

EPAMIN XR
FENITOINA
Cápsulas de liberación prolongada
Cápsulas, 37,5 mg
Cápsulas, 75 mg
Cápsulas, 150 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
INDICACIONES Y USO
Anticonvulsivante: EPAMIN® XR está indicado para el control de crisis epilépticas tonicoclónicas generalizadas (gran mal), parciales complejas (psicomotoras, del lóbulo temporal) y la prevención y tratamiento de crisis epilépticas que ocurren durante o después de neurocirugía.
Podrían requerirse determinaciones de las concentraciones sérica de fenitoína para ajustes óptimos de la dosis (ver secciones Dosis y administración y Farmacología clínica).
CONTRAINDICACIONES: La fenitoína está contraindicada en pacientes con antecedentes de hipersensibilidad a la fenitoína, sus excipientes u otras hidantoínas. Embarazo. Trastornos hepáticos o hematológicos y falla renal.
La coadministración de fenitoína está contraindicada con delavirdina debido a la posible pérdida de respuesta virológica y posible resistencia a delavirdina o a la clase de inhibidores no nucleosídicos de la transcriptasa inversa.
REACCIONES ADVERSAS
Generales: Se han observado reacciones alérgicas en forma de erupción y en formas raramente más serias (ver Piel y anexos) y RMESS (ver Advertencias). También se ha reportado anafilaxia.
Se han presentado también reportes de engrosamientos de los rasgos faciales, lupus eritematoso sistémico, periartritis nodosa y anomalías de la inmunoglobulina.
Sistema nervioso: Las reacciones adversas en este sistema corporal son frecuentes y usualmente relacionadas con la dosis. Las reacciones incluyen nistagmo, ataxia, dificultad para hablar, disminución de la coordinación, somnolencia y confusión mental. También se han observado mareo, vértigo, insomnio, nerviosismo transitorio, espasmos motores, parestesias y cefaleas. Se han reportado casos raros de discinesias inducidas por fenitoína, incluida córea, distonía, tremor y asterixis, similar a la inducida por fenotiacina y otros neurolépticos.
Una polineuropatía predominantemente sensorial periférica se ha observado en pacientes que han recibido terapia prolongada con fenitoína.
Sistema digestivo: Falla hepática aguda, hepatitis tóxica, daño hepático, vómito, náuseas, estreñimiento, agrandamiento de los labios e hiperplasia gingival.
Piel y anexos: Las manifestaciones dermatológicas algunas veces acompañadas por fiebre incluyen erupciones escarlatatiniformes o morbiliformes. La erupción morbiliforme (similar al sarampión) es la más frecuente; otros tipos de dermatitis se observan más raramente. Otras formas más serias que pueden ser fatales incluyen dermatitis bullosa, exfoliativa o purpúrica, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica. (ver Advertencias). También se han reportado casos de hipertricosis.
Sistema hematológico y linfático: Se han reportado ocasionalmente complicaciones hematopoyéticas, algunas fatales, asociadas con la administración de fenitoína. Estas han incluido trombocitopenia, leucopenia, granulocitopenia, agranulocitosis y pancitopenia con o sin supresión de la médula ósea. Aunque se ha presentado macrocitosis y anemia megaloblástica, estas condiciones usualmente responden a la terapia con ácido fólico. También se ha reportado linfadenopatía incluida hiperplasia benigna de nodo linfático, seudolinfoma, linfoma y enfermedad de Hodgkin (ver sección Advertencias).
Órganos de los sentidos: Alteración del sentido del gusto incluida sabor metálico.
Urogenitales: Enfermedad de Peyronie.
INTERACCIONES MEDICAMENTOSAS: La fenitoína se une extensamente a las proteínas plasmáticas séricas y está propensa a desplazamiento competitivo. La fenitoína se metaboliza por las enzimas hepáticas del citocromo P450, CYP2C9, CYP2C19 y son particularmente sensibles a interacciones medicamentosas inhibitorias debido a que están sujetas al metabolismo saturable. La inhibición del metabolismo puede producir aumentos significativos en las concentraciones circulantes de fenitoína y aumentar el riesgo de toxicidad al medicamento. La fenitoína es un inductor potente de enzimas hepáticas metabolizadores de medicamentos. Las determinaciones de las concentraciones séricas de fenitoína son especialmente útiles cuando se sospechan posibles interacciones medicamentosas.
Las interacciones medicamentosas más frecuentes son las siguientes
Medicamentos que afectan la concentración de fenitoína
• Los medicamentos que pueden aumentar las concentraciones séricas de fenitoína incluyen: Consumo prolongado de alcohol, amiodarona, antiepilépticos (etosuximida, felbamato, oxcarbazepina, metsuximida, topiramato), azoles (fluconazol, ketoconazol, itraconazol, voriconazol), capecitabina cloranfenicol, clordiazepóxido, diazepam, disulfiram, estrógenos, fluorouracilo, fluoxetina, fluvastatina, fluvoxamina, antagonistas de H2 (por ejemplo cimetidina), halotano, isoniazida, metilfenidato, omeprazol, fenotiazinas, salicilatos, sertralina, succinimidas, sulfonamidas (por ejemplo sulfametizol, sulfafenazol, sulfadiazina, sulfametoxazol-trimetroprim), tracrolimus, ticlopidina, tolbutamida, trazodona y warfarina.
• Medicamentos que pueden disminuir las concentraciones de fenitoína, incluyen: antineoplásicos usualmente en combinación (por ejemplo bleomicina, carboplatina, cisplatina, doxorubicina, metotrexate) usualmente como politerapia con carbamazepina, abuso crónico de alcohol, ácido fólico, fosamprenavir, nelfinavir, reserpina, ritonavir, hierba de San Juan, sucralfato y vigabatrina.
• La administración de fenitoína con preparaciones que aumentan el pH gástrico (por ejemplo suplementos o antiácidos que contienen carbonato de calcio, hidróxido de aluminio e hidróxido de magnesio) pueden afectar la absorción de la fenitoína. En la mayoría de los casos donde las interacciones fueron observadas, el efecto fue la disminución de las concentraciones de fenitoína cuando los medicamentos se toman al mismo tiempo. Cuando sea posible, la fenitoína y estos productos no deben tomarse a la misma hora del día.
• Los medicamentos que pueden aumentar o disminuir las concentraciones séricas de fenitoína incluyen: Fenobarbital, valproato sódico y ácido valproico. De manera similar, el efecto de la fenitoína sobre las concentraciones de fenobarbital, ácido valproico y valproato de sodio séricas son impredecibles
• La adición o el retiro de estos medicamentos en pacientes bajo terapia con fenitoína puede requerir ajuste de la dosis de fenitoína para lograr un desenlace clínico óptimo.
Medicamentos afectados por la fenitoína
• Medicamentos que no deben coadministrarse con fenitoína: Delavirdina (ver Contraindicaciones).
• Los medicamentos cuya eficacia se deteriora por la fenitoína incluyen: Azoles (fluconazol, ketoconazol, itraconazol, voriconazol, posaconazol), corticoesteroides, doxiciclina, estrógenos, furosemida, irinotecan, anticonceptivos orales, paclitaxel, paroxetina, quinidina, rifampina, sertralina, teniposida, teofilina y vitamina D.
• Se han reportado aumento y disminución de las respuestas PT/INR cuando la fenitoína se coadminstra con warfarina.
• La fenitoína disminuye las concentraciones plasmáticas de albendazol, algunos antivirales contra el VIH (efavirenz, lopinavir/ritonavir, indinavir, nelfinavir, ritonavir, saquinavir), antiepilépticos (felbamato, topiramato, oxcarbazepina, quetiapina) atorvastatina, ciclosporina, digoxina, fluvastatina, ácido fólico, mexiletina, nisoldipina, praziquantel y simvastatina.
• Cuando se administra la fenitoína con fosamprenavir solo puede disminuir la concentración de amprenavir, el metabolito activo. La fenitoína cuando se administra con la politerapia de fosamprenavir y ritonavir puede aumentar la concentración de amprenavir.
• La resistencia a la acción de bloqueo neuromuscular de los bloqueadores neuromusculares no despolarizantes pancuronio, vecuronio, rocuronio y cisatracurio se ha presentado en pacientes a los que se les ha administrado de manera crónica fenitoína. Se desconoce si la fenitoína tiene el mismo efecto sobre los no polarizantes. Los pacientes deben monitorearse de cerca para recuperación más rápida del bloqueo neuromuscular que la esperada y los requisitos de las tasas de infusión pueden ser mayores.
• La adición o el retiro de la fenitoína durante a terapia concomitante con estos agentes puede requerir ajuste de la dosis de estos agentes para lograr el resultado clínico óptimo.
Interacción con medicamentos y preparaciones de alimentación nutricionales/Enteral/: Los reportes de la literatura sugieren que los pacientes que han recibido preparaciones de nutrición enteral y/o suplementos nutricionales relacionados tienen concentraciones plasmáticas de fenitoínas menores de lo esperado. Por tanto se sugiere que la fenitoína no se debe administrar concomitantemente con una preparación de nutrición enteral. En estos pacientes podría ser necesario el monitoreo más frecuente de las concentraciones séricas de fenitoína.
Interacciones medicamentosas o con pruebas de laboratorio: La fenitoína puede disminuir las concentraciones séricas de T4. También puede producirse valores menores de lo normal para pruebas de dexametasona o metirapona. La fenitoína puede producir incremento de los niveles séricos de glucosa, fosfatasa alcalina, y gamma glutamil transpeptidasa (GGT).
Deberá tenerse cuidado cuando se utilizan métodos inmunoanalíticos para medir las concentraciones plasmáticas de fenitoína.
Carcinogénesis: Para información sobre carcinogénesis ver Advertencias.
Embarazo: Categoría de Embarazo D; ver Advertencias.
Madres lactantes: No se recomienda que las madres lacten a sus hijos si están tomando este medicamento debido a que la fenitoína parece distribuirse a bajas concentraciones en la leche materna.
Uso pediátrico: Ver Dosis y administración.
Utilización geriátrica: La depuración de fenitoína tiende a disminuir con el aumento de la edad (ver Farmacología clínica: Poblaciones especiales).
FARMACOLOGÍA CLÍNICA
Mecanismo de acción: La fenitoína es un antiepiléptico que puede utilizarse para el tratamiento de la epilepsia. El sitio principal de acción parece ser la corteza motora donde se inhibe la propagación de la actividad de las crisis epilépticas. Posiblemente mediante la estimulación del eflujo de sodio desde las neuronas, la fenitoína tiende a estabilizar el umbral contra la hiperexicitabilidad causada por la estimulación excesiva o los cambios ambientales capaces de reducir el gradiente de sodio de la membrana. Esto incluye la reducción de la potenciación postetánica en la sinapsis. La pérdida de la potenciación postetánica evita que el foco cortical de las crisis epilépticas detone en áreas corticales adyacentes. La fenitoína reduce la actividad máxima de los centros del tallo cerebral responsables de la fase tónica de las crisis epilépticas tonicoclónicas (gran mal).
Farmacocinética y metabolismos del medicamento: La vida media plasmática en el ser humano después de la administración oral de la fenitoína, promedia en 22 horas, con un intervalo de 7 a 42 horas. Los niveles terapéuticos en estado estacionario se alcanzan entre 7 a 10 días (5-7 vidas medias) después de iniciado el tratamiento con las dosis recomendadas de 300 mg/día.
Cuando es necesario determinar las concentraciones séricas, deberán obtenerse al menos 5-7 vidas medias después de iniciado el tratamiento, después del cambio de la dosis o de la adición o substracción de otro medicamento al régimen de modo que ya se haya alcanzado el equilibrio de estado estable. Por medio de las concentraciones plasmáticas se provee información acerca del rango de los niveles séricos y el cumplimiento terapéutico y son obtenidos antes de la siguiente dosis programada para el paciente. Las concentraciones máximas indican un umbral individual para la aparición de efectos secundarios relacionados con la dosis y se obtienen en el tiempo de concentración máxima esperada. Para EPAMIN® XR cápsulas, las concentraciones séricas máximas ocurren a las 4 a 12 horas después de la administración.
El control óptimo sin signos clínicos de toxicidad ocurren más a menudo con concentraciones séricas entre 10 y 20 mcg/ml, aunque algunos casos de epilepsia tónicoclónica (gran mal) pueden controlarse con concentraciones séricas más bajas de fenitoína.
En la mayoría de los pacientes que se mantienen con dosis estables, se pueden alcanzar concentraciones séricas estables de fenitoína. Puede presentarse gran variabilidad entre pacientes en las concentraciones séricas de fenitoína con dosis equivalentes. Los pacientes que presenten concentraciones bajas inusuales pueden ser pacientes que no cumplen con el tratamiento o son hipermetabolizadores de fenitoína. Inusualmente las altas concentraciones se deben a enfermedad hepática, variaciones en los alelos de CYP2C9 y CYP2C19 o interacciones medicamentosas que resultan en interferencia metabólica. El paciente con amplias variaciones en las concentraciones plasmáticas de fenitoína, a pesar de las dosis estándar, presentan un problema de dificultad clínica. Las determinaciones de las concentraciones séricas en dichos pacientes pueden ser particularmente útiles. Como la fenitoína presenta alta unión a proteínas, las concentraciones de fenitoína libre pueden alterarse en pacientes cuyas características de unión a las proteínas difieren de lo normal.
La mayoría del medicamento se elimina en la bilis como metabolitos inactivos que son posteriormente absorbidos desde el tubo intestinal y se eliminan en la orina. La eliminación urinaria de la fenitoína y sus metabolitos ocurren particularmente por filtración glomerular pero de manera más importante mediante secreción tubular. Debido a que la fenitoína se hidroliza en el hígado por un sistema de enzimas que se satura a concentraciones plasmáticas altas, dosis incrementales pequeñas pueden aumentar la vida media y producir aumentos muy sustanciales en las concentraciones séricas, cuando se encuentran en un rango superior. Al aumentar la dosis en un 10% podría causar intoxicación debido que la concentración en el estado estacionario puede aumentarse desproporcionadamente.
• Poblaciones especiales
Pacientes con enfermedad renal o hepática: Debido a un aumento en la fracción de la fenitoína no unida a proteína en pacientes con enfermedad renal, hepática o con hipoalbuminemia, la interpretación de la concentración plasmática de fenitoína total debe realizarse con precaución (ver Dosis y administración). Las concentraciones de fenitoína no unida a proteína pueden ser más útiles en estas poblaciones de pacientes.
Edad: La depuración de fenitoína tiende a disminuir a medida que aumenta la edad (20% menos en pacientes mayores de 70 años de edad con relación a la de los pacientes entre 20-30 años de edad). Los requerimientos de dosis de fenitoína son altamente variables y deben individualizarse (ver Dosis y administración).
Sexo y Raza: El sexo y la raza no tiene impacto significativo sobre la farmacocinética de la fenitoína.
Pacientes pediátricos: Inicialmente, 5 mg/kg/día divididos en dos o tres dosis iguales, con posterior individualización de la terapia hasta un máximo de 300 mg diarios. La dosis de mantenimiento diaria recomendada usualmente es de 4 a 8 mg/kg. Los niños mayores de 6 años y adolescentes pueden requerir la dosis mínima de los adultos (300 mg/día).
ADVERTENCIAS
Efectos de la suspensión abrupta: La suspensión abrupta de la fenitoína en pacientes epilépticos puede precipitar el estado epiléptico. Cuando, a criterio del médico, surja la necesidad de reducir o interrumpir la dosis, o sustituir el medicamento por un antiepiléptico alternativo, deberá realizarse de manera gradual. En caso de una reacción alérgica o de hipersensibilidad, podría requerirse una sustitución más rápida por la terapia alternativa. En este caso, la terapia alternativa debe ser un antiepiléptico que no pertenezca a la clase química de hidantoínas.
Conductas e ideas suicidas: Los antiepilépticos (AE), incluido el EPAMIN® XR, aumentan el riesgo de ideas o conductas suicidas en pacientes que toman estos medicamentos para cualquier indicación. Los pacientes tratados con AE para cualquier indicación deben controlarse con relación a la aparición o empeoramiento de la depresión, pensamientos o conductas suicidas o cambios inusuales en el estado de ánimo o la conducta.
Los análisis combinados de 199 ensayos clínicos controlados- placebo (monoterapias y terapias adyuvantes) de 11 diferentes AE mostraron que los pacientes aleatorizados a uno de los AE tenían aproximadamente el doble del riesgo (Riesgo Relativo ajustado 1,8, IC del 95%: 1,2, 2,7) de pensamientos o conductas suicidas en comparación con los pacientes asignados aleatoriamente a placebo. En estos ensayos, los cuales tenían una mediana de duración del tratamiento de 12 semanas, la tasa de incidencia estimada de conductas o ideas suicidas fue de 0.43% entre 27.863 pacientes tratados con un AE en comparación con 0,24% entre 16.029 pacientes tratados con placebo, lo que representó un aumento de aproximadamente un caso de pensamiento o conductas suicidas por cada 530 pacientes tratados. Se presentaron cuatro suicidios en pacientes tratados con el medicamento (AE) en los ensayos clínicos y en ninguno de los pacientes tratados con placebo. P ero el número es demasiado pequeño para permitir conclusiones sobre el efecto del medicamento sobre el suicidio. El aumento del riesgo de pensamientos o conductas suicidas con AE se observó una semana después del inicio del tratamiento con éste y persistió durante todo el tiempo que se evaluó el tratamiento Debido a que la mayoría de los ensayos incluidos en el análisis no se extendieron más allá de 24 semanas, el riesgo de pensamientos o conductas suicidas más allá de las 24 semanas no pudo ser evaluada.
El riesgo de pensamientos o ideas suicidas fue generalmente consistente entre los medicamentos en los datos analizados. El hallazgo del aumento de riesgo con AE de mecanismos de acción variables y sobre un intervalo de indicaciones, sugiere que el riesgo aplica a todos los AE utilizados para cualquier indicación. El riesgo no varía sustancialmente por edad. (5-100 años) en los ensayos clínicos analizados.
La Tabla 1 muestra el riesgo absoluto y relativo por indicación para todos los AE evaluados.
|
Tabla 1. Riesgo por indicación para antiepilépticos en el análisis combinado |
||||
|
Indicación |
Pacientes con Placebo con Eventos Por 1000 Pacientes |
Pacientes con el Medicamento con Eventos Por 1000 Pacientes |
Riesgo Relativo: Incidencia de Eventos en Pacientes con el Medicamento/Incidencia en Pacientes con Placebo |
Diferencia de Riesgos: Pacientes con Medicamento Adicional con Eventos Por 1000 Pacientes |
|
Epilepsia |
1,0 |
3,4 |
3,5 |
2,4 |
|
Siquiátrico |
5,7 |
8,5 |
1,5 |
2,9 |
|
Otro |
1,0 |
1,8 |
1,9 |
0,9 |
|
Total |
2,4 |
4,3 |
1,8 |
1,9 |
El riesgo relativo de pensamientos y conductas suicidas fue mayor en los ensayos clínicos de epilepsia que en los ensayos clínicos de condiciones psiquiátricas u otras condiciones, pero las diferencias de riesgos absolutos fueron similares para las indicaciones epilépticas y psiquiátricas.
Cualquiera que considere prescribir EPAMIN® XR u otro AE debe balancear el riesgo de tener pensamientos y/ o conductas suicidas con el riesgo de no tratar la enfermedad. La epilepsia y muchas otras enfermedades para las que los AE se prescriben están por sí mismas asociadas a morbilidad, mortalidad y aumento del riesgo de pensamientos y conductas suicidas. Si surgen pensamientos o conductas suicidas durante el tratamiento, el médico debe considerar si la aparición de estos síntomas en un determinado paciente se relaciona con la enfermedad que se está tratando.
Los pacientes, sus cuidadores y familiares deben estar informados que los AE aumentan el riesgo de pensamientos e ideas suicidas y se les debe recomendar estar alertas con relación a la aparición o empeoramiento de los signos y síntomas de depresión, cambios inusuales en el estado de ánimo o en la conducta o la aparición de pensamientos, conductas suicidas o pensamientos sobre daño auto infringido. Las conductas preocupantes deben reportarse de inmediato al médico.
Reacciones dermatológicas serias: Se han reportado reacciones dermatológicas serias y algunas veces mortales, incluida necrólisis epidérmica tóxica (NET) y síndrome de Stevens-Johnson (SSJ), con el tratamiento con fenitoína. La aparición de síntomas usualmente ocurre dentro de los primeros 28 días, pero puede ocurrir posteriormente. Deberá interrumpirse EPAMIN® XR cuando surja el primer signo de erupción, a menos que la erupción no esté claramente relacionada con el medicamento. Si los signos o síntomas sugieren SSJ/NET, la utilización del medicamento no deberá reiniciarse y deberá considerarse un tratamiento alterno. Si se presenta erupción, el paciente deberá ser evaluado con relación a los signos y síntomas sistémicos de Reacción Medicamentosa con Eosinofilia y Síntomas Sistémicos (ver Reacción Medicamentosa con Eosinofilia y Síntomas Sistémicos (RMESS)/Hipersensibilidad Multiorgánica).
Los estudios en pacientes con ascendencia China han encontrado una fuerte asociación entre el riesgo de desarrollar SSJ/NET y la presencia de HLA-B*1502, una variante alélica heredada del gen HLA B, en pacientes que utilizan carbamazepina. La evidencia limitada sugiere que HLA-B*1502 puede ser un factor de riesgo de desarrollar SSJ/NET en pacientes de ascendencia asiática que toman otros medicamentos antiepilépticos asociados con SSJ/NET, incluida fenitoína. Deberá considerarse evitar la fenitoína como una alternativa de la carbamazepina en pacientes positivos para HLA-B*1502.
La utilización de la genotipificación de HLA-B*1502 tiene limitaciones importantes y nunca debe sustituirse por la vigilancia clínica apropiada y el manejo de los pacientes. El papel de otros posibles factores en el desarrollo y la morbilidad del SSJ/NET, tales como la dosis del antiepiléptico (AE), el cumplimiento terapéutico, los medicamentos concomitantes, las coomorbilidades y el nivel de control dermatológico no se han estudiado.
Reacción Medicamentosa con Eosinofilia y Síntomas Sistémicos (RMESS)/Hipersensibilidad Multiorgánica: Se ha reportado Reacción Medicamentosa con Eosinofilia y Síntomas Sistémicos (RMESS) conocida también como hipersensibilidad multiorgánica en pacientes que están tomando antiepilépticos incluido EPAMIN® XR. Algunos de estos eventos han sido fatales o potencialmente mortales. La RMESS típicamente, aunque no exclusivamente, se presenta con fiebre, erupción y/o linfadenopatía, asociada con deterioro de otros sistemas u órganos, como hepatitis, nefritis, anomalías hematológicas, miocarditis o miositis, algunas veces semejándose a infección vírica aguda. A menudo se presenta Eosinofilia. Debido a que éste desorden varía en la forma como se presenta, podrían estar comprometidos otros sistemas u órganos no mencionados. Es importante tener en cuenta que las manifestaciones tempranas de hipersensibilidad, como fiebre o linfadenopatía, pueden estar presentes incluso si la erupción es leve o no es evidente. Si los signos y síntomas mencionados anteriormente están presentes, deberá evaluarse de inmediato al paciente. EPAMIN® XR no debe interrumpirse si no puede establecerse la etiología de los signos y síntomas.
Hipersensibilidad: EPAMIN® XR y otras hidantoínas están contraindicadas en pacientes que han experimentado hipersensibilidad a la fenitoína (ver Contraindicaciones). Adicionalmente deberán considerarse alternativas para los medicamentos estructuralmente similares tales como carboxamidas (p. ej., carbamazepina), barbituratos, succinatos y oxazolidindionas (p. ej., trimetadiona) en estos pacientes. De manera similar, si existen antecedentes de reacciones de hipersensibilidad a estos medicamentos estructuralmente similares en el paciente o en miembros cercanos de la familia, deberán considerarse como alternativas a EPAMIN® XR.
Lesión hepática: Se han reportado casos de hepatotoxicidad aguda, incluidos casos poco frecuentes de insuficiencia hepática aguda con el tratamiento de EPAMIN® XR. Estos eventos pueden hacer parte del espectro de RMESS o pueden ocurrir de manera aislada. Otras manifestaciones frecuentes incluyen ictericia, hepatomegalia, elevación de los niveles de transaminasa séricas, leucocitosis y eosinofilia. El curso clínico de la hepatotoxicidad aguda por fenitoína varía desde la recuperación inmediata hasta desenlaces fatales. En estos pacientes con hepatotoxicidad aguda EPAMIN® XR debe interrumpirse inmediatamente y no debe volverse a administrar.
Sistema hematopoyético: Las complicaciones hematopoyéticas, algunas fatales, se han reportado ocasionalmente asociadas con la administración de EPAMIN® XR. Esto ha incluido trombocitopenia, leucopenia, granulocitopenia, agranulocitosis y pancitopenia con o sin supresión de la médula ósea.
Se han reportado numerosos casos que sugieren una relación entre la fenitoína y el desarrollo de linfadenopatía (local o generalizada) incluida hiperplasia benigna en nodos linfáticos, seudolinfoma, linfoma y enfermedad de Hodgkin. Aunque una relación de causa y efecto no se ha establecido, la ocurrencia de linfadenopatía indica la necesidad de diferenciar dicha condición de otros tipos de patologías de nodos linfáticos. El compromiso de los nodos linfáticos puede ocurrir con o sin síntomas y signos de RMESS.
En todos los casos de linfadenopatía, está indicada la observación para seguimiento durante un periodo prolongado y deben realizarse todos los esfuerzos para controlar las crisis epilépticas usando antiepilépticos alternativos.
Efectos sobre la vitamina D y sobre los huesos: La utilización a largo plazo de fenitoína en pacientes con epilepsia se ha asociado con un incremento del riesgo de trastornos de la densidad mineral ósea, que puede conducir al desarrollo de osteopenia, osteoporosis, osteomalacia y fracturas óseas. Durante el tratamiento a largo plazo se recomienda monitorear la densidad mineral ósea del paciente.
La fenitoína induce enzimas metabolizadoras hepáticas. Esto puede mejorar el metabolismo de la vitamina D y disminuir las concentraciones de vitamina D que puedan conllevar a deficiencia de ésta vitamina, hipocalcemia e hipofosfatemia. Deberá considerarse la detección mediante pruebas de laboratorio radiológicas relacionadas con los huesos según sea apropiado e iniciar los planes de tratamiento de acuerdo con las directrices establecidas.
Efectos del consumo de alcohol sobre las concentraciones séricas de fenitoína: El consumo prolongado de alcohol puede aumentar las concentraciones séricas de fenitoína, aunque el consumo crónica del alcohol puede disminuir las concentraciones séricas.
Exacerbación de la porfiria: Debido a que sólo existen informes aislados que asocian la fenitoína con la exacerbación de porfiria, deberá tenerse precaución durante la utilización de este medicamento en pacientes que sufren esta enfermedad.
Utilización durante el embarazo
— Clínicas
Riesgo para la madre: Un aumento en la frecuencia de las crisis epilépticas puede ocurrir durante el embarazo debido a la alteración de la farmacocinética de la fenitoína. La medición periódica de las concentraciones plasmáticas de fenitoína puede ser valiosa para el manejo de las mujeres embarazadas como una guía para el ajuste apropiado de la dosis (ver Precauciones, Pruebas de Laboratorio). Sin embargo, probablemente sea indicado restaurar en el postparto la dosis original. Esto será únicamente, criterio del médico tratante Riesgo para el Feto. Si este medicamento se utiliza durante el embarazo o si la paciente queda embarazada mientras está tomando el medicamento, deberá advertírsele sobre el riesgo para el feto.
La exposición prenatal a la fenitoína puede aumentar el riesgo de malformaciones congénitas y otros desenlaces adversos relacionados con el desarrollo. El aumento de las frecuencias de malformaciones mayores (tales como hendiduras bucofaciales y defectos cardiacos), anomalías menores (características faciales dismórficas, hipoplasia de la uña y el dedo), anomalías del crecimiento (incluida microcefalia) y deficiencia mental se han reportado en niños nacidos de mujeres epilépticas que estaban tomando fenitoína sola o como politerapia con otros antiepilépticos durante el embarazo. Se han reportado varios casos de cánceres, incluido neuroblastoma, en niños cuyas madres recibieron fenitoína durante el embarazo. La incidencia global de malformaciones para niños de mujeres epilépticas tratados con medicamentos antiepilépticos (fenitoína u otros) durante el embarazo es aproximadamente 10% o dos a tres veces el de la población general. Sin embargo, las contribuciones relativas de medicamentos antiepilépticos y otros factores asociados con epilepsia al aumento del riesgo son inciertos y en la mayoría de los casos no se han podido atribuir anomalías específicas del desarrollo a antiepilépticos específicos.
Los pacientes deben consultar con sus médicos para balancear los riesgos y los beneficios de la fenitoína durante el embarazo.
Periodo postparto: Pueden presentarse trastornos hemorrágicos potencialmente mortales relacionados con la disminución de los niveles de factores de coagulación dependientes de la vitamina K en neonatos expuestos a fenitoína en el útero. Esta condición inducida por medicamentos puede prevenirse con la administración de vitamina K a la madre antes del nacimiento y al neonato después del nacimiento.
Preclínicos: Aumento de las tasas de resorción y malformación fueron reportadas después de la administración de dosis de fenitoína de 75 mg/kg o mayores (aproximadamente 120% de la dosis inicial máxima en humanos o mayor en mg/m2) a conejas gestantes.
PRECAUCIONES
Generales: El hígado es el lugar principal de biotransformación de fenitoína; los pacientes con deterioro de la función hepática, pacientes ancianos o los que están gravemente enfermos pueden presentar signos tempranos de toxicidad.
Un porcentaje pequeño de personas que se han tratado con fenitoína han demostrado metabolizar lentamente el medicamento. El metabolismo lento puede deberse a la disponibilidad limitada de enzimas y la falta de inducción; esto parece estar determinado genéticamente. Si los signos iniciales de toxicidad en el SNC relacionados con la dosis se desarrollan, deberán verificarse inmediatamente las concentraciones plasmáticas.
Se ha reportado hiperglicemia como resultado de los efectos inhibitorios del medicamento sobre la liberación de insulina. La fenitoína puede también aumentar el nivel de glucosa sérica en pacientes diabéticos.
La fenitoína no está indicada en crisis epilépticas debidas a causas hipoglucémicas u otras causas metabólicas. Deberán realizarse, según sean indicados, los procedimientos diagnósticos apropiados.
La fenitoína no es eficaz para crisis epilépticas de ausencia (petit mal). Si están presentes crisis epilépticas tonicoclónicas (gran mal) y de ausencia (petit mal), es necesaria la politerapia.
Las concentraciones séricas de fenitoína sostenidas por encima del intervalo óptimo pueden producir estados de confusión denominados “delirios”. “psicosis” o “encefalopatía”, o disfunción cerebelosa raramente irreversible. En consecuencia, ante el primer signo de toxicidad aguda, se recomienda la medición de las concentraciones plasmáticas. La reducción de la dosis de la terapia con fenitoína está indicada si son excesivas las concentraciones plasmáticas; si los síntomas persisten, se recomienda la suspensión (ver Advertencias).
Información para los pacientes: Deberá instruirse a los pacientes para que tomen EPAMIN® XR en la forma prescrita.
Deberá advertirse a los pacientes que están tomando fenitoína, la importancia de adherirse de manera estricta al régimen posológico prescrito y la importancia de informar al médico las condiciones clínicas en las cuales no sea posible tomar el medicamento vía oral de la forma prescrita, por ejemplo debido a cirugía, etc.
Los pacientes deben conocer los síntomas y signos tóxicos de posibles reacciones hematológicas, dermatológicas de hipersensibilidad o hepáticas. Estos síntomas pueden incluir, entre otros, fiebre, dolor de garganta, erupción, úlceras en la boca, facilidad para que se produzcan hematomas, linfadenopatía y hemorragia petequial o purpúrica y en el caso de reacciones hepáticas, anorexia, náuseas/vómito o ictericia. Se debe advertir al paciente que, debido a que los signos y síntomas pueden indicar una reacción seria, deben reportar la ocurrencia inmediatamente al médico. Además, debe advertírsele al paciente que éstos signos y síntomas deben reportarse incluso si son leves o si ocurren después de la utilización prolongada.
Deberá también advertirse a los pacientes sobre la utilización de otros medicamentos o bebidas alcohólicas sin que haya buscado primero asesoría médica.
La importancia de la buena higiene dental debe resaltarse para minimizar el desarrollo de hiperplasia gingival y sus complicaciones.
Deberá informarse a los pacientes, sus cuidadores y familiares que los AE, incluido EPAMIN® XR, pueden aumentar el riesgo de pensamientos y conductas suicidas y debe advertirse sobre la necesidad de estar alerta de aparición o empeoramiento de síntomas de depresión, cambios inusuales en el estado de ánimo o la conducta, o la aparición de ideas o conductas suicidas, o ideas de autoinfringirse daño. Las conductas preocupantes deberán reportarse inmediatamente a los médicos.
No utilice cápsulas que estén decoloradas
Pruebas de Laboratorio: Pueden ser necesarias determinaciones de las concentraciones séricas de fenitoína para obtener ajustes óptimos de la dosis. Las dosis de la fenitoína son usualmente seleccionadas para obtener concentraciones plasmáticas totales terapéuticas de 10 a 20 µg/mL (concentraciones de fenitoína no unida a proteína de 1 a 2µg/mL).
DOSIS Y ADMINISTRACIÓN: Deberán monitorearse las concentraciones séricas cuando se haga el cambio desde EPAMIN® XR (cápsulas de liberación prolongada de fenitoína sódica, USP) a cápsulas de liberación rápida de fenitoína sódica USP y desde sales de sodio a formas ácido libre.
EPAMIN® XR (cápsulas de liberación prolongada de fenitoína sódica, USP) está formulada con la sal de sodio de fenitoína. Debido a que existe aproximadamente un 8% de aumento en el contenido del medicamento con la forma de ácido libre sobre la de sal sódica, podrían requerirse ajustes de la dosis y control de la concentración sérica cuando se cambia desde un producto formulado con ácido libre a un producto formulado con sal sódica y viceversa.
Generales: La dosis debe individualizarse para proporcionar un beneficio máximo. En algunos casos, podrían ser necesarias determinaciones de las concentraciones séricas y sanguíneas para ajustes óptimos de la dosis -la concentración sérica clínicamente eficaz usualmente es 10-20 mcg/mL. Con la dosis recomendada, podría requerirse un periodo de siete a diez días para alcanzar las concentraciones sanguíneas de estado estable con fenitoína y cambios en la dosis (aumento o disminución) no deben realizarse a intervalos menores de siete a diez días.
Dosis en adultos
Dosis dividida diariamente: Los pacientes que no han recibido tratamiento previo pueden iniciar con EPAMIN® XR (cápsulas de liberación prolongada de fenitoína sódica USP) tres veces al día y después ajustar la dosis a los requisitos individuales. Para la mayoría de los adultos, la dosis de mantenimiento satisfactoria será una cápsula tres a cuatro veces al día. Un aumento a dos cápsulas tres veces al día puede realizarse, si se considera necesario.
Dosis una vez al día: En adultos, si se establece el control de las crisis epilépticas cuando se dividen con las dosis de tres veces al día de EPAMIN® XR 100 mg (cápsulas de liberación prolongada de fenitoína sódica USP), podría considerarse una dosis una vez al día de 300 mg de EPAMIN® XR (cápsulas de liberación prolongada de fenitoína sódica USP). Los estudios que comparan la dosis dividas de 300 mg con una única dosis diaria de esta cantidad indicaron que la absorción, concentraciones máximas plasmáticas, semivida biológica, diferencia entre los valores máximo y mínimo y la recuperación en la orina fueron equivalentes. La dosis una vez al día puede ser conveniente para algunos pacientes o para el personal de enfermería de pacientes en instituciones y se diseñó para utilización únicamente con pacientes que requieren esta cantidad diaria del medicamento. El problema mayor en relación con la motivación de los pacientes que no se adhieren al tratamiento puede superarse cuando el paciente puede tomar el medicamento una vez al día. Sin embargo debe recomendarse a los pacientes no olvidar tomar una dosis de manera inadvertida.
Únicamente EPAMIN® XR (cápsulas de liberación prolongada de fenitoína sódica USP) se recomienda para una dosis una vez al día. Las diferencias inherentes en las características de dilución y las tasas de absorción resultantes de fenitoína debido a los diferentes procedimientos de fabricación y/o formas farmacéuticas no permiten dicha recomendación para otros productos de fenitoína. Cuando un cambio en la forma farmacéutica o marca se prescribe, deberán monitorearse cuidadosamente las concentraciones séricas de fenitoína.
Dosis inicial: Algunas autoridades han defendido la utilización de una dosis inicial oral de fenitoína en los adultos que requieren concentraciones de estado estable rápidas y en los casos en que la administración intravenosa no es deseable. Este régimen posológico debe reservarse para pacientes en clínicas u hospitales donde las concentraciones séricas de fenitoína puedan controlarse cuidadosamente. Los pacientes con antecedentes de enfermedad renal o hepática no deben recibir el régimen de carga oral.
Inicialmente, un gramo de EPAMIN® XR (cápsulas de liberación prolongada de fenitoína sódica USP) se divide en tres dosis (400 mg, 300 mg, 300 mg) y administrado a intervalos de dos horas. La dosis de mantenimiento normal se establece dentro de las 24 horas después de la dosis inicial, con determinaciones frecuentes de la concentración sérica.
Posología en poblaciones especiales
Pacientes con enfermedad hepática o renal: Debido a aumento de la fracción de la fenitoína no unida a proteínas en pacientes con enfermedad hepática o renal o en aquellos pacientes con hipoalbuminemia, la interpretación de las concentraciones plasmáticas de fenitoína total, deben realizarse con precaución. La concentración de fenitoína no unida a proteínas puede ser más útil en estas poblaciones de pacientes.
Pacientes adultos mayores: La depuración de fenitoína se disminuye levemente en los pacientes adultos mayores y podrían requerirse dosis menores o menos frecuentes.
Pacientes pediátricos: Inicialmente, 5 mg/kg/día en dos o tres dosis igualmente dividas, con posología subsiguiente individualizada a un máximo de 300 mg diarios. Una dosis de mantenimiento diaria recomendada es usualmente 4 a 8 mg/kg. Los niños mayores de 6 años y adolescentes pueden requerir la dosis para adultos mínima (300 mg/día).
Es posible que la información de prescripción de este producto haya sido revisada y actualizada después de la fecha de impresión del PLM 2016. Para obtener información más actualizada comuníquese con la Dirección Médica de Pfizer S.A.S Teléfono: (1) 6002300 Ext. 2509 Bogotá – Colombia.
EPAMIN® XR 100 mg Cápsulas de Liberación Prolongada (Reg. San. INVIMA 2015M-0015854).
Título del Documento de Producto: EPAMIN® XR 100 mg cápsulas de liberación prolongada.
Versión USPI: LAB-0375-11.0
Fecha de revisión: Septiembre 2013
USPIvLAB-0375-11.0_Col_Sep2013
PFIZER S.A.S.
SOBREDOSIS: Se desconoce la dosis letal en los pacientes pediátricos. La dosis letal en los adultos se estima es de 2 a 5 gramos. Los síntomas iniciales son nistagmo, ataxia y disartria. Otros signos son tremor, hiperreflexia, letargia, dificultad para hablar, náuseas, vómito. El paciente puede tornarse comatoso e hipotenso. La muerte se produce debido a la depresión respiratoria y circulatoria.
Existen variaciones marcadas entre las personas con respecto a las concentraciones plasmáticas de fenitoína donde puede ocurrir toxicidad. El nistagmo o la lateralidad de la mirada, usualmente aparecen a los 20 mcg/mL, la ataxia a los 30 mcg/mL; la disartria y la letargia aparecen cuando la concentración plasmática es mayor de 40 mcg/mL, aunque a concentraciones de hasta 50 mcg/mL no se ha reportado ninguna evidencia de toxicidad. Cuando se ha ingerido hasta 25 veces la dosis terapéutica que produce una concentración sérica mayor de 100 mcg/mL con recuperación completa.
Tratamiento: No se especifica ningún tratamiento ya que no existe antídoto.
La adecuación de los sistemas respiratorio y circulatorio debe observarse cuidadosamente y emplearse medidas de soporte apropiadas. La hemodiálisis puede considerarse ya que la fenitoína no se une completamente a las proteínas plasmáticas. Se ha utilizado exanguinotransfusión en el tratamiento de la intoxicación severa de pacientes pediátricos.
En caso de sobredosis aguda, deberá considerarse la posibilidad de otros depresores del SNC, incluido el alcohol.
DESCRIPCIÓN: La fenitoína sódica es un antiepiléptico. La fenitoína sódica está relaciona con los barbitúricos en su estructura química, pero cuenta con un anillo de cinco miembros. El nombre químico es 5,5-difenil1-2,4-imidazolidinadiona sódica, que tiene la siguiente fórmula estructural:
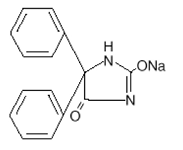
Cada 100 mg de EPAMIN® XR -100 mg (cápsulas de fenitoína sódica de liberación prolongada, USP)- contiene 100 mg de fenitoína sódica. El comportamiento del producto in vivo, se caracteriza por una tasa de absorción lenta y prolongada con una concentración máxima en sangre esperada entre 4 a 12 horas en contraste con las cápsulas de liberación rápida de fenitoína sódica, USP con una tasa de absorción rápida y una concentración máxima en sangre esperada a las 1 ½, a 3 horas.


