
REMINYL/ REMINYL ER
GALANTAMINA
Cápsulas de liberación prolongada
Frasco, Cápsulas de liberación prolongada, 8 Miligramos
Frasco, Cápsulas de liberación prolongada, 16 Miligramos
Frasco, Cápsulas de liberación prolongada, 24 Miligramos
CARACTERÍSTICAS FARMACÉUTICAS:
Lista de excipientes: Los ingredientes inactivos son gelatina, dietil ftalato, etilcelulosa, hipromelosa, polietilén glicol, dióxido de titanio (E171), sucrosa y almidón de maíz. La cápsula de 16 mg contiene también óxido férrico rojo (E172). La cápsula de 24 mg contiene también óxido férrico rojo (E172) y óxido férrico amarillo (E172).
Incompatibilidades: No aplica.
Precauciones especiales de almacenamiento: Cápsulas de liberación prolongada [extendida]: almacenar entre 15 ºC y 30 ºC.
Naturaleza y contenido del envase: La información será suplida localmente.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA: Las cápsulas de liberación prolongada (extendida) de REMINYL ER® contienen bromhidrato de galantamina equivalente a 8, 16 y 24 mg de galantamina base, respectivamente.
Para excipientes, ver Lista de excipientes.
FORMA FARMACÉUTICA: Cápsulas de liberación prolongada (extendida) para uso oral.
— Cápsulas de gelatina dura de 8 mg blanco opaco, tamaño 4, con la inscripción “GAL 8” [“G8”], que contienen gránulos de blancos a blancuzcos;
— Cápsulas de gelatina dura de 16 mg rosado opaco, tamaño 2, con la inscripción “GAL 16” [“G16”], que contienen gránulos de blancos a blancuzcos;
— Cápsulas de gelatina dura de 24 mg rosado opaco, tamaño 1, con la inscripción “GAL 24” [“G24”], que contienen gránulos de blancos a blancuzcos.
INSTRUCCIONES PARA USO Y MANIPULACIÓN <Y DESECHO>:
Abra el frasco y use la pipeta:
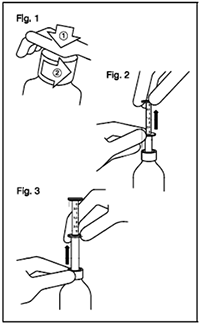
Fig. 1: El frasco viene con tapa resistente-a-niños y debe ser abierta de la manera siguiente:
— Empuje la tapa plástica de rosca hacia abajo mientras la desenrosca en contra de las agujas el reloj.
— Remueva la tapa desenroscada.
Fig. 2: Inserte la pipeta dentro del frasco.
Mientras sostiene el anillo inferior, hale hacia arriba el anillo superior hacia la marca correspondiente al número de milímetros que usted necesite administrar.
Fig.3: Sosteniendo el anillo de abajo, saque la pipeta completa del frasco.
Vacíe la pipeta en una bebida que no sea alcohólica deslizando hacia abajo el anillo superior y tómeselo inmediatamente.
Cierre el frasco.
Enjuague la pipeta con algo de agua.
Fecha de la revisión del texto: 1 de Febrero de 2.011
JANSSEN PHARMACEUTICAL COMPANIES
INDICACIONES TERAPÉUTICAS: REMINYL ER® está indicado para el tratamiento de demencia de leve a moderadamente severa del tipo Alzheimer, [incluyendo demencia Alzheimer con enfermedad cerebrovascular.
PROPIEDADES FARMACOLÓGICAS:
Propiedades farmacodinámicas: Grupo farmacoterapéutico: Drogas antidemencia; Código ATP: N06D A04.
Galantamina, alcaloide terciario, es un inhibidor de acetilcolinesterasa selectivo, competitivo y reversible. Además, galantamina mejora la acción intrínseca de acetilcolina sobre los receptores nicotínicos, probablemente a través de ligadura con un sitio alostérico del receptor. Como consecuencia, puede lograrse un aumento de la actividad del sistema colinérgico asociado con mejoría de la función cognoscitiva en pacientes con demencia tipo Alzheimer.
Estudios clínicos: La dosis de REMINYL ER® que mostraron ser efectivas en los estudios clínicos controlados en enfermedad de Alzheimer fueron 16, 24 y 32 mg/día. De estas dosis, se determinó que 16 y 24 mg/día tuvieron la mejor relación beneficio/riesgo y son las dosis recomendadas. La eficacia de galantamina ha sido estudiada usando cuatro mediciones específicas de resultado: ADAS-cog (medición de cognición basada en desempeño), CIBIC-plus (evaluación global hecha por un médico independiente basada en entrevista clínica con el paciente y con el médico tratante), varias mediciones de las actividades de la vida diaria y el Inventario Neuropsiquiátrico (NPI, escala que mide los trastornos de conducta).
En los estudios clínicos, el desempeño de los pacientes tratados con galantamina medido con ADAS-cog (ver Figura) y CIBIC-plus, fue de manera consistente estadísticamente significativo mejor que el de los pacientes que recibieron placebo. Los pacientes que fueron tratados durante 6 meses con galantamina tuvieron puntuaciones ADAS-cog que estuvieron significativamente mejoradas, comparado con sus puntuaciones basales. Al comparar con los pacientes no tratados, hubo un beneficio sustancial y sostenido en el funcionamiento cognoscitivo. El tratamiento con galantamina también preservó significativamente las actividades de la vida diaria, tales como vestirse, higiene, preparación de alimentos. Estas fueron evaluadas usando las evaluaciones hechas por el médico de la Evaluación de Incapacidad en demencia DAD) y el Inventario-ADL-Estudio Cooperativo de Enfermedad de Alzheimer (ADCS). Las dosis de galantamina de 16 y 24 mg/día mantuvieron la puntuación NPI a lo largo del período de observación, mientras que las puntuaciones de los pacientes con placebo se deterioraron claramente, como resultado del surgimiento de trastornos de conducta.
Figura 1: Cambio medio (±SE) desde la base en la puntuación ADAS-cog/11 a través del tiempo (datos observados) (datos agrupados GAL-USA-1 y GAL-INT-1)
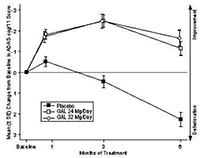
Figura 2: Cambio medio (±SE) desde la base en la puntuación ADAS-cog/11 a través del tiempo (todos los pacientes, datos observados) (GAL-USA-10)
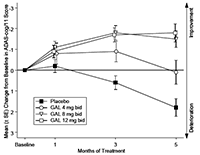
El tratamiento a largo-plazo (combinación de 6 meses doble-ciego seguido por 6 meses de tratamiento abierto) sugirió que el desempeño cognoscitivo y funcional fue mantenido por un año completo.
Se estudió la eficacia de REMINYL ER® en forma de cápsulas de liberación prolongada [extendida] en un estudio aleatorizado, doble-ciego, controlado con placebo, en enfermedad de Alzheimer. Los pacientes recibieron galantamina 8 mg/día durante 4 semanas, seguido por galantamina 16 mg/día por 4 semanas. En la semana 8, la dosis podía ser aumentada a 24 mg/día sobre la base de la seguridad y tolerabilidad, y podía ser reducida a 16 mg/día en la semana 12. La selección de dosis de la semana 12 fue fija para los 6 meses restantes. En el análisis de eficacia primaria especificado por el protocolo, con respecto a los dos puntos finales (ADAS-cog/11 y CIBIC-plus) en el Mes 6 simultáneamente, REMINYL ER® de liberación prolongada [extendida] fue de manera estadísticamente significativa mejor que el placebo en el mejoramiento de las actividades de la vida diaria (ADCS-ADL), que es una medición clave secundaria de eficacia. Los resultados de eficacia fueron similares con las cápsulas de Liberación Prolongada [Extendida) de REMINYL ER® y con las tabletas de REMINYL®, las cuales sirvieron como control activo en este estudio.
Enfermedad de Alzheimer con Enfermedad Cerebrovascular (AD+CVD): Se investigó la eficacia y seguridad de galantamina en sujetos con enfermedad de Alzheimer y enfermedad cerebrovascular significativa (AD+CVD), en un estudio doble-ciego, controlado con placebo. Hubo 282 sujetos, 48% de la población total del estudio (N=592) fueron los que llenaron los criterios de AD+CVD. Aunque la prueba clínica no tuvo poder para el análisis de subgrupo, los sujetos tratados con galantamina experimentaron una mejoría estadísticamente significativa al comparar con los sujetos tratados con placebo, en ambos resultados primarios [(cognición: ADAS-cog/11 [p<0,001]; evaluación clínica global: CIBIC-plus [p<0,001] y en la medición de las actividades de la vida diaria (DAD [p<0,003]. En general la seguridad y tolerabilidad de galantamina en sujetos con AD+CVD fueron similares a las vistas en estudios previos de galantamina en enfermedad de Alzheimer. El evento adverso reportado con mayor frecuencia en los sujetos fue náusea (19% de los sujetos con galantamina y 11% de los sujetos con placebo). Otros eventos, que ocurrieron en >5% de los sujetos con AD+CVD y reportados más frecuentemente en el grupo con galantamina que con placebo fueron mareos, vómitos, dolor abdominal, diarrea y fatiga. La incidencia de “trastornos cerebrovasculares” (por ej., apoplejía) fue más alta en el grupo con placebo (placebo, 5/96 [5%] sujetos: galantamina, 2/186 [1%] sujetos).
En general, el perfil de seguridad en AD+CVD fue consistente con el observado en estudios de galantamina en sujetos con enfermedad de Alzheimer.]
Daño cognoscitivo leve (MCI): Dos estudios de controlados de 2 años en sujetos con MCI no llenaron los resultados duales de eficacia primaria. Aunque la mortalidad fue baja (0,7%) inicialmente se registraron más muertes en los sujetos aleatorizados a galantamina (13/1.026) que a placebo (1/1.022), pero la incidencia de eventos adversos seros fue idéntica (19%) entre los grupos de tratamiento.
Cuando se incluyeron los datos recogidos de la gran proporción de pacientes de ambos grupos de tratamiento que descontinuaron antes de completar el período doble-ciego (GAL-COG-3002), se identificó un total de 102 muertes, 56 en el grupo con galantamina y 46 en el grupo con placebo (riesgo relativo [95% de CI] = 1,24 [0,84, 1,83]; p=0,274). El análisis de intención-de-tratar de 24 meses registró 20 muertes entre los sujetos aleatorizados a placebo comparado con 34 muertes registradas entre los sujetos aleatorizados a REMINYL ER® (riesgo relativo [95% de CI] = 1,70 [1,00, 2,90]; p=0,051). De los sujetos que fallecieron dentro del período especificado por protocolo de 30 días de descontinuación de la medicación del estudio doble-ciego, hubo 14 en el grupo con galantamina y 3 del grupo con placebo (riesgo relativo [95% de CI] = 4,08 [1,57, 10,57]; p=0,004).
Más sujetos tratados con placebo que tratados con galantamina descontinuaron antes de morir, lo cual puede contar para la diferencia en la mortalidad registrada inicialmente. Trece muertes en el grupo con placebo y 20 en el grupo con galantamina se encontraron como directamente relacionadas con los eventos adversos que ocurrieron mientras los sujetos estuvieron expuestos al estudio doble-ciego con la droga (riesgo relativo [95% de CI] = 1,54 [0,78, 3,04]; p=0,218).
Las muertes fueron debidas a varias causas que no so son inesperadas en una población de ancianos. Cerca de la mitad de las muertes, tanto en el grupo con placebo como con tratamiento activo fueron debidas a causas vasculares. No hubo evidencia de aumento del riesgo de muerte en los sujetos tratados con REMINYL®, a través del tiempo. Este patrón se observó de manera consistente en todos los análisis de la data.
Los resultados del estudio MCI discrepan de los observados en estudios de enfermedad de Alzheimer. En estudios agrupados de enfermedad de Alzheimer (n=4.614) la tasa de mortalidad fue numéricamente más alta en el grupo con placebo que en el grupo con REMINYL®. No hay evidencia de aumento de la mortalidad debida a REMINYL ER® en enfermedad de Alzheimer, incluyendo demencia por Alzheimer con enfermedad cerebrovascular].
Propiedades farmacocinéticas: Galantamina es una droga de baja-depuración (depuración plasmática de aproximadamente 300 ml/min) con un moderado volumen de distribución (promedio VDss de 175 lt). La eliminación de galantamina es bi-exponencial, con una vida media terminal del orden de 7-8 horas.
Después de la ingesta oral de una sola dosis de 8 mg de galantamina en forma de tabletas, la absorción es rápida, con una concentración pico en plasma de 43 ± 13 ng/ml, la cual se alcanza después de 1,2 horas, y AUC8 medio de 427 ± 102 ng.h/ml. La biodisponibilidad oral absoluta de galantamina es 88,5%. La ingesta oral de tabletas de galantamina con los alimentos enlentece su tasa de absorción (Cmáx reducida en ~ 25%) pero no afecta el grado en el que se absorbe (AUC).
Después de dosificación oral repetida de 1º2 mg de galantamina BID en forma de tabletas, las concentraciones medias pico y valle en plasma fluctúan entre 30 y 90 ng/ml. Las farmacocinéticas de galantamina son lineares en el rango de dosis de 4-16 mg BID.
Siete días después de una dosis única oral de 4 mg de galantamina-H3, se recuperó 90 – 97% de la radioactividad en la orina y 2,2 – 6,3% en las heces. Después de administración IV y oral, 18-22% de la dosis fue excretada en forma de galantamina inalterada en la orina en 24 horas, con una depuración renal de cerca de 65 ml/min, lo cual representa 20-25% de la depuración total plasmática.
Las vías metabólicas principales fueron N-oxidación, N-demetilación, O-demetilación, glucuronidización y epimerización. O-demetilación fue bastante más importante en los metabolizadores rápidos de CYP2D6. Los niveles de excreción de la radioactividad total en orina y heces no fueron diferentes entre los metabolizadores lentos y rápidos. Estudios in vitro confirmaron que citocromo P450, 2D6 y 3A4 fueron las principales isoenzimas citocromo P450 involucradas en el metabolismo de galantamina.
En el plasma de los metabolizadores lentos y rápidos, galantamina inalterada y su glucurónido fueron los que contaron para la mayor parte de la radioactividad de la muestra. En el plasma de los metabolizadores rápidos, el glucurónido O-desmetilgalantamina también fue importante.
Ninguno de los metabolitos activos de galantamina (norgalantamina, O-desmetilgalantamina y O-desmetil-norgalantamina) pudo ser detectado en su forma conjugada en el plasma de los metabolizadores lentos o rápidos, después de una dosis única. Norgalantamina fue detectable en el plasma de pacientes después de dosificación múltiple, pero no representó más de 10% de los niveles de galantamina.
Los datos de los estudios clínicos en pacientes, indican que las concentraciones plasmáticas de galantamina en pacientes con enfermedad de Alzheimer son 30 -40% más altas que en sujetos jóvenes saludables.
Las farmacocinéticas de galantamina en sujetos con daño hepático leve (puntuación Child-Pugh 5-6) fueron comparables a las de los sujetos saludables. En pacientes con daño hepático moderado (puntuación Child-Pugh de 7-9), AUC y vida media de galantamina aumentaron en cerca de 30% (ver Posología y métodos de administración).
Se estudió la disposición de galantamina es sujetos jóvenes con grado variables de función renal. La eliminación de galantamina disminuyó al disminuir la depuración de creatinina. Las concentraciones plasmáticas de galantamina aumentaron en los sujetos que tenían función renal dañada, en 38% con daño renal moderado (ClCR=52-1014 ml/min) o 67% con daño renal severo (ClCR=9-51 ml/min), al comparar con sujetos saludables compaginados por edad y peso (ClCR>=121 ml/min). Un análisis y simulaciones farmacocinéticas en la población indican que no se necesita ajustar la dosis en los pacientes con Alzheimer que tengan daño renal, dado que la CLCR es de por lo menos 9 ml/min (ver Posología y métodos de administración), ya que la depuración de creatinina es más baja en la población con Alzheimer.
Unión a las proteínas del plasma: La ligadura de galantamina a las proteínas del plasma es baja: 17,7 ± 0,8%. En sangre total, galantamina se distribuye principalmente en las células sanguíneas (52,5%) y en el agua del plasma (39,0%), mientras que la fracción de galantamina ligada a las proteínas del plasma es de solamente 8,4%. La relación de la concentración sangre-a plasma de galantamina es 1,17.
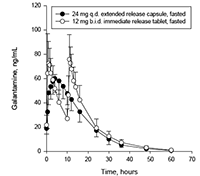
En un estudio de biodisponibilidad a niveles-constantes, se demostró que REMINYL ER® en forma de cápsulas de liberación prolongada [extendida], 24 mg una vez al día, es equivalente a 12 mg de tabletas de liberación inmediata 2 veces al día, con respecto a AUC24h y Cmín. El valor de Cmáx con 24 mg una vez al día de cápsula de liberación prolongada [extendida] el cual se alcanzó después de 4,4 horas, fue cerca de 24% más bajo que 12 mg dos veces al día de tableta de liberación inmediata. Los alimentos no tuvieron efecto sobre la biodisponibilidad a niveles-constantes de 24 mg de las cápsulas de liberación prolongada [extendida]. En un estudio de proporcionalidad de dosis de REMINYL ER® en forma de cápsulas de liberación prolongada [extendida], en sujetos adultos jóvenes y ancianos saludables, se lograron las concentraciones plasmáticas a niveles-constantes a los 6 días, con todas las dosis (8 mg, 16 mg y 24 mg) en ambos grupos de edad. Las farmacocinéticas a niveles-constantes fueron proporcionales a la dosis dentro del rango de dosis estudiado de 8 mg a 24 mg, en ambos grupos de edad.
Figura 3: Gráfico linear comparativo de los perfiles medios de concentración en plasma-tiempo de galantamina.
CONTRAINDICACIONES: No debe administrarse REMINYL ER® a pacientes que tengan hipersensibilidad conocida a bromhidrato de galantamina o a alguno de los excipientes usados en las formulaciones.
EMBARAZO Y LACTANCIA:
Uso durante el embarazo: Estudios de reproducción realizados en ratas embarazadas a dosis de hasta 16 mg/kg (cerca de 25 veces la dosis terapéutica humana) y en conejas embarazadas con hasta 40 mg/kg (cerca de 63 veces la dosis terapéutica humana) no mostraron evidencia alguna de potencial teratogénico. Se notó un aumento no significativo de la incidencia de anormalidades esqueléticas menores a la dosis de 16 mg/kg en las ratas.
No hay disponibles estudios sobre el uso de REMINYL ER® en mujeres embarazadas.
REMINYL ER® debe ser usado durante embarazo solamente si el beneficio potencial justifica el potencial riesgo al feto.
Uso durante lactancia: No se sabe si REMINYL ER® se excreta en la leche materna humana y no hay estudios en mujeres que se encuentren lactando. Por esta razón, las mujeres que reciban REMINYL ER® no deben lactar.
EFECTOS INDESEABLES:
Datos de estudios clínicos:
— Datos de doble-ciego – Reacciones adversas a la droga reportadas con frecuencia de ≥1%: Se evaluó la seguridad de REMINYL ER® en 4.457 sujetos con demencia de leve a moderadamente severa del tipo Alzheimer, quienes participaron en 7 estudios clínicos controlados con placebo, doble-ciego. La información presentada en esta sección se derivó de los datos agrupados.
Las Reacciones Adversas a la Droga (ADRs) reportadas por ≥1% de los sujetos tratados con REMINYL®, en estos estudios, se muestran en la Tabla 1.
|
Tabla 1: Reacciones Adversas a la Droga reportadas por ≥1% de sujetos tratados con REMINYL ER® en 7 estudios clínicos controlados con placebo, doble-ciego. |
||
|
Producto comercial |
Placebo |
|
|
Clase sistema/órgano |
(n=2932) |
(n=1525) |
|
Reacciones adversas |
% |
% |
|
Trastornos de metabolismo y nutrición |
||
|
Disminución de apetito |
5,2 |
1,4 |
|
Anorexia |
3,8 |
1,0 |
|
Trastorno psiquiátricos |
||
|
Depresión |
4,2 |
2,9 |
|
Trastornos del sistema nervioso |
||
|
Mareos |
8,9 |
4,6 |
|
Cefaleas |
7,6 |
5,4 |
|
Temblores |
2,0 |
0,8 |
|
Síncope |
1,8 |
0,7 |
|
Letargo |
1,7 |
0,7 |
|
Somnolencia |
1,7 |
0,8 |
|
Trastornos gastrointestinales |
||
|
Náuseas |
25,0 |
7,6 |
|
Vómitos |
12,8 |
3,1 |
|
Diarrea |
9,0 |
6,3 |
|
Dolor abdominal |
2,4 |
0,9 |
|
Dolor abdominal superior |
2,0 |
1,4 |
|
Dispepsia |
1,8 |
1,3 |
|
Malestar estomacal |
1,6 |
0,6 |
|
Malestar abdominal |
1,0 |
0,4 |
|
Trastornos de piel y tejido subcutáneo |
||
|
Hiperhidrosis |
1,2 |
0,7 |
|
Trastornos músculo-esqueléticos y del tejido conectivo |
||
|
Espasmos musculares |
1,5 |
0,8 |
|
Trastornos generales y condiciones en el sitio de administración |
||
|
Fatiga |
4,0 |
2,2 |
|
Astenia |
2,3 |
1,7 |
|
Malasia |
1,4 |
0,7 |
|
Investigaciones |
||
|
Disminución de peso |
5,1 |
1,4 |
En un estudio clínico aleatorizado, doble-ciego, controlado con placebo, el perfil de seguridad del tratamiento una vez al día con cápsulas de liberación prolongada [extendida] de REMINYL®, fue similar en frecuencia y naturaleza al visto con las tabletas.
Náuseas y vómitos, que son las reacciones adversas a la droga más frecuentes, ocurrieron principalmente durante los períodos de titulación, duraron menos de una semana en la mayoría de los casos y la mayoría de los pacientes tuvo un solo episodio. En estos casos puede ser útil prescribir anti-eméticos y garantizar un ingreso de líquidos adecuado.
Datos de diseño abierto - Reacciones adversas a la droga reportadas con frecuencia de ≥1%: Se evaluó la seguridad de REMINYL ER® en 1.454 sujetos con demencia de leve a moderadamente severa del tipo Alzheimer, quienes participaron en 5 estudios clínicos con diseño-abierto. La información presentada en esta sección se derivó de los datos agrupados.
Las Reacciones Adversas a la Droga (ADRs) reportadas por ≥1% de los sujetos tratados con REMINYL ER® en estos estudios y no enlistadas en la Tabla 1 incluyeron caída, la cual ocurrió a una tasa de 6,5% en los estudios con diseño-abierto.
Datos de doble-ciego y diseño-abierto - Reacciones adversas a la droga reportadas con frecuencia de ≥1%: Las ADRs adicionales que ocurrieron en <1% de los sujetos tratados con REMINYL®, en las bases de datos clínicas de doble-ciego y de diseño-abierto, están enlistados en la Tabla 2.
|
Tabla 2. Reacciones adversas a la droga reportadas por <1% de los sujetos tratados con REMINYL ER® en los estudios clínicos doble-ciego o con diseño-abierto. |
|
Metabolismo y trastornos nutricionales Deshidratación |
|
Trastornos del sistema nervioso Disgeusia, Hiperinsomnio, Parestesia |
|
Trastornos oculares Visión borrosa |
|
Trastornos cardiacos Bloqueo aurículo-ventricular de primer grado, Palpitaciones, Bradicardia sinus, Extrasístoles supraventriculares |
|
Trastornos vasculares Rubor, Hipotensión |
|
Trastornos gastrointestinales Náuseas |
|
Trastornos músculo-esqueléticos y del tejido conectivo Debilidad muscular |
En la Tabla 3 las ADRs se presentan por frecuencia basado en las tasas de reportes espontáneos.
En la Tabla 4 las ADRs se presentan por frecuencia basado en la incidencia en los estudios clínicos, cuando se conoce.
|
Tabla 3. Reacciones adversas a la droga identificadas durante la experiencia post-mercadeo con REMINYL ER® según frecuencia estimada a partir de los reportes espontáneos |
|
Trastornos psiquiátricos Muy raro: Alucinaciones, alucinaciones visuales, alucinaciones auditivas |
|
Trastornos del oído y laberinto Muy raro: Tinitus |
|
Trastornos vasculares Muy raro: Hipertensión |
|
Trastornos Hepatobiliares Muy raro: Hepatitis |
|
Investigaciones Muy raro: Aumento de enzima hepática |
|
Tabla 4. Reacciones adversas a la droga identificadas durante la experiencia post-mercadeo con REMINYL ER® según frecuencia estimada a partir de estudios clínicos |
|
Trastornos psiquiátricos Común: Alucinaciones No-común: Alucinaciones visuales, alucinaciones auditivas |
|
Trastornos del oído y laberinto No común: Tinitus |
|
Trastornos vasculares Común: Hipertensión |
|
Trastornos hepatobiliares Raro: Hepatitis |
|
Investigaciones No común: Aumento de enzima hepática |
EFECTOS SOBRE LA CAPACIDAD DE CONDUCIR VEHÍCULOS Y DE USAR MÁQUINAS: La enfermedad de Alzheimer puede ocasionar un daño gradual en la capacidad para manejar o puede comprometer la capacidad de utilizar maquinaria. Más aún, como sucede con otros colinomiméticos, REMINYL ER® puede producir mareos y somnolencia, lo cual podría afectar la capacidad de manejar o de usar maquinaria, especialmente durante las primeras semanas después de iniciar el tratamiento.
INTERACCIONES CON OTROS PRODUCTOS MEDICINALES Y OTRAS FORMAS DE INTERACCIÓN:
Interacciones farmacodinámicas: Por su mecanismo de acción, galantamina no debe ser administrada concomitantemente con otros colinomiméticos. Galantamina antagoniza el efecto de medicación anticolinérgica. Como se espera de los colinomiméticos, es posible que haya una interacción farmacodinámica con drogas que reduzcan significativamente la frecuencia cardiaca (por ej., digoxina y beta-bloqueadores).
Galantamina, como colinomimético, es probable que exagere la relajación muscular tipo-succinilcolina durante la anestesia.
Interacciones farmacocinéticas: En la eliminación de galantomina están implicadas múltiples vías metabólicas y la excreción renal. Basado en estudios in vitro, CYP2D6 y CYP3A4 fueron las principales enzimas implicadas en el metabolismo de galantamina.
La inhibición de secreción ácida gástrica no perturbará la absorción de galantamina.
Otras drogas que afectan el metabolismo de galantamina: Las drogas que son inhibidores potentes de CYP2D6 o CYP3A4 pueden aumentar el AUC de galantamina. Múltiples estudios farmacocinéticos de dosis demostraron que el AUC de galantamina aumentó 30% y 40%, respectivamente, durante la administración conjunta de ketoconazol y paroxetine. Co-administrada con eritromicina, que es otro inhibidor de CYP3A4, el AUC de galantamina sólo aumentó aproximadamente 10%. La población de análisis farmacocinético (PK) en pacientes con enfermedad de Alzheimer mostró que la depuración de galantamina disminuyó en cerca de 25-33% por la administración concurrente de amitriptilina, fluoxetina, fluvozamina, paroxetina y quinidina, que son inhibidores conocidos de CYP2D6.
De allí que, durante la iniciación de tratamiento con inhibidores potentes de CYP2D6 o CYP3A4, los pacientes pueden experimentar un aumento de incidencia de efectos colaterales colinérgicos, predominantemente náuseas y vómitos. Bajo estas circunstancias, basado en la tolerabilidad, puede considerarse una reducción de la dosis de mantenimiento de galantomina (ver Posología y métodos de administración).
Memantina, que es un antagonista de receptor N-metil-D-aspartato (NMDA), a dosis de 10 mg/diarios por 2 días, seguido por 10 mg BID por 12 días, no tuvo efecto sobre las farmacocinéticas de galantamina a razón de 16 mg/día a niveles constantes.
Efecto de galantamina sobre el metabolismo de otras drogas: Dosis terapéuticas de galantamina (12 mg BID) no tuvieron efecto sobre las cinéticas de digoxina y de warfarina. Galantamina no afectó el aumento de tiempo de protrombina inducido por warfarina.
Estudios in vitro indicaron que la potencial inhibición de galantamina con respecto a las principales formas de citocromo P450 humano es muy bajo.
DATOS DE SEGURIDAD PRE-CLÍNICA: Todos los otros datos de seguridad preclínica relevantes para el que prescribe han sido incluidos en las secciones apropiadas.
ADVERTENCIAS ESPECIALES Y PRECAUCIONES ESPECIALES DE USO: REMINYL ER® está indicado para pacientes con demencia de leve a moderada del tipo Alzheimer, [incluyendo demencia Alzheimer con enfermedad cerebrovascular]. No ha sido demostrado beneficio de REMINYL ER® en pacientes con otros tipos de demencia u otros tipos de trastornos de memoria.
Los pacientes con enfermedad de Alzheimer pierden peso. El tratamiento con inhibidores de colinesterasa, incluyendo galantamina, ha sido asociado con pérdida de peso en estos pacientes. Durante la terapia debe monitorearse el peso del paciente.
Como sucede con otros colinomiméticos, REMINYL ER® debe ser administrado con precaución en las siguientes condiciones:
Condiciones cardiovasculares: Por su acción farmacológica, los colinomiméticos pueden tener efectos vagotónicos sobre la frecuencia cardiaca (por ej., bradicardia). El potencial de esta acción puede ser particularmente importante en pacientes con “síndrome de seno enfermo” u otro trastorno de conducción cardiaca supraventricular, o en pacientes que usen drogas que reduzcan significativamente la frecuencia cardíaca concomitantemente, tales como digoxina y beta-bloqueadores. En las pruebas clínicas el uso de REMINYL ER® ha sido asociado con síncope y raramente con bradicardia severa.
Condiciones gastrointestinales: Los pacientes que tienen riesgo aumentado de desarrollar úlceras pépticas, por ej., los que tiene historia de enfermedad ulcerosa, o lo que están predispuestos a sufrir esta condición, incluyendo los que reciben concomitantemente drogas antiinflamatorias no esteroideas (NSAIDS), debe ser monitoreados con respecto a los síntomas. Sin embargo, estudios clínicos con REMINYL ER® no mostraron aumento, con relación al placebo, de la incidencia de enfermedad de úlcera péptica ni de sangrado gastrointestinal. No se recomienda usar REMINYL ER® en pacientes que tengan obstrucción gastrointestinal o que se estén recuperando de cirugía gastrointestinal.
Condiciones neurológicas: Aunque se cree que los colinomiméticos tienen algún potencial de producir convulsiones, la actividad convulsiva también puede ser una manifestación de la enfermedad de Alzheimer.
Condiciones pulmonares: En vista de sus acciones colinomiméticas, los colinomiméticos deben ser prescritos con cuidado en pacientes que tengan historia de asma severa u enfermedad pulmonar obstructiva.
Genitourinario: No se recomienda usar REMINYL ER® en pacientes que tengan obstrucción al flujo urinario o que se estén recuperando de cirugía de la vejiga urinaria.
Seguridad en sujetos con daño cognoscitivo leve (MCI): No está indicado REMINYL ER® para personas que tengan daño cognoscitivo leve (MCI), o sea, los que demuestren daño aislado de la memoria mayor del esperado para su edad y educación, pero no que no llenan los criterios para enfermedad de Alzheimer.
Dos estudios controlados de 2-años, en sujetos con MCI no cumplieron con los resultados de eficacia primaria dual. Aunque la mortalidad en ambos brazos de tratamiento fue baja, inicialmente se registraron más muerte en sujetos aleatorizados a galantamina que a placebo, pero la incidencia de eventos adversos serios fue idéntica entre los grupos de tratamiento. Las muertes fueron debidas a varias causas que no son inesperadas en una población de ancianos. Cuando se recogieron los datos de la gran proporción de pacientes que descontinuaron antes de incluir el completamiento del período doble-ciego, no hubo evidencia de aumento de riesgo de muerte en los sujetos tratados con REMINYL ER® a través del tiempo. Más sujetos del grupo de placebo que del grupo con galantomina descontinuaron antes de fallecer, lo cual puede contar para la diferencia en cuanto a la mortalidad registrada inicialmente.
Los resultados del estudio MCI discrepan de los observados en estudios de enfermedad de Alzheimer. En estudios agrupados de enfermedad de Alzheimer (n=4.614), la tasa de mortalidad fue numéricamente mayor en el grupo con placebo que en el grupo con REMINYL®.
NOMBRE DEL PRODUCTO MEDICINAL: REMINYL ER® (Bromhidrato de galantamina) Cápsulas de liberación prolongada [Extendida].
POSOLOGÍA Y MÉTODOS DE ADMINISTRACIÓN:
Adultos: REMINYL ER® en forma de cápsulas de liberación prolongada [extendida], debe ser administrado una vez al día en la mañana, preferiblemente con alimentos. La dosis de comienzo recomendada es 8 mg/día.
Los pacientes que están siendo tratados actualmente con REMINYL ER® en formulaciones de liberación inmediata (tabletas o solución oral) pueden cambiarse a REMINYL ER® en forma de cápsulas de liberación prolongada [extendida], tomando su última dosis de tabletas de liberación inmediata o solución oral de REMINYL ER® en la noche y comenzando tratamiento una vez al día con cápsulas de liberación prolongada [extendida] en la siguiente mañana. Al cambiarse de tabletas de liberación inmediata o solución oral de REMINYL ER® 2 veces al día a cápsulas de liberación prolongada [extendida] una vez al día, debe administrarse la misma dosis total diaria.
Debe asegurarse tomar una cantidad adecuada de líquidos durante el tratamiento.
Dosis de mantenimiento:
— La dosis inicial de mantenimiento es de 16 mg/día (8 mg dos veces al día con tabletas o solución oral, o 16 mg una vez al día con cápsulas) y los pacientes deben ser mantenidos con 16 mg/día por al menos 4 semanas.
— Debe considerase aumentar a la dosis máxima de mantenimiento recomendada de 24 mg/día (12 mg dos veces al día con tabletas o solución oral, o 24 mg una vez al día con cápsulas), después de una evaluación apropiada que incluya evaluación del beneficio clínico y de la tolerabilidad.
— No hay efecto de rebote después de descontinuar abruptamente el tratamiento (por ej., en preparación para cirugía).
Niños: No se recomienda el uso de REMINYL ER® en niños. No hay disponibles datos sobre el uso de REMINYL ER® en pacientes pediátricos.
Daño renal y hepático: Los niveles de galantamina en plasma pueden aumentar en pacientes con daño moderado a severo hepático o renal.
En pacientes que tienen función hepática moderadamente deteriorada, basado en el modelado farmacocinético, debe comenzarse la dosificación con 4 mg una vez al día con las tabletas e liberación inmediata, tomadas preferiblemente por la mañana, durante al menos una semana. Con las cápsulas de liberación prolongada [extendida], basado en el modelado farmacocinético, debe comenzarse la dosificación con 8 mg cada dos días durante al menos una semana, tomadas preferiblemente en la mañana. Posteriormente, los pacientes pueden continuar con 4 mg dos veces al día con las tabletas de liberación inmediata u 8 mg una vez al día con las cápsulas de liberación prolongada [extendida] durante al menos cuatro semanas. En estos pacientes las dosis totales diarias no deben exceder de 16 mg.
En pacientes con daño hepático severo no se recomienda REMINYL®.
En pacientes con depuración de creatinina mayor de 9 ml/min no se requiere ajustar la dosis.
En pacientes con daño renal severo (creatinina menor de 9 ml/min) no se recomienda REMINYL ER® en vista de que no hay información disponible.
Tratamiento concomitante: En pacientes tratados con inhibidores potentes de CYP2D6 o CYP3A4, puede considerarse reducir la dosis (ver Interacciones con otros productos medicinales y otras formas de interacción).
SOBREDOSIS:
Síntomas: Se pronostica que los signos y síntomas de una sobredosis significativa de galantamina son similares a los de sobredosis con otros colinomiméticos. Estos efectos generalmente involucran al sistema nervioso central, sistema nervioso parasimpático y unión neuromuscular. Además de debilidad muscular o fasciculaciones, pueden desarrollarse algunos o todos los signos de crisis colinérgica: náuseas severas, vómitos, calambres gastrointestinales, salivación, lagrimeo, ganas de orinar, de defecar, sudoración, bradicardia, hipotensión, colapso y convulsiones. El aumento de la debilidad muscular junto con hipersecreción traqueal y broncoespasmo pueden conducir a compromiso vital de las vías aéreas.
Ha habido reportes post-mercadeo de Torsade de Pointes, prolongación de QT, bradicardia, taquicardia ventricular y pérdida breve de conciencia, en asociación con sobre dosis inadvertidas de galantamina. En un caso donde se supo la dosis, fueron ingeridas 4 tabletas (32 mg de total) en un solo día.
Dos casos adicionales de ingestión accidental de 32 mg (náuseas, vómitos y boca seca; náuseas, vómitos y dolor toráxico subesternal) y uno con 40 mg (vómitos), resultaron en hospitalización breve para observación y se recuperaron completamente. Un paciente, a quien se prescribió 24 mg/día y tenía historia de alucinaciones en los dos años previos, recibió por error 24 mg dos veces al día durante 34 días y desarrolló alucinaciones que requirieron hospitalización. Otro paciente, a quien se prescribió 16 mg/día de solución oral, ingirió inadvertidamente 160 mg (40 ml) y experimentó sudoración, vómitos, bradicardia y casi síncope una hora más tarde, lo cual necesitó tratamiento hospitalario. Sus síntomas desaparecieron al cabo de 24 horas.
Tratamiento: Como en cualquier caso de sobredosis, deben usarse medidas generales de soporte. En los casos severos pueden usarse anticolinérgicos como atropina como antídoto general para colinomiméticos. Se recomienda una dosis inicial de 0,5 a 1,0 mg IV, con dosis subsecuentes basadas en la respuesta clínica.
En vista de que las estrategias para el manejo de sobredosis están evolucionando continuamente, es recomendable contactar un centro de control de envenenamiento para determinar las últimas recomendaciones para el manejo de una sobredosis.


