
XTANDI
ENZALUTAMIDA
Cápsulas
1 Caja, 120 Cápsulas, 40 Miligramos
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada CÁPSULA blanda de 40 mg contiene:
Enzalutamida 40 mg
Excipientes:
Macrogol-8 glicéridos de caprilocaproílo 905.81 mg
Butilhidroxianisol (E320) 0.095 mg
Butilhidroxitolueno (E321) 0.095 mg
INDICACIONES TERAPÉUTICAS: Tratamiento de hombres adultos con cáncer de próstata metastásico resistente a la castración que han recibido tratamiento con docetaxel. Tratamiento de hombres adultos con cáncer de próstata metastásico resistente a la castración que son asintomáticos o levemente sintomáticos después de no tener éxito con la terapia de deprivación de andrógenos, y a quienes aún no se indica clínicamente la quimioterapia.
FARMACOCINÉTICA Y FARMACODINAMIA:
Mecanismo de acción: Se sabe que el cáncer de próstata es sensible a los andrógenos y responde a la inhibición de la señalización de los receptores androgénicos. La señalización de los receptores androgénicos sigue favoreciendo la progresión de la enfermedad, aunque las concentraciones séricas de andrógenos sean bajas o incluso indetectables. La estimulación del crecimiento de la célula tumoral a través del receptor androgénico requiere localización nuclear y unión al DNA. Enzalutamida es un inhibidor potente de la señalización de los receptores androgénicos, que bloquea varios pasos en la vía de señalización del receptor androgénico. Enzalutamida inhibe de manera competitiva la unión de los andrógenos a los receptores androgénicos, inhibe la translocación nuclear de los receptores activados e inhibe la asociación del receptor androgénico activado al DNA, incluso en situación de sobreexpresión del receptor androgénico y de células de cáncer de próstata resistentes a los anti-andrógenos. El tratamiento con enzalutamida reduce el crecimiento de células cancerígenas de la próstata, y puede inducir la muerte de células cancerígenas y la regresión tumoral. En estudios preclínicos, enzalutamida carece de actividad agonista de los receptores androgénicos.
Efectos farmacodinámicos: En un ensayo clínico en fase 3, de pacientes que fracasaron con la quimioterapia previa con docetaxel, el 54% de los pacientes tratados con la enzalutamida, en comparación con el 1.5% de los pacientes que recibieron el placebo, presentó una disminución de las concentraciones de PSA de al menos un 50% con respecto a los valores iniciales.
Eficacia clínica y seguridad: La eficacia de la enzalutamida se estableció en dos estudios clínicos en fase 3 aleatorizados, multicéntricos, controlados con placebo [CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)] de pacientes con cáncer de próstata metastásico progresivo que habían fracasado con el tratamiento con supresión de andrógenos [análogo de la hormona liberadora de la hormona luteinizante (LHRH) o tras orquiectomía bilateral]. En el estudio PREVAIL, se inscribieron pacientes que no habían recibido quimioterapia; mientras que en el estudio AFFIRM, se inscribieron pacientes que habían recibido docetaxel anteriormente. Todos los pacientes continuaron con un análogo de LHRH o tuvieron una orquiectomía bilateral previa. En el grupo con tratamiento activo, enzalutamida fue administrada oralmente en una dosis de 160 mg diarios. En ambos ensayos clínicos, los pacientes recibieron placebo en el grupo de control y los pacientes podían, aunque no era requerido, tomar prednisona (dosis diaria máxima permitida fue de 10 mg de prednisona o equivalente).
Los cambios en la concentración sérica de PSA no siempre predicen independientemente un beneficio clínico. Por tanto, en ambos estudios fue recomendado que los pacientes fueran mantenidos con sus tratamientos de estudio hasta que los criterios de suspensión fueran satisfechos tal como se especifica a continuación para cada estudio.
Estudio MDV3100-03 (PREVAIL) (pacientes que nunca había recibido quimioterapia): Un total de 1717 pacientes asintomáticos o levemente sintomáticos que nunca habían recibido quimioterapia fueron aleatorizados 1:1 para recibir enzalutamida por vía oral en una dosis de 160 mg una vez al día (n=872) o placebo por vía oral una vez al día (n=845). Se admitieron pacientes con enfermedad visceral, pacientes con antecedentes de insuficiencia cardiaca leve a moderada (NYHA Clase 1 o 2) y pacientes que toman medicamentos asociados con la reducción del umbral de convulsiones. Se excluyeron los pacientes con antecedentes de convulsiones o una condición que pudiera predisponer a convulsiones y pacientes con dolor moderado o grave de cáncer de próstata. El tratamiento del estudio continuó hasta la progresión de la enfermedad (evidencia de la progresión radiográfica, un evento relacionado con el sistema óseo o la progresión clínica) y la iniciación de una quimioterapia citotóxica o de un agente en investigación o hasta que apareciera una toxicidad inaceptable.
Los datos demográficos y las características iniciales de la enfermedad de los pacientes estaban equilibrados entre los dos grupos de tratamiento. La mediana de la edad fue 71 años (rango 42-93) y la distribución racial fue de 77% caucásicos, 10% asiáticos, 2% negros y 11% de otras razas o razas desconocidas. Sesenta y ocho por ciento (68%) de los pacientes tuvo una puntuación del estado funcional del ECOG de 0, y 32% de los pacientes tuvo una puntuación del estado funcional del ECOG de 1. La evaluación del dolor inicial fue de 0-1 (asintomático) en 67% de los pacientes y de 2-3 (levemente sintomático) en 32% de los pacientes como se define en el Inventario breve del dolor – Cuestionario abreviado (dolor más intenso durante las últimas 24 horas en una escala de 0 a 10). Aproximadamente 45% de los pacientes tenía enfermedad mensurable del tejido blando al ingresar al estudio, y 12% de los pacientes tenía metástasis visceral (pulmón y/o hígado).
Los criterios de valoración coprincipales de la eficacia fueron la supervivencia global y la supervivencia sin progresión radiográfica (rPFS). Además de los criterios de valoración coprincipales, el beneficio también se evaluó usando las siguientes variables: tiempo hasta el inicio de la quimioterapia citotóxica, mejor respuesta general de tejido blando, tiempo hasta el primer evento relacionado con el sistema óseo, respuesta del PSA (≥50% de disminución con respecto al inicio), tiempo hasta la progresión del PSA y tiempo hasta la degradación de la puntuación total en el cuestionario FACT-P.
La progresión radiográfica se evaluó con el uso de estudios de imágenes secuenciales tal como se define en los criterios del Grupo de trabajo 2 sobre ensayos clínicos de cáncer de próstata (PCWG2) (para las lesiones óseas) y/o en los criterios de la Evaluación de la respuesta en tumores sólidos (RECIST v 1.1) (para las lesiones de tejidos blandos). El análisis del rPFS utilizó la evaluación radiográfica de la progresión, revisada centralmente.
En el análisis provisional especificado previamente para la supervivencia global cuando se observaron 540 muertes, el tratamiento con enzalutamida demostró una mejora estadísticamente significativa en la supervivencia global en comparación con el tratamiento con placebo, con una reducción de 29.4% del riesgo de muerte [cociente de riesgos instantáneos (HR) = 0.706, (CI del 95%: 0.596; 0.837), p <0.0001]. Se realizó un análisis de supervivencia actualizado cuando se observaron 784 muertes. Los resultados de este análisis concordaron con los del análisis provisional (tabla 1, figura 1). En el análisis actualizado, 52% de los pacientes tratados con enzalutamida y 81% de los pacientes tratados con placebo habían recibido tratamientos subsiguientes para el CRPC metastásico que podrían prolongar la supervivencia global.
Tabla 1: Supervivencia global de los pacientes tratados con enzalutamida o placebo en el estudio PREVAIL (análisis por intención de tratar)
|
Enzalutamida (N = 872) |
Placebo (N = 845) |
|
|
Análisis provisional pre-especificado |
||
|
Cantidad de muertes (%) |
241 (27.6%) |
299 (35.4%) |
|
Mediana de la supervivencia (meses) (CI del 95%) |
32.4 (30.1, NR) |
30.2 (28.0, NR) |
|
Valor pa |
< 0.0001 |
|
|
Cociente de riesgos instantáneos (CI del 95%)b |
0.71 (0.60, 0.84) |
|
|
Análisis de supervivencia actualizado |
||
|
Cantidad de muertes (%) |
368 (42.2%) |
416 (49.2%) |
|
Mediana de la supervivencia (meses) (CI del 95%) |
35.3 (32.2, NR) |
31.3 (28.8, 34.2) |
|
Valor pa |
0.0002 |
|
|
Cociente de riesgos instantáneos (CI del 95%)b |
0.77 (0.67, 0.88) |
|
a El valor p se obtiene de una prueba no estratificada de rango logarítmico.
b El cociente de riesgos instantáneos se obtiene a partir de un modelo de riesgos proporcionales no estratificado. Un cociente de riesgos instantáneos <1 favorece a enzalutamida.
NR, no alcanzado.
Figura 1: Curvas de supervivencia global de Kaplan-Meier basadas en el análisis de supervivencia actualizado del estudio PREVAIL (análisis por intención de tratar)
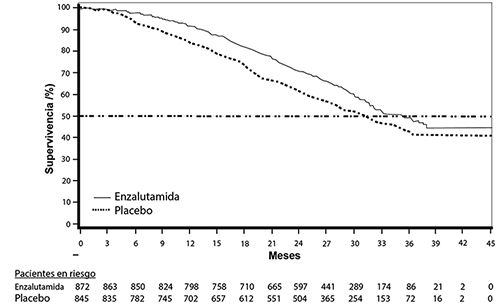
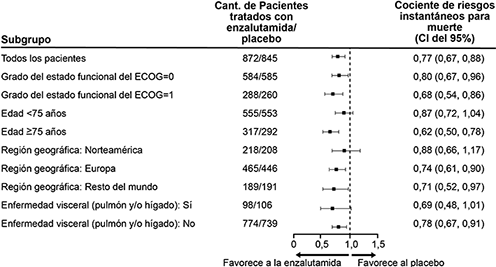
Figura 2: Actualización de supervivencia global por subgrupo; cociente de riesgos instantáneos e intervalo de confianza del 95 % en el estudio PREVAIL (análisis por intención de tratar)
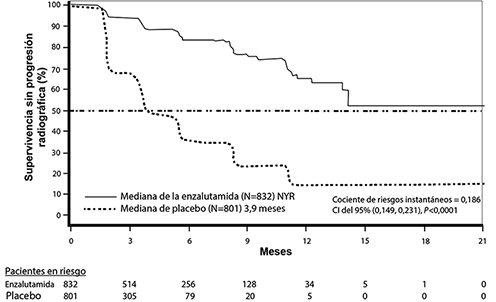
En el análisis rPFS pre-especificado, una mejora estadísticamente significativa se demostró entre los grupos de tratamiento con una reducción del 81.4% en el riesgo de progresión radiográfica o muerte [HR = 0.186 (CI del 95%: 0.149, 0.231), p < 0.0001]. Ciento dieciocho (14%) pacientes tratados con enzalutamida y 321 (40%) pacientes tratados con placebo tuvieron un evento. La mediana del rPFS no se alcanzó (CI del 95%: 13.8, no alcanzado) en el grupo tratado con enzalutamida y fue de 3.9 meses (CI del 95%: 3.7, 5.4) en el grupo tratado con placebo (figura 3). Se observó un beneficio uniforme de la rPFS en todos los subgrupos de pacientes especificados previamente (p. ej., edad, estado funcional inicial del ECOG, PSA y LDH iniciales, puntuación de Gleason en el diagnóstico y enfermedad visceral en la selección). Un análisis de la rPFS de seguimiento especificado previamente basado en la evaluación de la progresión radiográfica por el investigador demostró una mejora estadísticamente significativa entre los grupos de tratamiento, con una reducción del 69.3% del riesgo de progresión radiográfica o muerte [HR = 0.307 (CI del 95%: 0.267; 0.353), p <0.0001]. La mediana de la rPFS fue de 19.7 meses en el grupo con enzalutamida y 5.4 meses en el grupo placebo.
Figura 3: Curvas de supervivencia sin progresión radiográfica de Kaplan-Meier en el estudio PREVAIL (análisis por intención de tratar)
Al momento del análisis principal, se aleatorizó a 1633 pacientes.
Además de los criterios de valoración coprincipales de la eficacia, también se demostraron mejoras estadísticamente significativas en los siguientes criterios de valoración definidos de forma prospectiva.
La mediana de tiempo hasta el inicio de la quimioterapia citotóxica fue 28.0 meses para los pacientes que recibieron enzalutamida y 10.8 meses para los pacientes que recibieron placebo [HR = 0.350, CI 95%: (0.303, 0.403), p<0.0001].
La proporción de pacientes tratados con enzalutamida con enfermedad mensurable al inicio que tuvieron una respuesta objetiva de tejido blando fue 58.8% (CI del 95%: 53.8, 63.7) en comparación con 5.0% (CI del 95%: 3.0, 7.7) de los pacientes que recibían placebo. La diferencia absoluta en la respuesta objetiva del tejido blando entre los grupos de enzalutamida y placebo fue de 53.9% (CI del 95%: 48.5%, 59.1%, p <0.0001). Se informaron respuestas completas en 19.7% de los pacientes tratados con enzalutamida en comparación con 1.0% de los pacientes tratados con placebo, y se informaron respuestas parciales en 39.1% de los pacientes tratados con enzalutamida frente a 3.9% de los pacientes tratados con placebo.
La enzalutamida redujo significativamente el riesgo del primer evento relacionado con el sistema óseo en 28% [HR=0.718 (CI del 95%: 0.610, 0.844) valor de p <0.0001]. Un evento relacionado con el sistema óseo se definió como radioterapia o cirugía ósea por cáncer de próstata, fractura ósea patológica, compresión de la médula espinal, o cambio de la terapia antineoplásica para tratar el dolor óseo. El análisis incluyó 587 eventos relacionados con el sistema óseo, de los cuales 389 eventos (66.3%) fueron radiación ósea, 79 eventos (13.5%) fueron compresión de la médula espinal, 70 eventos (11.9%) fueron fractura ósea patológica, 45 eventos (7.6%) fueron cambio en la terapia antineoplásica para tratar el dolor óseo y 22 eventos (3.7%) fueron cirugía ósea.
Los pacientes que recibieron enzalutamida demostraron una tasa de respuesta total del PSA significativamente mayor (definida como una reducción ≥ 50% del valor inicial), en comparación con los pacientes que recibieron placebo, 78.0% frente a 3.5% (diferencia = 74.5%, p<0.0001).
La mediana del tiempo hasta la progresión del PSA según los criterios del PCWG2 fue de 11.2 meses para los pacientes tratados con enzalutamida y 2.8 meses para los pacientes que recibieron placebo [HR = 0.169, (CI del 95%: 0.147, 0.195), p <0.0001].
El tratamiento con enzalutamida disminuyó el riesgo de degradación según el FACT-P en un 37.5% en comparación con el placebo (p <0.001). La mediana del tiempo hasta la degradación en el FACT-P fue de 11.3 meses en el grupo con enzalutamida y de 5.6 meses en el grupo placebo.
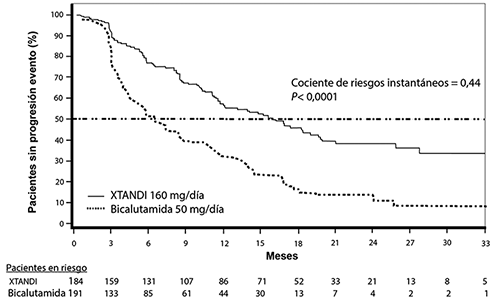
Estudio 9785-CL-0222 (TERRAIN) (pacientes que nunca había recibido quimioterapia): El estudio TERRAIN inscribió a 375 pacientes que nunca habían recibido quimioterapia ni terapia antiandrogénica, y estos fueron asignados aleatoriamente para recibir ya sea una dosis de 160 mg de enzalutamida una vez al día (N=184) o una dosis de 50 mg de bicalutamida una vez al día (N=191). La mediana de la PFS fue de 15.7 meses para pacientes tratados con enzalutamida y de 5.8 meses para pacientes que recibieron bicalutamida [HR=0.44 (CI del 95%: 0.34, 0.57); p <0.0001]. La supervivencia sin progresión (PFS) se definió como pruebas objetivas de la progresión radiográfica de la enfermedad a través de una revisión central independiente, eventos relacionados con el sistema óseo, inicio de una nueva terapia antineoplásica o fallecimiento por cualquier causa, lo que ocurriera primero. Se observó un beneficio uniforme de la PFS en todos los subgrupos de pacientes especificados previamente.
Figura 4: Curvas de supervivencia sin progresión de Kaplan-Meier en el estudio TERRAIN (análisis por intención de tratar)
Estudio CRPC2 (AFFIRM) (pacientes que recibieron quimioterapia previamente): En un ensayo clínico en fase 3, multicéntrico, aleatorizado y controlado con placebo, se evaluaron la eficacia y la seguridad de la enzalutamida en pacientes con cáncer de próstata metastásico resistente a la castración, que habían recibido docetaxel y estaban tomando un análogo de la LHRH o se habían sometido a orquiectomía. En total, se aleatorizó a 1199 pacientes en una proporción 2:1 para recibir enzalutamida por vía oral en una dosis de 160 mg una vez al día (N = 800) o placebo una vez al día (N=399). Los pacientes podían recibir prednisona, pero no era un requisito (la dosis diaria máxima permitida fue de 10 mg de prednisona o equivalente). Los pacientes aleatorizados a cualquiera de los grupos continuaron el tratamiento hasta la progresión de la enfermedad (definida como confirmación radiográfica de la progresión o la aparición de un evento relacionado con el sistema óseo) y el inicio de un nuevo tratamiento antineoplásico sistémico, una toxicidad inaceptable o el retiro.
Los siguientes datos demográficos y características iniciales de la enfermedad de los pacientes fueron equilibrados entre los dos grupos de tratamiento. La mediana de la edad fue de 69 años (intervalo: 41-92), y la distribución racial fue la siguiente: 93% blanca, 4% raza negra, 1% asiática y 2% otra. La puntuación del estado funcional del ECOG (Eastern Cooperative Oncology Group) fue de 0-1 en 91.5% de los pacientes y de 2 en 8.5% de los pacientes; 28% tuvo una puntuación media en el cuestionario breve del dolor (Brief Pain Questionnaire) ≥4 (media del dolor más intensos comunicado por el paciente durante las 24 horas previas, calculado durante los siete días previos a la aleatorización). La mayoría de los pacientes (91%) tenía metástasis en los huesos, y 23% tenía afectación visceral en pulmón y/o hígado. Al momento del ingreso al estudio, 41% de los pacientes aleatorizados tenía progresión del PSA solamente, mientras que 59% de los pacientes tenía progresión radiográfica. Cincuenta y uno por ciento (51%) de los pacientes estaba recibiendo bisfosfonatos en el inicio.
El estudio AFFIRM excluyó a pacientes con afecciones que pudieran predisponerlos a convulsiones y medicamentos que se sabe reducen el umbral convulsivo, así como también enfermedad cardiovascular clínicamente significativa, como hipertensión no controlada, antecedentes recientes de infarto de miocardio o angina inestable, insuficiencia cardiaca de clase III o IV según la New York Heart Association (a menos que la fracción de expulsión fuera ≥45%), arritmias ventriculares clínicamente significativas o bloqueo auriculoventricular (sin marcapasos permanente).
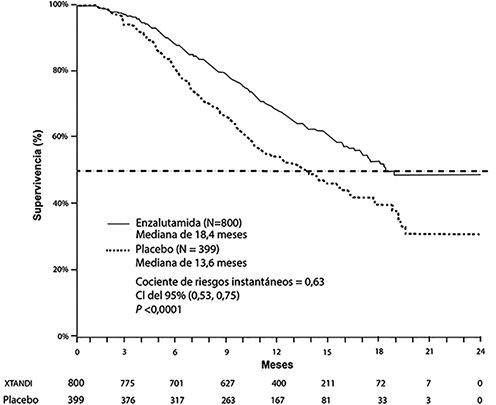
El análisis provisional especificado previamente en el protocolo demostró, luego de 520 muertes, una superioridad estadísticamente significativa en la mediana de la supervivencia global, en los pacientes tratados con enzalutamida en comparación con aquellos tratados con placebo (tabla 2 y figura 5).
Tabla 2. Supervivencia global de pacientes tratados con enzalutamida o placebo en el estudio AFFIRM (análisis por intención de tratar)
|
XTANDI (N=800) |
Placebo (N=399) |
|
|
Muertes (%) |
308 (38.5%) |
212 (53.1%) |
|
Mediana de la supervivencia (meses) (CI del 95%) |
18.4 (17.3; NR) |
13,6 (11.3; 15.8) |
|
Valor pa |
< 0,0001 |
|
|
Cociente de riesgos instantáneos (CI del 95%)b |
0.631 (0.529; 0.752) |
|
a El valor p se obtiene a partir de una prueba de orden logarítmico estratificada de acuerdo con la puntuación del estado funcional del ECOG (0-1 frente a 2) y la puntuación media para el dolor (puntuación < 4 frente a ≥4).
b El cociente de riesgos instantáneos se obtiene a partir de un modelo de riesgos proporcionales estratificado. Un cociente de riesgos instantáneos < 1 favorece a enzalutamida.
NR, no alcanzado.
Figura 5: Curvas de Kaplan-Meier para la supervivencia global en el estudio AFFIRM (análisis por intención de tratar)
El análisis de la supervivencia del subgrupo demostró un beneficio de supervivencia constante para el tratamiento con XTANDI (ver la figura 6).
Figura 6: Supervivencia global por subgrupos en el estudio AFFIRM: cociente de riesgos instantáneos e intervalo de confianza de 95%
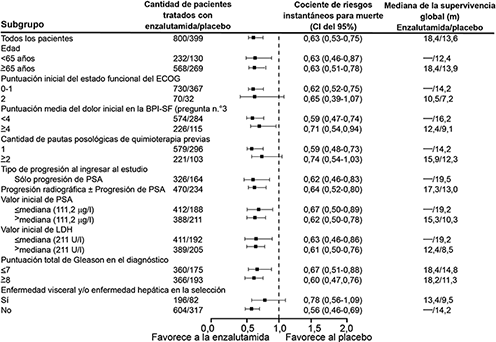
ECOG: Eastern Cooperative Oncology Group; BPI-SF: Inventario breve del dolor – Cuestionario abreviado (Brief Pain Inventory-Short Form); PSA: antígeno prostático específico.
Además de la mejora observada en la supervivencia global, los criterios de valoración secundarios clave (progresión de PSA, supervivencia sin progresión radiográfica y tiempo hasta el primer evento relacionado con el sistema óseo) favorecieron a la enzalutamida y fueron estadísticamente significativos luego de realizar los ajustes necesarios para los distintos análisis.
La supervivencia sin progresión radiográfica según evaluación del investigador usando RECIST v 1.1 para tejido blando y aparición de 2 o más lesiones óseas en la gammagrafía ósea fue de 8.3 meses para los pacientes tratados con enzalutamida y de 2.9 meses para los pacientes que recibieron el placebo (HR=0.404; CI del 95%: [0.350; 0.466]); p <0.0001). En análisis involucró 216 muertes sin progresión documentada y 645 eventos documentados de progresión, de los cuales 303 (47%) se debieron a progresión del tejido blando, 268 (42%) se debieron a progresión de lesión ósea y 74 (11%) se debieron a lesiones óseas y del tejido blando.
La disminución confirmada del PSA del 50% o 90% fue 54.0% y 24.8% respectivamente, para pacientes tratados con enzalutamida, y 1.5% y 0.9% respectivamente, para pacientes que recibieron el placebo (p <0.0001). La mediana de tiempo hasta la progresión del PSA fue de 8.3 meses para pacientes tratados con enzalutamida y de 3.0 meses para pacientes que recibieron placebo (HR=0.248, CI del 95%: [0.204, 0.303]; p <0.0001).
La mediana de tiempo hasta el primer evento relacionado con el sistema óseo fue de 16.7 meses para pacientes tratados con enzalutamida y de 13.3 meses para pacientes que recibieron placebo (HR=0.688; CI del 95%: [0.566; 0.835]; p <0.0001). Se definió evento relacionado con el sistema óseo como radioterapia o cirugía ósea, fractura ósea patológica, compresión medular o cambio de tratamiento antineoplásico para tratar el dolor óseo. El análisis incluyó 448 eventos relacionados con el sistema óseo, de los cuales 277 eventos (62%) fueron radiación ósea, 95 eventos (21%) fueron compresión medular, 47 eventos (10%) fueron fractura ósea patológica, 36 eventos (8%) fueron cambio de tratamiento antineoplásico para tratar el dolor óseo y 7 eventos (2%) fueron cirugía ósea.
La eficacia de enzalutamida en pacientes que han recibido previamente acetato de abiraterona no se ha estudiado.
La tasa de respuesta para la calidad de vida (cuestionario de evaluación funcional de la terapia del cáncer: Próstata; FACT-P) fue 43.2% para pacientes tratados con enzalutamida y 18.3% para pacientes que recibieron placebo (p <0.0001).
Propiedades farmacocinéticas: La enzalutamida es poco soluble en agua. En este producto, la solubilidad de la enzalutamida se aumenta mediante macrogolglicéridos de caprilocaproil como emulgente/surfactante. En estudios preclínicos, la absorción de la enzalutamida aumentó al disolverla en macrogolglicéridos de caprilocaproil.
Se evaluó la farmacocinética de la enzalutamida en pacientes con cáncer de próstata y en sujetos sanos de sexo masculino. La semivida terminal media (t1/2) para la enzalutamida en pacientes luego de una única dosis oral es de 5.8 días (intervalo de 2.8 a 10.2 días), y en aproximadamente un mes se alcanza un estado de equilibrio. Con la administración oral diaria, la enzalutamida se acumula aproximadamente 8.3 veces en relación con una dosis única. Las fluctuaciones diarias en las concentraciones plasmáticas son bajas (índice valle-pico de 1.25). La depuración de la enzalutamida se realiza, principalmente, a través del metabolismo hepático mediante la producción de un metabolito activo, igual de activo que la enzalutamida, que circula en aproximadamente la misma concentración plasmática que la enzalutamida. Absorción: Las concentraciones plasmáticas máximas (Cmáx) de enzalutamida en pacientes se observan entre 1 y 2 horas después de la administración. Según un estudio de equilibrio de masa en seres humanos, se calcula que la absorción oral de la enzalutamida es de al menos 84.2%. La enzalutamida no es un sustrato de los transportadores de eflujo P-gp o BCRP. En estado de equilibrio, los valores medios de Cmáx, para la enzalutamida y su metabolito activo son de 16.6 μg/ml (coeficiente de variación [CV] del 23%) y 12.7 μg/ml (CV del 30%), respectivamente.
Los alimentos no tienen un efecto de significancia clínica sobre el grado de absorción. En ensayos clínicos, XTANDI se administró independientemente de los alimentos.
Distribución: El volumen aparente de distribución (V/F) medio de la enzalutamida en pacientes luego de una dosis única oral es de 110 L (CV del 29%). El volumen de distribución de enzalutamida es mayor que el volumen de agua corporal total, lo que indica una amplia distribución extravascular. Los estudios realizados en roedores indican que la enzalutamida y su metabolito activo pueden atravesar la barrera hematoencefálica.
Entre 97% y 98% de la enzalutamida se une a las proteínas plasmáticas, principalmente la albúmina. El metabolito activo se une en 95% a las proteínas plasmáticas. No hubo desplazamiento de la unión de proteínas entre enzalutamida y otros fármacos con gran afinidad de unión (warfarina, ibuprofeno y ácido salicílico) in vitro.
Metabolismo: La enzalutamida se metaboliza ampliamente. En el plasma humano hay dos metabolitos principales: N-desmetil enzalutamida (activo) y un derivado del ácido carboxílico (inactivo). La enzalutamida es metabolizada por el CYP2C8 y, en menor grado, por el CYP3A4/5, los cuales participan en la formación del metabolito activo. In vitro, la N-desmetil enzalutamida se metaboliza al metabolito ácido carboxílico por la carboxilesterasa 1, que también desempeña una función menor en el metabolismo de la enzalutamida al metabolito ácido carboxílico. La N-desmetil enzalutamida no fue metabolizada por los CYP in vitro.
En condiciones de uso clínico, la enzalutamida es un inductor potente del CYP3A4, un inductor moderado del CYP2C9 y CYP2C19 y carece de efectos clínicamente significativos sobre CYP2C8.
Eliminación y excreción: La depuración aparente media (CL/F) de enzalutamida en pacientes oscila entre 0.520 y 0.564 l/h.
Luego de la administración oral de 14C-enzalutamida, 84.6% de la radiactividad se recuperó 77 días después de la administración de la dosis: 71.0% se recuperó en la orina (principalmente en forma de metabolito inactivo, con cantidades mínimas de enzalutamida y del metabolito activo) y 13.6%, en las heces (0.39% de la dosis en forma de enzalutamida intacta).
Los datos in vitro indican que la enzalutamida no es un sustrato para OATP1B1, OATP1B3, ni OCT1; y que N-desmetil enzalutamida no es un sustrato para P-gp o BCRP.
Los datos in vitro indican que la enzalutamida y sus metabolitos principales no inhiben los siguientes transportadores en concentraciones clínicamente pertinentes: OATP1B1, OATP1B3, OCT2 o OAT1.
Poblaciones especiales:
Población geriátrica: De los 1671 pacientes en los ensayos en fase 3 que recibieron enzalutamida, 1261 pacientes (75%) tenían 65 años o más y 516 pacientes (31%) tenían 75 años o más. No se observaron diferencias generales respecto a la seguridad o la eficacia entre estos pacientes geriátricos y los pacientes más jóvenes.
Población pediátrica: No ha habido uso relevante de enzalutamida en la población pediátrica en la indicación de tratamiento de hombres adultos con cáncer de próstata metastásico resistente a la castración.
Sexo: La enzalutamida no está indicada para su uso en mujeres. No se ha evaluado la farmacocinética de la enzalutamida en mujeres.
Raza: La mayoría de los pacientes en los ensayos clínicos (>84%) eran caucásicos. En base a los datos farmacocinéticos de un estudio en pacientes japoneses con cáncer de próstata, no hubo diferencias clínicamente relevantes en la exposición entre japoneses y caucásicos. Los datos son insuficientes para evaluar las posibles diferencias en la farmacocinética de la enzalutamida en otras razas.
Insuficiencia renal: No se han realizado estudios formales de la enzalutamida en pacientes con insuficiencia, renal. Se excluyó de los ensayos clínicos a los pacientes con una creatinina sérica >177 μmol/l (2 mg/dl). Según un análisis de farmacocinética poblacional, no es necesario ajustar la dosis en los pacientes con valores calculados de depuración de creatinina (CrCL) ≥30 ml/min (estimados mediante la fórmula de Cockcroft y Gault). No se ha evaluado el uso de la enzalutamida en pacientes con insuficiencia renal grave (CrCL <30 ml/min) ni con enfermedad renal en fase terminal, y se aconseja precaución al tratar a estos pacientes. Es poco probable que la enzalutamida se elimine significativamente mediante hemodiálisis intermitente o diálisis peritoneal ambulatoria continua.
Insuficiencia hepática: La insuficiencia hepática no tuvo un efecto marcado en la exposición total a la enzalutamida o a su metabolito activo. La semivida del fármaco, sin embargo, fue del doble en los pacientes con insuficiencia hepática grave en comparación con los controles sanos (10.4 días frente a 4.7 días), lo cual está posiblemente relacionado con un aumento en la distribución tisular.
La farmacocinética de la enzalutamida se evaluó en sujetos con insuficiencia hepática inicial leve (N=6), moderada (N=8) o grave (N=8) (clases A, B o C de Child-Pugh, respectivamente), comparados con 22 sujetos de control emparejados con una función hepática normal. Luego de administrar una dosis oral única de 160 mg de enzalutamida, el AUC y la Cmáx de la enzalutamida en sujetos con insuficiencia leve aumentaron 5% y 24%, respectivamente, el AUC y la Cmáx de la enzalutamida en sujetos con insuficiencia moderada aumentó 29% y disminuyó 11%, respectivamente, y el AUC y la Cmáx de la enzalutamida en sujetos con insuficiencia grave aumentó 5% y disminuyó 41%, respectivamente, en comparación con los sujetos de control sanos. Para la suma de enzalutamida libre más el metabolito activo libre, el AUC y la Cmáx en sujetos con insuficiencia leve aumentaron 14% y 19%, respectivamente, y el AUC aumentó 14% y la Cmáx disminuyó 17% en sujetos con insuficiencia moderada, y el AUC aumentó 34% y la Cmáx disminuyó 27%, respectivamente, en sujetos con insuficiencia hepática grave en comparación con los sujetos de control sanos.
CONTRAINDICACIONES: XTANDI está contraindicado en pacientes con hipersensibilidad al principio activo o a cualquiera de los excipientes y en mujeres que estén o puedan quedar embarazadas y que estén amamantando.
Este medicamento está contraindicado en personas menores de 18 años.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Anticoncepción en hombres y mujeres: Se desconoce si XTANDI o sus metabolitos están presentes en el semen. Si el paciente mantiene relaciones sexuales con una mujer embarazada, debe utilizar un preservativo durante el tratamiento con XTANDI y durante los 3 meses posteriores. Si el paciente mantiene relaciones sexuales con una mujer fértil, debe utilizar un preservativo y otro método anticonceptivo durante el tratamiento y en los 3 meses posteriores.
Embarazo: XTANDI no está indicado para mujeres. XTANDI está contraindicado en mujeres embarazadas o que puedan quedar embarazadas. No hay datos en seres humanos sobre el uso de enzalutamida en mujeres embarazadas.
Lactancia: XTANDI no está indicado para mujeres. Se desconoce si XTANDI o sus metabolitos se excretan en la leche materna.
REACCIONES SECUNDARIAS Y ADVERSAS: Las reacciones adversas más comunes son astenia/fatiga, sofocos, cefalea e hipertensión. Otras reacciones adversas importantes incluyen caídas, fracturas no patológicas, trastorno cognitivo y neutropenia.
Se produjeron convulsiones en 0.5% de los pacientes tratados con enzalutamida, en 0.1% de los pacientes tratados con placebo y en 0.3% de los pacientes tratados con bicalutamida.
Se han reportado casos infrecuentes de síndrome de encefalopatía posterior reversible en pacientes tratados con enzalutamida.
Las reacciones adversas observadas durante los ensayos clínicos y poscomercialización se enumeran a continuación por categoría de frecuencia.
Tabla 3. Reacciones adversas en el AFFIRM
|
XTANDI N = 800 |
Placebo N = 399 |
|||
|
Grado 1-4a (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
Trastornos generales |
||||
|
Trastornos asténicosb |
50.6 |
9.0 |
44.4 |
9.3 |
|
Edema periférico |
15.4 |
1.0 |
13.3 |
0.8 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor de espalda |
26.4 |
5.3 |
24.3 |
4.0 |
|
Artralgia |
20.5 |
2.5 |
17.3 |
1.8 |
|
Artromialgias |
15.0 |
1.3 |
11.5 |
0.3 |
|
Debilidad muscular |
9.8 |
1.5 |
6.8 |
1.8 |
|
Rigidez osteomuscular |
2.6 |
0.3 |
0.3 |
0.0 |
|
Trastornos gastrointestinales |
||||
|
Diarrea |
21.8 |
1.1 |
17.5 |
0.3 |
|
Trastornos vasculares |
||||
|
Sofoco |
20.3 |
0.0 |
10.3 |
0.0 |
|
Hipertensión |
6.4 |
2.1 |
2.8 |
1.3 |
|
Trastornos del sistema nervioso |
||||
|
Dolor de cabeza |
12.1 |
0.9 |
5.5 |
0.0 |
|
Mareosc |
9.5 |
0.5 |
7.5 |
0.5 |
|
Compresión de la médula espinal y síndrome de la cola de caballo |
7.4 |
6.6 |
4.5 |
3.8 |
|
Parestesia |
6.6 |
0.0 |
4.5 |
0.0 |
|
Trastornos de deterioro mentald |
4.3 |
0.3 |
1.8 |
0.0 |
|
Hipoestesia |
4.0 |
0.3 |
1.8 |
0.0 |
|
Infecciones e infestaciones |
||||
|
Infección de las vías respiratorias superiorese |
10.9 |
0.0 |
6.5 |
0.3 |
|
Infección pulmonar y de las vías respiratorias inferioresf |
8.5 |
2.4 |
4.8 |
1.3 |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
8.8 |
0.0 |
6.0 |
0.5 |
|
Ansiedad |
6.5 |
0.3 |
4.0 |
0.0 |
|
Trastornos renales y urinarios |
||||
|
Hematuria |
6.9 |
1.8 |
4.5 |
1.0 |
|
Polaquiuria |
4.8 |
0.0 |
2.5 |
0.0 |
|
Lesiones, envenenamientos y complicaciones de procedimiento |
||||
|
Caída |
4.6 |
0.3 |
1.3 |
0.0 |
|
Fracturas no patológicas |
4.0 |
1.4 |
0.8 |
0.3 |
|
Trastornos de la piel y del tejido subcutáneo |
||||
|
Prurito |
3.8 |
0.0 |
1.3 |
0.0 |
|
Piel seca |
3.5 |
0.0 |
1.3 |
0.0 |
|
Trastornos respiratorios |
||||
|
Epistaxis |
3.3 |
0.1 |
1.3 |
0.3 |
a CTCEA versión 4.
b Incluye astenia y fatiga.
c Incluye mareos y vértigo.
d Incluye amnesia, deterioro de la memoria, trastorno cognitivo y alteración de la atención.
e Incluye nasofaringitis, infección de las vías respiratorias superiores, sinusitis, rinitis, faringitis y laringitis.
f Incluye neumonía, infección de las vías respiratorias inferiores, bronquitis e infección pulmonar.
Las reacciones adversas del ensayo PREVAIL se enumeran a continuación por categoría de frecuencia.
Tabla 4. Reacciones adversas en el PREVAIL
|
XTANDI N = 871 |
Placebo N = 844 |
|||
|
Grado 1-4a (%) |
Grado 3-4 (%) |
Grado 1-4 (%) |
Grado 3-4 (%) |
|
|
Trastornos generales |
||||
|
Trastornos asténicosb |
46.9 |
3.4 |
33.0 |
2.8 |
|
Edema periférico |
11.5 |
0.2 |
8.2 |
0.4 |
|
Trastornos musculoesqueléticos y del tejido conjuntivo |
||||
|
Dolor de espalda |
28.6 |
2.5 |
22.4 |
3.0 |
|
Artralgia |
21.4 |
1.6 |
16.1 |
1.1 |
|
Trastornos gastrointestinales |
||||
|
Estreñimiento |
23.2 |
0.7 |
17.3 |
0.4 |
|
Diarrea |
16.8 |
0.3 |
14.3 |
0.4 |
|
Trastornos vasculares |
||||
|
Sofocos |
18.0 |
0.1 |
7.8 |
0.0 |
|
Hipertensión |
14.2 |
7.2 |
4.1 |
2.3 |
|
Trastornos del sistema nervioso |
||||
|
Mareosc |
11.3 |
0.3 |
7.1 |
0.0 |
|
Dolor de cabeza |
11.0 |
0.2 |
7.0 |
0.4 |
|
Disgeusia |
7.6 |
0.1 |
3.7 |
0.0 |
|
Trastorno cognitivod |
5.7 |
0.0 |
1.3 |
0.1 |
|
Síndrome de piernas inquietas |
2.1 |
0.1 |
0.4 |
0.0 |
|
Trastornos respiratorios |
||||
|
Disneae |
11.0 |
0.6 |
8.5 |
0.6 |
|
Infecciones e infestaciones |
||||
|
Infección de las vías respiratorias superioresf |
16.4 |
0.0 |
10.5 |
0.0 |
|
Infección pulmonar y de las vías respiratorias inferioresg |
7.9 |
1.5 |
4.7 |
1.1 |
|
Trastornos psiquiátricos |
||||
|
Insomnio |
8.2 |
0.1 |
5.7 |
0.0 |
|
Trastornos renales y urinarios |
||||
|
Hematuria |
8.8 |
1.3 |
5.8 |
1.3 |
|
Lesiones, envenenamientos y complicaciones de procedimiento |
||||
|
Caída |
12.7 |
1.6 |
5.3 |
0.7 |
|
Fractura no patológica |
8.8 |
2.1 |
3.0 |
1.1 |
|
Trastornos del metabolismo y la nutrición |
||||
|
Disminución del apetito |
18.9 |
0.3 |
16.4 |
0.7 |
|
Investigaciones |
||||
|
Pérdida de peso |
12.4 |
0.8 |
8.5 |
0.2 |
|
Trastornos torácicos y del sistema reproductivo |
||||
|
Ginecomastia |
3.4 |
0.0 |
1.4 |
0.0 |
a CTCEA versión 4.
b Incluye astenia y fatiga.
c Incluye mareos y vértigo.
d Incluye amnesia, deterioro de la memoria, trastorno cognitivo y alteración de la atención.
e Incluye disnea, disnea de esfuerzo y disnea en reposo.
f Incluye nasofaringitis, infección de las vías respiratorias superiores, sinusitis, rinitis, faringitis y laringitis.
g Incluye neumonía, infección de las vías respiratorias inferiores, bronquitis e infección pulmonar.
Convulsiones: En estudios clínicos controlados, 10 (0.5%) de los 2051 pacientes tratados con una dosis diaria de 160 mg de enzalutamida presentaron convulsiones, mientras que un paciente que recibió placebo (<0.1%) y un paciente (0.3%) que recibió bicalutamida presentaron una convulsión. La dosis parece ser un factor predictivo importante del riesgo de convulsiones, como se refleja en los datos preclínicos y en los datos de un estudio de incremento de la dosis. En los estudios clínicos controlados, se excluyeron los pacientes con factores de riesgo de convulsiones o con convulsiones previas.
En el ensayo AFFIRM, seis pacientes (0.8%) de los 800 pacientes de posquimioterapia tratados con una dosis diaria de 160 mg de enzalutamida experimentaron un ataque, mientras que ningún ataque ocurrió en pacientes que recibieron placebo. En varios de estos pacientes estuvieron presentes factores potencialmente contribuyentes que pueden haber aumentado independientemente su riesgo de convulsiones. En el ensayo PREVAIL, un paciente (0.1%) de los 871 pacientes que nunca habían recibido quimioterapia y tratados con una dosis diaria de 160 mg de enzalutamida, y un paciente (0.1%) que recibió placebo, experimentaron una convulsión. En ensayos controlados con bicalutamida, 3 pacientes (0.8%) de los 380 pacientes que nunca había recibido quimioterapia y tratados con enzalutamida y 1 paciente (0.3%) de los 387 que recibieron bicalutamida experimentaron una convulsión.
El mecanismo por el cual enzalutamida puede disminuir el umbral de convulsiones no se conoce, pero podría estar relacionado con datos obtenidos de estudios in vitro que muestran que enzalutamida y su metabolito activo se unen al canal de cloruro cerrado por GABA y pueden inhibir su actividad.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: No se han realizado estudios en animales a largo plazo para evaluar el potencial carcinogénico de enzalutamida.
Enzalutamida no indujo mutaciones en el ensayo de mutagénesis microbiana (Ames) y no fue clastogénico ni en el ensayo citogenético in vitro con células de linfoma de ratón ni en el ensayo in vivo de micronúcleos de ratón. No se han realizado estudios en animales a largo plazo para evaluar el potencial carcinogénico de enzalutamida. Enzalutamida no fue fototóxico in vitro.
Fertilidad: Los estudios en animales mostraron que enzalutamida afectó el sistema reproductivo de ratas y perros machos. El tratamiento con enzalutamida de ratonas preñadas produjo una mayor incidencia de muertes embriofetales y cambios externos y óseos. No se han llevado a cabo estudios de toxicología reproductiva con enzalutamida, pero en los estudios en ratas (4 y 26 semanas) y perros (4, 13 y 39 semanas) se observaron atrofia, aspermia/hipospermia e hipertrofia/hiperplasia en el sistema reproductivo, en consonancia con la actividad farmacológica de enzalutamida. En estudios en ratones (4 semanas), ratas (4 a 26 semanas) y perros (4, 13 y 39 semanas), los cambios en los órganos reproductores relacionados con la enzalutamida, fueron disminuciones del peso de los órganos con atrofia de la próstata y del epidídimo. Otros cambios en los tejidos reproductores fueron hipertrofia/hiperplasia de la hipófisis y atrofia de las vesículas seminales en ratas, e hipospermia testicular y degeneración de los túbulos seminíferos en perros. Se observaron diferencias sexuales en las glándulas mamarias de rata (atrofia en el macho e hiperplasia lobular en la hembra). Los cambios en los órganos reproductores en ambas especies fueron consistentes con la actividad farmacológica de enzalutamida y se revertieron o se resolvieron parcialmente después de un periodo de recuperación de 8 semanas. No hubo otros cambios importantes en la patología clínica o histopatología en ningún otro sistema de órganos, incluido el hígado, en cualquiera de las especies.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Posibilidad de que otros medicamentos modifiquen las exposiciones a la enzalutamida:
Inhibidores de CYP2C8: El CYP2C8 desempeña una función importante en la eliminación de la enzalutamida y en la formación de su metabolito activo. Luego de la administración oral a hombres sanos de gemfibrozilo (600 mg dos veces al día), un inhibidor potente del CYP2C8, el AUC de la enzalutamida aumentó 326%, mientras que la Cmáx de la enzalutamida disminuyó 18%. Para la suma de enzalutamida libre más el metabolito activo libre, el AUC aumentó 77%, mientras que la Cmáx disminuyó 19%. Se deben evitar o usar con precaución los inhibidores potentes de CYP2C8 (p. ej., gemfibrozilo) durante el tratamiento con XTANDI. Si se debe administrar de manera concomitante a los pacientes un inhibidor potente del CYP2C8, la dosis de enzalutamida se debe reducir a 80 mg una vez al día.
Inhibidores del CYP3A4: CYP3A4 tiene una función secundaria en el metabolismo de la enzalutamida. Luego de la administración oral a hombres sanos de itraconazol (200 mg una vez al día), un inhibidor potente del CYP3A4, el AUC de la enzalutamida aumentó 41%, mientras que la Cmáx, se mantuvo inalterada. Para la suma de enzalutamida libre más el metabolito activo libre, el AUC aumentó 27%, mientras que la Cmáx quedó nuevamente inalterada. No es necesario ajustar la dosis al administrar enzalutamida de manera concomitante con inhibidores del CYP3A4.
Inductores de CYP2C8 y CYP3A4: Luego de la administración oral a hombres sanos de rifampina (600 mg una vez al día), un inhibidor moderado del CYP2C8 y un inhibidor potente del CYP3A4, el AUC de la enzalutamida más el metabolito activo disminuyó 37%, mientras que la Cmáx permaneció inalterada. No es necesario ajustar la dosis al administrar XTANDI de manera concomitante con inductores de CYP2C8 o CYP3A4.
Posibilidad de que la enzalutamida modifique las exposiciones a otros medicamentos:
Inducción enzimática: La enzalutamida es un potente inductor enzimático y aumenta la síntesis de muchas enzimas y transportadores, por lo tanto, se prevé la interacción con muchos medicamentos habituales que son sustratos de enzimas o transportadores. La reducción de las concentraciones plasmáticas puede ser substancial y conducir a la pérdida del efecto clínico o a su reducción. Existe también un riesgo de formación incrementada de metabolitos activos. Las enzimas que pueden ser inducidas incluyen CYP3A en el hígado e intestino, CYP2B6, CYP2C9, CYP2C19 y uridina 5"-difosfo glucuronosiltransferasa (UGT, enzimas de conjugado glucurónido). La proteína de transporte P-gp podría también ser inducida, y probablemente otros transportadores también, por ejemplo, la proteína 2 asociada a la resistencia a multifármacos (MRP2), la proteína de resistencia al cáncer de mamas (BCRP) y el polipéptido de transporte de aniones orgánicos 1B1 (OATP1B1).
La enzalutamida es un inductor potente de la CYP3A4 y un inductor moderado de enzimas CYP2C9 y CYP2C19. La administración concomitante de la enzalutamida (160 mg una vez al día) con dosis orales únicas de sustratos sensibles de CYP a pacientes con cáncer de próstata produjo una disminución de 86% del AUC del midazolam (sustrato de CYP3A4), de 56% del AUC de la S-warfarina (sustrato de CYP2C9) y de 70% del AUC del omeprazol (sustrato de CYP2C19). También es posible que se haya producido la inducción de la uridina 5"-difosfo-glucuronosiltransferasa (UGT1A1).
En un estudio clínico en pacientes con CRPC metastásico, enzalutamida (160 mg una vez al día) no tuvo un efecto clínicamente relevante sobre la farmacocinética de docetaxel administrado intravenosamente (75 mg/m2 por infusión cada 3 semanas). El AUC del docetaxel disminuyó el 12% [cociente de las medias geométricas (GMR) = 0.882 (CI del 90%: 0.767, 1.02)], mientras que la Cmáx disminuyó el 4% [GMR = 0.963 (CI del 90%: 0.834, 1.11)].
Se prevén interacciones con determinados medicamentos que se eliminan a través del metabolismo o el transporte activo. Si su efecto terapéutico es de gran importancia para el paciente, y los ajustes de la dosis no son realizados con facilidad en base a la monitorización de la eficacia o de las concentraciones plasmáticas, estos medicamentos deben ser evitados o usados con precaución. Se sospecha que el riesgo de daño hepático después de la administración de paracetamol es superior en pacientes tratados concomitantemente con inductores enzimáticos.
Los medicamentos que pueden ser afectados incluyen, entre otros, los siguientes grupos:
• Analgésicos (p. ej., fentanilo, tramadol).
• Antibióticos (p. ej., claritromicina, doxiciclina).
• Fármacos antineoplásicos (p. ej., cabazitaxel).
• Anticoagulantes (p. ej., acenocumarol, warfarina).
• Antiepilépticos (p. ej., carbamazepina, clonazepam, fenitoína, primidona, ácido valproico).
• Antipsicóticos (p. ej., haloperidol).
• Betabloqueadores (p. ej., bisoprolol, propranolol).
• Bloqueadores del canal de calcio (p. ej., diltiazem, felodipina, nicardipina, nifedipina, verapamil).
• Glucósidos cardiacos (p. ej., digoxina).
• Corticoesteriodes (p. ej., dexametasona, prednisolona).
• Antivirales para el VIH (p. ej., indinavir, ritonavir).
• Moduladores de la respuesta inmunitaria (p. ej., ciclosporina, tacrolimús).
• Estatinas metabolizadas por CYP3A4 (p. ej., atorvastatina, simvastatina).
• Agentes tiroideos (p. ej., levotiroxina).
El potencial de inducción completa de enzalutamida no puede producirse hasta aproximadamente 1 mes después del inicio del tratamiento, cuando se alcanzan las concentraciones plasmáticas de estado de equilibrio de enzalutamida, aunque algunos efectos de inducción pueden ser evidentes antes. Se debe evaluar a los pacientes que toman medicamentos que son sustratos de CYP2B6, CYP3A4, CYP2C9, CYP2C19 o UGT1A1 para detectar la posible pérdida de efectos farmacológicos (o el aumento en los efectos en casos donde se formen metabolitos activos) durante el primer mes del tratamiento con enzalutamida, y se debe considerar el ajuste de la dosis cuando corresponda. Teniendo en cuenta la semivida prolongada de la enzalutamida, los efectos sobre las enzimas pueden persistir durante un mes o más después de interrumpir la administración de enzalutamida. Una reducción gradual de la dosis del medicamento concomitante puede ser necesaria cuando se interrumpe el tratamiento de enzalutamida.
Sustratos del CYP1A2 y del CYP2C8: La enzalutamida (160 mg una vez al día) no provocó un cambio clínicamente significativo en el AUC ni en la Cmáx de la cafeína (sustrato de CYP1A2) o de la pioglitazona (sustrato de CYP2C8). El AUC de la pioglitazona aumentó en 20%, mientras que la Cmáx se redujo en 18%. El AUC y la Cmáx de la cafeína disminuyeron 11% y 4%, respectivamente. No está indicado ajustar la dosis al administrar XTANDI de manera concomitante con sustratos de CYP1A2 o de CYP2C8.
Sustratos de la P-gp: Los datos in vitro indican que la enzalutamida puede ser un inhibidor del transportador de eflujo, la glucoproteína P (P-gp). El efecto de la enzalutamida sobre sustratos de la P-gp no se ha evaluado in vivo; sin embargo, en condiciones de uso clínico, la enzalutamida puede ser un inductor de la P-gp mediante la activación del receptor nuclear de pregnano (PXR). Los medicamentos con un estrecho margen terapéutico que sean sustratos de la P-gp (p. ej., colchicina, dabigatrán etexilato o digoxina) se deben usar con precaución cuando se administran de manera concomitante con la enzalutamida, y puede ser necesario ajustar la dosis para mantener concentraciones plasmáticas óptimas.
Sustratos de BCRP, MRP2, OAT3 y OCTI: Según los datos obtenidos in vitro, no se puede descartar la inhibición de BCRP y MRP2 (en el intestino) ni del transportador de aniones orgánicos 3 (OAT3) o el transportador de cationes orgánicos 1 (OCT1) (sistémicamente). En teoría, la inducción de estos transportadores también es posible, y el efecto neto se desconoce actualmente.
Efecto de los alimentos sobre la exposición a la enzalutamida: Los alimentos no tienen un efecto de significancia clínica sobre el grado de exposición a la enzalutamida. En ensayos clínicos, enzalutamida se administró independientemente de los alimentos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: En el ensayo clínico aleatorizado, se observaron casos de neutropenia de grado 1-4 (1% de grado 3-4) en el 15% de los pacientes tratados con XTANDI y en el 6% de los pacientes tratados con placebo (ninguno de grado 3-4). La incidencia de trombocitopenia de grado 1-4 fue similar en ambos grupos; el 0.5% de los pacientes tratados con XTANDI y el 1% de los que recibieron el placebo presentaron trombocitopenia de grado 3-4. Se produjeron elevaciones de grado 1-4 en los valores de ALT en el 10% de los pacientes tratados con XTANDI (0.3% de grado 3-4) y el 18% de los pacientes que recibieron el placebo (0.5% de grado 3-4). Se produjeron elevaciones de grado 1-4 en los valores de bilirrubina en el 3% de los pacientes tratados con XTANDI y el 2% de los pacientes que recibieron el placebo.
PRECAUCIONES GENERALES:
Riesgo de convulsiones: Se debe tener precaución al administrar XTANDI a pacientes con antecedentes de convulsiones u otros factores de predisposición, entre ellos, lesión cerebral subyacente, accidente cerebrovascular, tumores cerebrales primarios o metástasis cerebrales o alcoholismo. Además, el riesgo de convulsiones puede ser mayor en los pacientes que reciben medicamentos concomitantes que reducen el umbral convulsivo.
Síndrome de encefalopatía posterior reversible: Se han comunicado casos infrecuentes del síndrome de encefalopatía posterior reversible (PRES) en pacientes tratados con XTANDI. El PRES es un trastorno neurológico infrecuente y reversible que puede presentarse con síntomas de evolución rápida que incluyen convulsiones, cefalea, confusión, ceguera y otras alteraciones visuales y neurológicas, con o sin hipertensión asociada. El diagnóstico de PRES requiere la confirmación mediante gammagrafías cerebrales, preferentemente mediante resonancia magnética (MRI). Se recomienda suspender la administración de XTANDI en pacientes que presenten PRES.
Reacciones de hipersensibilidad: Se han observado reacciones de hipersensibilidad con enzalutamida, se manifiestan con síntomas que incluyen, pero no limitado se limitan a, edema de la lengua, edema de labios y edema faríngeo.
Uso concomitante con otros medicamentos: La enzalutamida es un potente inductor enzimático que puede producir la pérdida de eficacia de muchos medicamentos utilizados habitualmente (ver Interacción Farmacológica y otras interacciones). Por lo tanto, se debe realizar una revisión de los medicamentos concomitantes cuando se inicia el tratamiento con enzalutamida. En general, se debe evitar el uso concomitante de la enzalutamida con medicamentos que son sustratos sensibles de muchas enzimas metabolizadoras o transportadores (ver Interacción farmacológica y otras interacciones) si su efecto terapéutico es de gran importancia para el paciente y si no es posible realizar ajustes de dosis fácilmente en función de la supervisión de la eficacia o las concentraciones plasmáticas. Administración concomitante con cumarinas: Se debe evitar la administración concomitante con warfarina y anticoagulantes de tipo cumarínico. En caso de que XTANDI se administre de manera concomitante con un anticoagulante metabolizado por la CYP2C9 (como warfarina o acenocumarol), se debe realizar la monitorización adicional del índice internacional normalizado (INR).
Insuficiencia renal: Se requiere precaución en pacientes con insuficiencia renal grave, ya que XTANDI no se ha estudiado en esta población de pacientes.
Insuficiencia hepática grave: Se ha observado un aumento en la semivida en pacientes con insuficiencia hepática grave, lo cual está posiblemente relacionado con un aumento en la distribución tisular. Se desconoce la importancia clínica de esta observación. No obstante, se prevé un tiempo prolongado para alcanzar las concentraciones en estado de equilibrio, y posiblemente aumenten el tiempo hasta alcanzar el efecto farmacológico máximo así como el tiempo de inicio y disminución de la inducción enzimática.
Enfermedad cardiovascular reciente: Los estudios en fase 3 excluyeron a pacientes con infarto reciente de miocardio (en los últimos 6 meses) o angina inestable (en los últimos 3 meses), insuficiencia cardiaca clase III o IV de la New York Heart Association (NYHA), excepto si existe fracción de expulsión del ventrículo izquierdo (FEVI) ≥45%, bradicardia o hipertensión no controlada. Se debe tener en cuenta esto si se receta XTANDI en estos pacientes.
Uso con quimioterapia: No se han establecido la seguridad y la eficacia del uso concomitante de XTANDI con quimioterapia citotóxica. La administración concomitante de la enzalutamida no tiene un efecto clínicamente relevante en la farmacocinética del docetaxel intravenoso. Sin embargo, no puede excluirse un aumento de los casos de neutropenia inducida por el docetaxel.
Excipientes: XTANDI contiene sorbitol (E420). Los pacientes con problemas hereditarios raros de intolerancia a la fructosa no deben tomar XTANDI.
Efectos sobre la capacidad para manejar vehículos y máquinas: Debido al riesgo de convulsiones asociado al uso de XTANDI, se debe advertir a los pacientes sobre el riesgo de manejar vehículos o usar cualquier herramienta o máquina donde la pérdida repentina del conocimiento pudiera causar un daño grave a ellos mismos o a los demás.
DOSIS Y VÍA DE ADMINISTRACIÓN:
Posología: La dosis recomendada de XTANDI es 160 mg (cuatro cápsulas de 40 mg) en una dosis oral única diaria. XTANDI puede tomarse con o sin alimentos.
La castración médica con un análogo de LHRH debería continuarse durante el tratamiento de pacientes no castrados quirúrgicamente.
Si un paciente olvida tomar XTANDI a la hora habitual, debe tomar la dosis prescrita lo más cerca posible de la hora habitual. Si un paciente olvida tomar la dosis durante un día entero, el tratamiento se debe reanudar al día siguiente con la dosis diaria habitual.
Si un paciente experimenta una toxicidad ≥ grado 3 o una reacción adversa intolerable, la dosificación debería suspenderse por una semana o hasta que los síntomas mejoren hasta ≤ grado 2, y luego debería reiniciarse a la misma dosis o a una dosis reducida (120 mg o 80 mg), si es necesario.
Uso concomitante con inhibidores potentes del CYP2C8: Se debe evitar en lo posible el uso concomitante de inhibidores potentes de CYP2C8. Si se debe administrar de manera concomitante a un paciente un inhibidor potente del CYP2C8, la dosis de enzalutamida se debe reducir a 80 mg una vez al día. Si se suspende la administración concomitante del inhibidor potente de CYP2C8, se debe volver a la dosis de enzalutamida utilizada antes de empezar a administrar el inhibidor potente de CYP2C8.
Poblaciones especiales:
Pacientes geriátricos: No es necesario ajustar la dosis en pacientes geriátricos.
Pacientes con insuficiencia hepática: No es necesario ajustar la dosis en los pacientes con insuficiencia hepática leve, moderada o grave (clases A, B o C, respectivamente, de Child-Pugh).
Pacientes con insuficiencia renal: No es necesario ajustar la dosis en los pacientes con insuficiencia renal leve o moderada. Se recomienda precaución en los pacientes con insuficiencia renal grave o enfermedad renal en fase terminal.
Sexo: La enzalutamida no está indicada para su uso en mujeres.
Población pediátrica: La enzalutamida no está indicada para su uso en niños.
Forma de administración: Las cápsulas blandas de XTANDI se deben tragar enteras con agua y se pueden tomar con o sin alimentos.
Este medicamento no debe partirse, abrirse o masticarse.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL: No existe ningún antídoto para XTANDI. En caso de sobredosis, se debe interrumpir el tratamiento con XTANDI e iniciar medidas de apoyo generales teniendo en cuenta la semivida de 5.8 días. Los pacientes pueden estar en mayor riesgo de convulsiones después de una sobredosis.
En caso de sobredosis, dirigirse al hospital más cercano o comunicarse con los centros de toxicología.
PRESENTACIÓN: Caja de cartón con 120 cápsulas de 40 mg con instructivo anexo.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 30°C.
Consérvese la caja bien cerrada.
Proteger de la humedad.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. Trague las cápsulas completas, no mastique, disuelva o abra las cápsulas. Manténgase fuera del alcance de los niños. No administrar a mujeres y niños menores de 18 años. No administrar durante embarazo y lactancia. Debido al riesgo de convulsiones asociado al uso de XTANDI, no deberá conducir vehículos automotores ni maquinaria pesada durante su uso. Información sólo para médicos.
Reporte las sospechas de reacción adversa al correo:
farmacovigilancia@cofepris.gob.mx
Hecho en EUA por:
Catalent Pharma Solutions, LLC
Saint. Petersburg, FL 33716, EUA
Para:
Astellas Pharma US, Inc.
1 Astellas Way, Northbrook, Illinois (IL), 60062, EUA
Acondicionamiento primario por:
Packaging Coordinators, LLC.
2200 Lake Shore Dr. Woodstock, Illinois (IL), 60098, EUA
ó
AndersonBrecon Inc.
4545 Assembly Drive, Rockford, Illinois (IL) 61109, EUA
Acondicionamiento secundario por:
Adium Pharma, S.A.
Ruta 8 Km 17,500, local 320 y 801 Zona Franca
Montevideo, Uruguay
Distribuido por:
ASOFARMA DE MÉXICO, S.A. de C.V.
Calz. México Xochimilco No. 43,
Col. San Lorenzo Huipulco,
C.P. 14370, Deleg. Tlalpan, Ciudad de México, México
Reg. Núm. 466M2015, SSA IV
XTANDI® es una marca registrada de
Astellas Pharma Inc.


