BRILINTA
TICAGRELOR
Comprimidos recubiertos
1 Caja,30 Comprimidos recubiertos,90 mg
1 Caja,20 Comprimidos recubiertos,90 mg
Para visualizar el contenido completo de la IPPA (información para prescribir amplia), deberá iniciar sesión.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Cada COMPRIMIDO contiene 90 mg de ticagrelor.
Véase la sección “Lista de excipientes”.
INDICACIONES TERAPÉUTICAS: BRILINTA® está indicado para ser coadministrado con ácido acetilsalicílico (AAS) para la prevención de episodios atero-trombóticos en pacientes adultos con síndromes coronarios agudos (angina inestable, infarto de miocardio con o sin elevación del segmento ST), lo cual incluye a los pacientes que reciben un tratamiento médico y aquellos sometidos a angioplastia coronaria percutánea o a una intervención de revascularización coronaria por puenteo vascular.
MECANISMO DE ACCIÓN: BRILINTA® contiene ticagrelor, un miembro de la clase química ciclopentiltriazolopirimidinas (CPTP), el cual es un antagonista selectivo y de unión reversible a receptor P2Y12 oral, de acción directa, que impide la activación y agregación plaquetaria dependiente de P2Y12 mediada por adenosina difosfato (ADP). Ticagrelor no impide la unión de ADP pero cuando se enlaza al receptor P2Y12 impide la transducción de señal inducida por ADP. Dado que las plaquetas participan en el inicio y/o evolución de complicaciones trombóticas de la enfermedad aterosclerótica, se ha demostrado que la inhibición de la función plaquetaria reduce el riesgo de eventos cardiovasculares tales como la muerte, infarto de miocardio o accidente cerebrovascular.
Ticagrelor tiene un mecanismo de acción adicional, incrementando los niveles locales de adenosina endógena al inhibir el transportador-1 nucleósido equilibrante (ENT-1). La adenosina se forma localmente en sitios de hipoxia y daño tisular a través de degradación de adenosina tri- y difosfato (ATP y ADP). Como la degradación de la adenosina está esencialmente restringida al espacio intracelular, la inhibición de ENT-1 por ticagrelor prolonga la vida media de la adenosina y, por lo tanto, aumenta su concentración extracelular local proveyendo respuestas locales de adenosina aumentadas. Ticagrelor no tiene efecto directo clínicamente significativo sobre receptores de adenosina (A1, A2A, A2B, A3) y no es metabolizado a adenosina. Se ha documentado que la adenosina tiene varios efectos que incluyen: Vasodilatación, cardioprotección, inhibición plaquetaria, modulación de la inflamación e inducción de disnea, que pueden contribuir al perfil clínico de ticagrelor.
DATOS FARMACÉUTICOS
Lista de excipientes
Núcleo: Manitol (E421), Fosfato de calcio dibásico, Estearato de magnesio, Glicolato de almidón sódico, Hidroxipropilcelulosa
Recubrimiento: Talco, Dióxido de titanio (E171), Óxido de hierro amarillo, Polietilenglicol 400, Hipromelosa.
FORMA FARMACÉUTICA: Comprimidos recubiertos de 90 mg, redondos, biconvexos, de color amarillo, con la marca “90” arriba de una “T” en una cara y la otra cara lisa.
PROPIEDADES FARMACOLÓGICAS
Grupo farmacoterapéutico (código ATC): B01AC24
Grupo farmacéutico: Inhibidores de la agregación plaquetaria, excluyendo heparina.
PROPIEDADES FARMACOCINÉTICAS
Generalidades: La farmacocinética del ticagrelor es lineal y la exposición a BRILINTA® y a su metabolito activo (AR-C124910XX) es aproximadamente proporcional a la dosis.
Absorción: La absorción de BRILINTA® es rápida, con una mediana de tmáx de aproximadamente 1.5 horas. La formación del principal metabolito circulante AR-C124910XX (que también es activo) a partir de BRILINTA® es rápida, con una mediana de tmax de aproximadamente 2.5 horas. La Cmáx y el ABC de BRILINTA® y de su metabolito activo aumentaron de manera aproximadamente proporcional a la dosis en todo el intervalo de dosis examinado (de 30 mg a 1260 mg).
Se calculó que la media de la biodisponibilidad absoluta de BRILINTA® era del 36% (mínimo 25.4%, máximo 64.0%). La ingestión de una comida rica en grasas no tuvo ningún efecto en la Cmáx de BRILINTA® ni en el ABC de su metabolito activo, pero aumentó un 21% el ABC de BRILINTA® y redujo un 22% la Cmáx de su metabolito activo. Se considera que estos pequeños cambios revisten una importancia clínica mínima, por lo que BRILINTA® puede administrarse con o sin alimentos.
Ticagrelor como tabletas trituradas mezcladas en agua, administradas vía oral a través de una sonda nasogástrica al estómago, es bioequivalente a las tabletas enteras (AUC y Cmáx dentro de 80%-125% para ticagrelor y el metabolito activo). La exposición inicial (0.5 y 1 hora post-dosis) con tabletas trituradas de ticagrelor mezcladas en agua, fue mayor en comparación con las tabletas enteras, con un perfil de concentración generalmente idéntico de ahí en adelante (2 a 48 horas).
Distribución: El volumen de distribución de BRILINTA® en el estado de equilibrio es de 87.5 litros. BRILINTA® y su metabolito activo se unen extensamente a las proteínas plasmáticas humanas (>99.0%).
Metabolismo: El CYP3A es el principal sistema enzimático responsable del metabolismo de BRILINTA® y de la formación del metabolito activo, y sus interacciones con otros sustratos de CYP3A varían desde la activación hasta la inhibición de los mismos. BRILINTA® y su metabolito activo son inhibidores débiles de la glucoproteína P.
El principal metabolito de BRILINTA® es el AR-C124910XX, que también es activo puesto que se une in vitro al receptor P2Y12 de ADP plaquetario. La exposición sistémica al metabolito activo representa aproximadamente entre un 30% y un 40% de la exposición a BRILINTA®.
Excreción: La principal vía de eliminación de BRILINTA® es el metabolismo hepático. Tras la administración de BRILINTA® radiactivo, la recuperación media de radiactividad es de aproximadamente un 84% (57.8% en las heces y 26.5% en la orina). Las cantidades de BRILINTA® y de su metabolito activo recuperadas en la orina fueron inferiores al 1% de la dosis. La principal vía de eliminación del metabolito activo es la secreción biliar. La vida media de eliminación (t½) de BRILINTA® fue de aproximadamente 6.9 horas en promedio (mínimo 4.5 horas, máximo 12.8 horas) y la del metabolito activo de 8.6 horas (mínimo 6.5 horas, máximo 12.8 horas).
Poblaciones especiales:
Pacientes de edad avanzada: Con respecto a sujetos jóvenes, los sujetos de edad avanzada (=65 años) mostraron exposiciones más elevadas a BRILINTA® (aumento de aproximadamente un 60% de la Cmáx y del ABC) y a su metabolito activo (aumento de aproximadamente un 50% de la Cmáx y del ABC). Se considera que estas diferencias carecen de importancia clínica (véase la sección “Posología y forma de administración”).
Población pediátrica: BRILINTA® no se ha estudiado en la población pediátrica (véase la sección “Posología y forma de administración”).
Sexo: Con respecto a los varones, las mujeres presentan exposiciones más elevadas a BRILINTA® (aumentos de aproximadamente un 52% de la Cmáx y un 37% del ABC) y al metabolito activo (aumento de aproximadamente un 50% de la Cmáx y del ABC) Se considera que estas diferencias carecen de importancia clínica.
Insuficiencia renal: La exposición a BRILINTA® en los pacientes con insuficiencia renal grave fue aproximadamente un 20% más baja y la exposición a su metabolito activo fue aproximadamente un 17% más alta que en los sujetos con una función renal normal. Si bien el efecto inhibidor de la agregación plaquetaria de BRILINTA® fue similar entre los dos grupos, se observó una mayor variabilidad de las respuestas individuales en los pacientes con insuficiencia renal grave. No es necesario ajustar la dosis en los pacientes con insuficiencia renal. No hay información disponible sobre el tratamiento de pacientes sometidos a diálisis renal (véase la sección “Posología y forma de administración”).
Insuficiencia hepática: La Cmáx y el ABC de BRILINTA® aumentaron un 12% y un 23%, respectivamente, en pacientes con insuficiencia hepática leve frente a sujetos sanos equiparados, aunque el efecto inhibidor de la agregación plaquetaria de BRILINTA® fue similar entre los dos grupos. No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. BRILINTA® no se ha investigado en pacientes con insuficiencia hepática moderada o grave (véase la sección “Posología y forma de administración”).
Raza: Los pacientes de ascendencia asiática presentan una biodisponibilidad media un 39% mayor que los pacientes de raza blanca. En pacientes que se autodeclararon de raza negra, la biodisponibilidad de BRILINTA® fue un 18% menor que en los pacientes de raza blanca. En los estudios de farmacología clínica, la exposición a BRILINTA® (Cmáx y ABC) en sujetos japoneses fue aproximadamente un 40% mayor que en los de raza blanca (un 20% tras efectuar el ajuste correspondiente al peso corporal).
EXPERIENCIA POST-COMERCIALIZACIÓN: Las siguientes reacciones adversas han sido identificadas durante el uso posterior a la aprobación de BRILINTA®. Debido a que estas reacciones son voluntariamente reportadas a partir de una población de tamaño desconocido, no siempre es posible estimar de forma exacta su frecuencia.
Trastornos del sistema inmunológico: Reacciones de hipersensibilidad incluyendo angioedema (ver sección “Contraindicaciones”).
Trastornos de la piel y del tejido subcutáneo: Erupción (rash).
Descripción de reacciones adversas al medicamento selectas
Hemorragia: En el estudio PLATO se emplearon las siguientes definiciones:
“Hemorragia abundante mortal o potencialmente mortal”: Cualquiera de las siguientes condiciones: Hemorragia mortal; hemorragia intracraneana o intrapericárdica con taponamiento cardiaco; shock hipovolémico; hipotensión grave debido a una hemorragia que requiere la administración de fármacos presores o una intervención quirúrgica; hemorragia aparente o acompañada de manifestaciones clínicas, asociada con una disminución de la hemoglobina de más de 50 g/l; transfusión de 4 o más unidades (de sangre entera o de glóbulos rojos) debido a la hemorragia.
“Otro tipo de hemorragia abundante”: Cualquiera de las siguientes condiciones: Hemorragia que produce una incapacidad importante (por ejemplo, hemorragia intraocular con pérdida definitiva de la vista); hemorragia aparente o acompañada de manifestaciones clínicas, asociada con una disminución de la hemoglobina de 30 a 50 g/l; transfusión de 2 o 3 unidades (de sangre entera o de glóbulos rojos) debido a la hemorragia.
“Hemorragia menor”: Hemorragia que requiere una intervención médica para detenerla o tratarla (por ejemplo, epistaxis que requiere una consulta médica para realizar un taponamiento nasal).
“Hemorragia mínima”: Esta categoría incluía todos los demás tipos de hemorragia; estos acontecimientos se registraron pero no se clasificaron.
Las reacciones hemorrágicas notificadas durante el estudio PLATO también se clasificaron mediante la escala TIMI (Thrombolysis In Myocardial Infarction — Escala de evaluación de la trombólisis en el infarto de miocardio) para facilitar la comparación con otros estudios similares. Una “hemorragia abundante según la escala TIMI” se define como una hemorragia acompañada de manifestaciones clínicas y de una disminución >50 g/L de la hemoglobina o una hemorragia intracraneana, mientras que una “hemorragia menor según la escala TIMI” es una hemorragia acompañada de manifestaciones clínicas y de una disminución de la hemoglobina entre 30 y 50 g/L.
La Figura 1 y la Tabla 2 muestran una presentación general de las reacciones hemorrágicas notificadas durante el estudio PLATO.
FIGURA 1.Estimación de Kaplan-Meier del tiempo transcurrido hasta la primera reacción hemorrágica de la categoría “Hemorragias abundantes en total” según la definición del estudio PLATO

|
Tabla 2. Análisis de todos los eventos de sangrado |
|||
|
BRILINTA® 90 mg |
Clopidogrel 75 mg |
Valor de p |
|
|
Variable principal de seguridad Hemorragias abundantes en total |
11.6 |
11.2 |
0.4336 |
|
Variables secundarias Hemorragias mortales o potencialmente mortales |
5.8 |
5.8 |
0.6988 |
|
Combinación de hemorragias abundantes en total y hemorragias menores |
16.1 |
14.6 |
0.0084 |
|
Hemorragias abundantes no asociadas con una intervención de revascularización coronaria |
4.5 |
3.8 |
0.0264 |
|
Hemorragias abundantes sin relación con algún procedimiento |
3.1 |
2.3 |
0.0058 |
|
Hemorragias abundantes + menores sin relación con algún procedimiento |
5.9 |
4.3 |
<0.0001 |
|
Categorías de hemorragia según la escala TIMI |
|||
|
Hemorragias abundantes según la escala TIMI |
7.9 |
7.7 |
0.5669 |
|
Hemorragias abundantes + menores según la escala TIMI |
11.4 |
10.9 |
0.3272 |
En el estudio PLATO no hubo una diferencia significativa entre BRILINTA® y el clopidogrel en cuanto al tiempo hasta la primera reacción de la categoría “Hemorragias abundantes en total” según la definición del estudio PLATO. En el estudio se notificaron pocas reacciones hemorrágicas mortales: 20 (0.2%) con BRILINTA® 90 mg dos veces al día y 23 (0.3%) con el clopidogrel 75 mg una vez al día. Tras incluir las hemorragias menores, la incidencia de reacciones hemorrágicas abundantes y menores combinadas según la definición del estudio PLATO fue significativamente mayor con BRILINTA® que con el clopidogrel. No se observó una diferencia significativa entre BRILINTA® y el clopidogrel en lo que se refiere a la incidencia total de reacciones hemorrágicas según la escala TIMI.
Hemorragias relacionadas con una intervención de revascularización coronaria: En el estudio PLATO, 1584 pacientes (12%) se sometieron a una intervención de revascularización coronaria por puenteo vascular. La incidencia de hemorragia abundante mortal o potencialmente mortal fue de aproximadamente el 42% en ambos grupos de tratamiento. No hubo ninguna diferencia entre los grupos en cuanto al riesgo de “hemorragia abundante mortal o potencialmente mortal” asociada con la intervención de revascularización coronaria, en función del momento de la administración de la última dosis previa a la intervención. Las hemorragias mortales fueron poco frecuentes: 6 pacientes de cada grupo (0.8% y 0.7% de los pacientes sometidos a una intervención de revascularización coronaria de los grupos de BRILINTA® y del clopidogrel, respectivamente).
Hemorragias sin relación con una intervención de revascularización coronaria: Tras excluir del análisis las reacciones hemorrágicas relacionadas con una intervención de revascularización coronaria (véase la Tabla 3), las incidencias absolutas de hemorragia fueron más bajas en todas las categorías. No hubo ninguna diferencia entre los grupos en cuanto a las hemorragias abundantes mortales o potencialmente mortales sin relación con una intervención de revascularización coronaria según la definición del estudio PLATO, pero las “hemorragias abundantes en total” según la definición del estudio PLATO, las “hemorragias abundantes según la escala TIMI” y las “hemorragias abundantes + menores según la escala TIMI” fueron más frecuentes con BRILINTA®.
|
Tabla 3. Hemorragias abundantes sin relación con una intervención de revascularización coronaria según la definición del estudio PLATO y reacciones hemorrágicas según la escala TIMI |
|||
|
BRILINTA® |
Clopidogrel |
Valor de p |
|
|
Categorías de hemorragia según el estudio PLATO |
|||
|
Hemorragias abundantes en total |
4.5 |
3.8 |
0.0264 |
|
Hemorragias abundantes mortales o potencialmente mortales |
2.1 |
1.9 |
0.2516 |
|
Categorías de hemorragia según la escala TIMI |
|||
|
Hemorragias abundantes según la escala TIMI |
2.8 |
2.2 |
0.0246 |
|
Hemorragias abundantes + menores según la escala TIMI |
4.5 |
3.6 |
0.0093 |
Hemorragias sin relación con algún procedimiento: Como muestra la Tabla 1, las “hemorragias abundantes” y las “hemorragias abundantes + menores” sin relación con algún procedimiento según la definición del estudio PLATO fueron más frecuentes con BRILINTA®. Las suspensiones del tratamiento debido a una hemorragia sin relación con algún procedimiento fueron más frecuentes con BRILINTA® (2.9%) que con el clopidogrel (1.2%; p <0.001). Las “hemorragias abundantes + menores” de importancia clínica fueron de los siguientes tipos, en orden de frecuencia decreciente (BRILINTA®:Clopidogrel): Intracraneanas (27:14 casos), pericárdicas (11:11 casos), retroperitoneales (3:3 casos), intraoculares (2:4 casos) e intraarticulares (2:1 casos). En orden de frecuencia decreciente, otros tipos de hemorragias frecuentes fueron: Gastrointestinales (170:135 casos), epistaxis (116:61 casos), urinarias (45:37 casos), subcutáneas o dérmicas (43:38 casos) y hemoptisis (13:7 casos).
No hubo ninguna diferencia entre BRILINTA® y el clopidogrel en lo que respecta a la incidencia de hemorragias mortales sin relación con algún procedimiento. Las incidencias de “hemorragias gastrointestinales abundantes mortales o potencialmente mortales” fueron idénticas con BRILINTA® y con el clopidogrel, aunque el número de casos mortales fue mayor con el clopidogrel (5) que con BRILINTA® (ninguna). El número de hemorragias intracraneanas abundantes mortales o potencialmente mortales sin relación con algún procedimiento fue mayor con BRILINTA® (n=27 reacciones en 26 pacientes, 0.3%) que con el clopidogrel (n=14 reacciones, 0.2%); de estas reacciones hemorrágicas, 11 fueron mortales con BRILINTA® y 1 con el clopidogrel.
Se evaluaron algunas características iniciales como la edad, el sexo, el peso, la raza, la región geográfica, los antecedentes médicos, las enfermedades concurrentes y los tratamientos concomitantes, para explorar la posibilidad de que aumentara el riesgo hemorrágico con BRILINTA®. No se identificó ningún grupo con un riesgo particular de presentar alguna de las categorías de hemorragia.
Disnea: En el estudio PLATO, reacciones adversas de disnea fueron reportadas en 13.8% de los paciente del grupo del ticagrelor 90 mg dos veces al día y en 7.8% de los pacientes del grupo del clopidogrel 75 mg una vez al día. La mayoría de las RA reportadas de disnea fueron de intensidad leve a moderada y a menudo se resolvieron sin la necesidad de interrumpir el tratamiento. La disnea se observó generalmente durante la fase inicial del tratamiento. El 87% de los pacientes que presentaron disnea sufrieron un solo episodio. Reacciones adversas serias de disnea se presentaron en 0.7% del grupo tomando ticagrelor y 0.4% del grupo tomando clopidogel. Los pacientes que presentaron disnea tendieron a ser de edad más avanzada y a padecer con mayor frecuencia disnea, ICC, EPOC o asma antes del estudio. Los resultados del estudio PLATO no indican que la mayor frecuencia con BRILINTA® se deba a la aparición o al empeoramiento de una cardiopatía o neumopatía. No hubo indicación alguna de que BRILINTA® afectara la función pulmonar (véase la sección “Advertencias y precauciones especiales de uso”).
Valores anormales de laboratorio: En el estudio PLATO, la concentración sérica de ácido úrico aumentó por encima del límite superior normal en el 22% de los pacientes tratados con BRILINTA® frente al 13% de aquellos que recibieron el clopidogrel. La concentración sérica media de ácido úrico aumentó aproximadamente un 15% con BRILINTA® frente a un 7% aproximadamente con el clopidogrel, y disminuyó después de suspender el tratamiento. No se observó ninguna diferencia en la frecuencia de reacciones adversas clínicas.
En el estudio PLATO, la creatininemia aumentó más de un 50% en el 8% de los pacientes tratados con BRILINTA® frente al 7% de aquellos tratados con el clopidogrel. Los aumentos generalmente no progresaron y a menudo los valores disminuyeron al continuar el tratamiento. Se observaron signos de reversibilidad del efecto tras la suspensión del tratamiento, incluso en los pacientes que habían presentado los aumentos más importantes. No hubo diferencias entre los grupos de tratamiento del estudio PLATO en las reacciones adversas graves relacionadas con la creatinina.
CONTRAINDICACIONES: Hipersensibilidad al ticagrelor o a alguno de los excipientes (véase la sección “Lista de excipientes”).
Hemorragia patológica activa.
Antecedentes de hemorragia intracraneana.
Insuficiencia hepática grave.
EMBARAZO Y LACTANCIA: No se han realizado estudios clínicos durante el embarazo o la lactancia.
Existe información clínica limitada sobre la exposición a BRILINTA® durante el embarazo.
Los estudios en animales no indican efectos perjudiciales directos durante la gestación, el desarrollo embrionario y fetal, el parto o el desarrollo posnatal. El ticagrelor no tuvo ningún efecto en la fecundidad de animales machos o hembras (véase la sección “Datos preclínicos sobre toxicidad”).
Dado que los estudios sobre la reproducción en animales no siempre permiten pronosticar la respuesta humana, el ticagrelor solo debe usarse durante el embarazo si los posibles beneficios para la madre justifican los riesgos potenciales para el feto.
Lactancia: No se sabe si este medicamento pasa a la leche materna. Los estudios en ratas demostraron que el ticagrelor y sus metabolitos activos se excretan a la leche. Por lo tanto, no se recomienda usar BRILINTA® durante la lactancia.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y USAR MÁQUINAS: No se han realizado estudios sobre los efectos de BRILINTA® en la capacidad para conducir y usar máquinas. BRILINTA® no influye, o solo en un grado insignificante, en dicha capacidad. Durante el tratamiento de síndromes coronarios agudos se han notificado reacciones de mareo y confusión. En consecuencia, los pacientes que padecen estos síntomas deben ser prudentes al conducir o usar máquinas.
EFECTOS FARMACODINÁMICOS
Inicio de acción
Media (± error estándar) del grado final de inhibición de la agregación plaquetaria (IAP) tras la administración de dosis orales únicas de 180 mg de BRILINTA® o 600 mg de clopidogrel en pacientes con cardiopatía coronaria estable

En los pacientes con cardiopatía coronaria estable tratados con AAS, el efecto farmacológico de BRILINTA® comienza rápidamente, como muestra la media de la inhibición de la agregación plaquetaria (IAP) de aproximadamente un 41% media hora después de la administración de una dosis de carga de 180 mg de BRILINTA®, con un efecto máximo del 87.9% al 89.6% de 2 a 4 horas después de la dosis. El 90% de los pacientes consiguió un grado final de IAP >70% en las 2 primeras horas después de la dosis. El potente efecto inhibidor de la agregación plaquetaria de BRILINTA®, de entre el 87% y el 89%, se mantuvo durante un periodo de 2 a 8 horas.
Media (± error estándar) del grado final de inhibición de la agregación plaquetaria (IAP) tras la administración de la última dosis de mantenimiento del tratamiento con 90 mg dos veces al día de BRILINTA®, 75 mg de clopidogrel una vez al día, o un placebo
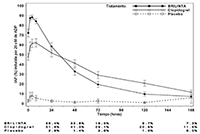
Desaparición del efecto: Una vez que las concentraciones de BRILINTA® y del metabolito activo pasan por debajo del nivel requerido para saturar los receptores, la IAP disminuye progresivamente a medida que bajan las concentraciones plasmáticas. Dado que la unión de BRILINTA® es reversible, la recuperación de la función plaquetaria no depende de la reposición de plaquetas. La desaparición de la IAP es más rápida con BRILINTA® que con el clopidogrel, como muestra la pendiente de la curva de desaparición del efecto de 4 a 72 horas después de la última dosis (véase la sección “Advertencias y precauciones especiales de uso”).
La mediana del grado final de IAP tras la última dosis de BRILINTA® es entre un 20% y un 30% mayor con BRILINTA® que con el clopidogrel. No obstante, la IAP (%) es similar con los dos fármacos 24 horas después de la dosis y es menor con BRILINTA® desde las 72 horas en adelante hasta el séptimo día. La media de la IAP (%) 72 horas después de la última dosis de BRILINTA® (día 3) fue comparable a la obtenida el día 5 con el clopidogrel, mientras que la media de la IAP (%) obtenida el día 5 con BRILINTA® fue comparable a la que se consiguió el día 7 con el clopidogrel, pero no fue significativamente diferente de la IAP producida por un placebo.
Pacientes que responden al tratamiento con BRILINTA®: En el momento de las concentraciones plasmáticas máximas de BRILINTA® y de su metabolito activo tras la administración de 90 mg dos veces al día, la IAP inducida por BRILINTA® muestra una menor variabilidad que con el clopidogrel. Los pacientes con cardiopatía coronaria estable que habían mostrado anteriormente una baja IAP con el clopidogrel (pacientes que no respondían al tratamiento) y que recibieron dosis concomitantes de AAS, presentaron una IAP media más potente después de la administración de BRILINTA® que con el clopidogrel. En los pacientes que no habían respondido al clopidogrel, se observó que la IAP era mayor y más uniforme con BRILINTA®. El tratamiento con BRILINTA® produjo un porcentaje de IAP sistemáticamente superior al que se consiguió con el clopidogrel, y este fenómeno se observó después de la administración tanto en los pacientes que respondieron como en los que no respondieron al clopidogrel.
Información sobre el cambio de tratamiento: El cambio del clopidogrel a BRILINTA® produce un aumento absoluto de la IAP del 26.4% y el cambio de BRILINTA® al clopidogrel da lugar a una disminución media de la IAP del 24.5%. Los pacientes pueden pasar del clopidogrel a BRILINTA® sin que se interrumpa el efecto antiplaquetario.
Mecanismo de la adenosina (ENT-1): Ticagrelor incrementó las concentraciones plasmáticas de adenosina en pacientes con SCA y se ha demostrado que aumenta varias respuestas fisiológicas a la adenosina. La adenosina es un vasodilatador; se ha demostrado que ticagrelor refuerza los incrementos del flujo sanguíneo coronario inducidos por adenosina en voluntarios sanos y pacientes con SCA. La adenosina es un inhibidor plaquetario endógeno; se ha demostrado que ticagrelor aumenta la inhibición de la agregación plaquetaria mediada por adenosina además de la inhibición plaquetaria causada por su antagonismo P2Y12. La adenosina se ha vinculado al efecto cardioprotector de preacondicionamiento; se ha demostrado que ticagrelor reduce el tamaño del infarto a través de un mecanismo mediado por adenosina en un modelo de lesión por reperfusión en ratas. La adenosina también induce disnea; se ha demostrado que ticagrelor aumenta la disnea inducida por adenosina en voluntarios sanos. Por consiguiente, la disnea observada en algunos pacientes que están tomando ticagrelor (véase sección “Reacciones adversas”) puede ser parcial o completamente mediada por adenosina.
REACCIONES ADVERSAS
Resumen del perfil de seguridad: El perfil de seguridad de BRILINTA® se ha evaluado en dos extensos estudios fase 3 de desenlace (PLATO y PEGASUS) que incluyeron más de 39.000 pacientes. A continuación se discuten las reacciones adversas del medicamento observadas en estos estudios.
La seguridad de BRILINTA® en pacientes con síndromes coronarios agudos (angina inestable, infarto de miocardio con o sin elevación del segmento ST) se evaluó en el estudio PLATO que comparó BRILINTA® 90 mg dos veces al día con el clopidogrel de 75 mg una vez al día, ambos asociados con AAS y otros tratamientos convencionales.
La mediana de la duración del tratamiento con BRILINTA® fue de 277.
En el PLATO, los pacientes tratados con BRILINTA® tuvieron una incidencia más alta de descontinuación debido a eventos adversos que con clopidogrel (7.4% vs. 5.4%).
La seguridad de BRILINTA® en pacientes con historia de IM (IM ocurrido por lo menos un año antes) y alto riesgo de desarrollar evento trombótico se evaluó en el estudio PEGASUS, el cual comparó pacientes tratados con BRILINTA® 60 mg dos veces al día o 90 mg dos veces al día combinado con AAS, con tratamiento con solo AAS y otras terapias estándar.
Las reacciones adversas al medicamento reportadas con mayor frecuencia en pacientes tratados con ticagrelor fueron sangrado y disnea.
Lista tabulada de reacciones adversas al medicamento: Las reacciones adversas observadas en los estudios clínicos con BRILINTA® se enumeran según la terminología MedDRA, por el sistema de clasificación de Órganos [System Organ Class (SOC)] y el tipo de frecuencia determinada por los estudios PEGASUS y PLATO en la Tabla 1. Dentro de cada categoría de sistema de clasificación de órganos y frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad. Las frecuencias se clasifican de acuerdo con las siguientes convenciones: Muy frecuentes (=1/10), frecuentes (=1/100 a <1/10), poco frecuentes (=1/1,000 a <1/100), raras (=1/10,000 a <1/1,000), muy raras (<1/10,000) y no conocida (no se pueden estimar a partir de los datos disponibles).
|
Tabla 1. Reacciones adversas al medicamento observadas en los estudios clínicos |
|||
|
Clasificación de sistema orgánico |
Muy frecuentes |
Frecuentes |
Poco frecuentes |
|
Neoplasias benignas, malignas y no especificadas (incluyendo quistes y pólipos) |
Sangrado de tumoresb |
||
|
Trastornos sanguíneos y del sistema linfático |
Trastornos sanguíneos, sangradoc |
||
|
Trastornos del metabolismo y nutrición |
Hiperuricemiaa |
Gota |
|
|
Trastornos psiquiáticos |
Confusión |
||
|
Trastornos del sistema nervioso |
Mareo Síncope |
Hemorragia intracraneal |
|
|
Trastornos oftalmológicos |
Hemorragia oculard |
||
|
Trastornos del oído y laberinto |
Vértigo |
Hermorragia del oído |
|
|
Trastornos vasculares |
Hipotensión |
||
|
Trastornos respiratorios, torácicos y mediastinales |
Disnea |
Sangrados del sistema respiratorioe |
|
|
Trastornos gastrointestinales |
Hemorragia gastrointestinalf Diarrea Náuseas |
Hermorragia retroperitoneal |
|
|
Trastornos de la piel y tejido subcutáneo |
Sangrado subcutáneo o dérmicog Prurito |
||
|
Trastornos musculoesqueléticos, del tejido conectivo y hueso |
Sangrado muscularh |
||
|
Trastornos renales y urinarios |
Sangrado del tracto urinarioi |
||
|
Trastornos del sistema reproductivo y del seno |
Sangrado del sistema reproductivoj |
||
|
Investigaciones |
Creatinina sérica aumentadaa |
||
|
Lesiones, intoxicación y complicaciones de procedimientos |
Hemorragia post-procedimiento traumáticask |
||
|
a Frecuencias derivadas de observaciones de laboratorio (ácido úrico hasta > ULN desde el nivel basal, por debajo o dentro del rango de referencia. Incrementos de la creatinina >50% desde el nivel basal) y frecuencia no cruda de reporte de eventos adversos). b por ej. sangrado por cáncer vesical, cáncer gástrico, cáncer de colon. c por ej. tendencia aumentada a equimosis, hematorma espontáneo, diátesis hermorrágica. d por ej. hemorragia conjuntival, retiniana, intraocular. e por ej. epistaxis, hemoptisis. f por ej. hemorragia gingival, hemorragia rectal, hemorragia de úlcera gástrica. g por ej. equimosis, hemorragia cutánea, petequias. h por ej. hemartrosis, hemorragia muscular. i por ej. hematuria, cistitis hemorrágica. j por ej. hemorragia vaginal, hematospermia, hemorragia postmenopáusica. k por ej. contusión, hematoma traumático, hemorragia traumática. |
|||
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN
Interacciones farmacológicas
Efectos de otros medicamentos en BRILINTA®
Fármacos metabolizados por la forma CYP3A4
Ketoconazol (potente inhibidor de la CYP3A4). La coadministración del ketoconazol con el ticagrelor produjo aumentos de 2.4 veces de la concentración plasmática máxima (Cmáx) del ticagrelor y de 7.3 veces de su área bajo la curva de concentraciones plasmáticas en función del tiempo (ABC). La Cmáx y el ABC del metabolito activo disminuyeron un 89% y un 56%, respectivamente. Se prevé que otros inhibidores potentes de la CYP3A4 (claritromicina, nefazodona, ritonavir y atazanavir) tendrán efectos similares, por lo que no deben coadministrarse con BRILINTA® (véase la sección “Advertencias y precauciones especiales de uso”).
Diltiazem (inhibidor moderado de la CYP3A4). La coadministración del diltiazem con el ticagrelor produjo aumentos del 69% y del 174%, de la Cmáx y el ABC del ticagrelor, respectivamente, así como una disminución del 38% de la Cmáx del metabolito activo, sin alterar su ABC. El ticagrelor no tuvo ningún efecto en las concentraciones plasmáticas del diltiazem. BRILINTA® también puede coadministrarse con otros inhibidores moderados de la CYP3A4 (por ejemplo, amprenavir, aprepitant, eritromicina, fluconazol y verapamilo).
Rifampicina y otros inductores de la CYP3A4: La coadministración de la rifampicina con el ticagrelor disminuyó un 73% la Cmáx del ticagrelor y un 86% su ABC. La Cmáx del metabolito activo no varió y su ABC disminuyó un 46%. Se prevé que otros inductores de la CYP3A4 (por ejemplo, fenitoína, carbamazepina y fenobarbital) también reducirán la exposición al ticagrelor y quizás la eficacia de BRILINTA®.
Ciclosporina (P-gp e inhibidor de CYP3A): La coadministración de ciclosporina (600 mg) con ticagrelor incrementó la Cmáx y AUC de ticagrelor 2.3 y 2.8 veces, respectivamente. El AUC del metabolito activo aumentó un 32% y la Cmáx se redujo un 15% en presencia de ciclosporina. No se observó efecto de ticagrelor sobre los niveles sanguíneos de ciclosporina.
Otros: Los estudios sobre interacciones farmacológicas clínicas mostraron que la coadministración del ticagrelor con la heparina, la enoxaparina y el ácido acetilsalicílico no tuvo ningún efecto en las concentraciones plasmáticas del ticagrelor ni en las de su metabolito activo. La coadministración del ticagrelor con la heparina no tuvo ningún efecto en la heparina, según las determinaciones del tiempo de tromboplastina parcial activado (TTPa) y del tiempo de coagulación activado (TCA). La coadministración del ticagrelor con la enoxaparina no tuvo ningún efecto en la enoxaparina según la determinación del factor Xa.
Efectos de BRILINTA® en otros medicamentos
Fármacos metabolizados por la forma CYP3A4
Simvastatina: La coadministración del ticagrelor con la simvastatina aumentó un 81% la Cmáx de la simvastatina y un 56% su ABC, y aumentó un 64% la Cmáx del metabolito ácido de la simvastatina y un 52% su ABC, con aumentos individuales de 2 a 3 veces. Se recomienda tener en cuenta la posible importancia clínica de la magnitud y amplitud de estos cambios de exposición en pacientes que requieren dosis superiores a 40 mg de simvastatina. La simvastatina no tuvo ningún efecto en las concentraciones plasmáticas del ticagrelor. BRILINTA® puede ejercer un efecto similar en la lovastatina, pero no se prevén efectos de importancia clínica en otras estatinas.
Atorvastatina: La coadministración de la atorvastatina con el ticagrelor aumentó un 23% la Cmáx de la atorvastatina (ácido) y un 36% su ABC. Se observaron aumentos similares del ABC y la Cmáx de todos los metabolitos ácidos de la atorvastatina. Se considera, sin embargo, que tales aumentos carecen de importancia clínica.
Fármacos metabolizados por la forma CYP2C9
Tolbutamida: La coadministración del ticagrelor con la tolbutamida no modificó las concentraciones plasmáticas de ninguno de los dos fármacos, lo cual indica que el ticagrelor no inhibe la CYP2C9 y que es improbable que altere el metabolismo mediado por la CYP2C9 de fármacos como la warfarina y la tolbutamida.
Anticonceptivos orales: La coadministración del ticagrelor con la asociación de levonorgestrel y etinilestradiol aumentó en aproximadamente un 20% la exposición al etinilestradiol, pero no modificó la farmacocinética del levonorgestrel. No se prevén efectos de importancia clínica en la eficacia del anticonceptivo oral tras la coadministración de BRILINTA® con la asociación de levonorgestrel y etinilestradiol.
Digoxina (sustrato de la P-gp): La coadministración del ticagrelor aumentó un 75% la Cmáx de la digoxina y un 28% su ABC. En consecuencia, se recomienda una supervisión clínica y de laboratorio adecuada al coadministrar BRILINTA® con fármacos dependientes de la PgP con un estrecho margen terapéutico, por ejemplo la digoxina.
Otros tratamientos concomitantes: En los estudios clínicos, BRILINTA® se administró frecuentemente con distintos medicamentos necesarios para tratar afecciones concomitantes tales como ácido acetilsalicílico, heparina, heparina de bajo peso molecular, inhibidores de la glucoproteína IIb/IIIa por vía intravenosa, inhibidores de la bomba de protones, estatinas, betabloqueadores, inhibidores de la enzima convertidora de angiotensina y bloqueadores de los receptores de angiotensina. Dichos estudios no revelaron interacciones adversas de importancia clínica.
EFICACIA CLÍNICA: Las pruebas clínicas de la eficacia de BRILINTA® provienen del estudio PLATO (PLATelet Inhibition and Patient Outcomes — Inhibición de la agregación plaquetaria y resultados de los pacientes) que comparó BRILINTA® con el clopidogrel, ambos asociados con ácido acetilsalicílico (AAS) y otro tratamiento convencional.
El estudio PLATO, de Fase III, con un diseño aleatorizado, doble ciego y de grupos paralelos, comparó la seguridad y la eficacia de BRILINTA® con las del clopidogrel para la prevención de accidentes vasculares en 18624 pacientes con síndromes coronarios agudos (angina inestable e infarto de miocardio con o sin elevación del segmento ST). En el estudio participaron pacientes que se presentaron en las 24 horas de la aparición del episodio más reciente de dolor torácico o de los síntomas. Los pacientes fueron distribuidos al azar entre el grupo del clopidogrel (75 mg una vez al día tras una dosis de carga inicial de 300 mg si no se había administrado un tratamiento anterior con una tienopiridina, permitiéndose una dosis de carga adicional de 300 mg a discreción del investigador), y un grupo tratado con una dosis de carga de 180 mg de BRILINTA® seguida por una dosis de mantenimiento de 90 mg dos veces al día. Los pacientes podían estar recibiendo un tratamiento médico, o haber sido sometidos a una angioplastia coronaria percutánea o a una intervención de revascularización coronaria por puenteo vascular.
FIGURA 2. Estimación del riesgo hasta la primera aparición de cualquier acontecimiento incluido en la variable compuesta de eficacia
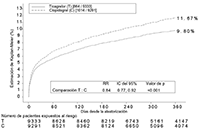
BRILINTA® redujo la incidencia de los episodios incluidos en la variable compuesta de eficacia frente al clopidogrel tanto en la población con angina inestable como en los pacientes con IM con o sin elevación del segmento ST.
|
Tabla 4. Variables examinadas en el estudio PLATO |
|||||
|
Porcentaje de pacientes con el episodio |
|||||
|
BRILINTA® |
Clopidogrel |
Reducción del riesgo relativo (RRR)a (%) |
Razón de riesgos (IC del 95%) |
Valor de p |
|
|
Variable principal |
|||||
|
Combinación de muerte CV + IM (excl. IM silencioso) + AVC |
9.3 |
10.9 |
16 |
0.84 (0.77, 0.92) |
p=0.0003 |
|
Muerte CV |
3.8 |
4.8 |
21 |
0.79 (0.69, 0.91) |
p=0.0013 |
|
IM (excl. IM silencioso)a |
5.4 |
6.4 |
16 |
0.84 (0.75, 0.95) |
p=0.0045 |
|
AVC |
1.3 |
1.1 |
-17 |
1.17 (0.91, 1.52) |
p=0.2249 |
|
Variables secundarias |
|||||
|
Muerte CV + IM (excl. IM silencioso) + AVC con intento de tratamiento invasivoa |
8.5 |
10.0 |
16 |
0.84 (0.75, 0.94) |
p=0.0025 |
|
Mortalidad por todas las causas + IM (excl. IM silencioso) + AVC |
9.7 |
11.5 |
16 |
0.84 (0.77, 0.92) |
p=0.0001 |
|
Mortalidad por todas las causas + Todos los IM + AVC + IRG + IR + AIT + Otros ETA |
13.8 |
15.7 |
12 |
0.88 (0.81, 0.95) |
p=0.0006 |
|
Mortalidad por todas las causas |
4.3 |
5.4 |
22 |
0.78 (0.69, 0.89) |
p=0.0003** |
|
aRRR = (1-Razón de riesgos) x 100%. Los valores asociados con una reducción del riesgo relativo negativa indican un aumento del riesgo relativo **Valor de p nominal Muerte CV = muerte de origen cardiovascular; IM = infarto de miocardio; AVC = accidente vascular cerebral; IRG = insuficiencia renal grave; IR = insuficiencia renal; AIT = ataque isquémico transitorio; ETA = episodio tromboembólico arterial |
|||||
BRILINTA® fue superior al clopidogrel (RRR=16%, relación de riesgos absoluta [RRA]=1.9%, NNT [número de pacientes que es necesario tratar]=54) para la prevención de los episodios trombóticos incluidos en la variable de eficacia compuesta (muerte de origen cardiovascular [CV], infarto de miocardio [IM] y accidente vascular cerebral [AVC]) durante 12 meses. La diferencia entre los tratamientos se explica por las muertes de origen cardiovascular y el infarto de miocardio, mientras que no hubo ninguna diferencia en la incidencia de accidentes vasculares cerebrales. BRILINTA® produjo reducciones estadísticamente significativas del 16% del riesgo relativo de IM (RRA=1.1%) y del 21% del riesgo relativo de muerte de origen CV (RRA=1.1%). El tratamiento de 91 pacientes con BRILINTA® en lugar del clopidogrel permitirá evitar 1 muerte de origen cardiovascular.
BRILINTA® demostró su superioridad sobre el clopidogrel para la prevención de los episodios incluidos en la variable compuesta (muerte CV + IM + AVC). Este resultado apareció temprano (reducción del riesgo absoluto [RRA] del 0.6% y reducción del riesgo relativo [RRR] del 12% después de 30 días), y el efecto del tratamiento fue constante durante el periodo entero de 12 meses, dando lugar a una RRA del 1.9% anual con una RRR del 16%. Esto indica que conviene administrar el tratamiento por lo menos 12 meses (véase la sección “Posología y forma de administración”).
En el estudio PLATO se llevaron a cabo numerosas comparaciones de la variable principal de eficacia en subgrupos con el fin de evaluar la robustez y la uniformidad del beneficio global. El efecto del tratamiento con BRILINTA® frente al clopidogrel parece ser uniforme en todos los subgrupos de pacientes definidos por características demográficas como el peso, el sexo, los antecedentes médicos, los tratamientos concomitantes y el diagnóstico del episodio final de referencia (IM con o sin elevación del segmento ST o angina inestable).
Se observó una interacción débil, pero significativa, entre el tratamiento y la región puesto que la razón de riesgos correspondiente a la variable principal resultó a favor de BRILINTA® en el resto del mundo, y a favor del clopidogrel en Norteamérica, región que representaba alrededor del 10% de la población total estudiada (valor de p de esta interacción = 0.045).
Esta interacción aparente entre el tratamiento y la región que se observó en el estudio PLATO bien podría atribuirse, al menos en parte, a la casualidad. Los análisis adicionales indican que la eficacia de BRILINTA® frente a la del clopidogrel depende de la dosis de mantenimiento de AAS. La información disponible indica que el ticagrelor es más eficaz que el clopidogrel en caso de asociación con una dosis de mantenimiento de AAS baja (75-150 mg), mientras que es dudosa la eficacia relativa del ticagrelor frente al clopidogrel cuando se asocia con dosis elevadas de AAS (>300 mg). Ante la relación que se observó entre la dosis de mantenimiento de AAS y la eficacia relativa del ticagrelor frente al clopidogrel, se recomienda usar BRILINTA® junto con una dosis de mantenimiento de AAS baja (entre 75 y 150 mg) (véanse las secciones “Posología y forma de administración” y “Advertencias y precauciones especiales de uso”).
Asimismo, los beneficios obtenidos con BRILINTA® fueron independientes del uso de otros tratamientos cardiovasculares a corto y largo plazos, incluido el tratamiento con heparina, heparina de bajo peso molecular, inhibidores de la glucoproteína IIb/IIIa intravenosos, hipolipemiantes, betabloqueadores, inhibidores de la enzima convertidora de angiotensina (ECA), antagonistas de los receptores de angiotensina II e inhibidores de la bomba de protones (véase la sección “Interacción con otros medicamentos y otras formas de interacción”).
BRILINTA® produjo una reducción estadísticamente significativa del riesgo relativo (RRR) correspondiente a la variable compuesta por muerte CV + IM + AVC en pacientes con síndromes coronarios agudos en quienes se había programado un tratamiento invasivo (RRR=16%, RRA=1.7%, p=0.0025). En un análisis exploratorio, BRILINTA® produjo una reducción del riesgo relativo correspondiente a la variable compuesta en pacientes con síndromes coronarios agudos en quienes se había programado un tratamiento médico (RRR=15%, RRA=2.3%, p nominal = 0.0444). Como en el caso de la variable principal del estudio, el efecto en estos dos grupos se explica básicamente por la muerte de origen cardiovascular y el IM, sin que se observara ningún efecto en los accidentes vasculares cerebrales. En los pacientes portadores de una endoprótesis vascular (stent), el número de trombosis manifiestas de la endoprótesis fue menor en el grupo del ticagrelor que en el del clopidogrel (73 frente a 107, RRR=32%, RRA=0.6%, p nominal = 0.0123).
BRILINTA® produjo una RRR estadísticamente significativa del 16% (RRA=2.1%) de la variable compuesta por la mortalidad por todas las causas + IM + AVC con respecto al clopidogrel.
La última variable secundaria evaluada fue la mortalidad por todas las causas. BRILINTA® produjo una RRR del 22% de la mortalidad por todas las causas con respecto al clopidogrel, con un nivel significativo nominal de p=0.0003 y una RRA del 1.4%.
Subestudio de supervisión con monitor de Holter: Con el fin de estudiar la incidencia de asistolias y otros episodios arrítmicos durante el estudio PLATO, los investigadores instauraron una supervisión electrocardiográfica con monitor de Holter en un subconjunto de unos 3.000 pacientes. En aproximadamente 2.000 de estos pacientes, se obtuvieron registros tanto durante la fase aguda del síndrome coronario agudo como al cabo de un mes. La variable principal de interés fue la aparición de asistolias = 3 segundos. La proporción de pacientes que presentaron asistolias fue mayor con BRILINTA® que con el clopidogrel (6.0% frente al 3.5% durante la fase aguda y 2.2% frente al 1.6% al cabo de 1 mes). Más pacientes presentaron asistolias con BRILINTA® que con el clopidogrel; no obstante, este desequilibrio no tuvo consecuencias clínicas adversas (por ejemplo, colocación de un marcapasos) en esta población de pacientes.
El ticagrelor ejerce su acción por vía oral. Al contrario del clopidogrel, no requiere la actividad de las enzimas del CYP450 para inhibir la agregación plaquetaria. El polimorfismo de la codificación genética de la forma CYP2C19 del citocromo P450 puede afectar la eficacia del clopidogrel. El polimorfismo de la codificación genética del transporte de glucoproteína P (ABCB1) puede afectar tanto la eficacia del clopidogrel como la del ticagrelor.
En el estudio PLATO se tomaron muestras genéticas de 10285 pacientes para la determinación del genotipo de los loci CYP2C19 y ABCB1. Un análisis realizado durante el estudio PLATO reveló las siguientes asociaciones entre agrupaciones de genotipos y la eficacia y seguridad:
• El genotipo de CYP2C19 del paciente no afecta de manera significativa la superioridad de BRILINTA® sobre el clopidogrel.
• BRILINTA® redujo la incidencia de episodios cardiovasculares graves con respecto al clopidogrel independientemente del genotipo de CYP2C19.
• Las incidencias de episodios con BRILINTA® no dependieron del genotipo de CYP2C19.
• En el grupo tratado con el clopidogrel, los portadores de un alelo de pérdida de función de CYP2C19 presentaron un aumento de la incidencia de los episodios incluidos en la variable compuesta principal con respecto a los no portadores.
• Al igual que en el estudio PLATO en general, la incidencia de hemorragia abundante en total no fue diferente entre BRILINTA® y el clopidogrel independientemente del genotipo de CYP2C19, aunque los pacientes con uno o varios alelos de ganancia de función de CYP2C19 tratados con el clopidogrel presentaron la máxima incidencia de hemorragia abundante.
• Al igual que en el estudio PLATO en general, la incidencia de hemorragia sin relación con una intervención de revascularización coronaria fue mayor con BRILINTA® que con el clopidogrel en los pacientes con un alelo de pérdida de función de CYP2C19.
• La incidencia de hemorragia sin relación con una intervención de revascularización coronaria fue similar entre BRILINTA® y el clopidogrel en los pacientes sin alelo de pérdida de función de CYP2C19.
Variable compuesta de eficacia y seguridad combinadas: El análisis de una variable compuesta que combinó eficacia y seguridad (muerte CV + IM + AVC + “hemorragia abundante en total” según la definición del estudio PLATO) confirmó el beneficio clínico del ticagrelor frente al clopidogrel (RRR = 8%, RRA = 1.4%, razón de riesgos (RR) = 0.92; p=0.0257) durante un periodo de 12 meses después de los episodios del síndrome coronario agudo.
DATOS PRECLÍNICOS SOBRE TOXICIDAD: Los resultados de los estudios preclínicos convencionales de toxicidad farmacológica, toxicidad tras dosis únicas y repetidas, y potencial genotóxico del ticagrelor y de su metabolito activo no han revelado un riesgo inaceptable de efectos adversos en el ser humano.
Las siguientes reacciones adversas, que no se han detectado durante los estudios clínicos, se observaron en animales tras exposiciones similares o superiores al grado de exposición clínica y podrían ser pertinentes para el uso clínico: Irritación gastrointestinal.
No se observaron tumores relacionados con el fármaco en un estudio de 2 años que se llevó a cabo en ratones tratados con dosis orales de hasta 250 mg/kg/día (>18 veces la exposición terapéutica humana). No aumentó la frecuencia de tumores en ratas machos que recibieron dosis orales de hasta 120 mg/kg/día (>15 veces la exposición terapéutica humana). Se observó un aumento de los adenocarcinomas uterinos y de los adenomas y adenocarcinomas hepatocelulares, así como una disminución de los adenomas hipofisarios y fibroadenomas mamarios solamente en las ratas hembras tratadas con dosis elevadas (>25 veces la exposición terapéutica humana). No se detectó ninguna modificación de la incidencia de tumores con la dosis de 60 mg/kg/día (diferencia >8 veces respecto a la dosis terapéutica humana). Se determinó que los tumores uterinos que se observaron únicamente en ratas se debían a un efecto endocrino no genotóxico de desequilibrio hormonal presente en ratas tratadas con dosis elevadas de ticagrelor. Se considera que los tumores hepáticos benignos son secundarios a la respuesta del hígado ante la carga metabólica provocada por las dosis elevadas de ticagrelor.
El ticagrelor se ha investigado en una serie de ensayos in vitro e in vivo y se demostró que no era genotóxico.
Se determinó que el ticagrelor no ejerce ningún efecto en la fecundidad de ratas hembras con dosis orales de hasta 200 mg/kg/día (aproximadamente 20 veces la exposición terapéutica humana) ni en la de ratas machos con dosis de hasta 180 mg/kg/día (15.7 veces la exposición terapéutica humana).
El ticagrelor no tuvo ningún efecto en el desarrollo fetal de ratas con dosis orales de hasta 100 mg/kg/día (5.1 veces la exposición terapéutica humana) y de hasta 42 mg/kg/día en conejos (equivalente a la exposición terapéutica humana). El ticagrelor no tuvo efectos en el parto ni en el desarrollo posnatal en ratas tratadas con dosis de hasta 60 mg/kg/día (4.6 veces la exposición terapéutica humana).
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE USO
Riesgo hemorrágico: Al igual que sucede con otros antiagregantes plaquetarios, el uso de BRILINTA® en pacientes con un riesgo hemorrágico elevado debe ponderarse frente a los beneficios esperados en materia de prevención de episodios trombóticos. Cuando exista una indicación clínica, BRILINTA® debe usarse con precaución en los siguientes grupos de pacientes, teniendo en cuenta los aspectos descritos a continuación:
• Pacientes propensos a sufrir hemorragias (por ejemplo por un traumatismo o una intervención quirúrgica recientes, por una hemorragia gastrointestinal activa o reciente o debido a insuficiencia hepática moderada). BRILINTA® está contraindicado en pacientes con hemorragia patológica activa y en aquellos con antecedentes de hemorragia intracraneana o insuficiencia hepática grave (véase la sección “Contraindicaciones”).
• Pacientes tratados en forma concomitante con medicamentos que pueden aumentar el riesgo hemorrágico (por ejemplo, antiinflamatorios no esteroides [AINE], fibrinolíticos y/o anticoagulantes orales en las 24 horas previas a la administración de BRILINTA®).
La transfusión de plaquetas no revirtió el efecto antiplaquetario de BRILINTA® en voluntarios sanos y es poco probable que sea de beneficio clínico en pacientes con sangrado. Dado que la coadministración de BRILINTA® y desmopresina no redujo el tiempo de sangrado de referencia, es improbable que la desmopresina sea eficaz para tratar hemorragias clínicas.
El tratamiento con antifibrinolíticos (ácido aminocaproico o ácido tranexámico) y con el factor VIIa recombinante puede aumentar la hemostasia. Una vez identificada y controlada la causa de la hemorragia, podrá reanudarse el tratamiento con BRILINTA®.
Cirugía
• Antes de una intervención quirúrgica, el médico debe tomar en cuenta el perfil clínico de cada paciente para determinar el momento idóneo para suspender el tratamiento con BRILINTA®, así como los beneficios y riesgos de continuar el tratamiento antiplaquetario.
• Debido a la unión reversible de BRILINTA®, el restablecimiento de la agregación plaquetaria es más rápido que con el clopidogrel. En el estudio OFFSET, la media de la inhibición de la agregación plaquetaria (IAP) conseguida con BRILINTA® 72 horas después de la administración fue comparable a la observada 120 horas después de la administración del clopidogrel. La desaparición más rápida del efecto podría reducir el riesgo de complicaciones hemorrágicas, por ejemplo en los cuadros en los que es preciso interrumpir temporalmente el tratamiento antiplaquetario debido a una intervención quirúrgica o un traumatismo.
• En los pacientes del estudio PLATO sometidos a una intervención de revascularización coronaria, BRILINTA® se asoció con una incidencia de hemorragias abundantes similar a la observada con el clopidogrel en cada uno de los días posteriores a la suspensión del tratamiento, salvo en el día 1 en el que la incidencia de hemorragia abundante fue mayor con BRILINTA®.
• Si se ha programado una cirugía y si no se desea el efecto antiplaquetario, el tratamiento con BRILINTA® debe suspenderse 5 días antes de la intervención (véase la sección “Propiedades farmacodinámicas”).
Pacientes con insuficiencia hepática moderada: Se recomienda precaución en caso de insuficiencia hepática moderada porque no se ha investigado el uso de BRILINTA® en este grupo de pacientes. BRILINTA® está contraindicado en los pacientes con insuficiencia hepática grave (véase la sección “Contraindicaciones”).
Pacientes con un riesgo elevado de episodios bradicárdicos: A raíz de la observación de asistolias generalmente asintomáticas en un estudio clínico anterior, el estudio principal que evaluó la seguridad y la eficacia de BRILINTA® excluyó a los pacientes con un riesgo elevado de episodios bradicárdicos (por ejemplo, pacientes sin marcapasos con síndrome de disfunción del nodo sinusal, bloqueo auriculoventricular de grado 2 o 3 o síncope asociado con bradicardia). En consecuencia, dada la experiencia clínica limitada, se recomienda precaución en estos pacientes (véase la sección “Propiedades Farmacodinámicas”).
Disnea: Se ha notificado disnea con BRILINTA®, generalmente de intensidad leve a moderada y que ha remitido sin necesidad de suspender el tratamiento, en alrededor del 13.8% de los pacientes (véase la sección “Reacciones adversas”). Aún no se ha dilucidado el mecanismo subyacente. En caso de aparición de disnea o de prolongación o recrudecimiento de una existente, se justifica una investigación completa y, si el paciente no tolera el tratamiento con BRILINTA®, éste debe suspenderse.
Otros: Dado que en el estudio PLATO se observó una relación entre la dosis de mantenimiento de AAS y la eficacia relativa del ticagrelor con respecto al clopidogrel, no se recomienda coadministrar el ticagrelor con dosis de mantenimiento de AAS elevadas (>300 mg) (véase la sección “Propiedades farmacodinámicas”).
Debe evitarse la coadministración de BRILINTA® con inhibidores potentes de la forma CYP3A4 (por ejemplo, ketoconazol, claritromicina, nefazadona, ritonavir y atazanavir) porque puede aumentar considerablemente la exposición a BRILINTA® (véase la sección “Interacción con otros medicamentos y otras formas de interacción”).
Suspensión del tratamiento: Los pacientes que deben suspender el tratamiento con BRILINTA® corren un mayor riesgo de sufrir accidentes cardiacos. Debe evitarse la suspensión prematura del tratamiento. Si es preciso interrumpir temporalmente el tratamiento con BRILINTA® a raíz de una reacción adversa, debe reanudarse a la brevedad posible cuando los beneficios esperados superen los riesgos asociados con la reacción adversa o una vez que se haya resuelto la reacción adversa (véase la sección “Posología y forma de administración”).
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN: El tratamiento con BRILINTA® debe iniciarse con una dosis de carga única de 180 mg (dos comprimidos de 90 mg) y continuarse con una dosis de 90 mg dos veces al día.
Los comprimidos BRILINTA® para administración oral pueden tomarse con o sin alimentos. Para pacientes que estén incapacitados para deglutir la(s) tableta(s) entera, las tabletas de BRILINTA® (90 mg y 2x90 mg) se pueden triturar para convertirlas en un polvo fino y mezclarlas en medio vaso de agua y beberlas inmediatamente. El vaso se debe enjuagar con medio vaso adicional de agua y beber el contenido. La mezcla también se puede administrar a través de una sonda nasogástrica (CH8 o mayor). Es importante purgar la sonda nasogástrica con agua después de administrar la mezcla.
A menos que exista una contraindicación específica, los pacientes tratados con BRILINTA® también deben tomar diariamente ácido acetil salicílico (AAS). Tras la dosis inicial de AAS, BRILINTA® debe asociarse con una dosis de mantenimiento de AAS de 75 a 150 mg (véase la sección “Propiedades farmacodinámicas”).
Deben evitarse las interrupciones del tratamiento. El paciente que haya omitido una dosis de BRILINTA® deberá tomar el comprimido de 90 mg a la hora programada para la siguiente dosis.
Los médicos que deseen reemplazar el clopidogrel en un paciente por un tratamiento con BRILINTA® deberán administrar la primera dosis de 90 mg de BRILINTA® 24 horas después de la última dosis de clopidogrel (véase la sección “Propiedades farmacodinámicas”).
Se recomienda administrar BRILINTA® durante un periodo mínimo de 12 meses a menos que la suspensión del tratamiento esté indicada por motivos clínicos (véase la sección “Propiedades farmacodinámicas”). En los pacientes con síndromes coronarios agudos (SCA), la suspensión prematura de cualquier tratamiento antiplaquetario, incluido BRILINTA®, podría aumentar el riesgo de muerte de origen cardiovascular o de infarto de miocardio debido a la enfermedad subyacente del paciente (véase la sección “Advertencias y precauciones especiales de uso”).
Poblaciones especiales
Pacientes pediátricos: No se han demostrado la inocuidad y la eficacia de BRILINTA® en pacientes menores de 18 años.
Pacientes de edad avanzada: No es necesario ajustar la dosis.
Pacientes con insuficiencia renal: No es necesario ajustar la dosis en los pacientes con insuficiencia renal (véase la sección “Propiedades farmacocinéticas”). Se carece de información sobre el tratamiento en pacientes sometidos a diálisis renal.
Pacientes con insuficiencia hepática: No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve. BRILINTA® no se ha investigado en pacientes con insuficiencia hepática moderada o grave (véase la sección “Propiedades farmacocinéticas”).
SOBREDOSIS: No se conoce actualmente ningún antídoto capaz de revertir los efectos de BRILINTA® y no se espera poder dializarlo (véase la sección “Advertencias y precauciones especiales de uso”). Para tratar una sobredosis conviene seguir la práctica médica convencional aplicable localmente. El efecto previsible de una sobredosis de BRILINTA® es una prolongación de la duración del riesgo hemorrágico asociado con la inhibición de la agregación plaquetaria. En caso de hemorragia deben tomarse medidas de apoyo adecuadas.
Las dosis únicas de ticagrelor de hasta 900 mg son bien toleradas. La toxicidad gastrointestinal limitó la dosis en un estudio sobre dosis únicas crecientes. Las otras reacciones adversas de importancia clínica que pueden producirse tras una sobredosis incluyen disnea y asistolias.
En caso de sobredosis deben vigilarse estos efectos adversos potenciales y considerarse la posibilidad de instaurar una supervisión del ECG.
PRESENTACIÓN: Comprimidos recubiertos de 90 mg. (Reg. San. INVIMA 2012M-0012872).
Venta con fórmula médica. Mantener fuera del alcance de los niños.
IPP Clave: 1-2016.
Fuente: Doc ID-001614189 V6.0.
Traducción de: Doc ID-001606805 V6.0.
Texto basado en: CDS Noviembre 4 de 2015.
Fecha de preparación de la versión: Enero 2016.
Mayor información Departamento Médico de
AstraZeneca Colombia S. A. S.
Bogotá, D.C., Colombia.