KEYTRUDA
PEMBROLIZUMAB
Solución inyectable para infusión I.V.
COMPOSICIÓN CUALITATIVA Y CUANTITATIVA:
Un vial de 4 mL de solución contiene 100 mg de pembrolizumab. Cada mL de solución contiene 25 mg de pembrolizumab.
Pembrolizumab es un anticuerpo monoclonal humanizado (IgG4/isotipo kappa con una alteración de la secuencia estabilizadora en la región Fc) frente a la muerte celular programada-1 (PD-1), producido en células de ovario de hámster chino mediante tecnología de ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
FORMA FARMACÉUTICA:
Solución para infusión intravenosa
Solución transparente a ligeramente opalescente, incolora a ligeramente amarilla con pH entre 5.2 y 5.8.
INDICACIONES TERAPÉUTICAS:
KEYTRUDA® en monoterapia está indicado para el tratamiento del melanoma avanzado (irresecable o metastásico) en adultos.
KEYTRUDA® en monoterapia está indicado para el tratamiento adyuvante en adultos con melanoma en estadio III y con afectación de los ganglios linfáticos que hayan sido sometidos a resección completa (ver Propiedades farmacodinámicas).
KEYTRUDA® en monoterapia está indicado para el tratamiento de primera línea del cáncer de pulmón no microcítico (CPNM) metastásico en adultos cuyos tumores expresen PD-L1 con una proporción de marcador tumoral (TPS, por sus siglas en inglés) ≥50% sin mutaciones tumorales positivas de EGFR o ALK.
KEYTRUDA®, en combinación con pemetrexed y quimioterapia basada en platino está indicado para el tratamiento de primera línea del CPNM no escamoso metastásico en adultos cuyos tumores no tengan mutaciones tumorales positivas de EGFR o ALK.
KEYTRUDA®, en combinación con carboplatino y paclitaxel o nab-paclitaxel, está indicado para el tratamiento de primera línea del CPNM escamoso metastásico en adultos.
KEYTRUDA® en monoterapia está indicado para el tratamiento del CPNM localmente avanzado o metastásico en adultos cuyos tumores expresen PD-L1con una TPS ≥1% y que hayan recibido al menos un tratamiento de quimioterapia previo. Los pacientes con mutaciones tumorales positivas de EGFR o ALK deben haber recibido también terapia dirigida antes de recibir KEYTRUDA®.
KEYTRUDA® en monoterapia está indicado para el tratamiento de pacientes adultos con linfoma de Hodgkin clásico (LHc) en recaída o refractario, que no han respondido a un trasplante autólogo de progenitores hematopoyéticos (TAPH) y a brentuximab vedotina (BV), o que no son candidatos a trasplante y no han respondido a BV.
KEYTRUDA® en monoterapia está indicado para el tratamiento del carcinoma urotelial localmente avanzado o metastásico en adultos que hayan recibido quimioterapia previa basada en platino (ver Propiedades farmacodinámicas).
KEYTRUDA® en monoterapia está indicado para el tratamiento del carcinoma urotelial localmente avanzado o metastásico en adultos que no son candidatos a quimioterapia basada en cisplatino y cuyos tumores expresen PD-L1 con una puntuación positiva combinada (CPS, por sus siglas en inglés) ≥ 10 (ver Propiedades farmacodinámicas).
KEYTRUDA® en monoterapia está indicado para el tratamiento del carcinoma de células escamosas de cabeza y cuello (CCECC) recurrente o metastásico en adultos cuyos tumores expresen PD-L1 con una TPS ≥ 50% y que progresen durante o después de quimioterapia basada en platino (ver Propiedades farmacodinámicas).
PROPIEDADES FARMACOLÓGICAS
Propiedades farmacodinámicas:
Grupo farmacoterapéutico: otros agentes antineoplásicos, anticuerpos monoclonales. Código ATC: L01XC18.
Mecanismo de acción:
KEYTRUDA® es un anticuerpo monoclonal humanizado, que se une al receptor de la muerte celular programada-1 (PD-1) y bloquea su interacción con los ligandos PD-L1 y PD-L2. El receptor PD-1 es un regulador negativo de la actividad de las células T que se ha demostrado que está involucrado en el control de las respuestas inmunitarias de las células T. KEYTRUDA® potencia las respuestas de las células T, incluyendo las respuestas antitumorales, mediante el bloqueo de PD-1, unido a PD-L1 y PD-L2, que se expresan en las células presentadoras de antígenos y que se pueden expresar por tumores u otras células en el microambiente tumoral.
Eficacia clínica y seguridad:
En ensayos clínicos en melanoma o CPNM tratado previamente, se evaluaron dosis de pembrolizumab de 2 mg/kg cada 3 semanas, 10 mg/kg cada 3 semanas y 10 mg/kg cada 2 semanas. De acuerdo a los modelos y simulación de las relaciones dosis/exposición de eficacia y seguridad de pembrolizumab, no hay diferencias clínicamente significativas en la eficacia o en la seguridad entre las dosis de 200 mg cada 3 semanas, de 2 mg/kg cada 3 semanas y, en monoterapia, de 400 mg cada 6 semanas (ver Posología y forma de administración).
Melanoma KEYNOTE-006: Ensayo controlado en pacientes con melanoma sin tratamiento previo con ipilimumab
La seguridad y eficacia de pembrolizumab se investigó en el ensayo KEYNOTE-006, un ensayo de Fase III, multicéntrico, controlado, para el tratamiento del melanoma avanzado en pacientes sin tratamiento previo con ipilimumab. Los pacientes fueron aleatorizados (1:1:1) a recibir pembrolizumab 10 mg/kg cada 2 (n=279) o 3 semanas (n=277) o ipilimumab 3 mg/kg cada 3 semanas (n=278). No se requirió que los pacientes con melanoma con mutación de BRAF V600E hubieran recibido tratamiento previo con un inhibidor de BRAF.
Los pacientes fueron tratados con pembrolizumab hasta progresión de la enfermedad o toxicidad inaceptable. A los pacientes clínicamente estables con evidencia inicial de progresión de la enfermedad se les permitió permanecer en tratamiento hasta que se confirmó la progresión de la enfermedad. Se realizó la evaluación del estado tumoral a las 12 semanas y luego cada 6 semanas hasta la semana 48, seguido posteriormente por la evaluación cada 12 semanas.
De los 834 pacientes, el 60% eran varones, el 44% eran ≥ 65 años (la mediana de edad era de 62 años [rango, 18-89]) y el 98% eran de raza blanca. El 65% de los pacientes tenían estadio M1c, el 9% tenían antecedentes de metástasis cerebral, el 66% no habían recibido tratamiento sistémico previo y el 34% habían recibido un tratamiento previo. El 31% tenían estado funcional ECOG de 1, el 69% tenían estado funcional ECOG de 0 y el 32% tenían LDH elevada. Se notificaron mutaciones de BRAF en 302 (36%) pacientes. Entre los pacientes con tumores con mutación de BRAF, 139 (46%) habían sido tratados previamente con un inhibidor de BRAF.
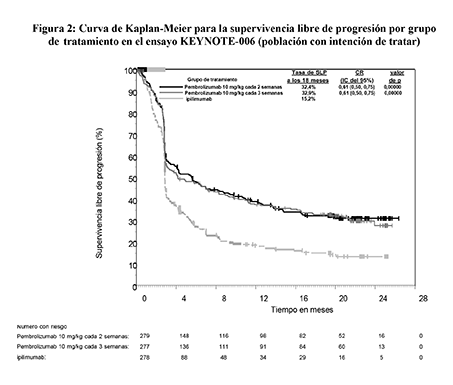
Las variables principales de eficacia fueron la supervivencia libre de progresión (SLP; evaluada mediante una revisión por Evaluación Radiológica y Oncológica Integrada [ROI] usando los Criterios de Evaluación de la Respuesta en Tumores Sólidos [RECIST], versión 1.1) y la supervivencia global (SG). Las variables secundarias de eficacia fueron la tasa de respuesta global (TRG) y la duración de la respuesta. La Tabla 3 resume las variables principales de eficacia en pacientes sin tratamiento previo con ipilimumab en el análisis final realizado tras un mínimo de 21 meses de seguimiento. En las Figuras 1 y 2 se muestran las curvas de Kaplan-Meier para la SG y la SLP de acuerdo al análisis final.
|
Tabla 3: Resultados de eficacia en el ensayo KEYNOTE-006 |
|||
|
Variable |
Pembrolizumab 10 mg/kg cada 3 semanas n=277 |
Pembrolizumab 10 mg/kg cada 2 semanas n=279 |
Ipilimumab 3 mg/kg cada 3 semanas n=278 |
|
SG |
|||
|
Número (%) de pacientes con acontecimiento |
119 (43%) |
122 (44%) |
142 (51%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.68 (0.53, 0.86) |
0.68 (0.53, 0.87) |
--- |
|
Valor de p† |
< 0.001 |
< 0.001 |
--- |
|
Mediana en meses (IC |
No alcanzada |
No alcanzada |
16 |
|
del 95%) |
(24, ND) |
(22, ND) |
(14, 22) |
|
SLP |
|||
|
Número (%) de pacientes con acontecimiento |
183 (66%) |
181 (65%) |
202 (73%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.61 (0.50, 0.75) |
0.61 (0.50, 0.75) |
--- |
|
Valor de p† |
< 0.001 |
< 0.001 |
--- |
|
Mediana en meses (IC |
4.1 |
5.6 |
2.8 |
|
del 95%) |
(2.9, 7.2) |
(3.4, 8.2) |
(2.8, 2.9) |
|
Mejor respuesta global |
|||
|
% de TRG (IC del 95%) |
36% |
37% |
13% |
|
(30, 42) |
(31, 43) |
(10, 18) |
|
|
% de respuesta completa |
13% |
12% |
5% |
|
% de respuesta parcial |
23% |
25% |
8% |
|
Duración de la respuesta‡ |
|||
|
Mediana en meses (rango) |
No alcanzada (2.0, 22.8+) |
No alcanzada (1.8, 22.8+) |
No alcanzada (1.1+, 23.8+) |
|
% que continúan a los 18 meses |
68%§ |
71%§ |
70%§ |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con ipilimumab) de acuerdo al modelo de riesgos proporcionales de Cox estratificado † Basado en la prueba de orden logarítmico estratificada ‡ Basado en pacientes con una mejor respuesta como respuesta completa o parcial confirmadas § Basado en la estimación de Kaplan- Meier ND = no disponible |
|||
Figura 1: Curva de Kaplan-Meier para la supervivencia global por grupo de tratamiento en el ensayo KEYNOTE-006 (población con intención de tratar)
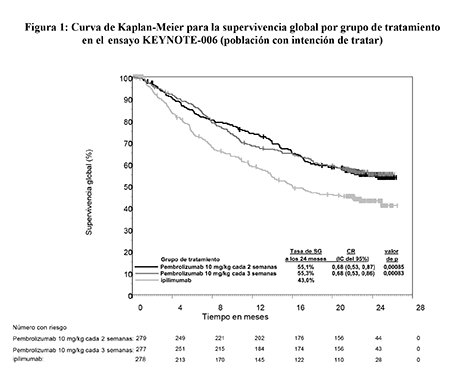
Figura 2: Curva de Kaplan-Meier para la supervivencia libre de progresión por grupo de tratamiento en el ensayo KEYNOTE-006 (población con intención de tratar)
KEYNOTE-002: Ensayo controlado en pacientes con melanoma tratados previamente con ipilimumab
La seguridad y eficacia de pembrolizumab se investigó en el ensayo KEYNOTE-002, un ensayo multicéntrico, controlado, para el tratamiento del melanoma avanzado en pacientes tratados previamente con ipilimumab y si eran positivos para la mutación de BRAF V600, con un inhibidor de BRAF o de MEK. Los pacientes fueron aleatorizados (1:1:1) a recibir pembrolizumab a una dosis de 2 (n=180) o 10 mg/kg (n=181) cada 3 semanas o quimioterapia (n=179; incluidas dacarbazina, temozolomida, carboplatino, paclitaxel o carboplatino+paclitaxel). El ensayo excluyó a pacientes con enfermedad autoinmune o a aquellos que recibían inmunosupresores; criterios adicionales de exclusión fueron antecedentes de reacciones adversas relacionadas con el sistema inmunitario graves o potencialmente mortales con el tratamiento con ipilimumab, definidas como cualquier toxicidad de Grado 4 o de Grado 3 que requería tratamiento con corticosteroides (dosis > 10 mg/día de prednisona o equivalente) durante más de 12 semanas; reacciones adversas en curso de Grado ≥ 2 por tratamientos previos con ipilimumab; hipersensibilidad grave previa a otros anticuerpos monoclonales; antecedentes de neumonitis o enfermedad pulmonar intersticial; infección por VIH, hepatitis B o hepatitis C y estado funcional ECOG ≥ 2.
Los pacientes fueron tratados con pembrolizumab hasta progresión de la enfermedad o toxicidad inaceptable. A los pacientes clínicamente estables con evidencia inicial de progresión de la enfermedad se les permitió permanecer en tratamiento hasta que se confirmó la progresión de la enfermedad. Se realizó la evaluación del estado tumoral a las 12 semanas y luego cada 6 semanas hasta la semana 48, seguida posteriormente por la evaluación cada 12 semanas. Los pacientes en quimioterapia que experimentaron progresión de la enfermedad, verificada de forma independiente después de la primera evaluación programada de la enfermedad, pudieron cambiar y recibir 2 mg/kg o 10 mg/kg de pembrolizumab cada 3 semanas, en un modelo a doble ciego.
De los 540 pacientes, el 61% eran varones, el 43% eran ≥ 65 años (la mediana de edad era de 62 años [rango, 15-89]) y el 98% eran de raza blanca. El 82% tenían estadio M1c, el 73% habían recibido al menos dos tratamientos sistémicos previos para el melanoma avanzado y el 32% de los pacientes habían recibido tres o más. El 45% tenían estado funcional ECOG de 1, el 40% tenían LDH elevada y el 23% tenían un tumor con mutación de BRAF.
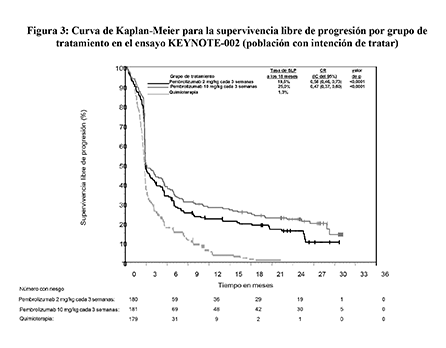
Las variables principales de eficacia fueron la SLP evaluada mediante ROI usando los criterios RECIST versión 1.1 y la SG. Las variables secundarias de eficacia fueron la TRG y la duración de la respuesta. La Tabla 4 resume las variables principales de eficacia en pacientes tratados previamente con ipilimumab y en la Figura 3 se muestra la curva de Kaplan-Meier para la SLP. Los dos grupos de tratamiento con pembrolizumab fueron superiores a la quimioterapia para la SLP, y no hubo diferencias entre las dosis de pembrolizumab. No hubo diferencia estadísticamente significativa entre pembrolizumab y la quimioterapia en el análisis preliminar de la SG, que no se ajustó en cuanto a los posibles efectos de confusión debidos al cambio de tratamiento. De los pacientes aleatorizados al grupo de quimioterapia, el 55% cambiaron de tratamiento y, posteriormente, recibieron tratamiento con pembrolizumab.
|
Tabla 4: Resultados de eficacia en el ensayo KEYNOTE-002 |
|||
|
Variable |
Pembrolizumab 2 mg/kg cada 3 semanas n=180 |
Pembrolizumab 10 mg/kg cada 3 semanas n=181 |
Quimioterapia n=179 |
|
SLP |
|||
|
Número (%) de pacientes con acontecimiento |
150 (83%) |
144 (80%) |
172 (96%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.58 (0.46, 0.73) |
0.47 (0.37, 0.60) |
--- |
|
Valor de p† |
< 0,001 |
< 0,001 |
--- |
|
Mediana en meses (IC del 95%) |
2.9 (2.8, 3,8) |
3.0 (2.8, 5.2) |
2.8 (2.6, 2.8) |
|
SG |
|||
|
Número (%) de pacientes con acontecimiento |
123 (68%) |
117 (65%) |
128 (72%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.86 (0.67, 1.10) |
0.74 (0.57, 0.96) |
--- |
|
Valor de p† |
0.1173 |
0.0106‡ |
--- |
|
Mediana en meses (IC del 95%) |
13.4 (11.0, 16.4) |
14.7 (11.3, 19.5) |
11.0 (8.9, 13.8) |
|
Mejor respuesta global |
|||
|
% de TRG (IC del 95%) |
22% (16, 29) |
28% (21, 35) |
5% (2, 9) |
|
% de respuesta completa |
3% |
7% |
0% |
|
% de respuesta parcial |
19% |
20% |
5% |
|
Duración de la respuesta§ |
|||
|
Mediana en meses (rango) |
22,8 (1.4+, 25.3+) |
No alcanzada (1.1+, 28.3+) |
6,8 (2.8, 11.3) |
|
% que continúan a los 12 meses |
73% ¶ |
79% ¶ |
0% ¶ |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con quimioterapia) de acuerdo al modelo de riesgos proporcionales de Cox estratificado † Basado en la prueba de orden logarítmico estratificada ‡ No estadísticamente significativo tras un ajuste por multiplicidad § Basado en pacientes con una mejor respuesta como respuesta completa o parcial confirmadas en el análisis final ¶ Basado en la estimación de Kaplan-Meier |
|||
Figura 3: Curva de Kaplan-Meier para la supervivencia libre de progresión por grupo de tratamiento en el ensayo KEYNOTE-002 (población con intención de tratar)
KEYNOTE-001: Ensayo no controlado en pacientes con melanoma sin tratamiento previo con ipilimumab y previamente tratados con ipilimumab
La seguridad y eficacia de pembrolizumab se investigó en pacientes con melanoma avanzado en un ensayo no controlado, abierto, KEYNOTE-001. Se evaluó la eficacia en 276 pacientes de dos
cohortes definidas, una que incluyó a pacientes tratados previamente con ipilimumab (y, si eran positivos para la mutación de BRAF V600, con un inhibidor de BRAF o de MEK) y, la otra, que incluyó a pacientes sin tratamiento previo con ipilimumab. Los pacientes fueron aleatorizados a recibir pembrolizumab a una dosis de 2 mg/kg cada 3 semanas o 10 mg/kg cada 3 semanas. Los pacientes fueron tratados con pembrolizumab hasta progresión de la enfermedad o toxicidad inaceptable. A los pacientes clínicamente estables con evidencia inicial de progresión de la enfermedad se les permitió permanecer en tratamiento hasta que se confirmó la progresión de la enfermedad. Los criterios de exclusión fueron similares a los del ensayo KEYNOTE-002.
De los 89 pacientes que recibieron 2 mg/kg de pembrolizumab que fueron tratados previamente con ipilimumab, el 53% eran varones, el 33% eran ≥ 65 años de edad y la mediana de edad era de 59 años (rango, 18-88). Todos, excepto dos pacientes, eran de raza blanca. El 84% tenían estadio M1c y el 8% de los pacientes tenían antecedentes de metástasis cerebral. El 70% habían recibido al menos dos tratamientos sistémicos previos para el melanoma avanzado y el 35% de los pacientes habían recibido tres o más. Se notificaron mutaciones de BRAF en el 13% de la población del ensayo. Todos los pacientes con tumores con mutación de BRAF fueron tratados previamente con un inhibidor de BRAF.
De los 51 pacientes que recibieron 2 mg/kg de pembrolizumab que eran sin tratamiento previo con ipilimumab, el 63% eran varones, el 35% tenían ≥ 65 años de edad y la mediana de edad era de 60 años (rango, 35-80). Todos, excepto uno de los pacientes, eran de raza blanca. El 63% tenían estadio M1c y el 2% de los pacientes tenían antecedentes de metástasis cerebral. El 45% no habían recibido tratamientos previos para el melanoma avanzado. Se notificaron mutaciones de BRAF en 20 (39%) pacientes. Entre los pacientes con tumores con mutación de BRAF, 10 (50%) fueron tratados previamente con un inhibidor de BRAF.
La variable principal de eficacia fue la TRG evaluada mediante una revisión independiente usando los criterios RECIST 1.1. Las variables secundarias de eficacia fueron la tasa de control de la enfermedad (TCE; incluyendo la respuesta completa, la respuesta parcial y la enfermedad estable), la duración de la respuesta, la SLP y la SG. La respuesta tumoral se evaluó a intervalos de 12 semanas. La Tabla 5 resume las variables principales de eficacia en pacientes tratados previamente o sin tratamiento previo con ipilimumab, que recibían pembrolizumab a la dosis recomendada, de acuerdo a un seguimiento mínimo de tiempo de 30 meses para todos los pacientes.
|
Tabla 5: Resultados de eficacia en el ensayo KEYNOTE-001 |
||
|
Variable |
Pembrolizumab 2 mg/kg cada 3 semanas en pacientes tratados previamente con ipilimumab n=89 |
Pembrolizumab 2 mg/kg cada 3 semanas en pacientes sin tratamiento previo con ipilimumab n=51 |
|
Mejor respuesta global* por ROI† |
||
|
% de TRG (IC del 95%) |
26% (17, 36) |
35% (22, 50) |
|
Respuesta completa |
7% |
12% |
|
Respuesta parcial |
19% |
24% |
|
% de tasa de control de la enfermedad‡ |
48% |
49% |
|
Duración de la respuesta § |
||
|
Mediana en meses (rango) |
30.5 (2.8+, 30.6+) |
27.4 (1.6+, 31.8+) |
|
% que continúan a los 24 meses¶ |
75% |
71%# |
|
SLP |
||
|
Mediana en meses (IC del 95%) |
4.9 (2.8, 8.3) |
4.7 (2.8, 13.8) |
|
Tasa de SLP a los 12 meses |
34% |
38% |
|
SG |
||
|
Mediana en meses (IC del 95%) |
18.9 (11, no disponible) |
28.0 (14, no disponible) |
|
Tasa de SG a los 24 meses |
44% |
56% |
|
* Incluye pacientes sin enfermedad medible en el estado basal mediante revisión radiológica independiente † ROI = Evaluación radiológica y oncológica integrada usando los criterios RECIST 1.1 ‡ Basado en la mejor respuesta de enfermedad estable o mejor § Basado en los pacientes con una respuesta confirmada mediante revisión independiente, comenzando desde la fecha en la que se registró por primera vez la respuesta; n=23 para pacientes tratados previamente con ipilimumab; n=18 para pacientes sin tratamiento previo con ipilimumab ¶ Basado en la estimación de Kaplan-Meier |
||
Los resultados de los pacientes tratados previamente con ipilimumab (n=84) y sin tratamiento previo con ipilimumab (n=52), que recibieron 10 mg/kg de pembrolizumab cada 3 semanas, fueron similares a los observados en pacientes que recibieron 2 mg/kg de pembrolizumab cada 3 semanas.
Análisis de subpoblaciones:
Estado de la mutación BRAF en melanoma
Se realizó un análisis de subgrupos como parte del análisis final del ensayo KEYNOTE-002 en pacientes que tenían BRAF sin mutación (n=414; 77%) o BRAF con mutación con tratamiento previo con un inhibidor de BRAF (n=126; 23%), como se resume en la Tabla 6.
|
Tabla 6: Resultados de eficacia por estado de la mutación BRAF en el ensayo KEYNOTE- 002 |
||||
|
BRAF sin mutación |
BRAF con mutación con tratamiento previo |
|||
|
Variable |
Pembrolizumab 2mg/kg cada 3 semanas (n=136) |
Quimioterapia (n=137) |
Pembrolizumab 2mg/kg cada 3 semanas (n=44) |
Quimioterapia (n=42) |
|
SLP Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.50 (0.39, 0.66) |
--- |
0.79 (0.50, 1.25) |
--- |
|
SG Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.78 (0.58, 1,04) |
--- |
1.07 (0.64, 1.78) |
--- |
|
% de TRG |
26% |
6% |
9% |
0% |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con quimioterapia) de acuerdo al modelo de riesgos proporcionales de Cox estratificado |
||||
Se realizó un análisis de subgrupos como parte del análisis final del ensayo KEYNOTE-006 en pacientes que tenían BRAF sin mutación (n=525; 63%), BRAF con mutación sin tratamiento previo con un inhibidor de BRAF (n=163; 20%) y BRAF con mutación con tratamiento previo con un inhibidor de BRAF (n=139; 17%), como se resume en la Tabla 7.
|
Tabla 7: Resultados de eficacia por estado de la mutación BRAF en el ensayo KEYNOTE-006 |
||||||
|
BRAF sin mutación |
BRAF con mutación sin tratamiento previo |
BRAF con mutación con tratamiento previo |
||||
|
Variable |
Pembrolizumab 10mg/kg cada 2 o 3 semanas (en conjunto) |
Ipilimumab (n=170) |
Pembrolizumab 10mg/kg cada 2 o 3 semanas (en conjunto) |
Ipilimumab (n=55) |
Pembrolizumab 10mg/kg cada 2 o 3 semanas (en conjunto) |
Ipilimumab (n=52) |
|
SLP Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.61 (0.49, 0.76) |
--- |
0.52 (0.35, 0.78) |
--- |
0.76 (0.51, 1.14) |
--- |
|
SG Cociente de riesgo (Hazard Ratio)* (IC de 95%) |
0.68 (0.52, 0.88) |
--- |
0.70 (0.40, 1.22) |
--- |
0.66 (0.41, 1.04) |
--- |
|
% de TRG |
38% |
14% |
41% |
15% |
24% |
10% |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con ipilimumab) de acuerdo al modelo de riesgos proporcionales de Cox estratificad |
||||||
Estado de PD-L1 en melanoma
Se realizó un análisis de subgrupos como parte del análisis final del ensayo KEYNOTE-002 en pacientes que eran PD-L1 positivo (expresión de PD-L1 en ≥ 1% de las células tumorales y las células inmunitarias asociadas al tumor, relativa a todas las células tumorales viables – puntuación de melanoma) frente a PD- L1 negativo. La expresión de PD-L1 se estudió de manera retrospectiva mediante un test de inmunohistoquímica con el anticuerpo anti PD-L1 22C3. Entre los pacientes que eran evaluables en cuanto a expresión de PD-L1 (79%), el 69% (n=294) eran PD-L1 positivo y el 31% (n=134) eran PD-L1 negativo. La Tabla 8 resume los resultados de eficacia por expresión de PD-L1.
|
Tabla 8: Resultados de eficacia por expresión de PD-L1 en el ensayo KEYNOTE-002 |
||||
|
Variable |
Pembrolizumab 2 mg/kg cada 3 semanas |
Quimioterapia |
Pembrolizumab 2 mg/kg cada 3 semanas |
Quimioterapia |
|
PD-L1 positivo |
PD-L1 negativo |
|||
|
SLP Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.55 (0.40, 0.76) |
--- |
0.81 (0.50, 1.31) |
--- |
|
SG Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.90 (0.63, 1.28) |
--- |
1.18 (0.70, 1.99) |
--- |
|
% de TRG |
25% |
4% |
10% |
8% |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con quimioterapia) de acuerdo al modelo de riesgos proporcionales de Cox estratificado |
||||
Se realizó un análisis de subgrupos como parte del análisis final del ensayo KEYNOTE-006 en pacientes que eran PD-L1 positivo (n=671; 80%) frente a PD-L1 negativo (n=150; 18%). Entre los pacientes que eran evaluables en cuanto a expresión de PD-L1 (98%), el 82% eran PD-L1 positivo y el 18% eran PD-L1 negativo. La Tabla 9 resume los resultados de eficacia por expresión de PD- L1.
|
Tabla 9: Resultados de eficacia por expresión de PD-L1 en el ensayo KEYNOTE-006 |
||||
|
Variable |
Pembrolizumab 10 mg/kg cada 2 o 3 semanas (en conjunto) |
Ipilimumab |
Pembrolizumab 10 mg/kg cada 2 o 3 semanas (en conjunto) |
Ipilimumab |
|
PD-L1 positivo |
PD-L1 negativo |
|||
|
SLP Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.53 (0.44, 0.65) |
--- |
0.87 (0.58, 1.30) |
--- |
|
SG Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.63 (0.50, 0.80) |
--- |
0.76 (0.48, 1.19) |
--- |
|
% de TRG |
40% |
14% |
24% |
13% |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con ipilimumab) de acuerdo al modelo de riesgos proporcionales de Cox estratificado |
||||
Melanoma ocular:
En 20 sujetos con melanoma ocular incluidos en el ensayo KEYNOTE-001, no se notificaron respuestas objetivas; se comunicó enfermedad estable en 6 pacientes.
KEYNOTE-054: Ensayo controlado con placebo para el tratamiento adyuvante de pacientes con resección completa del melanoma
La eficacia de pembrolizumab se investigó en el ensayo KEYNOTE-054, un ensayo multicéntrico, aleatorizado, doble ciego, controlado con placebo en pacientes con melanoma resecado completamente en estadio IIIA (metástasis a ganglios linfáticos > 1 mm), IIIB o IIIC. Un total de 1.019 pacientes adultos fueron aleatorizados (1:1) a recibir pembrolizumab 200 mg cada 3 semanas (n=514) o placebo (n=505), hasta un año hasta recidiva de la enfermedad o toxicidad inaceptable. La aleatorización se estratificó mediante el estadio de la 7ª edición del American Joint Committee on Cancer (AJCC, por sus siglas en inglés) (IIIA frente a IIIB frente a IIIC 1-3 ganglios linfáticos positivos frente a IIIC ≥ 4 ganglios linfáticos positivos) y por región geográfica (Norteamérica, países europeos, Australia y otros países designados). Los pacientes debían haber sido sometidos a disección de ganglios linfáticos y, si estaba indicada, a radioterapia dentro de las 13 semanas antes de comenzar el tratamiento. Se excluyeron los pacientes con enfermedad autoinmune activa o con una enfermedad que precisara immunosupresión o con melanoma ocular o de las mucosas. Se excluyeron los pacientes que recibieron tratamiento previo para el melanoma diferente de la cirugía o interferón para melanomas primarios gruesos sin evidencia de afectación de ganglios linfáticos. Los pacientes fueron sometidos a técnicas de imagen cada 12 semanas después de la primera dosis de pembrolizumab durante los dos primeros años, después cada 6 meses desde el año 3 al 5 y, a continuación, anualmente.
Entre los 1.019 pacientes, las características basales fueron: mediana de edad de 54 años (25% de 65 años o más); 62% varones; y con un estado funcional ECOG de 0 (94%) y de 1 (6%). El 16% tenía estadio IIIA; el 46% estadio IIIB; el 18% estadio IIIC (1-3 ganglios linfáticos positivos) y el 20% estadio IIIC (≥ 4 ganglios linfáticos positivos); el 50% era positivo para la mutación de BRAF V600 y el 44% no tenía mutación de BRAF. La expresión de PD-L1 se estudió de manera retrospectiva mediante un test de inmunohistoquímica con el anticuerpo anti PD-L1 22C3; el 84% de los pacientes presentaron melanoma PD-L1 positivo (expresión de PD-L1 en ≥ 1% de las células tumorales y las células inmunitarias asociadas al tumor, relativa a todas las células tumorales viables). Se usó el mismo sistema de puntuación para melanoma metastásico (puntuación de melanoma).
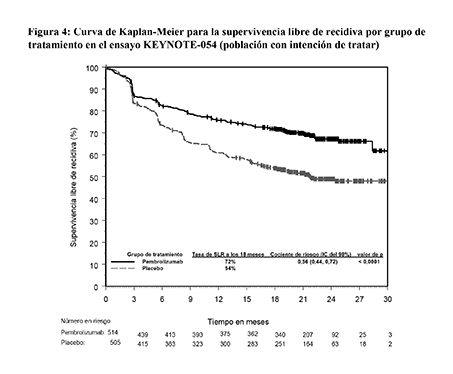
Las variables principales de eficacia fueron la supervivencia libre de recidiva (SLR) evaluada por el investigador en la población total y en la población con tumores PD-L1 positivos, donde la SLR fue definida como el tiempo entre la fecha de la aleatorización y la fecha de la primera recidiva (local, regional o metástasis a distancia) o la muerte, lo que ocurriera primero. El ensayo demostró una mejora estadísticamente significativa de la SLR en los pacientes aleatorizados al grupo de pembrolizumab comparado con placebo en el análisis intermedio preespecificado. Los resultados de eficacia basados en 7 meses adicionales de seguimiento se resumen en la Tabla 10 y en la Figura 4.
|
Tabla 10: Resultados de eficacia en el ensayo KEYNOTE-054 |
||
|
Variable |
KEYTRUDA® 200 mg cada 3 semanas n=514 |
Placebo n=505 |
|
Número (%) de pacientes con acontecimiento |
158 (31%) |
246 (49%) |
|
Mediana en meses (IC del |
NA |
21.7 |
|
95%) |
(17.1, NA) |
|
|
Cociente de riesgo (Hazard ratio)* (IC del 98%) |
0.56 (0.44, 0.72) |
|
|
Valor de p (orden logarítmico estratificado) |
< 0.0001 |
|
|
SLR a los 6 meses |
||
|
Tasa de SLR |
82% |
73% |
|
SLR a los 12 meses |
||
|
Tasa de SLR |
76% |
61% |
|
SLR a los 18 meses |
||
|
Tasa de SLR |
72% |
54% |
|
* De acuerdo al modelo de riesgos proporcionales de Cox estratificado NA = no alcanzada |
||
El ensayo KEYNOTE-054 incluyó a pacientes estratificados según la 7ª edición del AJCC y se realizó un análisis de subgrupos de la SLR según la 8ª edición del AJCC después de notificarse los resultados de SLR del ensayo. En la población total se demostró una mejora estadísticamente significativa de la SLR en los pacientes aleatorizados al grupo de pembrolizumab comparado con placebo en todos los tipos de melanoma en estadio III resecados según la 7ª edición del AJCC. El melanoma estadio IIIA de acuerdo con la 8ª edición del AJCC identifica a una población de pacientes con un mejor pronóstico comparado con el estadio IIIA de acuerdo con la 7ª edición del AJCC. Según la clasificación de la 8ª edición del AJCC, un total de 82 sujetos se clasificaron como estadio IIIA; 42 en el grupo de pembrolizumab y 40 en el grupo placebo; con un total de 13 acontecimientos de SLR; 6 en el grupo de pembrolizumab y 7 en el grupo placebo. Los datos son limitados en los sujetos con estadio IIIA de acuerdo con la 8ª edición del AJCC en el momento de este análisis de la SLR.
Figura 4: Curva de Kaplan-Meier para la supervivencia libre de recidiva por grupo de tratamiento en el ensayo KEYNOTE-054 (población con intención de tratar)
Si bien el análisis en pacientes con tumores PD-L1 positivos fue una variable co-primaria, se realizaron análisis de subgrupos predefinidos en pacientes cuyos tumores eran PD-L1 negativos, con mutación de BRAF positiva o negativa. La Tabla 11 resume los resultados de eficacia por expresión de PD-L1 y estado de mutación de BRAF.
|
Tabla 11: Resultados de eficacia por expresión de PD-L1 y por estado de mutación de BRAF en el ensayo KEYNOTE-054 |
||||
|
Variable |
Pembrolizumab 200 mg cada 3 semanas |
Placebo |
Pembrolizumab 200 mg cada 3 semanas |
Placebo |
|
PD-L1 positivo |
PD-L1 negativo |
|||
|
n=428 |
n=425 |
n=59 |
n=57 |
|
|
SLR Cociente de riesgo |
0.54 |
--- |
0.47 |
--- |
|
(Hazard ratio)* (IC del |
(0.42, 0.69) |
(0.26, 0.85) |
||
|
95%) |
||||
|
Tasa de SLR a los 6 meses |
84% |
75% |
81% |
64% |
|
Mutación de BRAF positiva |
Mutación de BRAF negativa |
|||
|
n=245 |
n=262 |
n=233 |
n=214 |
|
|
SLR Cociente de riesgo |
0.49 |
--- |
0.64 |
--- |
|
(Hazard ratio)* (IC del |
(0.36, 0.67) |
(0.47, 0.87) |
||
|
95%) |
||||
|
Tasa de SLR a los 6 meses |
83% |
73% |
80% |
72% |
|
* De acuerdo al modelo de riesgos proporcionales de Cox estratificado |
||||
CPNM KEYNOTE-024: Ensayo controlado en pacientes con CPNM sin tratamiento previo
La seguridad y la eficacia de pembrolizumab se investigó en el ensayo KEYNOTE-024, un ensayo multicéntrico, controlado, para el tratamiento del CPNM metastásico no tratado previamente. Los pacientes tenían expresión de PD-L1 con una proporción de marcador tumoral (TPS, por sus siglas en inglés) ≥50% de acuerdo al Kit de diagnóstico por inmunohistoquímica (IHQ) de PD-L1 22C3 pharmDxTM. Los pacientes fueron aleatorizados (1:1) a recibir pembrolizumab a una dosis de 200 mg cada 3 semanas (n=154) o quimioterapia con platino a elección del investigador (n=151; que incluyen pemetrexed+carboplatino, pemetrexed+cisplatino, gemcitabina+cisplatino, gemcitabina+carboplatino o paclitaxel+carboplatino. Los pacientes con CPNM no escamoso pudieron recibir mantenimiento con pemetrexed). Los pacientes fueron tratados con pembrolizumab hasta toxicidad inaceptable o progresión de la enfermedad. El tratamiento pudo continuar tras la progresión de la enfermedad, si el paciente estaba clínicamente estable y el investigador consideraba que estaba obteniendo beneficio clínico. Los pacientes sin progresión de la enfermedad pudieron ser tratados hasta 24 meses. El ensayo excluyó a los pacientes con mutaciones tumorales positivas de EGFR o ALK; enfermedad autoinmune que precisara tratamiento sistémico dentro de los 2 años de tratamiento; un problema médico que precisara inmunosupresión; o que hubieran recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. Se realizó evaluación del estado tumoral cada 9 semanas. Los pacientes en quimioterapia que experimentaron progresión de la enfermedad, verificada de forma independiente, pudieron cambiar y recibir pembrolizumab.
Entre los 305 pacientes del ensayo KEYNOTE-024, las características basales fueron: mediana de edad de 65 años (54% de 65 años o más); 61% varones; 82% de raza blanca; 15% asiáticos y 35% y 65% con un estado funcional ECOG de 0 y 1, respectivamente. Las características de la
enfermedad fueron CPNM escamoso (18%) y no escamoso (82%); M1 (99%) y metástasis cerebrales (9%).
La variable principal de eficacia fue la SLP evaluada mediante RCIE usando los criterios RECIST
Las variables secundarias de eficacia fueron la SG y la tasa de respuesta objetiva (TRO) (evaluadas mediante RCIE usando los criterios RECIST 1.1). La tabla 12 resume las variables principales de eficacia para toda la población con intención de tratar. Los resultados de SLP y TRO se notifican a partir de un análisis provisional, con una mediana de seguimiento de 11 meses. Los resultados de SG se notifican del análisis final, con una mediana de seguimiento de 25 meses.
|
Tabla 12: Resultados de eficacia en el ensayo KEYNOTE-024 |
||
|
Variable |
Pembrolizumab 200 mg cada 3 semanas n=154 |
Quimioterapia n=151 |
|
SLP |
||
|
Número (%) de pacientes con evento |
73 (47%) |
116 (77%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.50 (0.37, 0.68) |
|
|
Valor de p† |
<0.001 |
|
|
Mediana en meses (IC del 95%) |
10.3 (6.7, ND) |
6.0 (4.2, 6.2) |
|
SG |
||
|
Número (%) de pacientes con evento |
73 (47%) |
96 (64%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.63 (0.47, 0.86) |
|
|
Valor de p† |
0.002 |
|
|
Mediana en meses (IC del 95%) |
30 (18.3, ND) |
14.2 (9.8, 19.0) |
|
Tasa de respuesta objetiva |
||
|
% de TRO (IC del 95%) |
45% (37, 53) |
28% (21, 36) |
|
% de respuesta completa |
4% |
1% |
|
% de respuesta parcial |
41% |
27% |
|
Duración de la respuesta‡ |
||
|
Mediana en meses (rango) |
No alcanzada (1.9+, 14.5+) |
6.3 (2.1+, 12.6+) |
|
% con duración ≥ 6 meses |
88%§ |
59%¶ |
|
Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con quimioterapia) de acuerdo al modelo de riesgos proporcionales de Cox estratificado † Basado en la prueba de orden logarítmico estratificada ‡ Basado en pacientes con una mejor respuesta como respuesta completa o parcial confirmada § Basado en las estimaciones de Kaplan-Meier; incluye 43 pacientes con respuestas de 6 meses o más ¶ Basado en las estimaciones de Kaplan-Meier; incluye a 16 pacientes con respuestas de 6 meses o más ND = no disponible |
||
Figura 5: Curva de Kaplan-Meier para la supervivencia libre de progresión por grupo de tratamiento en el ensayo KEYNOTE-024 (población con intención de tratar)
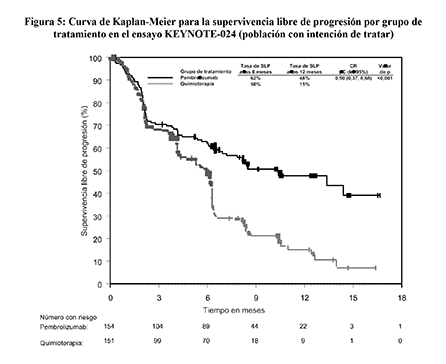
Figura 6: Curva de Kaplan-Meier para la supervivencia global por grupo de tratamiento en el ensayo KEYNOTE-024 (población con intención de tratar)
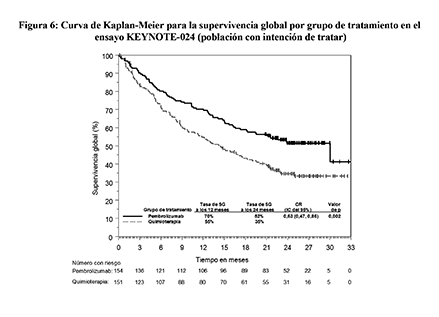
En el análisis de subgrupos, se observó un beneficio reducido de la supervivencia con pembrolizumab comparado con quimioterapia, en el bajo número de pacientes que nunca fueron fumadores; sin embargo, debido al bajo número de pacientes, no se pueden extraer conclusiones definitivas de estos datos.
KEYNOTE-189: Ensayo controlado del tratamiento en combinación en pacientes con CPNM no escamoso sin tratamiento previo
La eficacia de pembrolizumab en combinación con pemetrexed y quimioterapia basada en platino se investigó en el ensayo KEYNOTE-189, un ensayo multicéntrico, aleatorizado, controlado con principio activo, doble ciego. Los principales criterios de inclusión fueron pacientes con CPNM no escamoso metastásico, sin tratamiento sistémico previo para CPNM metastásico y sin mutaciones tumorales positivas de EGFR o ALK. El ensayo excluyó a los pacientes con enfermedad autoinmune que precisara tratamiento sistémico dentro de los 2 años de tratamiento; un problema médico que precisara inmunosupresión; o que hubieran recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. Los pacientes fueron aleatorizados (2:1) a recibir uno de las siguientes pautas de tratamiento:
Pembrolizumab 200 mg con pemetrexed 500 mg/m2 y, a elección del investigador, cisplatino.
75 mg/m2 o carboplatino AUC 5 mg/ml/min por vía intravenosa cada 3 semanas durante 4 ciclos seguido de pembrolizumab 200 mg y pemetrexed 500 mg/m2 por vía intravenosa cada 3 semanas (n=410).
Placebo con pemetrexed 500 mg/m2 y, a elección del investigador, cisplatino 75 mg/m2 o carboplatino AUC 5 mg/ml/min por vía intravenosa cada 3 semanas durante 4 ciclos seguido de placebo y pemetrexed 500 mg/m2 por vía intravenosa cada 3 semanas (n=206).
El tratamiento con pembrolizumab continuó hasta progresión de la enfermedad determinada por el investigador, definida según los criterios RECIST 1.1, toxicidad inaceptable o hasta un máximo de 24 meses. La administración de pembrolizumab podía continuar más allá de la progresión de la enfermedad definida según los criterios RECIST mediante RCIE o después de la suspensión de pemetrexed si el paciente estaba clínicamente estable y el investigador consideraba que estaba obteniendo beneficio clínico. En los pacientes que finalizaron los 24 meses de tratamiento o que mostraron respuesta completa, se pudo reiniciar el tratamiento con pembrolizumab para la progresión de la enfermedad y administrarse hasta 1 año adicional. Se realizó la evaluación del estado tumoral a las 6 y a las 12 semanas, seguido posteriormente por la evaluación cada 9 semanas. A los pacientes que recibían placebo y quimioterapia que experimentaron progresión de la enfermedad verificada independientemente se les ofreció pembrolizumab como monoterapia.
Entre los 616 pacientes del ensayo KEYNOTE-189, las características basales fueron: mediana de edad de 64 años (49% de 65 años o más); 59% varones; 94% de raza blanca y 3% de raza asiática; 43% y 56% con un estado funcional ECOG de 0 o 1, respectivamente; 31% PD-L1 negativo (TPS < 1%); y 18% con metástasis cerebrales tratadas o no tratadas en el estado basal. Un total de 67 pacientes del grupo de placebo más quimioterapia pasaron a recibir pembrolizumab en monoterapia en el momento de la progresión de la enfermedad y otros 18 pacientes más recibieron un inhibidor de puntos de control como tratamiento posterior.
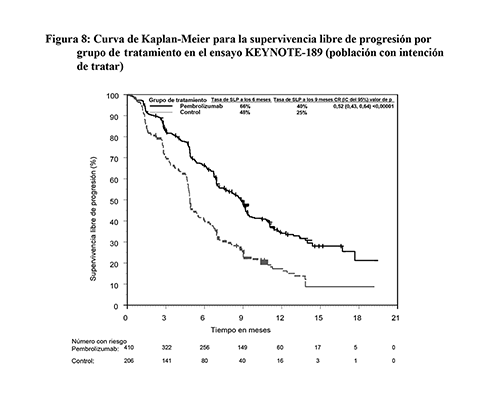
Las variables principales de eficacia fueron la SG y la SLP (evaluada mediante RCIE usando los criterios RECIST 1.1). Las variables secundarias de eficacia fueron la tasa de respuesta objetiva (TRO) y la duración de la respuesta, evaluada mediante RCIE usando los criterios RECIST 1.1. La mediana de seguimiento fue de 10.5 meses (rango: 0.2 a 20.4 meses). La Tabla 13 resume las variables principales de eficacia. En las Figuras 7 y 8 se muestran las curvas de Kaplan-Meier para la SG y la SLP.
|
Tabla 13: Resultados de eficacia en el ensayo KEYNOTE-189 |
||
|
Variable |
Pembrolizumab + Pemetrexed + Quimioterapia basada en platino n=410 |
Placebo + Pemetrexed + Quimioterapia basada en platino n=206 |
|
SG |
||
|
Número (%) de pacientes con acontecimiento |
127 (31%) |
108 (52%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.49 (0.38, 0.64) |
|
|
Valor de p† |
< 0.00001 |
|
|
Mediana en meses (IC del 95%) |
No alcanzada (ND, ND) |
11.3 (8.7, 15.1) |
|
SLP |
||
|
Número (%) de pacientes con acontecimiento |
244 (60%) |
166 (81%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.52 (0.43, 0.64) |
|
|
Valor de p† |
< 0.00001 |
|
|
Mediana en meses (IC del 95%) |
8.8 (7.6, 9.2) |
4.9 (4.7, 5.5) |
|
Tasa de respuesta objetiva |
||
|
% de TRO‡ (IC del 95%) |
48% (43, 53) |
19% (14, 25) |
|
% de respuesta completa |
0.5% |
0.5% |
|
% de respuesta parcial |
47% |
18% |
|
Valor de p§ |
< 0.0001 |
|
|
Duración de la respuesta |
||
|
Mediana en meses (rango) |
11.2 (1.1+, 18.0+) |
7.8 (2.1+, 16.4+) |
|
% con duración ≥ 6 meses¶ |
81% |
63% |
|
% con duración ≥ 9 meses¶ |
60% |
44% |
|
* Basado en el modelo de riesgos proporcionales de Cox estratificado † Basado en la prueba de orden logarítmico estratificada ‡ Basado en pacientes con una mejor respuesta global como respuesta completa o parcial confirmada § Basado en el método de Miettinen y Nurminen estratificado por estado de PD-L1, quimioterapia basada en platino y estado de tabaquismo ¶ Basado en la estimación de Kaplan-Meier ND = no disponible |
||
Figura 7: Curva de Kaplan-Meier para la supervivencia global por grupo de tratamiento en el ensayo KEYNOTE-189 (población con intención de tratar)
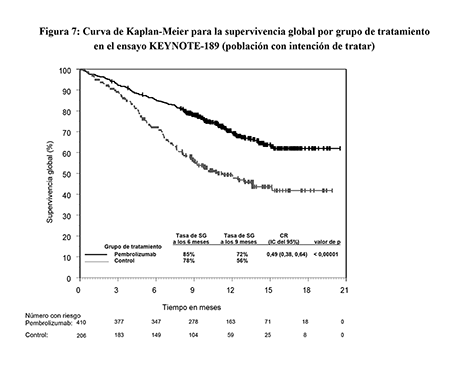
Figura 8: Curva de Kaplan-Meier para la supervivencia libre de progresión por grupo de tratamiento en el ensayo KEYNOTE-189 (población con intención de tratar)
En el ensayo KEYNOTE-189 se realizó un análisis en los pacientes que tenían expresión de PD-L1 con una TPS < 1% [pembrolizumab en combinación: n=127 (31%) frente a quimioterapia: n=63 (31%)], TPS 1-49% [pembrolizumab en combinación: n=128 (31%) frente a quimioterapia: n=58 (28%)] o TPS ≥ 50% [pembrolizumab en combinación: n=132 (32%) frente a quimioterapia: n=70 (34%)] (ver Tabla 14).
|
Tabla 14: Resultados de eficacia por expresión de PD-L1 en el ensayo KEYNOTE-189 |
||||||
|
Variable |
Tratamiento con pembrolizumab en combinación |
Quimioterapia |
Tratamiento con pembrolizumab en combinación |
Quimioterapia |
Tratamiento con pembrolizumab en combinación |
Quimioterapia |
|
TPS < 1% |
TPS 1 a 49% |
TPS ≥ 50% |
||||
|
SG Cociente de riesgo (Hazard ratio)* (IC del 95%) |
0.59 (0.38, 0.92) |
0.55 (0.34, 0.90) |
0.42 (0.26, 0.68) |
|||
|
SLP Cociente de riesgo (Hazard ratio)* (IC del 95%) |
0.75 (0.53, 1.05) |
0.55 (0.37, 0.81) |
0.36 (0.25, 0.52) |
|||
|
% de TRG |
32% |
14% |
48% |
21% |
61% |
23% |
|
* Cociente de riesgo (Hazard ratio) (tratamiento con pembrolizumab en combinación frente a quimioterapia) basado en el modelo de riesgos proporcionales Cox estratificados |
||||||
Se incluyó un total de 57 pacientes con CPNM de edad ≥ 75 años en el ensayo KEYNOTE-189 (35 en pembrolizumab en combinación y 22 en control). Dentro de este subgrupo de estudio el cociente de riesgo (Hazard ratio) fue HR=2.09 [IC del 95% 0.84, 5.23] en SG y el cociente de riesgo (Hazard ratio) fue HR=1.73 [IC del 95% 0.77, 3.90] en SLP para el tratamiento con pembrolizumab en combinación frente a quimioterapia. Los datos sobre eficacia y seguridad de pembrolizumab en combinación con quimioterapia basada en platino son limitados en esta población de pacientes.
KEYNOTE-407: Ensayo controlado del tratamiento en combinación en pacientes con CPNM escamoso sin tratamiento previo
La eficacia de pembrolizumab en combinación con carboplatino y paclitaxel o nab-paclitaxel se investigó en el ensayo KEYNOTE-407, un ensayo aleatorizado, doble ciego, multicéntrico, controlado con placebo. Los principales criterios de inclusión para este ensayo fueron CPNM escamoso metastásico, con independencia del estado de expresión tumoral de PD-L1 y sin tratamiento sistémico previo para enfermedad metastásica. Se excluyeron los pacientes con enfermedad autoinmune que precisara tratamiento sistémico dentro de los 2 años de tratamiento; pacientes con una enfermedad que precisara inmunosupresión; o que hubieran recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. La aleatorización se estratificó por expresión tumoral de PD-L1 (TPS < 1% [negativa] frente a TPS ≥ 1%), elección del investigador entre paclitaxel o nab-paclitaxel y región geográfica (Este asiático frente a lugares fuera del Este asiático). Los pacientes fueron aleatorizados (1:1) a uno de los siguientes grupos de tratamiento mediante infusión intravenosa:
• Pembrolizumab 200 mg y carboplatino AUC 6 mg/ml/min el Día 1 de cada ciclo de 21 días durante 4 ciclos y paclitaxel 200 mg/m2 el Día 1 de cada ciclo de 21 días durante 4 ciclos o nab-paclitaxel 100 mg/m2 los Días 1, 8 y 15 de cada ciclo de 21 días durante 4 ciclos, seguido por pembrolizumab 200 mg cada 3 semanas. Pembrolizumab se administró antes de la quimioterapia el Día 1.
• Placebo y carboplatino AUC 6 mg/ml/min el Día 1 de cada ciclo de 21 días durante 4 ciclos y paclitaxel 200 mg/m2 el Día 1 de cada ciclo de 21 días durante 4 ciclos o nab-paclitaxel 100 mg/m2 los Días 1, 8 y 15 de cada ciclo de 21 días durante 4 ciclos, seguido por placebo cada 3 semanas.
El tratamiento con pembrolizumab o placebo continuó hasta progresión de la enfermedad definida según los criterios RECIST 1.1, determinada mediante una Revisión Central Independiente Enmascarada (RCIE), toxicidad inaceptable o un máximo de 24 meses. Se permitió la administración de pembrolizumab más allá de la progresión de la enfermedad definida según los criterios RECIST si el paciente estaba clínicamente estable y el investigador consideraba que estaba obteniendo beneficio clínico.
A los pacientes del grupo placebo se les ofreció pembrolizumab como monoterapia en el momento de la progresión de la enfermedad.
Se realizó la evaluación del estado tumoral cada 6 semanas hasta la semana 18, cada 9 semanas hasta la semana 45 y posteriormente cada 12 semanas.
Se aleatorizó un total de 559 pacientes. Las características de la población del ensayo fueron: mediana de edad de 65 años (rango: 29 a 88); 55% de 65 años o más; 81% varones; 77% de raza blanca; estado funcional ECOG de 0 (29%) y 1 (71%); y un 8% con metástasis cerebrales tratadas en el estado basal. El 35% tenía una TPS de expresión tumoral de PD-L1 < 1% [negativa]; el 19% era del Este asiático y el 60% recibió paclitaxel.
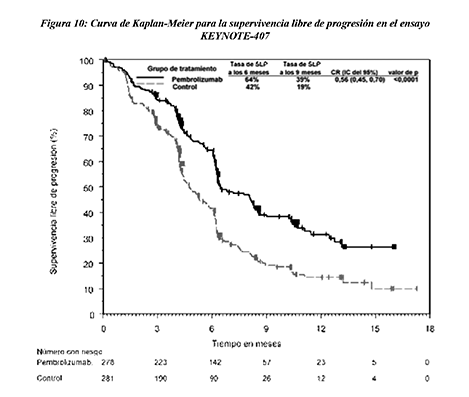
Las variables principales de eficacia fueron la SG y la SLP (evaluadas mediante RCIE según los criterios RECIST 1.1). Las variables secundarias de eficacia fueron la TRG y la duración de la respuesta, evaluadas mediante RCIE usando los criterios RECIST 1.1. La mediana de seguimiento fue de 7.8 meses (rango: 0.1 a 19.1 meses). La Tabla 15 resume las variables principales de eficacia. En las Figuras 9 y 10 se muestran las curvas de Kaplan-Meier para la SG y la SLP.
|
Tabla 15: Resultados de eficacia en el ensayo KEYNOTE-407 |
||
|
Variable |
Pembrolizumab carboplatino paclitaxel/nab-paclitaxel n=278 |
Placebo carboplatino paclitaxel/nab-paclitaxel n=281 |
|
SG |
||
|
Número de acontecimientos (%) |
85 (31%) |
120 (43%) |
|
Mediana en meses (IC del 95%) |
15.9 (13.2, ND) |
11.3 (9.5, 14.8) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.64 (0.49, 0.85) |
|
|
Valor de p† |
0.0008 |
|
|
SLP |
||
|
Número de acontecimientos (%) |
152 (55%) |
197 (70%) |
|
Mediana en meses (IC del 95%) |
6.4 (6.2, 8.3) |
4.8 (4.3, 5.7) |
|
Cociente de riesgo* (IC del 95%) |
0.56 (0.45, 0.70) |
|
|
Valor de p† |
< 0.0001 |
|
|
Tasa de respuesta global |
||
|
Tasa de respuesta global (IC del 95%) |
58% (52, 64) |
38% (33, 44) |
|
% de respuesta completa |
1.4% |
2.1% |
|
% de respuesta parcial |
57% |
36% |
|
Valor de p‡ |
< 0.0001 |
|
|
Duración de la respuesta |
||
|
Mediana de duración de la respuesta en meses (intervalo) |
7.7 (1.1+, 14.7+) |
4.8 (1.3+, 15.8+) |
|
% con duración ≥ 6 meses§ |
62% |
40% |
|
* Basado en el modelo de riesgos proporcionales de Cox estratificado † Basado en la prueba de orden logarítmico estratificada ‡ Basado en el método de Miettinen y Nurminen § Basado en la estimación de Kaplan-Meier ND = no disponible |
||
Figura 9: Curva de Kaplan-Meier para la supervivencia global en el ensayo KEYNOTE-407
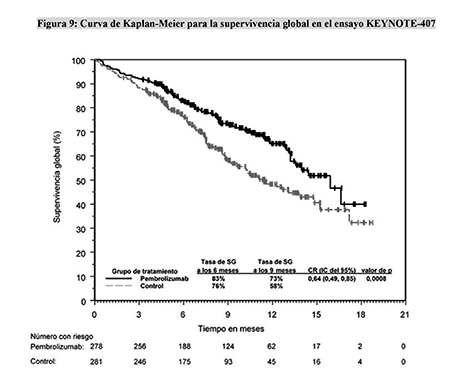
Figura 10: Curva de Kaplan-Meier para la supervivencia libre de progresión en el ensayo KEYNOTE-407
En el ensayo KEYNOTE-407 se realizó un análisis en pacientes que tenían expresión de PD-L1 con una TPS < 1% [grupo de pembrolizumab más quimioterapia: n=95 (34%) frente al grupo de placebo más quimioterapia: n=99 (35%)], TPS 1% a 49% [grupo de pembrolizumab más quimioterapia: n=103 (37%) frente a grupo de placebo más quimioterapia: n=104 (37%)] o TPS ≥ 50% [grupo de pembrolizumab más quimioterapia: n=73 (26%) frente a grupo de placebo más quimioterapia: n=73 (26%)] (ver Tabla 16).
|
Tabla 16: Resultados de eficacia por expresión de PD-L1 en el ensayo KEYNOTE-407 |
|||||||
|
Variable |
Tratamiento con pembrolizumab en combinación |
Quimioterapia |
Tratamiento con pembrolizumab en combinación |
Quimioterapia |
Tratamiento con pembrolizumab en combinación |
Quimioterapia |
|
|
TPS < 1% |
TPS 1 a 49% |
TPS ≥ 50% |
|||||
|
SG Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.61 (0.38, 0.98) |
0.57 (0.36, 0.90) |
0.64 (0.37, 1.10) |
||||
|
SLP Cociente de riesgo (Hazard ratio)* (IC del 95%) |
0.68 (0.47, 0.98) |
0.56 (0.39, 0.80) |
0.37 (0.24, 0.58) |
||||
|
% de TRG |
63% |
40% |
50% |
41% |
60% |
33% |
|
|
* Cociente de riesgo (Hazard ratio) (tratamiento con pembrolizumab en combinación frente a quimioterapia) basado en el modelo de riesgos proporcionales de Cox estratificado |
|||||||
Se incluyó un total de 65 pacientes con CPNM de edad ≥ 75 años en el ensayo KEYNOTE-407 (34 en el grupo de pembrolizumab en combinación y 31 en el control). Dentro de este subgrupo de estudio el cociente de riesgo (Hazard Ratio) fue HR=0.96 [IC del 95% 0.37, 2.52] en SG, un HR=0.60 [IC del 95% 0.29,1.21] en SLP y una TRG del 47% y el 42% para pembrolizumab en combinación frente a quimioterapia. Los datos de eficacia y seguridad de pembrolizumab en combinación con quimioterapia basada en platino son limitados en esta población de pacientes.
KEYNOTE-010: Ensayo controlado en pacientes con CPNM tratados previamente con quimioterapia La seguridad y la eficacia de pembrolizumab se investigó en el ensayo KEYNOTE-010, un ensayo multicéntrico, abierto, controlado, para el tratamiento del CPNM avanzado en pacientes tratados previamente con quimioterapia que incluyera platino. Los pacientes tenían expresión de PD-L1 con una TPS ≥ 1% de acuerdo al Kit de diagnóstico por inmunohistoquímica (IHQ) de PD-L1 22C3 pharmDxTM. Los pacientes con activación de las mutaciones de EGFR o translocación de ALK también tuvieron progresión de la enfermedad con el tratamiento aprobado para estas mutaciones antes de recibir pembrolizumab. Los pacientes fueron aleatorizados (1:1:1) a recibir pembrolizumab a una dosis de 2 mg (n=344) o 10 mg/kg (n=346) cada 3 semanas o docetaxel a una dosis de 75 mg/m2 cada 3 semanas (n=343) hasta progresión de la enfermedad o toxicidad inaceptable. El ensayo excluyó a los pacientes con enfermedad autoinmune; un problema médico que precisara inmunosupresión; o que hubieran recibido más de 30 Gy de radiación torácica dentro de las 26 semanas previas. Se realizó evaluación del estado tumoral cada 9 semanas.
Las características basales para esta población incluyeron: mediana de edad de 63 años (42% de 65 años o más); 61% varones; 72% de raza blanca y 21% asiáticos y 34% y 66% con un estado funcional ECOG de 0 y 1, respectivamente. Las características de la enfermedad fueron CPNM escamoso (21%) y no escamoso (70%); M1 (91%); metástasis cerebrales estables (15%) y la incidencia de mutaciones fue de EGFR (8%) o de ALK (1%). El tratamiento previo incluyó un régimen en doblete con platino (100%); los pacientes recibieron una (69%) o dos o más (29%) líneas de tratamiento.
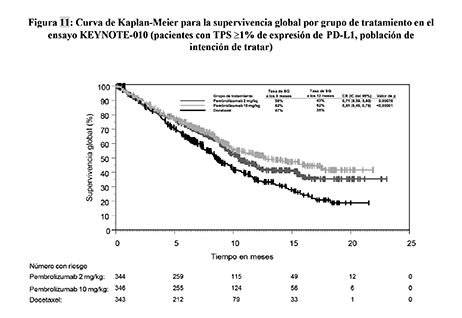
Las variables principales de eficacia fueron la SG y la SLP evaluadas mediante una Revisión Central Independiente Enmascarada (RCIE) usando los criterios RECIST 1.1. Las variables secundarias de resultados de eficacia fueron la TRG y la duración de la respuesta. La Tabla 17 resume las variables principales de eficacia para toda la población (TPS ≥ 1%) y para los pacientes con TPS ≥ 50% y en la Figura 11 se muestra la curva de Kaplan-Meier para la SG (TPS ≥ 1%).
|
Tabla 17: Respuesta a pembrolizumab 2 o 10 mg/kg cada 3 semanas en pacientes tratados previamente, con CPNM, en el ensayo KEYNOTE-010 |
|||
|
Variable principal |
Pembrolizumab 2 mg/kg cada 3 semanas |
Pembrolizumab 10 mg/kg cada 3 semanas |
Docetaxel 75 mg/m2 cada 3 semanas |
|
TPS ≥ 1% |
|||
|
Número de pacientes |
344 |
346 |
343 |
|
SG |
|||
|
Número (%) de los pacientes con acontecimiento |
172 (50%) |
156 (45%) |
193 (56%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.71 (0.58, 0.88) |
0.61 (0.49, 0.75) |
--- |
|
Valor de p† |
< 0.001‡ |
< 0.001‡ |
--- |
|
Mediana en meses (IC del 95%) |
10.4 (9.4, 11.9) |
12.7 (10.0, 17.3) |
8.5 (7.5, 9.8) |
|
SLP§ |
|||
|
Número (%) de pacientes con acontecimiento |
266 (77%) |
255 (74%) |
257 (75%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.88 (0.73, 1.04) |
0.79 (0.66, 0.94) |
--- |
|
Valor de p† |
0.068 |
0.005 |
--- |
|
Mediana en meses (IC del 95%) |
3.9 (3.1, 4.1) |
4.0 (2.6, 4.3) |
4.0 (3.1, 4.2) |
|
Tasa de respuesta global§ |
|||
|
% de TRG¶ (IC del 95%) |
18% (14, 23) |
18% (15, 23) |
9% (7, 13) |
|
Duración de la respuesta§,#,? |
|||
|
Mediana en meses (rango) |
No alcanzada (0.7+, 20.1+) |
No alcanzada (2.1+, 17.8+) |
6,2 (1.4+, 8.8+) |
|
% que continúan |
73% |
72% |
34% |
|
TPS ≥ 50% |
|||
|
Número de pacientes |
139 |
151 |
152 |
|
SG |
|||
|
Número (%) de los pacientes con acontecimiento |
58 (42%) |
60 (40%) |
86 (57%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.54 (0.38, 0.77) |
0.50 (0.36, 0.70) |
--- |
|
Valor de p† |
< 0.001‡ |
< 0.001‡ |
--- |
|
Mediana en meses (IC del 95%) |
14.9 (10.4, ND) |
17.3 (11.8, ND) |
8.2 (6.4, 10.7) |
|
SLP§ |
|||
|
Número (%) de pacientes con acontecimiento |
89 (64%) |
97 (64%) |
118 (78%) |
|
Cociente de riesgo (Hazard Ratio)* (IC del 95%) |
0.58 (0.43, 0.77) |
0.59 (0.45, 0.78) |
--- |
|
Valor de p† |
< 0.001‡ |
< 0.001‡ |
--- |
|
Mediana en meses (IC del 95%) |
5.2 (4.0, 6.5) |
5.2 (4.1, 8.1) |
4.1 (3.6, 4.3) |
|
Tasa de respuesta global§ |
|||
|
% de TRG¶ (IC del 95%) |
30% (23, 39) |
29% (22, 37) |
8% (4, 13) |
|
Duración de la respuesta§,#,ß |
|||
|
Mediana en meses (rango) |
No alcanzada (0.7+, 16.8+) |
No alcanzada (2.1+, 17.8+) |
8.1 (2.1+, 8.8+) |
|
% que continúa |
76% |
75% |
33% |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con docetaxel) de acuerdo al modelo de riesgos proporcionales de Cox estratificado † Basado en la prueba de orden logarítmico estratificada ‡ Estadísticamente significativo de acuerdo a un nivel de α pre especificado ajustado por multiplicidad § Evaluado mediante revisión central independiente enmascarada (RCIE) usando los criterios RECIST 1.1 ¶ Todas las respuestas fueron respuestas parciales # Basado en los pacientes con una mejor respuesta global de respuesta completa o parcial confirmadas Þ Incluye 30, 31 y 2 pacientes con respuestas que se mantienen de 6 meses o más en los grupos de pembrolizumab 2 mg/kg, pembrolizumab 10 mg/kg y docetaxel, respectivamente ß Incluye 22, 24 y 1 pacientes con respuestas que se mantienen de 6 meses o más en los grupos de pembrolizumab 2 mg/kg, pembrolizumab 10 mg/kg y docetaxel, respectivamente |
|||
Figura 11: Curva de Kaplan-Meier para la supervivencia global por grupo de tratamiento en el ensayo KEYNOTE-010 (pacientes con TPS ≥1% de expresión de PD-L1, población de intención de tratar)
Los resultados de eficacia fueron similares en los grupos de 2 mg/kg y 10 mg/kg de pembrolizumab. Los resultados de eficacia en la SG fueron coherentes independientemente de la antigüedad de la muestra tumoral (muestra nueva frente a muestra de archivo) de acuerdo a una comparación entre grupos.
En el análisis de subgrupo, se observó un beneficio reducido de la supervivencia con pembrolizumab comparado con docetaxel, en pacientes que nunca fueron fumadores o en pacientes con tumores que albergaban la activación de mutaciones de EGFR, que recibieron al menos quimioterapia basada en platino y un inhibidor de tirosina kinasa; sin embargo, debido al bajo número de pacientes, no se pueden extraer conclusiones definitivas de estos datos.
No se ha establecido la eficacia y seguridad de pembrolizumab en pacientes con tumores que no e xpresen PD-L1.
Linfoma de Hodgkin clásico:
KEYNOTE-087 y KEYNOTE-013: Ensayos abiertos en pacientes con linfoma de Hodgkin clásico (LHc) en recaída o refractario
La eficacia de pembrolizumab se investigó en los ensayos KEYNOTE-087 y KEYNOTE-013, dos ensayos multicéntricos, abiertos para el tratamiento de 241 pacientes con LHc. Estos ensayos incluyeron pacientes que no habían respondido a un TAPH y BV, que no eran candidatos a TAPH debido a que no fueron capaces de alcanzar una remisión completa o parcial con la quimioterapia de rescate y no habían respondido a BV, o que no habían respondido a un TAPH y no habían recibido BV. Cinco sujetos del ensayo no fueron candidatos a TAPH debido a razones distintas a la falta de respuesta a la quimioterapia de rescate. Ambos ensayos incluyeron pacientes con independencia de la expresión de PD-L1. Los pacientes con neumonitis activa no infecciosa, trasplante alogénico en los 5 años anteriores (o > 5 años, pero con EICH), enfermedad autoinmune activa o una afección que requiriera inmunosupresión, no fueron elegibles para ninguno de los ensayos. Los pacientes recibieron pembrolizumab 200 mg cada 3 semanas (n=210; KEYNOTE-087) o 10 mg/kg cada 2 semanas (n=31; KEYNOTE-013) hasta toxicidad inaceptable o progresión documentada de la enfermedad.
Entre los pacientes del ensayo KEYNOTE-087, las características basales fueron mediana de edad de 35
años (9% de edad igual o superior a 65 años); 54% varones; 88% de raza blanca; y 49% y 51% tenían un estado funcional ECOG de 0 y 1, respectivamente. La mediana del número de líneas de tratamiento previas administradas para el tratamiento de LHc fue de 4 (rango de 1 a 12). El ochenta y uno por ciento fueron refractarios al menos a una línea de tratamiento previa, incluido un 35% refractario al tratamiento de primera línea. El sesenta y uno por ciento de los pacientes había recibido un auto TPH, el 38% no era candidato a trasplante; el 17% no había usado brentuximab vedotina anteriormente; y el 36% de los pacientes se había sometido a radioterapia previa. Los subtipos de la enfermedad fueron 80% linfoma de esclerosis nodular, 11% linfoma de celularidad mixta, 4% linfoma con predominio linfocítico y 2% linfoma con depleción linfocítica.
Entre los pacientes del ensayo KEYNOTE-013, las características basales fueron mediana de edad de 32 años (7% de edad igual o superior a 65 años); 58% varones; 94% de raza blanca; y 45% y 55% tenían un estado funcional ECOG de 0 y 1, respectivamente. La mediana del número de líneas de tratamiento previas administradas para el tratamiento de LHc fue de 5 (rango de 2 a 15). El ochenta y siete por ciento fueron refractarios al menos a una línea de tratamiento previa, incluido un 39% refractario al tratamiento de primera línea. El setenta y cuatro por ciento de los pacientes habían recibido un auto TPH, el 26% no era candidato a trasplante y el 42% de los pacientes se había sometido a radioterapia previa. Los subtipos de la enfermedad fueron 97% linfoma de esclerosis nodular y 3% linfoma de celularidad mixta.
Las variables principales de eficacia (TRO y TRC) fueron evaluadas mediante una revisión central independiente enmascarada de acuerdo con los criterios del Grupo Internacional de Trabajo (IWG), revisados en 2007. Las variables secundarias de eficacia fueron la duración de la respuesta, la SLP y la SG. La respuesta se evaluó en los ensayos KN087 y KN013 cada 12 y 8 semanas, respectivamente, realizándose la primera evaluación planeada, posterior al momento basal, en la semana 12. La Tabla 18 resume los resultados de eficacia.
|
Tabla 18: Resultados de eficacia en los ensayos KEYNOTE-087 y KEYNOTE-013 |
||
|
KEYNOTE-087a |
KEYNOTE-013b |
|
|
Variable |
Pembrolizumab 200 mg cada 3 semanas n=210 |
Pembrolizumab 10 mg/kg cada 2 semanas n=31 |
|
Tasa de respuesta objetivac |
||
|
% de TRO (IC del 95%) |
69% (62.3, 75.2) |
58% (39.1, 75.5) |
|
Remisión completa |
22% |
19% |
|
Remisión parcial |
47% |
39% |
|
Duración de la respuestac |
||
|
Mediana en meses (rango) |
11.1 (0.0+, 11.1)d |
No alcanzada (0.0+, 26.1+)e |
|
% con duración ≥ 6 meses |
76%f |
80%g |
|
% con duración ≥ 12 meses |
--- |
70%h |
|
Tiempo hasta la respuesta |
||
|
Mediana en meses (rango) |
2.8 (2.1, 8.8)d |
2.8 (2.4, 8.6)e |
|
SLPc |
||
|
Número (%) de pacientes con acontecimiento |
70 (33%) |
18 (58%) |
|
Mediana en meses (IC del 95%) |
11.3 (10.8, No alcanzada) |
11.4 (4.9, 27.8) |
|
Tasa de SLP a los 6 meses |
72% |
66% |
|
Tasa de SLP a los 9 meses |
62% |
--- |
|
Tasa de SLP a los 12 meses |
--- |
48% |
|
SG |
||
|
Número (%) de pacientes con acontecimiento |
4 (2%) |
4 (13%) |
|
Tasa de SG a los 6 meses |
99.5% |
100% |
|
Tasa de SG a los 12 meses |
97.6% |
87.1% |
|
a Mediana del tiempo de seguimiento de 10.1 meses b Mediana del tiempo de seguimiento de 28.7 meses c Evaluada mediante una revisión central independiente enmascarada de acuerdo con los criterios del Grupo Internacional de Trabajo (IWG), revisados en 2007, mediante tomografía computarizada y tomografía por emisión de positrones d Basado en pacientes (n=145) con una respuesta evaluada mediante revisión independiente e Basado en pacientes (n=18) con una respuesta evaluada mediante revisión independiente f Basado en las estimaciones de Kaplan-Meier; incluye 31 pacientes con respuestas de 6 meses o más g Basado en las estimaciones de Kaplan-Meier; incluye 9 pacientes con respuestas de 6 meses o más h Basado en las estimaciones de Kaplan-Meier; incluye 7 pacientes con respuestas de 12 meses o más |
||
Seguridad y eficacia en pacientes de edad avanzada:
En total, 20 pacientes con LHc ≥ 65 años fueron tratados con pembrolizumab en los ensayos KEYNOTE- 087 y KEYNOTE-013. Los datos de estos pacientes son demasiado limitados como para extraer conclusiones sobre la seguridad o eficacia en esta población.
Carcinoma urotelial:
KEYNOTE-045: Ensayo controlado en pacientes con carcinoma urotelial que habían recibido quimioterapia previa basada en platino
La seguridad y la eficacia de pembrolizumab se investigaron en el ensayo KEYNOTE-045, un ensayo multicéntrico, aleatorizado (1:1), controlado, para el tratamiento del carcinoma urotelial localmente avanzado o metastásico en pacientes con progresión de la enfermedad durante o después de quimioterapia basada en platino. Los pacientes debían haber recibido una primera línea de tratamiento basado en platino en estadios localmente avanzados/metastásicos de la enfermedad o bien como tratamiento neoadyuvante/adyuvante, y haber tenido recidiva/progresión ≤12 meses después de haber completado el tratamiento. Los pacientes fueron aleatorizados (1:1) a recibir KEYTRUDA® 200 mg cada 3 semanas (n=270) o la terapia de elección del investigador, que incluía cualquiera de los siguientes regímenes de quimioterapia, todos ellos administrados por vía intravenosa cada 3 semanas (n=272): paclitaxel 175 mg/m2 (n=84), docetaxel 75 mg/m2 (n=84) o vinflunina 320 mg/m2 (n=87). Los pacientes recibieron pembrolizumab hasta toxicidad inaceptable o progresión de la enfermedad. El tratamiento podía continuar después de la progresión si el paciente estaba clínicamente estable y se consideraba que estaba obteniendo beneficio clínico según el investigador. Los pacientes sin progresión de la enfermedad podían recibir tratamiento hasta 24 meses. El ensayo excluyó a los pacientes con enfermedad autoinmune, una enfermedad que precisara inmunosupresión y a los pacientes que habían recibido más de dos líneas previas de quimioterapia sistémica para el tratamiento del cáncer urotelial metastásico. Los pacientes con un estado funcional ECOG de 2 tenían que tener una hemoglobina ≥10 g/dl, no podían tener metástasis hepáticas y debían haber recibido la última dosis de su pauta posológica de quimioterapia previa ≥3 meses antes de la entrada en el estudio. La evaluación del estado tumoral se realizó a las 9 semanas después de la primera dosis, luego cada 6 semanas durante el primer año, seguido posteriormente por la evaluación cada 12 semanas.
Entre los 542 pacientes aleatorizados en el ensayo KEYNOTE-045, las características basales fueron: mediana de edad de 66 años (rango: 26 a 88), 58% de 65 años o más; 74% varones; 72% de raza blanca y 23% de raza asiática; 56% con estado funcional ECOG de 1 y 1% con estado funcional ECOG de 2 y 96% con enfermedad M1 y 4% con enfermedad M0. El 87% de los pacientes tenían metástasis viscerales, incluido un 34% con metástasis hepáticas. El 86% tenía un tumor primario en las vías urinarias inferiores y el 14% tenía un tumor primario en las vías urinarias superiores. El 15% de los pacientes tuvo progresión de la enfermedad después de quimioterapia previa neoadyuvante o adyuvante con platino. El 21% había recibido dos tratamientos sistémicos previos en el contexto metastásico. El 76% de los pacientes había recibido cisplatino previamente, el 23% había recibido carboplatino previamente y el 1% había sido tratado con otros tratamientos basados en platino.
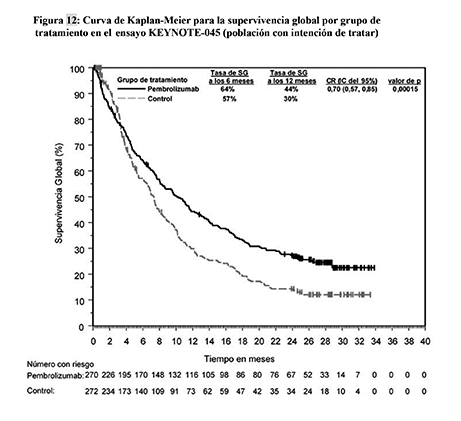
Las variables principales de eficacia fueron la SG y la SLP evaluadas mediante RCIE usando los criterios RECIST v1.1. Las variables secundarias fueron la TRO (evaluada mediante RCIE usando los RECIST v1.1) y la duración de la respuesta. La Tabla 19 resume las variables principales de eficacia para la población con intención de tratar. En la Figura 12 se muestra la curva de Kaplan-Meier para la SG. El ensayo demostró mejorías estadísticamente significativas en la SG y la TRO en los pacientes aleatorizados a pembrolizumab en comparación con los que recibieron quimioterapia. No hubo diferencia estadísticamente significativa entre pembrolizumab y la quimioterapia en la SLP.
|
Tabla 19: Respuesta a pembrolizumab 200 mg cada 3 semanas en pacientes con carcinoma urotelial tratado previamente con quimioterapia en el ensayo KEYNOTE-045 |
||
|
Variable |
Pembrolizumab 200 mg cada 3 semanas n=270 |
Quimioterapia n=272 |
|
SG |
||
|
Número (%) de pacientes con acontecimiento |
200 (74%) |
219 (81%) |
|
Cociente de riesgo (Hazard ratio)* (IC del 95%) |
0.70 (0.57, 0.85) |
|
|
Valor de p† |
< 0,001 |
|
|
Mediana en meses (IC del 95%) |
10.1 (8.0, 12.3) |
7.3 (6.1, 8.1) |
|
SLP‡ |
||
|
Número (%) de pacientes con acontecimiento |
233 (86%) |
237 (87%) |
|
Cociente de riesgo (Hazard ratio)* (IC del 95%) |
0.96 (0.79, 1.16) |
|
|
Valor de p † |
0.313 |
|
|
Mediana en meses (IC del 95%) |
2.1 (2.0, 2.2) |
3.3 (2.4, 3.6) |
|
Tasa de respuesta objetiva‡ |
||
|
% de TRO (IC del 95%) |
21% (16, 27) |
11% (8, 15) |
|
Valor de p§ |
< 0.001 |
|
|
Respuesta completa |
9% |
3% |
|
Respuesta parcial |
12% |
8% |
|
Enfermedad estable |
17% |
34% |
|
Duración de la respuesta ‡,¶ |
||
|
Mediana en meses (rango) |
No alcanzada (1.6+, 30.0+) |
4.4 (1.4+, 29.9+) |
|
Número (%#) de pacientes con duración ≥6 meses |
46 (84%) |
8 (47%) |
|
Número (%#) de pacientes con duración ≥12 meses |
35 (68%) |
5 (35%) |
|
* Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con quimioterapia) de acuerdo al modelo de riesgos proporcionales de Cox estratificado † Basado en la prueba de orden logarítmico estratificada ‡ Evaluada mediante revisión central independiente enmascarada (RCIE) usando los criterios RECIST 1.1 § Basado en el método de Miettinen y Nurminen ¶ Basado en los pacientes con una mejor respuesta global como respuesta completa o parcial confirmada # Basado en la estimación de Kaplan-Meier |
||
Figura 12: Curva de Kaplan-Meier para la supervivencia global por grupo de tratamiento en el ensayo KEYNOTE-045 (población con intención de tratar)
En el ensayo KEYNOTE-045 se realizó un análisis en los pacientes que tenían una CPS de PD-L1 <10 [pembrolizumab: n=186 (69%) frente a quimioterapia: n=176 (65%)] o ≥10 [pembrolizumab: n=74 (27%) frente a quimioterapia: n=90 (33%)] tanto en el grupo tratado con pembrolizumab como en el de quimioterapia (ver Tabla 20).
|
Tabla 20: SG por expresión de PD-L1 |
|||
|
Expresión de PD-L1 |
Pembrolizumab |
Quimioterapia |
|
|
SG por expresión de PD-L1 Número de acontecimientos (número de pacientes)* |
Cociente de riesgo (Hazard Ratio)? (IC del 95%) |
||
|
PPC <10 |
140 (186) |
144 (176) |
0.75 (0.59, 0.95) |
|
PPC ≥10 |
53 (74) |
72 (90) |
0.55 (0.37, 0.81) |
|
* De acuerdo al análisis final ? Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con quimioterapia) de acuerdo al modelo de riesgos proporcionales de Cox estratificados |
|||
Los resultados comunicados por los pacientes (RCP) se evaluaron usando el QLQ-C30 de la EORTC. Se observó un tiempo prolongado hasta el deterioro en el estado de salud global/CdV del QLQ-C30 de la EORTC en los pacientes tratados con pembrolizumab en comparación con la quimioterapia de elección del investigador (Cociente de riesgo 0.70; IC del 95% 0.55-0.90). A lo largo de 15 semanas de seguimiento, los pacientes tratados con pembrolizumab tuvieron un estado de salud global/CdV estable, mientras que los tratados con la quimioterapia de elección del investigador tuvieron una disminución del estado de salud global/CdV. Estos resultados se deben interpretar en el contexto del diseño de ensayo abierto y, por tanto, se deben tomar con precaución.
KEYNOTE-052: Ensayo abierto en pacientes con carcinoma urotelial no candidatos a quimioterapia basada en cisplatino
La seguridad y la eficacia de pembrolizumab se investigaron en el ensayo KEYNOTE-052, un ensayo multicéntrico, abierto, para el tratamiento del carcinoma urotelial localmente avanzado o metastásico en pacientes que no eran candidatos a quimioterapia basada en cisplatino. Los pacientes recibieron pembrolizumab a una dosis de 200 mg cada 3 semanas hasta toxicidad inaceptable o progresión de la enfermedad. El tratamiento podía continuar más allá de la progresión si el paciente estaba clínicamente estable y el investigador consideraba que estaba obteniendo beneficio clínico. Los pacientes sin progresión de la enfermedad podían tratarse hasta 24 meses. El ensayo excluyó a los pacientes con enfermedad autoinmune o una enfermedad que precisara inmunosupresión. La evaluación del estado tumoral se realizó a las 9 semanas después de la primera dosis, luego cada 6 semanas durante el primer año, seguido posteriormente por la evaluación cada 12 semanas.
Entre los 370 pacientes con carcinoma urotelial que no eran candidatos a quimioterapia basada en cisplatino, las características basales fueron: mediana de edad de 74 años (82% de 65 años o más); 77% varones y 89% de raza blanca y 7% de raza asiática. El 88% tenía enfermedad M1 y el 12% tenía enfermedad M0. El 85% de los pacientes tenía metástasis viscerales, incluido un 21% con metástasis hepáticas. Entre las razones para no ser candidatos a cisplatino estaban: aclaramiento basal de creatinina
<60 ml/min (50%), estado funcional ECOG de 2 (32%), estado funcional ECOG de 2 y aclaramiento basal de creatinina <60 ml/min (9%) y otros (insuficiencia cardíaca de clase III, neuropatía periférica de grado 2 o mayor y pérdida auditiva de grado 2 o mayor; 9%). El 90% de los pacientes nunca había recibido tratamiento y el 10% había recibido quimioterapia previa adyuvante o neoadyuvante basada en platino. El 81% tenía un tumor primario en las vías urinarias inferiores y el 19% de los pacientes tenía un tumor primario en las vías urinarias superiores.
La variable principal de eficacia fue la TRO evaluada mediante RCIE usando los criterios RECIST 1.1. Las variables secundarias de eficacia fueron la duración de la respuesta, la SLP y la SG. La Tabla 21 resume las variables principales de eficacia para la población del ensayo de acuerdo a una mediana de seguimiento de 11.5 meses en todos los pacientes.
|
Tabla 21: Respuesta a pembrolizumab 200 mg cada 3 semanas en pacientes con carcinoma urotelial no candidatos a quimioterapia con cisplatino en el ensayo KEYNOTE-052 |
|
|
Variable |
n=370 |
|
Tasa de respuesta objetiva* |
|
|
% de TRO (IC del 95%) |
29% (24, 34) |
|
Tasa de control de la enfermedad† |
47% |
|
Respuesta completa |
8% |
|
Respuesta parcial |
21% |
|
Enfermedad estable |
18% |
|
Duración de la respuesta |
|
|
Mediana en meses (rango) |
No alcanzada (1.4+, 27.9+) |
|
% con duración ≥ 6 meses |
82%‡ |
|
Tiempo hasta la respuesta |
|
|
Mediana en meses (rango) |
2.1 (1.3, 9.0) |
|
SLP* |
|
|
Mediana en meses (IC del 95%) |
2.3 (2.1, 3.4) |
|
Tasa de SLP a los 6 meses |
34% |
|
Tasa de SLP a los 12 meses |
22% |
|
SG* |
|
|
Mediana en meses (IC del 95%) |
11.5 (10.0, 13.3) |
|
Tasa de SG a los 6 meses |
67% |
|
Tasa de SG a los 12 meses |
48% |
|
* Evaluada mediante una revisión central independiente enmascarada (RCIE) usando los criterios RECIST 1.1 † Basada en la mejor respuesta de enfermedad estable o mejor ‡ Basado en las estimaciones de Kaplan-Meier; incluye 85 pacientes con respuesta de 6 meses o más |
|
Se realizó un análisis en el ensayo KEYNOTE-052 en pacientes que tenían una CPS de PD-L1 < 10 (n=251; 68%) o ≥ 10 (n=110; 30%) (ver Tabla 22).
|
Tabla 22: TRO y SG por expresión de PD-L1 |
||
|
Variable |
CPS < 10 N=251 |
CPS ≥ 10 N=110 |
|
Tasa de respuesta objetiva* |
||
|
% de TRO (IC del 95%) |
21% (16, 26) |
47% (38, 57) |
|
SG |
||
|
Mediana en meses (IC del 95%) |
10 (8, 12) |
19 (12, no alcanzada) |
|
Tasa de SG a los 12 meses |
42% |
61% |
|
* RCIE usando los criterios RECIST 1.1 |
||
El ensayo KEYNOTE-361 es un ensayo clínico de Fase III en curso, aleatorizado, controlado, abierto, de pembrolizumab con o sin quimioterapia en combinación basada en platino frente a quimioterapia en primera línea de tratamiento, en pacientes con carcinoma urotelial avanzado o metastásico.
Los datos preliminares de una revisión precoz mostraron una supervivencia reducida con pembrolizumab en monoterapia en pacientes cuyos tumores expresan PD-L1 con una CPS < 10 en comparación con la quimioterapia estándar.
De acuerdo con una recomendación emitida por un Comité de Monitorización de Datos externo, la entrada de pacientes en el grupo de pembrolizumab en monoterapia se detuvo para los pacientes cuyos tumores expresen PD-L1 con una CPS < 10. El grupo de pembrolizumab en monoterapia permanece abierto solo para los pacientes cuyos tumores expresen PD-L1 con una CPS ≥ 10. Los sujetos cuyos tumores expresen PD-L1 con una CPS < 10, ya incluidos dentro del grupo de pembrolizumab en monoterapia, pueden continuar el tratamiento. La aleatorización a los grupos de quimioterapia y de quimioterapia-pembrolizumab permanece abierta.
Carcinoma de células escamosas de cabeza y cuello
KEYNOTE-040: Ensayo controlado en pacientes con CCECC tratados previamente con quimioterapia basada en platino
La seguridad y la eficacia de pembrolizumab se investigaron en el ensayo KEYNOTE-040, un ensayo multicéntrico, abierto, aleatorizado, controlado, para el tratamiento del CCECC recurrente o metastásico en pacientes en los que la enfermedad ha progresado durante o después de la quimioterapia basada en platino administrada para el CCECC recurrente o metastásico, o después de la quimioterapia basada en platino administrada como tratamiento de inducción, concurrente o adyuvante y no candidatos a recibir tratamiento local con intención curativa. Los pacientes se estratificaron por expresión de PD-L1 (TPS ≥ 50%), estado de VPH y estado funcional ECOG y, a continuación, se aleatorizaron (1:1) para recibir pembrolizumab 200 mg cada 3 semanas (n=247) o uno de los tres tratamientos estándar (n=248): metotrexato 40 mg/m2 una vez a la semana (n=64), docetaxel 75 mg/m2 una vez cada 3 semanas (n=99) o cetuximab 400 mg/m2 como dosis de carga y, posteriormente, 250 mg/m2 una vez a la semana (n=71). El tratamiento podía continuar después de la progresión si el paciente estaba clínicamente estable y el investigador consideraba que se estaba obteniendo beneficio clínico. El ensayo excluyó a los pacientes con enfermedad autoinmune activa que precisara tratamiento sistémico dentro de los 2 años de tratamiento; pacientes con un problema médico que precisara inmunosupresión; o que hubieran sido previamente tratados con 3 o más tratamientos sistémicos para el CCECC recurrente y/o metastásico. Se realizó la evaluación del estado tumoral a las 9 semanas, luego cada 6 semanas hasta la semana 52, seguido por la evaluación cada 9 semanas hasta los 24 meses.
Entre los 495 pacientes en el ensayo KEYNOTE-040, 129 (26%) tenían tumores que expresaban PD-L1 con una TPS ≥ 50% de acuerdo al Kit de diagnóstico por inmunohistoquímica (IHQ) de PD-L1 22C3 pharmDxTM. Las características basales de estos 129 pacientes fueron: mediana de edad de 62 años (40% de 65 años o más); 81% varones; 78% de raza blanca, 11% de raza asiática y 2% de raza negra; 23% y 77% con un estado funcional ECOG de 0 o 1, respectivamente; y 19% con tumores VPH positivo. El 67% de los pacientes tenía enfermedad M1 y la mayoría tenía enfermedad en estadio IV (estadio IV 32%, estadio IVa 14%, estadio IVb 4% y estadio IVc 44%). El 16% presentaba progresión de la enfermedad tras la quimioterapia neoadyuvante o adyuvante basada en platino y el 84% había recibido 1-2 tratamientos sistémicos previos para la enfermedad metastásica.
La variable principal de eficacia fue la SG en la población con intención de tratar. El análisis inicial presentó un cociente de riesgo de 0.82 (IC del 95%: 0.67, 1.01) para la SG con un valor de p unidireccional de 0.0316. La mediana de la SG fue de 8.4 meses para pembrolizumab en comparación con los 7.1 meses para el tratamiento estándar. La Tabla 23 resume las variables principales de eficacia para la población con TPS ≥ 50%. En la Figura 13 se muestra la curva de Kaplan-Meier para la SG, para la población con TPS ≥ 50%.
|
Tabla 23: Eficacia de pembrolizumab 200 mg cada 3 semanas en pacientes con CCECC con TPS ≥ 50% que habían sido tratados previamente con quimioterapia basada en platino en el ensayo KEYNOTE- 040 |
||
|
Variable |
Pembrolizumab 200 mg cada 3 semanas n=64 |
Tratamiento estándar* n=65 |
|
SG |
||
|
Número (%) de pacientes con acontecimiento |
41 (64) |
56 (86) |
|
Cociente de riesgo (Hazard Ratio)† (IC del 95%) |
0.53 (0.35, 0.81) |
|
|
Valor de p‡ |
0.001 |
|
|
Mediana en meses (IC del 95%) |
11.6 (8.3, 19.5) |
6.6 (4.8, 9.2) |
|
SLP§ |
||
|
Número (%) de pacientes con acontecimiento |
52 (81) |
58 (89) |
|
Cociente de riesgo (Hazard ratio)† (IC del 95%) |
0.58 (0.39, 0.86) |
|
|
Valor de p‡ |
0.003 |
|
|
Mediana en meses (IC del 95%) |
3.5 (2.1, 6.3) |
2.1 (2.0, 2.4) |
|
Tasa (%) a los 6 meses (IC del 95%) |
40.1 (28.1, 51.9) |
17.1 (8.8, 27.7) |
|
Tasa de respuesta global§ |
||
|
% de TRG (IC del 95%) |
26.6 (16.3, 39.1) |
9.2 (3.5, 19.0) |
|
Valor de p ¶ |
0.0009 |
|
|
Respuesta completa |
5% |
2% |
|
Respuesta parcial |
22% |
8% |
|
Enfermedad estable |
23% |
23% |
|
Duración de la respuesta§,# |
||
|
Mediana en meses (rango) |
No alcanzado (2.7, 13.8+) |
6.9 (4.2, 18.8) |
|
Número (%Þ) de pacientes con duración ≥ 6 meses |
9 (66) |
2 (50) |
|
Metotrexato, docetaxel o cetuximab † Cociente de riesgo (Hazard Ratio) (pembrolizumab comparado con el tratamiento estándar) de acuerdo al modelo de riesgos proporcionales de Cox estratificado ‡ Valor de p unidireccional basado en la prueba de orden logarítmico estratificada § Evaluada mediante RCIE usando los criterios RECIST 1.1 ¶ Basado en el método de Miettinen y Nurminen # Basado en los pacientes con una mejor respuesta global como respuesta completa o parcial confirmada Þ Basado en la estimación de Kaplan-Meier |
||
Figura 13: Curva de Kaplan-Meier para la supervivencia global por grupo de tratamiento en los pacientes del ensayo KEYNOTE-040 con expresión de PD-L1 (TPS ≥ 50%)
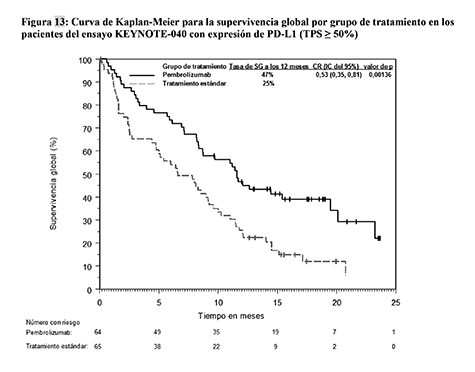
Población pediátrica:
La Agencia Europea de Medicamentos ha concedido al titular un aplazamiento para presentar los resultados de los ensayos realizados con pembrolizumab en uno o más grupos de la población pediátrica en el tratamiento de todas las enfermedades incluidas en la categoría de neoplasias malignas (excepto del sistema nervioso, hematopoyético y del tejido linfoide) (ver Posología y forma de administración para consultar la información sobre el uso en la población pediátrica).
DATOS FARMACÉUTICOS
Lista de excipientes
L-histidina
Hidrocloruro de L-histidina monohidrato Sacarosa
Polisorbato 80
Agua para inyección
Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros, excepto con los mencionados en la sección 6.6.
Periodo de validez
Vial no abierto 2 años
Después de la preparación de la infusión
Desde un punto de vista microbiológico, el producto una vez diluido, debe utilizarse inmediatamente. No congelar la solución diluida. Si no se usa inmediatamente, se ha demostrado la estabilidad química y física en uso de KEYTRUDA® durante 24 horas entre 2ºC y 8ºC. Este periodo de 24 horas puede incluir hasta 6
horas a temperatura ambiente (a o por debajo de 25°C). Si se refrigera, se debe dejar que los viales y/o las bolsas intravenosas alcancen la temperatura ambiente antes de su uso.
Precauciones especiales de conservación:
Conservar en refrigeración (entre 2°C y 8°C). No congelar.
Almacenar en su caja original para protegerlo de la luz.
Para las condiciones de conservación tras la dilución del medicamento, ver Periodo de validez
PROPIEDADES FARMACOCINÉTICAS
Se estudió la farmacocinética de pembrolizumab en 2993 pacientes con melanoma metastásico o irresecable, CPNM o carcinoma que recibieron dosis en el rango de 1 a 10 mg/kg cada 2 semanas, 2 a 10 mg/kg cada 3 semanas, o 200 mg cada 3 semanas.
Absorción:
Pembrolizumab se administra por vía intravenosa y, por lo tanto, tiene una biodisponibilidad inmediata y completa.
Distribución:
De forma coherente con una distribución extravascular limitada, el volumen de distribución de
pembrolizumab en el estado estacionario es pequeño (~6.0 1; CV: 20%). Como se esperaba de un anticuerpo, pembrolizumab no se une a las proteínas plasmáticas de una forma específica.
Biotransformación:
Pembrolizumab se cataboliza mediante vías inespecíficas; el metabolismo no contribuye a su eliminación.
Eliminación:
El aclaramiento de pembrolizumab es aproximadamente 23% menor (media geométrica, 195 ml/día [CV%: 40%]) después de alcanzar el cambio máximo en el estado estacionario comparado con la primera dosis (252 ml/día [CV%: 37%]); esta disminución en el aclaramiento con el tiempo no se considera clínicamente significativa. El valor de la media geométrica (CV%) para la semivida terminal es de 22 días (32%) en el estado estacionario.
Linealidad/No linealidad:
La exposición a pembrolizumab expresada mediante concentración máxima (Cmax) o área bajo la curva de tiempo-concentración plasmática (AUC) aumentó de forma proporcional a la dosis dentro del rango de dosis para la eficacia. Las concentraciones de pembrolizumab en el estado estacionario se alcanzaron a las 16 semanas de la administración repetida con un régimen de cada 3 semanas y la acumulación sistémica fue 2.1 veces mayor. La mediana de las concentraciones mínimas en el estado estacionario (Cmin) fue aproximadamente de 22 mcg/ml, a una dosis de 2 mg/kg cada 3 semanas y de 29 mcg/ml, a una dosis de 200 mg cada 3 semanas. La mediana del área bajo la curva de concentración-tiempo en el estado estacionario después de 3 semanas (AUC0-3semanas) fue de 794 mcg·día/ml a una dosis de 2 mg/kg cada 3 semanas y de 1.053 mcg·día/ml a una dosis de 200 mg cada 3 semanas.
Tras la administración de pembrolizumab 200 mg cada 3 semanas en pacientes con LHc, la mediana observada de la Cmin en el estado estacionario fue hasta un 40% mayor a la observada en otros tipos de tumores tratados con la misma dosis; no obstante, el intervalo de las concentraciones mínimas fue similar. No hay diferencias importantes en la mediana de la Cmax entre LHc y otros tipos de tumores. De acuerdo con los datos disponibles sobre seguridad en LHc y otros tipos de tumores, estas diferencias no son clínicamente significativas.
Poblaciones especiales:
Se evaluaron los efectos de diversas covariables sobre la farmacocinética de pembrolizumab mediante análisis de farmacocinética de poblaciones. Los siguientes factores no tuvieron un efecto clínicamente importante sobre el aclaramiento de pembrolizumab: edad (rango, 15-94 años), sexo, raza, insuficiencia renal leve o moderada, insuficiencia hepática leve y carga tumoral. La relación entre el peso corporal y el aclaramiento respalda el uso tanto de una dosis fija como de una dosis basada en el peso corporal para proporcionar un control adecuado y similar de la exposición.
Insuficiencia renal:
Se evaluó el efecto de la insuficiencia renal sobre el aclaramiento de pembrolizumab mediante análisis de farmacocinética de poblaciones en pacientes con insuficiencia renal leve o moderada en comparación con pacientes con función renal normal. No se observaron diferencias clínicamente importantes en el aclaramiento de pembrolizumab entre los pacientes con insuficiencia renal leve o moderada y los pacientes con función renal normal. No se ha estudiado pembrolizumab en pacientes con insuficiencia renal grave.
Insuficiencia hepática:
Se evaluó el efecto de la insuficiencia hepática sobre el aclaramiento de pembrolizumab mediante análisis de farmacocinética de poblaciones en pacientes con insuficiencia hepática leve (definida usando los criterios de disfunción hepática del National Cancer Institute de EE.UU.) en comparación con los pacientes con
función hepática normal. No se observaron diferencias clínicamente importantes en el aclaramiento de pembrolizumab entre los pacientes con insuficiencia hepática leve y los pacientes con función hepática normal. No se ha estudiado pembrolizumab en pacientes con insuficiencia hepática moderada o grave (ver Posología y forma de administración).
Datos preclínicos sobre seguridad:
Se evaluó la seguridad de pembrolizumab en un estudio de toxicidad a dosis repetidas de 1 mes y otro de 6 meses en monos Cynomolgus que recibieron dosis IV de 6, 40 o 200 mg/kg una vez a la semana en el estudio de 1 mes y una vez cada dos semanas en el estudio de 6 meses, seguido por un periodo de 4 meses sin tratamiento. No se observaron hallazgos de importancia toxicológica y el nivel sin efecto adverso observado (NOAEL) en ambos estudios fue ≥ 200 mg/kg, que produjo exposiciones múltiples de 19 y 94 veces la exposición en seres humanos a dosis de 10 y 2 mg/kg, respectivamente. La exposición múltiple entre el NOAEL y una dosis de 200 mg en seres humanos fue 74.
No se han realizado estudios de reproducción animal con pembrolizumab. La vía PD-1/PD-L1 se piensa que está involucrada en mantener la tolerancia al feto durante el embarazo. Se ha demostrado que el bloqueo de la señalización PD-L1 en modelos murinos de embarazo altera la tolerancia al feto y conduce a un aumento de pérdidas fetales.
No se han llevado a cabo estudios de fertilidad en animales con pembrolizumab. En estudios de toxicología a dosis repetidas de 1 mes y 6 meses, no hubo efectos notables en los órganos reproductores masculinos y femeninos; sin embargo, muchos animales de estos estudios no eran sexualmente maduros.
CONTRAINDICACIONES:
Hipersensibilidad al principio activo o a alguno de los excipientes incluidos en la sección 6.1.
ADVERTENCIAS Y PRECAUCIONES ESPECIALES DE EMPLEO:
Para mejorar la trazabilidad de los medicamentos biológicos, se debe registrar de forma clara el nombre y el número de lote del producto administrado.
Determinación de la expresión de PD-L1
Al evaluar el estado de PD-L1 del tumor, es importante que se elija una metodología bien validada y robusta para reducir al mínimo los resultados falsos negativos o falsos positivos.
Reacciones adversas relacionadas con el sistema inmunitario
En pacientes que reciben pembrolizumab, se han producido reacciones adversas relacionadas con el sistema inmunitario, incluidos casos graves y mortales. La mayoría de las reacciones adversas relacionadas con el sistema inmunitario que se produjeron durante el tratamiento con pembrolizumab fueron reversibles y se controlaron con interrupciones de pembrolizumab, la administración de corticosteroides y/o terapia de apoyo. También se produjeron reacciones adversas relacionadas con el sistema inmunitario después de la última dosis de pembrolizumab. Se pueden producir reacciones adversas relacionadas con el sistema inmunitario en más de un sistema/órgano simultáneamente.
En caso de sospecha de reacciones adversas relacionadas con el sistema inmunitario, se debe asegurar una evaluación adecuada para confirmar la etiología o descartar otras causas. De acuerdo con la gravedad de la reacción adversa, pembrolizumab se debe suspender temporalmente y se debe administrar corticosteroides. Tras la mejoría a Grado ≤ 1, se debe iniciar la reducción progresiva de los corticosteroides y continuar durante al menos 1 mes. De acuerdo a los datos limitados de ensayos clínicos en pacientes cuyas reacciones adversas relacionadas con el sistema inmunitario no pudieron ser controladas con el uso de corticosteroides, se puede evaluar la administración de otros inmunosupresores sistémicos.
Pembrolizumab se podría reiniciar en el plazo de 12 semanas después de la última dosis de KEYTRUDA®, si la reacción adversa se mantiene en Grado ≤ 1 y la dosis de corticosteroide se ha reducido a ≤ 10 mg de prednisona o equivalente al día.
Pembrolizumab se debe suspender definitivamente por cualquier reacción adversa relacionada con el sistema inmunitario recurrente de Grado 3 y por toxicidad de la reacción adversa relacionada con el sistema inmunitario de Grado 4, excepto por endocrinopatías que estén controladas con terapia hormonal sustitutiva (ver Posología y forma de administración y 4.8).
Neumonitis relacionada con el sistema inmunitario:
Se ha notificado neumonitis en pacientes que recibieron pembrolizumab (ver Reacciones adversas). Se debe vigilar a los pacientes en cuanto a signos y síntomas de neumonitis. Si se sospecha neumonitis, se debe confirmar con pruebas de imagen radiográficas y descartar otras causas. Se debe administrar corticosteroides en caso de acontecimientos de Grado ≥ 2 (dosis inicial de 1-2 mg/kg/día de prednisona o equivalente, seguida por una reducción progresiva); pembrolizumab se debe suspender temporalmente en caso de neumonitis de Grado 2 y se debe suspender definitivamente en caso de neumonitis de Grado 3, Grado 4 o recurrente de Grado 2 (ver Posología y forma de administración).
Colitis relacionada con el sistema inmunitario:
Se ha notificado colitis en pacientes que recibieron pembrolizumab (ver Reacciones adversas). Se debe vigilar a los pacientes en cuanto a signos y síntomas de colitis y descartar otras causas. Se debe administrar corticosteroides en caso de acontecimientos de Grado ≥ 2 (dosis inicial de 1-2 mg/kg/día de prednisona o equivalente, seguida por una reducción progresiva); pembrolizumab se debe suspender temporalmente en caso de colitis de Grado 2 o Grado 3 y se debe suspender definitivamente en caso de colitis de Grado 4 (ver Posología y forma de administración). Se debe tener en cuenta el riesgo potencial de perforación gastrointestinal.
Hepatitis relacionada con el sistema inmunitario:
Se ha notificado hepatitis en pacientes que recibieron pembrolizumab (ver Reacciones adversas). Se debe vigilar a los pacientes en cuanto a cambios en la función hepática (al comienzo del tratamiento, periódicamente durante el tratamiento y según esté indicado de acuerdo con la evaluación clínica) y en cuanto a síntomas de hepatitis y descartar otras causas. Se debe administrar corticosteroides (dosis inicial de 0.5-1 mg/kg/día (en caso de acontecimientos de Grado 2) y 1-2 mg/kg/día (en acontecimientos de Grado ≥ 3) de prednisona o equivalente, seguida por una reducción progresiva) y, de acuerdo con la intensidad de las elevaciones de las enzimas hepáticas, pembrolizumab se debe suspender temporalmente o suspender definitivamente (ver Posología y forma de administración).
Nefritis relacionada con el sistema inmunitario:
Se ha notificado nefritis en pacientes que recibieron pembrolizumab (ver Reacciones adversas). Se debe vigilar a los pacientes en cuanto a cambios en la función renal y descartar otras causas de disfunción renal. Se debe administrar corticosteroides en caso de acontecimientos de Grado ≥ 2 (dosis inicial de 1-2 mg/kg/día de prednisona o equivalente, seguida por una reducción progresiva) y, de acuerdo con la intensidad de las elevaciones de la creatinina, pembrolizumab se debe suspender temporalmente en caso de nefritis de Grado 2 y se debe suspender definitivamente en caso de nefritis de Grado 3 o Grado 4 (ver Posología y forma de administración).
Endocrinopatías relacionadas con el sistema inmunitario:
Con el tratamiento con pembrolizumab se han observado endocrinopatías graves, tales como hipofisitis, diabetes mellitus tipo 1, cetoacidosis diabética, hipotiroidismo e hipertiroidismo.
En casos de endocrinopatías relacionadas con el sistema inmunitario podría ser necesario el tratamiento a largo plazo con terapia hormonal sustitutiva.
Se ha notificado hipofisitis en pacientes que recibieron pembrolizumab (ver Reacciones adversas). Se debe vigilar a los pacientes en cuanto a signos y síntomas de hipofisitis (incluido hipopituitarismo e insuficiencia suprarrenal secundaria) y descartar otras causas. Se debe administrar corticosteroides para tratar la insuficiencia suprarrenal secundaria y otras terapias hormonales sustitutivas según esté clínicamente indicado, y pembrolizumab se debe suspender temporalmente por hipofisitis sintomática hasta que el acontecimiento se ha controlado con terapia hormonal sustitutiva. Se puede valorar la continuación del tratamiento con pembrolizumab, después de la reducción progresiva del corticosteoride, si es necesario (ver Posología y forma de administración). Se debe vigilar la función hipofisaria y los niveles hormonales para asegurar que la terapia hormonal sustitutiva es la adecuada.
Se ha notificado diabetes mellitus tipo 1, incluida cetoacidosis diabética, en pacientes que recibieron pembrolizumab (ver Reacciones adversas). Se debe vigilar a los pacientes en cuanto a hiperglucemia u otros signos y síntomas de diabetes. Se debe administrar insulina para la diabetes tipo 1 y se debe suspender temporalmente pembrolizumab en casos de hiperglucemia de Grado 3, hasta que se alcance el control metabólico (ver Posología y forma de administración).
Se han notificado trastornos del tiroides, como hipotiroidismo, hipertiroidismo y tiroiditis, en pacientes que recibieron pembrolizumab y se pueden producir en cualquier momento durante el tratamiento. El hipotiroidismo se ha notificado más frecuentemente en pacientes con CCECC que se han sometido a radioterapia previa. Se debe vigilar a los pacientes en cuanto a cambios en la función tiroidea (al comienzo del tratamiento, periódicamente durante el tratamiento y según esté indicado de acuerdo con la evaluación clínica) y en cuanto a signos y síntomas clínicos de trastornos tiroideos. El hipotiroidismo se puede manejar con tratamiento sustitutivo sin interrupción del tratamiento y sin corticosteroides. El hipertiroidismo se puede manejar sintomáticamente. Pembrolizumab se debe suspender temporalmente por hipertiroidismo de Grado ≥ 3 hasta que se recupere a Grado ≤ 1. Se puede valorar la continuación del tratamiento con pembrolizumab, después de la reducción progresiva del corticosteroide, si es necesario, en pacientes con hipertiroidismo de Grado 3 o Grado 4 que mejore a Grado 2 o menor (ver Posología y forma de administración y Reacciones adversas). Se debe vigilar la función tiroidea y los niveles hormonales para asegurar que la terapia hormonal sustitutiva es la adecuada.
Reacciones adversas cutáneas relacionadas con el sistema inmunitario:
Se han notificado reacciones cutáneas graves relacionadas con el sistema inmunitario, en pacientes que recibieron pembrolizumab (ver Reacciones adversas). Se debe vigilar a los pacientes en caso de sospecha de reacciones cutáneas graves y se deben descartar otras causas. De acuerdo con la gravedad de la reacción adversa, pembrolizumab se debe suspender temporalmente o suspender definitivamente y se deben administrar corticosteroides (ver Posología y forma de administración).
Se han notificado casos de síndrome de Stevens-Johnson (SSJ) y necrólisis epidérmica tóxica (NET) en pacientes que recibieron pembrolizumab (ver Reacciones adversas). En cuanto haya signos o síntomas de SSJ o NET, pembrolizumab se debe suspender temporalmente y se debe derivar al paciente a una unidad especializada para su evaluación y tratamiento. Si se confirma SSJ o NET, pembrolizumab se debe suspender definitivamente (ver Posología y forma de administración).
Se debe tener precaución cuando se considere el uso de pembrolizumab en un paciente que haya tenido previamente reacciones adversas cutáneas graves o potencialmente mortales con un tratamiento previo con otros medicamentos anticancerígenos inmunoestimuladores.
Otras reacciones adversas relacionadas con el sistema inmunitario:
En ensayos clínicos o en la experiencia tras la comercialización, se han notificado las siguientes reacciones adversas adicionales clínicamente significativas, relacionadas con el sistema inmunitario: uveítis, artritis, miositis, miocarditis, pancreatitis, síndrome de Guillain-Barré, síndrome miasténico, anemia hemolítica, sarcoidosis y encefalitis (ver Posología y forma de administración y Reacciones adversas).
De acuerdo a la gravedad de la reacción adversa, pembrolizumab se debe suspender temporalmente y administrar corticosteroides.
Pembrolizumab se puede reiniciar en el plazo de 12 semanas después de la última dosis de KEYTRUDA®, si la reacción adversa se mantiene en Grado ≤ 1 y la dosis del corticosteoride se ha reducido a ≤ 10 mg de prednisona o equivalente al día.
Pembrolizumab se debe suspender definitivamente por cualquier reacción adversa relacionada con el sistema inmunitario recurrente de Grado 3 y por cualquier reacción adversa relacionada con el sistema inmunitario de Grado 4 (ver Posología y forma de administración y Reacciones adversas).
Tras la comercialización se ha notificado el rechazo de trasplantes de órganos sólidos en pacientes tratados con inhibidores de la PD-1. El tratamiento con pembrolizumab puede aumentar el riesgo de rechazo en receptores de trasplantes de órganos sólidos. En estos pacientes se debe tener en cuenta el beneficio del tratamiento con pembrolizumab frente al riesgo de un posible rechazo del órgano.
Complicaciones del trasplante alogénico de progenitores hematopoyéticos (TPH):
TPH alogénico después del tratamiento con pembrolizumab:
Se han observado casos de enfermedad del injerto contra el huésped (EICH) y de enfermedad veno- oclusiva hepática en pacientes con linfoma de Hodgkin clásico sometidos a un TPH alogénico tras la exposición previa a pembrolizumab. Hasta que se disponga de nuevos datos, se deben valorar de forma detenida, caso por caso, los posibles beneficios del TPH y el posible aumento del riesgo de complicaciones relacionadas con el trasplante (ver Reacciones adversas).
TPH alogénico previo al tratamiento con pembrolizumab:
En pacientes con antecedentes de TPH alogénico, tras el tratamiento con pembrolizumab se ha notificado EICH aguda, incluida EICH mortal. Los pacientes que experimentaron EICH después de su trasplante, pueden tener un mayor riesgo de EICH después del tratamiento con pembrolizumab. Se debe valorar el beneficio del tratamiento con pembrolizumab frente al riesgo de posible EICH en pacientes con antecedentes de TPH alogénico.
Reacciones asociadas a la infusión:
Se han notificado reacciones graves asociadas a la infusión, incluyendo hipersensibilidad y anafilaxia, en pacientes que recibieron pembrolizumab (ver Reacciones adversas). En las reacciones graves a la infusión, se debe detener la infusión y pembrolizumab se debe suspender definitivamente (ver Posología y forma de administración). Los pacientes con reacción a la infusión leve o moderada pueden seguir recibiendo pembrolizumab con una vigilancia estrecha; se puede valorar la medicación previa con antipiréticos y antihistamínicos.
Precauciones específicas de cada enfermedad:
Uso de pembrolizumab en pacientes con carcinoma urotelial que han recibido quimioterapia previa basada en platino.
Antes de iniciar el tratamiento, los médicos deben valorar el retraso del inicio del efecto de pembrolizumab en pacientes con peores características pronósticas y/o enfermedad agresiva. En el cáncer urotelial, se observó un mayor número de muertes en el plazo de 2 meses con pembrolizumab en comparación con la quimioterapia (ver Propiedades farmacodinámicas). Los factores asociados con las muertes tempranas fueron la progresión rápida de la enfermedad con el tratamiento previo con platino y las metástasis hepáticas.
Uso de pembrolizumab en cáncer urotelial en pacientes que no son candidatos a quimioterapia basada en cisplatino y cuyos tumores expresen PD-L1 con una CPS ≥ 10
Las características basales y las características pronósticas de la enfermedad en la población del ensayo KEYNOTE-052 incluyeron una proporción de pacientes candidatos a una combinación basada en carboplatino, en los que el beneficio se está evaluando en un ensayo comparativo, y pacientes candidatos a quimioterapia en monoterapia, para los que no hay aleatorizados disponibles. Además, no se dispone de datos de seguridad y eficacia en pacientes más debilitados (p. ej., estado funcional ECOG 3), no considerados candidatos a quimioterapia. En ausencia de estos datos, pembrolizumab se debe usar con precaución en esta población después de la valoración cuidadosa de la posible relación riesgo-beneficio de forma individual.
Uso de pembrolizumab para el tratamiento de primera línea de pacientes con CPNM
En general, la frecuencia observada de reacciones adversas del tratamiento con pembrolizumab en combinación es mayor que la de pembrolizumab en monoterapia o quimioterapia sola, lo que es un reflejo de la contribución de cada uno de estos componentes (ver Posología y forma de administración y Reacciones adversas). No se dispone de una comparación directa de pembrolizumab cuando se usa en combinación con quimioterapia frente a pembrolizumab en monoterapia.
Antes de iniciar el tratamiento, los médicos deben valorar el balance beneficio/riesgo de las opciones de tratamiento disponibles (pembrolizumab en monoterapia o pembrolizumab en combinación con quimioterapia) en pacientes con cáncer de pulmón no microcítico no tratados previamente cuyos tumores expresen PD-L1.
Los datos de eficacia y seguridad de pacientes ≥ 75 años son limitados. Para pacientes ≥ 75 años, el tratamiento con pembrolizumab en combinación debe usarse con precaución después de la valoración cuidadosa de la posible relación riesgo-beneficio de forma individual (ver Propiedades farmacodinámicas).
Uso de pembrolizumab para el tratamiento adyuvante de pacientes con melanoma:
Se observó una tendencia al aumento de la frecuencia de reacciones adversas graves en pacientes de ≥ 75 años. Los datos de seguridad de pembrolizumab en el contexto adyuvante del melanoma en pacientes ≥ 75 años son limitados.
Pacientes excluidos de los ensayos clínicos:
Se excluyeron de los ensayos clínicos los pacientes con los siguientes problemas: metástasis activas en el SNC; estado funcional ECOG ≥ 2 (excepto para carcinoma urotelial); infección por VIH, hepatitis B o hepatitis C; enfermedad autoinmune sistémica activa; enfermedad pulmonar intersticial; neumonitis previa que precisa tratamiento con corticosteroides sistémicos; antecedentes de hipersensibilidad grave a otros anticuerpos monoclonales; que estén recibiendo tratamiento inmunosupresor y antecedentes de reacciones adversas graves relacionadas con el sistema inmunitario, debidas al tratamiento con ipilimumab, definidas como cualquier toxicidad de Grado 4 o de Grado 3 que requirió tratamiento con corticosteroides (> 10 mg/día de prednisona o equivalente) durante más de 12 semanas. Los pacientes con infecciones activas fueron excluidos de los ensayos clínicos y se requirió el tratamiento de la infección antes de recibir pembrolizumab. Los pacientes con infecciones activas que se produjeron durante el tratamiento con pembrolizumab fueron controlados con tratamiento médico adecuado. Los pacientes con anomalías, clínicamente significativas en el estado basal, renales (creatinina > 1.5 x LSN) o hepáticas (bilirrubina > 1.5 x LSN, ALT, AST > 2.5 x LSN en ausencia de metástasis hepáticas) fueron excluidos de los ensayos clínicos, por lo que la información en pacientes con insuficiencia renal grave e insuficiencia hepática de moderada a grave es limitada.
En sujetos con linfoma de Hodgkin clásico en recaída o refractario, los datos clínicos relativos al uso de pembrolizumab en pacientes no candidatos a TAPH debido a razones distintas al fracaso a la quimioterapia de rescate, son limitados (ver Propiedades farmacodinámicas).
Después de considerar cuidadosamente el potencial aumento del riesgo, se puede usar pembrolizumab con un control médico adecuado en estos pacientes.
Tarjeta de información para el paciente:
Todos los prescriptores de KEYTRUDA® deben estar familiarizados con la Información para el médico y las Directrices de manejo. El prescriptor debe explicar al paciente los riesgos del tratamiento con KEYTRUDA®. Se le debe proporcionar al paciente la tarjeta de información para el paciente con cada prescripción.
FERTILIDAD, EMBARAZO Y LACTANCIA
Mujeres en edad fértil:
Las mujeres en edad fértil deben utilizar métodos anticonceptivos efectivos durante el tratamiento con pembrolizumab y hasta al menos 4 meses después de la última dosis de pembrolizumab.
Embarazo:
No hay datos relativos al uso de pembrolizumab en mujeres embarazadas. No se han realizado estudios de reproducción animal con pembrolizumab; sin embargo, en modelos murinos de gestación se ha demostrado que el bloqueo de la señalización de PD-L1 altera la tolerancia al feto y conduce a un aumento de la pérdida fetal (ver Datos preclínicos sobre seguridad). Estos resultados indican un posible riesgo, de acuerdo a su mecanismo de acción, de que la administración de pembrolizumab durante el embarazo pudiera causar daño fetal, incluido aumento de las tasas de abortos o de fetos nacidos muertos. Se sabe que las inmunoglobulinas humanas G4 (IgG4) atraviesan la barrera placentaria; por lo tanto, siendo una IgG4, pembrolizumab tiene el potencial de transmitirse de la madre al feto en desarrollo. Pembrolizumab no se debe usar durante el embarazo a menos que el estado clínico de la mujer precise tratamiento con pembrolizumab.
Lactancia:
Se desconoce si pembrolizumab se excreta en la leche materna. Dado que se sabe que los anticuerpos se pueden excretar en la leche materna, no se puede excluir el riesgo en recién nacidos/niños. Se debe decidir si es necesario interrumpir la lactancia o interrumpir el tratamiento con pembrolizumab, tras considerar el beneficio de la lactancia para el niño y el beneficio del tratamiento con pembrolizumab para la madre.
Fertilidad:
No se dispone de datos clínicos relativos a los posibles efectos de pembrolizumab sobre la fertilidad. No hubo efectos notables en los órganos reproductores masculinos y femeninos en monos, de acuerdo a los estudios de toxicidad a dosis repetidas de 1 mes y 6 meses (ver Datos preclínicos sobre seguridad).
EFECTOS SOBRE LA CAPACIDAD PARA CONDUCIR Y UTILIZAR MÁQUINAS
Pembrolizumab puede tener una influencia pequeña sobre la capacidad para conducir y utilizar máquinas. Se ha notificado fatiga después de la administración de pembrolizumab (ver Reacciones adversas).
REACCIONES ADVERSAS:
Resumen del perfil de seguridad:
Pembrolizumab se asocia más frecuentemente a reacciones adversas relacionadas con el sistema inmunitario. La mayoría de éstas, incluyendo las reacciones graves, se resolvieron después de iniciar el tratamiento médico adecuado o de suspender definitivamente el tratamiento con pembrolizumab (ver “Descripción de reacciones adversas seleccionadas” más abajo).
Se ha evaluado la seguridad de pembrolizumab en monoterapia en ensayos clínicos en 4.948 pacientes con melanoma avanzado, melanoma en estadio III resecado (tratamiento adyuvante), CPNM, LHc, carcinoma urotelial o CCECC en cuatro dosis (2 mg/kg cada 3 semanas, 200 mg cada 3 semanas o 10 mg/kg cada 2 o 3 semanas). Las frecuencias incluidas a continuación y en la Tabla 2 se basan en todas las reacciones adversas medicamentosas notificadas, con independencia de la evaluación de causalidad por el investigador. En esta población de pacientes, la mediana del tiempo de observación fue de 7,3 meses (rango: 1 día a 31 meses) y las reacciones adversas más frecuentes con pembrolizumab fueron fatiga (34.1%), erupción (22.7%), náuseas (21.7%), diarrea (21.5%) y prurito (20.2%). La mayoría de las reacciones adversas notificadas para monoterapia fueron de intensidad de Grado 1 o 2. Las reacciones adversas más graves fueron reacciones adversas relacionadas con el sistema inmunitario y reacciones graves asociadas a la infusión (ver Advertencias y precauciones especiales de empleo).
Se ha evaluado la seguridad de pembrolizumab en combinación con quimioterapia en ensayos clínicos en 791 pacientes con CPNM que recibieron 200 mg, 2 mg/kg o 10 mg/kg de pembrolizumab cada 3 semanas. Las frecuencias incluidas a continuación y en la Tabla 2 se basan en todas las reacciones adversas notificadas, con independencia de la evaluación de causalidad por el investigador. En esta población de pacientes, las reacciones adversas más frecuentes fueron náuseas (49%), anemia (48%), fatiga (38%), estreñimiento (34%), diarrea (31%), neutropenia (29%) y apetito disminuido (28%). Las incidencias de reacciones adversas de Grado 3-5 fueron del 67% con el tratamiento de pembrolizumab en combinación y del 66% con la quimioterapia sola.
Tabla de reacciones adversas
Las reacciones adversas observadas en ensayos clínicos de pembrolizumab en monoterapia o en combinación con quimioterapia, o notificadas con el uso de pembrolizumab tras la comercialización se incluyen en la Tabla 2. Se sabe que las reacciones adversas que se producen con pembrolizumab o quimioterapias administradas en monoterapia pueden aparecer durante el tratamiento con estos medicamentos en combinación, incluso si esas reacciones adversas no se notificaron en ensayos clínicos con el tratamiento en combinación. Estas reacciones se presentan según el sistema de clasificación de órganos y por frecuencia. Las frecuencias se definen como: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000); muy raras (< 1/10.000); y frecuencia no conocida (no puede estimarse a partir de los datos disponibles). Dentro de
cada grupo de frecuencias, se presentan las reacciones adversas en orden decreciente de gravedad.
|
Tabla 2: Reacciones adversas en pacientes tratados con pembrolizumab |
||
|
Monoterapia |
Combinación con quimioterapia |
|
|
Infecciones e infestaciones |
||
|
Frecuentes |
neumonía |
neumonía |
|
Trastornos de la sangre y del sistema linfático |
||
|
Muy Frecuentes |
anemia |
neutropenia, anemia, trombocitopenia |
|
Frecuentes |
trombocitopenia, linfopenia |
neutropenia febril, leucopenia, linfopenia |
|
Poco frecuentes |
neutropenia, leucopenia, eosinofilia |
eosinofilia |
|
Raras |
púrpura trombocitopénica inmune, anemia hemolítica, aplasia eritrocitaria pura |
|
|
Trastornos del sistema inmunológico |
||
|
Frecuentes |
reacción asociada a infusióna |
reacción asociada a infusióna |
|
Poco frecuentes |
sarcoidosis |
|
|
Frecuencia no conocida |
rechazo de trasplantes de órganos sólidos |
|
|
Trastornos endocrinos |
||
|
Muy frecuentes |
hipotiroidismob |
|
|
Frecuentes |
hipertiroidismo |
hipertiroidismo, hipotiroidismo |
|
Poco frecuentes |
hipofisitisc, tiroiditisd, insuficiencia suprarrenal |
hipofisitisc, tiroiditis, insuficiencia suprarrenal |
|
Trastornos del metabolismo y de la nutrición |
||
|
Muy frecuentes |
apetito disminuido |
apetito disminuido |
|
Frecuentes |
hiponatremia, hipocalemia, hipocalcemia |
hiponatremia, hipocalemia, hipocalcemia |
|
Poco frecuentes |
diabetes mellitus tipo 1e |
diabetes mellitus tipo 1 |
|
Trastornos psiquiátricos |
||
|
Frecuentes |
insomnio |
insomnio |
|
Trastornos del sistema nervioso |
||
|
Muy frecuentes |
cefalea |
mareo, neuropatía periférica, disgeusia, cefalea |
|
Frecuentes |
mareo, neuropatía periférica, letargia, disgeusia |
letargia |
|
Poco frecuentes |
epilepsia |
epilepsia |
|
Raras |
síndrome de Guillain-Barréf, síndrome miasténicog, meningitis (aséptica), encefalitis |
|
|
Trastornos oculares |
||
|
Frecuentes |
ojo seco |
ojo seco |
|
Poco frecuentes |
uveítish |
|
|
Raras |
síndrome de Vogt-Koyanagi-Harada |
|
|
Trastornos cardiacos |
||
|
Poco frecuentes |
derrame pericárdico, pericarditis |
derrame pericárdico, pericarditis |
|
Raras |
miocarditis |
|
|
Trastornos vasculares |
||
|
Frecuentes |
hipertensión |
hipertensión |
|
Trastornos respiratorios, torácicos y mediastínicos |
||
|
Muy Frecuentes |
disnea, tos |
disnea, tos |
|
Frecuentes |
neumonitisi |
neumonitisi |
|
Trastornos gastrointestinales |
||
|
Muy frecuentes |
diarrea, dolor abdominalj, náuseas, vómitos, estreñimiento |
diarrea, náuseas, vómitos, estreñimiento, dolor abdominalj |
|
Frecuentes |
colitisk, boca seca |
colitisk, boca seca |
|
Poco frecuentes |
pancreatitisl |
pancreatitisl |
|
Raras |
perforación de intestino delgado |
|
|
Trastornos hepatobiliares |
||
|
Frecuentes |
hepatitism |
|
|
Poco frecuentes |
hepatitism |
|
|
Trastornos de la piel y del tejido subcutáneo |
||
|
Muy frecuentes |
erupciónn, pruritoo |
erupciónn, alopecia, pruritoo |
|
Frecuentes |
reacciones cutáneas gravesp, eritema, vitíligoq, piel seca, alopecia, eczema, dermatitis acneiforme |
reacciones cutáneas gravesp, eritema, dermatitis acneiforme, piel seca |
|
Poco frecuentes |
queratosis liquenoider, psoriasis, alopecia, dermatitis, pápula, cambios de color del pelo |
psoriasis, dermatitis, eczema, cambios de color del pelo, queratosis liquenoider, pápula, vitíligoq |
|
Raras |
necrólisis epidérmica tóxica, síndrome de Stevens-Johnson, eritema nudoso |
|
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||
|
Muy frecuentes |
dolor musculoesqueléticos, artralgia |
dolor musculoesqueléticos, artralgia |
|
Frecuentes |
dolor en una extremidad, miositist, artritisu |
miositist, dolor en una extremidad, artritisu |
|
Poco frecuentes |
tenosinovitisv |
tenosinovitisv |
|
Trastornos renales y urinarios |
||
|
Frecuentes |
nefritisw, lesión renal aguda |
|
|
Poco frecuentes |
nefritisw |
|
|
Trastornos generales y alteraciones en el lugar de administración |
||
|
Muy frecuentes |
Fatiga, astenia, edemax, pirexia |
fatiga, astenia, edemax, pirexia |
|
Frecuentes |
enfermedad de tipo gripal, escalofríos |
escalofríos, enfermedad de tipo gripal |
|
Exploraciones complementarias |
||
|
Muy frecuentes |
alanina aminotransferasa elevada, creatinina en sangre elevada |
|
|
Frecuentes |
aspartato aminotransferasa elevada, alanina aminotransferasa elevada, hipercalcemia, fosfatasa alcalina en sangre aumentada, bilirrubina elevada en sangre, creatinina en sangre elevada |
aspartato aminotransferasa elevada, hipercalcemia, fosfatasa alcalina en sangre aumentada |
|
Poco frecuentes |
amilasa elevada |
amilasa elevada, bilirrubina en sangre elevada |
|
*Las frecuencias de las reacciones adversas que se presentan en la Tabla 2 pueden no ser totalmente atribuibles a pembrolizumab en monoterapia, sino también incluir contribuciones de la enfermedad subyacente o de otros medicamentos usados en combinación. |
||
Los siguientes términos representan a un grupo de acontecimientos relacionados que describen a una enfermedad más que a un acontecimiento aislado.
a. reacción asociada a infusión (hipersensibilidad a fármaco, reacción anafiláctica, reacción anafilactoide, hipersensibilidad y síndrome de liberación de citocina)
b. hipotiroidismo (mixedema)
c. hipofisitis (hipopituitarismo)
d. tiroiditis (tiroiditis autoinmune y trastorno de tiroides)
e. diabetes mellitus tipo 1 (cetoacidosis diabética)
f. síndrome de Guillain-Barré (neuropatía axonal y polineuropatía desmielinizante)
g. síndrome miasténico (miastenia gravis, incluida la exacerbación)
h. uveítis (iritis e iridociclitis)
i. neumonitis (enfermedad pulmonar intersticial)
j. dolor abdominal (molestia abdominal, dolor en la zona superior del abdomen y dolor en la zona inferior del abdomen)
k. colitis (colitis microscópica, enterocolitis y colitis autoinmune)
l. pancreatitis (pancreatitis autoinmune y pancreatitis aguda)
m. hepatitis (hepatitis autoinmunitaria y lesión hepática inducida por fármacos)
n. erupción (erupción eritematosa, erupción folicular, erupción generalizada, erupción macular, erupción maculopapular, erupción papular, erupción prurítica, erupción vesicular y erupción genital)
o. prurito (urticaria, urticaria papular, prurito generalizado y prurito genital)
p. reacciones cutáneas graves (dermatitis bullosa, dermatitis exfoliativa, eritema multiforme, erupción exfoliativa, necrosis de la piel, erupción cutánea tóxica y las siguientes reacciones de Grado ≥ 3: dermatosis neutropénica aguda febril, contusión, úlcera de decúbito, dermatitis psoriasiforme, erupción medicamentosa, ictericia, penfigoide, prurito, prurito generalizado, erupción, erupción eritematosa, erupción generalizada, erupción maculopapular, erupción prurítica, erupción pustular y lesión de la piel)
q, vitíligo (despigmentación de la piel, hipopigmentación de la piel e hipopigmentación del párpado)
r. queratosis liquenoide (liquen plano y liquen escleroso)
s. dolor musculoesquelético (molestia musculoesquelética, dolor de espalda, rigidez musculoesquelética, dolor torácico musculoesquelético y tortícolis)
t. miositis (mialgia, miopatía, polimialgia reumática y rabdomiólisis)
u. artritis (hinchazón articular, poliartritis y derrame articular)
v. tenosinovitis (tendinitis, sinovitis y dolor tendinoso)
w. nefritis (nefritis autoinmune, nefritis tubulointersticial y fallo renal, fallo renal agudo o lesión renal aguda con evidencia de nefritis, síndrome nefrótico)
x. edema (edema periférico, edema generalizado, sobrecarga de líquido, retención de líquidos, edema palpebral y edema de labio, edema de cara, edema localizado y edema periorbital)
DESCRIPCIÓN DE REACCIONES ADVERSAS SELECCIONADAS
Los datos de las siguientes reacciones adversas relacionadas con el sistema inmunitario se basan en pacientes que recibieron pembrolizumab en cuatro dosis (2 mg/kg cada 3 semanas, 10 mg/kg cada 2 o 3 semanas o 200 mg cada 3 semanas) en ensayos clínicos (ver Propiedades farmacodinámicas). Las directrices para el manejo de estas reacciones adversas se describen en la Advertencias y precauciones especiales de empleo.
Reacciones adversas relacionadas con el sistema inmunitario (ver Advertencias y precauciones especiales de empleo)
Neumonitis relacionada con el sistema inmunitario:
Se produjo neumonitis en 182 (3.7%) pacientes, incluidos casos de Grado 2, 3, 4 o 5 en 78 (1.6%), 48 (1.0%), 9 (0.2%) y 7 (0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de neumonitis fue de 3.7 meses (rango, 2 días a 21.3 meses). La mediana de duración fue de 1.9 meses (rango, 1 día a 17.2+ meses). La neumonitis se produjo más frecuentemente en pacientes con antecedentes de radiación torácica previa (8.1%) que en pacientes que no recibieron radiación torácica previa (3.3%). La neumonitis condujo a la suspensión definitiva de pembrolizumab en 75 (1.5%) pacientes y se resolvió en 101 pacientes, 2 con secuelas.
En pacientes con CPNM, se produjo neumonitis en 107 (4.9%), incluidos casos de Grado 2, 3, 4 o 5 en 39 (1.8%), 30 (1.4%), 10 (0.5%) y 9 (0.4%) pacientes, respectivamente. En pacientes con CPNM, se produjo neumonitis en el 8.1% con antecedentes de radiación torácica previa.
Colitis relacionada con el sistema inmunitario:
Se produjo colitis en 97 (2.0%) pacientes, incluidos casos de Grado 2, 3 o 4 en 28 (0.6%), 56 (1.1%) y 3 (<0,1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de colitis fue de 3.8 meses (rango, 7 días a 20.2 meses). La mediana de duración fue de
1.2 meses (rango, 1 día a 8.7+ meses). La colitis condujo a la suspensión definitiva de pembrolizumab en 28 (0.6%) pacientes y se resolvió en 75 pacientes, 1 con secuelas.
Hepatitis relacionada con el sistema inmunitario:
Se produjo hepatitis en 39 (0.8%) pacientes, incluidos casos de Grado 2, 3 o 4 en 7 (0.1%), 26 (0.5%) y 4 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hepatitis fue de 2.8 meses (rango, 8 días a 21.4 meses). La mediana de duración fue de
1.1 meses (rango, 1 días a 20.9+ meses). La hepatitis condujo a la suspensión definitiva de pembrolizumab en 14 (0.3%) pacientes y se resolvió en 27 pacientes.
Nefritis relacionada con el sistema inmunitario:
Se produjo nefritis en 17 (0.3%) pacientes, incluidos casos de Grado 2, 3 o 4 en 3 (0.1%), 12 (0.2%) y 1 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab en monoterapia. La mediana de tiempo hasta la aparición de nefritis fue de 5.1 meses (rango, 12 días a 12,8 meses). La mediana de duración fue de 1.8 meses (rango, 6 días a 10.5+ meses). La nefritis condujo a la suspensión definitiva de pembrolizumab en 7 (0.1%) pacientes y se resolvió en 9 pacientes, 1 con secuelas. En pacientes con CPNM no escamoso tratados con pembrolizumab en combinación con pemetrexed y quimioterapia basada en platino (n=488), la incidencia de nefritis fue de 1,4% (todos los Grados) con 0.8% de Grado 3 y 0.4% de Grado 4.
Endocrinopatías relacionadas con el sistema inmunitario:
Se produjo hipofisitis en 32 (0.6%) pacientes, incluidos casos de Grado 2, 3 o 4 en 13 (0.3%), 15 (0.3%) y 1 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hipofisitis fue de 5.3 meses (rango, 1 día a 17.7 meses). La mediana de duración fue de 1.7 meses (rango, 3 días a 18.1+ meses). La hipofisitis condujo a la suspensión definitiva de pembrolizumab en 8 (0.2%) pacientes y se resolvió en 9 pacientes, 7 con secuelas.
Se produjo hipertiroidismo en 197 (4.0%) pacientes, incluidos casos de Grado 2 o 3 en 52 (1.1%) y 5 (0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hipertiroidismo fue de 1.4 meses (rango, 1 día a 21.9 meses). La mediana de duración fue de 1.7 meses (rango, 4 días a 15.5+ meses). El hipertiroidismo condujo a la suspensión definitiva de pembrolizumab en 3 (0.1%) pacientes y se resolvió en 152 (77.2%) pacientes, 1 con secuelas.
Se produjo hipotiroidismo en 514 (10.4%) pacientes, incluidos casos de Grado 2 o 3 en 377 (7.6%) y 7 (0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de hipotiroidismo fue de 3.5 meses (rango, 1 día a 18.9 meses). La mediana de duración no se alcanzó (rango, 2 días a 29.9+ meses). Dos pacientes (<0.1%) descontinuaron pembrolizumab debido a hipotiroidismo. El hipotiroidismo se resolvió en 107 (20.8%) pacientes, 9 con secuelas. En pacientes con LHc (n=241) la incidencia de hipotiroidismo fue de 14.1% (todos los Grados) con 0.4% de Grado 3. En pacientes con CCECC (n=609), la incidencia de hipotiroidismo fue de 15.1% (todos los Grados), con 0.5% de Grado 3.
Reacciones adversas cutáneas relacionadas con el sistema inmunitario:
Se produjeron reacciones adversas cutáneas graves relacionadas con el sistema inmunitario en 66 (1.3%) pacientes, incluidos casos de Grado 2, 3 o 5 en 6 (0.1%), 48 (1.0%) y 1 (<0.1%) pacientes, respectivamente, que recibieron pembrolizumab. La mediana de tiempo hasta la aparición de reacciones cutáneas graves fue de 3.2 meses (rango, 4 días a 19.4 meses). La mediana de duración fue de 1.6 meses (rango, 1 día a 16.1+ meses). Las reacciones cutáneas graves condujeron a la suspensión definitiva de pembrolizumab en 5 (0.2%) pacientes. Las reacciones cutáneas graves se resolvieron en 46 pacientes.
Se han observado casos raros de SSJ y NET, algunos de ellos con desenlace mortal (ver Posología y forma de administración y Advertencias y precauciones especiales de empleo).
Complicaciones del TPH alogénico en linfoma de Hodgkin clásico:
De 23 pacientes con LHc que se sometieron a un TPH alogénico después del tratamiento con pembrolizumab, 6 pacientes (26%) desarrollaron EICH, siendo uno de los casos mortal, y 2 pacientes (9%), desarrollaron enfermedad veno-oclusiva hepática grave tras un acondicionamiento de intensidad reducida, siendo uno de los casos mortal. Los 23 pacientes tuvieron una mediana de seguimiento, a partir del TPH alogénico posterior, de 5.1 meses (rango: 0-26.2 meses).
Anomalías de laboratorio:
En pacientes tratados con pembrolizumab en monoterapia, el porcentaje de pacientes que experimentaron un cambio desde el estado basal hasta una anomalía de laboratorio de Grado 3 o 4 fue el siguiente: 10.8% para linfocitos disminuidos, 7.6% para sodio disminuido, 6.5% para hemoglobina disminuida, 5.2% para fosfato disminuido, 5.2% para glucosa elevada, 2.9% para fosfatasa alcalina elevada, 2.6% para AST elevada, 2.3% para ALT elevada, 2% para potasio disminuido, 1.8% para bilirrubina elevada, 1.6% para potasio elevado, 1.5% para albúmina disminuida, 1.5% para calcio elevado, 1.4% para creatinina elevada, 1.4% para plaquetas disminuidas, 1.4% para neutrófilos disminuidos, 1.2% para calcio disminuido, 0.8% para magnesio elevado, 0.6% para leucocitos disminuidos, 0.5% para glucosa disminuida, 0.2% para magnesio disminuido y 0.2% para sodio elevado.
En pacientes tratados con pembrolizumab en combinación con quimioterapia, el porcentaje de pacientes que experimentaron un cambio desde el estado basal hasta una anomalía de laboratorio de Grado 3 o 4 fue el siguiente: 23.8% para neutrófilos disminuidos, 20.2% para linfocitos disminuidos, 16.2% para hemoglobina disminuida, 14.6% para leucocitos disminuidos, 10.3% para plaquetas disminuidas, 7.9% para glucosa elevada, 7.8% para fosfato disminuido, 7.4% para sodio disminuido, 4.6% para potasio disminuido, 3.7% para ALT elevada, 3.6% para creatinina elevada, 3.5% para AST elevada, 2.9% para calcio disminuido, 2.6% para potasio elevado, 2.5% para albúmina elevada, 1.7% para calcio elevado, 1.2% para fosfatasa alcalina elevada, 0.9% para glucosa disminuida, 0.7% para bilirrubina elevada y 0.1% para sodio elevado.
Inmunogenicidad:
En ensayos clínicos en pacientes tratados con pembrolizumab 2 mg/kg cada tres semanas, 200 mg cada tres semanas o 10 mg/kg cada dos o tres semanas, 36 (1.8%) de 2.034 pacientes evaluables dio positivo a anticuerpos generados por el organismo frente a pembrolizumab, de los cuales 9 (0.4%) pacientes tuvieron anticuerpos neutralizantes contra pembrolizumab. No hubo evidencia de alteración de la farmacocinética o del perfil de seguridad con el desarrollo de anticuerpos de unión anti- pembrolizumab o anticuerpos neutralizantes.
Notificación de sospechas de reacciones adversas:
Es importante notificar sospechas de reacciones adversas al medicamento tras su autorización. Ello permite una supervisión continuada de la relación beneficio/riesgo del medicamento. Se invita a los profesionales sanitarios a notificar las sospechas de reacciones adversas a través del sistema nacional de notificación.
INTERACCIÓN CON OTROS MEDICAMENTOS Y OTRAS FORMAS DE INTERACCIÓN:
No se han realizado estudios formales de interacciones medicamentosas farmacocinéticas con pembrolizumab. Como pembrolizumab se elimina de la circulación mediante catabolismo, no se esperan interacciones medicamentosas metabólicas.
Se debe evitar el uso de corticosteroides sistémicos o de inmunosupresores antes de comenzar el tratamiento con pembrolizumab, debido a su posible interferencia con la actividad farmacodinámica y la eficacia de pembrolizumab. Sin embargo, se puede usar corticosteroides sistémicos u otros inmunosupresores después de comenzar el tratamiento con pembrolizumab, para tratar las reacciones adversas relacionadas con el sistema inmunitario (ver Advertencias y precauciones especiales de empleo).
POSOLOGÍA Y FORMA DE ADMINISTRACIÓN:
El tratamiento se debe iniciar y supervisar por médicos especialistas con experiencia en el tratamiento del cáncer.
Pruebas de PD-L1 en pacientes con CPNM, con carcinoma urotelial o con CCECC
En los pacientes con CPNM se recomienda la realización de pruebas de PD-L1 para determinar la expresión tumoral utilizando una prueba validada. En pacientes con CPNM cuyos tumores tengan un alto nivel de expresión de PD-L1, debe considerarse el riesgo de reacciones adversas con el tratamiento en combinación con respecto a pembrolizumab en monoterapia, y evaluarse la relación riesgo- beneficio del tratamiento combinado de forma individual (ver las Indicaciones terapéuticas, Advertencias y precauciones especiales de empleo, Reacciones adversas, Propiedades farmacodinámicas).
Se debe seleccionar a los pacientes con carcinoma urotelial no tratado previamente o con CCECC para el tratamiento, de acuerdo a la expresión tumoral de PD-L1 confirmada mediante una prueba validada (ver Propiedades farmacodinámicas).
Posología:
La dosis recomendada de KEYTRUDA® en monoterapia es 200 mg cada 3 semanas o 400 mg cada 6 semanas, administrada mediante infusión intravenosa durante 30 minutos.
La dosis recomendada de KEYTRUDA® como parte de un tratamiento en combinación es 200 mg cada 3 semanas, administrada mediante infusión intravenosa durante 30 minutos.
Los pacientes deben recibir tratamiento con KEYTRUDA® hasta progresión de la enfermedad o toxicidad inaceptable. Se han observado respuestas atípicas (por ej., un aumento transitorio inicial del tamaño tumoral o nuevas lesiones pequeñas dentro de los primeros meses, seguido por una reducción del tamaño del tumor). Se recomienda continuar el tratamiento en pacientes clínicamente estables con evidencia inicial de progresión de la enfermedad hasta que se confirme la progresión de la enfermedad.
Para el tratamiento adyuvante del melanoma, KEYTRUDA® se debe administrar hasta recidiva de la enfermedad, toxicidad inaceptable o una duración del tratamiento de hasta un año.
Suspensión temporal de la dosis o suspensión definitiva del tratamiento (ver también Advertencias y precauciones especiales de empleo).
|
Tabla 1: Modificaciones del tratamiento recomendadas para KEYTRUDA® |
||
|
Reacciones adversas relacionadas con el sistema inmunitario |
Gravedad |
Modificación del tratamiento |
|
Neumonitis |
Grado 2 |
Suspender temporalmente hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Grado 3 o 4 o recurrente de Grado 2 |
Suspender definitivamente |
|
|
Colitis |
Grado 2 o 3 |
Suspender temporalmente hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Grado 4 o recurrente de Grado 3 |
Suspender definitivamente |
|
|
Nefritis |
Grado 2 con creatinina > 1.5 a 3 veces el límite superior de la normalidad (LSN) |
Suspender temporalmente hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Grado ≥ 3 con creatinina >3 veces el LSN |
Suspender definitivamente |
|
|
Endocrinopatías |
Hipofisitis sintomática Diabetes tipo 1 asociada a hiperglucemia de Grado ≥ 3 (glucosa > 250 mg/dL o > 13.9 mmol/L) o asociada a cetoacidosis Hipertiroidismo de Grado ≥ 3 |
Suspender temporalmente hasta que las reacciones adversas se recuperen a Grado 0-1* Se puede valorar la continuación del tratamiento con pembrolizumab después de la reducción progresiva de los corticosteroides, si es necesario, en pacientes con endocrinopatía de Grado 3 o Grado 4 que mejora a Grado 2 o menor y se controla con terapia hormonal sustitutiva, si está indicado. En caso contrario, el tratamiento se debe suspender definitivamente. El hipotiroidismo se puede controlar con terapia hormonal sustitutiva sin interrumpir el tratamiento. |
|
Hepatitis |
Grado 2 con la aspartato aminotransferasa (AST) o la alanina aminotransferasa (ALT) > 3 a 5 veces el LSN o la bilirrubina total > 1.5 a 3 veces el LSN |
Suspender temporalmente hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Grado ≥ 3 con la AST o la ALT > 5 veces el LSN o la bilirrubina total > 3 veces el LSN |
Suspender definitivamente |
|
|
En caso de metástasis hepática en estado basal con elevación de Grado 2 de la AST o la ALT, hepatitis con la AST o la ALT elevadas ≥ 50% y ésta dura ≥ 1 semana |
Suspender definitivamente |
|
|
Reacciones cutáneas |
Grado 3 o sospecha de síndrome de Stevens-Johnson (SSJ) o necrólisis epidérmica tóxica (NET) |
Suspender temporalmente hasta que las reacciones adversas se recuperen a Grado 0-1* |
|
Grado 4 o confirmación de SSJ o NET |
Suspender definitivamente |
|
|
Otras reacciones adversas relacionadas con el sistema inmunitario |
De acuerdo con la gravedad y tipo de la reacción (Grado 2 o Grado 3) Miocarditis de Grado 3 o 4 Encefalitis de Grado 3 o 4 Síndrome de Guillain-Barré de Grado 3 o 4 Grado 4 o recurrente de Grado 3 |
Suspender temporalmente hasta que las reacciones adversas se recuperen a Grado 0-1* Suspender definitivamente Suspender definitivamente |
|
Reacciones adversas asociadas a la infusión |
Grado 3 o 4 |
Suspender definitivamente |
|
Nota: los grados de toxicidad son coherentes con los Criterios de Terminología Comunes para Acontecimientos Adversos del National Cancer Institute versión 4 (CTCAE del NCI v.4). * Si la toxicidad relacionada con el tratamiento no se recupera a Grado 0-1 en el plazo de 12 semanas después de la última dosis de KEYTRUDA® o si no se puede reducir la dosis de corticosteroide a ≤ 10 mg de prednisona o equivalente al día en el plazo de 12 semanas, KEYTRUDA® se debe suspender definitivamente. |
||
Se desconoce la seguridad de reiniciar el tratamiento con pembrolizumab en pacientes que han experimentado previamente miocarditis relacionada con el sistema inmunitario.
KEYTRUDA® se debe suspender definitivamente por reacciones adversas de Grado 4 o recurrentes de Grado 3, a menos que se especifique lo contrario en la Tabla 1.
Solo en pacientes con LHc, KEYTRUDA® se debe suspender temporalmente por toxicidad hematológica de Grado 4, hasta que las reacciones adversas se recuperen a Grado 0-1.
Se debe dar a los pacientes tratados con KEYTRUDA® la tarjeta de información para el paciente y se les debe informar sobre los riesgos de KEYTRUDA® (ver también el prospecto).
Poblaciones especiales Pacientes de edad avanzada:
No es necesario un ajuste de la dosis en pacientes ≥ 65 años (ver Propiedades farmacodinámicas). Los datos de pacientes ≥ 65 años son demasiado limitados como para extraer conclusiones en la población con LHc (ver Propiedades farmacodinámicas). Los datos de pembrolizumab en monoterapia en pacientes con melanoma en estadio III resecado y de la combinación con quimioterapia en pacientes con CPNM metastásico ≥ 75 años son limitados (ver las Advertencias y precauciones especiales de empleo y Propiedades farmacodinámicas ).
Insuficiencia renal:
No es necesario un ajuste de la dosis en pacientes con insuficiencia renal leve o moderada. No se ha estudiado KEYTRUDA® en pacientes con insuficiencia renal grave (ver Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas).
Insuficiencia hepática:
No es necesario un ajuste de la dosis en pacientes con insuficiencia hepática leve. No se ha estudiado KEYTRUDA® en pacientes con insuficiencia hepática moderada o grave (ver Advertencias y precauciones especiales de empleo y Propiedades farmacocinéticas).
Melanoma ocular:
Los datos sobre la seguridad y eficacia de KEYTRUDA® en pacientes con melanoma ocular son limitados (ver Propiedades farmacodinámicas).
Puntuación de estado funcional Eastern Cooperative Oncology Group (ECOG) ≥ 2
Se excluyeron de los ensayos clínicos de melanoma, CPNM, LHc y CCECC, los pacientes con puntuación de estado funcional ECOG ≥ 2 (ver las Advertencias y precauciones especiales de empleo y Propiedades farmacodinámicas ).
Población pediátrica:
No se ha establecido todavía la seguridad y eficacia de KEYTRUDA® en niños menores de 18 años. No se dispone de datos.
Forma de administración:
KEYTRUDA® se debe administrar mediante infusión intravenosa durante 30 minutos. KEYTRUDA® no se debe administrar como inyección intravenosa rápida (bolus).
Cuando KEYTRUDA® se administra en combinación con quimioterapia, se debe administrar primero KEYTRUDA®. Ver también la ficha técnica de la quimioterapia administrada en combinación.
Para consultar las instrucciones de dilución del medicamento antes de la administración, ver sección 6.6.
SOBREDOSIS:
No hay información acerca de la sobredosis con pembrolizumab.
En caso de sobredosis, los pacientes deben ser vigilados estrechamente en cuanto a signos o síntomas de reacciones adversas y se debe instaurar tratamiento sintomático adecuado.
PRESENTACIÓN: Naturaleza y contenido del envase: 4 mL de solución en un vial de vidrio de tipo I de 10 mL con un tapón de clorobutilo de color gris y sello de aluminio con una tapa desprendible de color azul oscuro, que contiene 100 mg de pembrolizumab.
Cada caja contiene un vial.
Precauciones especiales de eliminación y otras manipulaciones
Preparación y administración de la infusión
No agitar el vial.
Mantener el vial a temperatura ambiente (a o por debajo de 25°C).
Antes de la dilución, el vial de líquido puede estar fuera de refrigeración (temperaturas a o por debajo de 25°C) hasta 24 horas.
Los medicamentos parenterales deben ser inspeccionados visualmente por si tienen partículas extrañas y cambio de color antes de la administración. KEYTRUDA® es una solución transparente a ligeramente opalescente, incolora a ligeramente amarilla. Desechar el vial si se observan partículas visibles.
Extraer el volumen requerido hasta 4 mL (100 mg) de KEYTRUDA® y transferir a una bolsa de infusión intravenosa que contenga 9 mg/mL de cloruro sódico (0,9%) o 50 mg/mL de glucosa (5%) para preparar una solución diluida con una concentración final que varía entre 1 a 10 mg/mL. Cada vial contiene un exceso de llenado de 0.25 mL (contenido total por vial 4.25 mL) para asegurar la recuperación de los 4 mL de concentrado. Mezclar la solución diluida mediante una inversión suave.
Desde un punto de vista microbiológico, el producto una vez diluido, debe utilizarse inmediatamente. No congelar la solución diluida. Si no se usa inmediatamente, se ha demostrado la estabilidad química y física en uso de KEYTRUDA® durante 24 horas entre 2ºC y 8ºC. Este periodo de 24 horas puede incluir hasta 6 horas a temperatura ambiente (a o por debajo de 25°C). Si se refrigera, se debe dejar que los viales y/o las bolsas intravenosas alcancen la temperatura ambiente antes de su uso. Administrar la solución de infusión por vía intravenosa durante 30 minutos utilizando un filtro de 0.2 a 5 μm, estéril, no pirogénico, de baja unión a proteínas, conectado en línea o añadido.
No administrar con otros medicamentos a través de la misma línea de infusión.
KEYTRUDA® es para un solo uso. Desechar cualquier porción no utilizada que quede en el vial.
La eliminación del medicamento no utilizado y de todos los materiales que hayan estado en contacto con
él se realizará de acuerdo con la normativa local.
FECHA DE REVISIÓN DEL TEXTO: Abril 2019
REFERENCIAS:
European Medicines Agency. KEYTRUDA® Summary of Product Characteristics
https://www.ema.europa.eu/en/documents/product-information/keytruda-epar-product-information_en.pdf
MK3475-CENA-2019-019920
Fabricante:
MSD Ireland (Carlow), Irlanda.
Titular:
Merck Sharp & Dohme Corp., Estados Unidos
Acondicionador secundario: Schering-Plough Labo N.V., Bélgica

