† |
|
HbA1c (%) |
||
|
Valor inicial (media) |
8.07 |
8.15 |
|
Variación frente al valor inicial (media ajustada‡) |
-0.82 |
-0.13 |
|
Diferencia con respecto al placebo (media ajustada‡) |
-0.68§ |
|
|
Porcentaje de pacientes que alcanzaron un valor de HbA1c <7% (tras un ajuste en función del valor inicial) |
31.7%§ |
13.0% |
|
Glucemia en ayunas (mg/dl) |
||
|
Valor inicial (media) |
172.4 |
172.7 |
|
Variación frente al valor inicial (media ajustada‡) |
-28.5 |
-2.0 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-26.5§ |
|
|
Glucemia posprandial de 2 horas¶ (mg/dl) |
||
|
Valor inicial (media) |
329.6 |
324.1 |
|
Variación frente al valor inicial (media ajustada‡) |
-60.6 |
-11.5 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-49.1§ |
|
|
Peso corporal (kg) |
||
|
Valor inicial (media) |
80.56 |
80.94 |
|
Variación frente al valor inicial (media ajustada‡) |
-2.26 |
-0.72 |
|
Diferencia con respecto al placebo (media ajustada‡) |
-1.54§ |
|
|
Asociación con insulina y hasta 2 antidiabéticos orales o sin ellos |
||
|
Población de intención de tratar |
N=194† |
N=193† |
|
HbA1c (%) |
||
|
Valor inicial (media) |
8.58 |
8.46 |
|
Variación frente al valor inicial (media ajustada‡) |
-0.90 |
-0.30 |
|
Diferencia con respecto al placebo (media ajustada‡) |
-0.60§ |
|
|
Dosis diaria media de insulina (UI)†† |
||
|
Valor inicial (media) |
77.96 |
73.96 |
|
Variación frente al valor inicial (media ajustada‡) |
-1.16 |
5.08 |
|
Diferencia con respecto al placebo |
-6.23§ |
|
|
Porcentaje de pacientes que consiguieron una reducción de al menos un 10% de la dosis diaria media de insulina (tras un ajuste en función del valor inicial) |
19.6%** |
11.0% |
|
Glucemia en ayunas (mg/dl) |
||
|
Valor inicial (media) |
173.7 |
170.0 |
|
Variación frente al valor inicial (media ajustada‡) |
-21.7 |
3.3 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-25.0§ |
|
|
Peso corporal (kg) |
||
|
Valor inicial (media) |
94.63 |
94.21 |
|
Variación frente al valor inicial (media ajustada‡) |
-1.67 |
0.02 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-1.68§ |
|
|
* LOCF: Método de reutilización del resultado de la última observación (antes del tratamiento de rescate, en su caso). † Pacientes aleatorizados y tratados que tuvieron una evaluación inicial de la eficacia y al menos una evaluación posterior. ‡ Media de mínimos cuadrados ajustada en función del valor inicial. § p < 0.0001 frente al placebo. ¶ Glucemia posprandial de 2 horas en respuesta a una prueba de tolerancia a 75 mg de glucosa oral. † Todos los pacientes aleatorizados que tomaron al menos una dosis del medicamento en investigación durante el periodo con un diseño doble ciego a corto plazo. ** p <0.05 frente al placebo. †† LOCF: Método de reutilización del resultado de la última observación (después del tratamiento de rescate). |
||
FIGURA 8. Variación media ajustada de la HbA1c (%) frente al valor inicial en función del tiempo en un estudio de 24 semanas controlado con placebo sobre la adición de FORXIGA® al tratamiento con una sulfonilurea (glimepirida).

FIGURA 9. Variación media ajustada de la HbA1c (%) frente al valor inicial en función del tiempo en un estudio de 24 semanas controlado con placebo sobre la adición de FORXIGA® al tratamiento con una tiazolidinediona (pioglitazona).

FIGURA 10. Variación media ajustada de la HbA1c (%) frente al valor inicial en función del tiempo en un estudio de 104 semanas controlado con placebo sobre la adición de FORXIGA® al tratamiento con insulina y hasta 2 antidiabéticos orales o sin ellos, excluyendo los datos obtenidos después del aumento de la dosis de insulina

Adición de FORXIGA® al tratamiento con sitagliptina sola o combinada con metformina: Un estudio controlado con placebo de 24 semanas y un periodo de extensión de 24 semanas incluyó un total de 452 pacientes con diabetes tipo 2 con un control glucémico inadecuado (HbA1c =7.0% y =10.0% en el momento de la aleatorización), no tratados anteriormente con antidiabéticos o que estaban recibiendo en el momento de su admisión un tratamiento con o sin metformina o con un inhibidor de la DPP4. Este estudio evaluó la adición de FORXIGA® al tratamiento con sitagliptina (un inhibidor de la DPP4), sola o combinada con metformina.
Los pacientes elegibles fueron estratificados en función de si ya estaban recibiendo o no metformina (=1500 mg al día) y, dentro de cada estrato, fueron aleatorizados a un tratamiento con 10 mg de FORXIGA® + 100 mg de sitagliptina una vez al día o bien a un tratamiento con un placebo + 100 mg de sitagliptina una vez al día. Las variables se analizaron comparando 10 mg de FORXIGA® frente al placebo considerando la totalidad del grupo estudiado (sitagliptina con o sin metformina) y considerando cada estrato (sitagliptina sola o sitagliptina con metformina). El 37% de los pacientes no habían recibido un tratamiento antidiabético anteriormente, el 32% estaban recibiendo metformina sola, el 13% un inhibidor de la DPP4 solo y el 18% un inhibidor de la DPP4 + metformina. No se permitió modificar la dosis de FORXIGA®, sitagliptina o metformina durante este estudio.
En asociación con la sitagliptina (con o sin metformina), el tratamiento con 10 mg de FORXIGA® produjo en la semana 24 mejoras significativas de la HbA1c, de la HbA1c en pacientes con valores iniciales =8%, y de la GPA y una reducción significativa del peso corporal frente al grupo que recibió un placebo + sitagliptina (con o sin metformina) (Tabla 7). Estas mejoras también ser observaron en el estrato de pacientes que recibieron 10 mg de FORXIGA® + sitagliptina sola (n=110) frente al grupo tratado con placebo + sitagliptina sola (n=111), y en el estrato que recibió 10 mg de FORXIGA® + sitagliptina y metformina (n=113) frente al grupo placebo + sitagliptina con metformina (n=113) (Tabla 8).
En la semana 48, la variación media ajustada con respecto al valor inicial de la HbA1c, de la HbA1c en pacientes con valores iniciales =8%, de la GPA, de la GPP y del peso corporal fueron -0.30%, -0.72%, -19.7 mg/dl, -43.0 mg/dl and -2.03 kg, respectivamente, en los pacientes tratados con 10 mg de FORXIGA® + sitagliptina con o sin metformina, y de 0.38%, 0.26%, 13.4 mg/dl,-12.1 mg/dl y 0.18 kg en aquellos que recibieron el placebo + sitagliptina con o sin metformina, según el análisis longitudinal de mediciones repetidas que excluyó los datos obtenidos después del rescate.238 En la semana 48, en el estrato sin metformina, la variación media ajustada de la HbA1c frente al valor inicial fue del 0.00% en los pacientes tratados con 10 mg de FORXIGA® + sitagliptina, frente al 0.85% con el placebo + sitagliptina. En el estrato con metformina, la variación media ajustada de la HbA1c frente al valor inicial fue de -0.44% en los tratados con 10 mg de FORXIGA® + sitagliptina frente al 0.15% con el placebo + sitagliptina, según el análisis longitudinal de mediciones repetidas que excluyó los datos posteriores al rescate.
En las semanas 24 y 48, la proporción de pacientes que recibieron un tratamiento de rescate o que suspendieron el tratamiento por un control glucémico deficiente (ajustado en función de la HbA1c inicial) fue mayor con la sitagliptina con o sin metformina (41.5% y 56.6%, respectivamente) que con FORXIGA® con o sin metformina (18.8% y 32.7%, respectivamente).
|
Tabla 7. Resultados de un estudio de 24 semanas controlado con placebo (método LOCF*) sobre la adición de FORXIGA® al tratamiento con sitagliptina, con o sin metformina (población total analizada y estratos con o sin metformina) |
||||||
|
Parámetro de eficacia |
FORXIGA® |
Placebo + sitagliptina con o sin metformina |
FORXIGA® |
Placebo + sitagliptina |
FORXIGA® + metformina |
Placebo |
|
N=223† |
N=224† |
N=110† |
N=111† |
N=113† |
N=113† |
|
|
HbA1c (%) |
||||||
|
Valor inicial (media) |
7.90 |
7.97 |
7.99 |
8.07 |
7.80 |
7.87 |
|
Variación frente al valor inicial (media ajustada‡) |
-0.45 |
0.04 |
-0.47 |
0.10 |
-0.43 |
-0.02 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-0.48§ |
-0.56§ (-0.79, -0.34) |
-0.40§ (-0.58, -0.23) |
|||
|
Variación de la HbA1c en pacientes con una HbA1c inicial =8% (media ajustada‡) |
-0.80§ (N= 94) |
0.03 (N= 99) |
-0.81§ |
0.06 |
-0.79§ |
0.0 |
|
GPA (mg/dl) |
||||||
|
Valor inicial (media) |
161.7 |
163.1 |
157.3 |
161.5 |
165.9 |
164.7 |
|
Variación frente al valor inicial (media ajustada‡) |
-24.1 |
3.8 |
-22.0 |
4.6 |
-26.2 |
3.0 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-27.9§ |
-26.6§ (-36.3, -16.85) |
-29.2§ (-38.0, -20.4) |
|||
|
Peso corporal (kg) |
||||||
|
Valor inicial (media) |
91.02 |
89.23 |
88.01 |
84.20 |
93.95 |
94.17 |
|
Variación frente al valor inicial (media ajustada‡) |
-2.14 |
-0.26 |
-1.91 |
-0.06 |
-2.35 |
-0.47 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-1.89§ |
-1.85§ (-2.47, -1.23) |
-1.87§ (-2.61, -1.13) |
|||
|
PAS en posición sentada en la semana 8 en pacientes con una PAS inicial =130 mmHg |
||||||
|
Valor inicial (media) |
140.5 (N=111) |
139.3 (N=101) |
138.5 |
137.9 |
141.9 |
140.3 |
|
Variación frente al valor inicial (media ajustada‡) |
-6.0 |
-5.1 |
-6.6 |
-4.2 |
-5.3 |
-5.5 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-0.86 (-3.8, 2.0) |
-2.4 (-6.4, 1.7) |
0.2 (-3.85, 4.32) |
|||
|
GPP después de 2 horas ¶ (mg/dl) |
||||||
|
Valor inicial (media) |
227.8 |
226.3 |
225.3 |
231.2 |
230.2 |
221.0 |
|
Variación frente al valor inicial (media ajustada‡) |
-47.7 |
-4.8 |
-46.3 |
-2.6 |
-48.9 |
-7.2 |
|
Diferencia con respecto al placebo (media ajustada‡) (IC del 95%) |
-42.9 |
-43.7 |
-41.6 |
|||
|
Pacientes con una disminución de la HbA1c =0.7% (ajustada,%) |
35.4% |
16.6% |
42.8 |
17.2 |
28.0 |
16.0 |
|
* LOCF: Método de reutilización del resultado de la última observación (antes del tratamiento de rescate, en su caso). † Pacientes aleatorizados y tratados que tuvieron una evaluación inicial de la eficacia y al menos una evaluación posterior. ‡ Media de mínimos cuadrados ajustada en función del valor inicial. § p < 0.0001 frente al placebo. ¶ Glucemia posprandial de 2 horas en respuesta a una prueba de tolerancia a 75 mg de glucosa oral. |
||||||
ADVERTENCIAS Y PRECAUCIONES DE USO
Generalidades: FORXIGA® no debe usarse en pacientes con diabetes mellitus tipo 1 ni para tratar la cetoacidosis diabética.
Uso en pacientes con insuficiencia renal: La eficacia de FORXIGA® depende de la función renal.
FORXIGA® no debe usarse en pacientes con insuficiencia renal moderada a grave (definida como una TFGe <45 ml/min/1.73 m2 persistente calculada con la fórmula MDRD o una DEPCr <60 ml/min persistente calculada con la fórmula de Cockcroft-Gault) o enfermedad renal en etapa terminal (ERET). En consecuencia, como en todos los pacientes diabéticos, se debe evaluar la función renal antes de iniciar el tratamiento con FORXIGA® y luego periódicamente durante el mismo (véanse los apartados Posología y formas de administración: Insuficiencia renal, Insuficiencia renal y Experiencia adquirida durante los estudios clínicos).
FORXIGA® no se ha investigado en pacientes con insuficiencia renal grave (TFGe <30 ml/min/1.73 m2 calculada con la fórmula MDRD o DEPCr =30 ml/min calculada con la fórmula de Cockcroft-Gault) o enfermedad renal en etapa terminal (ETET), por lo que no debe usarse en esta población.
Uso en pacientes con un riesgo de hipovolemia: El efecto diurético de FORXIGA® reduce el volumen intravascular. En los pacientes expuestos a un riesgo de hipovolemia debido a enfermedades concurrentes, puede ser adecuado usar una dosis inicial de FORXIGA® de 5 mg. En los pacientes que desarrollan hipovolemia, debe considerarse la interrupción temporal de FORXIGA® (véase el apartado Experiencia adquirida durante los estudios clínicos).
Uso con medicamentos con efectos hipoglucemiantes conocidos: La insulina y los secretagogos de insulina, como las sulfonilureas, causan hipoglucemia. En consecuencia, en caso de coadministración con FORXIGA®, puede ser necesario reducir la dosis de insulina o del secretagogo de insulina para limitar el riesgo de hipoglucemia (véase el apartado Experiencia adquirida durante los estudios clínicos).
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
Dosis recomendada: La dosis recomendada de FORXIGA® es de 10 mg una vez al día y puede tomarse a cualquier hora del día independientemente del horario de las comidas.
Monoterapia y adición al tratamiento con otros fármacos: La dosis recomendada de FORXIGA® es de 10 mg una vez al día administrada en monoterapia o adicionada al tratamiento con metformina, una sulfonilurea, un inhibidor de la DPP4 (con o sin metformina) o insulina (asociada o no con un tratamiento antidiabético oral que puede ser una terapia dual de metformina + insulina o una triple terapia de metformina + una sulfonilurea + insulina).
Tratamiento combinado inicial: En el marco de un tratamiento combinado inicial, la dosis inicial recomendada es de 10 mg de FORXIGA® + 500 mg de metformina una vez al día. En los pacientes cuyo control glucémico sigue siendo inadecuado con esta dosis, puede aumentarse la dosis de metformina conforme a la información de prescripción local aprobada.
Insuficiencia renal: No es necesario ajustar la dosis de FORXIGA® en función del grado de insuficiencia renal.
La eficacia de FORXIGA® depende de la función renal. FORXIGA® no debe usarse en pacientes con insuficiencia renal moderada a grave (definida como una TFGe <45 mL/min/1.73 m2 persistente calculada con la fórmula MDRD o una DEPCr < 60 mL/min persistente calculada con la fórmula de Cockcroft-Gault) o con enfermedad renal en etapa terminal (ERET) (véanse los apartados Uso en pacientes con insuficiencia renal, Insuficiencia renal y Experiencia adquirida durante los estudios clínicos).
Insuficiencia hepática: No es necesario ajustar la dosis de FORXIGA® en los pacientes con insuficiencia hepática leve, moderada o grave (véase el apartado Farmacocinética).
Niños y adolescentes: No se han establecido la seguridad y la eficacia de FORXIGA® en niños y adolescentes.
Pacientes de edad avanzada: No es necesario ajustar la dosis de FORXIGA® en función de la edad (véase el apartado Uso geriátrico).
Pacientes expuestos a un riesgo de hipovolemia: En los pacientes expuestos a un riesgo de hipovolemia debido a enfermedades concurrentes, puede ser adecuado usar una dosis inicial de FORXIGA® de 5 mg (véanse los apartados Uso en pacientes con riesgo de hipovolemia y Experiencia adquirida durante los estudios clínicos).
DATOS PRECLÍNICOS DE TOXICOLOGÍA
Potencial cancerígeno, potencial mutágeno y alteración de la fecundidad: En estudios de 2 años sobre el poder cancerígeno, la dapagliflozina no indujo tumores ni en ratones ni en ratas con ninguna de las dosis evaluadas. Las dosis orales administradas a los ratones fueron de 5, 15 y 40 mg/kg/día en los machos y de 2, 10 y 20 mg/kg/día en las hembras, mientras que las dosis orales administradas a las ratas fueron de 0.5, 2 y 10 mg/kg/día tanto en los machos como en las hembras. Las dosis máximas evaluadas en ratones fueron equivalentes a una exposición (ABC) de 72 veces (machos) y 105 veces (hembras) el ABC humana correspondiente a la DMRH de 10 mg al día. En ratas, las exposiciones fueron de aproximadamente 131 veces (machos) y 186 veces (hembras) el ABC humana correspondiente a la DMRH.
La dapagliflozina dio resultados negativos en el ensayo de mutagénesis de Ames, pero resultados positivos en un ensayo de clastogénesis in vitro, aunque solamente en presencia del sistema de activación metabólica S9 y con concentraciones = 100 µg/mL. Cabe notar que la dapagliflozina no mostró efectos clastógenos in vivo en una serie de ensayos que evaluaron la formación de micronúcleos o la reparación del ADN en ratas sujetas a una exposición > 2100 veces la exposición humana correspondiente a la DMRH. Tales ensayos, junto con la ausencia de hallazgos tumorales en los estudios sobre el poder cancerígeno en ratas y ratones, demuestran que la dapagliflozina no plantea un riesgo genotóxico para el ser humano.
Se evaluaron los cambios en la transcripción, relacionados con dapagliflozina, en riñones, hígado, tejido adiposo y músculo esquelético de ratas Zucker Diabetic Fatty (ZDF) tratadas con dapagliflozina por 5 semanas. Estos órganos fueron específicamente seleccionados puesto que representan órganos blanco en el tratamiento de la diabetes. No hubo evidencia de que dapagliflozina causara cambios transcripcionales que sean predictivos de promotores tumorales.
Dapagliflozina y su metabolito humano primario (3-O-glucurónido) no incrementaron el crecimiento in vitro de líneas celulares de seis carcinomas de células transicionales (TCC, por sus siglas en inglés) de vejiga urinaria humana a concentraciones =100× Cmáx humana de la DMRH. En un estudio de xenoinjerto, dapagliflozina administrada a ratones machos y hembras desnudos a los cuales se implantaron tumores TCC humanos, no aumentó significativamente el tamaño de los tumores a exposiciones de hasta 75× y hasta 0.9× las exposiciones clínicas de la DMRH de dapagliflozina y su metabolito 3-O-glucurónido, respectivamente. Estos estudios proveen evidencia de que dapagliflozina y su metabolito humano primario no acentúan el crecimiento de tumores de la vejiga urinaria.
En un estudio de fenotipificación de 15 meses, no hubo evidencia de ninguna diferencia en la supervivencia, peso corporal, parámetros de patología clínica, o hallazgos histopatológicos observados entre ratones SGLT2 KO y sus contrapartes de tipo silvestre (WT). Los ratones SGLT2 KO tenían glucosuria, a diferencia de los ratones WT. A pesar de glucosuria durante toda la vida, no hubo evidencia de ninguna alteración de la función renal o de cambios proliferativos observados en los riñones o vejiga urinaria de ratones SGLT2 KO. Estos datos sugieren fuertemente que los niveles elevados de glucosa urinaria no inducen tumores del tracto urinario ni aceleran la patología del tracto urinario relacionada con la edad.
En un estudio sobre la fecundidad y el desarrollo embrionario inicial en ratas, se administraron dosis de 15, 75 o 300/210 mg/kg/día de dapagliflozina a los machos (la dosis de 300 mg/kg/día se redujo a 210 mg/kg/día después de 4 días) y dosis de 3, 15 o 75 mg/kg/día a las hembras. La dapagliflozina no tuvo ningún efecto en el apareamiento, la fecundidad o el desarrollo embrionario inicial de los animales, tanto machos como hembras, tratados con cualquiera de las dosis examinadas (exposiciones = 1708 y 998 veces la exposición correspondiente a la DMRH en los machos y en las hembras, respectivamente). No obstante, con la dosis de 300/210 mg/kg/día se observaron disminuciones del peso de las vesículas seminales y del epidídimo, disminuciones de los recuentos y de la motilidad de los espermatozoides, además de pequeñas cantidades de espermatozoides morfológicamente anormales.
Teratogénesis y alteraciones del desarrollo inicial: La administración directa de la dapagliflozina a ratas jóvenes recién destetadas y la exposición indirecta durante la última fase del embarazo y la lactancia (periodo correspondiente al segundo y tercer trimestres de la gestación con respecto a la maduración renal humana) se asocian, cada una, con un aumento de la incidencia y/o intensidad de dilataciones pélvicas y tubulares en las crías.
En un estudio de toxicidad en ratas jóvenes en el que se administraron dosis de dapagliflozina de 1, 15 y 75 mg/kg/día directamente desde el día posnatal 21 (DPN 21) hasta el DPN 90, se observaron dilataciones pélvicas y tubulares renales con todas las dosis; con la dosis más baja examinada, las exposiciones de las crías fueron =15x DMRH. Estos hallazgos se acompañaron de aumentos del peso de los riñones y de hipertrofia renal macroscópica, en función de la dosis, con todos los niveles de dosis examinados. Las dilataciones pélvicas y tubulares renales observadas en animales jóvenes no fueron totalmente reversibles durante el periodo de recuperación de aproximadamente 1 mes.
En un estudio separado sobre el desarrollo prenatal y posnatal también se administraron dosis de 1, 15 o 75 mg/kg/día a ratas hembras desde el día 6 de la gestación (DG 6) hasta el DPN 21 y las crías fueron expuestas indirectamente in utero y a través de la lactancia. (Se llevó a cabo un estudio satélite para evaluar la excreción de la dapagliflozina en la leche y la exposición de las crías). Nuevamente se observó un aumento de la incidencia o intensidad de dilataciones pélvicas renales en la progenie adulta de las ratas tratadas, aunque solo con la dosis de 75 mg/kg/día (las exposiciones asociadas de las madres y de las crías fueron, respectivamente, de 1415 y 137 veces la exposición humana correspondiente a la DMRH). Los demás efectos tóxicos en el desarrollo se limitaron a reducciones del peso corporal de las crías en función de la dosis, pero solamente con dosis = 15 mg/kg/día (asociadas con exposiciones de las crías = 29 veces la exposición humana correspondiente a la DMRH). La toxicidad materna, que se observó únicamente con la dosis de 75 mg/kg/día, se limitó a reducciones pasajeras del peso corporal y del consumo de alimentos al principio del tratamiento. El grado de exposición (NOAEL) en el que no se observaron efectos tóxicos en el desarrollo (1 mg/kg/día) se asocia con una exposición sistémica de las madres de aproximadamente 19 veces la exposición humana correspondiente a la DMRH.
En otros estudios sobre el desarrollo embrionario y fetal en ratas y conejos, la dapagliflozina se administró durante los intervalos correspondientes a los periodos de organogénesis más importantes de cada especie. En conejos no se observaron efectos tóxicos ni en las madres ni en el desarrollo con ninguna de las dosis examinadas (20, 60 y 180 mg/kg/día); la dosis de 180 mg/kg/día se asoció con una exposición sistémica de aproximadamente 1191 veces la exposición humana correspondiente a la DMRH. En ratas, la dapagliflozina no tuvo efectos embrioletales ni teratógenos con dosis de hasta 75 mg/kg/día (1441 veces la DMRH). El tratamiento con dosis = 150 mg/kg/día (= 2344 veces la exposición humana correspondiente a la DMRH) produjo efectos tóxicos en la madre y en el desarrollo. La toxicidad materna consistió en mortalidad, signos clínicos adversos y disminuciones del peso corporal y del consumo de alimentos. Los efectos tóxicos en el desarrollo consistieron en un aumento de la mortalidad embrionaria y fetal, un aumento de la incidencia de malformaciones y alteraciones esqueléticas fetales, y reducciones del peso corporal de los fetos. Entre las malformaciones observadas, hubo una baja incidencia de malformaciones arteriales, costillas fusionadas y centros vertebrales malformados, duplicación del manubrio esternal y centros esternales malformados. Las alteraciones esqueléticas consistieron básicamente en reducciones de la osificación.
Toxicología en animales: Se consideró que la mayoría de los efectos observados en los estudios fundamentales de toxicidad tras dosis repetidas, tanto en ratas como en perros, fueron secundarios a los aumentos de la glucosa urinaria mediados por la actividad farmacológica de la dapagliflozina e incluyeron disminuciones o aumentos del peso corporal, aumentos del consumo de alimentos y aumentos del volumen de orina debidos a la diuresis osmótica. La dapagliflozina fue bien tolerada tras la administración oral a ratas durante un periodo de hasta 6 meses, usando dosis = 25 mg/kg/día (= 346 veces la exposición humana correspondiente a la DMRH), y a perros durante un periodo de hasta 12 meses con dosis = 120 mg/kg/día (= 3200 veces la exposición humana correspondiente a la DMRH). Además, los estudios realizados con dosis únicas de dapagliflozina indicaron que, en los estudios de toxicidad en ratas y perros, el metabolito 3-O-glucurónido de dapagliflozina se habría formado con grados de exposición (ABC) superiores o aproximadamente iguales a la exposición humana prevista a este metabolito tras la administración de la DMRH. En ratas, el hallazgo de toxicología preclínica más importante fue un aumento de la mineralización del tejido y hueso trabecular (acompañado de un aumento de la concentración sérica de calcio), que se observó únicamente con grados de exposición elevados (= 2100 veces la exposición humana correspondiente a la DMRH). A pesar de exposiciones = 3200 veces la exposición humana correspondiente a la DMRH, no se identificaron efectos tóxicos en los órganos afectados ni efectos que limitaran la dosis durante el estudio de 12 meses en perros.
SOBREDOSIS: Se ha demostrado que la dapagliflozina es inocua y bien tolerada por vía oral en voluntarios sanos, con dosis únicas de hasta 500 mg (50 veces la dosis máxima recomendada en el ser humano [DMRH]). Se detectó la presencia de glucosa en la orina de estos sujetos durante un periodo que dependió de la dosis administrada (al menos 5 días con la dosis de 500 mg), sin que se notificaran casos de deshidratación, hipotensión o desequilibrio electrolítico, y sin efectos de importancia clínica en el intervalo QTc. La incidencia de hipoglucemia en pacientes tratados con dapaglifozina fue similar a la registrada con un placebo. En los estudios clínicos en los que se administraron dosis de hasta 100 mg de dapaglifozina una vez al día (10 x DMRH) durante 2 semanas a voluntarios sanos y a pacientes con diabetes tipo 2, la incidencia de hipoglucemia en pacientes que recibieron dapagliflozina fue ligeramente mayor que con el placebo y no dependió de la dosis. Las incidencias de reacciones adversas de deshidratación o hipotensión en pacientes tratados con dapaglifozina fueron similares a las registradas con el placebo, y no hubo variaciones de importancia clínica en función de la dosis en los parámetros de laboratorio tales como las concentraciones séricas de electrolitos y biomarcadores de la función renal.
En caso de sobredosis, debe administrarse un tratamiento de apoyo adecuado que depende del estado clínico del paciente. No se ha investigado la eliminación de la dapagliflozina mediante hemodiálisis.
DESCRIPCIÓN: FORXIGA® (dapagliflozina propanodiol) es un inhibidor potente y extremadamente selectivo del cotransportador 2 de sodio-glucosa renal humano (SGLT2), que es el principal transportador responsable de la reabsorción renal de glucosa, de administración oral.
La descripción química de la dapagliflozina propanodiol es: D-glucitol, 1,5-anhidro-1-C-[4-cloro-3-[(4-etoxifenil)metil]fenilo]-, (1S)-, compuesto con (2S)-1,2-propanodiol, hidrato (1:1:1). Su fórmula empírica es C21H25ClO6 • C3H8O2 • H2O, su masa molecular de 502.98, y su fórmula estructural:
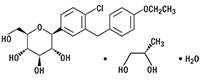
FORXIGA® está disponible en COMPRIMIDOS RECUBIERTOS para administración oral que contienen una cantidad de dapagliflozina propanodiol equivalente a 5 mg o 10 mg de dapagliflozina.
ESTUDIOS COMPLEMENTARIOS
Uso en pacientes con diabetes tipo 2 e hipertensión: En dos estudios de 12 semanas, placebo-controlados, un total de 1062 pacientes con diabetes tipo 2 e hipertensión inadecuadamente controladas fueron tratados con FORXIGA® 10 mg o placebo. Fueron elegibles para estos estudios los pacientes con hipertensión inadecuadamente controlada (presión arterial sistólica en posición sentada =140 y <165 mmHg, presión arterial diastólica en posición sentada =85 y <105 mmHg, y una presión arterial media de 24 horas =130/80 mmHg) a pesar de tratamiento estable preexistente con un inhibidor de la enzima convertidora de angiotensina (IECA) o bloqueador de receptor de angiotensina (BRA) (solo [Estudio 1] o en combinación con un antihipertensivo adicional [Estudio 2]), como también un control glucémico inadecuado (HbA1c =7.0% y =10.5%) a pesar de tratamiento estable preexisten con ADOs o insulina (solo o en combinación) previamente al ingresos. Durante los estudios, no se permitieron ajustes en los medicamentos antidiabéticos y antihipertensivos. A través de los 2 estudios, 527 pacientes fueron tratados con FORXIGA® 10 mg y 535 con placebo. Los pacientes tratados con FORXIGA® 10 mg o placebo también recibieron los siguientes medicamentos para controlar la presión arterial, los cuales fueron equilibrados entre los grupos de tratamiento: IECAs (64%), BRAs (36%), diuréticos tiazídicos (16%), calcioantagonistas (9%), y betabloqueadores (6%).
A la semana 12 en ambos estudios, FORXIGA® 10 mg más el tratamiento usual proporcionaron mejoría significativa en la HbA1c y reducción significativa en la presión arterial sistólica en posición sentada comparados con placebo más tratamiento usual (véase Tabla 8). Se observaron reducciones consistentes en la presión arterial sistólica media de 24 horas ambulatoria en pacientes tratados con FORXIGA® 10 mg comparado con placebo. Se produjo una pequeña reducción en la presión arterial diastólica media en posición sentada en pacientes tratados con FORXIGA® 10 mg, que no fue estadísticamente significativa en comparación con el placebo.
|
Tabla 8. Resultados a la semana 12 en 2 estudios placebo-Controlados de FORXIGA® en pacientes con diabetes tipo 2 e hipertensión |
||||
|
Estudio 1 |
Estudio 2 |
|||
|
Parámetro de eficacia |
FORXIGA® 10 mg + tratamiento usual N=302† |
Placebo + tratamiento usual N=311† |
FORXIGA® 10 mg + tratamiento usual N=225† |
Placebo |
|
HbA1c (%) (LRM)* |
||||
|
Nivel basal (media) |
8.1 |
8.0 |
8.1 |
8.0 |
|
Cambio desde el nivel basal (media ajustada‡) |
-0.6 |
-0.1 |
-0.6 |
0.0 |
|
Diferencia con el placebo (media ajustada‡) |
-0.5§ |
-0.6§ |
||
|
Presión arterial sistólica en posición sentada (mmHg) (LRM) * |
||||
|
Nivel basal (media) |
149.8 |
149.5 |
151.0 |
151.3 |
|
Cambio desde el nivel basal (media ajustada‡) |
-10.4 |
-7.3 |
-11.9 |
-7.6 |
|
Diferencia con el placebo (media ajustada‡) |
-3.1¶ |
-4.3¶ |
||
|
* LRM: Siglas en inglés para análisis de mediciones longitudinales repetidas. † Todos los pacientes aleatorizados que tomaron por lo menos una dosis de medicamento doble-ciego del estudio durante el periodo doble-ciego, a corto plazo. ‡ Media de mínimos cuadrados ajustada en función del valor inicial. § valor de p <0.0001. ¶ valor de p <0.05. # LOCF: Sigla en inglés para última observación llevada a cabo. |
||||
Uso en pacientes con diabetes tipo 2 y enfermedad cardiovascular: En el marco de dos estudios controlados con placebo de 24 semanas de duración y periodos de extensión de 80 semanas, un total de 1887 pacientes con diabetes tipo 2 y afecciones cardiovasculares (CV) recibieron un tratamiento con 10 mg de FORXIGA® o un placebo.
En estos estudios participaron pacientes con una afección CV confirmada y un control glucémico inadecuado (HbA1c =7.0% y =10.0%), pese a un tratamiento estable con antidiabéticos orales (ADO) o insulina (sola o combinada) antes de su admisión en el estudio. Los pacientes fueron estratificados en función de su edad (<65 o =65 años), del uso o no de insulina y del tiempo transcurrido desde el acontecimiento cardiovascular cualificador más reciente (>1 año o <1 año antes de su admisión en el estudio). Sumando los dos estudios, 942 pacientes recibieron FORXIGA® 10 mg y 945 el placebo. El 96% de aquellos tratados con FORXIGA® en los dos estudios eran hipertensos en el momento de su admisión, la mayoría desde hace más de 10 años. El acontecimiento cardiovascular cualificador más frecuente fue la cardiopatía coronaria (75%) o el accidente cerebrovascular (22%). Aproximadamente el 19% de los pacientes tomaban diuréticos de asa en el momento de la admisión y el 15% padecían insuficiencia cardiaca congestiva (el 2% de clase III según la NYHA). Aproximadamente el 37% de los pacientes tratados con FORXIGA® 10 mg también recibían una combinación de metformina + un antidiabético oral en el momento de su admisión (sulfonilurea, tiazolidinediona, inhibidor de la DPP4 u otro antidiabético asociado o no con insulina), el 39% recibían insulina + al menos un antibiabético oral y el 18% solamente insulina.
En estos dos estudios, la adición de 10 mg de FORXIGA® al tratamiento antidiabético existente produjo una mejora significativa durante el periodo de 24 semanas de las covariables principales de HbA1c y beneficio clínico compuesto comparado con placebo. Éste último se definió como la proporción de pacientes con una disminución absoluta de la HbA1c del 0.5% frente al valor inicial, una pérdida relativa del peso corporal total de al menos el 3% frente al peso inicial, y una caída absoluta de al menos 3 mmHg de la presión arterial sistólica en posición sentada frente al valor inicial (Tabla 9). También se observaron reducciones significativas del peso corporal total y de la presión arterial sistólica en posición sentada en pacientes tratados con FORXIGA® 10 mg en comparación con los pacientes tratados con placebo.
A la semana 52 y semana 104 del Estudio 1, el cambio en la media ajustada desde el nivel basal en la HbA1c, presión arterial sistólica en posición sentada, y cambio en el porcentaje ajustado desde el nivel basal en el peso corporal fueron de -0.44% and -0.41%, -3.40 mmHg y -2.64 mmHg, y -2.89% and -3.53%, respectivamente, para pacientes tratados con FORXIGA® 10 mg más tratamiento usual, con base en el análisis de mediciones longitudinales repetidas. Las cifras correspondientes de pacientes tratados con placebo más tratamiento usual fueron de 0.22% y 0.50%, 0.18 mmHg y 1.54 mmHg, y -0.29% y -0.02%. A la semana 52 y semana 104, el porcentaje de beneficio clínico combinado fue todavía más alto en el grupo tratado con FORXIGA® 10 mg (6.6% y 3.8%) que en el grupo placebo (0.7% y 0.5%).
A la semana 24, semana 52, y semana 104 del Estudio 1, la proporción de pacientes que fueron rescatados por ausencia de control glucémico (ajustado en función de la HbA1c inicial) fue más alta en el grupo placebo más tratamiento usual (24.0%, 51.8%, y 57.3%, respectivamente) que en el grupo tratado con FORXIGA® 10 mg más tratamiento usual (7.8%, 24.6%, y 31.8%, respectivamente).
A la semana 52 y semana 104 del Estudio 2, el cambio en la media ajustada desde el nivel basal en la HbA1c, presión arterial sistólica en posición sentada, y cambio en el porcentaje ajustado desde el nivel basal en el peso corporal fueron de -0.47% y -0.37%, -3.56 mmHg y -1.96 mmHg, y -3.20% y -3.51%, respectivamente, para pacientes tratados con FORXIGA® 10 mg más tratamiento usual, con base en el análisis de mediciones longitudinales repetidas. Las cifras correspondientes de pacientes tratados con placebo más tratamiento usual fueron de 0.03% y -0.18%, -0.91 mmHg y -0.37 mmHg, y -1.12% y -0.65%. A la semana 52 y semana 104, el porcentaje de beneficio clínico combinado fue todavía más alto en el grupo tratado con FORXIGA® 10 mg (10.6% y 4.2%) que en el grupo placebo (3.1% y 1.1%).
A la semana 24, semana 52, y semana 104 del Estudio 2, la proporción de pacientes que fueron rescatados por ausencia de control glucémico (ajustado en función de la HbA1c inicial) fue más alta en el grupo placebo más tratamiento usual (22.3%, 43.6%, y 50.5%, respectivamente) que en el grupo tratado con FORXIGA® 10 mg más tratamiento usual (7.6%, 18.7%, y 27.5%, respectivamente).
|
Tabla 9. Resultados obtenidos en la semana 24 (método LOCF*) en dos estudios controlados con placebo que compararon el tratamiento con FORXIGA® y un placebo en pacientes con diabetes tipo 2 y con afecciones cardiovasculares |
||||
|
Estudio 1 |
Estudio 2 |
|||
|
Parámetro de eficacia |
FORXIGA® 10 mg + tratamiento habitual |
Placebo + tratamiento habitual |
FORXIGA® 10 mg + tratamiento habitual |
Placebo + tratamiento habitual |
|
HbA1c (%) |
||||
|
Valor inicial (media) |
8.18 |
8.08 |
8.04 |
8.07 |
|
Variación frente al valor inicial (media ajustada‡) |
-0.38 |
0.08 |
-0.33 |
0.07 |
|
Diferencia con respecto al placebo (media ajustada ‡) (IC del 95%) |
-0.46§ (-0.56, -0.37) |
-0.40§ (-0.50, -0.30) |
||
|
Pacientes que obtuvieron un beneficio clínico compuesto (%) |
11.7 |
0.9 |
10.0 |
1.9 |
|
Diferencia con respecto al placebo (porcentaje ajustado) |
9.9§ |
7.0§ |
||
|
Componentes de la variable compuesta (%)250,251 |
||||
|
Pacientes con una disminución absoluta de la HbA1c =0.5% (porcentaje ajustado) |
46.2 |
19.7 |
42.2 |
21.2 |
|
Pacientes con una pérdida de peso corporal de al menos 3% frente al peso inicial (porcentaje ajustado) |
40.0 |
13.9 |
41.3 |
15.4 |
|
Pacientes con una disminución absoluta de la PAS =3 mmHg (porcentaje ajustado) |
49.1 |
41.6 |
46.2 |
40.9 |
|
Peso corporal (kg) |
||||
|
Valor inicial (media) |
92.63 |
93.59 |
94.53 |
93.22 |
|
Variación frente al valor inicial (porcentaje ajustado‡) |
-2.56 |
-0.30 |
-2.53 |
-0.61 |
|
Diferencia con respecto al placebo (porcentaje ajustado‡) (IC del 95%) |
-2.27§ (-2.64, -1.89) |
-1.93§ (-2.31, -1.54) |
||
|
Pérdida de peso de al menos el 5% en pacientes con un IMC inicial =27 kg/m2 (%) |
16.5§ |
4.0 |
18.4§ |
4.8 |
|
Presión arterial sistólica en posición sentada (mmHg) |
||||
|
Variación entre el inicio y la semana 24 (media ajustada‡) |
-2.99 |
-1.03 |
-2.70 |
0.32 |
|
Diferencia con respecto al placebo (media ajustada ‡) (IC del 95%) |
-1.95¶ (-3.56, -0.34) |
-3.02¶ (-4.59, -1.46) |
||
|
Variación de la PAS en posición sentada durante 8 semanas en pacientes con una PAS inicial =130 mmHg (media ajustada‡) |
- |
- |
-5.33¶ |
-1.89 |
|
* LOCF: Método de reutilización del resultado de la última observación. |
||||
Después de 24 semanas, en los grupos predefinidos en función de la edad (<65 y =65 años), los pacientes tratados con 10 mg de FORXIGA® también mostraron mejoras significativas de las covariables principales (HbA1c y beneficio clínico compuesto) frente al placebo en los dos estudios. En estos dos grupos de edad, en los pacientes <65 años tratados con 10 mg de FORXIGA® se registró asimismo frente al placebo una reducción significativa del peso corporal total y una disminución significativa de la presión arterial sistólica en posición sentada después de 24 semanas. Estos efectos se mantuvieron a la semana 52 y semana 104.
Absorciometría de rayos X de doble energía en pacientes diabéticos tipo 2: Debido al modo de acción de FORXIGA®, se llevó a cabo un estudio para evaluar la composición del organismo y la densidad mineral ósea en 182 pacientes con diabetes tipo 2. La adición de 10 mg de FORXIGA® al tratamiento con metformina durante un periodo de 24 semanas produjo mejorías significativas frente al placebo + metformina, respectivamente, del peso corporal (variación media con respecto al valor inicial: 2.96 kg frente a -0.88 kg), de la circunferencia de la cintura (variación media con respecto al valor inicial: -2.51 cm frente a -0.99 cm) y de la masa adiposa del organismo medida por DXA (variación media con respecto al valor inicial: -2.22 kg frente a -0.74 kg) en lugar de una pérdida de tejido magro o de líquidos. En un subestudio que incluyó análisis por RMN, el tratamiento con la asociación de FORXIGA® + metformina produjo una disminución numérica de la masa adiposa visceral frente al tratamiento con placebo + metformina (variación con respecto al valor inicial: -322.6 cm3 frente a -8.7 cm3). La semana 24 se analizó usando análisis de la última observación llevada a cabo (LOCF), incluyendo datos posteriores al rescate.
A la semana 24, 2 pacientes (2.2%) en el grupo placebo más metformina y ningún paciente en el grupo tratado con FORXIGA® 10 mg más metformina fueron rescatados por ausencia de control glucémico.
A la semana 50 y semana 102, las mejorías se mantuvieron en el grupo tratado con FORXIGA® 10 mg adicionado a metformina comparado con el grupo que recibió placebo más metformina en cuanto al peso corporal (cambio en la media ajustada desde el nivel basal hasta la semana 50: -4.39 kg vs. -2.03 kg; cambio en la media ajustada desde el nivel basal hasta la semana 102: -4.54 kg vs. -2.12 kg), circunferencia de la cintura (cambio en la media ajustada desde el nivel basal hasta la semana 50: -5.0 cm vs. -3.0 cm; cambio en la media ajustada desde el nivel basal hasta la semana 102: -5.0 cm vs. -2.9 cm ), y masa grasa corporal según lo medido por DXA a la semana 102 (cambio promedio desde el nivel basal: -2.80 kg vs. -1.46 kg), con base en el análisis de mediciones longitudinales repetidas incluyendo datos posteriores al rescate. En un subestudio de MRI a las semanas 50 y 102, el tratamiento con FORXIGA® más metformina mostró una reducción numérica en el tejido adiposo visceral comparado con el tratamiento con placebo más metformina (cambio en la media ajustada desde el nivel basal hasta la semana 50: -120.0 cm3 vs. 61.5 cm3; cambio en la media ajustada desde el nivel basal hasta la semana 102: -214.9 cm3 vs. -22.3 cm3).
La proporción de pacientes a la semana 50 (sin ajuste en función de la HbA1c inicial) y a la semana 102 (ajustado en función de la HbA1c inicial) que fueron rescatados o que descontinuaron por ausencia de control glucémico fue más alta en el grupo placebo más metformina (6.6% y 33.2%, respectivamente) que en el grupo tratado con FORXIGA® 10 mg más metformina (2.2% y 13.5%, respectivamente).
En una extensión de este estudio hasta la semana 50, ninguno de los grupos presentó variaciones de la densidad minera ósea de la columna lumbar, el cuello femoral o la cadera total (el decrecimiento medio frente al valor inicial fue <0.5% en todas las zonas anatómicas). Tampoco hubo cambio en el IMC en ningún grupo de tratamiento hasta la semana 102 (reducción media desde el nivel basal para todas las regiones anatómicas <1.0%). No hubo cambios clínicamente representativos en los marcadores de resorción ósea o formación ósea.
Uso en pacientes con diabetes tipo 2 e insuficiencia renal moderada: Se llevó a cabo un estudio en pacientes con diabetes tipo 2 e insuficiencia renal moderada para evaluar distintos parámetros relacionados con el control glucémico y la seguridad en esta población. El tratamiento con FORXIGA® no produjo mejorías de importancia clínica o estadísticamente significativas de la HbA1c frente al placebo en la población total del estudio después de 24 semanas. Se observaron resultados similares después de la semana 104 (véase el apartado Uso en pacientes con diabetes y afecciones cardiovasculares).
Presión arterial: Después de 24 semanas, tras 11 estudios clínicos, el tratamiento con FORXIGA® 10 mg redujo la presión arterial sistólica un promedio de 1.3 a 5.3 mmHg (tras la corrección del efecto placebo) entre el inicio del estudio y todos los estudios sobre la monoterapia y en los estudios controlados con placebo sobre la adición de FORXIGA® a otros tratamientos.
En los dos estudios específicos en pacientes con diabetes tipo 2 y afecciones cardiovasculares, el tratamiento con 10 mg de FORXIGA® redujo la presión arterial sistólica un promedio de 2.0 a 3.0 mmHg (tras la corrección del efecto placebo) entre el inicio del estudio y la semana 24 (véase la Tabla 9) y se mantuvo hasta la semana 104.
Para los dos estudios asignados en pacientes con diabetes tipo 2 e hipertensión, el tratamiento con FORXIGA® 10 mg redujo significativamente la presión arterial sistólica placebo-corregida, en promedio 3.0 a 4.0 mmHg desde el nivel basal hasta la semana 12 (véase Tabla 8).
PRESENTACIONES COMERCIALES: FORXIGA® 10 mg: Caja por 14 y 28 comprimidos en blíster. (Reg. San. INVIMA 2015M-0015725).
Mantener este y todos los medicamentos en su envase original y fuera del alcance de los niños. Venta con fórmula médica.
IPP Clave 2-2014(4).
Texto basado en: CDS 10 de Junio de 2013.
Fuente: CV.000-804-209.2.0. Traducción de CV.000-804-208.2.0
Fecha de preparación de la versión: Julio de 2014.
Fecha de revisión del texto: Julio de 2014.
Mayor Información Departamento Médico de
AstraZeneca Colombia S.A.S.
Bogotá, D.C., Colombia


