DACOGEN
DECITABINA
Vial
Caja, 1 Vial,
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN: Cada frasco-ampolla de vidrio de dosis única de 20 ml contiene 50 mg de decitabina. Después de reconstituir asépticamente 10 mL de agua estéril para inyección, cada mL de la SOLUCIÓN concentrada para infusión contiene 5 mg de decitabina.
PROPIEDADES FARMACOLÓGICAS/PROPIEDADES FARMACODINÁMICAS:
Grupo farmacoterapéutico: Agente antineoplásico e inmunomodulador, Análogo a la pirimidina.
Código ATC: L01BC08.
Mecanismo de acción: La decitabina (5-Aza-2’-desoxicitidina) es un análogo del nucleósido citocina que inhibe selectivamente las metiltransferasas del ADN en dosis bajas, dando lugar a una hipometilación del promotor del gen que puede traducirse en reactivación de los genes supresores tumorales, inducción de diferenciación celular o senescencia celular seguido de muerte celular programada.
Estudios clínicos:
Estudios clínicos en SMD:
Estudio de Fase 2 (DACO-020): Régimen de dosificación de 5 días.
Un estudio a rótulo abierto, de un solo grupo, multicéntrico (DACO-20) se llevó a cabo para evaluar la eficacia del DACOGEN® en pacientes con MDS con cualquiera de los subtipos FAB. En este estudio, 99 pacientes con puntuaciones pronósticas IPSS Intermedia-1, Intermedia-2, o de alto riesgo recibieron DACOGEN® en el régimen de dosificación de 5 días en infusión intravenosa 20 mg/m2 20 mg/m2 en 1hora una vez al día, en los Días 1 a 5 cada 4 semanas (1 ciclo). Los resultados concordaron con los del estudio de Fase 3 y se resumen en la Tabla 2.
|
Tabla2: Eficacia de DACOGEN®en el estudio de fase 2 DACO-020 |
|
|
Parámetro |
DACOGEN®(N=99) |
|
Tasa de respuesta global (CR+mCR+PR) |
33 (33%) |
|
Remisión completa (CR) |
17 (17%) |
|
Remisión medular completa (mCR) |
16 (16%) |
|
Tasa general de mejoría (CR+mCR+PR+HI) |
51 (52%) |
|
HI: Mejoría hematológica |
|
Estudio de Fase 3 (D-0007): Régimen de dosificación de 3 días: En un estudio aleatorizado, a rótulo abierto multicéntrico, controlado (D-0007) se evaluó el DACOGEN® en 170 sujetos con MDS que cumplían los criterios de clasificación FAB y puntuaciones pronósticas IPSS de alto riesgo, Intermedia-2, e Intermedia-1. DACOGEN® se administró en el régimen de dosificación de 3 días en dosis de 15 mg/m2, en infusión intravenosa continua durante 3 horas repetida cada 8 horas durante 3 días consecutivos de cada ciclo de 6 semanas.
En el estudio clínico de Fase 3, las remisiones CR y PR se observaron en todos los subgrupos IPSS. Sin embargo, se hizo evidente un mayor efecto beneficioso en los subgrupos de pacientes clasificados como Int-2 y de alto riesgo; ver la Tabla 3.
|
Tabla 3: Eficacia por IPSS Subgrupo en Estudio D-0007 |
||||
|
Subgrupo IPSS |
DACOGEN® |
Cuidados de soporte |
||
|
Tasa de respuesta global (CR + PR) |
Mediana del tiempo (días) hasta LMA o Muerte |
Tasa de respuesta global (CR + PR) |
Mediana del tiempo (días) hasta LMA o Muerte |
|
|
Todos los pacientes |
15/89 (17%) |
340 |
0/81 |
219 |
|
Int-2 & Alto riesgo |
11/61 (18%) |
335 |
0/57 |
189 |
|
Int-2 |
7/38 (18%) |
371 |
0/36 |
263 |
|
Alto riesgo |
4/23 (17%) |
260 |
0/21 |
79 |
|
LMA= leucemia mieloide aguda; CR= remisión completa; IPSS= Sistema internacional de puntuación pronóstica; Int-2= Intermedia-2; PR= remisión parcial Fuente: D-0007 CSR |
||||
Estudios clínicos en AML: El uso de DACOGEN fue analizado en un estudio clínico abierto, aleatorizado, multicéntrico Fase 3 (DACO-016) en sujetos recientemente diagnosticado de novo o secundario AML de acuerdo a la clasificación de la OMS. DACOGEN (n=242) fue comparado al tratamiento de elección (TC, n=243) el cual consistió en seleccionar pacientes quienes tenía el suporte médico adecuado (n=28, 11.5%) o 20 mg/m2 citarabina subcutánea una vez al día por 10 días consecutivos repetidos cada 4 semanas (n=215, 88.5%). DACOGEN fue administrado como una infusión intravenosa de 1 hora de 20 mg/m2 una vez al día por 5 días consecutivos repetidos cada 4 semanas. El promedio de edad de la población con intención a ser tratada (ITT) fue 73 años (rango 64 a 91 años). El treinta y seis por ciento de los sujetos tenían citogenética de riesgo bajo al inicio del estudio. El resto de los sujetos tenían citogenética. El criterio de valoración principal del estudio fue la supervivencia global. El criterio de valoración secundario fue la tasa de remisión completa que fue evaluada por examen de expertos independientes. La supervivencia libre de progresión y la supervivencia libre de eventos fueron variables terciarias.
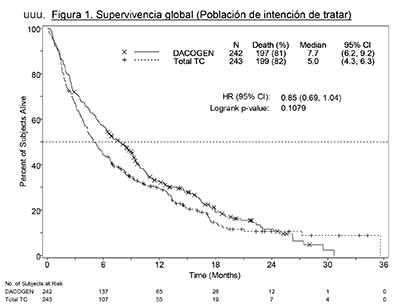
La mediana de la supervivencia global en la población de IDT fue de 7,7 meses en sujetos tratados con DACOGEN® comparada con 5,0 meses para sujetos en el grupo de TC (razón de riesgo [HR] 0,85; IC 95%: 0,69, 1,04, p = 0,1079). Sin embargo, la diferencia no alcanzó significancia estadística; hubo una tendencia hacia la mejoría de la supervivencia con una reducción de 15% en el riesgo de muerte entre los sujetos del grupo de DACOGEN® (Figura 1). Cuando se hizo la censura para terapia subsiguiente potencialmente modificadora de la enfermedad (es decir, quimioterapia de inducción o agente hipometilante) el análisis de la supervivencia global mostró una reducción del 20% en el riesgo de muerte entre los sujetos del grupo de DACOGEN® (HR = 0,80; IC 95%: 0,64; 0,99, valor de p = 0,0437).
Figura 1. Supervivencia globral (Población de intención de tratar)
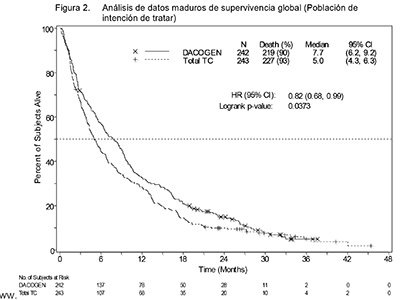
En un análisis con 1 año adicional de datos maduros de supervivencia, el efecto de DACOGEN® sobre la supervivencia global demostró una mejoría clínicamente significativa comparada con el grupo de TC (7,7 meses frente a 5,0 meses, respectivamente, HR = 0,82; IC 95%: 0,68, 0,99, valor nominal de p = 0,0373, Figura 2).
Figura 2. Análisis de datos maduros de supervivencia global
(Población de intención de tratar)
Con base en el análisis inicial en la población de intención de tratar, se alcanzó una diferencia estadísticamente significativa en la tasa de remisión completa (CR+CRp) en favor de los sujetos del grupo de DACOGEN®, 17,8% (43/242) comparada con el grupo de TC, 7,8% (19/243); diferencia del tratamiento 9,9% (IC 95%: 4,07; 15,83), p = 0,0011, La mediana del tiempo hasta la mejor respuesta y la mediana de la duración de la mejor respuesta en pacientes que alcanzaron una CR o CRp fueron de 4,3 meses y 8,3 meses, respectivamente. La supervivencia libre de avance fue significativamente más larga en los sujetos del grupo de DACOGEN®, 3,7 meses (IC 95%: 2,7; 4,6) comparados con los sujetos del grupo de TC, 2.1 meses (IC 95%: 1,9; 3,1); HR = 0,75; IC 95%: 0,62; 0,91, p = 0,0031, Estos resultados, así como otros puntos finales se muestran en la Tabla 4.
|
Tabla 4: Otros puntos finales de eficacia para el estudio DACO-016 (Población de IDT) |
|||
|
Desenlaces |
DACOGEN® |
TC (grupo combinado) |
Valor de p |
|
CR + CRp |
43 (17,8%) |
19 (7,8%) |
0,0011 |
|
OR = 2,5 (1,40; 4,78)b |
|||
|
CR |
38 (15,7%) |
18 (7,4%) |
— |
|
EFSa |
3,5 (2,5; 4,1)b |
2,1 (1,9; 2,8)b |
0,0025 |
|
HR = 0,75 (0,62; 0,90) b |
|||
|
PFSa |
3,7 (2,7; 4,6)b |
2,1 (1,9; 3,1)b |
0,0031 |
|
HR = 0,75 |
|||
|
CR = Remisión completa; CRp = Remisión completa con recuperación incompleta de las plaquetas, EFS = Supervivencia libre de evento, PFS = Supervivencia libre de avance, o = Razón de momio, HR = Razón de riesgo. - = No evaluable. a Informada como mediana de los meses. b Intervalos de confianza del 95%. |
|||
La supervivencia global y la tasa de remisión completa en subgrupos previamente especificados relacionados con la enfermedad (es decir, riesgo genético, puntuación del Eastern Cooperative Oncology Group [ECOG], edad, tipo de LMA, y recuento inicial de blastos en la médula ósea) concordaron con los resultados de la población general del estudio.
El uso de DACOGEN® como terapia inicial también fue evaluado en un estudio a rótulo abierto, de un solo grupo, de Fase 2 (DACO-017) en 55 sujetos >60 años con LMA de acuerdo con la clasificación de la OMS. El punto final primario fue la tasa de CR valorada por revisión de experto independiente. El punto final secundario del estudio fue la supervivencia global. El DACOGEN® se administró como infusión intravenosa en 1 hora de 20 mg/m2 una vez al día durante 5 días consecutivos repetida cada 4 semanas. En el análisis de la población de IDT se observó una tasa de CR de 23,6% (IC 95%: 13,2% a 37%) en 13/55 sujetos tratados con DACOGEN®. La mediana del tiempo hasta la CR fue de 4,1 meses, y la mediana de la duración de la CR fue de 18,2 meses. La mediana de la supervivencia global en la población de intención de tratar fue de 7,6 meses (IC 95%: 5,7, 11,5).
INDICACIONES TERAPÉUTICAS:
DACOGEN® está indicado para: Tratamiento de pacientes adultos con síndromes mielodisplásicos (SMD) de riesgo intermedio-1 y 2 y de alto riesgo, no candidatos a trasplante de médula ósea o a quimioterapia intensiva. Tratamiento de pacientes adultos de 65 y mas años con leucemia mieloide aguda (LMA) de novo o secundaria quienes no son candidatos para quimioterapia de inducción estándar de reciente diagnóstico, de acuerdo con la clasificación de la OMS.
FORMA FARMACÉUTICA: DACOGEN® (decitabina) inyectable es un polvo liofilizado estéril blanco a casi blanco.
PROPIEDADES FARMACOCINÉTICAS: Los parámetros poblacionales de farmacocinética (FC) de la decitabina se obtuvieron de tres estudios clínicos [DACO-017 (n=11), DACO-020 (n=11) y DACO-016 (n=23)] utilizando el régimen de 5 días (20 mg/m2 x 1-hora x 5 días cada 4 semanas) y 1 estudio, DACO-018 (n=12), utilizando el régimen de 3 días (15 mg/m2 x 3-horas cada 8 horas x 3 días cada 6 semanas) en pacientes con MDS o AML. En el régimen de 5 días, la farmacocinética (FC) fue evaluada en el quinto día del primer ciclo de tratamiento. La dosis total por ciclo, fue 100 mg/m2. En el régimen de 3 días, la farmacocinética (FC) de decitabina fue evaluado después de la primera dosis cada día del primer ciclo de tratamiento. La dosis total por ciclo fue 135 mg/m2.
Distribución: La farmacocinética de la decitabina después de la administración intravenosa de una hora (Régimen de 5 días) o 3 horas de infusión (Régimen de 3 días) fue descrita por un modelo lineal de dos compartimentos, caracterizada por la rápida eliminación del fármaco desde el compartimento central y por la distribución relativamente lenta desde el compartimento periférico.
Para un paciente tipo (peso 70 kg / área de superficie corporal 1,73m2) los parámetros FC de la decitabina se enumeran en la tabla 5.
|
Tabla 5: Resumen del análisis poblacional de FC de un paciente tipo (Régimen de 5 y 3 días) |
||||
|
Parámetro |
Régimen de 5 días |
Régimen de 3 días |
||
|
Valor establecido |
95% CI |
Valor establecido |
95% CI |
|
|
Cmáx (ng/mL) |
107 |
88.5 - 129 |
42.3 |
35.2 - 50.6 |
|
Cmáx (ng/mL) |
107 |
88.5 - 129 |
42.3 |
35.2 - 50.6 |
|
AUCcum (ng.h/mL) |
580 |
480 - 695 |
1161 |
972 - 1390 |
|
t1/2 (min) |
68.2 |
54.2 - 79.6 |
67.5 |
53.6 - 78.8 |
|
Vdss (L) |
116 |
84.1 - 153 |
49.6 |
34.9 - 65.5 |
|
CL (L/h) |
298 |
249 - 359 |
201 |
168 - 241 |
|
AUC= área bajo la curva concentración plasmática-tiempo. |
||||
La decitabina presenta FC lineal después de la infusión intravenosa, en estado estacionario las concentraciones se alcanzan en 0,5 horas. Basado en el modelo de simulación, los parámetros FC fueron independientes del tiempo (e.d., no ha cambiado de ciclo a ciclo) y no se observó acumulación con este régimen de dosificación. La unión a proteínas plasmáticas de decitabina es insignificante (<1%). La El Vdss de la decitabina en pacientes con cáncer es un indicativo de la gran distribución de la droga en los tejidos periféricos. No hubo evidencia de la dependencia de la edad, depuración de creatinina, bilirrubina total o enfermedad.
Metabolismo: Dentro de las células, la decitabina se activa a través de fosforilación mediante actividades secuencial de fosfoquinasa al correspondiente trifosfato, el cual luego es incorporado por la polimerasa del ADN. A la luz de los datos de metabolismo in vitro, los resultados del estudio de balance de masas en humanos indicaron que el sistema del citocromo P450 no está involucrado en el metabolismo de la decitabina. La vía primaria del metabolismo probablemente pasa por la desaminación por la desaminasa de citidina en el hígado, riñón, epitelio intestinal y sangre.
Los resultados del estudio de balance de masas en humanos mostraron que la decitabina intacta en el plasma representada aproximadamente por el 2,4% del total radiactivo en plasma no parece ser farmacológicamente activo. Los principales metabolitos circulantes no se cree que sean farmacológicamente activos. La presencia de estos metabolitos en orina junto con la alta depuración corporal total y la baja excreción urinaria de medicamento intacto en la orina (~4% de la dosis) indican que la decitabina se metaboliza en grado apreciable in vivo. Además, los datos in vitro muestran que decitabina es un pobre sustrato P-gp.
Eliminación: La depuración plasmática promedio luego de la administración intravenosa en sujetos con cáncer fue de 200 L/h con moderada variabilidad inter-sujeto (Coeficiente de variación [CV] es aproximadamente 50%). La excreción de medicamento intacto parece desempeñar tan solo un papel menor en la eliminación de la decitabina.
Los resultados de un estudio de balance de masas con 14C-decitabine radiactiva en pacientes con cáncer mostraron que 90% de la dosis de decitabina (4% de droga intacta) es excretada en la orina.
Poblaciones especiales: Los efectos de la insuficiencia renal o hepatica, género, edad o raza sobre la farmacocinética de la decitabina no se han estudiado formalmente. La información en poblaciones especiales se deriva de los datos farmacocinéticos de 4 estudios señalados.
Ancianos: El análisis farmacocinético poblacional mostró que la FC de la decitabina no depende de la edad (rango estudiado de 40 a 87 años, promedio 70 años).
Insuficiencia hepática: La farmacocinética de la decitabina no se ha estudiado formalmente en pacientes con insuficiencia hepática. Los resultados de un estudio de balance de masas en humanos y los experimentos in vitro como se mencionó anteriormente, indicaron que las enzimas CYP es poco probable que participen en el metabolismo de la decitabina. Además, la escasez de datos a partir del análisis farmacocinético poblacional indicaron que no había dependencia FC significativa sobre la concentración de bilirrubina total a pesar de una amplio rango gama de niveles de bilirrubina total.
En consecuencia, la exposición a la decitabina no parece que sea afectada en pacientes con insuficiencia hepática.
Insuficiencia renal: La FC de la decitabina no ha sido formalmente estudiada en pacientes con insuficiencia renal. El análisis FC poblacional sobre el límite de decitabina indicaron que no hay dependencia significativa del parámetro de FC sobre la depuración normalizada de creatinina, un indicador de la función renal.Por lo tanto, la exposición a la decitabina no parece ser afectada en pacientes con insuficiencia renal
Otras poblaciones:
Género: El análisis FC poblacional de la decitabina no mostró diferencias clínicamente relevantes entre hombres y mujeres
Raza: La mayoría de los pacientes estudiados eran de raza blanca. Sin embargo, el análisis farmacocinético poblacional de la decitabina indica que la raza no tuvo ningún efecto aparente sobre la exposición a la decitabina.
CONTRAINDICACIONES: Hipersensibilidad conocida a la decitabina o alguno de los excipientes.
EMBARAZO Y LACTANCIA:
Advertencias especiales y precauciones para el uso:
Mielosupresión: La mielosupresión y complicaciones de mielosupresión, incluyen infecciones y sangrado que se presentan en pacientes con SMD o LMA se pueden exacerbar con el tratamiento. La mielosupresión causada por DACOGEN® es reversible. Se deben realizar regularmente recuentos completos de sangre y plaquetas según la indicación clínica y previo a cada ciclo de tratamiento. En la presencia de mielosupresión o sus complicaciones, el tratamiento con DACOGEN® se debe interrumpir, reducir la dosis o instaurar medidas de soporte como se recomienda en las secciones: Posología y Método de Administración y Efectos indeseables.
Disfunción hepática: No se ha establecido el uso de DACOGEN® en pacientes con deterioro de la función hepática. Se deberá tener precaución con la administración de DACOGEN® a los pacientes que tienen deterioro de la función hepática y se deberá ejercer una estrecha vigilancia (ver Posología y Método de Administración y Propiedades Farmacocinéticas).
Insuficiencia renal: No se ha estudiado el uso de DACOGEN en pacientes con insuficiencia renal grave. Se debe tener cuidado en la administración de DACOGEN a pacientes con insuficiencia renal grave (depuración de creatinina [CrCl] <30 ml/min) y estos pacientes se deben monitorear de cerca (ver Posología).
Enfermedad cardiáca: Pacientes con historia de falla cardiaca severa congestiva o enfermedad cardiaca clínicamente inestable fueron excluidos de los estudios clínicos y por lo tanto la seguridad y eficacia de DACOGEN® en estos pacientes no han sido establecidas.
EMBARAZO Y LACTANCIA:
Uso durante el embarazo: Se deberá insistir a las mujeres con posibilidades de concebir que usen efectivas medidas anticonceptivas y que eviten quedar embarazadas durante el tratamiento con DACOGEN®. Se desconoce el periodo después de tratamiento con DACOGEN® donde es seguro quedar embarazada No hay datos adecuados sobre el uso de DACOGEN® en mujeres embarazadas. Estudios han mostrado que la decitabina es teratogénica en ratas y ratones. El riesgo potencial en humanos es desconocido. Basados en resultados de estudios realizados en animales y su mecanismo de acción, DACOGEN® no debe ser usado durante el embarazo, a menos que sea claramente necesario. Si esta droga es usada durante el embarazo, o si una paciente queda embarazada mientras recibe DACOGEN®, la paciente debe ser advertida sobre los posibles riesgos para el feto.
Uso en hombres: Se deberá advertir a los varones que no conciban mientras que reciben el DACOGEN®, y después de 3 meses de la terminación del tratamiento.
Fertilidad: Se debe advertir a pacientes mujeres con potencial para dar a luz que busquen una consulta respecto a la criopreservación de ovicitos antes de iniciar el tratamiento con DACOGEN®. A causa de la posibilidad de infertilidad como consecuencia de la terapia con DACOGEN®, los varones deben ser aconsejados de consultar sobre la conservación de espermatozoides antes del tratamiento.
Uso durante la lactancia: Se desconoce si decitabina o sus metabolitos se excretan por la leche materna. El DACOGEN® está contraindicado durante la lactancia; por eso, si se necesita el tratamiento con DACOGEN®, se deberá suspender la lactancia (ver Contraindicaciones).
EFECTOS SOBRE LA HABILIDAD PARA CONDUCIR:
No se han realizado estudios de los efectos sobre la habilidad para conducir o usar máquinas con DACOGEN®. Los pacientes deben ser advertidos que ellos pueden experimentar efectos indeseables, como anemia, durante el tratamiento. Por lo tanto, se recomienda precaución cuando se conduzca un carro u opere una máquina
REACCIONES ADVERSAS:
INCOMPATIBILIDADES: En ausencia de estudios de compatibilidad, este producto medicinal no se debe mezclar con otros productos medicinales. El DACOGEN® no se debe infundir por la misma vía intravenosa con otros productos medicinales.
INTERACCIONES CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN: No han sido realizados estudios formales de interacciones medicamentosas con decitabina.
Existe el potencial para una interacción fármaco-fármaco con otros agentes que también son activados por fosforilación secuencial (por actividades de fosfoquinasa intracelular) y/o metabolizados por enzimas implicadas en la inactivación de decitabina (ej. citidina deaminasa). Por lo tanto, se debe tener precaución si estos fármacos se combinan con DACOGEN®.
Impacto de los fármacos administrados conjuntamente con decitabina: Las interacciones medicamentosas no están mediadas por el metabolismo de la CYP-450 como el metabolismo de la decitabina no está mediado por este sistema sino por la deaminación oxidativa.
El desplazamiento de decitabina de su unión a las proteínas plasmáticas por fármacos administrados conjuntamente es improbable dado que la unión a las proteínas plasmáticas in vitro no es significativa (< 1%) de decitabina.
Los datos in vitro indicaron que la decitabina es un mal sustrato de la glucoproteína P (P-gp) y por tanto no está predispuesto a interacciones con inhibidores de la P-gp.
Impacto de decitabina sobre los fármacos administrados conjuntamente: Dada su baja unión a las proteínas plasmáticas in vitro (< 1%), es poco probable que la decitabina desplace fármacos administrados conjuntamente de su unión a las proteínas plasmáticas. Los estudios in vitro muestran que decitabina no inhibe ni induce las enzimas CYP 450 más de 20 veces la concentración plasmática máxima (Cmax) terapéutica observada. Por lo tanto, no se esperan interacciones medicamentosas mediadas por CYP y es improbable que haya interacción con agentes metabolizados por estas vías.
La decitabina es un inhibidor débil del transporte mediado por P-gp in vitro y por tanto tampoco se espera que afecte el transporte mediado por P-gp de los medicamentos administrados conjuntamente (ver la sección de Propiedades Farmacocinéticas).
DATOS DE ESTUDIOS CLÍNICOS: A lo largo de esta sección, se informaron reacciones adversas. Las reacciones adversas son eventos adversos que se consideraron razonablemente relacionados con el uso de DACOGEN® en base a la evaluación integral de la información de eventos adversos disponible. No se puede establecer una relación causal con DACOGEN® de manera confiable en casos individuales. Además, debido a que los ensayos clínicos se realizan bajo condiciones ampliamente variables, los índices de reacciones adversas observadas en los ensayos clínicos de una droga no se pueden comparar directamente con los índices en los ensayos clínicos de otra droga y pueden no reflejar los índices observados en la práctica clínica.
Las reacciones adversas más importantes y frecuentes en los dos regímenes de 5 días y de 3 días son la mielosupresión y las que sobrevienen como consecuencia de la mielosupresión.
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN: DACOGEN® debe ser administrado bajo la supervisión de médicos experimentados en el uso de agentes quimioterapéuticos.
Posología: Hay 2 regímenes recomendados para la administración de DACOGEN®. El régimen de dosificación en el tratamiento de SMD de 3 o 5 días Con cualquiera de los dos se recomienda que los pacientes sean tratados por un mínimo de 4 ciclos; sin embargo, puede tardar más de 4 ciclos para obtenerse una respuesta. En el SMD el tiempo promedio de respuesta (RC+RP) en estudios de Fase 2 con el régimen de dosificación de 5 días, fue de 3.5 ciclos. En estudio de Fase 3 en el régimen de dosificación de 3 días, el tiempo promedio para respuesta fue de 3 ciclos de tratamiento. El tratamiento se puede continuar tanto tiempo como el paciente muestre respuesta, continúe beneficiándose o presente enfermedad estable, e.d., en ausencia de avance manifiesto.
Si después de 4 ciclos, los valores hematológicos del paciente (p.ej., recuentos de plaquetas o recuento absoluto de neutrófilos, no han regresado a los niveles previos al tratamiento o si se presenta el avance de la enfermedad (el recuento de blastos periféricos va en aumento o recuento de blastos en la médula ósea está empeorando), se puede considerar que el paciente no es un respondedor y se deberán ponderar opciones terapéuticas alternativas al DACOGEN®.
La pre-medicación para la prevención de náuseas y vómito no es recomendada rutinariamente, pero puede ser administrada si se requiere
Régimen de tratamiento en Leucemia Mieloide Aguda: En un ciclo de tratamiento, DACOGEN® se administra a una dosis de 20 mg/m2 de superficie corporal en infusión intravenosa en 1 hora repetida diariamente durante 5 dias consecutivos (es decir, un total de 5 dosis por ciclo). La dosis diaria total no puede superar los 20 mg/m2 y la dosis total por ciclo de tratamiento no puede superar los 100 mg/m2. El ciclo se debe repetir cada 4 semanas dependiendo de la respuesta clínica del paciente y la toxicidad observada. Si se omite una dosis, se deberá reanudar el tratamiento lo más pronto posible. Este régimen se puede administrar en modalidad ambulatoria.
Régimen de tratamiento en síndrome mielodisplásico:
Régimen de dosificación de 3 días: En un ciclo único de tratamiento, DACOGEN® se administra por 3 días consecutivos a una dosis fija de 15 mg/m2 de superficie corporal durante un periodo de 3 horas cada 8 horas (es decir, un total de 9 dosis por ciclo de tratamiento). Este ciclo se repite cada 6 semanas aproximadamente dependiendo de la respuesta clínica del paciente y la toxicidad observada. La dosis diaria total no debe exceder 45 mg/m2 y la dosis total por ciclo de tratamiento no debe exceder 135 mg/m2. Si una dosis se olvida, el tratamiento se debe reanudar tan pronto como sea posible.
Régimen de dosificación de 5 días en SMD: En un ciclo de tratamiento, DACOGEN® se administra a una dosis de 20 mg/m2 de superficie corporal en infusión intravenosa en 1 hora repetida diariamente durante 5 dias consecutivos (es decir, un total de 5 dosis por ciclo). La dosis diaria total no puede superar los 20 mg/m2 y la dosis total por ciclo de tratamiento no puede superar los 100 mg/m2. El ciclo se debe repetir cada 4 semanas dependiendo de la respuesta clínica del paciente y la toxicidad observada. Si se omite una dosis, se deberá reanudar el tratamiento lo más pronto posible. Este régimen se puede administrar en modalidad ambulatoria.
Manejo de la mielosupresión y complicaciones asociadas: La mielosupresión y eventos adversos relacionados a la mielosupresión (trombocitopenia, anemia, neutropenia, y neutropenia febril) son frecuentes en pacientes con SMD tratados y no tratados. Las complicaciones de la mielosupresión incluyen infecciones y sangrado. El tratamiento puede ser modificado en pacientes que experimenten mielosupresión y las complicaciones asociadas que se describen abajo:
En LMA: El tratamiento se puede retrasar a discreción del médico tratante si el paciente experimenta complicaciones asociadas con la mielosupresión, como las que se describen a continuación:
• Neutropenia febril (temperatura ≥38.5°C y recuento absoluto de neutrófilos <1,000/µL).
• Infección viral, bacteriana o micótica activa (es decir, que requiere anti-infecciosos por vía intravenosa o cuidados de soporte costosos).
• Hemorragia (gastrointestinal, genitourinaria, pulmonar con plaquetas <25,000/ µL o cualquier hemorragia del sistema nervioso central).
En SMD:
Régimen de dosificación de 5 días: No es recomienda la reducción de la dosis en este escenario clínico para optimizar el beneficio para el paciente, las dosis deben ser retrasadas como sigue:
Modificaciones del régimen de dosificación en los primeros 3 ciclos: Durante los primeros ciclos de tratamiento las citopenias Grados 3 y 4 son frecuentes y pueden no representar el avance de la MDS. Las citopenias previas al tratamiento pueden no mejorar hasta después del ciclo 3.
Durante los primeros 3 ciclos, para optimizar el beneficio para el paciente en el escenario de neutropenia moderada (recuento absoluto de neutrófilos < 1000/µL), se debe hacer todo lo posible por mantener el tratamiento con dosis completas con el intervalo estándar entre ciclos de tratamiento. La profilaxis antimicrobiana concomitante de acuerdo con los lineamientos institucionales se puede administrar hasta la recuperación de los granulocitos hasta arriba de 500/µL. Los médicos también deben considerar la necesidad de la administración precoz de factores de crecimiento durante este tiempo para la prevención o el tratamiento de las infecciones en pacientes con MDS.
De manera similar, para optimizar el beneficio para el paciente en el escenario de trombocitopenia moderada (recuento de plaquetas <25.000/µL), se deberá hacer todo lo posible por mantener el tratamiento con dosis completas con el intervalo estándar entre ciclos de tratamiento con administración concomitante de transfusiones de plaquetas en caso de eventos hemorrágicos.
Modificaciones del régimen de dosificación después del ciclo 3: Se deberá retrasar la dosis en caso de que se considere que las siguientes toxicidades están por lo menos posiblemente relacionadas con el tratamiento:
• Complicaciones asociadas con mielosupresión grave (infecciones que no se resuelven con terapia antiinfecciosa adecuada, hemorragia que no se resuelve con tratamiento adecuado)
• Mielosupresión prolongada definida como una médula hipocelular (celularidad de 5% o menos) sin evidencia de avance de la enfermedad durante 6 semanas o más después del comienzo de un curso de terapia.
Si la recuperación (recuento absoluto de neutrófilos >1.000/µL y plaquetas >50.000/µL) toma más de 8 semanas, el paciente deberá suspender el tratamiento con el medicamento y valorar el avance de la enfermedad (mediante mielograma) dentro de los 7 días siguientes al final de las 8 semanas. En pacientes que han recibido tratamiento durante por lo menos 6 ciclos, y que siguen derivando beneficio de la terapia, se puede permitir una demora prolongada más allá de 8 semanas, en ausencia de avance, a discreción del médico tratante.
Régimen de dosificación de 3 días:
Modificaciones del régimen de dosificación en los primeros 3 ciclos: Durante los primeros ciclos de tratamiento las citopenias Grados 3 y 4 son frecuentes y pueden no representar el avance de la MDS. Las citopenias previas al tratamiento pueden no mejorar hasta después del ciclo 3.
Durante los primeros 3 ciclos, para optimizar el beneficio para el paciente en el escenario de neutropenia moderada (recuento absoluto de neutrófilos < 1000/µL), se debe hacer todo lo posible por mantener el tratamiento con dosis completas con el intervalo estándar entre ciclos de tratamiento. La profilaxis antimicrobiana concomitante de acuerdo con los lineamientos institucionales se puede administrar hasta la recuperación de los granulocitos hasta arriba de 500/µL. Los médicos también deben considerar la necesidad de la administración precoz de factores de crecimiento durante este tiempo para la prevención o el tratamiento de las infecciones en pacientes con MDS.
De manera similar, para optimizar el beneficio para el paciente en el escenario de trombocitopenia moderada (recuento de plaquetas <25.000/µL), se deberá hacer todo lo posible por mantener el tratamiento con dosis completas con el intervalo estándar entre ciclos de tratamiento con administración concomitante de transfusiones de plaquetas en caso de eventos hemorrágicos.
Modificaciones de la dosis después del ciclo 3:
• Si recuperación hematológica (recuento absoluto de neutrófilos >1.000/µL y plaquetas >50.000/µL) de un ciclo de previo de tratamiento con DACOGEN®, en donde la citopenia persistente se considera relacionada con la administración del medicamento, requiere más de 6 semanas, entonces el siguiente ciclo de la terapia con DACOGEN® se debe demorar y reducir la dosis de acuerdo con el algoritmo que se presenta más adelante. Todas las reducciones de la dosis que se hagan deberán permanecer válidas durante toda la quimioterapia; no debe haber nuevo aumento de la dosis.
• Si la recuperación toma más de 6 semanas, pero menos de 8 semanas - la dosis de DACOGEN® se debe demorar hasta por 2 semanas y reducir la dosis a 11 mg/m2 cada 8 horas (33 mg/m2/día, 99 mg/m2/ciclo) una vez se reanuda la terapia.
• Si la recuperación toma más de 8 semanas, pero menos de 10 semanas - la dosis de DACOGEN® se debe demorar hasta por 2 semanas más y reducir la dosis a 11 mg/m2 cada 8 horas (33 mg/m2/día, 99 mg/m2/ciclo) una vez se reanuda la terapia, luego mantenerla en los siguientes ciclos según indicación clínica.
• Si la recuperación toma más de 10 semanas: Se deberá suspender el tratamiento con el medicamento y valorar el avance de la enfermedad (mediante mielograma) dentro de los 7 días siguientes al final de las 10 semanas. Sin embargo, en pacientes que han recibido tratamiento durante al menos 6 ciclos, y que siguen derivando beneficio de la terapia, se puede permitir una demora prolongada más allá de las 10 semanas, en ausencia de avance, a discreción del médico tratante.
Poblaciones especiales:
Pacientes pediátricos: No ha sido establecida la seguridad y efectividad en pacientes pediátricos.
Disfunción hepática: No se han realizado estudios en pacientes con disfunción hepática.
La necesidad de ajuste de la dosis en pacientes con enfermedad hepática no ha sido evaluada. Si ocurre empeoramiento de la función, los pacientes deben ser monitorizados cuidadosamente. (ver Advertencias especiales y precauciones para el uso y Propiedades Farmacocinéticas).
Disfunción renal: No se han realizado estudios en pacientes con disfunción renal, sin embargo, los datos de los estudios clínicos que incluyeron pacientes con insuficiencia leve-moderada no indicaron necesidad de ajuste de la dosis. Los pacientes con insuficiencia renal grave fueron excluidos de estas pruebas (ver Propiedades Farmacocinéticas).
Método de administración: DACOGEN® es administrado por infusión intravenosa. No se requiere catéter central venoso. Para instrucciones sobre reconstitución y dilución del producto medicinal antes de la administración, ver Instrucciones para uso, manejo y disposición final.
SOBREDOSIS: No hay experiencia directa con sobredosis en seres humanos ni existe un antídoto específico. Sin embargo, los datos de los estudios clínicos iniciales en la bibliografía publicada en dosis más de 20 veces que las dosis terapéuticas actuales, informan un aumento de la mielosupresión incluyendo neutropenia y trombocitopenia prolongadas. Es probable que la toxicidad s manifieste como exacerbaciones de reacciones adversas, sobre todo mielosupresión. El tratamiento de la sobredosis debe ser de soporte.
PRESENTACIÓN: DACOGEN® (decitabina) inyectable se suministra como un polvo estéril liofilizado blanco a casi blanco, en un frasco ampolla de dosis única, acondicionado en cajas de 1 frasco ampolla, caja por 1 vial (Reg. San No. INVIMA 2008M-0008815).
JANSSEN-CILAG, S. A.
Av. Calle 26 No. 69-76 - Edificio Elemento
Torre 2 - Piso 11 - PBX: (+57) 1 9271200 - Bogotá, D. C.
PRECAUCIONES ESPECIALES PARA EL ALMACENAMIENTO: Almacenar en su envase y empaque original a una temperatura entre 2°C-8°C. Después de la reconstitución: A menos que se use dentro de los 15 minutos de la reconstitución, la solución diluida se tiene que preparar usando líquidos de infusión fríos (2°C a 8°C) y almacenarla a temperatura de 2°C a 8°C durante máximo 4 horas hasta su administración.
Manténgase fuera del alcance de los niños
Instrucciones para uso, manejo y disposición final: Este producto medicinal es para un solo uso.
Se debe evitar el contacto de la piel con la solución y usar guantes protectores. Se deben seguir los procedimientos estándar para el manejo de agentes anticancerosos.
El DACOGEN® se debe reconstituir asépticamente con 10 mL de agua estéril para inyección. Luego de la reconstitución, cada mL contiene aproximadamente 5.0 mg de decitabina a pH 6,7 a 7,3.
Inmediatamente después de la reconstitución, la solución se debe diluir aún más con solución de cloruro de sodio al 0,9% para inyección o dextrosa al 5% para inyección hasta una concentración final del medicamento de 0,1 a 1,0 mg/mL.
A menos que se use dentro de los 15 minutes siguientes a la reconstitución, la solución diluida se tiene que preparar usando líquidos de infusión fríos (2°C a 8°C) y almacenarla a 2°C a 8°C durante un máximo de 4 horas hasta su administración.
Todo producto sin usar o material de desecho se tiene que disponer de conformidad con los requisitos locales.