
FASLODEX 250 MG
FULVESTRANT
Solución inyectable
2 Jeringa(s) prellenada(s) , Solución inyectable , 5 Mililitros
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Una JERINGA PRELLENADA contiene 250 mg de fulvestrant en 5 ml de solución.
Los excipientes se detallan en la sección “Lista de excipientes”.
INDICACIONES TERAPÉUTICAS: FASLODEX® está indicado para el tratamiento de mujeres posmenopáusicas con cáncer de mama avanzado local o metastásico, con receptores de estrógeno positivos, que presentan una recidiva durante o después del tratamiento antiestrogénico adyuvante o bien una progresión de la enfermedad durante el tratamiento con un antiestrógeno.
FORMA FARMACÉUTICA: Solución inyectable.
Solución viscosa límpida, de incolora a amarilla.
PROPIEDADES FARMACOCINÉTICAS
Absorción: Después de la administración de una inyección intramuscular de acción prolongada de FASLODEX®, el fulvestrant se absorbe lentamente y alcanza sus concentraciones plasmáticas máximas (Cmáx) después de aproximadamente 5 días. El tratamiento con 500 mg de FASLODEX® permite alcanzar grados de exposición equivalentes o cercanos al estado de equilibrio desde el primer mes (media [CV]: ABC 475 [33.4%] ng.días/ml, Cmáx = 25.1 [35.3%] ng/ml, Cmín = 16.3 [25.9%] ng/ml, respectivamente). En el estado de equilibrio, las concentraciones plasmáticas de fulvestrant se mantienen en un intervalo relativamente estrecho, con una diferencia de hasta aproximadamente 3 veces entre las concentraciones máximas y mínimas. Después de la administración intramuscular, la exposición es aproximadamente proporcional a la dosis entre 50 y 500 mg.
Distribución: El fulvestrant es objeto de una distribución extensa y rápida. Su amplio volumen de distribución aparente en el estado de equilibrio (Vdss), de aproximadamente 3 a 5 litros/kg, indica que la distribución es en gran medida extravascular. Es considerable la unión del fulvestrant a las proteínas plasmáticas (99%). Las principales fracciones a las que se une el fulvestrant son las lipoproteínas de muy baja densidad (VLDL), las lipoproteínas de baja densidad (LDL) y las lipoproteínas de alta densidad (HDL). No se efectuaron estudios sobre las posibles interacciones del medicamento causadas por la unión competitiva a las proteínas. No se ha determinado el papel de la globulina transportadora de hormonas sexuales (SHBG).
Metabolismo: No se ha evaluado por completo el metabolismo del fulvestrant pero se sabe que implica combinaciones de varias vías de biotransformación posibles, similares a las de los esteroides endógenos. Los metabolitos identificados (que incluyen los metabolitos 17-cetona, sulfona, 3-sulfato, 3- y 17-glucurónido) muestran una actividad igual o menor que la del fulvestrant en modelos de antiestrógenos. Los estudios con preparaciones de hígado humano y enzimas humanas recombinantes indican que la CYP 3A4 es la única isoenzima del citocromo P450 que interviene en la oxidación del fulvestrant, mientras que, in vivo, parecen predominar otras vías sin relación con el P450. Los datos in vitro sugieren que el fulvestrant no inhibe las isoenzimas del citocromo P450.
Eliminación: El fulvestrant se elimina principalmente en forma metabolizada. La principal vía de excreción es la vía fecal y menos del 1% del producto se excreta en la orina. La depuración del fulvestrant es considerable (11±1.7 ml/min/kg), lo cual sugiere una elevada relación de extracción hepática. La vida media terminal (t½) después de la administración intramuscular depende de la velocidad de absorción y se estima que es de 50 días.
Grupos de pacientes especiales: En un análisis farmacocinético poblacional de los resultados de los estudios de Fase III no se detectó ninguna diferencia en el perfil farmacocinético del fulvestrant en función de la edad (de 33 a 89 años), del peso (de 40 a 127 kg) o de la raza.
Insuficiencia renal: La insuficiencia renal leve a moderada no tiene un efecto de importancia clínica en la farmacocinética del fulvestrant.
Insuficiencia hepática: La farmacocinética del fulvestrant se evaluó en un estudio clínico de dosis únicas en sujetos con insuficiencia hepática leve a moderada (clases A y B de la escala Child-Pugh). Se empleó una dosis elevada de una formulación de acción más corta administrada por inyección intramuscular. Con respecto a los voluntarios sanos, el área bajo la curva de concentraciones plasmáticas en función del tiempo (ABC) de los sujetos con insuficiencia hepática aumentó aproximadamente 2.5 veces. En los pacientes que reciben FASLODEX®, se prevé que un aumento de la exposición de esta magnitud será bien tolerado. No se evaluaron sujetos con insuficiencia hepática grave (clase C de la escala Child-Pugh).
PROPIEDADES FARMACODINÁMICAS:
Grupo farmacoterapéutico: Tratamiento endocrino, antiestrógeno. Código ATC: L02BA03.
CONTRAINDICACIONES: Hipersensibilidad al principio activo o a uno de los excipientes. Embarazo y lactancia (véase la sección “Embarazo y lactancia”). Insuficiencia hepática grave (véanse las secciones “Advertencias y precauciones especiales de empleo” y “Propiedades farmacocinéticas”).
FECUNDIDAD, EMBARAZO Y LACTANCIA
Mujeres en edad de procrear: Se debe aconsejar a las pacientes en edad de procrear que utilicen un método anticonceptivo eficaz durante el tratamiento.
Embarazo: FASLODEX® está contraindicado durante el embarazo (véase la sección “Contraindicaciones”). En ratas y conejos se demostró que el fulvestrant atraviesa la barrera placentaria después de la administración de dosis únicas por vía intramuscular. Los estudios en animales mostraron efectos tóxicos en la reproducción, por ejemplo un aumento de la frecuencia de anomalías y muertes fetales (véase la sección “Datos preclínicos sobre la seguridad”). Si una paciente se embaraza durante el tratamiento con FASLODEX®, debe recibir información sobre los posibles riesgos de aborto y para el feto.
Lactancia: La lactancia debe interrumpirse durante el tratamiento con FASLODEX®. El fulvestrant se secreta en la leche de ratas. No se sabe si el fulvestrant se secreta en la leche humana. Tomándo en cuenta el riesgo de reacciones adversas graves causadas por el fulvestrant en los lactantes, está contraindicado su uso durante la lactancia (véase la sección “Contraindicaciones”).
Fecundidad: No se han estudiado los efectos de FASLODEX® en la fecundidad humana.
EFECTOS EN LA CAPACIDAD PARA CONDUCIR O UTILIZAR MÁQUINAS: FASLODEX® no tiene influencia, o solo una insignificante, en la capacidad para conducir o utilizar máquinas. Sin embargo, como se han observado casos muy frecuentes de astenia con FASLODEX®, deben tener precaución las pacientes que presenten esta reacción adversa al conducir o utilizar máquinas.
REACCIONES ADVERSAS: La información presentada en esta sección se basa en todas las reacciones adversas registradas durante los estudios clínicos y los estudios de farmacovigilancia, o bien, notificadas espontáneamente. Las reacciones adversas más frecuentes consisten en reacciones en el lugar de la inyección, astenia, náuseas y elevaciones de las enzimas hepáticas (ALT, AST, fosfatasa alcalina).
Para asignar las siguientes categorías de frecuencia a las reacciones adversas, el cálculo se basó en el grupo de tratamiento con 500 mg de FASLODEX® de los análisis combinados de la seguridad de los estudios CONFIRM (estudio D6997C00002), FINDER 1 (estudio D6997C00004), FINDER 2 (estudio D6997C00006) y NEWEST (estudio D6997C00003), que compararon dosis de FASLODEX® de 500 mg y 250 mg. Las frecuencias que figuran en la siguiente tabla se basaron en todas las reacciones adversas notificadas, independientemente de su relación causal con el medicamento según la evaluación del investigador.
Las reacciones adversas se presentan a continuación por frecuencia y por sistema/órgano. Las categorías de frecuencia se definen de la siguiente manera: Muy frecuentes (=1/10), frecuentes (=1/100 y <1/10) y poco frecuentes (=1/1000 y <1/100). En cada categoría, las reacciones adversas se presentan en orden de gravedad decreciente.
|
Tabla 1. Reacciones adversas |
|||
|
Órgano o sistema |
Muy frecuentes (=10% ) |
Frecuentes ( =1%-<10%) |
Poco frecuentes ( =0.1%-<1%) |
|
Trastornos del sistema nervioso |
Cefalea |
||
|
Trastornos gastrointestinales |
Náuseas |
Vómito, diarrea |
|
|
Infecciones e infestaciones |
Infecciones urinarias |
||
|
Trastornos de la piel y del tejido subcutáneo |
Exantema |
||
|
Trastornos osteomusculares y del tejido conjuntivo |
Lumbalgiaa |
||
|
Trastornos del metabolismo y de la nutrición |
Anorexiaa |
||
|
Trastornos vasculares |
Tromboembolia venosaa, bochornos |
||
|
Trastornos generales y reacciones en el sitio de administración |
Asteniaa, reacciones en el lugar de la inyecciónb |
Hemorragia y hematoma en el lugar de la inyección |
|
|
Trastornos del sistema inmunitario |
Reacciones de hipersensibilidad |
||
|
Trastornos hepatobiliares |
Elevaciones de las enzimas hepáticas (ALT, AST, fosfatasa alcalina)a |
Elevaciones de la bilirrubinaa |
Insuficiencia hepáticac, hepatitisc, elevaciones de gamma-GT |
|
Trastornos del sistema reproductor y de la mama |
Candidiasis vaginal, leucorrea, hemorragia vaginal |
||
|
Sistema sanguíneo y linfático |
Recuento reducido de plaquetas |
||
|
a Incluye las reacciones adversas en las que la enfermedad subyacente impide evaluar la contribución exacta de FASLODEX®. b El término “reacciones en el lugar de la inyección” no incluye los términos “hemorragia y hematoma en el lugar de la inyección”. c Esta reacción no se observó en los estudios clínicos más importantes (CONFIRM, FINDER1, FINDER2, NEWEST). El cálculo de la frecuencia se basó en el límite superior del intervalo de confianza del 95% de la estimación puntual. El resultado es 3/560 (donde 560 es el número de pacientes incluidas en los estudios clínicos más importantes), lo que corresponde a la categoría “poco frecuentes”. |
|||
INTERACCIONES CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: Un estudio clínico sobre la interacción con el midazolam (sustrato de la enzima CYP 3A4) demostró que el fulvestrant no inhibe esta enzima. Los estudios clínicos sobre las interacciones con la rifampicina (inductor de la CYP 3A4) y el ketoconazol (inhibidor de la CYP 3A4) no revelaron cambios de importancia clínica en la depuración del fulvestrant. Por lo tanto, no es necesario ajustar la dosis en pacientes que reciben el fulvestrant en forma concomitante con inhibidores o inductores de la CYP 3A4.
DATOS PRECLÍNICOS SOBRE LA SEGURIDAD: Es baja la toxicidad aguda del fulvestrant.
FASLODEX® y otras formulaciones del fulvestrant fueron bien tolerados en las especies animales utilizadas en los estudios sobre la administración de dosis múltiples. Aunque las reacciones locales como miositis y granulomas en el sitio de inyección se atribuyeron al vehículo, la gravedad de la miositis en conejos aumentó con el fulvestrant con respecto a la administración de una solución salina de control. En los estudios de toxicidad con dosis intramusculares múltiples de fulvestrant en ratas y perros, la actividad antiestrogénica del fulvestrant fue responsable de la mayoría de los efectos observados, en particular en el aparato reproductor de las hembras, pero también en otros órganos sensibles a las hormonas de ambos sexos. En algunos perros que recibieron dosis crónicas (durante 12 meses) se observó arteritis que implicó varios tejidos.
En los estudios en perros después de la administración oral e intravenosa, se observaron efectos en el sistema cardiovascular (ligeras elevaciones del segmento S-T del ECG [vía oral] y paro sinusal en un perro [vía intravenosa]). Ya que estos efectos se presentaron con niveles de exposición mayores que los registrados en pacientes (Cmáx >15 veces), es probable que tengan una importancia limitada para la seguridad de las mujeres tratadas con dosis clínicas.
El fulvestrant no mostró ningún potencial genotóxico.
Con dosis similares a la dosis clínica, los efectos del fulvestrant en la reproducción y el desarrollo embriofetal fueron compatibles con su actividad antiestrogénica. En ratas se observaron una disminución reversible de la fecundidad de las hembras y de la supervivencia de los embriones, así como distocia y un aumento de la incidencia de anomalías fetales tales como flexión tarsal permanente. Conejas tratadas con el fulvestrant abortaron. Se observaron aumentos del peso de la placenta y de la pérdida de fetos después de la implantación y se registró una mayor incidencia de defectos fetales en conejos (desplazamiento hacia atrás de la cintura pélvica y 27 vértebras presacras).
Un estudio de oncogenicidad de dos años en ratas (con la administración intramuscular de FASLODEX®) mostró un aumento de la incidencia de tumores benignos de las células granulosas ováricas en las ratas hembras tratadas con la dosis alta (10 mg/rata/15 días) y una mayor incidencia de tumores de las células de Leydig testiculares en los machos. En un estudio de oncogenicidad de dos años en ratones (administración oral diaria), se observó un aumento de la incidencia de tumores del estroma de los cordones sexuales ováricos (tanto benignos como malignos), con dosis de 150 y 500 mg/kg/día. En el nivel de dosis que no produjo efectos relacionados con estos hallazgos, los grados de exposición sistémica (ABC) fueron de aproximadamente 1.5 veces la exposición prevista en el ser humano en las ratas hembras y de 0.8 veces en los machos, y de aproximadamente 0.8 veces la exposición prevista en el ser humano tanto en los ratones machos como hembras. La inducción de este tipo de tumores es compatible con las alteraciones de las concentraciones de gonadotropinas relacionadas con los efectos endocrinos del fármaco, causados por sus propiedades antiestrogénicas en animales con ciclo estral. En consecuencia, se considera que estos hallazgos carecen de importancia para la utilización del fulvestrant en mujeres posmenopáusicas con cáncer de mama avanzado.
Lista de excipientes
• Etanol al 96%
• Alcohol bencílico
• Benzoato de bencilo
• Aceite de ricino
Incompatibilidades: Dado que no se han realizado estudios de compatibilidad, este medicamento no debe mezclarse con otros productos farmacéuticos.
Plazo de caducidad: Véase la fecha de caducidad en la etiqueta o en la caja de cartón externa.
Precauciones especiales de conservación: Conservar entre 2 °C y 8 °C (en un refrigerador).
Conservar la jeringa prellenada en el envase original para proteger el producto de la luz.
Tamaño del envase: Véase en la caja de cartón externa.
Instrucciones de administración y precauciones especiales de desecho y manipulación
Advertencia: La aguja de seguridad (aguja hipodérmica de seguridad SafetyGlide de BD con funda protectora) no debe esterilizarse en la autoclave antes de usarla. Mantener en todo momento las manos detrás de la aguja durante su uso y desecho.
Con cada una de las dos jeringas:
1. Extraer de la bandeja el cilindro de vidrio de la jeringa y comprobar que no esté dañado.
2. Romper el sello de la cubierta de plástico blanca del conector Luer-Lok de la jeringa para retirar la cubierta junto con la tapa de caucho.
FIGURA 1
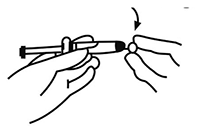
3. Abrir el envase externo de la aguja de seguridad (SafetyGlide de BD) desprendiendo la membrana de protección. Fijar la aguja de seguridad al conector Luer-Lok.
4. Enroscar la aguja hasta que quede firme.
5. Enroscar la aguja para fijarla al conector Luer.
6. Retirar la funda protectora de la aguja tirando con un movimiento recto para no dañar la punta.
FIGURA 2
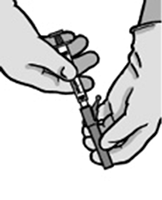
7. Llevar la jeringa llena al lugar de administración.
8. Retirar el capuchón de la aguja.
9. Antes de administrar una solución parenteral, ésta siempre debe examinarse a simple vista para comprobar la ausencia de partículas sólidas y decoloración.
10. Eliminar el exceso de aire de la jeringa.
11. Administrar lentamente (1-2 minutos por inyección) por vía intramuscular en el glúteo. Para mayor comodidad del usuario, el lado de la aguja con el bisel hacia arriba debe dirigirse hacia la palanca.
FIGURA 3
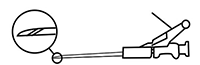
12. Después de la inyección, dar inmediatamente un impulso a la palanca con un solo dedo para accionar el mecanismo de protección. NOTA: Al accionar el mecanismo, mantener siempre la aguja alejada de sí mismo y de los demás. Tras escuchar un chasquido, comprobar a simple vista que la punta de la aguja esté totalmente cubierta.
FIGURA 4
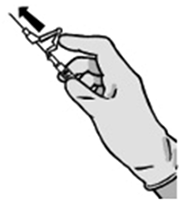
Desecho: Las jeringas prellenadas son para un solo uso.
Los residuos del producto farmacéutico o de los materiales deben desecharse de acuerdo con la reglamentación local.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO: FASLODEX® debe usarse con precaución en pacientes con insuficiencia hepática leve a moderada (véanse las secciones “Posología y forma de administración”, “Contraindicaciones” y “Propiedades farmacocinéticas”).
FASLODEX® debe usarse con precaución en pacientes con insuficiencia renal grave (depuración de creatinina <30 ml/min) (véase la sección “Propiedades farmacocinéticas”).
Debido a su vía de administración intramuscular, FASLODEX® debe usarse con precaución en pacientes con diátesis hemorrágicas o trombocitopenia y en las que reciben un tratamiento anticoagulante.
En las mujeres con cáncer de mama avanzado son frecuentes los accidentes tromboembólicos y estos se han registrado en los estudios clínicos de FASLODEX® (véase la sección “Reacciones adversas”). Esto debe tomarse en cuenta al prescribir FASLODEX® a pacientes de alto riesgo.
Se carece de información sobre los efectos a largo plazo del fulvestrant en los huesos. En vista del mecanismo de acción del fulvestrant, existe un posible riesgo de osteoporosis.
MECANISMO DE ACCIÓN Y EFECTOS FARMACODINÁMICOS: El fulvestrant es un antagonista competitivo de los receptores de estrógeno (RE) con una afinidad comparable a la del estradiol. El fulvestrant bloquea las acciones tróficas de los estrógenos sin ejercer una acción agonista parcial (estrogénica). Su mecanismo de acción es la regulación a la baja de la concentración de proteína receptora de estrógeno (RE). Los estudios clínicos en mujeres posmenopáusicas con cáncer de mama primario demostraron que, comparado con un placebo, el fulvestrant produce una regulación a la baja significativa de la proteína RE en tumores con RE positivos. También se observó una disminución significativa de la expresión de receptores de progesterona, lo que confirma que el fulvestrant carece de efectos agonistas estrogénicos intrínsecos. También se ha demostrado que, en el contexto del tratamiento neoadyuvante del cáncer de mama posmenopáusico, la regulación a la baja de los receptores de estrógeno y del marcador de la proliferación celular Ki67 es mayor con la dosis de 500 mg de fulvestrant que con la de 250 mg.
Seguridad y eficacia clínicas en el cáncer de mama avanzado: Se realizó un estudio clínico de Fase III en 736 mujeres posmenopáusicas con cáncer de mama avanzado que presentaron una recidiva durante o después del tratamiento endocrino adyuvante, o una progresión de la enfermedad tras el tratamiento endocrino de un cáncer avanzado. El estudio incluyó 423 pacientes con una recidiva o progresión de la enfermedad durante el tratamiento antiestrogénico (subgrupo AE) y 313 pacientes con una recidiva o progresión de la enfermedad durante el tratamiento con un inhibidor de la aromatasa (subgrupo IA). Este estudio comparó la eficacia y la seguridad de las dosis de FASLODEX® de 500 mg (n=362) y 250 mg (n=374). La variable principal fue la supervivencia sin progresión del cáncer (SSP), mientras que las variables secundarias más importantes fueron la tasa de respuesta objetiva (TRO), la tasa de beneficio clínico (TBC) y la supervivencia total (ST). La Tabla 2 resume los resultados de eficacia del estudio CONFIRM.
|
Tabla 2. Resumen de los resultados de la variable principal de eficacia (SSP) y de las variables secundarias más importantes del estudio CONFIRM |
||||||
|
Variable |
Tipo de estimación; comparación entre los tratamientos |
FASLODEX® 500 mg |
FASLODEX® 250 mg |
Comparación entre los grupos |
||
|
Razón de riesgos |
IC del 95% |
Valor de p |
||||
|
SSP |
Mediana de K-M en meses; razón de riesgos |
|||||
|
Todas las pacientes |
6.5 |
5.5 |
0.80 |
0.68, 0.94 |
0.006 |
|
|
- Subgrupo AE (n=423) |
8.6 |
5.8 |
0.76 |
0.62, 0.94 |
0.013 |
|
|
- Subgrupo IA (n=313)a |
5.4 |
4.1 |
0.85 |
0.67, 1.08 |
0.195 |
|
|
STb |
Mediana de K-M en meses; razón de riesgos |
|||||
|
Todas las pacientes |
26.4 |
22.3 |
0.81 |
0.69, 0.96 |
0.016c |
|
|
-Subgrupo AE (n=423) |
30.6 |
23.9 |
0.79 |
0.63, 0.99 |
0.038c |
|
|
-Subgrupo IA (n=313)a |
24.1 |
20.8 |
0.86 |
0.67, 1.11 |
0.241c |
|
|
Variable |
Tipo de estimación; comparación entre los tratamientos |
FASLODEX® 500 mg |
FASLODEX® 250 mg |
Comparación entre los grupos (FASLODEX® 500 mg/FASLODEX® 250 mg) |
||
|
Diferencia absoluta (%) |
IC del 95% |
|||||
|
TROd |
% de pacientes con RO; diferencia absoluta (%) |
|||||
|
Todas las pacientes |
13.8 |
14.6 |
-0.8 |
-5.8, 6.3 |
||
|
-Subgrupo AE (n=296) |
18.1 |
19.1 |
-1.0 |
-8.2, 9.3 |
||
|
-Subgrupo IA (n=205)a |
7.3 |
8.3 |
-1.0 |
-5.5, 9.8 |
||
|
TBCe |
% de pacientes con BC; diferencia absoluta (%) |
|||||
|
Todas las pacientes |
45.6 |
39.6 |
6.0 |
-1.1, 13.3 |
||
|
-Subgrupo AE (n=423) |
52.4 |
45.1 |
7.3 |
-2.2, 16.6 |
||
|
-Subgrupo IA (n=313)a |
36.2 |
32.3 |
3.9 |
-6.1, 15.2 |
||
|
a FASLODEX® está indicado para pacientes que presentan una recidiva o progresión de la enfermedad mientras reciben un tratamiento antiestrogénico. No fueron concluyentes los resultados del subgrupo AI. b Se presenta la supervivencia total (ST) evaluada en los análisis finales de la supervivencia que se efectuaron tras alcanzar un 75% de madurez de los datos. c Valor de p nominal (sin el ajuste correspondiente a comparaciones múltiples) entre los análisis iniciales de la supervivencia total que se efectuaron tras alcanzar un 50% de madurez de los datos y los análisis actualizados que se efectuaron tras alcanzar un 75% de madurez de los datos. d La TRO se evaluó en las pacientes con una respuesta inicial evaluable (es decir, las pacientes cuyo tumor pudo medirse inicialmente: 240 pacientes del grupo de FASLODEX® 500 mg y 261 pacientes del grupo de FASLODEX® 250 mg). e Pacientes cuya mejor respuesta objetiva fue: Respuesta completa, respuesta parcial o enfermedad estable = 24 semanas. SSP = Supervivencia sin progresión del cáncer; TRO = Tasa de respuesta objetiva; RO = Respuesta objetiva; TBC = Tasa de beneficio clínico; BC = Beneficio clínico; ST = Supervivencia total; K-M = Kaplan-Meier; IC = Intervalo de confianza. IA = Inhibidor de la aromatasa; AE = Antiestrógeno. |
||||||
Se llevaron a cabo dos estudios clínicos de Fase III en un total de 851 mujeres posmenopáusicas con cáncer de mama avanzado que presentaron recidivas de la enfermedad durante o después del tratamiento endocrino adyuvante o bien una progresión de la enfermedad después del tratamiento endocrino del cáncer avanzado. El 77% de la población incluida en el estudio tenía cáncer de mama con receptores de estrógeno positivos. Dichos estudios compararon la seguridad y la eficacia de 250 mg de FASLODEX® al mes con respecto a la administración diaria de 1 mg de anastrozol (un inhibidor de la aromatasa). Globalmente, la dosis mensual de FASLODEX® de 250 mg tuvo efectos por lo menos equivalentes a los del anastrozol en la supervivencia sin progresión del cáncer, la tasa de respuesta objetiva y el tiempo de supervivencia. No se observaron diferencias estadísticamente significativas entre los dos grupos de tratamiento en ninguno de estos criterios de valoración. La variable principal fue la supervivencia sin progresión del cáncer. El análisis combinado de los dos estudios reveló que el 83% de las pacientes que recibieron FASLODEX® mostraron una progresión de la enfermedad, contra el 85% de las pacientes tratadas con el anastrozol. El análisis combinado de los dos estudios mostró que la relación de riesgos entre FASLODEX® 250 mg y el anastrozol en cuanto a la supervivencia sin progresión del cáncer fue de 0.95 (IC del 95%: 0.82-1.10). Se registró un porcentaje de respuesta objetiva del 19.2% con FASLODEX® 250 mg, frente al 16.5% con el anastrozol. La mediana del tiempo de supervivencia fue de 27.4 meses en las pacientes tratadas con FASLODEX® y de 27.6 meses en aquellas tratadas con el anastrozol. La relación de riesgos entre FASLODEX® 250 mg y el anastrozol en cuanto al tiempo de supervivencia fue de 1.01 (IC del 95%: 0.86-1.19).
Efectos en el endometrio posmenopáusico: Los resultados de los estudios preclínicos no indican un efecto estimulante del fulvestrant en el endometrio de las mujeres posmenopáusicas (véase la sección “Datos preclínicos sobre la seguridad”). Un estudio de 2 semanas en voluntarias sanas posmenopáusicas tratadas con 20 µg al día de etinilestradiol demostró que, con respecto a un placebo, el tratamiento previo con 250 mg de FASLODEX® produjo una reducción significativa de la estimulación del endometrio posmenopáusico, determinada midiendo el espesor del endometrio por ecografía.
El tratamiento neoadyuvante de pacientes con cáncer de mama durante un periodo de hasta 16 semanas con dosis de FASLODEX® de 500 mg o 250 mg no produjo variaciones de importancia clínica del espesor del endometrio, lo cual indica la ausencia de un efecto agonista. No hubo pruebas de efectos adversos en el endometrio en las pacientes con cáncer de mama estudiadas. Se carece de información sobre la morfología del endometrio.
En dos estudios a corto plazo (1 y 12 semanas) en mujeres premenopáusicas con afecciones ginecológicas benignas, no se observaron diferencias significativas entre los grupos del fulvestrant y del placebo en cuanto al espesor del endometrio medido por ecografía.
Efectos en el esqueleto: Se carece de información sobre los efectos a largo plazo del fulvestrant en los huesos. El tratamiento neoadyuvante de pacientes con cáncer de mama durante un periodo de hasta 16 semanas con dosis de FASLODEX® de 500 mg o 250 mg no produjo variaciones de importancia clínica de los marcadores séricos de la remodelación ósea.
MODO DE ADMINISTRACIÓN: FASLODEX® debe administrarse en forma de dos inyecciones intramusculares lentas consecutivas de 5 ml (de 1 a 2 minutos por inyección), una en cada glúteo.
Las instrucciones completas de administración se encuentran en la sección “Instrucciones de administración y precauciones especiales de desecho y manipulación”.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN
Posología
Mujeres adultas (incluyendo las de edad avanzada): La dosis recomendada es de 500 mg cada mes, con una dosis adicional de 500 mg administrada dos semanas después de la dosis inicial.
Poblaciones especiales
Pacientes pediátricos: Dado que no se han establecido la seguridad y la eficacia de FASLODEX® en niños y adolescentes, no se recomienda en este grupo de edad.
Insuficiencia renal: No es necesario ajustar la dosis en pacientes con insuficiencia renal leve a moderada (depuración de creatinina = 30 ml/min). Dado que no se han evaluado la seguridad y la eficacia de FASLODEX® en pacientes con insuficiencia renal grave (depuración de creatinina <30 ml/min), se recomienda tener precaución en este grupo de pacientes (véase la sección “Advertencias y precauciones especiales de empleo”).
Insuficiencia hepática: No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve a moderada; sin embargo, en vista de que puede aumentar la exposición al fulvestrant, FASLODEX® debe utilizarse con precaución en estos pacientes. No se dispone de datos en la insuficiencia hepática grave (véanse las secciones “Contraindicaciones”, “Advertencias y precauciones especiales de empleo” y “Propiedades farmacocinéticas”).
SOBREDOSIS: Se carece de experiencia sobre las sobredosis en el ser humano. Los estudios en animales sugieren que dosis altas de fulvestrant no producen otros efectos aparte de los directa o indirectamente relacionados con la actividad antiestrogénica del medicamento. En caso de sobredosis, se recomienda un tratamiento de apoyo sintomático.
PRESENTACIÓN: FASLODEX® 250 mg. Jeringa prellenada. (Reg. San. INVIMA 2005M-0004795).
Fecha de revisión del texto: Abril de 2015
Fecha de preparación de la versión: Enero de 2016
Clave: 1-2016. Fuente: Doc ID-000162581 V 7.0
FASLODEX® es una marca registrada del grupo AstraZeneca.
© AstraZeneca 2006-2015
Fabricación y acondicionamiento primario por Vetter Pharma-Fertigung GmbH & Co., KG, Schützenstrasse 87, 88212 Ravensburg, Alemania. Acondicionamiento secundario por AstraZeneca UK Limited, Silk Road Business Park, Macclesfield, Cheshire, SK10 2NA, Reino Unido.
Mayor información Departamento Médico de
AstraZeneca Colombia S.A.S.
Bogotá, D.C., Colombia.

