OFEV COSTA RICA
Cápsula blanda
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA:
OFEV® 100 mg cápsulas blandas:
Una cápsula blanda contiene 100 mg de nintedanib (como esilato).
Excipiente con efecto conocido.
Cada CÁPSULA blanda de 100 mg contiene 1.2 mg de lecitina de soja.
OFEV® 150 mg cápsulas blandas:
Una cápsula blanda contiene 150 mg de nintedanib (como esilato).
Excipiente con efecto conocido
Cada CÁPSULA blanda de 150 mg contiene 1.8 mg de lecitina de soja.
Para consultar la lista completa de excipientes, ver sección 6.1
FORMA FARMACÉUTICA: Cápsula blanda (cápsula) OFEV® 100 mg cápsulas blandas
OFEV® 100 mg cápsulas blandas son cápsulas de gelatina blanda oblongas, opacas y de color durazno, grabadas en un lado en color negro con el logotipo de la empresa Boehringer Ingelheim y “100”.
OFEV® 150 mg cápsulas blandas
OFEV® 150 mg cápsulas blandas son cápsulas de gelatina blanda oblongas, opacas y de color marrón, grabadas en un lado en color negro con el logotipo de la empresa Boehringer Ingelheim y “150”.
INDICACIONES TERAPÉUTICAS:
OFEV® está indicado en adultos para tratar la fibrosis pulmonar idiopática (FPI).
OFEV® también está indicado en adultos para el tratamiento de otras enfermedades pulmonares intersticiales (EPI) fibrosantes crónicas con un fenotipo progresivo (ver sección 5.1).
OFEV® está indicado en adultos para tratar la enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES)
OFEV® está indicado en combinación con docetaxel para el tratamiento de pacientes adultos con cáncer de pulmón no microcítico (CPNM) localmente avanzado, metastásico o localmente recurrente con histología tumoral de adenocarcinoma después de la quimioterapia de primera línea.
PROPIEDADES FARMACOLÓGICAS:
• Propiedades farmacodinámicas: Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
Grupo farmacoterapéutico: Agentes antineoplásicos, inhibidores de la proteína cinasa.
Código ATC: L01XE31.
Mecanismo de acción: Nintedanib es un inhibidor de molécula pequeña de la tirosina cinasa, incluidos los receptores del factor de crecimiento derivados de plaquetas (PDGFR) α y β, los receptores del factor de crecimiento de fibroblastos (FGFR) 1-3 y los receptores del factor de crecimiento del endotelio vascular (VEGFR) 1-3. Además, nintedanib inhibe las cinasas Lck (proteína tirosina cinasa específica de los linfocitos), Lyn (proteína tirosina cinasa lyn), Src (proteína tirosina cinasa proto-oncogénica Src) y CSF1R (receptor del factor estimulante de colonias de tipo 1). Nintedanib se une de forma competitiva al sitio de unión de adenosina trifosfato (ATP) de estas cinasas y bloquea las cascadas de señalización intracelular que se ha demostrado que participan en la patogenia de la remodelación del tejido fibrótico en las enfermedades pulmonares intersticiales.
Efectos farmacodinámicos: En los estudios in vitro con células humanas, se ha demostrado que nintedanib inhibe los procesos que se supone que participan en el inicio de la patogenia fibrótica, la liberación de mediadores profibróticos de las células monocíticas de sangre periférica y la polarización de los macrófagos a macrófagos activados de forma alternativa. Se ha demostrado que nintedanib inhibe los procesos fundamentales en la fibrosis de los órganos, la proliferación y la migración de los fibroblastos y la transformación al fenotipo de miofibroblasto activo y la secreción de matriz extracelular. En los estudios realizados en animales con múltiples modelos de FPI, ES/EPI-ES, EPI asociada a artritis reumatoide (AR) y otras fibrosis de órganos, nintedanib ha demostrado efectos antiinflamatorios y antifibróticos en el pulmón, la piel, el corazón, el riñón y el hígado. Nintedanib también ejerció actividad vascular. Redujo la apoptosis de las células endoteliales microvasculares dérmicas y atenuó la remodelación vascular pulmonar reduciendo la proliferación de las células de músculo liso vascular, el grosor de las paredes de los vasos pulmonares y el porcentaje de vasos pulmonares ocluidos.
Eficacia clínica y seguridad:
Fibrosis pulmonar idiopática (FPI): La eficacia clínica de nintedanib se ha estudiado en pacientes con FPI en dos ensayos con diseño idéntico fase III, aleatorizados, doble ciego y controlados con placebo (INPULSIS-1 (1199.32) e INPULSIS-2 (1199.34)). Los pacientes con una CVF basal prevista inferior al 50% o un factor de transferencia de monóxido de carbono (DLCO, corregido para la hemoglobina) prevista inferior al 30% en el nivel basal se excluyeron de los ensayos. Los pacientes se aleatorizaron en una proporción de 3:2 entre el tratamiento con OFEV® 150 mg y el tratamiento con placebo dos veces al día durante 52 semanas.
La variable principal fue la pérdida anual de la capacidad vital forzada (CVF). Los criterios de valoración secundarios principales fueron el cambio respecto al valor basal en la puntuación obtenida a las 52 semanas en el Saint George’s Respiratory Questionnaire (CRSG) y el tiempo hasta que se produjo la primera exacerbación aguda de la FPI.
Pérdida anual de la CVF: La pérdida anual de la CVF (en ml) se redujo significativamente en los pacientes que recibieron nintedanib en comparación con los pacientes que recibieron placebo. El efecto del tratamiento fue coincidente en ambos ensayos. Ver los resultados del estudio individual y conjunto en la Tabla 6.
|
Tabla 6 Pérdida anual de la CVF (ml) en los ensayos INPULSIS-1, INPULSIS-2 y sus datos conjuntos: grupo tratado |
||||||
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSI-2 conjuntos |
||||
|
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número de pacientes analizados |
204 |
309 |
219 |
329 |
423 |
638 |
|
Pérdida1 (error estándar) en el transcurso de 52 semanas |
-239.9 |
-114.7 |
-207.3 |
-113.6 |
-223.5 |
-113.6 |
|
(18.71) |
(15.33) |
(19.31) |
(15.73) |
(13.45) |
(10.98) |
|
|
Comparación con el placebo |
||||||
|
Diferencia1 |
125.3 |
93.7 |
109.9 |
|||
|
95% de IC |
(77.7, |
(44.8, |
(75.9, |
|||
|
172.8) |
142.7) |
144.0) |
||||
|
valor p |
<0.0001 |
0.0002 |
<0.0001 |
|||
|
1 Calculado basándose en un modelo de regresión de coeficientes aleatorios. IC: intervalo de confianza |
||||||
En un análisis de sensibilidad que supuso que, en el caso de los pacientes en los que faltan datos en la semana 52, la pérdida de la CVF después del último valor observado sería la misma que en todos los pacientes tratados con placebo, la diferencia ajustada en la pérdida anual entre nintedanib y placebo fue de 113.9 ml/año (95% de IC: 69.2, 158.5) en el ensayo INPULSIS-1 y de 83.3 ml/año (95% de IC: 37.6, 129.0) en el ensayo INPULSIS-2.
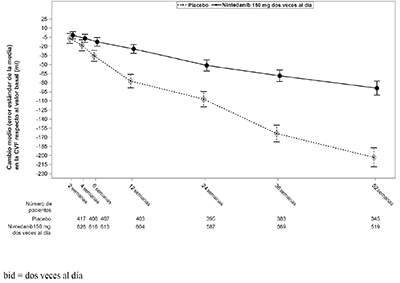
Ver en la figura 1 la evolución del cambio respecto al valor basal a lo largo del tiempo en los dos grupos de tratamiento basándose en el análisis conjunto de los estudios INPULSIS-1 e INPULSIS- 2.
Figura 1: Cambio medio (error estándar de la media (SEM)) respecto al valor basal (ml) observado en la CVF a lo largo del tiempo; estudios INPULSIS-1 e INPULSIS-2 conjuntos
Análisis de los pacientes que responden en términos de CVF: En ambos ensayos INPULSIS, la proporción de pacientes que respondieron en términos de CVF, definidos como los pacientes con una pérdida absoluta en la CVF prevista inferior al 5% (un umbral indicador del aumento del riesgo de mortalidad en la FPI), fue significativamente más alta en el grupo tratado con nintedanib que en el grupo tratado con placebo. En los análisis que utilizaron un umbral conservador del 10% se observaron resultados similares. Ver los resultados del estudio individual y conjunto en la Tabla 7
|
Tabla 7 Proporción de pacientes que respondieron en términos de CVF a las 52 semanas en los ensayos INPULSIS-1, INPULSIS-2 y sus datos conjuntos: grupo tratado |
||||||
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2 conjunto |
||||
|
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número de pacientes analizados |
204 |
309 |
219 |
329 |
423 |
638 |
|
Umbral del 5% |
||||||
|
Número (%) de pacientes que respondieron en términos de CVF1 |
78 (38.2) |
163 (52.8) |
86 (39.3) |
175 (53.2) |
164 (38.8) |
338 (53.0) |
|
Comparación con el placebo |
||||||
|
Cociente de posibilidades (odds ratio) |
1.85 |
1.79 |
1.84 |
|||
|
95% de IC |
(1.28,2.66) |
(1.26,2.55) |
(1.43,2.36) |
|||
|
valor p2 |
0.0010 |
0.0011 |
<0.0001 |
|||
|
Umbral del 10% |
||||||
|
Número (%) de pacientes que respondieron en términos de CVF1 |
116 (56.9) |
218 (70.6) |
140 (63.9) |
229 (69.6) |
256 (60.5) |
447 (70.1) |
|
Comparación con el placebo |
||||||
|
Cociente de posibilidades (odds ratio) |
.91 |
1.29 |
1.58 |
|||
|
95% de IC |
(1.32,2.79) |
(0.89,1.86) |
(1.21,2.05) |
|||
|
valorp2 |
0,0007 |
0,1833 |
0.0007 |
|||
|
1 Los pacientes que responden son aquellos que no presentan una pérdida absoluta superior al 5% o superior al 10% en el% de CVF previsto, dependiendo del umbral y con una evaluación de la CVF a las 52 semanas. 2 Basándose en una regresión logística. |
||||||
Tiempo hasta la progresión (≥ 10% de pérdida absoluta de la CVF prevista o muerte): Desde el punto de vista estadístico, en ambos ensayos INPULSIS, el riesgo de progresión fue significativamente menor en los pacientes tratados con nintedanib que en los tratados con placebo. En el análisis conjunto, el cociente de riesgos (hazard ratio) fue de 0.60, lo que indica una reducción de un 40% del riesgo de progresión en los pacientes tratados con nintedanib en comparación con los tratados con placebo.
|
Tabla 8 Frecuencia de pacientes con ≥ 10% de pérdida absoluta de la CVF prevista o muerte en el transcurso de 52 semanas y tiempo hasta la progresión en los ensayos INPULSIS-1, INPULSIS-2 y sus datos conjuntos: grupo tratado |
||||||
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2 conjunto |
||||
|
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número en riesgo |
204 |
309 |
219 |
329 |
423 |
638 |
|
Pacientes con episodios, N (%) |
83 (40.7) |
75 (24.3) |
92 (42.0) |
98 (29.8) |
175 (41.4) |
173 (27.1) |
|
Comparación con el placebo1 |
||||||
|
valor p2 |
0.0001 |
0.0054 |
<0.0001 |
|||
|
Cociente de riesgos (hazard ratio)3 |
0.53 |
0.67 |
0.60 |
|||
|
95% de IC |
(0.39,0.72) |
(0.51,0.89) |
(0.49,0.74) |
|||
|
1 Basándose en los datos recopilados durante un máximo de 372 días (52 semanas + 7 días de margen). 2 Basándose en una prueba de orden logarítmico. 3 Basándose en un modelo de regresión de Cox. |
||||||
Cambio respecto al valor basal en la puntuación total del CRSG en la semana 52: En el análisis conjunto de los ensayos INPULSIS, el valor basal de la puntuación del CRSG fue de 39.51 en el grupo tratado con nintedanib y de 39.58 en el grupo tratado con placebo. El cambio medio calculado desde el valor basal hasta la semana 52 en la puntuación total del CRSG fue menor en el grupo tratado con nintedanib (3.53) que en el grupo tratado con placebo (4.96), con una diferencia entre los grupos de tratamiento de -1.43 (95% de IC: -3.09, 0.23; p=0.0923). En conjunto, el efecto de nintedanib sobre la calidad de vida relacionada con la salud medida por la puntuación total del CRSG es moderado, lo que indica que empeora menos la situación que el placebo.
Tiempo hasta la primera exacerbación aguda de la FPI: En el análisis conjunto de los ensayos INPULSIS, los pacientes que recibieron nintedanib presentaron un riesgo de una primera exacerbación aguda numéricamente menor que los que fueron tratados con placebo. Ver los resultados del estudio individual y conjunto en la Tabla 9.
|
Tabla 9 Frecuencia de pacientes con exacerbaciones agudas de la FPI en el transcurso de 52 semanas y análisis del tiempo hasta la primera exacerbación basándose en los episodios descritos por el investigador en los ensayos INPULSIS-1, INPULSIS-2 y sus datos conjuntos: grupo tratado |
||||||
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2 conjuntos |
||||
|
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número en riesgo |
204 |
309 |
219 |
329 |
423 |
638 |
|
Pacientes con episodios, N (%) |
11(5.4) |
19(6.1) |
21(9.6) |
12(3.6) |
32(7.6) |
31(4.9) |
|
Comparación con el placebo1 |
||||||
|
valor p2 |
0.6728 |
0.0050 |
0.0823 |
|||
|
Cociente de riesgos (hazard ratio)3 |
1.15 |
0.38 |
0.64 |
|||
|
95% de IC |
(0.54, 2.42) |
(0.19, 0.77) |
(0.39, 1.05) |
|||
|
1 Basándose en los datos recopilados durante un máximo de 372 días (52 semanas + 7 días de margen). 2 Basándose en una prueba de orden logarítmico. 3 Basándose en un modelo de regresión de Cox. |
||||||
En un análisis preespecificado de sensibilidad la frecuencia de pacientes con al menos una exacerbación adjudicada en el transcurso de 52 semanas fue inferior en el grupo tratado con nintedanib (1.9% de los pacientes) que en el grupo tratado con placebo (5.7% de los pacientes). El análisis del tiempo transcurrido hasta los episodios de exacerbación adjudicados que utilizó los datos conjuntos obtuvo un cociente de riesgos (hazard ratio) de 0.32 (95% de IC: 0.16, 0.65; p=0.0010).
Análisis de supervivencia: En el análisis conjunto preespecificado de los datos de supervivencia de los ensayos INPULSIS, la mortalidad global en el transcurso de 52 semanas fue inferior en el grupo tratado con nintedanib (5.5%) que en el grupo tratado con placebo (7.8%). El análisis del tiempo transcurrido hasta la muerte dio lugar a un cociente de riesgos (hazard ratio) de 0,70 (95% de IC: 0.43, 1.12; p=0.1399).
Los resultados de todos los criterios de valoración de supervivencia (tales como la mortalidad durante el tratamiento o la mortalidad respiratoria) mostraron una diferencia numérica homogénea en favor de nintedanib.
|
Tabla 10 Mortalidad por todas las causas en el transcurso de 52 semanas en los ensayos INPULSIS- 1, INPULSIS-2 y sus datos conjuntos: grupo tratado |
||||||
|
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2 conjunto |
||||
|
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número en riesgo |
204 |
309 |
219 |
329 |
423 |
638 |
|
Pacientes con episodios, N (%) |
13(6.4) |
13(4.2) |
20(9.1) |
22(6.7) |
33(7.8) |
35(5.5) |
|
Comparación con el placebo1 |
||||||
|
valor p2 |
0.2880 |
0.2995 |
0.1399 |
|||
|
Cociente de riesgos (hazard ratio)3 |
0.63 |
0.74 |
0.70 |
|||
|
95% de IC |
(0.29,1.36) |
(0.40,1.35) |
(0.43,1.12) |
|||
|
1 Basándose en los datos recopilados durante un máximo de 372 días (52 semanas + 7 días de margen). 2 Basándose en una prueba de orden logarítmico. 3 Basándose en un modelo de regresión de Cox. |
||||||
Tratamiento a largo plazo con OFEV® en pacientes con FPI (INPULSIS-ON): En un estudio de extensión abierto de OFEV® se incluyó a 734 pacientes con FPI. Los pacientes que completaron el periodo de tratamiento de 52 semanas en un ensayo INPULSIS recibieron tratamiento abierto con OFEV® en el ensayo de extensión INPULSIS-ON. La mediana del tiempo de exposición en los pacientes tratados con OFEV® en los ensayos INPULSIS e INPULSIS-ON fue de 44.7 meses (intervalo 11.9-68.3). Entre los criterios de valoración exploratorios de la eficacia se encuentra la tasa anual de pérdida de la CVF durante 192 semanas, que fue de -135.1 (5.8) ml/año en todos los pacientes tratados y que fue consistente con la tasa anual de pérdida de la CVF en los pacientes tratados con OFEV® en los ensayos de fase III INPULSIS (-113.6 ml al año). El perfil de acontecimientos adversos de OFEV® en el ensayo INPULSIS-ON fue consistente con el observado en los ensayos de fase III INPULSIS.
Pacientes con FPI y afectación avanzada de la función pulmonar (INSTAGE): INSTAGE fue un ensayo clínico prospectivo, doble ciego, aleatorizado, multicéntrico, multinacional y de grupos paralelos en pacientes con FPI y afectación avanzada de la función pulmonar (DLCO prevista ≤ 35%) durante 24 semanas. Fueron tratados con OFEV® en monoterapia 136 pacientes. El resultado de la variable principal mostró una reducción en la puntuación total del cuestionario respiratorio de St George (SGRQ) de -0.77 unidades en la semana 12, basada en el cambio medio ajustado respecto al valor basal. Una comparación post hoc demostró que la disminución de la CVF en estos pacientes fue consistente con la disminución de la CVF en los pacientes con enfermedad menos avanzada y tratados con OFEV® en los ensayos INPULSIS de fase III.
El perfil de seguridad y tolerabilidad de OFEV® en los pacientes con FPI y afectación avanzada de la función pulmonar fue consistente con el observado en los ensayos INPULSIS de fase III.
Datos adicionales del ensayo de fase IV INJOURNEY con OFEV® 150 mg dos veces al día y pirfenidona como tratamiento complementario: Se ha investigado el tratamiento conjunto con nintedanib y pirfenidona en un ensayo exploratorio, abierto, aleatorizado de nintedanib 150 mg dos veces al día con pirfenidona como tratamiento complementario (ajustada a 801 mg tres veces al día) en comparación con nintedanib 150 mg dos veces al día en monoterapia en 105 pacientes aleatorizados durante 12 semanas. La variable principal fue el porcentaje de pacientes con reacciones adversas gastrointestinales desde el inicio del estudio y hasta la semana 12. Las reacciones adversas gastrointestinales fueron frecuentes y en línea con el perfil de seguridad establecido de cada componente. Las reacciones adversas más frecuentes fueron diarrea, náuseas y vómitos, notificadas en pacientes tratados con pirfenidona añadida a nintedanib frente a nintedanib en monoterapia, respectivamente.
La media (EE) de los cambios absolutos de la CVF con respecto al valor basal en la semana 12 fue de -13.3 (17.4) ml en los pacientes tratados con nintedanib con pirfenidona como tratamiento complementario (n = 48) en comparación con -40.9 (31.4) ml en los pacientes tratados con nintedanib en monoterapia (n = 44).
Otras enfermedades pulmonares intersticiales (EPI) fibrosantes crónicas con un fenotipo progresivo: La eficacia clínica de OFEV® se ha estudiado en pacientes con otras EPI fibrosantes crónicas con un fenotipo progresivo en un ensayo de fase III, aleatorizado, doble ciego y controlado con placebo (INBUILD). Se excluyó a pacientes con FPI. Se seleccionó a pacientes con un diagnóstico clínico de EPI fibrosante crónica si presentaban fibrosis relevante (> 10% de características fibróticas) en la TCAR y signos clínicos de progresión (definida como pérdida de la CVF ≥ 10%, pérdida de la CVF ≥ 5% y < 10% junto con un empeoramiento de los síntomas o de los estudios de imagen torácica, o empeoramiento de los síntomas de los estudios de imagen torácica, en los 24 meses previos a la selección). Los pacientes tenían que tener una CVF igual o superior al 45% del valor previsto y una DLCO de entre el 30% y menos del 80% del valor previsto. Los pacientes tenían que haber experimentado progresión a pesar del tratamiento considerado apropiado en la práctica clínica para la EPI relevante del paciente.
Se aleatorizó a un total de 663 pacientes en una relación de 1:1 para recibir OFEV® 150 mg dos veces al día o un placebo equivalente durante al menos 52 semanas. La mediana de la exposición a OFEV® durante todo el ensayo fue de 17.4 meses y la media de la exposición a OFEV® durante todo el ensayo fue de 15.6 meses. La aleatorización se estratificó en función del patrón fibrótico observado en la TCAR valorado por evaluadores centrales. Se aleatorizó a 412 pacientes con un patrón fibrótico de tipo neumonía intersticial usual (NIU) en la TCAR y a 251 pacientes con otros patrones fibróticos en la TCAR. Se definieron dos poblaciones coprincipales para los análisis de este ensayo: todos los pacientes (la población global) y los pacientes con un patrón fibrótico de tipo NIU en la TCAR. Los pacientes con otros patrones fibróticos en la TCAR representaban la población “complementaria”.
El criterio de valoración principal fue la tasa anual de pérdida de la capacidad vital forzada (CVF) (en ml) en el transcurso de 52 semanas. Los criterios de valoración secundarios principales fueron el cambio absoluto respecto al valor basal en la puntuación total obtenida a las 52 semanas en el cuestionario King’s Brief Interstitial Lung Disease (K-BILD), el tiempo hasta la primera exacerbación aguda de la EPI o la muerte en el transcurso de 52 semanas, y el tiempo hasta la muerte en el transcurso de 52 semanas.
Los pacientes tenían una media (desviación estándar [DE, mín-máx]) de edad de 65.8 (9.8, 27-87) años y un porcentaje medio de CVF previsto del 69.0% (15.6, 42-137). Los diagnósticos clínicos de EPI subyacente en los grupos representados en el ensayo fueron neumonitis por hipersensibilidad (26.1%), EPI autoinmunes (25.6%), neumonía intersticial idiopática inespecífica (18.9%), neumonía intersticial idiopática inclasificable (17.2%) y otras EPI (12.2%).
El ensayo INBUILD no estaba diseñado ni tenía la potencia estadística adecuada para proporcionar evidencias de un beneficio de nintedanib en subgrupos específicos de diagnóstico. Se demostraron efectos homogéneos en los subgrupos basados en los diagnósticos de EPI. La experiencia con nintedanib en EPI fibrosantes progresivas muy raras es limitada.
Pérdida anual de la CVF: La pérdida anual de la CVF (en ml) en el transcurso de 52 semanas disminuyó de forma significativa en 107.0 ml en los pacientes que recibieron OFEV® en comparación con los pacientes que recibieron placebo (Tabla 11), lo que corresponde a un efecto relativo del tratamiento del 57.0%.
|
Tabla 11: Pérdida anual de la CFV (ml) en el transcurso de 52 semanas |
||
|
Tabla 11: Pérdida anual de la CFV (ml) en el transcurso de 52 semanas |
Placebo |
OFEV® 150 mg dos veces al día |
|
Número de pacientes analizados |
331 |
332 |
|
Pérdida1 (EE) en el transcurso de 52 semanas |
–187.8(14.8) |
–80.8(15.1) |
|
Comparación con el placebo |
||
|
Diferencia1 |
107.0 |
|
|
95% de IC |
(65.4, 148.5) |
|
|
Valor p |
<0.0001 |
|
|
1Basándose en un modelo de regresión de coeficientes aleatorios con efectos categóricos fijos del tratamiento, patrón en la TCAR, efectos continuos fijos del tiempo, CVF basal [ml] e incluyendo las interacciones de tratamiento por tiempo y de momento basal por tiempo. |
||
Se observaron resultados similares en la población coprincipal de pacientes con un patrón fibrótico de tipo NIU en la TCAR. El efecto del tratamiento fue homogéneo en la población complementaria de pacientes con otros patrones fibróticos en la TCAR (valor de p de la interacción 0.2268) (Figura 2).
Figura 2. Gráfico de bosque de la pérdida anual de la CVF (ml) en el transcurso de 52 semanas en las poblaciones de pacientes
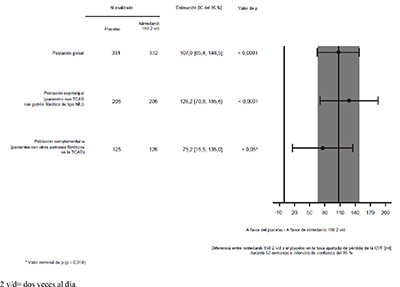
Los resultados del efecto de OFEV® en la reducción de la pérdida anual de la CVF se confirmaron en todos los análisis preespecificados de sensibilidad y se observaron resultados homogéneos en los subgrupos preespecificados de eficacia: sexo, grupo de edad, raza, porcentaje de CVF basal previsto y diagnóstico clínico de EPI subyacente original en los grupos.
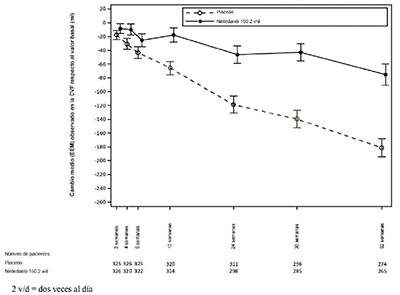
En la Figura 3 se muestra la evolución del cambio de la CVF respecto al valor basal a lo largo del tiempo en los grupos de tratamiento.
Figura 3. Cambio medio (EEM) observado en la CVF respecto al valor basal (ml) en el transcurso de 52 semanas
Además, se observaron efectos favorables de OFEV® sobre el cambio medio absoluto ajustado respecto al valor basal en el porcentaje de CVF previsto en la semana 52. El cambio medio absoluto ajustado respecto al valor basal en el porcentaje de CVF previsto en la semana 52 fue menor en el grupo tratado con nintedanib (–2.62%) que en el grupo tratado con placebo (–5.86%). La diferencia media ajustada entre los grupos de tratamiento fue de 3.24 (IC del 95%: 2.09, 4.40, valor nominal de p < 0.0001).
Análisis de los pacientes que responden en términos de CVF: La proporción de pacientes que responden en términos de CVF, definidos como los pacientes con una pérdida relativa del porcentaje de CVF previsto no superior al 5%, fue mayor en el grupo tratado con OFEV® que en el grupo tratado con placebo. Se observaron resultados similares en los análisis utilizando un umbral del 10% (Tabla 12).
|
Tabla 12 Proporción de pacientes que respondieron en términos de CVF a las 52 semanas en el ensayo INBUILD |
||
|
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número de pacientes analizados |
331 |
332 |
|
Umbral del 5% |
||
|
Número (%) de pacientes que respondieron en términos de CVF1 |
104 (31.4) |
158 (47.6) |
|
Comparación con el placebo |
||
|
Cociente de posibilidades (odds ratio)2 |
2.01 |
|
|
95% de IC |
(1.46,2.76) |
|
|
Valor nominal de p |
<0.0001 |
|
|
Umbral del 10% |
||
|
Número (%) de pacientes que respondieron en términos de CVF |
169 (51.1) |
197 (59.3) |
|
Comparación con el placebo |
||
|
Cociente de posibilidades (odds ratio)2 |
1.42 |
|
|
95% de IC |
(1.04, 1.94) |
|
|
Valor nominal de p |
0.0268 |
|
|
1Los pacientes que responden son aquellos que no presentan una pérdida relativa superior al 5% o superior al 10% en el% de CVF previsto, dependiendo del umbral y con una evaluación de la CVF a las 52 semanas (los pacientes de los que no se disponía de datos a las 52 semanas se consideraron pacientes que no respondieron). 2Basándose en un modelo de regresión logística con el% de CVF basal previsto como covariable continua y el patrón en la TCAR como covariable binaria. |
||
Tiempo hasta la primera exacerbación aguda de la EPI o la muerte: Durante todo el ensayo, la proporción de pacientes con al menos un acontecimiento de primera exacerbación aguda de la EPI o la muerte fue del 13.9% en el grupo tratado con OFEV® y del 19.6% en el grupo tratado con placebo. El cociente de riesgos (hazard ratio) fue de 0.67 (IC del 95%: 0.46, 0.98; valor nominal de p = 0.0387), que indica una reducción del 33% del riesgo de una primera exacerbación aguda de la EPI o de muerte en los pacientes tratados con OFEV® en comparación con los tratados con placebo (Figura 4).
Figura 4. Gráfico de Kaplan–Meier del tiempo hasta la primera exacerbación aguda de la EPI o la muerte durante todo el ensayo
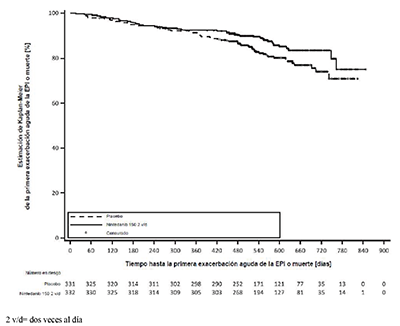
Análisis de supervivencia: El riesgo de muerte fue menor en el grupo tratado con OFEV® que en el grupo tratado con placebo. El cociente de riesgos (hazard ratio) fue 0.78 (IC del 95%: 0.50, 1.21; valor nominal de p = 0.2594), que indica una reducción del 22% del riesgo de muerte en los pacientes tratados con OFEV® en comparación con los tratados con placebo.
Tiempo hasta la progresión (≥ 10% de pérdida absoluta del porcentaje de CVF previsto) o muerte: En el ensayo INBUILD, el riesgo de progresión (≥ 10% de pérdida absoluta del porcentaje de CVF previsto) o muerte disminuyó en los pacientes tratados con OFEV®. La proporción de pacientes con un acontecimiento fue del 40.4% en el grupo tratado con OFEV® y del 54.7% en el grupo tratado con placebo. El cociente de riesgos (hazard ratio) fue de 0.66 (IC del 95%: 0.53, 0.83; p = 0.0003), que indica una reducción del 34% del riesgo de progresión (≥ 10% de pérdida absoluta del porcentaje de CVF previsto) o muerte en los pacientes tratados con OFEV® en comparación con los tratados con placebo.
Calidad de vida: El cambio medio ajustado respecto al valor basal en la puntuación total del K-BILD a las 52 semanas fue de –0.79 unidades en el grupo tratado con placebo y de 0.55 en el grupo tratado con OFEV®. La diferencia entre los grupos de tratamiento fue de 1.34 (IC del 95%: –0.31, 2.98; valor nominal de p = 0.1115).
El cambio absoluto medio ajustado respecto al valor basal en la puntuación del dominio de disnea del cuestionario de síntomas Living with Pulmonary Fibrosis (L-PF) a las 52 semanas fue de 4.28 en el grupo tratado con OFEV® en comparación con 7.81 en el grupo tratado con placebo. La diferencia media ajustada entre los grupos a favor de OFEV® fue de –3.53 (IC del 95%: –6.14, –0.92; valor nominal de p = 0.0081). El cambio absoluto medio ajustado respecto al valor basal en la puntuación del dominio de tos del cuestionario de síntomas L-PF a las 52 semanas fue de -1.84 en el grupo tratado con OFEV® en comparación con 4.25 en el grupo tratado con placebo. La diferencia media ajustada entre los grupos a favor de OFEV® fue de –6.09 (IC del 95%: –9.65, –2.53; valor nominal de p = 0.0008).
Enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES): Se ha estudiado la eficacia clínica de OFEV® en pacientes con EPI-ES en un ensayo fase III doble ciego, aleatorizado y controlado con placebo (SENSCIS). Los pacientes habían sido diagnosticados de EPIES de acuerdo con los criterios de clasificación de ES del American College of Rheumatology/European League Against Rheumatism de 2013 y una tomografía computarizada de alta resolución (TCAR) torácica realizada dentro de los 12 meses previos. Un total de 580 pacientes fueron aleatorizados en una proporción 1:1 a recibir OFEV® 150 mg dos veces al día o bien un placebo equivalente durante al menos 52 semanas, de los cuales 576 fueron tratados. La aleatorización se estratificó por el estado de anticuerpos antitopoisomerasa (ATA). Los pacientes individuales permanecieron con el tratamiento del ensayo ciego hasta 100 semanas (mediana de exposición a OFEV® de 15.4 meses; media de exposición a OFEV® de 14.5 meses).
El criterio de valoración principal fue la tasa anual de pérdida de la CVF a lo largo de 52 semanas. Los criterios de valoración secundarios clave fueron el cambio absoluto respecto al valor basal en la puntuación de la escala cutánea de Rodnan modificada (mRSS) en la semana 52 y el cambio absoluto respecto al valor basal en la puntuación total del cuestionario respiratorio de Saint George (SGRQ) en la semana 52.
En la población global, el 75.2% de los pacientes eran mujeres. La media (desviación estándar [DE, mín- máx]) de edad era de 54.0 (12.2, 20-79) años. En conjunto, el 51.9% de los pacientes tenían esclerosis sistémica (ES) cutánea difusa y el 48.1% tenían ES cutánea limitada. La media (DE) de tiempo desde la primera aparición de un síntoma no-Raynaud fue de 3.49 (1.7) años. El 49.0% de los pacientes estaban en tratamiento estable con micofenolato en el momento basal. El perfil de seguridad en pacientes con o sin micofenolato en el momento basal fue comparable.
Pérdida anual de la CVF: La pérdida anual de la CVF (ml) en el transcurso de 52 semanas se redujo significativamente en 41.0 ml en los pacientes que recibieron OFEV® en comparación con los pacientes que recibieron placebo (Tabla 13), lo que corresponde a un efecto relativo del tratamiento del 43.8%.
|
Tabla 13: Pérdida anual de la CFV (ml) en el transcurso de 52 semanas |
||
|
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número de pacientes analizados |
288 |
287 |
|
Pérdida1 (EE) en el transcurso de 52 semanas |
-93.3 (13.5) |
–52.4 (13.8) |
|
Comparación con el placebo |
||
|
Diferencia1 |
41.0 |
|
|
95% de IC |
(2.9,79.0) |
|
|
Valor p |
<0.05 |
|
|
1Basándose en un modelo de regresión de coeficientes aleatorios con efectos categóricos fijos del tratamiento, estado de ATA, sexo, efectos continuos fijos del tiempo, CVF basal [ml], edad, estatura e incluyendo las interacciones de tratamiento por tiempo y de momento basal por tiempo. Se incluyó el efecto aleatorio para la intersección específica del paciente y el tiempo. Los errores en un mismo paciente se modelizaron mediante una matriz de varianzas-covarianzas no estructurada. La variabilidad interindividual se modelizó mediante una matriz de varianzas-covarianzas de componentes de varianza. |
||
El efecto de OFEV® para reducir la pérdida anual de la CVF fue similar en los distintos análisis de sensibilidad preespecificados y no se detectó heterogeneidad en los subgrupos preespecificados (p. ej., por edad, sexo y uso de micofenolato).
Además, se observaron efectos similares en otros criterios de valoración de la función pulmonar, p. ej., el cambio absoluto respecto al valor basal en la CVF en ml en la semana 52 (Figura 5 y Tabla 14) y la pérdida de la CVF en% prevista en el transcurso de 52 semanas (Tabla 15), lo que corrobora aún más los efectos de OFEV® a la hora de enlentecer la progresión de la EPI-ES. Además, hubo menos pacientes en el grupo de OFEV® que tuvieron una pérdida absoluta de la CVF prevista > 5% (20.6% en el grupo de OFEV® frente a 28.5% en el grupo de placebo, CP=0.65, p=0.0287). La pérdida relativa de la CVF en ml > 10% fue comparable entre ambos grupos (16.7% en el grupo de OFEV® frente a 18.1% en el grupo de placebo, CP=0.91, p=0.6842). En estos análisis, los valores de CVF ausentes en la semana 52 se imputaron con el peor valor del paciente en el tratamiento.
Un análisis exploratorio de los datos de hasta 100 semanas (duración máxima del tratamiento en SENSCIS) sugirió que el efecto de OFEV® durante el tratamiento a la hora de enlentecer la progresión de la EPI-ES persistió pasadas las 52 semanas.
Figura 5. Cambio medio (EEM) observado en la CVF respecto al valor basal (ml) en el transcurso de 52 semanas
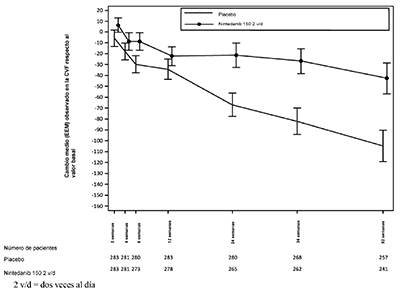
|
Tabla 14 Cambio absoluto respecto al valor basal en la CVF (ml) en la semana 52 |
||
|
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número de pacientes analizados |
288 |
288 |
|
Media (DE) en el momento basal |
2541.0(815.5) |
2458.5 (735.9) |
|
Cambio medio1 (EE) respecto al valor basal en la semana 52 |
-101.0(13.6) |
-54.6(13.9) |
|
Comparación con el placebo |
||
|
Media1 |
46.4 |
|
|
95% de IC |
(8.1,84.7) |
|
|
Valor p |
<0.05 |
|
|
1Basándose en un modelo mixto de medidas repetidas (MMMR), con efectos categóricos fijos de estado de ATA, visita, interacción de tratamiento por visita, interacción de momento basal por visita, edad, sexo y estatura. La visita fue la medida repetida. Los errores dentro de un mismo paciente se modelizaron mediante estructura de varianzas-covarianzas no estructurada. La media ajustada se basó en todos los pacientes analizados en el modelo (no solo los pacientes con una medición basal y una medición en la semana 52). |
||
|
Tabla 15: Pérdida anual de la CVF (% previsto) en el transcurso de 52 semanas |
||
|
Placebo |
OFEV® 150 mg dos veces al día |
|
|
Número de pacientes analizados |
288 |
287 |
|
Perdida1 (EE) en el transcurso de 52 semanas |
-2.6(0.4) |
-1.4(0.4) |
|
Comparación con el placebo |
||
|
Diferencia1 |
1.15 |
|
|
95% de IC |
(0.09, 2.21) |
|
|
Valor p |
<0.05 |
|
|
1Basándose en un modelo de regresión de coeficientes aleatorios con efectos categóricos fijos de tratamiento, estado de ATA, efectos continuos fijos del tiempo, CVF basal [% previsto] e incluyendo las interacciones de tratamiento por tiempo y de momento basal por tiempo. Se incluyó el efecto aleatorio para la coordenada específica del paciente y el tiempo. Los errores dentro de un mismo paciente se modelizaron mediante una matriz de varianzas-covarianzas no estructurada. La variabilidad interindividual se modelizó mediante una matriz de varianzas-covarianzas con componentes de varianza. |
||
Cambio respecto al valor basal en la puntuación de la escala cutánea de Rodnan modificada (mRSS)en la semana 52: El cambio absoluto medio ajustado respecto al valor basal en la escala mRSS en la semana 52 fue comparable entre el grupo de OFEV® (-2.17 [IC del 95% -2.69, -1.65]) y el grupo de placebo (-1.96 [IC del 95% -2.48, -1.45]). La diferencia media ajustada entre los grupos de tratamiento fue de -0.21 (IC del 95% -0.94, 0.53; p=0.5785).
Cambio respecto al valor basal en la puntuación total del cuestionario respiratorio de St. George (SGRQ) en la semana 52: El cambio absoluto medio ajustado respecto al valor basal en la puntuación total del SGRQ en la semana 52 fue comparable entre el grupo de OFEV® (0.81 [IC del 95% -0.92, 2.55]) y el grupo de placebo (-0.88 [IC del 95% -2.58, 0.82]). La diferencia media ajustada entre los grupos de tratamiento fue de 1.69 (IC del 95% -0.73, 4.12; p=0.1711).
Análisis de la supervivencia: La mortalidad a lo largo de todo el ensayo fue comparable entre el grupo de OFEV® (N=10; 3.5%) y el grupo de placebo (N=9; 3.1%). El análisis del tiempo hasta la muerte durante todo el ensayo condujo a un CR de 1.16 (IC del 95% 0.47, 2.84; p=0.7535).
Intervalo QT: En un estudio específico realizado en pacientes con cáncer de las células renales, se registraron las mediciones de los intervalos QT/QTc y se demostró que una dosis oral única de 200 mg de nintedanib, así como dosis orales múltiples de 200 mg de nintedanib administradas dos veces al día durante 15 días no prolongaron el intervalo QTcF.
Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con OFEV® en todos los grupos de la población pediátrica en la indicación de FPI (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
Cáncer de pulmón no microcítico (CPNM):
Grupo farmacoterapéutico: Antineoplásicos, inhibidores de la proteína cinasa, código ATC: L01XE31
Mecanismo de acción: Nintedanib es un triple inhibidor de la angiocinasa que bloquea los receptores del factor de crecimiento del endotelio vascular (VEGFR -1-3), los receptores del factor de crecimiento derivados de plaquetas (PDGFR α y ß) y los receptores del factor de crecimiento de fibroblastos (FGFR 1-3). Nintedanib se une de forma competitiva al sitio de unión de adenosina trifosfato (ATP) de estos receptores y bloquea la señalización intracelular, que es crucial para la proliferación y la supervivencia de las células endoteliales y perivasculares (pericitos y células del músculo liso vasculares). Además, se inhiben la proteína tirosina cinasa 3 similar a Fms (Flt), la proteína tirosina cinasa específica de linfocitos (Lck) y la proteína tirosina cinasa proto-oncogénica Src (Src).
Efectos farmacodinámicos: La angiogénesis tumoral es una característica fundamental que contribuye al crecimiento y la progresión tumoral y a la formación de metástasis, y se activa predominantemente por la liberación de factores pro- angiogénicos secretados por la célula tumoral (es decir, VEGF y bFGF) para atraer a las células endoteliales del huésped así como a las células perivasculares con el fin de facilitar el suministro de oxígeno y nutrientes a través del sistema vascular del huésped. En modelos preclínicos de la enfermedad, nintedanib en monoterapia interfirió de forma eficaz en la formación y el mantenimiento del sistema vascular tumoral, lo que dio lugar a una inhibición del crecimiento tumoral y a una estasis tumoral. En particular, el tratamiento de xenoinjertos tumorales con nintedanib dio lugar a una rápida reducción en la densidad de los microvasos tumorales, la cobertura de los vasos pericitos y la perfusión tumoral.
Las mediciones realizadas por resonancia magnética dinámica realizada con contraste (DCE-MRI) mostraron un efecto anti-angiogénico de nintedanib en humanos. Este no fue claramente dependiente de la dosis, pero la mayor parte de las respuestas se observaron a dosis ≥ 200 mg. La regresión logística reveló una asociación estadísticamente significativa entre el efecto anti-angiogénico y la exposición a nintedanib. Los efectos de la DCE-MRI se observaron de 24 a 48 horas después de la primera toma del medicamento y se mantuvieron o incluso aumentaron después del tratamiento continuo durante varias semanas. No se encontró ninguna correlación entre la respuesta a la DCE-MRI y la reducción posterior clínicamente significativa del tamaño de la lesión diana, pero la respuesta a la DCE-MRI se asoció a la estabilización de la enfermedad.
Eficacia clínica y seguridad:
Eficacia en el ensayo pivotal fase 3 LUME-Lung 1:
La eficacia y la seguridad de OFEV® se evaluaron en 1.314 pacientes adultos con CPNM localmente avanzado, metastásico o recurrente después de una primera línea de quimioterapia. “Localmente recurrente” se definió como una recidiva local del tumor sin metástasis al comienzo del estudio. El ensayo incluyó a 658 pacientes (50.1%) con adenocarcinoma, 555 pacientes (42.2%) con carcinoma epidermoide y 101 pacientes (7.7%) con otras histologías tumorales.
Los pacientes se aleatorizaron (1:1) para recibir nintedanib 200 mg por vía oral dos veces al día en combinación con 75 mg/m² de docetaxel intravenoso cada 21 días (n = 655) o placebo por vía oral dos veces al día en combinación con 75 mg/m² de docetaxel cada 21 días (n = 659). La aleatorización se estratificó según el estado ECOG (Eastern Cooperative Oncology Group) (0 frente a 1), el tratamiento anterior con bevacizumab (sí frente a no), la metástasis cerebral (sí frente a no) y la histología del tumor (histología escamosa frente a no escamosa).
Las características de los pacientes estaban bien distribuidas entre los grupos de tratamiento dentro de la población global y dentro de los subgrupos de acuerdo con la histología. En la población global, el 72.7% de los pacientes eran varones. La mayoría de los pacientes era de raza no asiática (81.6%), la mediana de edad era de 60.0 años y el estado ECOG basal era 0 (28.6%) o 1 (71.3%); un paciente tenía un estado ECOG basal de 2. El 5.8% de los pacientes presentó una metástasis cerebral estable al comienzo del estudio y el 3.8% había recibido un tratamiento anterior con bevacizumab. El estadio de la enfermedad se determinó en el momento del diagnóstico utilizando la edición 6 o 7 del sistema de clasificación UICC (Union Internationale Conte le Cancer)/AJCC (American Joint Committee on Cancer).
En la población global, el 16.0% de los pacientes tenían un estadio de la enfermedad < IIIB/IV, el 22.4% tenían un estadio de la enfermedad IIIB y el 61.6% tenían un estadio de la enfermedad IV. El 9.2% de los pacientes entraron en el estudio con enfermedad localmente recurrente evaluada en el nivel basal. En el caso de pacientes con histología tumoral de adenocarcinoma, el 15.8% tenían un estadio de la enfermedad < IIIB/IV, el 15.2% tenían un estadio de la enfermedad IIIB y el 69.0% tenían un estadio de la enfermedad IV.
El 5.8% de los pacientes con adenocarcinoma entraron en el estudio con enfermedad localmente recurrente evaluada en el nivel basal.
La variable principal del estudio fue la supervivencia libre de progresión (SLP), evaluada por un comité de revisión independiente (IRC), en la población global del estudio con intención de tratar (ITT) y examinado según histología. La supervivencia global (SG) fue la variable clave secundaria. Otras variables relativas a la eficacia incluyeron la respuesta objetiva, el control de la enfermedad, el cambio en el tamaño del tumor y la calidad de vida de los pacientes.
La adición de nintedanib a docetaxel produjo una reducción estadísticamente significativa del 21% en el riesgo de progresión o muerte en el caso de la población global (cociente de riesgos (hazard ratio) 0.79; 95% de intervalo de confianza (IC): 0.68 – 0.92; p = 0.0019), tal como determinó el comité de revisión independiente (IRC). Este resultado se confirmó en el estudio de seguimiento de la SLP (cociente de riesgos (hazard ratio) 0.85, 95% de IC: 0.75 – 0.96; p = 0.0070), que incluyó todos los episodios recopilados en el momento del análisis final de la SG. El análisis de la supervivencia global en la población de pacientes global no alcanzó una significación estadística (cociente de riesgos (hazard ratio): 0.94; 95% de IC: 0.83-1.05).
Cabe reseñar que los análisis planificados previamente según la histología presentaron una diferencia estadísticamente significativa en la SG entre los grupos de tratamiento, solamente en la población con adenocarcinoma (ver Tabla 16).
Como se muestra en la Tabla 16, la adición de nintedanib a docetaxel produjo una reducción estadísticamente significativa del 23% en el riesgo de progresión o muerte en el caso de la población con adenocarcinoma (cociente de riesgos (hazard ratio) 0.77; 95% de IC: 0.62 – 0.96). En línea con estas observaciones, las otras variables de valoración relacionadas con el estudio, tales como el control de la enfermedad y el cambio en el tamaño del tumor, mostraron una mejoría significativa.
|
Tabla 16 Resultados de eficacia del estudio LUME-Lung 1 para pacientes con una histología tumoral de adenocarcinoma |
||
|
OFEV® + docetaxel |
Placebo + docetaxel |
|
|
Supervivencia libre de progresión (SLP)*: análisis principal |
||
|
Pacientes, n |
277 |
285 |
|
Número de muertes o progresiones, n (%) |
152 (54.9) |
180 (63.2) |
|
Mediana de SLP (meses) |
4.0 |
2.8 |
|
Cociente de riesgos (hazard ratio) (95% de IC) |
0.77 (0.62;0.96) |
|
|
Valor p del Log-Rank Test estratificado ** |
0.0193 |
|
|
Supervivencia libre de progresión (SLP)***: análisis de seguimiento |
||
|
Pacientes, n |
322 |
336 |
|
Número de muertes o progresiones, n (%) |
255 (79.2) |
267 (79.5) |
|
Mediana de SLP (meses) |
4.2 |
2.8 |
|
Cociente de riesgos (hazard ratio) (95% de IC) |
0.84 (0.71;1.00) |
|
|
Valor p del Log-Rank Test estratificado ** |
0.0485 |
|
|
Control de la enfermedad (%) |
60.2 |
44.0 |
|
Cociente de posibilidades (odds ratio) (95% de IC)+ |
1.93 (1.42; 2.64) |
|
|
valor p+ |
< 0,0001 |
|
|
Respuesta objetiva (%) |
4.7 |
3.6 |
|
Cociente de posibilidades (odds ratio) (95% de IC)+ |
1.32 (0.61; 2.93) |
|
|
valor p+ |
0.4770 |
|
|
Reducción del tamaño tumoral (%) ° |
-7.76 |
-0.97 |
|
valor p° |
0.0002 |
|
|
Supervivencia global (SG)*** |
||
|
Pacientes, n |
322 |
336 |
|
Número de muertes,n (%) |
259 (80.4) |
276 (82.1) |
|
Mediana de SG (meses) |
12.6 |
10.3 |
|
Cociente de riesgos (hazard ratio) (95% de IC) |
0.83 (0.70; 0.99) |
|
|
Valor p del Log-Rank Test estratificado * |
0.0359 |
|
|
HR: cociente de riesgos (hazard ratio); IC: intervalo de confianza * Análisis principal de la SLP, cuando se habían observado 713 episodios de SLP basándose en la evaluación realizada por el IRC en la población ITT global (332 episodios en pacientes con adenocarcinoma). ** Estratificado por el estado funcional ECOG basal (0 frente a 1), las metástasis cerebrales en el nivel basal (sí frente a no) y el tra°tamiento anterior con bevacizumab (sí frente a no). *** Análisis de la SG y análisis de seguimiento de la SLP realizados cuando se observaron 1121 casos de muerte en la población ITT global (535 episodios en pacientes con adenocarcinoma) + El cociente de posibilidades (odds ratio) y el valor p se obtuvieron a partir de un modelo de regresión logística ajustado para la puntuación ECOG basal (0 frente a 1). ° Media ajustada del mejor porcentaje de cambio respecto al valor basal y valor p generado de un modelo ANOVA que realiza un ajuste de la puntuación ECOG basal (0 frente a 1), las metástasis cerebrales en el nivel basal (sí frente a no) y el tratamiento anterior con bevacizumab (sí frente a no). |
||
En pacientes con adenocarcinoma se demostró una mejoría estadísticamente significativa en la SG, que corrobora la eficacia del tratamiento con nintedanib más docetaxel, con un 17 % de reducción en el riesgo de muerte (cociente de riesgos (hazard ratio) 0.83, p = 0.0359) y una mediana de mejoría en la SG de 2.3 meses (10.3 frente a 1.6 meses, Figura 6).
Figura 6: Curva de Kaplan-Meier relativa a la supervivencia global en pacientes con histología tumoral de adenocarcinoma por grupo de tratamiento en el ensayo LUME-Lung 1
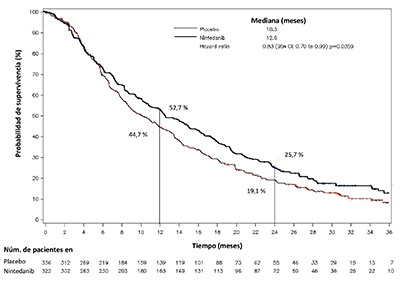
Se realizó una evaluación preespecificada en la población de pacientes con adenocarcinoma en los que se consideraba que habían entrado en el estudio con un especial mal pronóstico para el tratamiento, es decir, pacientes que habían experimentado una progresión de la enfermedad durante el tratamiento de primera línea o poco después del inicio del mismo antes de comenzar el estudio. Esta población incluyó a los pacientes con un adenocarcinoma que habían experimentado una progresión de la enfermedad y fueron incluidos en el estudio con un intervalo inferior a 9 meses después de iniciar el tratamiento de primera línea. El tratamiento de estos pacientes con nintedanib en combinación con docetaxel redujo el riesgo de muerte en un 25% en comparación con placebo más docetaxel (cociente de riesgos (hazard ratio) 0.75; 95% de IC: 0.60 a 0.92; p = 0.0073). La mediana de SG mejoró en 3 meses (nintedanib: 10.9 meses; placebo: 7.9 meses). En un análisis posterior en pacientes con adenocarcinoma que habían experimentado una progresión de la enfermedad y fueron incluidos en el estudio con un intervalo ≥ 9 meses después de iniciar el tratamiento de primera línea, la diferencia no fue estadísticamente significativa (cociente de riesgos (hazard ratio) para la SG: 0.89; 95% de IC: 0.66-1.19).
La proporción de pacientes con adenocarcinoma en estadio < IIIB/IV en el momento del diagnóstico fue reducida y estuvo equilibrada entre los grupos de tratamiento (placebo: 54 pacientes (16.1%); nintedanib: 50 pacientes (15.5%)). El cociente de riesgos (hazard ratio) de estos pacientes para la SLP y la SG fue de 1.24 (95% de IC: 0.68, 2.28) y 1.09 (95% de IC: 0.70, 1.70) respectivamente. No obstante, el tamaño de la muestra era pequeño, no hubo una interacción significativa y el IC fue amplio e incluyó el cociente de riesgos (hazard ratio) para la SG de la población global con adenocarcinoma.
Calidad de vida: El tratamiento con nintedanib no produjo un cambio significativo en el tiempo hasta el deterioro de los síntomas preespecificados, tos, disnea y dolor, pero dio lugar a un deterioro significativo en la escala de síntomas de diarrea. Sin embargo, la mejoría global proporcionada por el tratamiento con nintedanib no afectó negativamente a la calidad de vida global descrita por los propios pacientes.
Efectos en el intervalo QT: Las mediciones de QT/ QTc se registraron y analizaron a partir de un estudio específico que comparó nintedanib en monoterapia con sunitinib en monoterapia en pacientes con carcinoma de células renales. En este estudio, dosis orales únicas de 200 mg de nintedanib, así como dosis orales múltiples de 200 mg de nintedanib administradas dos veces al día durante 15 días no prolongaron el intervalo QTcF. No obstante, no se realizó ningún ensayo del QT completo con nintedanib administrado en combinación con docetaxel.
Población pediátrica: La Agencia Europea de Medicamentos ha eximido al titular de la obligación de presentar los resultados de los ensayos realizados con OFEV® en todos los grupos de la población pediátrica en la indicación de cáncer de pulmón no microcítico (ver sección 4.2 para consultar la información sobre el uso en la población pediátrica).
DATOS FARMACÉUTICOS:
Lista de excipientes:
Contenido de la cápsula:
triglicéridos de cadena media grasa dura
lecitina (soja) (E322)
Cubierta exterior de la cápsula gelatina:
glicerol (85%)
dióxido de titanio (E171)
óxido de hierro rojo (E172)
óxido de hierro amarillo (E172)
Tinta de impresión:
barniz de goma laca
óxido de hierro negro (E172)
propilenglicol (E1520)
Incompatibilidades: No procede.
Periodo de validez: 3 años
Precauciones especiales de conservación:
Su venta requiere receta médica.
Léase instructivo anexo.
Consérvese en refrigeración (2-8°C). No se deje al alcance de los niños.
No se use en el embarazo ni en la lactancia ni en menores de 18 años.
Contiene lecitina.
Este medicamento deberá ser prescrito por médicos especialistas con experiencia en terapia antineoplástica.
PROPIEDADES FARMACOCINÉTICAS:
Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
Absorción: Nintedanib alcanzó la concentración plasmática máxima aproximadamente de 2 a 4 horas después de la administración oral como cápsula de gelatina blanda junto con alimentos (rango de 0.5 a 8 horas). La biodisponibilidad absoluta de una dosis de 100 mg fue del 4.69% (90% de IC: 3.615 a 6.078) en voluntarios sanos. La absorción y la biodisponibilidad disminuyen por los efectos de los transportadores y por el metabolismo sustancial de primer paso. La proporcionalidad de la dosis se demostró mediante un aumento de la exposición a nintedanib (rango de dosis de 50 a 450 mg una vez al día y de 150 a 300 mg dos veces al día). Las concentraciones plasmáticas en estado estacionario se lograron como muy tarde en el plazo de una semana de dosificación.
Después de la ingesta de alimentos, la exposición a nintedanib aumentó en aproximadamente el 20% en comparación con la administración en ayunas (IC: 95.3 a 152.5%) y la absorción se retrasó (mediana de tmax en ayunas: 2.00 h; con alimentos: 3.98 h).
Distribución: Nintedanib sigue al menos una cinética de disposición bifásica. Después de una perfusión intravenosa, se observó un alto volumen de distribución (Vss: 1.050 l, 45.0% de gCV).
La unión a proteínas in vitro de nintedanib en el plasma humano fue alta, con una fracción unida del 97.8%. Se considera que la albúmina sérica es la proteína de unión más importante. Nintedanib se distribuye preferentemente en el plasma, con una relación sangre/plasma de 0.869.
Biotransformación: La reacción metabólica prevalente para nintedanib es la ruptura hidrolítica mediante esterasas que dan lugar a la fracción de ácido libre BIBF 1202. A continuación, BIBF 1202 se glucuronida mediante enzimas uridina 5’- difosfo-glucurosiltransferasa (UGT), concretamente UGT 1A1, UGT 1A7, UGT 1A8 y UGT 1A10, a glucurónido de BIBF 1202.
Tan solo una pequeña proporción de la biotransformación de nintedanib se realiza a través de las vías CYP, siendo la CYP 3A4 la enzima predominante implicada. El principal metabolito dependiente de CYP no pudo detectarse en el plasma en el estudio ADME realizado con humanos. In vitro, el metabolismo dependiente de CYP representó aproximadamente un 5% en comparación con aproximadamente un 25% de ruptura de ésteres. Nintedanib, BIBF 1202 y el glucurónido BIBF 1202 tampoco inhibieron ni indujeron las enzimas CYP en estudios preclínicos con animales. Por lo tanto, no cabe esperar interacciones farmacológicas entre nintedanib y sustratos de CYP, inhibidores de CYP o inductores de CYP.
Eliminación: El aclaramiento plasmático total después de la perfusión intravenosa fue alto (aclaramiento: 1.390 ml/min, 28.8% de gCV). La eliminación por la orina del principio activo inalterado en el plazo de 48 horas fue de aproximadamente el 0.05% de la dosis (31.5% de gCV) después de la administración oral, y de aproximadamente 1.4% de la dosis (24.2% de gCV) después de la administración intravenosa; el aclaramiento renal fue de 20 ml/min (32.6% de gCV). La principal vía de eliminación del fármaco marcado radioactivamente después de la administración oral de [14C] nintedanib fue la excreción biliar/fecal (93.4% de la dosis, 2.61% de gCV). La contribución de la eliminación renal al aclaramiento total fue baja (0.649% de la dosis, 26.3% de gCV). La recuperación total se consideró completa (por encima del 90%) en los cuatro días posteriores a la dosificación. La semivida terminal de nintedanib fue de entre 10 y 15 horas (gCV de aproximadamente el 50%).
Linealidad/No linealidad: La farmacocinética de nintedanib se puede considerar lineal respecto al tiempo (es decir, los datos de una sola dosis pueden extrapolarse a los datos de múltiples dosis). La acumulación en el caso de múltiples administraciones fue de 1.04 veces para la Cmax y de 1.38 veces para el AUCτ. Las concentraciones mínimas de nintedanib permanecieron estables durante más de un año.
Transporte: Nintedanib es un sustrato de la gp-P. Para obtener más información sobre el potencial de interacción de nintedanib con este transportador, ver sección 4.5. Se demostró que nintedanib no es un sustrato ni un inhibidor de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 o MRP-2 in vitro. Nintedanib tampoco resultó ser un sustrato de la BCRP. Solo se observó in vitro un leve potencial inhibidor en el OCT-1, la BCRP y la gp-P, pero se considera que esto tiene una baja relevancia clínica. Lo mismo se aplica a nintedanib como sustrato del OCT-1.
Análisis farmacocinético poblacional en poblaciones especiales: Las propiedades farmacocinéticas de nintedanib fueron similares en los voluntarios sanos, los pacientes con FPI, los pacientes con otras EPI fibrosantes crónicas con un fenotipo progresivo, los pacientes con EPI-ES y los pacientes con cáncer. Basándose en los resultados de un análisis farmacocinético poblacional en pacientes con FPI y cáncer de pulmón no microcítico (CPNM) (N=1.191) y en las investigaciones descriptivas, la exposición a nintedanib no se vio afectada por el sexo (corregido por el peso corporal), la insuficiencia renal leve y moderada (calculada según el aclaramiento de creatinina), el consumo de alcohol ni por el genotipo de la gp-P. Los análisis farmacocinéticos poblacionales indicaron efectos moderados en la exposición a nintedanib dependiendo de la edad, el peso corporal y la raza (ver más abajo). Teniendo en cuenta la alta variabilidad de la exposición entre individuos, los efectos moderados observados no se consideran clínicamente relevantes (ver sección 4.4).
Edad: La exposición a nintedanib aumentó linealmente con la edad. El AUCτ,ss disminuyó en un 16% en el caso de un paciente de 45 años y aumentó en un 13% en el caso de un paciente de 76 años en comparación con un paciente con la mediana de edad de 62 años. El rango de edad cubierto por el análisis fue de 29 a 85 años; aproximadamente el 5% de la población era mayor de 75 años. Basándose en un modelo farmacocinético poblacional, se observó un aumento en la exposición a nintedanib de aproximadamente entre el 20 y el 25% en pacientes de 75 años o más, en comparación con pacientes menores de 65 años.
No se han realizado estudios con poblaciones pediátricas.
Peso corporal: Se observó una correlación inversa entre el peso corporal y la exposición a nintedanib. El AUCτ,ss aumentó en un 25% en el caso de un paciente de 50 kg (percentil 5) y disminuyó en un 19% en el caso de un paciente de 100 kg (percentil 95) en comparación con un paciente con la mediana de peso de 71.5 kg.
Raza: La exposición media de la población a nintedanib fue un 33-50% superior en pacientes chinos, taiwaneses e indios y un 16% superior en pacientes japoneses, mientras que fue un 16-22% inferior en el caso de pacientes coreanos comparados con los caucásicos (corregido por peso corporal). Los datos procedentes de individuos de raza negra eran muy limitados, pero se encuentran en el mismo rango que en el caso de los caucásicos.
Insuficiencia hepática: En un estudio específico de fase I de dosis única y comparado con individuos sanos, la exposición a nintedanib basándose en la Cmax y el AUC fue 2.2 veces más alta en voluntarios con insuficiencia hepática leve (Child Pugh A; IC del 90%: 1.3 - 3,7 para la Cmax y 1.2 – 3.8 para el AUC, respectivamente). En voluntarios con insuficiencia hepática moderada (Child Pugh B), la exposición fue 7.6 veces más alta basándose en la Cmax (IC del 90%: 4.4 – 13.2) y 8,7 veces más alta (IC del 90%: 5.7 – 13.1) basándose en el AUC, respectivamente, comparado con los voluntarios sanos. No se han realizado estudios en individuos con insuficiencia hepática grave (Child Pugh C).
Tratamiento conjunto con pirfenidona: En un estudio farmacocinético específico, se investigó el tratamiento conjunto de nintedanib con pirfenidona en pacientes con FPI. El grupo 1 recibió una dosis única de 150 mg de nintedanib antes y después del ajuste a 801 mg de pirfenidona tres veces al día en estado estacionario (n = 20 pacientes tratados). El grupo 2 recibió tratamiento en estado estacionario con 801 mg de pirfenidona tres veces al día y fue objeto de la elaboración de un perfil farmacocinético antes y después de al menos 7 días de tratamiento conjunto con 150 mg de nintedanib dos veces al día (n = 17 pacientes tratados). En el grupo 1, las relaciones ajustadas de las medias geométricas (intervalo de confianza [IC] del 90%) fueron del 93% (57%-151%) y del 96% (70%-131%) para la Cmax y el AUC0-tz de nintedanib, respectivamente (n = 12 para la comparación intraindividual). En el grupo 2, las relaciones ajustadas de las medias geométricas (IC del 90%) fueron del 97% (86% -110%) y del 95% (86% -106%) para la Cmax,ss y el AUCτ,ss de pirfenidona, respectivamente (n = 12 para la comparación intraindividual). De acuerdo con estos resultados, no existe evidencia de una interacción farmacocinética relevante entre nintedanib y pirfenidona cuando se administran en combinación (ver sección 4.4).
Tratamiento concomitante con bosentán: En un estudio de farmacocinética específico, se investigó el tratamiento concomitante de OFEV® con bosentán en voluntarios sanos. Los sujetos recibieron una dosis única de 150 mg de OFEV® antes y después de administraciones múltiples de 125 mg de bosentán dos veces al día en estado estacionario. Los cocientes ajustados de media geométrica (intervalo de confianza [IC] del 90% fueron del 103% [86-124%] y del 99% [91%-107%] para la Cmáx y el AUC0-tz de nintedanib, respectivamente (n=13), lo que indica que la administración conjunta de nintedanib con bosentán no alteró la farmacocinética de nintedanib.
Tratamiento concomitante con anticonceptivos hormonales orales: En un estudio farmacocinético específico, las pacientes con EPI-ES recibieron una dosis única de una combinación de 30 µg de etinilestradiol y 150 µg de levonorgestrel antes y después de una pauta de dos veces al día de 150 mg de nintedanib durante al menos 10 días. Los cocientes ajustados de media geométrica (intervalo de confianza [IC] del 90%) fueron del 117% (108%-127%; Cmáx) y del 101% (93%-111%; AUC0-tz) para etinilestradiol y del 101% (90%-113%; Cmáx) y del 96% (91%-102%; AUC0-tz ) para levonorgestrel, respectivamente (n=15), lo que indica que la administración conjunta de nintedanib no tiene ningún efecto relevante sobre la exposición plasmática de etinilestradiol y levonorgestrel.
Relación exposición-respuesta: Los análisis de la relación exposición-respuesta de pacientes con FPI y otras EPI fibrosantes crónicas con un fenotipo progresivo indicaron una relación débil entre la exposición plasmática a nintedanib y las elevaciones de la ALT y/o de la AST. La dosis administrada real podría ser el mejor factor predictivo del riesgo de aparición de diarrea de cualquier intensidad, aunque no pueda descartarse la exposición plasmática como factor determinante del riesgo (ver sección 4.4).
Cáncer de pulmón no microcítico (CPNM):
Absorción: Nintedanib alcanzó la concentración plasmática máxima aproximadamente de 2 a 4 horas después de la administración oral como cápsula de gelatina blanda junto con alimentos (rango de 0.5 a 8 horas). La biodisponibilidad absoluta de una dosis de 100 mg fue del 4.69% (90% de IC: 3.615 a 6.078) en voluntarios sanos. La absorción y la biodisponibilidad disminuyen por los efectos de los transportadores y por el metabolismo sustancial de primer paso. La proporcionalidad de la dosis se demostró mediante un aumento de la exposición a nintedanib (rango de dosis de 50 a 450 mg una vez al día y de 150 a 300 mg dos veces al día). Las concentraciones plasmáticas en estado estacionario se lograron como muy tarde en el plazo de una semana de dosificación.
Después de la ingesta de alimentos, la exposición a nintedanib aumentó en aprox imadamente el 20% en comparación con la administración en ayunas (IC: 95.3 a 152.5%) y la absorción se retrasó (mediana de tmax en ayunas: 2.00 horas; con alimentos: 3.98 horas).
Distribución: Nintedanib sigue al menos una cinética de disposición bifásica. Después de una perfusión intravenosa, se observó un alto volumen de distribución (Vss: 1.050 l, 45.0% de gCV).
La unión a proteínas in vitro de nintedanib en el plasma humano fue alta, con una fracción unida del 97.8%. Se considera que la albúmina sérica es la proteína de unión más importante. Nintedanib se distribuye preferentemente en el plasma, con una relación sangre/plasma de 0.869.
Biotransformación: La reacción metabólica prevalente para nintedanib es la ruptura hidrolítica mediante esterasas que dan lugar a la fracción de ácido libre BIBF 1202. A continuación, BIBF 1202 se glucuronida mediante enzimas UGT, concretamente UGT 1A1, UGT 1A7, UGT 1A8 y UGT 1A10, a glucurónido de BIBF 1202.
Tan solo una pequeña proporción de la biotransformación de nintedanib se realiza a través de las vías CYP, siendo la CYP 3A4 la enzima predominante implicada. El principal metabolito dependiente de CYP no pudo detectarse en el plasma en el estudio ADME realizado con humanos. In vitro, el metabolismo dependiente de CYP representó aproximadamente un 5% en comparación con aproximadamente un 25% de ruptura de ésteres. En experimentos preclínicos in vivo, BIBF 1202 no mostró eficacia a pesar de su actividad en los receptores diana de la sustancia.
Eliminación: El aclaramiento plasmático total después de la perfusión intravenosa fue alto (aclaramiento: 1390 ml/min, 28.8 % de gCV). La eliminación por la orina del principio activo inalterado en el plazo de 48 horas fue de aproximadamente el 0.05% de la dosis (31.5% de gCV) después de la administración oral, y de aproximadamente 1.4% de la dosis (24.2% de gCV) después de la administración intravenosa; el aclaramiento renal fue de 20 ml/min (32.6% de gCV). La principal vía de eliminación del fármaco marcado radioactivamente después de la administración oral de [14C] nintedanib fue la excreción biliar/fecal (93.4% de la dosis, 2.61% de gCV).
La contribución de la eliminación renal al aclaramiento total fue baja (0.649% de la dosis, 26.3% de gCV).
La recuperación total se consideró completa (por encima del 90%) en los cuatro días posteriores a la dosificación. La semivida terminal de nintedanib fue de entre 10 y 15 horas (gCV de aproximadamente el 50%).
Linealidad/No linealidad: La farmacocinética de nintedanib se puede considerar lineal respecto al tiempo (es decir, los datos de una sola dosis pueden extrapolarse a los datos de múltiples dosis). La acumulación en el caso de múltiples administraciones fue de 1,04 veces para la Cmax y de 1.38 veces para el AUCτ. Las concentraciones mínimas de nintedanib permanecieron estables durante más de un año.
Información adicional sobre interacciones farmacológicas:
Metabolismo: No cabe esperar interacciones farmacológicas entre nintedanib y sustratos de CYP, inhibidores de CYP o inductores de CYP, puesto que nintedanib, BIBF 1202 y el glucurónido de BIBF 1202 no inhibieron ni indujeron las enzimas CYP en estudios preclínicos y nintedanib tampoco fue metabolizado por las enzimas CYP de forma significativa.
Transporte: Nintedanib es un sustrato de la gp-P. Para obtener más información sobre el potencial de interacción de nintedanib con este transportador, ver sección 4.5. Se demostró que nintedanib no es un sustrato ni un inhibidor de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 o MRP-2 in vitro. Nintedanib tampoco resultó ser un sustrato de la BCRP. Solo se observó in vitro un leve potencial inhibidor en el OCT-1, la BCRP y la gp-P, pero se considera que esto tiene una baja relevancia clínica. Lo mismo aplica a nintedanib como sustrato del OCT-1.
Relación(es) farmacocinética(s)/farmacodinámica(s): En los análisis farmacocinéticos exploratorios de efectos adversos, una exposición más alta a nintedanib tendió a estar asociada a un aumento en las enzimas hepáticas, pero no a efectos adversos gastrointestinales.
No se realizaron análisis farmacocinéticos de eficacia para los criterios de valoración clínicos. La regresión logística reveló una asociación estadísticamente significativa entre la exposición a nintedanib y la respuesta a la DCE-MRI.
Análisis farmacocinético poblacional en poblaciones especiales: Las propiedades farmacocinéticas de nintedanib fueron similares en los voluntarios sanos, los pacientes con cáncer y los pacientes objeto de estudio. La exposición a nintedanib no se vio afectada por el sexo (corregido por el peso corporal), la insuficiencia renal leve y moderada (calculada según el aclaramiento de creatinina), las metástasis hepáticas, el estado general medido por ECOG, el consumo de alcohol ni por el genotipo de la gp-P.
Los análisis farmacocinéticos poblacionales indicaron unos efectos moderados en la exposición a nintedanib dependiendo de la edad, el peso corporal y la raza (ver más abajo). Teniendo en cuenta la alta variabilidad de la exposición entre individuos que se observó en el ensayo clínico LUME-Lung 1, estos efectos no se consideran clínicamente relevantes. No obstante, se recomienda realizar un control estrecho en el caso de pacientes con varios de estos factores de riesgo (ver sección 4.4).
Edad: La exposición a nintedanib aumentó linealmente con la edad. El AUCτ,ss disminuyó en un 16% en el caso de un paciente de 45 años (percentil 5) y aumentó en un 13% en el caso de un paciente de 76 años (percentil 95) en comparación con un paciente con la mediana de edad de 62 años. El rango de edad cubierto por el análisis fue de 29 a 85 años; aproximadamente el 5% de la población era mayor de 75 años.
No se han realizado estudios con poblaciones pediátricas.
Peso corporal: Se observó una correlación inversa entre el peso corporal y la exposición a nintedanib. El AUCτ,ss aumentó en un 25% en el caso de un paciente de 50 kg (percentil 5) y disminuyó en un 19% en el caso de un paciente de 100 kg (percentil 95) en comparación con un paciente con la mediana de peso de 71.5 kg.
Raza: La exposición media de la población a nintedanib fue un 33 - 50% superior en pacientes chinos, taiwaneses e indios y un 16% superior en pacientes japoneses, mientras que fue un 16 - 22% inferior en el caso de pacientes coreanos comparados con los caucásicos (corregido por peso corporal). Teniendo en cuenta la alta variabilidad de la exposición entre individuos, estos efectos no se consideran clínicamente relevantes. Los datos procedentes de individuos de raza negra eran muy limitados, pero se encuentran en el mismo rango que en el caso de los caucásicos.
Insuficiencia hepática: En un estudio específico de fase I de dosis única y comparado con individuos sanos, la exposición a nintedanib basándose en la Cmax y el AUC fue 2.2 veces más alta en voluntarios con insuficiencia hepática leve (Child Pugh A; IC del 90%: 1.3 – 3.7 para la Cmax y 1.2 – 3.8 para el AUC, respectivamente). En voluntarios con insuficiencia hepática moderada (Child Pugh B), la exposición fue 7.6 veces más alta basándose en la Cmax (IC del 90%: 4.4 – 13.2) y 8.7 veces más alta (IC del 90%: 5.7 – 13.1) basándose en el AUC, respectivamente, comparado con los voluntarios sanos. No se han realizado estudios en individuos con insuficiencia hepática grave (Child Pugh C).
Tratamiento concomitante con anticonceptivos hormonales orales: En un estudio farmacocinético específico, las pacientes con EPI-ES recibieron una dosis única de una combinación de 30 µg de etinilestradiol y 150 µg de levonorgestrel antes y después de una pauta de dos veces al día de 150 mg de nintedanib durante al menos 10 días. Los cocientes ajustados de media geométrica (intervalo de confianza [IC] del 90%) fueron del 117% (108%-127%; Cmáx) y del 101% (93%-111%; AUC0-tz) para etinilestradiol y del 101% (90%-113%; Cmáx) y del 96% (91%-102%; AUC0-tz) para levonorgestrel, respectivamente (n=15), lo que indica que la administración conjunta de nintedanib no tiene ningún efecto relevante sobre la exposición plasmática de etinilestradiol y levonorgestrel.
CONTRAINDICACIONES: Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
• Embarazo (ver sección 4.6)
• Hipersensibilidad a nintedanib, a los cacahuetes, a la soja o a alguno de los excipientes incluidos en la sección 6.1
Cáncer de pulmón no microcítico (CPNM):
• Hipersensibilidad a nintedanib, a los cacahuetes, a la soja o a alguno de los excipientes incluidos en la sección 6.1.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
• Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
Trastornos gastrointestinales:
Diarrea: En los ensayos clínicos (ver sección 5.1), la diarrea fue la reacción adversa gastrointestinal más frecuente descrita (ver sección 4.8). En la mayoría de los pacientes, la reacción adversa fue de intensidad leve a moderada y se produjo en los primeros 3 meses de tratamiento.
En el periodo de poscomercialización, se han notificado casos graves de diarrea causantes de deshidratación y trastornos electrolíticos. Se debe tratar a los pacientes en cuanto aparezcan los primeros síntomas con una adecuada hidratación y la administración de medicamentos antidiarreicos, como la loperamida y puede requerir la reducción de la dosis o la interrupción del tratamiento. El tratamiento con OFEV® puede reanudarse a una dosis reducida (100 mg dos veces al día) o a la dosis completa (150 mg dos veces al día). Si se produce una diarrea grave y persistente a pesar de seguir un tratamiento sintomático, el tratamiento con OFEV® se debe suspender.
Náuseas y vómitos: Las náuseas y los vómitos fueron reacciones adversas gastrointestinales descritas con frecuencia (ver sección 4.8). En la mayoría de los pacientes con náuseas y vómitos, el episodio presentó una intensidad de leve a moderada. En ensayos clínicos, las náuseas dieron lugar a la suspensión del tratamiento con OFEV® en hasta el 2.1% de los pacientes y los vómitos dieron lugar a la suspensión del tratamiento con OFEV® en hasta el 1.4% de los pacientes.
Si los síntomas persisten a pesar de recibir un tratamiento de soporte adecuado (incluido un tratamiento antiemético), puede que sea necesario reducir la dosis o interrumpir el tratamiento. El tratamiento puede reanudarse a una dosis reducida (100 mg dos veces al día) o a la dosis completa (150 mg dos veces al día). Si persisten los síntomas graves, el tratamiento con OFEV® se debe suspender.
Función hepática: La seguridad y la eficacia de OFEV® no se han estudiado en pacientes con insuficiencia hepática moderada (Child Pugh B) o grave (Child Pugh C). Por lo tanto, el tratamiento con OFEV® no se recomienda en dichos pacientes (ver sección 4.2). Teniendo en cuenta el aumento de la exposición, el riesgo de reacciones adversas puede aumentar en pacientes con insuficiencia hepática leve (Child Pugh A). Los pacientes con insuficiencia hepática leve (Child Pugh A) deben recibir tratamiento con una dosis reducida de OFEV® (ver las secciones 4.2 y 5.2).
Se han observado casos de daño hepático inducido por el fármaco con el tratamiento con nintedanib, incluido daño hepático grave con desenlace mortal. La mayoría de los episodios hepáticos ocurren en los tres primeros meses de tratamiento. Por lo tanto, los niveles de transaminasas hepáticas y bilirrubina se deben evaluar antes de iniciar el tratamiento con OFEV® y durante el primer mes de tratamiento con OFEV®. Después, se debe controlar a los pacientes a intervalos regulares durante los siguientes dos meses de tratamiento y de ahí en adelante de forma periódica, por ejemplo, en cada visita del paciente o siempre que esté clínicamente indicado.
Los aumentos de las enzimas hepáticas (ALT, AST, fosfatasa alcalina (FA) en sangre, gammaglutamiltransferasa (GGT), ver sección 4.8) y de la bilirrubina fueron reversibles en la mayoría de los casos al reducir la dosis o interrumpir el tratamiento. Si se detectan aumentos de las transaminasas (AST o ALT) > 3 veces el LSN, se recomienda reducir la dosis o interrumpir el tratamiento con OFEV®, así como vigilar al paciente de forma estrecha. Una vez que las transaminasas han recuperado los valores basales, el tratamiento con OFEV® se puede reanudar a la dosis completa (150 mg dos veces al día) o reiniciar a una dosis reducida (100 mg dos veces al día), que después se podrá aumentar a la dosis completa (ver sección 4.2). Si se detecta algún aumento en las pruebas hepáticas asociado a signos o síntomas clínicos de daño hepático, como es la ictericia, el tratamiento con OFEV® se debe suspender de forma permanente. Asimismo, es preciso investigar otras posibles causas de los aumentos de las enzimas hepáticas.
Los pacientes con bajo peso corporal (< 65 kg), raza asiática y las mujeres tienen un mayor riesgo de aumento de las enzimas hepáticas. La exposición a nintedanib aumentó de manera lineal con la edad del paciente, lo cual puede también aumentar el riesgo de presentar un aumento de las enzimas hepáticas (ver sección 5.2). Se recomienda realizar un control estrecho en pacientes con estos factores de riesgo.
Función renal: Se han notificado casos de insuficiencia/fallo renal, algunos de ellos con un desenlace mortal, con el uso de nintedanib (ver sección 4.8).
Los pacientes deben ser controlados durante el tratamiento con nintedanib, con especial atención aquellos pacientes que presenten factores de riesgo para insuficiencia/fallo renal. En caso de insuficiencia/fallo renal, se debe considerar el ajuste del tratamiento (ver sección 4.2, Ajustes de la dosis).
Hemorragia: La inhibición del receptor del factor de crecimiento endotelial vascular (VEGFR) puede estar asociada a un aumento del riesgo de hemorragia.
Los pacientes con riesgo conocido de presentar sangrado, incluidos los pacientes con una predisposición hereditaria al sangrado o los pacientes que recibían una dosis completa de anticoagulante, no se incluyeron en los ensayos clínicos. Se han notificado episodios de sangrado no graves y graves, algunos de ellos mortales, en el periodo de poscomercialización (incluidos pacientes con o sin tratamiento anticoagulante u otros medicamentos que podrían causar sangrado). Por lo tanto, estos pacientes solo deben ser tratados con OFEV® si los beneficios esperados superan el riesgo potencial.
Episodios tromboembólicos arteriales: Los pacientes con antecedentes recientes de infarto de miocardio o ictus se excluyeron de los ensayos clínicos.
En los ensayos clínicos, los episodios tromboembólicos arteriales se describieron con poca frecuencia (2.5% con OFEV® frente al 0.7% con placebo en los ensayos INPULSIS; 0.9% con OFEV® frente al 0.9% con placebo en el ensayo INBUILD; 0.7% con OFEV® frente al 0.7% con placebo en el ensayo SENSCIS). En los ensayos INPULSIS, el porcentaje de pacientes que sufrió un infarto de miocardio fue mayor en el grupo tratado con OFEV® (1.6%) que en el grupo tratado con placebo (0.5%), mientras que los efectos adversos que reflejaban una cardiopatía isquémica estuvieron equilibrados entre los grupos de OFEV® y de placebo. En el ensayo INBUILD, se observó infarto de miocardio con una frecuencia baja: 0.9% con OFEV® y 0.9% con placebo.
En el ensayo SENSCIS, se observó infarto de miocardio con una baja frecuencia en el grupo de placebo (0.7%) y no se observó en el grupo de OFEV®. Se deben tomar las debidas precauciones cuando se trate a pacientes con un alto riesgo cardiovascular, incluida una enfermedad de las arterias coronarias conocida. En pacientes que desarrollan signos o síntomas de isquemia miocárdica aguda, se debe valorar la necesidad de interrumpir el tratamiento.
Aneurismas y disecciones arteriales: El uso de inhibidores de la vía VEGF en pacientes con o sin hipertensión puede promover la formación de aneurismas y/o disecciones arteriales. Antes de iniciar el tratamiento con OFEV®, este riesgo se debe evaluar de forma cuidadosa en pacientes con factores de riesgo como hipertensión o antecedentes de aneurisma.
Tromboembolismo venoso: En los ensayos clínicos no se observó ningún aumento del riesgo de sufrir tromboembolismo venoso en los pacientes tratados con nintedanib. Debido al mecanismo de acción de nintedanib, los pacientes pueden presentar un riesgo más alto de sufrir episodios tromboembólicos.
Perforaciones gastrointestinales y colitis isquémica: En los ensayos clínicos, la frecuencia de los casos de perforación fue de hasta el 0.3% en ambos grupos de tratamiento. Debido al mecanismo de acción de nintedanib, los pacientes pueden presentar un riesgo más alto de sufrir perforaciones gastrointestinales. Se han notificado casos de perforaciones gastrointestinales y casos de colitis isquémica, algunos de ellos mortales, durante el periodo de poscomercialización. Se deben tomar las debidas precauciones cuando se trate a pacientes que se hayan sometido anteriormente a una cirugía abdominal, tengan antecedentes de úlceras pépticas o enfermedad diverticular o reciban tratamiento concomitante con corticosteroides o AINEs. El tratamiento con OFEV® se debe iniciar como mínimo 4 semanas después de una cirugía abdominal. El tratamiento con OFEV® se debe suspender de forma permanente en el caso de pacientes que desarrollen una perforación gastrointestinal o una colitis isquémica. De forma excepcional, se puede reanudar el tratamiento con OFEV® tras la resolución completa de la colitis isquémica y tras una evaluación meticulosa del estado del paciente y de otros factores de riesgo.
Proteinuria en rango nefrótico y microangiopatía trombótica: En el periodo de poscomercialización, se han notificado muy pocos casos de proteinuria en rango nefrótico con o sin disfunción renal. Los hallazgos histológicos en casos individuales eran compatibles con microangiopatía glomerular con o sin trombos renales. Se ha observado la desaparición de los síntomas tras suspender el tratamiento con OFEV®, con proteinuria residual en algunos casos.
En pacientes que desarrollan signos o síntomas de síndrome nefrótico se debe valorar la necesidad de interrumpir el tratamiento. Los inhibidores de la vía del VEGF se han asociado a microangiopatía trombótica (MAT), incluido un número muy bajo de informes de casos en relacionados con nintedanib. Si se observan hallazgos analíticos o clínicos asociados a MAT en un paciente tratado con nintedanib, se debe interrumpir el tratamiento con nintedanib y se debe llevar a cabo una evaluación meticulosa relacionada con la MAT.
Hipertensión: La administración de OFEV® puede aumentar la tensión arterial. La tensión arterial sistémica se debe medir de forma periódica y siempre que esté indicado clínicamente.
Hipertensión pulmonar: Los datos sobre el uso de OFEV® en pacientes con hipertensión pulmonar son limitados.
Los pacientes con hipertensión pulmonar significativa (índice cardiaco ≤ 2 l/min/m² o epoprostenol/treprostinilo parenteral o insuficiencia cardiaca derecha significativa) fueron excluidos de los ensayos INBUILD y SENSCIS.
OFEV® no debe usarse en pacientes con hipertensión pulmonar grave. Se recomienda una vigilancia estrecha en pacientes con hipertensión pulmonar leve y moderada.
Complicaciones en la cicatrización de las heridas: En los ensayos clínicos, no se observó un aumento de la frecuencia de complicaciones en la cicatrización de las heridas. Teniendo en cuenta su mecanismo de acción, nintedanib puede dificultar la cicatrización de las heridas. No se realizaron estudios específicos para evaluar el efecto de nintedanib sobre la curación de las heridas. Así pues, el tratamiento con OFEV® solo se debe iniciar o, en el caso de una interrupción perioperatoria, reanudar basándose en la evaluación clínica de una adecuada cicatrización de las heridas.
Administración conjunta con pirfenidona: En un estudio farmacocinético específico, se investigó el tratamiento conjunto de nintedanib con pirfenidona en pacientes con FPI. De acuerdo con estos resultados, no existe evidencia de una interacción farmacocinética relevante entre nintedanib y pirfenidona cuando se administran en combinación (ver sección 5.2). Dada la similitud de los perfiles de seguridad de ambos medicamentos, cabe prever reacciones adversas aditivas, incluidas reacciones adversas gastrointestinales y hepáticas. No se ha establecido el balance beneficio -riesgo del tratamiento conjunto con pirfenidona.
Efecto en el intervalo QT: En el programa de ensayos clínicos realizados, no se observó ninguna evidencia de una prolongación del intervalo QT al administrar nintedanib (ver sección 5.1). Como se sabe que algunos otros inhibidores de la tirosina cinasa ejercen un efecto sobre el intervalo QT, se debe tomar precaución cuando se administre nintedanib a pacientes que puedan desarrollar una prolongación del intervalo QTc.
Reacción alérgica: Se sabe que los productos alimenticios con soja provocan reacciones alérgicas, incluido un choque anafiláctico grave en personas con alergia a la soja. Los pacientes con una alergia conocida a la proteína del cacahuete presentan un mayor riesgo de desarrollar reacciones graves a los preparados de soja.
• Cáncer de pulmón no microcítico (CPNM):
Trastornos gastrointestinales: La diarrea fue la reacción adversa gastrointestinal descrita con más frecuencia y se produjo en estrecha relación temporal con la administración de docetaxel (ver sección 4.8). En el ensayo clínico LUME- Lung 1 (ver sección 5.1), la mayoría de los pacientes presentó diarrea de leve a moderada.
En el periodo de poscomercialización, se han notificado casos graves de diarrea causantes de deshidratación y trastornos electrolíticos con nintedanib. La diarrea se debe tratar en cuanto aparezcan los primeros síntomas con una adecuada hidratación y la administración de medicamentos antidiarreicos, como la loperamida; además, puede requerir la interrupción, la reducción de la dosis o la suspensión del tratamiento con OFEV® (ver sección 4.2).
Las náuseas y los vómitos, en su mayoría de grado leve a moderado, fueron reacciones adversas gastrointestinales descritas con frecuencia (ver sección 4.8). A pesar de administrar un tratamiento de soporte adecuado, puede ser necesario interrumpir, reducir la dosis o suspender el tratamiento con OFEV® (ver sección 4.2). Los tratamientos de soporte para las náuseas y los vómitos incluyen, entre otros, medicamentos antieméticos como son los glucocorticoides, los antihistamínicos o los antagonistas del receptor 5-HT3, así como una hidratación adecuada.
En el caso de producirse una deshidratación, es preciso administrar electrolitos y líquidos. Si se producen efectos adversos gastrointestinales relevantes, es necesario controlar los niveles plasmáticos de electrolitos. Puede requerir la interrupción, la reducción de la dosis o la suspensión del tratamiento con OFEV® (ver sección 4.2).
Neutropenia y sepsis: En los pacientes tratados con OFEV® en combinación con docetaxel la frecuencia de neutropenia de grado ≥ 3 según los CTCAE fue mayor que en los pacientes tratados con docetaxel en monoterapia. Se han observado complicaciones asociadas, como la sepsis o la neutropenia febril (incluidos casos mortales).
Los recuentos celulares sanguíneos se deben controlar durante el tratamiento, en particular durante el tratamiento combinado con docetaxel. En el caso de pacientes que reciben el tratamiento de nintedanib en combinación con docetaxel es necesario realizar un control frecuente de los recuentos celulares sanguíneos completos al principio de cada ciclo de tratamiento y alrededor de la fecha nadir, así como siempre que esté clínicamente indicado después de administrar el último ciclo de la combinación.
Función hepática: Teniendo en cuenta el aumento de la exposición, el riesgo de efectos adversos puede aumentar en pacientes con insuficiencia hepática leve (Child Pugh A; ver las secciones 4.2 y 5.2). Se dispone de datos limitados de seguridad de 9 pacientes con carcinoma hepatocelular e insuficiencia hepática moderada clasificados como Child Pugh B. Aunque no se notificaron hallazgos inesperados de seguridad en estos pacientes, los datos son insuficientes para respaldar una recomendación para el tratamiento de pacientes con insuficiencia hepática moderada. La eficacia de nintedanib no se ha investigado en pacientes con insuficiencia hepática moderada (Child Pugh B). La seguridad, la eficacia y la farmacocinética de nintedanib no se han estudiado en pacientes con insuficiencia hepática grave (Child Pugh C). El tratamiento con OFEV® no se recomienda en pacientes con insuficiencia hepática moderada o grave (ver sección 4.2).
Se han observado casos de daño hepático inducido por el fármaco con el tratamiento con nintedanib, incluido daño hepático grave con desenlace mortal. Los aumentos de las enzimas hepáticas (ALT, AST, FA, gamma-glutamiltransferasa (GGT)) y de la bilirrubina fueron reversibles en la mayoría de los casos al reducir la dosis o interrumpir el tratamiento.
Los niveles de transaminasas, FA y bilirrubina se deben evaluar antes de iniciar el tratamiento combinado de OFEV® más docetaxel. Los valores se deben controlar cuando esté clínicamente indicado o periódicamente durante el tratamiento, es decir, en la fase de combinación con docetaxel al principio de cada ciclo de tratamiento y una vez al mes en el caso de que el tratamiento con OFEV® se continúe como monoterapia después de suspender el tratamiento con docetaxel.
Si se detectan aumentos relevantes de las enzimas hepáticas, puede ser preciso reducir la dosis, interrumpir o suspender el tratamiento con OFEV® (ver sección 4.2). Asimismo, es preciso investigar otras posibles causas de los aumentos de las enzimas hepáticas y tomar las medidas correspondientes que sean necesarias. Si se producen cambios específicos en los valores hepáticos (AST/ALT > 3 x LSN; bilirrubina total ≥ 2 x LSN y FA < 2 x LSN), el tratamiento con OFEV® se debe interrumpir. A menos que se haya constatado que existe otra causa para ello, OFEV® se debe suspender de forma permanente (ver sección 4.2).
Los pacientes con bajo peso corporal (< 65 kg), raza asiática y las mujeres tienen un mayor riesgo de aumento de las enzimas hepáticas. La exposición a nintedanib aumentó de manera lineal con la edad del paciente, lo cual puede también aumentar el riesgo de presentar un aumento de las enzimas hepáticas (ver sección 5.2). Se recomienda realizar un control estrecho en pacientes con estos factores de riesgo.
Función renal: Se han notificado casos de insuficiencia/fallo renal, algunos de ellos con un desenlace mortal, con el uso de nintedanib (ver sección 4.8).
Los pacientes deben ser controlados durante el tratamiento con nintedanib, con especial atención aquellos que presenten factores de riesgo para insuficiencia/fallo renal. En caso de insuficiencia/fallo renal, se debe considerar el ajuste del tratamiento (ver sección 4.2 Ajustes de la dosis).
Hemorragia: La inhibición del receptor del factor de crecimiento endotelial vascular (VEGFR) puede estar asociada a un aumento del riesgo de hemorragia. En el ensayo clínico (LUME-Lung 1; ver sección 5.1) realizado con OFEV®, la frecuencia de hemorragia fue similar en los dos grupos de tratamiento (ver sección 4.8). La epistaxis de leve a moderada representó el episodio hemorrágico más frecuente. La mayoría de los episodios hemorrágicos mortales estuvieron asociados al tumor. No se produjeron desequilibrios de hemorragias respiratorias o mortales y no se comunicaron hemorragias intracraneales.
Los pacientes con hemorragia pulmonar reciente (> 2.5 ml de sangre roja), así como los pacientes con tumores centralmente localizados con signos radiográficos de invasión local de los vasos sanguíneos principales o signos radiográficos de tumores cavitados o necróticos, se excluyeron de los ensayos clínicos. Por lo tanto, no se recomienda tratar a estos pacientes con OFEV®.
Se han notificado episodios de sangrado no graves y graves, algunos de ellos mortales, en el periodo de poscomercialización, incluidos pacientes con o sin tratamiento anticoagulante u otros medicamentos que podrían causar sangrado (para los datos de ensayos clínicos, ver también “Anticoagulación terapéutica” más abajo). En caso de sangrado, se debe considerar el ajuste de la dosis, la interrupción o la suspensión del tratamiento basándose en la evaluación clínica (ver sección 4.2). Los episodios de sangrado durante la poscomercialización afectan, entre otros, a órganos de los sistemas gastrointestinal, respiratorio y nervioso central, si bien los más frecuentes son respiratorios.
Anticoagulación terapéutica: No existen datos de ensayos clínicos relativos a pacientes con una predisposición hereditaria al sangrado ni relativos a pacientes que reciben una dosis completa de anticoagulante antes de iniciar el tratamiento con OFEV® (para la experiencia de poscomercialización, ver “Hemorragia” más arriba). En el caso de pacientes que seguían un tratamiento crónico con bajas dosis de heparinas de bajo peso molecular o de ácido acetilsalicílico, no se observó un aumento de la frecuencia de hemorragias. Los pacientes que desarrollaron episodios tromboembólicos durante el tratamiento y que requirieron un tratamiento anticoagulante pudieron continuar el tratamiento con OFEV® y no mostraron un aumento en la frecuencia de episodios hemorrágicos. Los pacientes que tomen anticoagulantes de forma conjunta, tales como la warfarina o el fenprocumón, se deben controlar de forma regular para ver si se producen cambios en el tiempo de protrombina, el Ratio Internacional Normalizado (INR) y los episodios hemorrágicos clínicos.
Metástasis cerebral:
Metástasis cerebral estable: No se observó un aumento de la frecuencia de hemorragia cerebral en pacientes con metástasis cerebrales
pretratadas de forma adecuada que se habían mantenido estables durante ≥ 4 semanas antes de comenzar el tratamiento con OFEV®. No obstante, es preciso controlar de forma estrecha a tales pacientes para ver si se producen signos y síntomas de hemorragia cerebral.
Metástasis cerebral activa: Los pacientes con metástasis cerebral activa se excluyeron de los ensayos clínicos, por lo que no se recomienda tratarlos con OFEV®.
Tromboembolismo venoso: Los pacientes tratados con OFEV® presentan un mayor riesgo de sufrir un tromboembolismo venoso, incluidas la embolia pulmonar y la trombosis venosa profunda. Es preciso controlar de forma estrecha a tales pacientes para ver si se producen episodios tromboembólicos. Se debe tener precaución especialmente en pacientes con factores de riesgo adicionales de episodios tromboembólicos. El tratamiento con OFEV® se debe suspender en el caso de pacientes con reacciones tromboembólicas venosas que puedan poner en peligro su vida.
Episodios tromboembólicos arteriales: La frecuencia de episodios tromboembólicos arteriales fue similar entre los dos grupos de tratamiento en el ensayo fase 3 1199.13 (LUME-Lung 1). Los pacientes con antecedentes recientes de infarto de miocardio o ictus se excluyeron de este ensayo. No obstante, se observó un aumento de la frecuencia de episodios tromboembólicos arteriales en pacientes con fibrosis pulmonar idiopática (FPI) tratados con nintedanib en monoterapia. Tome las debidas precauciones cuando trate a pacientes con un alto riesgo cardiovascular, incluida una enfermedad de las arterias coronarias conocida. En pacientes que desarrollan signos o síntomas de isquemia miocárdica aguda, se debe valorar la necesidad de interrumpir el tratamiento.
Aneurismas y disecciones arteriales: El uso de inhibidores de la vía VEGF en pacientes con o sin hipertensión puede promover la formación de aneurismas y/o disecciones arteriales. Antes de iniciar el tratamiento con OFEV®, este riesgo se debe evaluar de forma cuidadosa en pacientes con factores de riesgo como hipertensión o antecedentes de aneurisma.
Perforaciones gastrointestinales: La frecuencia de perforaciones gastrointestinales fue similar entre los grupos de tratamiento del ensayo clínico. No obstante, teniendo en cuenta el mecanismo de acción, los pacientes tratados con OFEV® pueden presentar un mayor riesgo de sufrir perforaciones gastrointestinales. Se han notificado casos de perforaciones gastrointestinales, algunas de ellas mortales, durante el periodo de poscomercialización. Se deberán tomar las debidas precauciones cuando se trate a pacientes que se hayan sometido anteriormente a una cirugía abdominal o que presenten antecedentes recientes de perforación de las vísceras huecas. Así pues, el tratamiento con OFEV® se debe iniciar como mínimo 4 semanas después de una cirugía mayor. El tratamiento con OFEV® se debe suspender de forma permanente en el caso de pacientes que desarrollen una perforación gastrointestinal.
Proteinuria en rango nefrótico: En el periodo de poscomercialización, se han notificado muy pocos casos de proteinuria en el rango nefrótico. Los hallazgos histológicos en casos individuales eran compatibles con microangiopatía glomerular con o sin trombos renales. Se ha observado la desaparición de los síntomas tras suspender el tratamiento con OFEV®. En pacientes que desarrollan signos o síntomas de síndrome nefrótico se debe valorar la necesidad de interrumpir el tratamiento.
Complicaciones en la cicatrización de las heridas: Teniendo en cuenta su mecanismo de acción, nintedanib puede dificultar la cicatrización de las heridas. En el ensayo LUME-Lung 1 no se observó un aumento de la frecuencia de complicaciones en la cicatrización de las heridas. No se realizaron ensayos específicos para evaluar el efecto de nintedanib sobre la curación de las heridas. Así pues, el tratamiento con OFEV® solo se debe iniciar o, en el caso de una interrupción perioperatoria, reanudar basándose en la evaluación clínica de una adecuada cicatrización de las heridas.
Efectos en el intervalo QT: No se observó ninguna prolongación del intervalo QT para nintedanib en el programa del ensayo clínico (ver sección 5.1).
Como se sabe que otros inhibidores de la tirosina cinasa ejercen un efecto sobre el intervalo QT, se deberán tomar las debidas precauciones cuando se administre nintedanib a pacientes que puedan desarrollar una prolongación del intervalo QTc.
Reacción alérgica: Se sabe que los productos alimenticios con soja provocan reacciones alérgicas, incluido un choque anafiláctico grave en personas con alergia a la soja. Los pacientes con una alergia conocida a la proteína del cacahuete presentan un mayor riesgo de desarrollar reacciones graves a los preparados de soja.
Poblaciones especiales: En el ensayo 1199.13 (LUME-Lung 1), la frecuencia de efectos adversos graves en pacientes tratados con nintedanib más docetaxel fue mayor en los pacientes con un peso corporal inferior a 50 kg que en los pacientes con un peso ≥ 50 kg; no obstante, el número de pacientes con un peso corporal inferior a 50 kg fue reducido. Así pues, se recomienda realizar un control estrecho en el caso de pacientes que pesen < 50 kg.
FERTILIDAD, EMBARAZO Y LACTANCIA:
• Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
Mujeres en edad fértil / anticoncepción: Nintedanib puede causar daño fetal en humanos (ver sección 5.3). Se debe advertir a las mujeres en edad fértil que estén siendo tratadas con OFEV® que eviten quedarse embarazadas mientras reciban dicho tratamiento y que utilicen métodos anticonceptivos altamente efectivos al inicio del tratamiento, durante el mismo y al menos 3 meses después de la última dosis de OFEV®. Nintedanib no afecta de manera relevante a la exposición plasmática de etinilestradiol y levonorgestrel (ver sección 5.2). La eficacia de los anticonceptivos hormonales orales puede verse reducida por los vómitos y/o la diarrea u otras situaciones en las que la absorción pueda verse afectada. Se debe advertir a las mujeres que tomen anticonceptivos hormonales orales y que experimenten estas situaciones que utilicen un método anticonceptivo alternativo altamente efectivo.
Embarazo: No existe información sobre el uso de OFEV® en mujeres embarazadas, pero los estudios preclínicos de este principio activo en animales han mostrado toxicidad para la reproducción (ver sección 5.3). Como nintedanib puede causar daño fetal también en humanos, no se debe utilizar durante el embarazo (ver sección 4.3) y se deben realizar pruebas de embarazo antes y durante el tratamiento con OFEV®, según proceda.
Se debe aconsejar a las pacientes que informen a su médico o farmacéutico si se quedan embarazadas durante el tratamiento con OFEV®.
Si una paciente se queda embarazada mientras está recibiendo tratamiento con OFEV®, se debe suspender el tratamiento y la paciente debe recibir la información correspondiente sobre el riesgo potencial que existe para el feto.
Lactancia: No se dispone de información relativa a la excreción de nintedanib y sus metabolitos en la leche materna. Los estudios preclínicos mostraron que pequeñas cantidades de nintedanib y sus metabolitos (≤ 0.5% de la dosis administrada) se excretaban en la leche de las ratas lactantes. No se puede excluir el riesgo para los recién nacidos/ lactantes. Debe interrumpirse la lactancia durante el tratamiento con OFEV®.
Fertilidad: Basándose en las investigaciones preclínicas, no hay evidencia de que afecte a la fertilidad masculina (ver sección 5.3). Teniendo en cuenta los estudios de toxicidad subcrónica y crónica, no hay evidencia de que la fertilidad femenina en ratas se vea afectada a un nivel de exposición sistémica similar al de la dosis humana máxima recomendada de 150 mg dos veces al día (ver sección 5.3).
• Cáncer de pulmón no microcítico (CPNM):
Mujeres en edad fértil / anticoncepción: Nintedanib puede causar daño fetal en humanos (ver sección 5.3). Se debe advertir a las mujeres en edad fértil para que eviten quedarse embarazadas mientras reciban tratamiento con OFEV® y para que utilicen métodos anticonceptivos altamente efectivos al inicio del tratamiento, durante el mismo y al menos 3 meses después de la última dosis de OFEV®. Nintedanib no afecta de manera relevante a la exposición plasmática de etinilestradiol y levonorgestrel (ver sección 5.2). La eficacia de los anticonceptivos hormonales orales puede verse reducida por los vómitos y/o la diarrea u otras situaciones en las que la absorción pueda verse afectada. Se debe advertir a las mujeres que tomen anticonceptivos hormonales orales y que experimenten estas situaciones que utilicen un método anticonceptivo alternativo altamente efectivo.
Embarazo: No existe información sobre el uso de OFEV® en mujeres embarazadas, pero los estudios preclínicos de este fármaco con animales han mostrado toxicidad para la reproducción (ver sección 5.3). Como nintedanib puede causar daño fetal también en humanos, no se debe utilizar durante el embarazo a menos que el estado clínico requiera dicho tratamiento. Es necesario realizar una prueba de embarazo, como mínimo, antes del tratamiento con OFEV®.
Se debe aconsejar a las pacientes que informen a su médico o farmacéutico si se quedan embarazadas durante el tratamiento con OFEV®.
Si una paciente se queda embarazada mientras está recibiendo tratamiento con OFEV®, debe recibir la información correspondiente sobre el riesgo potencial que existe para el feto. En este caso, se debe valorar la necesidad de finalizar el tratamiento con OFEV®.
Lactancia: No se dispone de información relativa a la excreción de nintedanib y sus metabolitos en la leche materna. Los estudios preclínicos mostraron que pequeñas cantidades de nintedanib y sus metabolitos (≤ 0.5% de la dosis administrada) se secretaban en la leche de las ratas lactantes. No se puede excluir el riesgo para los niños lactantes. Se debe interrumpir la lactancia durante el tratamiento con OFEV®.
Fertilidad: Basándose en las investigaciones preclínicas, no hay pruebas de que afecte a la fertilidad masculina (ver sección 5.3). No se dispone de datos con humanos o animales sobre los potenciales efectos de nintedanib en la fertilidad femenina.
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS: Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES), enfermedades pulmonares intersticiales (EPI) y Cáncer de pulmón no microcítico (CPNM)
La influencia de OFEV® sobre la capacidad para conducir y utilizar máquinas es pequeña. Se aconsejará a los pacientes que tomen las debidas precauciones cuando conduzcan o utilicen máquinas si están siguiendo un tratamiento con OFEV®.
REACCIONES ADVERSAS:
Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
Resumen del perfil de seguridad: En los ensayos clínicos y durante la experiencia de poscomercialización, las reacciones adversas a medicamentos (RAMs) asociadas al uso de nintedanib descritas con más frecuencia incluyeron diarrea, náuseas y vómitos, dolor abdominal, apetito disminuido, pérdida de peso y aumento de las enzimas hepáticas.
Para obtener información sobre el manejo de reacciones adversas específicas, ver sección 4.4.
Tabla de reacciones adversas: La tabla 3 incluye un resumen de las reacciones adversas a medicamentos (RAM) según la clasificación de órganos del sistema MedDRA y la categoría de frecuencia., usando la siguiente convención: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000), muy raras (< 1/10.000), frecuencia no conocida (no se puede estimar a partir de los datos disponibles).
|
Tabla 3 Resumen de las RAMs por categoría de frecuencia |
|||
|
Frecuencia |
|||
|
Término preferido del sistema de clasificación de órganos |
Fibrosis pulmonar idiopática |
Otras EPI fibrosantes crónicas con un fenotipo progresivo |
Enfermedad pulmonar intersticial asociada a esclerosis sistémica |
|
Trastornos de la sangre y del sistema linfático |
|||
|
Trombocitopenia |
Poco frecuente |
Poco frecuente |
Poco frecuente |
|
Trastornos del metabolismo y de la nutrición |
|||
|
Pérdida de peso |
Frecuente |
Frecuente |
Frecuente |
|
Apetito disminuido |
Frecuente |
Muy frecuente |
Frecuente |
|
Deshidratación |
Poco frecuente |
Poco frecuente |
Frecuencia no conocida |
|
Trastornos cardiacos |
|||
|
Infarto de miocardio |
Poco frecuente |
Poco frecuente |
Frecuencia no conocida |
|
Trastornos vasculares |
|||
|
Sangrado (ver sección 4.4) |
Frecuente |
Frecuente |
Frecuente |
|
Hipertensión |
Poco frecuente |
Frecuente |
Frecuente |
|
Aneurismas y disecciones arteriales |
Frecuencia no conocida |
Frecuencia no conocida |
Frecuencia no conocida |
|
Trastornos gastrointestinales |
|||
|
Diarrea |
Muy frecuente |
Muy frecuente |
Muy frecuente |
|
Náuseas |
Muy frecuente |
Muy frecuente |
Muy frecuente |
|
Dolor abdominal |
Muy frecuente |
Muy frecuente |
Muy frecuente |
|
Vómitos |
Frecuente |
Muy frecuente |
Muy frecuente |
|
Pancreatitis |
Poco frecuente |
Poco frecuente |
Frecuencia no conocida |
|
Colitis |
Poco frecuente |
Poco frecuente |
Poco frecuente |
|
Trastornos hepatobiliares |
|||
|
Daño hepático inducido por el fármaco |
Poco frecuente |
Frecuente |
Poco frecuente |
|
Aumento de las enzimas hepáticas |
Muy frecuente |
Muy frecuente |
Muy frecuente |
|
Aumento de la alanina aminotransferase (ALT) |
Frecuente |
Muy frecuente |
Frecuente |
|
Aumento de la aspartato aminotransferasa (AST) |
Frecuente |
Frecuente |
Frecuente |
|
Aumento de la gamma-glutamil-transferasa (GGT) |
Frecuente |
Frecuente |
Frecuente |
|
Hiperbilirrubinemia |
Poco frecuente |
Poco frecuente |
Frecuencia no conocida |
|
Aumento de la fosfatasa alcalina (FA) en sangre |
Poco frecuente |
Frecuente |
Frecuente |
|
Trastornos de la piel y del tejido subcutáneo |
|||
|
Erupción |
Frecuente |
Frecuente |
Poco frecuente |
|
Prurito |
Poco frecuente |
Poco frecuente |
Poco frecuente |
|
Alopecia |
Poco frecuente |
Poco frecuente |
Frecuencia no conocida |
|
Trastornos renales y urinarios |
|||
|
Insuficiencia renal (ver sección 4.4) |
Frecuencia no conocida |
Frecuencia no conocida |
Poco frecuente |
|
Proteinuria |
Poco frecuente |
Poco frecuente |
Frecuencia no conocida |
|
Trastornos del sistema nervioso |
|||
|
Cefalea |
Frecuente |
Frecuente |
Frecuente |
Descripción de reacciones adversas específicas:
Diarrea: En los ensayos clínicos (ver sección 5.1), la diarrea fue el acontecimiento gastrointestinal más frecuente notificado. En la mayoría de los pacientes, el episodio fue de intensidad leve a moderada. Más de dos tercios de los pacientes que sufrieron diarrea describieron que ésta había aparecido por primera vez durante los primeros tres meses de tratamiento. En la mayoría de los pacientes, los episodios se trataron con un tratamiento antidiarreico, una reducción de la dosis o la interrupción del tratamiento (ver sección 4.4). En la Tabla 4 se muestra una descripción general de los episodios de diarrea notificados en los ensayos clínicos:
|
Tabla 4 Diarrea en los ensayos clínicos a lo largo de 52 semanas |
||||||
|
INPULSIS |
INBUILD |
SENSCIS |
||||
|
Placebo |
OFEV® |
Placebo |
OFEV® |
Placebo |
OFEV® |
|
|
Diarrea |
18.4% |
62.4% |
23.9% |
66.9% |
31.6% |
75.7% |
|
Diarrea grave |
0.5% |
3.3.% |
0.9% |
2.4% |
1.0% |
4.2% |
|
Diarrea que da lugar a la reducción de la dosis de OFEV® |
0% |
10.7% |
0.9% |
16.0% |
1.0% |
22.2% |
|
Diarrea que da lugar a la suspensión de OFEV® |
0.2% |
4.4% |
0.3% |
5.7% |
0.3% |
6.9% |
Aumento de las enzimas hepáticas: En los ensayos INPULSIS, el aumento de las enzimas hepáticas (ver sección 4.4) se describió en el 13.6% frente al 2.6% de los pacientes tratados con OFEV® y placebo, respectivamente. En el ensayo INBUILD, se notificó aumento de las enzimas hepáticas en el 22.6% frente al 5.7% de los pacientes tratados con OFEV® y placebo, respectivamente. En el ensayo SENSCIS, se notificó aumento de las enzimas hepáticas en el 13.2% frente al 3.1% de los pacientes tratados con OFEV® y placebo, respectivamente. Los aumentos de las enzimas hepáticas fueron reversibles y no estuvieron asociados a una enfermedad hepática clínicamente manifiesta.
Para obtener más información sobre las poblaciones especiales y sobre las medidas y ajustes de dosis recomendados en caso de diarrea y aumento de las enzimas hepáticas, ver las secciones 4.4 y 4.2 respectivamente.
Sangrado: En los ensayos clínicos, la frecuencia de pacientes que experimentaron sangrado fue ligeramente mayor en los pacientes tratados con OFEV® o similar entre los grupos de tratamiento (10.3% con OFEV® frente al 7.8% con placebo en los ensayos INPULSIS; 11.1% con OFEV® frente al 12.7% con placebo en el ensayo INBUILD; 11.1% con OFEV® frente al 8.3% con placebo en el ensayo SENSCIS). El acontecimiento de sangrado notificado más frecuente fue epistaxis no grave. Se produjeron acontecimientos de sangrado graves con una frecuencia baja en los dos grupos de tratamiento (1.3% con OFEV® frente al 1.4% con placebo en los ensayos INPULSIS; 0.9% con OFEV® frente al 1.5% con placebo en el ensayo INBUILD; 1.4% con OFEV® frente al 0.7% con placebo en el ensayo SENSCIS). Los acontecimientos de sangrado del periodo de poscomercialización afectan, entre otros, al aparato gastrointestinal, al aparato respiratorio y al sistema nervioso central, siendo los más frecuentes los acontecimientos gastrointestinales (ver sección 4.4).
Proteinuria: En los ensayos clínicos, la frecuencia de pacientes que experimentaron proteinuria fue baja y similar entre los grupos de tratamiento (0.8% con OFEV® frente al 0.5% con placebo en los ensayos INPULSIS; 1.5% con OFEV® frente al 1.8% con placebo en el ensayo INBUILD; 1.0% con OFEV® frente al 0.0% con placebo en el ensayo SENSCIS). No se ha notificado síndrome nefrótico en los ensayos clínicos. En el periodo de poscomercialización, Se han notificado muy pocos casos de proteinuria en rango nefrótico con o sin disfunción renal. Los hallazgos histológicos en casos individuales eran compatibles con microangiopatía glomerular con o sin trombos renales. Se ha observado la desaparición de los síntomas tras suspender el tratamiento con de OFEV®, con proteinuria residual en algunos casos. En pacientes que desarrollan signos o síntomas de síndrome nefrótico se debe valorar la necesidad de interrumpir el tratamiento (ver sección 4.4).
Cáncer de pulmón no microcítico (CPNM):
Resumen del perfil de seguridad: Los datos de seguridad que se incluyen en los apartados siguientes se basan en el ensayo pivotal fase 3, aleatorizado, doble ciego y global 1199.13 (LUME-Lung 1), que comparó el tratamiento con nintedanib más docetaxel con el de placebo más docetaxel en pacientes con un CPNM localmente avanzado, metastásico o recurrente después de una quimioterapia de primera línea, y en los datos observados durante el periodo de poscomercialización. Las reacciones adversas a medicamentos (RAMs) específicas de nintedanib que se describieron con más frecuencia fueron diarrea, aumento de los valores de las enzimas hepáticas (ALT y AST) y vómitos. La Tabla 5 incluye un resumen de las reacciones adversas según el sistema de clasificación de órganos. Para obtener información sobre el manejo de reacciones adversas específicas, ver sección 4.4. A continuación se incluye información sobre reacciones adversas específicas observadas en el ensayo LUME-Lung 1.
Tabla de reacciones adversas: La Tabla 5 resume la frecuencia de las reacciones adversas que se describieron en el ensayo pivotal LUME-Lung 1 para pacientes con un CPNM de histología adenocarcinoma (n = 320) o a partir del periodo de poscomercialización.Para clasificar las RAMs por frecuencia se utilizan los siguientes términos: muy frecuentes (≥ 1/10), frecuentes (≥ 1/100 a < 1/10), poco frecuentes (≥ 1/1.000 a < 1/100), raras (≥ 1/10.000 a < 1/1.000), muy raras (< 1/10.000), frecuencia no conocida (no se puede estimar a partir de los datos disponibles).
Dentro de cada grupo de frecuencia las reacciones adversas se presentan por orden descendente de gravedad.
|
Tabla 5 Resumen de RAMs por categoría de frecuencia |
||||
|
Sistema de clasificación de órganos |
Muy frecuentes (≥1/10) |
Frecuentes (≥1/100<1/10) |
Poco frecuentes (≥1/1.000 <1/100) |
Frecuencia no conocida |
|
Infecciones e infestaciones |
Neutropenia febril, abscesos, Sepsis |
|||
|
Trastornos de la sangre y del sistema linfático |
Neutropenia (incluye neutropenia febril) |
Trombocitopenia |
||
|
Trastornos del metabolismo y de la nutrición |
Apetito disminuido, Desequilibrio electrolítico |
Deshidratación, Disminución del peso |
||
|
Trastornos del sistema nervioso |
Neuropatía periférica |
Cefalea1) |
||
|
Trastornos cardiacos |
Infarto de miocardio (ver sección 4.4) |
|||
|
Trastornos vasculares |
Hemorragia1) (ver sección 4.4) |
Tromboembolia venosa3) Hipertensión |
Aneurismas y disecciones arteriales |
|
|
Trastornos gastrointestinales |
Diarrea, Vómitos, Náuseas, Dolor abdominal |
Perforación1) Pancreatitis2) |
Colitis |
|
|
Trastornos hepatobiliares |
Alanina aminotransferasa elevada (ALT) Aspartato aminotransferasa (AST) elevada Aumento de la fosfatasa alcalina (FA) en sangre |
Hiperbilirrubinemia, Gamma- glutamiltransferasa (GGT) elevada |
Daño hepático inducido por el fármaco |
|
|
Trastornos de la piel y del tejido subcutáneo |
Mucositis (incluida estomatitis), Erupción Alopecia1) |
Prurito |
||
|
Trastornos renales y urinarios |
Proteinuria1) |
Fallo renal (ver sección 4.4) |
||
|
1) En los ensayos clínicos, la frecuencia no fue mayor en los pacientes tratados con nintedanib más docetaxel que en los pacientes tratados con placebo más docetaxel. 2) Se han notificado episodios de pancreatitis en pacientes que tomaban nintedanib para el tratamiento de la FPI y el CPNM. La mayoría de estos episodios se notificaron en pacientes con la indicación de FPI. 3) Se han notificado casos de embolia pulmonar |
||||
Descripción de reacciones adversas seleccionadas:
Diarrea: La diarrea se produjo en el 43.4% (≥ grado 3: 6.3%) de los pacientes con adenocarcinoma del grupo tratado con nintedanib. La mayoría de las reacciones adversas se produjeron en estrecha relación temporal con la administración de docetaxel. La mayor parte de los pacientes se recuperó de la diarrea después de la interrupción del tratamiento, un tratamiento antidiarreico y una reducción de la dosis de nintedanib.
Para conocer las medidas y los ajustes de la dosis recomendados en los casos de diarrea, ver las secciones 4.4 y 4.2 respectivamente.
Aumento de las enzimas hepáticas e hiperbilirrubinemia: Las reacciones adversas relacionadas con el hígado se produjeron en el 42.8% de los pacientes tratados con nintedanib. Aproximadamente un tercio de estos pacientes presentaron reacciones adversas relacionadas con el hígado con una intensidad de grado ≥ 3. En pacientes con parámetros hepáticos elevados, la reducción gradual de la dosis fue la medida adecuada y la suspensión del tratamiento solo fue necesaria en el 2.2% de los pacientes. Los aumentos de los parámetros hepáticos fueron reversibles en la mayoría de los pacientes.
Para obtener información sobre las poblaciones especiales y sobre las medidas y ajustes de la dosis recomendados en los casos de aumento de las enzimas hepáticas y de la bilirrubina, ver las secciones 4.4 y 4.2 respectivamente.
Neutropenia, neutropenia febril y sepsis: La sepsis y la neutropenia febril se han descrito como complicaciones asociadas a la neutropenia. Los índices de sepsis (1.3%) y de neutropenia febril (7.5%) aumentaron más en el grupo tratado con nintedanib que en el grupo tratado con placebo. Es importante controlar los recuentos celulares sanguíneos del paciente durante el tratamiento, en particular durante el tratamiento combinado con docetaxel (ver sección 4.4).
Hemorragia: En el periodo de poscomercialización, se han notificado episodios de sangrado no graves y graves, algunos de ellos mortales, incluidos pacientes con o sin tratamiento anticoagulante u otros medicamentos que podrían causar sangrado. Los episodios de sangrado durante la poscomercialización afectan, entre otros, a órganos de los sistemas gastrointestinal, respiratorio y nervioso central, si bien los más frecuentes son respiratorios (ver también sección 4.4).
Perforación: Como cabe esperar por su mecanismo de acción, en pacientes tratados con nintedanib se puede producir una perforación. Sin embargo, la frecuencia de pacientes con perforación gastrointestinal fue baja.
Neuropatía periférica: También se sabe que la neuropatía periférica se produce en el tratamiento con docetaxel. La neuropatía periférica se describió en el 16.5% de los pacientes del grupo tratado con placebo y en el 19.1% de los pacientes del grupo de nintedanib.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN:
Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
Glicoproteína-P (gp-P): Nintedanib es un sustrato de la gp-P (ver sección 5.2). La administración conjunta con ketoconazol, un potente inhibidor de la gp-P, aumentó la exposición a nintedanib 1.61 veces basándose en el AUC y 1.83 veces basándose en la Cmax en un estudio específico de interacción farmacológica. En un estudio de interacción farmacológica con rifampicina, un potente inductor de la gp-P, la exposición a nintedanib disminuyó al 50.3% basándose en el AUC y al 60.3% basándose en la Cmax en la administración conjunta con rifampicina en comparación con la administración de nintedanib en monoterapia. Si se administran de forma conjunta con OFEV®, los inhibidores potentes de la gp-P (por ejemplo, ketoconazol, eritromicina o ciclosporina) pueden aumentar la exposición a nintedanib. En tales casos, es preciso controlar estrechamente a los pacientes para evaluar la tolerabilidad a nintedanib. El tratamiento de los efectos adversos puede requerir la interrupción, la reducción de la dosis o la suspensión del tratamiento con OFEV® (ver sección 4.2).
Los inductores potentes de la gp-P (por ejemplo, rifampicina, carbamazepina, fenitoína y la hierba de San Juan) pueden disminuir la exposición a nintedanib. En este caso se debe valorar la selección de un medicamento concomitante alternativo que no tenga potencial de inducción de la gp-P o en el que dicho potencial sea mínimo.
Enzimas del citocromo (CYP): Tan solo una pequeña proporción de la biotransformación de nintedanib se produce a través de las vías del CYP. Nintedanib y sus metabolitos, la porción de ácido libre BIBF 1202 y su glucurónido BIBF 1202, no inhibieron ni indujeron las enzimas CYP en estudios preclínicos con animales (ver sección 5.2). Así pues, se considera que hay pocas probabilidades de que se produzcan interacciones farmacológicas con nintedanib basándose en el metabolismo del CYP.
Administración conjunta con otros medicamentos: La administración conjunta de nintedanib con anticonceptivos hormonales orales no alteró la farmacocinética de los anticonceptivos hormonales orales de forma significativa (ver sección 5.2).
La administración conjunta de nintedanib y bosentán no alteró la farmacocinética de nintedanib (ver sección 5.2).
• Cáncer de pulmón no microcítico (CPNM):
Los estudios de interacción se han realizado solo en adultos
Glicoproteína-P (gp-P): Nintedanib es un sustrato de la gp-P (ver sección 5.2). Su administración conjunta con ketoconazol, un potente inhibidor de la gp-P, aumentó la exposición a nintedanib 1.61 veces basándose en el AUC y 1.83 veces basándose en la Cmax en un estudio de interacción farmacológica. En un estudio de interacción farmacológica con rifampicina, un potente inductor de la gp-P, la exposición a nintedanib disminuyó al 50.3% basándose en el AUC y al 60.3% basándose en la Cmax en la administración conjunta con rifampicina en comparación con la administración de nintedanib en monoterapia. Si se administran de forma conjunta con nintedanib, los inhibidores potentes de la gp-P (por ejemplo ketoconazol o eritromicina) pueden aumentar la exposición a nintedanib. En tales casos, es preciso controlar estrechamente a los pacientes para evaluar la tolerabilidad a nintedanib. El tratamiento de las reacciones adversas puede requerir la interrupción, la reducción de la dosis o la suspensión del tratamiento con OFEV® (ver sección 4.2).
Los inductores potentes de la gp-P (por ejemplo, rifampicina, carbamazepina, fenitoína y la hierba de San Juan) pueden disminuir la exposición a nintedanib. La administración conjunta con nintedanib se debe valorar cuidadosamente.
Enzimas del citocromo (CYP): Tan solo una pequeña proporción de la biotransformación de nintedanib se produce a través de las vías del CYP. Nintedanib y sus metabolitos, la porción de ácido libre BIBF 1202 y su glucurónido BIBF 1202, no inhibieron ni indujeron las enzimas CYP en estudios preclínicos con animales (ver sección 5.2). Así pues, se considera que hay pocas probabilidades de que se produzcan interacciones farmacológicas con nintedanib basándose en el metabolismo del CYP.
Administración conjunta con otros medicamentos: La administración conjunta de nintedanib con docetaxel (75 mg/m²) no afectó significativamente a la farmacocinética de estos medicamentos.
La administración conjunta de nintedanib con anticonceptivos hormonales orales no alteró la farmacocinética de los anticonceptivos hormonales orales de forma significativa (ver sección 5.2).
DATOS PRECLÍNICOS SOBRE SEGURIDAD:
Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI).
Toxicología general: Los estudios de toxicidad de una sola dosis realizados en ratas y ratones indicaron un bajo potencial de toxicidad aguda de nintedanib. En los estudios de toxicología con dosis repetidas realizados en ratas, los efectos adversos (como el engrosamiento de las placas epifisarias o las lesiones de los incisivos) estuvieron relacionados en su mayoría con el mecanismo de acción (es decir, la inhibición del VEGFR-2) de nintedanib. Estos cambios se conocen de otros inhibidores del VEGFR-2 y se pueden considerar efectos de clase.
La diarrea y los vómitos, acompañados de una reducción en la ingesta de alimentos y una pérdida del peso corporal, se observaron en estudios de toxicidad con no roedores.
No se produjeron signos de aumentos en las enzimas hepáticas en ratas, perros y monos cynomolgus.
Los aumentos leves en las enzimas hepáticas que no se debían a efectos adversos graves, como la diarrea, se observaron únicamente en monos rhesus.
Toxicidad para la reproducción: En el caso de las ratas, la mortalidad embriofetal y los efectos teratogénicos se observaron a una exposición inferior a la exposición humana a la dosis humana máxima recomendada de 150 mg dos veces al día. También se observaron efectos en el desarrollo del esqueleto axial y en el desarrollo de las grandes arterias a niveles de exposición subterapéuticos.
En el caso de los conejos, la mortalidad embriofetal y los efectos teratogénicos se observaron a una exposición aproximadamente 3 veces superior a la dosis humana máxima recomendada, pero se observaron efectos equívocos en el desarrollo embriofetal del esqueleto axial y del corazón a una exposición inferior a la dosis humana máxima recomendada de 150 mg dos veces al día.
En un estudio de desarrollo prenatal y posnatal realizado en ratas, los efectos sobre el desarrollo prenatal y posnatal se observaron a una exposición inferior a la dosis humana máxima recomendada.
Un estudio de la fertilidad en varones y del desarrollo embrionario temprano hasta la implantación en ratas no reveló efectos en el sistema reproductivo ni en la fertilidad de los varones.
En el caso de las ratas, pequeñas cantidades de nintedanib marcado radiactivamente y/o sus metabolitos se excretaron en la leche (≤ 0.5% de la dosis administrada).
A partir de los estudios de carcinogénesis de dos años en ratones y ratas, no se obtuvieron pruebas de un potencial carcinógeno de nintedanib.
Los estudios de genotoxicidad no indicaron potencial mutágeno en el caso de nintedanib.
Cáncer de pulmón no microcítico (CPNM):
Toxicología general: Los estudios de toxicidad de una sola dosis realizados en ratas y ratones indicaron un bajo potencial de toxicidad aguda de nintedanib. En los estudios de toxicología con dosis repetidas realizados en ratas, los efectos adversos (como el engrosamiento de las placas epifisarias o las lesiones de los incisivos) estuvieron relacionados en su mayoría con el mecanismo de acción (es decir, la inhibición del VEGFR-2) de nintedanib. Estos cambios se conocen de otros inhibidores del VEGFR -2 y se pueden considerar efectos de clase.
La diarrea y los vómitos, acompañados de una reducción en la ingesta de alimentos y una pérdida del peso corporal, se observaron en estudios de toxicidad con no roedores.
No se produjeron signos de aumentos en las enzimas hepáticas en ratas, perros y monos Cynomolgus. Los aumentos leves en las enzimas hepáticas que no se debían a efectos adversos graves, como la diarrea, se observaron únicamente en monos Rhesus.
Toxicidad para la reproducción: Un estudio de la fertilidad en machos y del desarrollo embrionario temprano hasta la implantación en ratas no reveló efectos en el sistema reproductivo ni en la fertilidad de los machos.
En el caso de las ratas, la mortalidad embriofetal y los efectos teratogénicos se observaron a niveles de exposición por debajo de la exposición humana, a la dosis humana máxima recomendada de 200 mg dos veces al día. También se observaron efectos en el desarrollo del esqueleto axial y en el desarrollo de las grandes arterias a niveles de exposición subterapéuticos.
En el caso de los conejos, la mortalidad embriofetal se observó a una exposición aproximadamente 8 veces superior a la dosis humana máxima recomendada. Se constataron efectos teratogénicos en los arcos aórticos en combinación con el corazón y el sistema urogenital a una exposición 4 veces superior a la dosis humana máxima recomendada y en el desarrollo embriofetal del esqueleto axial a una exposición 3 veces superior a la dosis humana máxima recomendada.
En el caso de las ratas, pequeñas cantidades de nintedanib marcado radioactivamente y/o sus metabolitos se excretaron en la leche (≤ 0.5% de la dosis administrada).
Los estudios de genotoxicidad no indicaron potencial mutágeno en el caso de nintedanib.
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
• Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI)
El tratamiento debe ser iniciado por médicos con experiencia en el manejo de enfermedades para las que está aprobado OFEV®.
Posología: La dosis recomendada es de 150 mg de nintedanib dos veces al día, administrado aproximadamente con 12 horas de diferencia.
La dosis diaria de 100 mg dos veces al día solo se recomienda en pacientes que no toleran la dosis diaria de 150 mg dos veces al día.
Si se olvida una dosis, la administración se debe reanudar a la dosis recomendada a la siguiente hora del programa establecido. Si se olvida una dosis, el paciente no debe tomar una dosis adicional. No se debe superar la dosis diaria máxima recomendada de 300 mg.
Ajustes de la dosis: Además del tratamiento sintomático si procede, el manejo de las reacciones adversas de OFEV® (ver las secciones 4.4 y 4.8) puede incluir la reducción de la dosis y la interrupción temporal del tratamiento hasta que la reacción adversa específica haya alcanzado de nuevo niveles que permitan la continuación del tratamiento. El tratamiento con OFEV® se puede reanudar a la dosis completa (150 mg dos veces al día) o a una dosis reducida (100 mg dos veces al día). Si un paciente no tolera 100 mg dos veces al día, el tratamiento con OFEV® se debe suspender.
Si la diarrea, las náuseas y/o los vómitos persisten a pesar de un tratamiento de apoyo adecuado (que incluye tratamiento antiemético), puede ser necesario reducir la dosis o interrumpir el tratamiento. El tratamiento se puede reanudar a una dosis reducida (100 mg dos veces al día) o a la dosis completa (150 mg dos veces al día). En caso de diarrea, náuseas y/o vómitos intensos persistentes a pesar del tratamiento sintomático, se debe suspender el tratamiento con OFEV® (ver sección 4.4).
En caso de interrumpir el tratamiento debido al aumento de la aspartato aminotransferasa (AST) o la alanina aminotransferasa (ALT) por encima de 3 veces el límite superior de la normalidad (LSN), una vez que las transaminasas hayan recuperado los valores basales, el tratamiento con OFEV® se puede reanudar a una dosis reducida (100 mg dos veces al día), que posteriormente podrá aumentarse a la dosis completa (150 mg dos veces al día) (ver las secciones 4.4 y 4.8).
• Poblaciones especiales
Pacientes de edad avanzada (≥ 65 años): No se observaron diferencias globales en la seguridad y la eficacia en pacientes de edad avanzada. A priori, no es necesario un ajuste de la dosis en función de la edad del paciente. Los pacientes con una edad igual o superior a 75 años pueden tener más probabilidades de necesitar una reducción de la dosis para tratar los efectos adversos (ver sección 5.2).
Insuficiencia renal: No es necesario ajustar la dosis inicial en pacientes con insuficiencia renal de leve a moderada. La seguridad, la eficacia y la farmacocinética de nintedanib no se han estudiado en pacientes con insuficiencia renal grave (< 30 ml/min de aclaramiento de creatinina).
Insuficiencia hepática: En pacientes con insuficiencia hepática leve (Child Pugh A), la dosis recomendada de OFEV® es de 100 mg dos veces al día con aproximadamente 12 horas de intervalo entre la administración de las dos dosis. En pacientes con insuficiencia hepática leve (Child Pugh A), se debe considerar la interrupción o la suspensión del tratamiento para el manejo de las reacciones adversas. La seguridad y la eficacia de nintedanib no se han estudiado en pacientes con insuficiencia hepática clasificada como Child Pugh B y C. No se recomienda tratar con OFEV® a pacientes con insuficiencia hepática moderada (Child Pugh B) y grave (Child Pugh C) (ver sección 5.2).
Población pediátrica: No se ha establecido la seguridad y la eficacia de OFEV® en niños de 0 a 18 años. No se dispone de datos.
Forma de administración: OFEV® se administra por vía oral. Las cápsulas se deben tomar con alimentos, y tragarse enteras con agua, sin masticarlas. La cápsula no se debe abrir ni partir (ver sección 6.6)
Cáncer de pulmón no microcítico (CPNM): El tratamiento con OFEV® debe ser iniciado y supervisado por un médico con experiencia en el empleo de terapias antineoplásicas.
Posología: La dosis recomendada de nintedanib es de 200 mg dos veces al día, administrado aproximadamente con 12 horas de diferencia los días del 2 al 21 de un ciclo de tratamiento estándar con docetaxel cada 21 días.
OFEV® no se debe tomar el mismo día en el que se administre la quimioterapia con docetaxel (= día 1).
Si se olvida una dosis de nintedanib, la administración se debe reanudar a la dosis recomendada a la siguiente hora del programa establecido. Las dosis diarias individuales de nintedanib no deben aumentarse por encima de la dosis recomendada para compensar una dosis olvidada. No se debe superar la dosis diaria máxima recomendada de 400 mg.
Los pacientes pueden continuar el tratamiento con nintedanib después de suspender el tratamiento con docetaxel mientras se observen ventajas clínicas o hasta que se produzca una toxicidad inaceptable.
Para obtener información sobre la posología, las formas de administración y las modificaciones de la dosis de docetaxel, consulte la ficha técnica correspondiente a este fármaco.
Ajustes de la dosis: Como medida inicial para el manejo de posibles reacciones adversas (ver Tabla 1 y 2) se debe interrumpir temporalmente el tratamiento con nintedanib hasta que la reacción adversa específica haya alcanzado de nuevo niveles que permitan la continuación del tratamiento (grado 1 o nivel basal).
El tratamiento con nintedanib puede reanudarse a una dosis reducida. Se recomiendan ajustes de la dosis en pasos de 100 mg al día (es decir, una reducción de 50 mg en cada dosis) basándose en la seguridad y la tolerabilidad individual, tal como se describe en la Tabla 1 y en la Tabla 2.
En el caso de que la reacción o reacciones adversas persistan, es decir, si un paciente no tolera 100 mg dos veces al día, el tratamiento con OFEV® debe suspenderse de forma permanente. Si se producen aumentos específicos de los valores de aspartato aminotransferasa (AST) / alanina aminotransferasa (ALT) a > 3 x del límite superior de la normalidad (LSN) en combinación con un aumento de la bilirrubina total a ≥ 2 x LSN y de la fosfatasa alcalina (FA) < 2 x LSN (ver Tabla 2), el tratamiento con OFEV® se debe interrumpir. A menos que se haya constatado que existe otra causa para ello, OFEV® se debe suspender de forma permanente (ver también sección 4.4).
|
Tabla 1 Ajustes de la dosis recomendados para OFEV® (nintedanib) en el caso de diarrea, vómitos y otras reacciones adversas no hematológicas o hematológicas |
|
|
Reacción adversa según los CTCAE* |
Ajuste de la dosis |
|
Diarrea ≥ grado 2 durante más de 7 días consecutivos a pesar de seguir un tratamiento antidiarreico O Diarrea ≥ grado 3 a pesar de seguir un tratamiento antidiarreico |
Después de la interrupción del tratamiento y de la recuperación al grado 1 o al nivel basal, reducción de la dosis de 200 mg dos veces al día a 150 mg dos veces al día y, - si se considera necesaria una 2a reducción de la dosis, de 150 mg dos veces al día a 100 mg dos veces al día. |
|
Vómitos ≥ grado 2 Y/O Náuseas ≥ grado 3 a pesar de seguir un tratamiento antiemético |
|
|
Otras reacciones adversas no hematológicas o hematológicas de ≥ grado 3 |
|
|
* CTCAE: Criterios terminológicos comunes para acontecimientos adversos |
|
|
Tabla 2 Ajustes de la dosis recomendados para OFEV® (nintedanib) en el caso de aumentos en los niveles de AST y/o ALT y bilirrubina |
|
|
Aumento de los niveles de AST/ALT y bilirrubina |
Ajuste de la dosis |
|
Aumento de los valores de AST y/o ALT a > 2.5 x LSN en combinación con un aumento de la bilirrubina total a ≥ 1.5 x LSN O Aumento de los valores de AST y/o ALT a > 5 x LSN |
Después de la interrupción del tratamiento y de la recuperación de los niveles de transaminasas a ≤ 2.5 x LSN en combinación con una recuperación de los niveles de bilirrubina a los valores normales, reducción de la dosis de 200 mg dos veces al día a 150 mg dos veces al día y, si se considera necesaria una 2a reducción de la dosis, de 150 mg dos veces al día a 100 mg dos veces al día. |
|
Aumento de los valores de AST y/o ALT a > 3 x LSN en combinación con un aumento de la bilirrubina total a ≥ 2 x LSN y FA < 2 x LSN |
A menos que se haya constatado que existe otra causa para ello, OFEV® se debe suspender de forma permanente. |
|
AST: Aspartato aminotransferasa; ALT: Alanina aminotransferasa FA: Fosfatasa alcalina; LSN: Límite superior de la normalidad |
|
• Poblaciones especiales:
Población pediátrica: No se ha establecido la seguridad y la eficacia de OFEV® en niños de 0 a 18 años. No se dispone de datos.
Pacientes de edad avanzada (≥ 65 años): No se observaron diferencias globales en la seguridad y la eficacia en pacientes de edad avanzada.
En el estudio pivotal 1199.13, 85 pacientes (12.9% de ellos con histología de adenocarcinoma) tenían una edad ≥ 70 años (mediana de edad: 72 años, rango: 70 - 80 años) (ver sección 5.1).
No es preciso realizar un ajuste de la dosis inicial basándose en la edad del paciente (ver sección 5.2).
Raza y peso corporal: Basándose en los análisis farmacocinéticos (FC) poblacionales, a priori no es necesario realizar ajustes de la dosis de OFEV® (ver sección 5.2). Los datos de seguridad para pacientes de raza negra y afroamericana son limitados.
Insuficiencia renal: Menos del 1% de una dosis única de nintedanib se elimina a través del riñón (ver sección 5.2). No es necesario ajustar la dosis inicial en pacientes con insuficiencia renal de leve a moderada. La seguridad, la eficacia y la farmacocinética de nintedanib no se han estudiado en pacientes con insuficiencia renal grave (<30 ml/min de aclaramiento de creatinina).
Insuficiencia hepática: Nintedanib se elimina sobre todo a través de la excreción biliar y fecal (> 90%). Aumento de la exposición en pacientes con insuficiencia hepática (Child Pugh A, Child Pugh B; ver sección 5.2). Teniendo en cuenta los datos clínicos, no es necesario realizar un ajuste de la dosis inicial en el caso de pacientes con insuficiencia hepática leve (Child Pugh A). Los datos de seguridad disponibles de 9 pacientes con insuficiencia hepática moderada (Child Pugh B) son limitados y resultan insuficientes para caracterizar esta población. La seguridad, la eficacia y la farmacocinética de nintedanib no se han investigado en pacientes con insuficiencia hepática grave (Child Pugh C). No se recomienda tratar con OFEV® a pacientes con insuficiencia hepática moderada (Child Pugh B) y grave (Child Pugh C) (ver las secciones 4.4 y 5.2).
Forma de administración: Las cápsulas de OFEV® se deben tomar por vía oral, preferiblemente con alimentos, y tragarse enteras con agua, sin masticarlas. La cápsula no se debe abrir ni partir (ver sección 6.6)
SOBREDOSIS:
Fibrosis pulmonar idiopática (FPI), enfermedad pulmonar intersticial asociada a la esclerosis sistémica (EPI-ES) y enfermedades pulmonares intersticiales (EPI): No hay ningún antídoto ni tratamiento específico en el caso de producirse una sobredosis de OFEV®. Dos pacientes del programa de oncología presentaron una sobredosis de un máximo de 600 mg dos veces al día durante un máximo de ocho días. Las reacciones adversas observadas coincidieron con el perfil de seguridad conocido de nintedanib, es decir, aumento de las enzimas hepáticas y síntomas gastrointestinales. Ambos pacientes se recuperaron de estas reacciones adversas. En los ensayos INPULSIS, un paciente se expuso de forma accidental a una dosis de 600 mg al día durante un total de 21 días. Durante el período de dosificación incorrecta, se produjo y se resolvió un efecto adverso no grave (rinofaringitis), pero no se informó de la aparición de otros episodios. En el caso de producirse una sobredosis, es preciso interrumpir el tratamiento e iniciar las medidas de apoyo generales que resulten adecuadas.
Cáncer de pulmón no microcítico (CPNM): No hay ningún antídoto ni tratamiento específico en el caso de producirse una sobredosis de nintedanib. La dosis única más alta de nintedanib administrada en estudios fase I fue de 450 mg una vez al día.
Además, 2 pacientes presentaron una sobredosis de un máximo de 600 mg dos veces al día durante un máximo de ocho días. Los efectos adversos observados coincidieron con el perfil de seguridad conocido de nintedanib, es decir, aumento de las enzimas hepáticas y síntomas gastrointestinales. Ambos pacientes se recuperaron de estas reacciones adversas. En el caso de producirse una sobredosis, es preciso interrumpir el tratamiento e iniciar las medidas de apoyo generales que resulten adecuadas.
DESCRIPCIÓN: OFEV® 100 mg CÁPSULAS Blandas OFEV® 150 mg cápsulas blandas.
PRESENTACIÓN:
Naturaleza y contenido del envase:
OFEV® 100 mg cápsulas blandas / OFEV® 150 mg cápsulas blandas se encuentra disponible en el siguiente tamaño de envase:
Caja con 60 cápsulas blandas de 100 mg.
Caja con 60 cápsulas blandas de 150 mg
Precauciones especiales de eliminación y otras manipulaciones: En caso de entrar en contacto con el contenido de la cápsula, se deben lavar las manos inmediatamente con abundante agua (ver sección 4.2).
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con él se realizará de acuerdo con la normativa local.
Nombre y domicilio del laboratorio
Hecho en Alemania por:
Catalent Germany Eberbach GmbH Gammelsbacher Str. 2, 69412 Eberbach, Alemania
Acondicionado por:
Boehringer Ingelheim Pharma GmbH & Co KG Binger Straβe 173, D-55216 Ingelheim, Alemania
Para:
BOEHRINGER INGELHEIM PROMECO S.A DE C.V.
Calle Del Maíz No. 49, Barrio Xaltocan,
Deleg. Xochimilco C.P. 16090, Ciudad de México.
®Marca registrada
Fecha de revisión de la monografía: Agosto 2021

