VALDUREIM
PARECOXIB
Solución inyectable
Vial(es), 40 mg Polvo y disolvente para solución inyectable,
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Vial de 40 mg: Cada VIAL contiene 40 mg de parecoxib (presente como 42,36 mg de parecoxib sódico) para reconstitución con 2 mL de solvente. Después de la reconstitución, la concentración final de parecoxib es 20 mg/mL.
PARTICULARIDADES FARMACÉUTICAS:
Incompatibilidades: Después de la reconstitución con un diluente aceptado, el parecoxib sódico puede ser inyectado a través de una línea IV infundiendo solución de cloruro de sodio 0,9% para inyección, solución de dextrosa 5% para inyección, solución-ringer lactato para inyección o solución de dextrosa 5% y cloruro de sodio 0,45% para inyección. No se recomienda usar una línea IV que esté entregando solución de dextrosa 5% en ringer-lactosa, ni ninguna otra solución IV que no esté entre las mencionadas, ya que podría causar precipitación desde la solución.
El parecoxib sódico no se debe mezclar para inyectarlo con ninguna otra droga.
No inyecte el parecoxib a través de una línea IV que esté entregando alguna otra droga. La línea IV debe ser lavada antes y después de la inyección del parecoxib adecuadamente, con una solución de compatibilidad conocida (ver Instrucciones).
Instrucciones para su uso y manipulación: El parecoxib sódico para inyección, es un polvo liofilizado sin agentes conservadores. El parecoxib sódico se debe reconstituir con 1 mL (vial de 20 mg) o 2 mL (vial de 40 mg) de solución de cloruro de sodio para inyección (0,9%).
Alternativamente, el parecoxib sódico se puede reconstituir con solución bacteriostática de cloruro de sodio 0,9% para inyección, solución de dextrosa 5% para inyección o solución de dextrosa 5% y cloruro de sodio 0,45% para inyección.
Para la reconstitución no se recomienda usar solución ringer-lactosa para inyección o solución de dextrosa 5% en ringer-lactosa para inyección, ya que podrían causar precipitación de la droga desde la solución. Tampoco se recomienda usar agua para Inyección para reconstituir el parecoxib sódico, ya que resultaría una solución que no es isotónica.
No refrigere ni congele el producto reconstituído.
Condiciones de almacenamiento: Conservar a una temperatura menor de 30 °C. Proteger de la luz. Una vez recosntituido posee una vida útil de 48 horas cuando se almacena a una temperatura inferior a 30 °C. Este producto es monodosis.
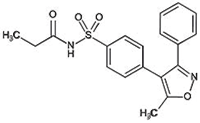
Estructura y nombre químico del principio activo: N-[[4-(5-Metil-3-fenil-4-isoxazolil)fenil]sulfonil]propanamida.
PFIZER
® Marca registrada
PROPIEDADES FARMACODINÁMICAS: El parecoxib es una prodroga del valdecoxib. El valdecoxib es un medicamento antiinflamatorio no-esteroideo (AINE), que exhibe propiedades antiinflamatorias, analgésicas y antipiréticas en modelos animales. Se cree que el mecanismo de acción se explica por la inhibición de la síntesis de prostaglandi-nas, principalmente por inhibición de la ciclooxigenasa-2 (COX-2). En concentraciones plasmáticas terapéuticas en humanos, el valdecoxib no inhibe la ciclooxigenasa-1 (COX-1).
La eficacia del parecoxib se estableció en estudios de dolor en cirugías dental, ginecológica (histerec-tomía), ortopédica (reemplazo de rodilla y cadera) e implante de derivación coronaria (ver Contraindicaciones). El primer efecto analgésico perceptible se observó en 7-13 minutos, con demostración de analgesia clínicamente significativa a los 23-39 minutos y un efecto pico dentro de las 2 horas, después de la administración IV o IM de dosis únicas de 40 mg de parecoxib. La magnitud del efecto analgésico de la dosis de 40 mg, fue comparable al de 60 mg IM o 30 mg IV de ketorolac. Después de la administración de una sola dosis, la duración de la analgesia fue dependiente de la dosis y del modelo clínico de dolor y varió desde 6 hasta más de 12 horas.
Efectos ahorrador del opioides: En las dosis recomendadas, el parecoxib redujo significativamente el consumo de opioides y los efectos adversos relacionados con estos (fatiga, somnolencia, confusión, incapacidad para concentrarse, mareo, náusea, estreñimiento, dificultad para orinar, prurito, arcadas/vómito) reportados por el paciente, proporcionando a la vez alivio del dolor, en comparación con el uso de opioides solos.
En un estudio controlado con placebo de ortopedia y cirugía general (n =1050), los pacientes recibieron una dosis parenteral inicial de parecoxib de 40 mg IV seguido por 20 mg dos veces al día por un mínimo de 72 horas además de recibir cuidados estándar incluyendo suplemento de opioides controlados (sulfato de morfina IV). La disminución en el uso del opioide con el tratamien-to con parecoxib en los días 2 y 3, fue de 7.2 mg y 2.8 mg (37% y 28% respectivamente). Esta reducción en el uso de opioides se acompañó de una disminución significativa del reporte de los efectos adversos desagradables relacionados con los opioides, así como un mejor alivio del dolor en comparación con los opioides solos. Estudios adicionales en otros escenarios quirúrgicos, arrojaron resultados similares.
Plaquetas: En estudios clínicos con adultos jóvenes (18-55 años) y de edad avanzada (65-83 años), la dosificación hasta por 7 días con dosis únicas y múltiples de 20 mg y 40 mg de parecoxib dos veces al día, no tuvo ningún efecto sobre la agregación plaquetaria o el tiempo de sangrado. En comparación, 15 mg y 30 mg de ketorolac en dosis únicas o después de 5 días de tratamiento, disminuyeron significativamente la agregación plaquetaria y aumentaron el tiempo de sangrado. El parecoxib (40 mg dos veces al día) no tuvo ningún efecto clínicamente significativo sobre la inhibición de la función plaquetaria mediada por la aspirina, ni tampoco alteró los efectos farmacodinámicos de la heparina sobre el tiempo parcial de tromboplastina activada (TPTa) o las plaquetas, en comparación con placebo.
Estudios gastrointestinales: En estudios de corto-plazo (7 días), la incidencia de úlceras gastroduodenales o erosiones observadas endoscópicamente en sujetos jóvenes y de edad avanzada ( 65 años) saludables que recibieron parecoxib (5-21%) fue, aunque mayor que la del placebo (5-12%), significativamente más baja que la incidencia observada con DAINEs (66-90%).
Estudios de seguridad post-cirugía de implante de bypass coronario: Además de los reportes de los eventos adversos rutinarios, categorías de eventos prespecificadas, adjudicadas por un comité independiente de expertos, fueron examinadas en dos estudios de seguridad controlados con placebo, en los cuales los pacientes recibieron el parecoxib sódico durante 3 días por lo menos y luego se les hizo la transición a valdecoxib oral para una duración total de 10-14 días. Todos los pacientes recibieron la atención estándar de analgesia durante el tratamiento.
Los pacientes recibieron una dosis baja de ácido acetilsalicílico desde antes de la aleatorización y a lo largo de los dos estudios de cirugía de implante de bypass coronario.
El primer estudio doble ciego, controlado con placebo de cirugía de bypass coronario, evaluó pacientes tratados con 40 mg de parecoxib sódico IV dos veces al día, por un mínimo de 3 días, seguido por un tratamiento con 40 mg de valdecoxib dos veces al día (grupo parecoxib sódico/valdecoxib) (n=311) o con placebo/placebo (n=151). Se evaluaron nueve categorías de eventos adversos preespecificadas (eventos tromboembólicos cardiovasculares; pericarditis; nuevo inicio o exacerbación de insuficiencia cardiaca congestiva; insuficiencia/disfunción renal; complicaciones de úlceras del tracto GI superior; hemorragias mayores no-GI, infecciones; complicaciones pulmonares no-infecciosas; muerte). Se detectó una incidencia significativamente mayor (p<0,05) de eventos tromboembólicos/cardiovasculares (infarto miocárdico, isquemia, accidente cerebrovascular, trombosis venosa profunda y embolismo pulmonar) en el grupo de tratamiento parecoxib/valdecoxib, en comparación con la del placebo/placebo, durante el periodo de la dosificación IV (2,2% y 0,0%, respectivamente), así como durante todo el periodo del estudio (4,8% y 1,3%, respectivamente). Con el tratamiento parecoxib sódico/valdecoxib se observó un incremento de la incidencia de complicaciones de la herida quirúrgica (la mayoría involucrando la herida esternal).
En el segundo estudio de cirugía de bypass coronario, se evaluaron cuatro categorías de eventos predefinidos (tromboembólicos/cardiovasculares; insuficiencia/deterioro renal; úlceras/hemorragias del tracto GI superior; complicación de la herida quirúrgica).
Los pacientes fueron aleatorizados durante las 24 horas posteriores a la cirugía de derivación coronaria, a los siguientes 3 grupos de tratamiento: Dosis inicial de 40 mg de parecoxib IV, luego 20 mg de parecoxib IV cada 12 horas durante un periodo mínimo de 3 días, siguiendo con 20 mg de valdecoxib PO cada 12 horas (n=544) hasta completar los 10 días del tratamiento; o placebo IV, seguido por valdecoxib PO cada 12 horas (n=544); o placebo IV, seguido por placebo oral (n=548). Se detectó una incidencia significativamente mayor (p=0,033) en la categoría de eventos tromboembólicos/cardiovasculares en el grupo de tratamiento parecoxib/valdecoxib (2,0%), en comparación con el grupo de tratamiento placebo/placebo (0,5%). Asimismo, el grupo de tratamiento placebo/valdecoxib se asoció con una mayor incidencia en la categoría de eventos tromboembólicos cardiovasculares, versus el tratamiento placebo, si bien esta diferencia no alcanzó significación estadística. Tres de los seis eventos tromboembólicos cardiovasculares detectados en el grupo de tratamiento placebo/valdecoxib, ocurrieron durante el periodo de tratamiento con placebo; estos pacientes no recibieron valdecoxib. Los eventos predefinidos que ocurrieron con mayor incidencia en todos los tres grupos de tratamiento, fueron los incluidos en la categoría de complicaciones de la herida quirúrgica, incluyendo infecciones quirúrgicas profundas y eventos relacionados con la cicatrización de la herida esternal.
No hubo diferencias significativas entre los tratamientos activos y el placebo en ninguna de las demás categorías de eventos predeterminados (insuficiencia/deterioro renal, complicaciones ulcerosas del tracto digestivo superior o complicaciones de la herida quirúrgica).
El parecoxib no ha sido estudiado en procedimientos de revascularización cardiovascular diferentes al bypass coronario.
En un análisis de 17 estudios controlados en cirugía mayor no-cardiaca, donde la mayoría de los pacientes se trataron durante 2 días, los pacientes que recibieron parecoxib no experimentaron un aumento en el riesgo de eventos adversos cardiovasculares, en comparación con los pacientes que recibieron placebo. Esto incluyó a pacientes con ningún, uno o dos factores de riesgo cardiovascular. Este análisis tuvo un poder de alrededor del 77% para detectar una duplicación en la tasa basal de eventos adversos cardiovasculares, en los pacientes tratados con parecoxib.
Cirugía general: En un estudio grande (N=1.050) de cirugía ortopédica/general mayor, los pacientes recibieron una dosis inicial de 40 mg IV de parecoxib, luego 20 mg IV cada 12 horas por un mínimo de 3 días, seguido por valdecoxib PO (20 mg cada 12 horas) (n=525) durante el resto del periodo de tratamiento de 10 días, o placebo IV seguido por placebo PO (n=525). En estos pacientes post-quirúrgicos no hubo diferencias significativas en el perfil de seguridad global, incluyendo las cuatro categorías preespecificadas de eventos descritas antes para el segundo estudio de cirugía de bypass coronario, para el parecoxib sódico/valdecoxib en comparación con el tratamiento placebo.
PROPIEDADES FARMACOCINÉTICAS: Posterior a la administración de una inyección IV o IM, el parecoxib es convertido rápidamente a valdecoxib, la sustancia farmacológicamente activa, por hidrólisis enzimática en el hígado.
Absorción: La exposición del valdecoxib después de dosis únicas de parecoxib, medida tanto por el área bajo la curva de concentración plasmática vs. tiempo (ABC) y la concentración máxima (Cmáx), es aproximada-mente linear en el rango de las dosis clínicas. El ABC y la Cmáx después de la administración dos veces al día, tienen comportamiento lineal hasta 50 mg IV y 20 mg IM. Las concentraciones plasmáticas de valdecoxib en estado estacionario, se obtuvieron a los 4 días con dosis dos veces al día.
Después de dosis IV e IM únicas de 20 mg de parecoxib sódico, la Cmáx del valdecoxib se alcanza en aproximadamente 30 minutos y aproximadamente 1 hora, respectivamente. En términos de ABC y Cmáx, la exposición al valdecoxib fue similar, después de la administración IV e IM. La Cmáx promedio del parecoxib después de la administración IM, fue más baja que la obtenida con la dosificación por bolo IV, hecho que se atribuye a una absorción extravascular más lenta después de la administración IM. Estas disminuciones no se consideran clínicamente relevantes, ya que la Cmáx del valdecoxib es comparable después de la administración IM e IV de parecoxib sódico.
Distribución: El volumen de distribución de valdecoxib después de su administración IV, es aproximadamente 55 litros. La unión a proteínas plasmáticas es aproximadamente de 98%, dentro del rango de concentraciones alcanzadas con la mayor dosis recomendada, 80 mg/día. El valdecoxib, pero no el parecoxib, se distribuye extensamente en el interior de los eritrocitos.
Metabolismo: El parecoxib es convertido rápida y casi completamente in vivo a valdecoxib y ácido propiónico, con una vida media plasmática de aproximadamente 22 minutos. La eliminación del valdecoxib ocurre a través de un metabolismo hepático extenso, involucrando diferentes vías que incluyen las isoenzimas del citocromo P-450 (CYP) 3A4 y CYP2C9 y glucuronidación independiente del CYP (cerca del 20%) de la fracción sulfonamida.
En el plasma humano se ha identificado un metabolito hidroxilado del valdecoxib (vía CYP), que es activo como inhibidor de la COX-2. Éste representa aproximadamente 10% de la concentración de valdecoxib; debido a la baja concentración de este metabolito, no se espera que contribuya significativamente al efecto clínico subsiguiente a la administración de dosis terapéuticas de parecoxib sódico.
Eliminación: El valdecoxib se elimina vía metabolismo hepático, con menos del 5% de la droga recuperado en forma inalterada en la orina. No se ha detectado parecoxib inalterado en la orina y solamente cantidades trazas en las heces. Cerca del 70% de la dosis se excreta en la orina como metabolitos inactivos. La depuración plasmática (CLp) del valdecoxib, es de alrededor de 6 L/h. Después de la dosificación IV o IM del parecoxib sódico, la vida media de eliminación (t½) del valdecoxib es de aproximadamente 8 horas.
CONTRAINDICACIONES:
El parecoxib está contraindicado en:
• Pacientes con antecedentes de hipersensibilidad al parecoxib o a algún otro ingrediente del producto.
• Pacientes que tengan reacciones de tipo alérgico demostradas a las sulfonamidas.
• Pacientes que hayan experimentado asma, urticaria o reacciones de tipo alérgico, después de tomar ácido acetilsalicílico (aspirina) o drogas antiinflamatorias no-esteroideas (AINEs), incluyendo a los otros inhibidores específicos de la ciclooxigenasa-2 (COX-2).
• El parecoxib está contraindicado para el tratamiento del dolor postoperatorio, subsiguiente a una cirugía de implante de derivación (bypass) coronaria y no se debe usar en este escenario.
• Úlcera péptica activa o hemorragia gastrointestinal.
• Disfunción hepática grave (albúmina sérica <25 g/l o puntuación ≥10 en la escala Child-Pugh).
• Enfermedad inflamatoria intestinal.
• Insuficiencia cardiaca congestiva (clases funcionales II-IV según la clasificación de la Asociación Cardiaca de Nueva York, NYHA).
• Cardiopatía isquémica o enfermedad cerebrovascular establecida.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES PARA SU USO:
Efectos cardiovasculares: Los inhibidores de la COX-2, de los cuales el parecoxib es uno, se han asociado con un mayor riesgo de eventos adversos cardiovasculares y trombóticos, cuando se usan a largo-plazo. No se ha determinado la magnitud exacta del riesgo asociado con una sola dosis, ni la duración exacta del tratamiento asociada con incremento del riesgo.
Dos estudios realizados por separado de cirugía de implante de bypass coronario, evidenció que los pacientes que recibieron parecoxib durante un mínimo de 3 días, seguido por valdecoxib oral (el metabolito activo del parecoxib) durante 7-14 días, tuvieron un aumento en la incidencia de eventos cardiovasculares/tromboembólicos (p. ej., infarto miocárdico y accidente cerebrovascular), en comparación con los que recibieron placebo (ver Propiedades farmacodinámicas). Por consiguiente, el parecoxib está contraindicado para el tratamiento del dolor postoperatorio subsiguiente a la cirugía de bypass coronario.
Efectos gastrointestinales (GIs): En pacientes tratados con parecoxib, se han presentado perforaciones, úlceras o hemorragias gastrointes-tinales (GIs) superiores. Los pacientes con mayor riesgo de desarrollar ese tipo de complicaciones GIs con los AINEs, son los de edad avanzada, los pacientes que tienen una enfermedad cardiovascular, los pacientes que usan concomitantemente aspirina o los pacientes con antecedentes, o enfermedad GI activa, tal como ulceración, sangrado o condiciones inflamatorias.
Efectos en la piel: El valdecoxib, que es el metabolito activo de de la molécula del parecoxib, contiene una fracción sulfonamida y los pacientes con antecedentes conocidos de alergia a las sulfonamidas, pueden tener un mayor riesgo de presentar reacciones en la piel. Los pacientes sin antecedentes de alergia a sulfonamidas, también podrían estar en riesgo de reacciones severas de la piel.
Durante la vigilancia posterior a la comercialización se han reportado reacciones serias de la piel, incluyendo eritema multiforme y Sindrome de Stevens-Johnson, en pacientes recibiendo el parecoxib. Además del eritema multiforme y del Sindrome de Stevens-Johnson,durante la vigilancia posterior a la comercialización se ha reportado la necrólisis epidérmica tóxica en pacientes recibiendo el valdecoxib. Con el valdecoxib se han reportado fallecimientos debidos al síndrome de Stevens-Johnson y a la necrolisis epidérmica tóxica y no se puede excluir la posibilidad para el parecoxib. Los pacientes estarían aparentemente en mayor riesgo de sufrir dichos eventos al principio del tratamiento, ya que en la mayoría de los casos el inicio del evento se produce durante las dos primeras semanas del tratamiento. El parecoxib se debe descontinuar al producirse la primera manifestación de erupción en la piel, lesiones en las mucosas u otros signos de hipersensibilidad. Durante la experiencia posterior a la comercialización, se han reportado reacciones dermatológicas serias con otros inhibidores de la COX-2. Las tasas de reportes de esos eventos, son aparentemente mayores para el valdecoxib, en comparación con los demás agentes COX-2.
Reacciones anafilactoides: Durante la experiencia posterior a la comercialización con el valdecoxib y el parecoxib45, se han reportado reacciones de hipersensibilidad (reacciones anafilácticas y angiodema) (ver Eventos adversos durante la vigilancia posterior a la comercialización). Estas reacciones han ocurrido en pacientes con y sin anteceden-tes de reacciones de tipo alérgico a las sulfonamidas (ver Contraindicaciones).
Uso con la warfarina o agentes similares: La coadministración del parecoxib con warfarina, causó un pequeño aumento en el área bajo la curva (ABC) de la warfarina y también en el tiempo de protrombina (medido por el Índice Normalizado Internacional [INI]). Aunque los valores del INI aumentaron tan solo levemente con la coadministración del parecoxib, la variabilidad día-a-día en los valores individuales aumentó. Se debe monitorear la actividad anticoagulante, particularmente durante los primeros días siguientes a la iniciación del parecoxib, en los pacientes que estén recibiendo warfarina o agentes similares, ya que estos pacientes pueden estar en mayor riesgo de complicaciones hemorrágicas.
Hipertensión: Así como el resto de los AINEs, el parecoxib puede provocar el inicio de hipertensión o empeorar una hipertensión ya existente, ambas pueden contribuir a un aumento en la incidencia de eventos cardiovasculares. Los AINEs, incluyendo el parecoxib, deben ser utilizados con precaución en pacientes con hipertensión. La presión arterial debe ser monitoreada de cerca durante el inicio y transcurso de la terapia con parecoxib.
Retención de líquidos y edema: Al igual que con otros medicamentos conocidos por inhibir la síntesis de las prostaglandinas, se han observado retención y edema en pacientes recibiendo parecoxib. Por lo tanto, el parecoxib se debe usar con precaución en pacientes con compromiso de la función cardiaca, edema preexistente u otras condiciones que predispongan, o empeoren, la retención de líquidos inclusive quienes utilicen tratamiento con diuréticos o en riesgo de hipovolemia.
Efectos renales: Durante la vigilancia post-comercialización se ha reportado insuficiencia renal aguda en pacientes recibiendo parecoxib (ver Efectos indeseables). La función renal debe ser monitoreada de cerca en los pacientes con enfermedad renal avanzada, que reciben parecoxib. (Ver Posología y administración).
Se debe tener precaución cuando se inicie un tratamiento en pacientes con deshidratación. Es recomendable prehidratar a los pacientes e iniciar después el tratamiento con parecoxib.
Efectos hepáticos: Los pacientes con insuficiencia hepática severa (Clase C Child-Pugh) no han sido estudiados. No se recomienda el uso del parecoxib en pacientes con insuficiencia hepática severa. El parecoxib se debe usar con precaución, cuando se traten pacientes con insuficiencia hepática moderada (Clase B Child-Pugh), iniciándolos en la dosis recomendada más baja. (Ver Posología y administración).
Un paciente con síntomas y/o signos de disfunción hepática, o en quien haya ocurrido una prueba anormal de la función hepática, debe ser monitoreado con cuidado para evidenciar el desarrollo de una reacción hepática más severa, mientras esté en tratamiento con parecoxib.
General: Al reducir la inflamación el parecoxib puede disminuir la utilidad de signos de diagnóstico, tal como la fiebre, en la detección de infecciones. El uso concomitante de parecoxib con otros AINEs no específicos se debe evitar.
Los pacientes que presenten factores de riesgo relevantes para el desarrollo de acontecimientos cardiovasculares (p. ej., pacientes con hipertensión, hiperlipidemia, diabetes mellitus, fumadores), sólo podrán ser tratados con parecoxib sódico, después de una cuidadosa valoración.
EMBARAZO Y LACTANCIA: No hubo hallazgos de teratogenicidad en los estudios en ratas y conejos. Los estudios en ratas con dosis tóxicas para la madre y en conejos con la dosis máxima evaluable, no revelaron efectos embriotóxicos distintos a la pérdida post-implantación, que también se ha observado con otros medicamentos que inhiben la síntesis de las prostaglandinas.
No se han efectuado estudios en mujeres embarazadas.
El parecoxib se debe usar en el embarazo, solamente si el beneficio potencial para la madre, justifica el riesgo potencial para el feto.
Al igual que con otros medicamentos que se sabe que inhiben la síntesis de las prostaglandinas, el uso del parecoxib durante el tercer trimestre del embarazo se debe evitar, ya que puede provocar inercia uterina y cierre prematuro del ducto arterioso.
El parecoxib y su metabolito activo se excretan en la leche de ratas amamantando. No se sabe si se excretan en la leche humana. Como muchos medicamentos se excretan en la leche humana y debido a la posibilidad de que ocurran las reacciones adversas del parecoxib en infantes alimentados con leche materna, se debe tomar la decisión de descontinuar la lactancia o descontinuar el medicamento, tomando en cuenta la importancia del medicamento para la madre.
EFECTOS INDESEABLES:
Estudios clínicos: Los siguientes eventos adversos fueron reportados en pacientes que recibieron parecoxib (N = 5,402) en 28 estudios controlados con placebo.
Eventos que ocurrieron ≥ 10%:
Trastornos gastrointestinales: Náusea.
Eventos que ocurrieron en ≥ 1% y <10%:
Trastornos gastrointestinales: Dolor abdominal estreñimiento, dispepsia, vómito.
Trastornos generales y del sitio de administración: Edema periférico.
Infecciones e infestaciones: Osteitis alveolar (alveolo seco).
Trastornos del sistema nervioso: Mareo.
Trastornos psiquiátricos: Insomnio.
Trastornos renales y urinarios: Oliguria.
Trastornos de piel y tejido subcutáneo: Sudoración aumentada, prurito.
Trastornos vasculares: Hipotensión.
Eventos que ocurrieron en ≥ 0,5% y < 1%:
Trastornos gastrointestinales: Boca seca, flatulencia
Trastornos musculoesqueléticos y del tejido conectivo: Dolor de espalda.
Trastornos cardiacos: Bradicardia.
Infecciones e infestaciones: Faringitis.
Trastornos de Piel y tejido subcutáneo: Erupción.
Trastornos vasculares: Hipertensión.
Eventos que ocurrieron en < 0.5%:
Trastornos cardiacos: Infarto al miocardio.
Trastorno auditivo y vestibular: Dolor de oído.
Trastornos gastrointestinales: Esofagitis, reflujo gastroesofágico, sonidos de intestino hipoactivo, pancreatitis, sudoración perioral.
Trastornos generales y del sitio de administración: Dolor en el sitio de la inyección, reacción en el sitio de la inyección, astenia.
Trastornos del sistema inmune: Reacción anafilactoide.
Exámenes de laboratorio: Aumento del nitrógeno ureico sérico, aumento de la creatin fosfoqui-nasa, aumento de creatinina, aumento de la DHL.
Lesión, toxicidad y complicaciones de procedimiento: Complicaciones post operatorias en piel.
Trastornos del metabolismo y nutrición: Anorexia, hiperglicemia.
Trastornos musculo esqueléticos y del tejido conectivo: Artralgia.
Trastornos del sistema nervioso: Trastorno cerebrovascular.
Trastornos psiquiátricos: Agitación.
Trastornos renales y urinarios: Insuficiencia renal aguda.
Trastornos respiratorios, torácicos y mediastinales: Embolismo pulmonar.
Trastornos en piel y tejido conectivo: Equimosis, urticaria.
Trastornos vasculares: Hipertensión agravada, hipotensión postural.
Después de la cirugía de bypass coronario, los pacientes que recibieron parecoxib presentaron un mayor riesgo de eventos adversos, tales como eventos tromboembólicos cardiovasculares (p. ej., infarto miocárdico y accidente cerebrovascular), infecciones quirúrgicas profundas o complicaciones de la cicatrización de heridas esternales.
Vigilancia post-comercialización:
En la experiencia post-comercialización, se han reportado los siguientes eventos adversos serios, raros, en asociación con el uso del parecoxib: Eritema multiforme, síndrome de Stevens-Johnson, insufi-ciencia renal y reacciones de hipersensibilidad, incluyendo anafilaxis y angiodema (ver Advertencias y precauciones especiales de uso).
Además de las reacciones severas cutáneas como eritema multiforme y síndrome de Stevens- Johnson, durante la vigilancia post-comercialización se ha reportado necrolisis epidérmica tóxica en asociación con el uso del valdecoxib y no se puede excluir para el parecoxib.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: El efecto del parecoxib sobre la capacidad para conducir y utilizar máquinas, no se ha estudiado.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN:
General: Los estudios de interacciones de medicamentos se realizaron con el parecoxib o con su metabolito activo (valdecoxib).
En humanos, el parecoxib sufre metabolismo hepático extenso, involucrando las isoenzimas del P450 3A4 y 2C9 y vías no-dependientes del P450 (p. ej., glucuronidación). La administración concomitante del parecoxib con inhibidores conocidos de las CYP 3A4 y 2C9, puede resultar en un aumento del área bajo la curva (ABC) del parecoxib.
Medicamentos específicos:
Interacción del parecoxib con warfarina o agentes similares: Ver Advertencias y precauciones.
Fluconazol y ketoconazol: La coadministración del fluconazol, un inhibidor de la CYP2C9, y ketocona-zol, un inhibidor de la CYP3A4, aumentó el ABC del valdecoxib en 62% y 38%, respectivamente. Cuando se coadministre el parecoxib con fluconazol, se debe usar la dosis recomendada más baja. No es necesario ajustar la dosificación cuando se coadministre el parecoxib con ketoconazol. (Ver Posología y administración).
Inhibidores de la ECA: La inhibición de las prostaglandinas puede disminuir el efecto antihipertensivo de los inhibidores de la enzima convertidora de la angiotensina (ACE). Esta interacción debe ser tomada en cuenta en pacientes que reciban concomitantemente parecoxib con inhibidores de la ECA.
Diuréticos: Estudios clínicos han demostrado que los AINEs pueden disminuir, en algunos pacientes, el efecto natriurético de la furosemida y tiazidas, por inhibición de la síntesis de las prostaglandinas renales.
Litio: El valdecoxib produjo disminuciones significativas en la depuración sérica (25%) y la depuración renal (30%) del litio, resultantes en un ABC 34% mayor, comparada con la del litio solo. Se deben monitorear de cerca las concentraciones séricas de litio, cuando se inicie o se cambie el tratamiento con parecoxib, en pacientes que reciben litio.
Otras: Se condujeron estudios de interacción entre el parecoxib y el midazolam oral o I.V., heparina, propofol, fentanilo y alfentanilo. También se realizaron estudios de interacción entre el valdecoxib y glibenclamida (gliburida), metotrexato, anticonceptivos orales (etinil estradiol/noretindrona), fenitoína, omeprazol y diazepam. En dichos estudios no se observaron interacciones clínicamente relevantes.
Parecoxib puede ser coadministrado con opioides analgésicos. En estudios clínicos, el requerimiento de dosis según la necesidad de opioides (PRN) fue reducido significativamente cuando se coadministró con parecoxib.
No se han efectuado estudios formales de interacción entre el parecoxib y agentes anestésicos por inhalación, tales como el óxido nitroso y el isoflurano; sin embargo, en los estudios clínicos no hubo evidencia de alguna interacción medicamentosa.
El parecoxib no interfiere con el efecto antiplaquetario de una dosis baja de aspirina. Debido a esta carencia de efectos plaquetarios, el parecoxib no es un reemplazo de la aspirina en el tratamiento profiláctico de enfermedades cardiovasculares.
PARTICULARIDADES CLÍNICAS:
Indicaciones terapéuticas:
El parecoxib está indicado para: Manejo del dolor agudo1. En el preoperatorio (analgesia preventiva), para prevenir o disminuir el dolor postoperatorio. Concomitantemente con analgésicos opioides, para disminuir los requerimientos de estos.
POSOLOGÍA Y MÉTODO DE ADMINISTRACIÓN: El parecoxib se puede administrar en dosis I.V. o I.M. únicas o múltiples, en forma regular o cuando se necesite. Después de iniciar el tratamiento, la dosis se debe ajustar con base en la respuesta del paciente. Los estudios clínicos del parecoxib se condujeron usando hasta 7 días de tratamiento. El parecoxib está indicado solamente para pacientes que necesitan tratamiento parenteral y en quienes no se puede obtener un beneficio similar con un tratamiento oral alternativo. Se recomienda la transición de los pacientes a un tratamiento oral alternativo lo más pronto posible, al estar indicado clínicamente.
Como el riesgo cardiovascular de los inhibidores específicos de la ciclooxigenasa-2 (COX-2) puede aumentar con la dosis y la duración de la exposición, se debe usar la duración más corta posible y la dosis efectiva diaria más baja. Sin embargo, la relevancia de esos hallazgos para el uso a corto-plazo del parecoxib en el escenario postoperatorio, no se ha evaluado.
— Manejo del dolor agudo: La dosis única o inicial recomendada para el tratamiento del dolor agudo es 40 mg, administrada I.V. o I.M., seguida por 20 o 40 mg cada 6 a 12 horas, según lo requerido, hasta una dosis diaria máxima de 80 mg. La inyección por bolo IV se puede administrar directamente en la vena o a través de una vía I.V. existente. La inyección IM se debe aplicar lenta y profundamente en el músculo.
— Prevención o disminución del dolor postoperatorio: La dosis recomendada es 40 mg, administrados I.V. o IM (aunque preferiblemente I.V.), 30-45 minutos antes de la incisión quirúrgica. Para obtener un efecto analgésico adecuado, podría necesitarse medicación postoperatoria continua.
— Uso concomitante con analgésicos opioides: Los analgésicos opioides se pueden usar concomitante-mente con el parecoxib, en su dosis descrita. En estudios clínicos, los requerimientos diarios de opioides se vieron significativamente disminuidos (20-40%), cuando se coadministraron con el parecoxib. El efecto óptimo se obtuvo cuando se administró el parecoxib previamente al opioide. En todos los estudios clínicos el parecoxib se administró en un intervalo de tiempo fijo, mientras que los opiáceos se administraron según la necesidad (PRN).
Edad avanzada: Generalmente no se necesitan ajustes de la dosificación. Sin embargo, sería aconsejable disminuir en 50% la dosis inicial de parecoxib, en los pacientes de edad avanzada que pesen menos de de 50 kg. La dosis máxima diaria en los pacientes que pesen menos de 50 kg, debería ser disminuida a 40 mg.
Insuficiencia hepática: No se requieren ajustes de dosis en pacientes con insuficiencia hepática leve (Clase A Child-Pugh). El tratamiento con parecoxib se debe iniciar en la dosis más baja recomendada, en pacientes con insuficiencia hepática moderada (Clase B Child-Pugh).
Los pacientes con insuficiencia hepática severa (Clase C Child-Pugh), no han sido estudiados. No se recomienda el uso del parecoxib en esos pacientes.
Insuficiencia renal: En pacientes con insuficiencia renal severa (depuración de creatinina <30 mL/minute), o en pacientes con predisposición a retención de líquidos, el parecoxib se debe iniciar en la dosis recomendada más baja, con monitoreo a fondo de la función renal del paciente.
Coadministración con fluconazol: Cuando se coadministre el parecoxib con fluconazol, se debe usar la dosis recomendada más baja de parecoxib.
Pacientes pediátricos: No se han establecido la seguridad y eficacia en niños menores de 18 años.
SOBREDOSIS: La experiencia clínica con sobredosis es limitada. Se han administrado dosis únicas I.V. de parecoxib de hasta 20 mg a sujetos saludables, sin efectos adversos clínicamente significativos. Dosis de parecoxib de 50 mg I.V. dos veces al día (100 mg/día) por 7 días, no resultaron en ningún síntoma de toxicidad.
En caso de sospechar una sobredosis, se debe prestar la atención médica de soporte apropiada. Es improbable que la diálisis represente un método eficiente para remover el medicamento, debido a su alto grado de unión a las proteínas plasmáticas.

