

FARMORUBICINA
EPIRRUBICINA
Solución estéril inyectable
Caja , 1 Vial(es) , Solución inyectable , 25 Mililitros
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Cada vial (frasco ampolla) con 25 mL de SOLUCIÓN inyectable contienen clorhidrato de epirubicina 50 mg.
INDICACIONES TERAPÉUTICAS: Tratamiento de leucemias agudas, linfomas malignos, sarcomas de partes blandas, carcinoma gástrico, carcinoma del hígado, pancreas, recto sigmoideo, carcinomas cervifaciales, carcinoma pulmonar, carcinoma ovárico.
DETALLES FARMACÉUTICOS
Incompatibilidades: No se debe mezclar la epirubicina con otros medicamentos. Ha de evitarse el contacto con cualquier solución de pH alcalino ya que dará lugar a la hidrólisis de la epirubicina. La epirubicina no se debe mezclar con heparina a causa de una incompatibilidad química que puede conducir a la precipitación.
Precauciones especiales para el almacenamiento
Solución para inyección lista para usar: El almacenamiento de la solución para inyección en condiciones de refrigeración (2-8 ºC) puede dar lugar a la formación de un producto en forma de gel. Este producto en forma de gel regresará a una solución de ligeramente viscosa a móvil después de dos, hasta máximo cuatro horas de equilibrio a temperatura ambiente controlada (15 – 25°C).
Precauciones especiales para la eliminación y otro manejo
Medidas de protección: Se dan las siguientes recomendaciones de protección dada la naturaleza tóxica de esta sustancia
• El personal debe tener entrenamiento en las técnicas adecuadas de reconstitución y manejo;
• Las mujeres embarazadas deben ser excluidas del trabajo con este fármaco;
• El personal que maneje epirubicina debe usar ropa protectora: gafas, batas, guantes desechables y tapabocas;
• Se debe definir un área designada para la reconstitución (de preferencia bajo un sistema de flujo laminar); la superficie de trabajo se debe proteger con papel desechable, absorbente, con forro de plástico;
• Todos los artículos utilizados para la reconstitución, administración o limpieza, incluidos los guantes, deben disponerse en bolsas de basura de alto riesgo, para incineración a alta temperatura;
• Los derrames o escapes se deben tratar con hipoclorito de sodio diluido (1% de cloro disponible), de preferencia con remojo, y luego agua;
• Todos los materiales de limpieza se deben desechar como se indicó antes;
• En caso de contacto con la piel debe lavarse bien el área afectada con agua, jabón o una solución de bicarbonato de sodio. Sin embargo, no se debe lesionar la piel usando un cepillo de lavado.
• En caso de contacto con los ojos, se debe mantener retraído el párpado y lavar con copiosas cantidades de agua durante cuando menos 15 minutos. Luego hay que buscar la evaluación de un médico.
• Siempre hay que lavarse las manos después de quitarse los guantes.
Condiciones de almacenamiento: Almacénese entre 2°C y 8°C, en su envase y empaque original.
Vida útil: 36 meses.
Naturaleza y contenido del recipiente: Frasco vial.
Es posible que la información de prescripción de este producto haya sido revisada y actualizada después de la fecha de impresión del PLM 2016. Para obtener información más actualizada comuníquese con la Dirección Médica de Pfizer S.A.S Teléfono: (1) 6002300 Ext. 2509 Bogotá – Colombia.
Farmorubicina 50 mg/25 ml solución inyectable (Reg. San. INVIMA 2008M -010231-R1).
Título del documento del producto: Clorhidrato de epirubicina.
Fecha última revisión: 21 de julio de 2011
CDS Versión: 6.0
Fecha efectiva: 6 de marzo de 2014
LLD_Col_CDSv6.0_6Marzo2014_v1.0_Aprobado por Resol. 2014038685_03Dic2014
PFIZER S.A.S.
PROPIEDADES FARMACOCINÉTICAS: La farmacocinética de la epirubicina es lineal sobre el intervalo de dosis de 60 a 150 mg/m2 y la depuración del plasma no se ve afectada por la duración de la infusión o el esquema de administración.
Distribución: Luego de la administración intravenosa, la epirubicina se distribuye rápida y ampliamente en los tejidos. La unión de la epirubicina a las proteínas plasmáticas, sobre todo la albúmina, es de cerca de 77% y no resulta afectada por la concentración del fármaco. La epirubicina también parece concentrarse en los glóbulos rojos; las concentraciones en sangre total son aproximadamente dos veces las del plasma.
Metabolismo: La epirubicina es metabolizada de forma extensa y rápida por el hígado y también por otros órganos y células, incluidos los glóbulos rojos.
Se han identificado cuatro vías metabólicas principales:
1. Reducción del grupo C-13 ceto con formación del derivado 13(S)-dihidro, epirrubicinol;
2. Conjugación tanto del fármaco no modificado como del epirrubicinol con ácido glucurónico;
3. Pérdida del residuo de amino azúcar a través de un proceso hidrolítico con formación de agluconas de doxorrubicina y doxorubicinol; y
4. Pérdida del residuo de amino azúcar a través de un proceso redox con formación de la aglucona 7-desoxi-doxorrubicina y la aglucona 7-desoxi-doxorrubicinol. El epirrubicinol tiene una actividad citotóxica in vitro que es una décima parte de la de la epirrubicina. Como los niveles plasmáticos de epirrubicinol son más bajos que los del fármaco inmodificado, es poco probable que alcancen concentraciones in vivo suficientes para la citotoxicidad. No se ha informado de actividad o toxicidad significativas de los otros metabolitos.
Excreción: La epirubicina y sus principales metabolitos se eliminan por excreción biliar y, en menor grado, por excreción urinaria. Los datos de balance de masa de 1 paciente muestran cerca de 60% de la dosis radioactiva total en las heces (34%) y la orina (27%). Estos datos concuerdan con los obtenidos en 3 pacientes con obstrucción extrahepática y drenaje percutáneo, en quienes aproximadamente 35% y 20% de la dosis administrada se recuperaron como epirubicina o sus principales metabolitos en la bilis y la orina, respectivamente, 4 días después del tratamiento.
Farmacocinética en poblaciones especiales
Deterioro de la función hepática: La epirubicina se elimina tanto por metabolismo hepático como por excreción biliar y la depuración está reducida en pacientes que tienen disfunción hepática. En un estudio sobre el efecto de la disfunción hepática, se clasificó en tres grupos a pacientes con tumores sólidos. Los pacientes del Grupo 1 (n = 22) tenían valores de AST (SGOT) en suero por encima del límite superior normal (mediana: 93 IU/L) y noveles normales de bilirrubina en suero (mediana: 0,5 mg/dL) y se les administró epirubicina en dosis de 12,5 a 90 mg/m2. Los pacientes del Grupo 2 tenían alteraciones tanto de los niveles séricos de AST (mediana: 175 IU/L) y como de bilirrubina (mediana: 2,7 mg/dL) y fueron tratados con una dosis de epirubicina de 25 mg/m2 (n = 8). Su farmacocinética se comparó con la de los pacientes que tenían valores normales de AST y bilirrubina en suero, que recibieron epirubicina en dosis de 12,5 a 120 mg/m2. La mediana de la depuración del plasma de la epirubicina se redujo en comparación con la de los pacientes que tenían función hepática normal en cerca de 30% de los pacientes del Grupo 1 y en 50% de los pacientes del Grupo 2. No se han evaluado pacientes que acusan un deterioro más grave de la función hepática (véase la Sección Posología y método de administración).
Deterioro de la función renal: No se han observado alteraciones significativas de la farmacocinética de la epirubicina o su principal metabolito, el epirrubicinol, en pacientes con creatinina sérica < 5 mg/dL. Se informó una reducción de 50% en la depuración del plasma en cuatro pacientes con creatinina sérica > 5 mg/dL (véanse las secciones Posología y método de administración y Advertencias y precauciones especiales para el uso). No se han estudiado pacientes en diálisis.
PROPIEDADES FARMACODINÁMICAS: La epirubicina es un agente citotóxico de la familia de las antraciclinas. Aunque se sabe que las antraciclinas pueden alterar diversas funciones bioquímicas y biológicas de las células eucarióticas, aún no se han terminado de dilucidar los mecanismos exactos de las propiedades citotóxicas o antiproliferadoras de la epirubicina.
La epirubicina forma un complejo con el ADN mediante la intercalación de sus anillos planares entre pares de bases de nucleótidos, con la consiguiente inhibición de la síntesis de los ácidos nucleicos (ADN y ARN) y de las proteínas. Esta intercalación desencadena la fisión del ADN por parte de la topoisomerasa II, lo cual da lugar a la actividad citocida. La epirubicina también inhibe la actividad de la helicasa del ADN, lo cual impide la separación enzimática del ADN de doble cadena y altera la replicación y la transcripción. La epirubicina también está implicada en reacciones de oxidación/reducción mediante la generación de radicales libres citotóxicos. Se piensa que la actividad antiproliferadora y citotóxica de la epirubicina es el resultado de estos u otros posibles mecanismos.
La epirubicina is citotóxica in vitro para diversas líneas celulares murinas y humanas establecidas y para cultivos primarios de tumores humanos. También tiene actividad in vivo contra diversos tumores murinos y xenoinjertos humanos en ratones atímicos, incluidos los tumores de mama.
• Estudios clínicos
Tratamiento adyuvante en pacientes con Cáncer Mamario Temprano: Dos estudios multicéntricos abiertos aleatorios evaluaron el uso de epirubicin 100 a 120 mg/m2 en combinación con ciclofosfamide y fluorouracilo para el tratamiento adyuvante de pacientes con cáncer mamario nódulo axilar positivo y sin evidencia de enfermedad metastásica distante (etapa II o III). El estudio MA-5 evaluó 120 mg/m2 de epirubicin por curso en combinación con ciclofosfamide y fluorouracilo (Régimen CEF-120). Este estudio asignó aleatoriamente a mujeres premenopáusicas y perimenopáusicas con uno o más nódulos linfáticos positivos a un régimen CEF-120 conteniendo epirubicin o un régimen CMF. El estudio GFEA-05 evaluó el uso de 100 mg/m2 de epirubicin por curso de tratamiento en combinación con fluorouracil y ciclofosfamide (FEC-100). Este estudio distribuyó aleatoriamente mujeres pre y post menopausicas al régimen FEC-100 o a un régimen de dosis más bajas FEC-50. En el estudio GFEA-05, las pacientes elegibles fueron aquellas que requerian tener = 4 nódulos involucrados con el tumor o si solo tenian 1 a 3 nódulos positivos, tener receptores de progesterona y estrógenos negativos y un grado histológico de tumor de grado 2 o 3. Un total de 1281 mujeres participaron en estos estudios. Las pacientes con tumores T4 no fueron elegibles para ningún estudio.
La Tabla 1 muestra los regímenes de tratamiento que los pacientes recibieron. El objetivo final primario fue supervivencia libre de recurrencia, por ejemplo, tiempo de ocurrencia de recurrencia local, regional o distante, o muerte relacionada con la enfermedad. Las pacientes con cáncer de mama contralateral, segunda malignidad primaria o muerte por causas distintas al cáncer de mama fueron censurados al momento de la última visita previa a estos eventos.
|
Tabla 1. Regímenes de Tratamiento Usados en los estudios de Fase 3 de Pacientes con Cáncer Mamario Temprano |
|||
|
Grupos de Tratamiento |
Agente |
Régimen |
|
|
MA-51 N=716 |
CEF-120 (total, 6 ciclos)2 N=356 CMF (total, 6 ciclos) N=360 |
Ciclofosfamida Epirubicina Fluorouracilo Ciclofosfamida Metotrexato Fluorouracilo |
75 mg/m2 PO, d 1-14, c 28 días 60 mg/m2 IV, d 1 & 8, c 28 días 500 mg/m2 IV, d 1 & 8, c 28 días 100 mg/m2 PO, d 1-14, c 28 días 40 mg/m2 IV, d 1 & 8, c 28 días 600 mg/m2 IV, d 1 & 8, c 28 días |
|
GFEA-053 N=565 |
FEC-100 (total, 6 ciclos) N=276 FEC-50 (total, 6 ciclos) N=289 Tamoxifen 30 mg diariamente x 3 años, mujeres posmenopáusicas, cualquier estatus de receptor. |
Fluorouracilo Epirubicina Ciclofosfamida Fluorouracilo Epirubicina Ciclofosfamida |
500 mg/m2 IV, d 1, c 21 días 100 mg/m2 IV, d 1, c 21 días 500 mg/m2 IV, d 1, c 21 días 500 mg/m2 IV, d 1, c 21 días 50 mg/m2 IV, d 1, c 21 días 500 mg/m2 IV, d 1, c 21 días |
|
1 En mujeres que fueron llevadas a lupectomía, se administró irradiación en el seno después de terminada la quimioterapia del estudio. 2 Pacientes también recibieron terapia antibiótica profiláctica con trimetoprim-sulfametoxazole o fluoroquinolone durante la duración de su quimioterapia. 3 Todas las mujeres recibieron irradiación del seno después de completar la quimioterapia. |
|||
En el ensayo MA-5 la mediana de la edad de la población de estudio fue 45 años. Aproximadamente 60% de los pacientes tenían 1 o 3 nódulos involucrados y aproximadamente 40% tuvo = 4 nódulos involucrados con tumor. En el estudio GFEA-05, la mediana de la edad fue 51 años y aproximadamente la mitad de las pacientes fueron post menopausicas. Aproximadamente 17% de la población del estudio tenía 1 a 3 nódulos positivos y 80% de las pacientes tenían = 4 nódulos linfáticos involucrados. Las características demográficas y del tumor estuvieron bien balanceadas entre los brazos de tratamiento en cada estudio.
Los objetivos finales de eficacia supervivencia libre de recurrencia (Relapse Free Survival - RFS) y la supervivencia globall (Overall Survival - OS) fueron analizados usando los métodos Kaplan-Meier en las poblaciones con intención de tratamiento (intent –to-treat - ITT), Los resultados de los puntos finales fueron analizados inicialmente hasta los 5 años de seguimiento y estos resultados se presentan en el texto a continuación y en la tabla 2. Los resultados hasta 10 años de seguimiento se presentaron en la tabla 2. En el estudio MA-5, la terapia de combinación conteniendo epirubicin (CEF-120) mostraron significativamente una supervivencia libre de recurrencia mas duradera que CMF (los estimados de 5 años fueron 62% versus 53%, logrank estratificado para el RFS total p=0.013). La reducción estimada en el riesgo de recurrencia fue de un 24% en 5 años. La OS fue también mayor para con el régimen CEF-120 con epirubicin que para el régimen CMF (estimado de 5 años 77% versus 70%; logrank estratificado para la sobrevivencia global p=0.043; logrank no estratificado p=0.13). La reducción estimada en el riesgo de muerte fue 29% a 5 años.
En el estudio GFEA-05, los pacientes tratados con el régimen de dosis altas de epirubicin (FEC-100) tuvieron una Supervivencia Libre de Recurencia (RFS)de 5 años significativamente más prolongada (estimado 65% versus 52%, logrank para RFS total p=0.007) y OS (estimado 76% versus 65%, logrank para la sobrevivencia global p=0.007) que en los pacientes con régimen de dosis más bajos (FEC-50). La reducción estimada en el riesgo de recurrencia fue de 32% en 5 años. La reducción estimada en el riesgo de muerte fue 31% en 5 años.
Los resultados de seguimiento hasta 10 años (media del seguimiento = 8.8 años y 8.3 años, respectivamente para el estudio MA-5 y el estudio GFEA-05) se presentaron en la tabla 2.
Aunque las pruebas no fueron potenciadas para el análisis de subgrupo, en el estudio MA-5, se observaron mejoras a favor de CEF-120 vs. CMF, en RFS y OS en pacientes con 1 a 3 nódulos positivos y en aquellos con = 4 nódulos positivos con tumor. En el estudio GFEA-05 se observaron mejoras en RFS y OS en mujeres pre y post menopáusicas tratadas con FEC-100 comparado con FEC-50.
|
Tabla 2. Resultados de Eficacia de los estudios Fase 3 de pacientes con Cáncer de Seno Temprano * |
||||
|
Estudio MA-5 |
Estudio GFEA-05 |
|||
|
CEF-120 N=356 |
CMF N=360 |
FEC-100 N=276 |
FEC-50 N=289 |
|
|
RFS a 5 años (%) |
62 |
53 |
65 |
52 |
|
Hazard ratio† |
0.76 |
0.68 |
||
|
Bilateral 95% CI |
(0.60, 0.96) |
(0.52, 0.89) |
||
|
Prueba rango log estratificada** |
(p = 0.013) |
(p = 0.007) |
||
|
OS a 5 años (%) |
77 |
70 |
76 |
65 |
|
Hazard ratio |
0.71 |
0.69 |
||
|
Bilateral 95% CI |
(0.52, 0.98) |
(0.51, 0.92) |
||
|
Prueba Log Rank estratificada** |
(p = 0.043) (no estratificado p = 0.13) |
(p = 0.007) |
||
|
RFS a 10 años (%) |
51 |
44 |
49 |
43 |
|
Hazard ratio |
0.78 |
0.78 |
||
|
Bilateral 95% CI |
(0.63, 0.95) |
(0.62, 0.99) |
||
|
Prueba rango log estratificada** |
(p = 0.017) (no estratificado p = 0.023) |
(p = 0.040) (no estratificado p = 0.09) |
||
|
OS a 10 años (%) |
61 |
57 |
56 |
50 |
|
Hazard ratio† |
0.82 |
0.75 |
||
|
Bilateral 95% CI |
(0.65, 1.04) |
(0.58, 0.96) |
||
|
Prueba rango log estratificada** |
(p = 0.100) (no estratificado p = 0.18) |
(p = 0.023) (no estratificado p = 0.039) |
||
|
* Basado en estimados Kaplan-Meier ** Los paciente en el estudio MA-5 fueron estratificados por estatus nodal (1-3, 4-10, y >10 nódulos positivos), tipo de cirugía inicial (lumpectomía versus mastectomía), y por estatus de receptor de hormonas (ER o PR positivo (=10 fmol), ambos negativos (<10 fmol), o estatus desconocido). Los pacientes en GFEA-05 fueron estratificados por estatus nodal (1-3, 4-10, y >10 nódulos positivos). † Hazard ratio: CMF:CEF-120 en MA-5, FEC-50:FEC-100 en GFEA-05 |
||||
Las curvas Kaplan-Meier para RFS y OS del estudio MA-5 se muestran en las figuras 1 y 2 y los del estudio GFEA-05 se muestran en las figuras 3 y 4.
Figura 1. Sobrevivencia Libre de Recurrencia en el Estudio MA-5
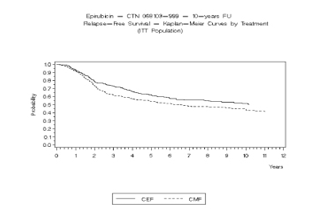
Figura 2. Sobrevivencia Global en el Estudio MA-5
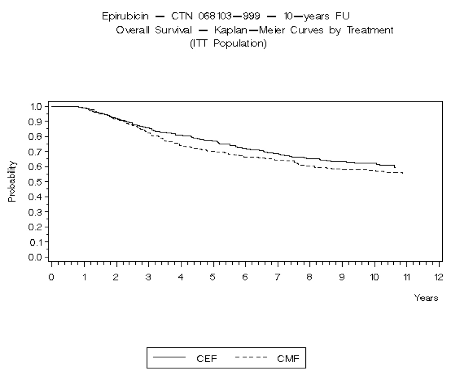
Figura 3. Sobrevivencia Libre de Recurrencia en el Estudio GFEA-05
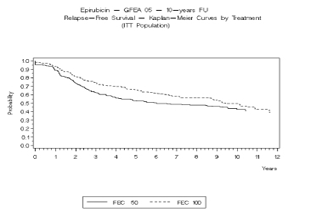
Figura 4. Sobrevivencia Global en el Estudio GFEA-05
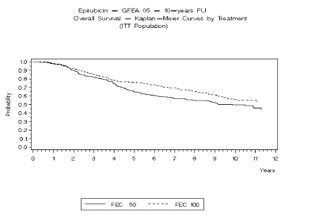
Ver Tabla 2 para las estadísticas de los análisis de 5 y 10 años.
Función cardiaca
En una encuesta retrospectiva, en la cual se incluyeron 9144 pacientes, casi todos con tumores sólidos en estadios avanzados, la probabilidad de aparición de ICC se incrementó al aumentar las dosis acumulativas de epirubicina (Figura 5). El riesgo estimado de aparición de ICC clínicamente evidente en pacientes tratados con epirubicina fue de 0,9% con una dosis acumulativa de 550 mg/m2, 1,6% con 700 mg/m2, y 3,3% con 900 mg/m2. El riesgo de que sobrevenga una ICC en ausencia de otros factores de riesgo cardiaco aumentó de forma aguda después de una dosis acumulativa de epirubicina de 900 mg/m2.
Figura 5. Riesgo de ICC en 9144 pacientes tratados con epirubicina
Probabilidad
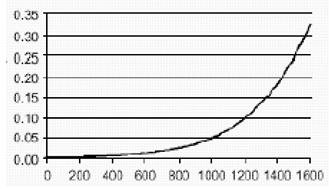
Dosis acumulativa de epirubicina (mg/m²)
En otra encuesta retrospectiva en 469 pacientes tratados con epirubicina con cáncer de mama en etapa incipiente o metastásico, el riesgo informado de ICC fue comparable al observado en el estudio más grande con más de 9000 pacientes.
Leucemia secundaria: Un análisis de 7110 pacientes que recibieron tratamiento adyuvante con epirubicina en estudios clínicos controlados como componente de regímenes de poli-quimioterapia para el cáncer de mama en etapa incipiente, demostró un riesgo acumulativo de leucemia mieloide aguda secundaria o síndrome mielodisplásico (AML/MDS) de cerca de 0,27% (IC 95% aproximado, 0,14-0,40) a los 3 años, 0,46% (IC 95% aproximado, 0,28-0,65) a los 5 años y 0,55% (IC 95% aproximado, 0,33-0,78) a los 8 años. El riesgo de que sobrevenga AML/MDS aumenta al aumentar la dosis acumulativa de epirubicina como se muestra en la Figura 6.
Figura 6. Riesgo de AML/MDS en 7110 pacientes tratados con epirubicina
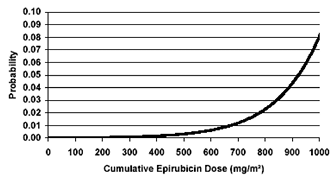
Las tasas de AML/MDS aumentaron con las dosis de epirubicina por ciclo, y dosis acumulativa. Por ejemplo, en el estudio MA-5, en pacientes que recibieron dosis intensivas de epirubicina (120 mg/m2), la incidencia de AML/MDS fue de 1,1 % a los 5 años sin observarse casos adicionales observados durante los segundos 5 años (años 6-10) de seguimiento.
Se encontró que la probabilidad acumulativa de que sobrevenga AML/MDS aumentó especialmente en los pacientes que recibieron más de la máxima dosis acumulativa recomendada de epirrubicina (720 mg/m2) o de ciclofosfamida (6.300 mg/m2), como se muestra en la Tabla 3.
|
Tabla 3. Probabilidad acumulativa de AML/MDS en relación con la dosis acumulativa de epirubicina y ciclofosfamida |
||||
|
Años desde el inicio del tratamiento |
Probabilidad acumulativa de que sobrevenga AML/MDS % (IC 95%) |
|||
|
Dosis acumulativa de ciclofosfamida < 6,300 mg/m2 |
Dosis acumulativa de ciclofosfamida > 6,300 mg/m2 |
|||
|
Dosis acumulativa de epirubicina < 720 mg/m2 N = 4760 |
Dosis acumulativa de epirubicina >720 mg/m2 N = 111 |
Dosis acumulativa de epirubicina < 720 mg/m2 N = 890 |
Dosis acumulativa de epirubicina > 720 mg/m2 N = 261 |
|
|
3 |
0,12 (0,01-0,22) |
0,00 (0,00-0,00) |
0,12 (0,00-0,37) |
4,37 (1,69-7,05) |
|
5 |
0,25 (0,08-0,42) |
2,38 (0,00-6,99) |
0,31 (0,00-0,75) |
4,97 (2,06-7,87) |
|
8 |
0,37 (0,13-0,61) |
2,38 (0,00-6,99) |
0,31 (0,00-0,75) |
4,97 (2,06-7,87) |
PROPIEDADES FARMACODINÁMICAS
Estudios clínicos
Gastrointestinal: La epirubicina causa vómito. La mucositis/estomatitis suele aparecer poco después de la administración del medicamento y, si es severa, puede avanzar en unos pocos días hasta la formación de ulceraciones en la mucosa. La mayoría de los pacientes se recupera de este evento adverso hacia la tercera semana de terapia.
Función hepática: La principal vía de eliminación de la epirubicina es el sistema hepatobiliar. Se deben valorar los niveles de bilirrubina sérica total y AST antes y durante el tratamiento con epirubicina. Los pacientes que tienen niveles elevados de bilirrubinas o AST pueden experimentar una depuración más lenta del medicamento con un aumento de la toxicidad general. En estos pacientes se recomienda usar dosis más bajas (véanse las Secciones Posología y método de administración y Propiedades farmacocinéticas). Los pacientes que tienen deterioro severo de la función hepática no deben recibir epirubicina (véase la Sección Contraindicaciones).
Función renal: Se debe valorar la creatinina sérica antes y durante la terapia. Es preciso ajustar la dosificación en pacientes que tienen creatinina sérica > 5 mg/dL (véase la Sección Posología y método de administración).
Efectos en el lugar de la inyección: Se puede presentar una fleboesclerosis como resultado de una inyección en un vaso de pequeño calibre o por las inyecciones repetidas en la misma vena. Si se siguen los procedimientos recomendados para la administración se puede reducir al mínimo el riesgo de flebitis/tromboflebitis en el lugar de la inyección (véase la Sección Posología y método de administración).
Extravasación: La extravasación de epirubicina durante la inyección intravenosa puede producir dolor local, lesiones tisulares severas (ampollas, celulitis grave) y necrosis. En caso de que se observen signos o síntomas de extravasación durante la administración intravenosa de epirubicina, se deberá interrumpir de inmediato la infusión.
Otros: Igual que sucede con otros agentes citotóxicos, se ha informado la aparición coincidencial con el uso de la epirubicina de tromboflebitis y fenómenos tromboembólicos, incluido embolismo pulmonar (en algunos casos mortal).
Síndrome de lisis tumoral: La epirubicina puede inducir hiperuricemia a causa del extenso catabolismo de las purinas que acompaña la rápida lisis de las células neoplásicas inducida por el medicamento (síndrome de lisis tumoral). Se deben evaluar los niveles sanguíneos de ácido úrico, potasio, fosfato de calcio y creatinina después del tratamiento inicial. Las posibles complicaciones del síndrome de lisis tumoral se pueden reducir al mínimo con hidratación, alcalinización de la orina y profilaxis con allopurinol para prevenir la hiperuricemia.
Efectos Inmunosupresores / incremento de susceptibilidad a infecciones: La administración de vacunas vivas o vacunas vivas atenuadas en pacientes con inmunocompromiso por agentes quimioterapéuticos incluyendo epirubicina, pueden resultar en infecciones serias o fatales. La vacunación con una vacuna viva debe ser evitada en pacientes con que estén recibiendo epirubicina. Vacunas muertas o inactivadas pueden ser administradas; sin embargo, la respuesta a tales vacunas puede estar disminuida.
CONTRAINDICACIONES: Hipersensibilidad a la epirrubicina o a cualquier otro componente del producto, otras antraciclinas o antracenoides.
Mielosupresión persistente, deterioro grave de la función hepática, miocardiopatía, infarto de miocardio reciente, arritmias severas, tratamientos previos con dosis máximas acumulativas de epirrubicina y/o otras antraciclinas y antracenoides. (Véase la Sección Advertencias y precauciones especiales para el uso).
FERTILIDAD, EMBARAZO Y LACTANCIA: (Véase la Sección Datos preclínicos de seguridad).
Afectación de la fertilidad: La epirubicina podría inducir daños cromosómicos en los espermatozoides humanos. Los varones sometidos a tratamiento con epirubicina deben usar métodos anticonceptivos eficaces.
La epirubicina puede causar amenorrea o menopausia precoz en mujeres premenopáusicas.
Embarazo: Las mujeres en edad fértil deben evitar quedar embarazadas durante el tratamiento y deben utilizar métodos anticonceptivos efectivos.
Los datos experimentales en animales sugieren que la epirubicina puede causar daños al feto cuando se administra a una mujer embarazada. Si se usa la epirubicina durante el embarazo o si la paciente queda embarazada mientras toma el medicamento, deberá ser puesta al corriente de los posibles riesgos para el feto.
No se han hecho estudios en mujeres embarazadas. Sólo se usará la epirubicina durante el embarazo si el beneficio potencial justifica el riesgo posible para el feto.
Lactancia: No se sabe si la epirubicina se excreta por la leche humana. Dado que muchos medicamentos, incluidas otras antraciclinas, se excretan por la leche humana y debido al potencial de reacciones adversas serias en bebés lactantes por la epirubicina, las madres deben suspender la lactancia antes de tomar este medicamento.
EFECTOS SOBRE LA CAPACIDAD DE CONDUCIR VEHÍCULOS Y OPERAR MAQUINARIAS: No se ha hecho una evaluación sistemática del efecto de la epirubicina sobre la capacidad de conducir vehículos y operar maquinarias.
EFECTOS INDESEABLES
Estudios clínicos: Se ha realizado un gran número de estudios clínicos con epirubicina, administrada tanto en dosis convencionales como en dosis altas en diferentes indicaciones. A continuación se enumeran los eventos adversos serios relacionados con el medicamento que se presentaron durante los estudios clínicos.
Infecciones e infestaciones: Infección.
Neoplasias benignas y malignas: Leucemia linfocítica aguda, leucemia mieloide aguda.
Trastornos de la sangre y el sistema linfático: Anemia, trombocitopenia, neutropenia febril, neutropenia, leucopenia.
Trastornos del metabolismo y la nutrición: Anorexia.
Trastornos oculares: Conjuntivitis/queratitis.
Trastornos cardiacos: Insuficiencia cardiaca congestiva, taquicardia ventricular, bloqueo AV, bloqueo completo de rama, bradicardia.
Trastornos vasculares: Oleadas de calor, tromboembolismo.
Trastornos gastrointestinales: Náuseas/vómito, mucositis/estomatitis, diarrea.
Trastornos de piel y tejido celular subcutáneo: Alopecia, toxicidad local, erupción/prurito, cambios de la piel.
Trastornos del sistema reproductor y la mama: Amenorrea.
Trastornos generales y afecciones del lugar de administración: Malestar general/astenia, fiebre.
Investigaciones: Caídas asintomáticas de la fracción de eyección del ventrículo izquierdo, cambios de los niveles de transaminasas.
• Vigilancia posterior al mercadeo
Infecciones e infestaciones: Sepsis, neumonía.
Trastornos del sistema inmunológico: Anafilaxia.
Trastornos del metabolismo y nutrición: Deshidratación, hiperuricemia.
Trastornos vasculares: Shock, hemorragia, embolismo arterial, tromboflebitis, flebitis.
Trastornos respiratorios, torácicos y mediastínicos: Embolismo pulmonar.
Trastornos gastrointestinales: Erosiones, ulceraciones, dolor o sensación de quemadura, hemorragia, hiperpigmentación de la mucosa oral.
Trastornos de la piel y el tejido celular subcutáneo: Eritema, enrojecimiento, hiperpigmentación de piel y uñas, fotosensibilidad, hipersensibilidad de la piel irradiada (reacción de recuerdo de la radiación), urticaria.
Trastornos renales y urinarios: Coloración roja de la orina durante 1 a 2 días después de la administración.
Trastornos generales y afecciones del lugar de administración: Fiebre, escalofríos.
INTERACCIÓN CON OTROS PRODUCTOS MEDICINALES Y OTRAS FORMAS DE INTERACCIÓN: La epirubicina se usa principalmente en combinación con otros fármacos citotóxicos. Puede sobrevenir toxicidad aditiva, en especial en lo que respecta a los efectos sobre la médula ósea/hematógicos y gastrointestinales (véase la Sección Advertencias y precauciones especiales para el uso). El uso de epirubicina en quimioterapia combinada con otros medicamentos potencialmente cardiotóxicos, así como el uso concomitante de otros compuestos cardioactivos (p.ej., bloqueadores de los canales del calcio), exige la vigilancia de la función cardiaca durante todo el tratamiento.
La epirubicina es extensamente metabolizada por el hígado. Los cambios en la función hepática inducidos por las terapias concomitantes pueden afectar el metabolismo, la farmacocinética, eficacia terapéutica o la toxicidad de la epirubicina (véase la Sección Advertencias y precauciones especiales para el uso).
La cimetidina aumentó el ABC de la epirubicina en 50% y se debe suspender durante el tratamiento con epirubicina.
Cuando se da antes de la epirubicina, el paclitaxel puede causar aumento de las concentraciones plasmáticas de epirubicina no modificada y sus metabolitos, no siendo estos últimos, sin embargo, ni tóxicos ni activos. La administración conjunta de paclitaxel o docetaxel no afectó la farmacocinética de la epirubicina cuando ésta se administró antes del taxane.
DATOS PRECLÍNICOS DE SEGURIDAD: La epirubicina es mutagénica, clastogénica, y carcinogénica en animales.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES PARA EL USO
Vigilancia del paciente durante el tratamiento, monitoreo de glóbulos blancos, rojos y plaquetas. Estricta vigilancia de la función cardiaca.
General: La epirubicina sólo se administrará bajo la supervisión de médicos calificados con experiencia en el uso de terapia citotóxica.
Los pacientes se deben recuperar de las toxicidades agudas (como estomatitis, neutropenia, trombocitopenia e infecciones generalizadas) del tratamiento citotóxico previo antes de comenzar el tratamiento con epirubicina.
Aunque el tratamiento con dosis altas de epirubicina (p.ej., > 90 mg/m2 cada 3 a 4 semanas) causa eventos adversos que en general son similares a los que se observan con las dosis estándar (< 90 mg/m2 cada 3 a 4 semanas), la severidad de la neutropenia y la estomatitis/mucositis puede verse aumentada. El tratamiento con dosis altas de epirubicina sí exige prestar atención especial a las posibles complicaciones clínicas debidas a la mielosupresión profunda.
Función cardiaca: La cardiotoxicidad es uno de los riesgos del tratamiento con antraciclinas que se puede manifestar por eventos de aparición precoz (es decir, agudos) o de aparición tardía (es decir, retardados).
Eventos de aparición precoz (es decir, agudos): La cardiotoxicidad temprana de la epirubicina consiste principalmente en taquicardia sinusal o anomalías electrocardiográficas (EKG) como cambios inespecíficos en la onda ST-T. También se ha informado de taquiarritmias, incluidas contracciones ventriculares prematuras, taquicardia ventricular y bradicardia, así como bloqueos aurículo-ventriculares y de rama. Estos efectos no suelen predecir la aparición posterior de cardiotoxicidad tardía, rara vez revisten importancia clínica y por lo general no constituyen una consideración para el abandono del tratamiento con epirubicina.
Eventos de aparición tardía (es decir, retardados): La cardiotoxicidad tardía suele sobrevenir tarde en el curso de la terapia con epirubicina o dentro de los 2 a 3 meses siguientes a la terminación del tratamiento, pero también se han informado eventos más tardíos (varios meses a años después de terminado el tratamiento). La cardiomiopatía tardía se manifiesta como una reducción de la fracción de eyección del ventrículo izquierdo (FEVI) o signos y síntomas de insuficiencia cardiaca congestiva (ICC) como disnea, edema pulmonar, edema de zonas declives, cardiomegalia y hepatomegalia, oliguria, ascitis, derrame pleural y ritmo de galope. La ICC potencialmente mortal es la forma más severa de cardiomiopatía inducida por antraciclinas y representa la toxicidad acumulativa limitante de la dosis del fármaco.
El riesgo de que sobrevenga una ICC aumenta rápidamente conforme aumentan las dosis totales acumulativas de epirubicina por encima de 900 mg/m2; esta dosis acumulativa sólo se excederá con extrema precaución (véase la Sección Propiedades farmacodinámicas).
Se debe valorar la función cardiaca antes que el paciente se someta al tratamiento con epirubicina y se vigilará a lo largo de toda la terapia a fin de reducir al mínimo el riesgo que sobrevenga un deterioro severo de la función cardiaca. Es posible disminuir el riesgo por medio de la vigilancia regular de la FEVI durante el curso del tratamiento, con el pronto abandono del tratamiento de epirubicina ante la primera señal de deterioro de la función. El método cuantitativo adecuado para hacer la valoración repetida de la función cardiaca (evaluación de la FEVI) incluye angiografía de múltiples puertas con radioisótopos (MUGA) o ecocardiografía (ECO). Se recomienda hacer la evaluación cardiaca inicial con un EKG y bien sea una gammagrafía MUGA o una ECO, sobre todo en pacientes que tienen factores de riesgo de aumento de la cardiotoxicidad. Se repetirán las determinaciones de la FEVI con MUGA o ECO, en especial cuando se usan dosis altas acumulativas de la antraciclina. La técnica empleada para la valoración debe ser la misma durante todo el seguimiento.
Dado el riesgo de cardiomiopatía, la dosis acumulativa de 900 mg/m2 de epirubicina sólo se excederá tomando extremas precauciones.
Los factores de riesgo de toxicidad cardiaca incluyen la enfermedad cardiovascular activa o latente, la radioterapia previa o concomitante en el área del mediastino/pericardio, la terapia previa con otras antraciclinas o antracenedionas y el uso concomitante de otros fármacos que pueden suprimir la contractilidad del miocardio o fármacos cardiotóxicos, (p. ej., trastuzumab). Las antraciclinas incluyendo epirubicin no deben ser administradas en combinación con otros agentes cardiotóxicos a menos que la función cardiaca del pacientes sea cercanamente monitoreada (véase la Sección Interacción con otros productos medicinales y otras formas de interacción). Los pacientes que estén recibiendo antraciclinas después de parar el tratamiento con otros agentes cardiotóxicos, especialmente aquellos con vidas medias prolongadas como el trastuzumab, pueden también tener un riesgo incrementado de desarrollar cardiotoxicidad. La vida media informada de trastuzumab es aproximadamente 28-38 días y puede persistir en la circulación por hasta 27 semanas. Por lo tanto, los médicos deben evitar terapia basada en antraciclina por hasta 27 semanas después de suspender el trastuzumab cuando sea posible. Si se usan antraciclinas antes de este tiempo, se recomienda un monitoreo cuidadoso de la función cardiaca.
La vigilancia de la función cardiaca tiene que ser especialmente estricta en pacientes que reciben dosis altas acumulativas y en los que tienen factores de riesgo. Sin embargo, la cardiotoxicidad puede sobrevenir con dosis acumulativas más bajas de epirubicina ya sea que haya o no presencia de factores de riesgo cardiovascular.
Es probable que la toxicidad de la epirrubicina y de otras antraciclinas o antracenedionas sea aditiva.
Toxicidad hematológica: Igual que sucede con otros agentes citotóxicos, la epirrubicina puede producir mielosupresión. Se deberán valorar los perfiles hematológicos antes y durante cada ciclo de terapia con epirubicina, incluido el recuento diferencial de los glóbulos blancos (WBC). Una leucopenia o granulocitopenia (neutropenia) reversible dependiente de la dosis es la manifestación predominante de la toxicidad hematológica de la epirubicina y es la más frecuente toxicidad aguda limitante de la dosis de este fármaco. La leucopenia y la neutropenia suelen ser más graves con los esquemas de dosis altas, alcanzando el nadir en la mayoría de los casos entre los días 10 y 14 después de la administración del medicamento; suelen ser transitorias y los recuentos de GB/neutrófilos regresan a los valores normales en casi todos los casos hacia el día 21. También pueden presentarse trombocitopenia y anemia. Las consecuencias clínicas de la mielosupresión grave incluyen fiebre, infección, sepsis/septicemia, choque séptico, hemorragia, hipoxia tisular o muerte.
Leucemia secundaria: Se ha informado de leucemia secundaria, con o sin una fase preleucémica, en pacientes tratados con antraciclinas, incluida la epirubicina. La leucemia secundaria es más frecuente cuando estos medicamentos se dan en combinación con agentes antineoplásicos que dañan el ADN, cuando los pacientes han sido previamente tratados intensamente con fármacos citotóxicos, o cuando se han aumentado progresivamente las dosis de las antraciclinas. Estas leucemias pueden tener un periodo de latencia de 1 a 3 años. (Véase la Sección Propiedades farmacodinámicas).
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN: La epirubicina por lo general se administra en inyección intravenosa. Se ha encontrado que la administración intravesical en el tratamiento de cáncer de vejiga superficial así como en la profilaxis de la recurrencia del tumor después de la resección transuretral es benéfica. La vía de administración intra-arterial también se ha usado para producir intensa actividad local con menor toxicidad general (véase la Sección Advertencias y precauciones especiales para el uso).
Administración intravenosa (IV): La dosis total de epirubicina por ciclo puede variar conforme a su uso dentro de un régimen específico de tratamiento (p.ej., dado como agente único o en combinación con otros fármacos citotóxicos) y conforme a la indicación.
La epirubicina se debe administrar en equipo de venoclisis (solución salina normal 0,9% o dextrosa al 5%). A fin de reducir al mínimo el riesgo de trombosis o extravasación, el tiempo usual de infusión oscila entre 3 y 20 minutos dependiendo de dosificación y el volumen de la solución para infusión. No se recomienda la inyección en bolo directo debido al riesgo de extravasación, la cual puede sobrevenir aún en presencia de un retorno venoso adecuado cuando se succiona con la aguja (véase la Sección Advertencias y precauciones especiales para el uso).
Regímenes estándar de dosificación inicial: En forma de agente único, la dosis inicial estándar recomendada de epirubicina por ciclo en adultos es de 60-120 mg/m2 de área de superficie corporal. La dosis de inicio recomendada de epirubicina cuando se use como componente de terapia adyuvante en pacientes con cáncer mamario nódulo axilar positivo es de 100 a 120 mg/m2. La dosis total de inicio por ciclo se puede dar en una dosis única o dividida en 2 - 3 días sucesivos. En condiciones de recuperación normal de la toxicidad inducida por fármacos (en especial depresión de la médula ósea y estomatitis), los ciclos de tratamiento se podrían repetir cada 3 a 4 semanas. Si se usa la epirubicina en combinación con otros fármacos citotóxicos cuyas toxicidades posiblemente se superponen, la dosis recomendada por ciclo se debe reducir de conformidad (véase la referencia a la indicación específica).
Regímenes con dosis de inicio altas: Se pueden usar altas dosis de inicio de epirubicina en el tratamiento de cáncer mamario y de pulmón. Cuando se usa como agente único, la dosis alta de inicio recomendada de epirubicina por ciclo en adultos (hasta 135 mg/m2) se debe administrar en el día 1 o en dosis divididas en los días 1, 2, 3, cada 3 a 4 semanas. En la terapia combinada, la dosis alta de inicio recomendada (hasta 120 mg/m2) se debe administrar en el día 1, cada 3 a 4 semanas.
Modificaciones de las dosis
Disfunción renal: Aunque no se pueden hacer recomendaciones específicas de dosificación debido a lo limitado de la información disponible proveniente de pacientes con deterioro de la función renal, se debe considerar el uso de dosis de inicio más bajas en los pacientes que tienen un grado alto de deterioro de la función renal (creatinina sérica >5 mg/dL).
Disfunción hepática: Se recomienda reducir la dosis en pacientes que tienen los siguientes valores en suero:
• Bilirrubina 1,2 a 3 mg/dL o AST 2 a 4 veces el límite superior normal: ½ de la dosis de inicio recomendada
• Bilirrubina > 3 mg/dL o AST > 4 veces el límite superior normal: ¼ de la dosis de inicio recomendada.
Otras poblaciones especiales: Es posible que haya que considerar dar dosis de inicio más bajas o dejar intervalos más largos entre ciclos en el caso de pacientes que han sido previamente tratados de forma intensa o en pacientes que tienen infiltración neoplásica de la médula ósea (véase la Sección Advertencias y precauciones especiales para el uso). En los ancianos se han usado dosis de inicio y regímenes estándar.
SOBREDOSIS: La sobredosificación aguda con epirubicina dará lugar a una mielosupresión severa (principalmente leucopenia y trombocitopenia), efectos tóxicos gastrointestinales (sobre todo mucositis) y complicaciones cardiacas agudas.


