

CELEBREX
CELECOXIB
Cápsulas duras
1 Caja, 10 Cápsulas, 200 mg
1 Caja, 20 Cápsulas, 200 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN:
Cada CÁPSULA de 200 mg contiene:
Celecoxib 200 mg
INDICACIONES TERAPÉUTICAS:
Particularidades clínicas:
Analgésico y antiinflamatorio.
PROPIEDADES FARMACÉUTICAS:
Propiedades farmacológicas:
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: Coxibs M01AH.
El mecanismo de acción de celecoxib es por inhibición de la síntesis de prostaglandina, sobre todo mediante la inhibición de la COX-2. En concentraciones terapéuticas en humanos, celecoxib no inhibe la ciclooxigenasa 1 (COX-1). COX-2 se induce como respuesta a los estímulos inflamatorios. Esto conduce a la síntesis y acumulación de prostanoides inflamatorios, en especial prostaglandina E2, que causa inflamación, edema y dolor. Celecoxib actúa como agente antiinflamatorio, analgésico y antipirético en modelos animales mediante el bloqueo de la producción de prostanoides inflamatorios a través de la inhibición de COX-2. En modelos tumorales de colon en animales, celecoxib redujo la incidencia y multiplicidad de tumores.
Estudios in vivo y ex vivo muestran que celecoxib tiene una afinidad muy baja por la enzima COX-1 que se expresa en forma constitutiva. Por consiguiente, en dosis terapéuticas celecoxib no tiene efecto en los prostanoides sintetizados por la activación de COX-1, por lo que no interfiere con los procesos fisiológicos normales relacionados con COX-1 en los tejidos, sobre todo en el estómago, intestino y plaquetas.
Estudios clínicos:
Osteoartritis (OA): Celecoxib mostró una disminución significativa del dolor articular en comparación con el placebo. Celecoxib se evaluó para el tratamiento de los signos y síntomas de OA de rodilla y cadera en cerca de 4200 pacientes en estudios clínicos controlados con placebo y con producto activo de hasta 12 semanas de duración. En los pacientes con OA, el tratamiento con celecoxib 100 mg dos veces al día y/o 200 mg una vez al día produjo mejoría en el índice de osteoartritis WOMAC (Western Ontario and McMaster Universities), un conjunto de dolor, rigidez y medidas funcionales en la OA. En tres estudios de 12 semanas sobre el dolor que acompaña a la exacerbación de OA, celecoxib en dosis de 100 mg dos veces al día o 200 mg dos veces al día produjeron una disminución significativa del dolor 24 a 48 horas después de iniciar la dosis. Con dosis de 100 mg dos veces al día o 200 mg dos veces al día la eficacia de celecoxib fue similar a la de naproxeno 500 mg dos veces al día. Las dosis de 200 mg dos veces al día no brindan beneficio adicional al observado con 100 mg dos veces al día. Se mostró que una dosis diaria total de 200 mg tiene la misma efectividad sin importar que se administre como 100 mg dos veces al día o 200 mg una vez al día.
Artritis reumatoide (RA): Celecoxib mostró una disminución significativa en la sensibilidad /dolor articular y en la inflamación articular en comparación con el placebo. Celecoxib se evaluó en el tratamiento de los signos y síntomas de RA con cerca de 2100 pacientes en estudios clínicos controlados con placebo y con compuesto activo de hasta 24 semanas de duración. En estos estudios se mostró que celecoxib es superior al placebo mediante el uso del Índice ACR20, una herramienta compuesta de medidas clínicas, de laboratorio y funcionales en RA. Las dosis de celecoxib 100 mg dos veces al día y 200 mg dos veces al día tuvieron eficacia similar y ambas fueron comparables con naproxeno 500 mg dos veces al día.
Aunque celecoxib 100 mg dos veces al día y 200 mg dos veces al día tienen eficacia general similar, algunos pacientes obtuvieron un beneficio adicional del esquema con 200 mg dos veces al día. Las dosis de 400 mg dos veces al día no brindan beneficio adicional al observado con 100 mg a 200 mg dos veces al día.
Analgesia, incluida dismenorrea primaria: En los modelos analgésicos agudos de dolor posterior a cirugía oral, dolor posoperatorio ortopédico y dismenorrea primaria, celecoxib alivió el dolor calificado como moderado a intenso por los pacientes. Las dosis únicas (véase sección Posología y método de administración) de celecoxib produjeron alivio del dolor en 60 minutos.
Espondilitis anquilosante (EA): Celecoxib se evaluó en pacientes con EA en dos estudios clínicos controlados con placebo y con producto activo (naproxeno o ketoprofeno) de 6 y 12 semanas de duración. En dosis de 100 mg dos veces al día, 200 mg una vez al día y 400 mg una vez al día, se mostró que celecoxib era estadísticamente superior al placebo en estos estudios para las tres medidas de eficacia primarias que valoraban la intensidad global del dolor (Escala Visual Análoga), la actividad global de la enfermedad (Escala Visual Análoga) y el daño funcional (Índice Funcional de Espondilitis anquilosante de Bath). En el estudio de 12 semanas no hubo diferencia en el grado de mejoría entre las dosis de 200 mg y 400 mg de celecoxib en una comparación del cambio promedio con respecto a los parámetros basales, pero hubo un mayor porcentaje de pacientes con respuesta a celecoxib 400 mg, 53%, que a celecoxib 200 mg, 44%, según los criterios de respuesta de la Valoración de Espondilitis anquilosante (ASAS 20). La ASAS 20 define al sujeto con respuesta como aquel con mejoría de por lo menos 20% con respecto al valor basal y una mejoría absoluta de por lo menos 10 mm en una escala de 0 a 100 mm, por lo menos en tres de los cuatro dominios siguientes: dolor global del paciente, Índice Funcional de Bath para Espondilitis anquilosante e inflamación. El análisis de sujetos con respuesta tampoco demostró cambios en los índices de respuesta después de 6 semanas.
Dolor de espalda baja (LBP): Celecoxib se utilizó para tratar pacientes que habían presentado LBP no neuropático preexistente durante 12 semanas o más. En la siguiente tabla, se presentan los resultados para eficacia en cinco ensayos clínicos utilizando la Evaluación de Intensidad del Dolor del Paciente (escala visual análoga de 100 mm) desde el inicio hasta el final del tratamiento:
Tabla 4. Evaluación de la Intensidad del Dolor del Paciente en Ensayos Clínicos de Dolor de espalda Baja
|
Identificación del estudio (duración) Tratamiento (TDD) |
N |
Intensidad del dolor al iniciod |
Cambio en la intensidad del dolord |
Valor P para diferencia entre tratamientosd |
|
Estudio 244 (12 semanas)a |
||||
|
Placebo Celecoxib 200 mg |
177 183 |
76.6 73.6 |
-30.1 -35.9 |
- 0.0503 |
|
Estudio 245 (12 semanas)a |
||||
|
Placebo Celecoxib 200 mg |
191 183 |
75.7 72.8 |
-26.2 -32.2 |
- 0.0427 |
|
Estudio 1165 (6 semanas)b |
||||
|
Celecoxib 400 mg Tramadol 200 mg |
402 389 |
65.5 66.1 |
-34.6 -30.4 |
0.008 - |
|
Estudio 1338 (6 semanas)b |
||||
|
Celecoxib 400 mg Tramadol 200 mg |
386 385 |
65.9 66.6 |
-34.8 -34.4 |
0.595 - |
|
Estudio 1174 (4 semanas) |
||||
|
Placebo Celecoxib 400 mg Loxoprofeno 180 mg |
410 410 407 |
65.1 65.0 65.6 |
-26.2 -31.7 -29.3 |
- <0.001 No evaluada |
N = Número de pacientes que proporcionan datos al inicio y al final del tratamiento. TDD = Total dosis diaria.
a La Evaluación de la Intensidad del Dolor del Paciente es un criterio coprimario de valoración de la eficacia en estos estudios, junto con la Evaluación Global de Dolor de espalda Baja del Paciente (las diferencias entre tratamientos favorecen significativamente a celecoxib por encima del placebo en los Estudios 244 y 245) y el Cuestionario de Discapacidad de Roland-Morris (las diferencias entre tratamientos favorecen significativamente a celecoxib por encima del placebo en el Estudio 244).
b El criterio primario de valoración de la eficacia en estos estudios fue el porcentaje de pacientes que experimentaron al menos un 30% de mejoría sobre la Evaluación del Dolor de la Escala de Calificación numérica (NRS), para este criterio los resultados en ambos estudios muestran superioridad estadística de celecoxib sobre tramadol.
d Con base en las medias de mínimos cuadrados de los modelos de Análisis de Covarianza, con cambios en la intensidad del dolor calculada mediante la resta del valor inicial al valor al final del tratamiento; los valores p fueron calculados con base en las diferencias de medias de mínimos cuadrados entre los grupos de tratamiento.
Información adicional de estudios clínicos:
Estudios endoscópicos:
Se realizaron evaluaciones endoscópicas GI superiores programadas en más de 4,500 pacientes con artritis que se reclutaron en cinco estudios aleatorios controlados de 12 a 24 semanas. Con comparadores activos, dos de los cuales también incluyeron controles con placebo. No hubo una relación consistente entre la incidencia de úlceras gastroduodenales y la dosis de celecoxib en el rango estudiado.
La Tabla 5 resume la incidencia de úlceras endoscópicas en dos estudios de 12 semanas que reclutaron pacientes en los que las endoscopias basales no revelaron úlceras.
Tabla 5. Incidencia de úlceras gastroduodenales en estudios endoscópicos en pacientes con OA y RA
|
Estudios de 3 meses |
||
|
Estudio 1 (n = 1108) |
Estudio 2 (n = 1049) |
|
|
Placebo |
2.3% (5/217) |
2.0% (4/200) |
|
Celecoxib 50 mg dos veces al día |
3.4% (8/233) |
- |
|
Celecoxib 100 mg dos veces al día |
3.1% (7/227) |
4.0% (9/223) |
|
Celecoxib 200 mg dos veces al día |
5.9% (13/221) |
2.7% (6/219) |
|
Celecoxib 400 mg dos veces al día |
- |
4.1% (8/197) |
|
Naproxeno 500 mg dos veces al día |
16.2% (34/210)* |
17.6% (37/210)* |
* p ≤ 0.05 vs. todos los demás tratamientos.
La Tabla 6 resume los datos de dos estudios de 12 semanas que reclutaron pacientes en los que las endoscopias basales no revelaron úlceras. Los pacientes se sometieron a endoscopias a intervalos de 4 semanas para obtener información sobre el riesgo de úlcera con el paso del tiempo.
Tabla 6. Incidencia de úlceras gastroduodenales de una serie de estudios endoscópicos de 3 meses en pacientes con OA y AR
|
Semana 4 |
Semana 8 |
Semana 12 |
Final |
|
|
Estudio 3 (n = 523) |
||||
|
CELECOXIB 200 mg dos veces al día |
4.0% (10/252)* |
2.2% (5/227)* |
1.5% (3/196)* |
7.5% (20/266)* |
|
Naproxeno 500 mg dos veces al día |
19.0% (47/247) |
14.2% (26/182) |
9.9% (14/141) |
34.6% (89/257) |
|
Estudio 4 (n = 1062) |
||||
|
CELECOXIB 200 mg dos veces al día |
3.9% (13/337)† |
2.4% (7/296)† |
1.8% (5/274)† |
7.0% (25/356)† |
|
Diclofenaco 75 mg dos veces al día |
5.1% (18/350) |
3.3% (10/306) |
2.9% (8/278) |
9.7% (36/372) |
|
Ibuprofeno 800 mg tres veces al día |
13.0% (42/323) |
6.2% (15/241) |
9.6% (21/219) |
23.3% (78/334) |
* p ≤ 0.05 celecoxib vs. naproxeno con base en análisis de intervalo y acumulativos.
† p ≤ 0.05 celecoxib vs. ibuprofeno con base en análisis de intervalo y acumulativos.
Se realizó un estudio aleatorio y doble ciego de 6 meses en 430 pacientes con AR en el que se practicó un examen endoscópico a los 6 meses. La incidencia de úlceras endoscópicas en pacientes que toman celecoxib 200 mg dos veces al día fue 4% vs. 15% en pacientes que tomaron diclofenaco SR 75 mg dos veces al día (p <0.001).
En 4 de los 5 estudios endoscópicos, cerca del 11% de los pacientes (440/4,000) tomaba ácido acetilsalicílico (≤ 325 mg/día). En los grupos con celecoxib, el índice de úlceras endoscópicas pareció más alto entre los usuarios de ácido acetilsalicílico que entre los no usuarios. Sin embargo, el aumento en el índice de úlceras entre estos usuarios de ácido acetilsalicílico fue menor al observado en los grupos con comparadores activos, con o sin ácido acetilsalicílico.
La correlación entre los hallazgos de los estudios endoscópicos y la incidencia relativa de eventos GI superiores serios de importancia clínica no está bien establecida. Se ha observado sangrado GI superior de importancia clínica en pacientes que reciben celecoxib en estudios abiertos y controlados; aunque con poca frecuencia (véase sección Advertencias especiales y precauciones para uso, Efectos gastrointestinales (GI)).
Metaanálisis de seguridad gastrointestinal de estudios en osteoartritis y artritis reumatoide: Un análisis de 31 estudios clínicos aleatorios controlados en osteoartritis y artritis reumatoide que incluyó 39,605 pacientes con osteoartritis (N = 25,903), artritis reumatoide (N = 3,232) o pacientes con cualquiera de estos trastornos (N = 10,470) comparó la incidencia de eventos adversos GI en los pacientes tratados con celecoxib con los que recibieron placebo o AINE (incluidos naproxeno, diclofenaco e ibuprofeno). La incidencia de úlceras clínicas y sangrados ulcerosos con la dosis total diaria de 200 a 400 mg de celecoxib fue 0.2%, comparada con una incidencia de 0.6% con AINE (RR = 0.35; 95% IC 0.22 - 0.56).
Estudio de Seguridad a Largo Plazo en Artritis con Celecoxib (CLASS) incluyendo uso con ácido acetilsalicílico:
En un estudio prospectivo de resultados de seguridad a largo plazo que se realizó después de la comercialización en cerca de 5,800 pacientes con OA y 2,200 pacientes con RA. Los pacientes recibieron celecoxib 400 mg dos veces al día (4 y 2 veces más de la dosis recomendada para OA y RA, respectivamente), ibuprofeno 800 mg tres veces al día o diclofenaco 75 mg dos veces al día (dosis terapéuticas frecuentes). La mediana de exposición a celecoxib (n = 3,987) y diclofenaco (n = 1,996) fue de 9 meses, mientras que para ibuprofeno (n = 1,985) fue de 6 meses. Se presentan los índices acumulativos Kaplan-Meier a los 9 meses para todos los análisis. El parámetro de valoración primario de este estudio de resultado fue la incidencia de úlceras complicadas (sangrado gastrointestinal, perforación u obstrucción). Se permitió que los pacientes tomaran una dosis baja concomitante de ácido acetilsalicílico (ASA) (≤ 325 mg/día) como profilaxis CV (subgrupos con ASA: celecoxib, n = 882; diclofenaco, n = 445; ibuprofeno, n = 412). Las diferencias en la incidencia de úlceras complicadas entre celecoxib y el grupo combinado de ibuprofeno y diclofenaco no alcanzaron significancia estadística. Los pacientes con celecoxib y dosis baja concomitante de ASA tuvieron índices 4 veces más altos de úlceras complicadas que aquellos sin ASA (véase sección Advertencias especiales y precauciones para uso, Efectos gastrointestinales [G]). Los resultados para celecoxib se presentan en la Tabla 7.
Tabla 7. Efectos de la administración concomitante de dosis bajas de ASA en los índices de úlcera complicada con celecoxib 400 mg dos veces al día (índices Kaplan-Meier a los 9 meses [%])
|
No usuarios de ASA n = 3105 |
Usuarios de ASA n = 882 |
|
|
Úlceras complicadas |
0.32 |
1.12 |
Función plaquetaria:
En voluntarios sanos, las dosis terapéuticas de celecoxib y las dosis múltiples de 600 mg dos veces al día (tres veces la dosis más alta recomendada) no tuvieron efecto en la agregación plaquetaria ni en el tiempo de sangrado en comparación con el placebo.
Todos los controles activos (inhibidores inespecíficos de COX) presentaron disminución significativa de la agregación plaquetaria y prolongación del tiempo de sangrado (ver figura 1).
Figura 1. Efecto de una dosis alta de celecoxib (600 mg dos veces al día) en la agregación plaquetaria y el tiempo de hemorragia en pacientes sanos
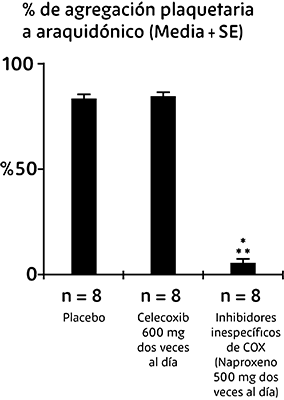
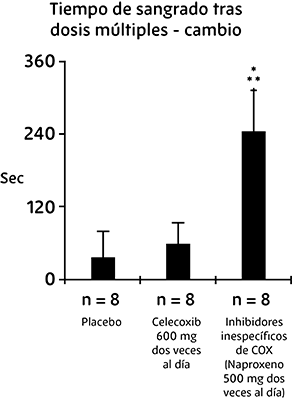
* Significativamente diferente del placebo; p <0.05.
** Significativamente diferente de celecoxib; p <0.05.
Estudio de celecoxib versus omeprazol y diclofenaco en pacientes con riesgo de osteoartritis y artritis reumatoidea. (CONDOR):
En esta prospectiva, un estudio de 24 semanas en pacientes mayores de 60 años o con historia de úlceras gastroduodenales (usuarios de bajas dosis de ácido acetilsalicílico fueron excluidos), el porcentaje de pacientes con eventos GI clínicamente significativos (Composición del Criterio principal de valoración) fue menor en pacientes tratados con celecoxib 200 mg dos veces al día, comparado con pacientes tratados con diclofenaco SR 75 mg dos veces al día + omeprazol 20 mg una vez al día. Esta diferencia fue manejada por el decrecimiento clínicamente significativo en hemoglobina (≥ 2 g/dL) y /o hematocrito (≥ 10%) de origen GI definido o presumido. Los resultados para los componentes individuales para estos criterios de valoración se muestran a continuación:
|
Composición criterios de valoración GI predefinidos |
Celecoxib 200 mg dos veces al día (n = 2238) |
Diclofenaco SR 75 mg dos veces al día + omeprazol 20 mg una vez al día (n = 2246) |
|---|---|---|
|
Componentes |
n (%) de pacientes |
|
|
Hemorragia gastroduodenal |
3 (0.1) |
3 (0.1) |
|
Hemorragia en intestino grueso |
1 (<0.1) |
1 (<0.1) |
|
Hemorragia GI aguda de origen desconocido |
1 (< 0.1) |
0 (< 0.0) |
|
Decrecimiento clínicamente significativo de hemoglobina (≥ 2 g/dL) y/o hematocrito (≥ 10%) de origen GI definido |
5 (0.2) |
24 (1.1) |
|
Decrecimiento clínicamente significativo de hemoglobina (≥ 2 g/dL) y/o hematocrito (≥ 10%) de origen GI oculto |
10 (0.4) |
53 (2.3) |
|
Total* |
20 (0.9) |
81 (3.6) |
Para los siguientes componentes del punto final compuesto GI predefinido, no se encontraron ninguno de los siguientes eventos en los grupos tratados: obstrucción de la salida gástrica; perforaciones gastroduodenales, de intestino delgado o de intestino grueso; hemorragias en intestino delgado. Todos los eventos comprendidos como criterios de valoración fueron juzgados por un panel de expertos independiente y ciego con asignaciones de tratamiento al azar.
* En un tiempo de análisis de eventos mediante técnicas de tablas de vida, p < 0,0001 para la comparación entre el grupo de tratamiento con celecoxib y el grupo de tratamiento con diclofenaco más omeprazol para los criterios de valoración.
Seguridad cardiovascular – estudios a largo plazo en pacientes con pólipos adenomatosos esporádicos:
Se llevaron a cabo dos estudios con celecoxib en pacientes con pólipos adenomatosos esporádicos, el estudio APC (Prevención de Adenomas con Celecoxib) y el estudio PreSAP (Prevención de Pólipos Adenomatosos Espontáneos). En el estudio APC hubo un aumento relacionado con la dosis en el parámetro de valoración compuesto de muerte CV, infarto miocárdico y eventos cerebrovasculares (adjudicada) con celecoxib, en comparación con el placebo durante 3 años de tratamiento. El estudio PreSAP no mostró un aumento estadísticamente significativo en el riesgo para el mismo parámetro de valoración compuesto.
En el estudio APC, los índices de riesgo comparados con el placebo con respecto a un parámetro de valoración compuesto de muerte CV, infarto del miocardio o eventos cerebrovasculares (adjudicados) fueron 3.4 (95% IC 1.4 - 8.5) con celecoxib 400 mg dos veces al día y 2.8 (95% IC 1.1 - 7.2) con celecoxib 200 mg dos veces al día. Los índices acumulativos para este parámetro de valoración compuesto durante 3 años fueron 3.0% (20/671) y 2.5% (17/685) para grupos de tratamiento con celecoxib 200 o 400 mg dos veces al día respectivamente, en comparación con 0.9% (6/679) del grupo placebo. Los aumentos para ambos grupos con dosis de celecoxib contra placebo fueron impulsados sobre todo por el infarto del miocardio.
En el estudio PreSAP, índices de riesgo comparado con el placebo para este mismo parámetro de valoración compuesto fue 1.2 (95% IC 0.6 - 2.4) con celecoxib 400 mg una vez al día. Los índices acumulativos para este parámetro de valoración compuesto durante 3 años fueron 2.3% (21/933), comparado con 1.9% (12/628) para el grupo con placebo.
Estudio a largo plazo de Prevención Antiinflamatoria en la Enfermedad de Alzheimer (ADAPT, por sus siglas en inglés) – seguridad cardiovascular:
Los datos del estudio ADAPT, no mostraron un riesgo CV significativamente mayor con celecoxib 200 mg dos veces al día en comparación con placebo. El riesgo relativo, en comparación con placebo, para un punto final compuesto similar (muerte CV, IM, evento vascular cerebral) fue del 1.14 (IC 95% 0.61 - 2.12) con celecoxib 200 mg dos veces al día.
Seguridad cardiovascular – Metaanálisis de estudios con uso crónico:
Se realizó un metaanálisis de los datos de seguridad (eventos adversos serios adjudicados, reportados por el investigador) de 39 estudios clínicos terminados con celecoxib de hasta 65 semanas de duración, representa 41,077 pacientes [23,030 (56.1 %) pacientes expuestos a celecoxib 200 mg a 800 mg dosis diaria total (TDD), 13,990 (34.1 %) pacientes expuestos a AINE no selectivos y 4,057 (9.9 %) pacientes expuestos a placebo].
En el análisis, el índice de eventos adjudicados para el parámetro de valoración compuesto de muerte CV, infarto miocárdico no mortal y apoplejía no mortal fue similar entre el tratamiento con celecoxib (N = 19,773; 0.96 eventos/100 años-paciente) y el AINE no selectivo (N = 13,990; 1.12 eventos/100 años-paciente) (RR = 0.90, 95% IC 0.60 - 1.33). Este patrón de efecto se mantuvo con o sin el uso de ASA (≤ 325mg). El índice de eventos adjudicados de infarto miocárdico no mortal tendió a ser más alto (RR = 1.76, 95% IC 0.93 - 3.35); sin embargo, el de apoplejía no mortal tendió a ser menor (RR = 0.51, 95% IC 0.23 - 1.10), el de muerte CV fue comparable (RR = 0.57, 95% IC 0.28 - 1.14) para celecoxib y los AINE combinados no selectivos.
En este análisis, el índice de eventos adjudicados para el parámetro de valoración compuesto de muerte CV, infarto miocárdico no mortal y apoplejía no mortal fue 1.42/100 años-paciente para el tratamiento con celecoxib (N = 7,462) y 1.20/100 años-paciente para el placebo (N = 4,057) (RR = 1.11, 95% IC 0.47 - 2.67). Este patrón de efecto se mantuvo con o sin el uso de ASA (≤ 325mg). La incidencia de infarto miocárdico no mortal tendió a ser más alta (RR = 1.56, 95% IC 0.21 - 11.90), al igual que la de muerte CV (RR = 1.26, 95% IC 0.33 - 4.77); la de apoplejía no mortal fue similar (RR = 0.80, 95% IC 0.19 - 3.31) para celecoxib y para placebo.
Seguridad cardiovascular:
Los resultados de seguridad CV se evaluaron en el estudio CLASS (véase arriba la descripción del estudio). Los índices acumulativos de Kaplan-Meier para los eventos adversos tromboembólicos CV serios reportados por el investigador (incluidos MI, embolia pulmonar, trombosis venosa profunda, angina inestable, ataques isquémicos transitorios y accidentes vasculares cerebrales isquémicos) no mostraron diferencia entre los grupos terapéuticos con celecoxib, diclofenaco e ibuprofeno. Los índices acumulativos en todos los pacientes a los nueve meses para celecoxib, diclofenaco e ibuprofeno fueron 1.2%, 1.4% y 1.1%, respectivamente. Los índices acumulativos en los no usuarios de ASA a los nueve meses de cada uno de los tres grupos terapéuticos fueron menores de 1%. Los índices acumulativos para infarto miocárdico en los no usuarios de ASA a los nueve meses en cada uno de los tres grupos terapéuticos fueron menores de 0.2%. No hubo grupo placebo en el estudio CLASS, lo cual limita la capacidad para establecer si los tres fármacos evaluados no tuvieron aumento en el riesgo de eventos CV o si todos elevaron el riesgo en grado similar.
Evaluación Prospectiva Aleatorizada de la Seguridad Integrada de celecoxib frente a ibuprofeno o naproxeno (PRECISIÓN):
Diseño:
El estudio PRECISION fue un estudio doble ciego de seguridad cardiovascular en pacientes que padecen OA o AR con un alto riesgo de enfermedad cardiovascular que comparó celecoxib (200 mg a 400 mg diarios) con naproxeno (750 mg a 1000 mg diarios) e ibuprofeno (1800 mg a 2400 mg diarios). El desenlace primario, Colaboración de los Autores de Ensayos Antiplaquetarios (APTC), fue un compuesto adjudicado de forma independiente de muerte cardiovascular (incluida la muerte debido a hemorragia), infarto de miocardio no mortal o accidente cerebrovascular no mortal. El estudio fue planeado con una potencia de un 80% para evaluar no-inferioridad. A todos los pacientes se les prescribió esomeprazol de forma abierta (20 mg a 40 mg) para protección gástrica. A los pacientes que estaban tomando una dosis baja de aspirina se les permitió continuar con el tratamiento.
Otros desenlaces secundarios y terciarios adjudicados de forma independiente incluyeron resultados cardiovasculares, gastrointestinales y renales. Además, hubo un subestudio de 4 meses que se enfocó en los efectos de los tres medicamentos sobre la presión arterial medida a través del monitoreo ambulatorio (ABPM).
Resultados:
Tabla 8. Población y dosis de tratamiento
|
Conjunto de análisis |
Celecoxib 100 mg-200 mg bid |
Ibuprofeno 600 mg-800 mg tid |
Naproxeno 375 mg-500 mg bid |
Total |
|
Aleatorizado (IDT) |
8072 |
8040 |
7969 |
24,081 |
|
En tratamiento (mIDT) |
8030 |
7990 |
7933 |
23,953 |
|
Dosis promedio1 (mg/día) |
209 ± 37 |
2045 ± 246 |
852 ± 103 |
NA |
1 Dosis promedio dispensada.
IDT: con intención de tratar; todos los sujetos aleatorizados.
mIDT: con intención de tratar modificada; Todos los sujetos aleatorizados con al menos una dosis del medicamento del estudio y una visita posterior al periodo inicial.
bid: dos veces al día.
tid: tres veces al día.
NA: no aplicable.
Desenlace primario de valoración:
• Celecoxib, ya sea comparado con naproxeno o ibuprofeno, cumplió con los cuatro requisitos de no-inferioridad previamente especificados (p < 0.001 para la no-inferioridad en ambas comparaciones). No-inferioridad se establece cuando la razón de tasas instantáneas (HR) es ≤ 1.12 tanto en el análisis de IDT como en el análisis de mIDT, y el IC superior del 95% es ≤ 1.33 para el análisis de IDT y ≤ 1.40 para el análisis de mIDT.
Los análisis primarios para IDT y mIDT se describen a continuación en la Tabla 9.
Tabla 9. Análisis primario del desenlace compuesto adjudicado APTC
|
Análisis de intención de tratar (IDT, hasta el mes 30) |
|||
|
Celecoxib 100 mg200 mg bid |
Ibuprofeno 600 mg800 mg tid |
Naproxeno 375 mg500 mg bid |
|
|
N |
8072 |
8040 |
7969 |
|
Sujetos con eventos |
188 (2.3%) |
218 (2.7%) |
201 (2.5%) |
|
Comparación en pares |
Celecoxib frente a naproxeno |
Celecoxib frente a ibuprofeno |
Ibuprofeno frente a naproxeno |
|
HR (IC del 95%) |
0.93 |
0.86 |
1.08 |
|
Análisis de intención de tratar modificada (mIDT, en tratamiento hasta el mes 43) |
|||
|
Celecoxib 100 mg200 mg bid |
Ibuprofeno 600 mg800 mg tid |
Naproxeno 375 mg500 mg bid |
|
|
N |
8030 |
7990 |
7933 |
|
Sujetos con eventos |
134 (1.7%) |
155 (1.9%) |
144 (1.8%) |
|
Comparación en pares |
Celecoxib frente a naproxeno |
Celecoxib frente a ibuprofeno |
Ibuprofeno frente a naproxeno |
|
HR (IC del 95%) |
0.90 |
0.81 |
1.12 |
Desenlaces secundarios y terciarios claves:
El análisis de los Eventos adversos cardiovasculares mayores (MACE)* para mIDT se describe a continuación en la Tabla 10.
Tabla 10. Eventos adversos CV mayores adjudicados en tratamiento
|
Celecoxib 100 mg-200 mg bid |
Ibuprofeno 600 mg-800 mg tid |
Naproxeno 375 mg-500 mg bid |
|
|---|---|---|---|
|
N |
8030 |
7990 |
7933 |
|
Cantidad de sujetos con eventos (%) |
|||
|
MACE |
247 (3.1%) |
284 (3.6%) |
253 (3.2%) |
|
Muerte CV |
35 (0.4%) |
51 (0.6%) |
49 (0.6%) |
|
IM no mortal |
58 (0.7%) |
76 (1.0%) |
53 (0.7%) |
|
Accidente cerebrovascular no mortal |
43 (0.5%) |
32 (0.4%) |
45 (0.6%) |
|
Hospitalización por angina inestable |
46 (0.6%) |
49 (0.6%) |
44 (0.6%) |
|
Revascularización |
132 (1.6%) |
158 (2.0%) |
122 (1.5%) |
|
Hospitalización por AIT |
12 (0.1%) |
21 (0.3%) |
16 (0.2%) |
|
Comparación por pares HR (IC del 95%) |
Celecoxib frente a naproxeno |
Celecoxib frente a ibuprofeno |
Ibuprofeno frente a Naproxeno |
|
MACE |
0.95 (0.80; 1.13) |
0.82 (0.69; 0.97) |
1.17 (0.98; 1.38) |
|
Muerte CV |
0.69 (0.45; 1.07) |
0.64 (0.42; 0.99) |
1.08 (0.73; 1.60) |
|
IM no mortal |
1.06 (0.73; 1.54) |
0.72 (0.51; 1.01) |
1.48 (1.04; 2.11) |
|
Accidente cerebrovascular no mortal |
0.93 (0.61; 1.42) |
1.26 (0.79; 1.98) |
0.74 (0.47; 1.16) |
|
Hospitalización por angina inestable |
1.02 (0.67; 1.54) |
0.89 (0.59; 1.33) |
1.16 (0.77; 1.74) |
|
Revascularización |
1.06 (0.83; 1.35) |
0.78 (0.62. 0.99) |
1.35 (1.07; 1.72) |
|
Hospitalización por AIT |
0.73 (0.35; 1.55) |
0.54 (0.26; 1.09) |
1.38 (0.72; 2.64) |
* MACE = Desenlace compuesto de APTC más revascularización coronaria u hospitalización por angina inestable o ataque isquémico transitorio en la población con IDT para el desenlace de MACE no hubo diferencias significativas en las comparaciones por pares entre los regímenes de tratamiento.
El análisis de eventos gastrointestinales para mIDT se describe a continuación en la Tabla 11.
Tabla 11. Desenlaces gastrointestinales adjudicados en tratamiento
|
Celecoxib 100 mg200 mg bid |
Ibuprofeno 600 mg800 mg tid |
Naproxeno 375 mg500 mg bid |
|
|
N |
8030 |
7990 |
7933 |
|
Cantidad de sujetos con eventos, n (%) |
|||
|
CSGIE |
27 (0.3%) |
59 (0.7%) |
52 (0.7%) |
|
ADH de origen GI |
27 (0.3%) |
58 (0.7%) |
66 (0.8%) |
|
Comparación por pares, HR (IC del 95%) |
Celecoxib frente a naproxeno |
Celecoxib frente a ibuprofeno |
Ibuprofeno frente a naproxeno |
|
CSGIE |
0.51 |
0.43 |
1.16 |
|
ADH de origen GI |
0.39 |
0.43 |
0.91 |
* CSGIE (Eventos gastrointestinales significativos a nivel clínico) = compuestos de lo siguiente: hemorragia gastroduodenal; obstrucción de la salida gástrica; perforación gastroduodenal del intestino delgado o intestino grueso; hemorragia del intestino grueso; hemorragia del intestino delgado; hemorragia GI aguda de origen desconocido, incluida la presunta hemorragia del intestino delgado; úlcera duodenal o gástrica sintomática.
** ADH (anemia por deficiencia de hierro)= anemia por deficiencia de hierro significativa a nivel clínico de origen GI o disminución en Hct (hematocrito) y/o Hgb (hemoglobina) (definida como Hct > 10 puntos y/o Hgb > 2 g/dL desde el periodo inicial).
En la población con IDT para el desenlace CSGIE no hubo diferencias significativas en las comparaciones por pares entre los regímenes de tratamiento (no se muestran los datos). Con relación al desenlace de anemia por deficiencia de hierro de origen GI, se observaron diferencias significativas (celecoxib frente a naproxeno; celecoxib frente a ibuprofeno) y diferencias no significativas (ibuprofeno frente a naproxeno) de forma coherente con los datos presentados anteriormente.
El análisis de eventos renales significativos a nivel clínico*, hospitalización por ICC e hipertensión para mIDT se describe a continuación en la Tabla 12.
Tabla 12. Eventos renales adjudicados en tratamiento, hospitalización por ICC e hipertensión
|
Celecoxib 100 mg200 mg bid |
Ibuprofeno 600 mg800 mg tid |
Naproxeno 375 mg500 mg bid |
|
|---|---|---|---|
|
N |
8030 |
7990 |
7933 |
|
Cantidad de sujetos con eventos, n (%) |
|||
|
Eventos renales |
42 (0.5%) |
73 (0.9%) |
62 (0.8%) |
|
Hospitalización por ICC |
28 (0.3%) |
38 (0.5%) |
35 (0.4%) |
|
Hospitalización por hipertensión |
25 (0.3%) |
37 (0.5%) |
32 (0.4%) |
|
Cualquiera de las anteriores |
89 (1.1%) |
139 (1.7%) |
120 (1.5%) |
|
Comparación por pares. HR (IC del 95%) |
Celecoxib frente a naproxeno |
Celecoxib frente a ibuprofeno |
Ibuprofeno frente a naproxeno |
|
Eventos renales |
0.66 |
0.54 |
1.21 |
|
Hospitalización por ICC |
0.77 |
0.70 |
1.12 |
|
Hospitalización por hipertensión |
0.76 |
0.64 |
1.18 |
|
Cualquiera de las anteriores |
0.72 |
0.60 |
1.19 |
* N.B: Los eventos renales incluyeron aumento predefinido en los niveles de creatinina (creatinina sérica verificada de ≥ 2.0 mg/dL [177 μmol/L] y un aumento de ≥ 0.7 mg/mL [62 μmol/L]). u hospitalización por insuficiencia renal aguda (definida como la duplicación de la creatinina sérica o la confirmación de hipercalemia con ≥ 50% de elevación en la creatinina sérica) o el inicio de hemodiálisis o diálisis peritoneal.
En la población con IDT para el desenlace de eventos renales significativos a nivel clínico, solamente la comparación por pares entre celecoxib e ibuprofeno fue significativa, HR 0.61 (0.44; 0.85), no se observaron diferencias significativas entre los regímenes de tratamiento en la incidencia de hospitalización por insuficiencia cardiaca congestiva, y se observó una incidencia significativamente más baja de hospitalización por hipertensión entre celecoxib e ibuprofeno, HR 0.59 (0.36; 0.99).
Mortalidad por cualquier causa:
En las poblaciones con mIDT, celecoxib, naproxeno e ibuprofeno se asociaron con 53 (0.7%), 79 (1.0%) y 73 (0.9%) muertes, respectivamente. Se observaron diferencias significativas en las comparaciones por pares ya sea entre celecoxib y naproxeno HR 0.65 (0.46; 0.92) o entre celecoxib e ibuprofeno HR 0.68 (0.48; 0.97). En la población con IDT, celecoxib, naproxeno e ibuprofeno se asociaron con 132 (1.6%), 163 (2.0%) y 142 (1.8%) muertes, respectivamente. No se observaron diferencias significativas en las comparaciones por pares entre los tratamientos.
Subestudio ABPM:
En el subestudio PRECISION-ABPM, entre el total de 444 pacientes analizables, en el cuarto mes, los pacientes tratados con celecoxib presentaron el cambio más pequeño en la presión arterial sistólica (PAS) ambulatoria de 24 horas en comparación con ibuprofeno y naproxeno: celecoxib produjo una leve reducción de 0.3 mmHg mientras que ibuprofeno y naproxeno aumentaron la PAS media de 24 horas en 3.7 mmHg y 1.6 mmHg, respectivamente. Estos cambios dieron como resultado una diferencia significativa a nivel estadístico y clínico de -3.9 mmHg (p=0.0009) entre celecoxib e ibuprofeno; una diferencia no significativa de -1.8 mmHg (p=0.119) entre celecoxib y naproxeno; y una diferencia no significativa de -2.1 mmHg (p=0.0787) entre naproxeno e ibuprofeno.
Propiedades farmacocinéticas:
Absorción:
La farmacocinética de celecoxib se ha evaluado en cerca de 1500 individuos. Cuando se administra en condiciones de ayuno, celecoxib se absorbe bien y alcanza concentraciones plasmáticas máximas después de aproximadamente 2-3 horas. La biodisponibilidad oral de las cápsulas es cercana a 99% en relación con la administración en suspensión (forma de dosificación oral con disponibilidad óptima). En condiciones de ayuno, tanto los niveles plasmáticos máximos (Cmáx) como el área bajo la curva (ABC) son más o menos proporcionales a la dosis hasta 200 mg dos veces al día; con dosis más altas se producen aumentos menores a los proporcionales en Cmáx y ABC.
Distribución:
La unión con proteínas plasmáticas, que es independiente de la concentración, es cercana al 97% con concentraciones plasmáticas terapéuticas; celecoxib no se une en forma preferencial con los eritrocitos en la sangre.
Metabolismo:
El metabolismo de celecoxib está mediado principalmente por el citocromo P450 2C9. Se han identificado en el plasma humano tres metabolitos, sin actividad inhibidora de COX-1 o COX-2; éstos son un alcohol primario, el ácido carboxílico correspondiente y su conjugado glucurónido.
La actividad del citocromo P450 2C9 es baja en personas con polimorfismos genéticos que reducen la actividad enzimática, como los homocigotos para el polimorfismo CYP2C9*3.
En un estudio farmacocinético de celecoxib 200 mg administrado una vez al día en voluntarios sanos con genotipo CYP2C9*1/*1, CYP2C9*1/*3 o CYP2C9*3/*3, la mediana de Cmáx y ABC0-24 de celecoxib el día 7 fue aproximadamente 4 y 7 veces más alta en sujetos con genotipo CYP2C9*3/*3 en comparación con otros genotipos. En tres estudios de dosis única que incluyeron un total de 5 sujetos con genotipo CYP2C9*3/*3, el ABC0-24 de la dosis única aumentó casi 3 veces en comparación con los sujetos de metabolismo normal. Se estima que la frecuencia del genotipo homocigótico *3/*3 es 0.3 – 1.0% entre diferentes grupos étnicos.
Celecoxib se debe usar con cuidado en los pacientes con certeza o sospecha de metabolismo deficiente mediante CYP2C9 con base en los antecedentes o experiencias previas con otros sustratos de CYP2C9. Considere iniciar el tratamiento con la mitad de la dosis más baja recomendada. (Véanse secciones Posología y método de administración y Interacción con otros medicamentos y otras formas de interacción).
Excreción:
La depuración de celecoxib es principalmente por metabolismo hepático, menos del 1% de la dosis se excreta sin cambios en la orina. Después de dosis múltiples, la vida media de depuración es de 8 a 12 horas y la velocidad de depuración es cercana a 500 mL/min.
Con las dosis múltiples, las concentraciones plasmáticas del estado estable se alcanzan antes del día 5. La variabilidad entre sujetos en los principales parámetros farmacocinéticos (ABC, Cmáx y vida media de depuración) es cercana al 30%. El volumen de distribución medio del estado estable es cercano a 500 L/70 kg en adultos jóvenes sanos, lo que indica una amplia distribución de celecoxib en los tejidos. Los estudios preclínicos indican que el fármaco cruza la barrera hematoencefálica.
Efectos de los alimentos:
Su administración con alimentos (comida rica en grasas) retrasa la absorción de celecoxib que resulta en un Tmáx de aproximadamente 4 horas y aumenta la biodisponibilidad en aproximadamente un 20%. (Véase sección Posología y forma de administración).
La coadministración de CELEBREX con un antiácido que contiene magnesio y aluminio dio como resultado una reducción en las concentraciones plasmáticas de celecoxib con una disminución del 37% en Cmáx y 10% en ABC.
En voluntarios adultos sanos, la exposición sistémica global (ABC) de celecoxib fue equivalente cuando celecoxib se administró en cápsulas o cápsulas contenido intacto espolvoreado en puré de manzana. No hubo alteraciones significativas en Cmáx, Tmáx y T1/2 después de la administración del contenido de la cápsula en puré de manzana.
Poblaciones especiales:
Adultos mayores:
En la población > 65 años se observa un aumento de 1.5 a 2 veces en el promedio de Cmáx y ABC para celecoxib. Éste es un cambio relacionado sobre todo con el peso, más que con la edad, los niveles de celecoxib son más altos en individuos con peso bajo y por consiguiente, más altos en la población geriátrica, que suele tener peso promedio más bajo que la población más joven. Por lo tanto, las mujeres adultas mayores tienden a tener concentraciones plasmáticas más elevadas del fármaco que los varones adultos mayores. Por lo general no es necesario ajustar la dosis. Sin embargo, para los pacientes adultos mayores con peso corporal menor al promedio (< 50 kg), se debe iniciar la administración con la dosis más baja recomendada.
Raza:
Un metaanálisis de los estudios farmacocinéticos sugirió un ABC casi 40% mayor de celecoxib en la población negra con respecto a la caucásica. Se desconoce la causa y la importancia clínica de este hallazgo.
Daño hepático: Las concentraciones plasmáticas de celecoxib en pacientes con daño hepático leve (clase A Child-Pugh) no son significativamente diferentes a las de los controles de edad y sexo equiparables. En pacientes con daño hepático moderado (clase B Child-Pugh) las concentraciones plasmáticas de celecoxib son casi dos veces mayores a las de controles equiparables. (Véase sección Posología y método de administración).
Daño renal: En voluntarios adultos mayores con reducciones del tasa de filtración glomerular (TFG) (TFG promedio > 65 mL/min/1.73 m2) relacionadas con la edad y en pacientes con insuficiencia renal crónica estable (TFG 35-60 mL/min/1.73 m2), la farmacocinética de celecoxib era comparable a la observada en pacientes con función renal normal. No se encontró una relación significativa entre la creatinina sérica (o depuración de creatinina) y la depuración de celecoxib. No se esperaría que la insuficiencia renal grave alterara la depuración de celecoxib, ya que la principal vía de depuración es el metabolismo hepático hasta metabolitos inactivos.
Efectos renales: No se comprenden por completo las funciones relativas de COX-1 y COX-2 en la fisiología renal. Celecoxib disminuye la excreción urinaria de PGE2 y 6-ceto-PGF1∞ (un metabolito de la prostaciclina), pero no afecta el nivel sérico de tromboxano B2 (TXB2) ni la excreción urinaria de 11-dehidro-TXB2), un metabolito del tromboxano (ambos productos de COX-1). Estudios específicos muestran que celecoxib no produce descensos en la TFG en los adultos mayores ni en personas con insuficiencia renal crónica. Estos estudios también muestran disminuciones transitorias en la excreción fraccional de sodio. En estudios de pacientes con artritis, se observó una incidencia comparable de edema periférico a la que se observa con inhibidores inespecíficos de COX (que también tienen actividad inhibidora COX-2). Esto resultó más evidente en pacientes que reciben tratamiento diurético concomitante. Sin embargo, no se ha observado aumento en la incidencia de hipertensión ni insuficiencia cardiaca, y el edema periférico ha sido leve y auto limitado.
CONTRAINDICACIONES:
Hipersensibilidad al principio activo, o alguno de sus excipientes.
Hipersensibilidad a sulfonamidas.
Ulceración péptica activa o hemorragia gastrointestinal, antecedente de enfermedad ácido péptica.
Broncoespasmo, rinitis aguda, pólipos nasales y edema angioneurótico.
Reacciones alérgicas a ácido acetilsalicílico (aspirina) o AINE, incluyendo inhibidores específicos de la COX-2.
Disfunción hepática severa (albúmina sérica < 25 g/L o Child-Pugh ≥ 10).
Disfunción ventricular izquierda.
Insuficiencia cardiaca congestiva severa (clase funcional IV según la clasificación de la Asociación Cardiaca de Nueva York, NYHA).
Cirugía de derivación arterial coronaria (Bypass) (ver sección Advertencias especiales y precauciones para uso).
Insuficiencia renal severa (pacientes con aclaramiento de creatinina < 30 mL/min).
Enfermedad inflamatoria intestinal.
Embarazo.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Fertilidad, embarazo y lactancia:
Fertilidad:
Con base en el mecanismo de acción, la utilización de AINE, incluido el celecoxib, puede retrasar o evitar la rotura de los folículos ováricos, lo que a su vez se ha asociado con infertilidad reversible en algunas mujeres. En mujeres con dificultad para concebir o que se encuentran bajo exámenes por infertilidad, deberá considerarse el retiro de los AINE, incluido celecoxib.
Embarazo:
No hay estudios en mujeres embarazadas. Los estudios con animales muestran toxicidad reproductiva (véase sección Datos preclínicos de seguridad). Se desconoce la relevancia de estos datos para los humanos.
Como otros fármacos que inhiben la síntesis de prostaglandinas, celecoxib puede causar inercia uterina y cierre prematuro del conducto arterioso, por lo que se debe evitar en el tercer trimestre del embarazo.
Celecoxib se debe usar durante el embarazo sólo si el beneficio potencial para la madre justifica el riesgo para el feto.
La inhibición de la síntesis de prostaglandina puede afectar adversamente el embarazo.
Los datos de los estudios epidemiológicos sugieren aumento del riesgo de aborto espontáneo después de la utilización de inhibidores de la síntesis de prostaglandina al inicio del embarazo. En animales, la administración de inhibidores de síntesis de prostaglandina ha demostrado producir aumento de la pérdida pre y posimplantación.
Si se usan durante el segundo o tercer trimestre del embarazo, los AINE pueden causar disfunción renal fetal que pueden resultar en la reducción del volumen de líquido amniótico u oligohidramnios en casos severos. Tales efectos pueden ocurrir poco después del inicio del tratamiento y son usualmente reversibles. Las mujeres embarazadas tratadas con celecoxib deben ser monitoreadas de cerca en cuanto al volumen del líquido amniótico.
Lactancia:
Estudios en ratas muestran que celecoxib se excreta en la leche en concentraciones similares a las plasmáticas. La administración de celecoxib a mujeres lactantes muestra transferencia muy baja del fármaco a la leche materna. Por la posibilidad de reacciones adversas en lactantes por el celecoxib, se debe decidir si se suspende la lactancia o se suspende el fármaco, después de considerar el beneficio esperado del fármaco para la madre.
EFECTO EN LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS:
Efectos en la capacidad para conducir y usar maquinaria:
No se ha estudiado el efecto de celecoxib en la capacidad para conducir o usar maquinaria, Sin embargo, los pacientes que experimenten mareos, vértigo o somnolencia mientras toman celecoxib deben abstenerse de conducir o manejar maquinaria.
REACCIONES ADVERSAS:
Efectos indeseables:
Experiencia de los estudios clínicos:
Las siguientes reacciones adversas del medicamento de la Tabla 1 se identificaron con una incidencia mayor a 0.01% en el grupo de celecoxib y superiores a los resultados en el grupo placebo durante 12 estudios clínicos controlados con placebo y/o productos activos con duración de tratamiento de hasta 12 semanas y dosis diarias de 100 mg a 800 mg en adultos.
Las frecuencias de las reacciones adversas del medicamento en la Tabla 1 están actualizadas según los datos de 89 estudios clínicos aleatorizados controlados más recientes, que representan la exposición clínica de 38.102 pacientes que tomaron celecoxib. Las frecuencias de los eventos adversos se definen como: muy común (≥ 10%), común (≥ 1% y < 10%), poco común (≥ 0,1% y < 1%), raro (≥ 0,01% y < 0,1%), muy raro (<0,01%). Las reacciones adversas se listan por sistema orgánico y se ordenan por frecuencia en la tabla 1.
Tabla 1. Reacciones adversas al medicamento en 12 ensayos clínicos controlados con placebo y/o activos y frecuencia de reacciones adversas al medicamento de 89 estudios clínicos de dolor e inflamación aleatorizados y controlados con dosis diarias de 25 mg - 800 mg en poblaciones adultas
|
Clase de sistema orgánico Frecuencia |
Reacción farmacológica adversa |
|---|---|
|
Infecciones e infestaciones |
|
|
Común |
Bronquitis, sinusitis, infección del tracto respiratorio superior, infección del tracto urinario |
|
Poco común |
Faringitis, rinitis |
|
Trastornos sanguíneos y del sistema linfático |
|
|
Poco común |
Anemia |
|
Raro |
Trombocitopenia |
|
Trastornos del sistema inmunitario |
|
|
Poco común |
Hipersensibilidad |
|
Trastornos psiquiátricos |
|
|
Común |
Insomnio |
|
Poco común |
Ansiedad |
|
Raro |
Estado confusional |
|
Trastornos del sistema nervioso |
|
|
Común |
Mareo |
|
Poco común |
Hipertonía, somnolencia |
|
Trastornos oculares |
|
|
Poco común |
Visión borrosa |
|
Trastornos óticos y laberínticos |
|
|
Poco común |
Tinnitus |
|
Trastornos cardiacos |
|
|
Poco común |
Palpitaciones |
|
Raro |
Insuficiencia cardiaca congestiva, arritmia, taquicardia |
|
Trastornos vasculares |
|
|
Común Raro |
Hipertensión (incluida la hipertensión agravada) Rubefacción |
|
Trastornos respiratorios, torácicos y mediastinales |
|
|
Común |
Tos |
|
Trastornos gastrointestinales |
|
|
Común |
Vómito, dolor abdominal, diarrea, dispepsia, flatulencia |
|
Poco común |
Úlcera gástrica, trastorno dental |
|
Raro |
Úlcera duodenal, úlcera esofágica |
|
Muy raro |
Perforación intestinal, pancreatitis |
|
Trastornos hepatobiliares |
|
|
Poco común |
Aumento de enzimas hepáticas (incluye aumento de la alanina-aminotransferasa y aumento de la aspartato-aminotransferasa) |
|
Trastornos de piel y tejido subcutáneo |
|
|
Común |
Prurito (incluye prurito generalizado), erupción |
|
Poco común |
Urticaria, equimosis |
|
Raro |
Angioedema, alopecia |
|
Muy raro |
Dermatitis ampollosa |
|
Trastornos generales y alteraciones en sitio de administración |
|
|
Común |
Edema periférico |
|
Poco común |
Edema facial, enfermedad similar a gripe (enfermedad pseudogripal) |
|
Lesión, intoxicación y alteraciones en los procedimientos |
|
|
Poco común |
Lesión |
Las siguientes reacciones adversas al medicamento adicionales* de la Tabla 2 fueron identificadas con una incidencia mayor a la del placebo en estudios a largo plazo sobre prevención de pólipos, que tuvieron una duración de hasta 3 años con dosis diarias entre 400 mg hasta 800 mg (véase sección Propiedades farmacodinámicas, Seguridad cardiovascular, Estudios a largo plazo en pacientes con pólipos adenomatosos esporádicos). Las frecuencias se definen como: muy común (≥ 10%), común (≥ 1% y < 10%), poco común (≥ 0.1% y < 1%). Las reacciones adversas al medicamento en la Tabla 2 se listan por clase de sistema orgánico y se ordenan por frecuencia en orden descendiente.
Tabla 2. Reacciones adversas de estudios de prevención de pólipos con una duración hasta de 3 años y dosis diarias de 400 mg – 800 mg
|
Clase de sistema orgánico Frecuencia |
Reacción farmacológica adversa |
|---|---|
|
Infecciones e infestaciones |
|
|
Común |
Infección ótica, infección micótica** |
|
Poco común |
Infección por Helicobacter, herpes zóster, erisipela, infección de herida, gingivitis, laberintitis, infección bacteriana |
|
Neoplasmas benignos, malignos y no especificados |
|
|
Poco común |
Lipoma |
|
Trastornos psiquiátricos |
|
|
Poco común |
Trastorno del sueño |
|
Trastornos del sistema nervioso |
|
|
Poco común |
Infarto cerebral |
|
Trastornos oculares |
|
|
Poco común |
Hemorragia conjuntival, flotantes en vítreo |
|
Trastornos óticos y laberínticos |
|
|
Poco común |
Hipoacusia |
|
Trastornos cardiacos |
|
|
Común |
Infarto miocárdico, angina de pecho |
|
Poco común |
Angina inestable, insuficiencia valvular aórtica, arterioesclerosis arterial coronaria, bradicardia sinusal, hipertrofia ventricular |
|
Trastornos vasculares |
|
|
Muy común |
Hipertensión*, (incluyendo empeoramiento de la hipertensión) |
|
Poco común |
Trombosis venosa profunda, hematoma |
|
Trastornos respiratorios, torácicos y mediastinales |
|
|
Común |
Disnea |
|
Poco común |
Disfonía |
|
Trastornos gastrointestinales |
|
|
Muy común |
Diarrea* |
|
Común |
Vómito, disfagia, síndrome de intestino irritable, enfermedad por reflujo gastroesofágico, náusea, divertículo. |
|
Poco común |
Hemorragia hemorroidal, evacuaciones intestinales frecuentes, ulceración bucal, estomatitis |
|
Trastornos hepatobiliares |
|
|
Común |
Elevación de enzimas hepáticas (como la alanina-aminotransferasa y aumento de la aspartato-aminotransferasa)* |
|
Trastornos de piel y tejido subcutáneo |
|
|
Poco común |
Dermatitis alérgica |
|
Trastornos musculoesqueléticos y de tejido conectivo |
|
|
Común |
Espasmos musculares |
|
Poco común |
Quiste sinovial |
|
Trastornos renales y urinarios |
|
|
Común |
Nefrolitiasis |
|
Poco común |
Nicturia |
|
Trastornos mamarios y del sistema reproductor |
|
|
Común |
Hemorragia vaginal, prostatitis, hiperplasia prostática benigna |
|
Poco común |
Quiste ovárico, síntomas menopáusicos, sensibilidad mamaria, dismenorrea |
|
Trastornos generales y alteraciones en sitio de administración |
|
|
Poco común |
Edema |
|
Investigaciones |
|
|
Común |
Aumento de creatinina sanguínea, aumento de antígeno prostático específico, aumento de peso |
|
Poco común |
Aumento de potasio sanguíneo, aumento de sodio sanguíneo, descenso de testosterona sanguínea, descenso de hematocrito, aumento de hemoglobina |
|
Lesión, intoxicación y complicaciones de procedimientos |
|
|
Poco común |
Fractura de pie, fractura de extremidad inferior, fractura, epicondilitis, rotura tendinosa |
* La hipertensión, vómito, diarrea y aumento de enzimas hepáticas se incluyen en la Tabla 2 porque se informaron con mayor frecuencia en estos estudios, que tuvieron una duración de 3 años, en comparación con la Tabla 1, que incluye las reacciones adversas de estudios de 12 semanas de duración.
** Las infecciones micóticas fueron principalmente no sistémicas.
Tabla 3. Otras reacciones adversas en estudios clínicos y poscomercialización
|
Clase de sistema orgánico Frecuencia |
Reacción farmacológica adversa |
|
Trastornos psiquiátricos |
|
|
Poco común |
Depresión, fatiga |
|
Trastornos respiratorios |
|
|
Común |
Rinitis |
|
Trastornos renales |
|
|
Poco común |
Creatinina elevada en sangre, urea elevada en sangre |
|
Trastornos del sistema nervioso |
|
|
Común |
Hipertonía, cefalea |
|
Poco común |
Parestesia |
|
Trastornos cardiacos |
|
|
Poco común |
Insuficiencia cardiaca, taquicardia |
Experiencia posterior a la comercialización:
A continuación se presentan las reacciones adversas identificadas por la experiencia posterior a la comercialización. A pesar de que fueron identificadas como reacciones de los informes de poscomercialización, se consultaron los datos de los ensayos para estimar la frecuencia. Como se citó previamente, las frecuencias se basan en una agrupación de ensayos que representan la exposición en 38.102 pacientes. Las frecuencias se definen como: muy común (≥ 10%), común (≥ 1% y < 10%), poco común (≥ 0,1% y < 1%), raro (≥ 0,01% y < 0,1%), muy raro (< 0,01%), no conocida (no puede estimarse a partir de los datos disponibles).
Trastornos del sistema inmunitario: Muy raro: reacción anafiláctica
Trastornos psiquiátricos: Raro: alucinaciones.
Trastornos del sistema nervioso: Muy raro: hemorragia cerebral, meningitis aséptica, ageusia, anosmia.
Trastornos oculares: Poco común: conjuntivitis.
Trastornos vasculares: Muy raro: vasculitis.
Trastornos respiratorios, torácicos y mediastinales: Raro: embolismo pulmonar, neumonitis.
Trastornos gastrointestinales: Raro: hemorragia gastrointestinal.
Trastornos hepatobiliares: Raro: hepatitis, Muy raro: insuficiencia hepática, hepatitis fulminante, necrosis hepática (véase sección Advertencias especiales y precauciones para uso; Efectos hepáticos), colestasis, hepatitis colestásica, ictericia.
Trastornos de piel y tejido subcutáneo: Raro: reacción por fotosensibilidad; Muy raro: síndrome de Stevens-Johnson, eritema multiforme, necrólisis epidérmica tóxica, reacción del medicamento con eosinofilia y síntomas sistémicos (DRESS o síndrome de hipersensibilidad), pustulosis exantemática aguda generalizada (AGEP), dermatitis exfoliativa.
Trastornos renales y urinarios: Raro: insuficiencia renal aguda (véase sección Advertencias especiales y precauciones para su uso, Efectos renales), hiponatremia; Muy raro: nefritis tubulointersticial, síndrome nefrótico, glomerulonefritis de lesión mínima.
Trastornos mamarios y del sistema reproductor: Raro: trastorno menstrual desconocido:
Infertilidad femenina (disminución de la fertilidad femenina) (ver sección Fertilidad, embarazo y lactancia)†.
Trastornos generales y condiciones en el sitio de administración: Poco común: dolor en el pecho.
† Se excluyeron de todos los ensayos a las mujeres que deseaban quedar embarazadas, por lo tanto no tenía sentido consultar la base de datos de los ensayos respecto de la frecuencia de este evento.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacción con otros productos medicinales y otras formas de interacción:
General:
El metabolismo de celecoxib está mediado predominantemente por el citocromo P450 (CYP) 2C9 en el hígado. Celecoxib se debe usar con cuidado en los pacientes con certeza o sospecha de metabolismo deficiente por CYP2C9 con base en antecedentes o experiencias previas con otros sustratos de CYP2C9, ya que pueden alcanzar niveles plasmáticos más altos de lo normal por la menor depuración metabólica. Considerar el inicio del tratamiento con la dosis más baja recomendada (Véanse ecciones Posología y método de administración; y Metabolismo).
La administración concomitante de celecoxib con inhibidores de CYP2C9 puede conducir a un aumento de las concentraciones plasmáticas de celecoxib. Por lo tanto, una reducción de la dosis de celecoxib puede ser necesaria cuando celecoxib se coadministra con inhibidores del CYP2C9.
La administración concomitante de celecoxib con inductores del CYP2C9 como rifampicina, carbamazepina y barbitúricos puede conducir a una disminución en las concentraciones plasmáticas de celecoxib. Por lo que un aumento de la dosis de celecoxib puede ser necesaria cuando celecoxib está coadministrado con inductores del CYP2C9.
Estudios de farmacocinética clínica e in vitro indican que aunque celecoxib no es un sustrato, es inhibidor de CYP2D6, existe la posibilidad de una interacción farmacológica in vivo con medicamentos metabolizados por CYP2D6.
Específicas por fármaco:
Interacción de celecoxib con warfarina o agentes similares: (Véase sección Advertencias especiales y precauciones para uso, Uso con anticoagulantes orales).
Litio: En sujetos sanos, los niveles plasmáticos de litio aumentaron aproximadamente 17% en pacientes que recibieron litio junto con celecoxib. Los pacientes bajo tratamiento con litio deben ser vigilados muy de cerca cuando se inicie o se suspenda el tratamiento con celecoxib.
Ácido acetilsalicílico: El celecoxib no interfiere con el efecto antiplaquetario del ácido acetilsalicílico a dosis baja. (Ver sección Advertencias especiales y precauciones para uso, efectos gastrointestinales (GI). Debido a su falta de efectos plaquetarios, celecoxib no es un sustituto del ácido acetilsalicílico en el tratamiento profiláctico de la enfermedad CV. La administración concomitante de la aspirina con CELEBREX puede ocasionar un aumento en la tasa de ulceración gastrointestinal u otras complicaciones, en comparación con el uso de celecoxib solo.
Antihipertensivos incluyendo los inhibidores de enzimas convertidoras de la angiotensina (IECA), los antagonistas de la angiotensina II (conocidos también como bloqueadores de los receptores de angiotensina, ARAII), diuréticos y beta-bloqueadores: La inhibición de prostaglandinas, puede disminuir el efecto de antihipertensivos como los IECA y/o los ARAII, los diuréticos y los beta-bloqueadores. Esta interacción se debe considerar en pacientes que toman celecoxib al mismo tiempo que los IECA y/o los ARA II, los diuréticos y los beta-bloqueadores T.
En pacientes ancianos, con volumen disminuido (incluidos los que están bajo tratamiento diurético) o con compromiso de la función renal, la coadministración de AINE, entre ellos los inhibidores selectivos de COX-2, con antagonistas de angiotensina II, diuréticos o con inhibidores de la ECA, puede conllevar a deterioro de la función renal, incluida posible insuficiencia renal aguda. Usualmente estos efectos son reversibles. Por lo tanto, la administración concomitante de estos medicamentos debe realizarse con precaución. Los pacientes deben estar adecuadamente hidratados y se debe evaluar la necesidad clínica de monitorear la función renal al inicio del tratamiento concomitante y periódicamente después.
Los resultados del estudio lisinopril: En un estudio clínico controlado con lisinopril de 28 días en pacientes con hipertensión estadio I y II, la administración de 200 mg de celecoxib dos veces al día no produjo aumentos clínicamente significativos, en comparación con el tratamiento con placebo, de la presión arterial sistólica o diastólica media diaria tal como se determina mediante el control de la presión arterial ambulatoria de 24 horas. Entre los pacientes coadministrados con celecoxib 200 mg dos veces al día, 48% se consideraron que no respondían a lisinopril en la visita clínica final (definido como presión arterial diastólica con esfigmomanómetro de presión arterial > 90 mmHg o aumento de > 10% en la presión arterial diastólica con con esfigmomanómetro de presión arterial en comparación con el valor basal), en comparación con 27% de los pacientes coadministrados con placebo; esta diferencia fue estadísticamente significativa.
Ciclosporina: Debido al efecto que tienen en las prostaglandinas renales, los AINE pueden incrementar el riesgo de nefrotoxicidad si se administran con ciclosporina.
Fluconazol y ketoconazol: La administración concomitante de fluconazol, a dosis de 200 mg una vez al día, provocó un incremento del doble en la concentración plasmática de celecoxib. Este incremento se debe a la inhibición, por parte de fluconazol, del metabolismo de celecoxib a través del CYP2C9. La administración de celecoxib debe iniciarse a la mitad de la dosis recomendada en pacientes que están recibiendo el inhibidor del CYP2C9, fluconazol (ver sección Posología y método de administración). Ketoconazol, un inhibidor del CYP3A4, no demostró una inhibición clínicamente relevante en el metabolismo de celecoxib.
Dextrometorfano y metoprolol: La administración concomitante de 200 mg de celecoxib dos veces al día resultó en un aumento de 2,6 veces y un aumento de 1,5 veces de la concentración plasmática de dextrometorfano y metoprolol (sustratos del CYP2D6), respectivamente. Estos incrementos se deben a la inhibición de celecoxib para el metabolismo del sustrato de la CYP2D6 vía CYP2D6. Por lo tanto, la dosis de los fármacos, como los sustratos de CYP2D6 puede necesitar una reducción cuando el tratamiento con celecoxib se ha iniciado o un incremento cuando el tratamiento con celecoxib se termine. (Vea sección Advertencias especiales y precauciones para uso, Uso con anticoagulantes orales).
Diuréticos: Los estudios clínicos han mostrado que en algunos pacientes los AINE disminuyen el efecto natriurético de la furosemida y las tiazidas por inhibición de la síntesis renal de prostaglandinas.
Metotrexato: No se han observado interacciones farmacocinéticas ni clínicamente importantes en un estudio clínico entre celecoxib y metotrexato. El uso concomitante de AINE y metotrexato puede aumentar el riesgo de toxicidad por metotrexato (p. ej., neutropenia, trombocitopenia, disfunción renal). Durante el uso concomitante de CELEBREX y metotrexato, los pacientes deben ser monitoreados para detectar toxicidad por metotrexato.
Anticonceptivos orales: En un estudio de interacción, el celecoxib no tuvo efectos de relevancia clínica en la farmacocinética de una combinación prototípica de anticonceptivo oral (1 mg de noretindrona /0,035 mg de etinilestradiol).
Antiácidos (de aluminio y magnesio): Ver sección Propiedades farmacocinéticas.
Otros medicamentos: No se han observado interacciones de importancia clínica entre celecoxib y antiácidos (de aluminio y magnesio), omeprazol, glibenclamida (gliburida), fenitoína o tolbutamida.
HALLAZGOS DE LABORATORIO CLÍNICO:
Datos preclínicos de seguridad:
Los datos de seguridad obtenidos en estudios preclínicos no revelan peligros particulares para los humanos con base en estudios convencionales de toxicidad con dosis repetidas, mutagenicidad o carcinogenicidad.
Celecoxib en dosis orales ≥ 150 mg/kg/día (aproximadamente 2 veces la exposición habitual en humanos con dosis de 200 mg dos veces al día, según el ABC0-24 provocó un aumento en la incidencia de defectos del septo ventricular, un evento raro, así como alteraciones fetales como la fusión de costillas y fusión o deformación del cuerpo del esternón, cuando se usó como tratamiento en conejos durante la organogénesis. Se observó un aumento de hernias diafragmáticas dependiente de la dosis de celecoxib cuando se administró a ratas en periodo de organogénesis, en dosis orales de ≥ 30 mg/kg/día (aproximadamente 6 veces la exposición habitual en humanos a dosis de 200 mg dos veces al día, según el ABC0-24). Estos efectos son esperables después de la inhibición de la síntesis de prostaglandina. En ratas, la exposición al celecoxib durante el desarrollo embrionario temprano provocó pérdidas pre y posimplantación y redujo la supervivencia embrionaria/fetal.
Toxicología animal: Se observó un incremento en la incidencia de hallazgos de base de espermatocele con o sin cambios secundarios como hipospermia epididimal, así como dilatación mínima a leve de los túbulos seminíferos en la rata joven. Estos hallazgos reproductivos, aunque aparentemente estaban relacionados con el tratamiento, no se incrementaron en cuanto a incidencia ni a severidad con la dosis y pueden indicar una exacerbación de una condición espontánea. No se observaron hallazgos reproductivos similares en estudios de perros jóvenes o adultos o en ratas adultas que recibieron celecoxib. Se desconoce la significación clínica de esta observación.
PRECAUCIONES Y ADVERTENCIAS:
Advertencias especiales y precauciones para uso:
Lactancia, insuficiencia hepática moderada, hiperlipidemia, diabetes, fumadores, enfermedad arterial periférica. Se recomienda iniciar el tratamiento con las dosis más bajas.
El uso concomitante con el ácido acetilsalicílico (ASA) incrementa el riesgo de úlcera gastrointestinal y sus complicaciones. Adminístrese con precaución a pacientes tratados con warfarina por cuanto los mismos tienen mayor riesgo de complicaciones por sangrado.
Efectos cardiovasculares:
Eventos trombóticos cardiovasculares: Celecoxib puede causar un incremento en el riesgo de eventos trombóticos CV serios, infarto miocárdico (IM) y eventos cerebrovasculares, los cuales pueden ser fatales. Todos los AINE tienen un riesgo similar. El riesgo se puede incrementar con la dosis y duración del uso. El aumento relativo de este riesgo parece ser similar en aquellos con o sin enfermedad CV conocida o factores de riesgo CV. Sin embargo, los pacientes con enfermedad CV o factores de riesgo CV tienen un mayor riesgo en términos de incidencia absoluta, debido a su tasa de referencia incrementada. Para minimizar el riesgo potencial de un evento CV adverso en pacientes tratados con celecoxib, se debe usar la menor dosis efectiva durante el menor tiempo posible. Los médicos y los pacientes se deben mantener alerta ante el desarrollo de tales eventos, aún en ausencia de síntomas CV previos. Los pacientes deben estar informados sobre los signos y síntomas de toxicidad CV seria y los pasos a tomar en caso que aparezcan (Véase sección, Propiedades farmacodinámicas).
Dos estudios clínicos controlados de diferentes AINE selectivos de la COX-2 para el tratamiento del dolor durante los primeros 10 a 14 días después de un procedimiento CABG encontraron una mayor incidencia de infarto miocárdico y eventos cerebrovasculares. (Véase Sección Contraindicaciones).
Los pacientes con cardiopatía isquémica establecida, enfermedad arterial periférica y/o enfermedad cerebrovascular, o con factores de riesgo significativos de eventos cardiovasculares (p. ej., hipertensión, hiperlipidemia, diabetes mellitus, tabaquismo) solo deben tratarse con celecoxib después de una cuidadosa consideración (ver sección Propiedades farmacodinámicas).
Celecoxib no es un sustituto del ácido acetilsalicílico para la prevención de enfermedades tromboembólicas cardiovasculares por la falta de efecto en la función plaquetaria. Como celecoxib no inhibe la agregación plaquetaria, no se deben suspender los tratamientos antiplaquetarios (p. ej., ácido acetilsalicílico).
Hipertensión: Al igual que con todos los AINE, celecoxib puede provocar el inicio de hipertensión o empeorar la hipertensión preexistente; cualquiera de los dos casos puede contribuir a una mayor incidencia de eventos cardiovasculares CV. Los AINE, incluyendo celecoxib, se deben usar con precaución en pacientes con hipertensión. Se debe vigilar la presión arterial muy de cerca al iniciar y durante el curso del tratamiento con celecoxib. (Ver sección Propiedades farmacodinámicas – Estudios clínicos –Subestudio ABPM).
Retención de fluidos y edema: Al igual que con otros medicamentos que se sabe inhiben la síntesis de prostaglandinas, se ha observado retención de fluidos y edema en algunos pacientes que toman celecoxib. Por lo tanto, los pacientes con insuficiencia cardiaca congestiva (ICC) preexistente se deben vigilar de cerca. Celecoxib se debe usar con precaución en pacientes con una función cardiaca comprometida, edema preexistente u otras condiciones que predispongan a, o que se empeoran por retención de fluidos, incluyendo aquellos que toman diuréticos o que de otra manera están en riesgo de hipovolemia.
Efectos gastrointestinales (GI):
Ha habido casos de perforaciones (GI) superiores e inferiores, úlceras o sangrados en pacientes tratados con celecoxib. Los pacientes con mayor riesgo de desarrollar estos tipos de complicaciones GI con los AINE son los adultos mayores, pacientes con enfermedad CV, personas tomando concomitantemente medicamentos antiplaquetarios (como aspirina), glucocorticoides, u otros AINE, que beben alcohol al mismo tiempo y con antecedente o con alguna enfermedad GI activa, como ulceración, sangrado GI o trastornos inflamatorios. La mayoría de los reportes espontáneos de eventos GI mortales han sido en adultos mayores o pacientes debilitados.
Efectos renales:
Los AINE, incluyendo celecoxib, pueden causar toxicidad renal. Los estudios clínicos con celecoxib han demostrado efectos renales similares a los observados con AINE comparadores. Los pacientes en mayor riesgo de desarrollar toxicidad renal son aquellos con una función renal alterada, insuficiencia cardiaca, disfunción hepática y las personas de edad avanzada. Estos pacientes se deben vigilar cuidadosamente mientras reciben tratamiento con celecoxib.
Se debe tener precaución al iniciar tratamiento en pacientes deshidratados. Se recomienda rehidratar a los pacientes primero y después iniciar el tratamiento con celecoxib.
Enfermedad renal avanzada:
La función renal se debe vigilar de cerca en pacientes con enfermedad renal avanzada que reciben celecoxib. (Ver sección Posología y metodo de administración.) En los pacientes con insuficiencia renal severa, el medicamento se encuentra contraindicado. (Ver sección Contraindicaciones.)
Reacciones anafilactoides:
Al igual que con los AINE en general, se han reportado reacciones anafilactoides en pacientes expuestos a celecoxib (ver sección Contraindicaciones).
Reacciones cutáneas serias:
Muy rara vez se han reportado reacciones cutáneas serias, algunas de ellas fatales, incluyendo reacción por medicamentos con eosinofilia y síntomas sistémicos (síndrome DRESS, por sus siglas en inglés), dermatitis exfoliativa, síndrome de Stevens-Johnson y necrólisis epidérmica tóxica en asociación con el uso de celecoxib. Los pacientes parecen estar en mayor riesgo de desarrollar estos eventos tempranamente durante el tratamiento: en la mayoría de los casos, el evento inicia en el primer mes de tratamiento. Celecoxib se debe suspender a la primera aparición de exantema cutáneo, lesiones mucosales o de cualquier otro signo de hipersensibilidad.
Efectos hepáticos:
No se han estudiado pacientes con daño hepático severo (clase C de Child-Pugh). El celecoxib se debe usar con precaución en el tratamiento de pacientes con daño hepático moderado (clase B de Child-Pugh) e iniciar con la mitad de la dosis recomendada. (Véase sección Posología y método de administración).
Rara vez se han reportado casos de reacciones hepáticas severas, incluyendo hepatitis fulminante (algunos con resultados fatales), necrosis hepática, insuficiencia hepática (algunos con resultado fatal o con la necesidad de un trasplante de hígado) con celecoxib.
Un paciente con síntomas y/o signos de disfunción hepática o aquel con resultados anormales en las pruebas de función hepática se debe vigilar de cerca para detectar la evidencia del desarrollo de una reacción hepática más grave durante el tratamiento con celecoxib.
Uso con anticoagulantes orales:
El uso concomitante de AINE con anticoagulantes orales incrementa el riesgo de sangrado y deben ser administrados con precaución. Los anticoagulantes orales incluyen warfarina/tipo cumarina y anticoagulantes orales nuevos (p. ej., apixabán, dabigatrán y rivaroxabán). En pacientes a los que se les administre concomitantemente con AINE, warfarina o agentes similares, han reportado eventos hemorrágicos serios, algunos de ellos mortales. Como hay informes de aumento en el tiempo de protrombina (INR), la actividad anticoagulante/INR en pacientes que toman anticoagulantes como warfarina/tipo cumarina se debe vigilar después de iniciar el tratamiento con celecoxib o cuando se modifica la dosis (ver sección Interacción con otros productos medicinales y otras formas de interacción).
General:
Al reducir la inflamación, celecoxib puede reducir la utilidad de los signos diagnósticos, como fiebre, para detectar infecciones.
Se debe evitar el uso concomitante de celecoxib y de un AINE que no sea ácido acetilsalicílico.
Inhibición del CYP2D6:
Celecoxib ha mostrado ser un inhibidor moderadamente potente del CYP2D6. Para fármacos que son metabolizados por el CYP2D6 puede ser necesaria una reducción de la dosis durante el inicio del tratamiento con celecoxib o un incremento de la dosis hacia el término del tratamiento con celecoxib (ver sección Interacción con otros productos medicinales y otras formas de interacción).
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología y método de administración:
Forma de administración: Vía oral.
Se puede tomar celecoxib cápsulas, en dosis de hasta 200 mg dos veces al día, con o sin alimentos.
Dado que los riesgos cardiovasculares (CV) de celecoxib pueden incrementarse con la dosis y la duración del tratamiento, se debe utilizar por la duración más corta posible y en la dosis diaria efectiva más baja. Se debe reevaluar periódicamente la necesidad de alivio sintomático y la respuesta al tratamiento, especialmente en pacientes con artrosis.
Adultos:
Artrosis (osteoartritis-OA): La dosis recomendada de celecoxib es 200 mg por día, administrada como dosis única o en dos tomas. En ocasiones si se requiere se puede administrar 200 mg cada 12 horas. Si no se observa beneficio terapéutico a las 2 semanas. Se debe considerar otras alternativas terapéuticas.
Artritis reumatoide (AR): La dosis diaria inicial recomendada es de 200 mg administrados en dos tomas. Posteriormente, si fuera necesario, la dosis puede ser incrementada a 200 mg dos veces al día. Si transcurridas 2 semanas, no se observara un incremento del beneficio terapéutico, se deben considerar otras alternativas terapéuticas.
Espondilitis anquilosante (EA): La dosis diaria recomendada es de 200 mg, administrados como dosis única o en dos tomas. En casos de alivio insuficiente de los síntomas, puede incrementarse la dosis a 400 mg de forma ocasional, administrada una vez al día o dividida en dos tomas. Si transcurridas 2 semanas, no se observara un incremento del beneficio terapéutico, se deben considerar otras alternativas terapéuticas.
Tratamiento del dolor agudo: La dosis de celecoxib recomendada es 400 mg el primer día. Seguida por 200 mg una vez al día en los días subsiguientes hasta un máximo de 7 días. Se puede indicar a los pacientes que tomen una dosis adicional de 200 mg en un día determinado, si es necesario. Dosis máxima = 400 mg al día hasta 7 días.
Tratamiento de dismenorrea primaria: La dosis de celecoxib recomendada es 400 mg el primer día. Seguida por 200 mg una vez al día en los días subsiguientes hasta un máximo de 7 días. Se puede indicar a los pacientes que tomen una dosis adicional de 200 mg en un día determinado, si es necesario. Dosis máxima = 400 mg al día hasta 7 días.
Dolor de espalda baja (LBP): La dosis de celecoxib recomendada de celecoxib es 200 o 400 mg diarios, administrados como una única dosis de 200 mg o 200 mg dos veces al día. Algunos pacientes pueden beneficiarse de una dosis diaria total de 400 mg.
La dosis diaria máxima recomendada es de 400 mg para todas las indicaciones.
Poblaciones especiales:
Metabolización lenta por el citocromo CYP2C9: celecoxib se debe administrar con precaución en aquellos pacientes que presenten, o se sospeche que pueda presentar una metabolización lenta por el citocromo CYP2C9 con base en los genotipos o antecedentes/experiencias previas con otros sustratos de CYP2C9, dado que aumenta el riesgo de presentar reacciones adversas dosis-dependientes, debe administrar celecoxib con precaución. Considere iniciar el tratamiento con la mitad de la dosis más baja recomendada. (Véanse secciones, Interacción con otros productos medicinales y otras formas de interacción; y Metabolismo).
Pacientes de edad avanzada (con una edad superior a 65 años): como en el caso de adultos más jóvenes, se debe utilizar inicialmente la dosis de 200 mg al día. Si fuera necesario, la dosis puede incrementarse posteriormente a 200 mg dos veces al día. Generalmente no es necesario el ajuste de la dosis. Sin embargo, se deberá tener especial precaución con aquellos pacientes de edad avanzada con un peso inferior a 50 kg, es aconsejable iniciar la terapia a la dosis más baja recomendada.
Población pediátrica: No está indicado el uso de celecoxib en niños.
Método de administración:
Para los pacientes con dificultad para deglutir las cápsulas, el contenido de una cápsula de celecoxib se puede añadir a una papilla de manzana, colada de arroz, yogurt o a una papilla de banano. Para hacer esto, es necesario vaciar con cuidado el contenido completo de la cápsula en una cucharadita de papilla de manzana, cereal de arroz, yogurt o papilla de banano fría o a temperatura ambiente y se debe ingerir de inmediato con agua. El contenido de la cápsula mezclado con papilla de manzana, colada de arroz o yogurt es estable hasta por 6 horas en refrigeración (2-8° C/35-45° F). El contenido de la cápsula mezclado en la papilla de banano no se debe refrigerar y se debe ingerir de inmediato.
Insuficiencia hepática: En pacientes con insuficiencia hepática moderada establecida (albúmina sérica de 25 a 35 g/L), (Child-Pugh B) el tratamiento debe iniciarse con la mitad de la dosis recomendada. En estos pacientes la experiencia está limitada a cirróticos. Los pacientes con daño hepático severo (clase C de Child-Pugh) no se han estudiado. (Véase la sección Advertencias especiales y precauciones para uso, Efectos hepáticos.)
Insuficiencia renal: Se dispone de experiencia limitada en la administración de celecoxib a los pacientes con insuficiencia renal leve o moderada. Por lo tanto, estos pacientes deben ser tratados con precaución. No hay evidencia de seguridad ni eficacia en pacientes con insuficiencia renal severa por lo que está contraindicado. (Véase la sección Advertencias especiales y precauciones para uso, Efectos renales.)
Administración concomitante con fluconazol: Celecoxib se debe introducir en la mitad de la dosis recomendada en pacientes que reciben fluconazol, un inhibidor de CYP2C9. Se recomienda tener precaución cuando se administra celecoxib al mismo tiempo que otros inhibidores de CYP2C9. (Véase sección, Interacción con otros productos medicinales y otras formas de interacción).
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Sobredosis:
La experiencia clínica con sobredosis es limitada. Se han administrado dosis únicas de hasta 1200 mg y repetidas de hasta 1200 mg dos veces al día a sujetos sanos sin efectos adversos de importancia clínica. En caso de sospecha de sobredosis, se debe brindar la atención médica de apoyo apropiada. Es improbable que la diálisis sea un método efectivo para eliminar el fármaco por el alto porcentaje de unión con proteínas de este fármaco.
PRESENTACIÓN:
Caja con 10 y 20 cápsulas de 200 mg.
ASPEN LABS


