
XEOMEEN
TOXINA BOTULÍNICA TIPO A
Solución inyectable
1 Caja, 1 Frasco(s) ámpula, 100 U
1 Caja, 1 Frasco(s) ámpula, 50 U
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada frasco ámpula con polvo contiene:
Toxina botulínica tipo A 100 U
Vehículo cbp
Cada frasco ámpula con polvo contiene:
Toxina botulínica tipo A 50 U
Vehículo cbp
INDICACIONES TERAPÉUTICAS: XEOMEEN® está indicado en casos de hiperactividad muscular por ejemplo en:
Dermatología: Líneas faciales hiperquinéticas.
Neurología: Parálisis cerebral, tremor, mioclonías, distonías focales, espasticidad.
Oftalmología: Blefaroespasmo, estrabismo y distonía focal.
Traumatología/Ortopedia: Distonía cervical idiopática de forma predominantemente rotacional (tortícolis espasmódica).
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacodinámicas: La neurotoxina botulínica tipo A bloquea la transmisión colinérgica en la unión neuromuscular, evitando la liberación de acetilcolina. Las terminaciones nerviosas de la unión neuromuscular dejan de responder a los impulsos nerviosos y se evita la secreción del neurotransmisor (denervación química). La recuperación de la transmisión de los impulsos se restablece por neoformación de las terminales nerviosas y las placas motoras.
El mecanismo de acción mediante el cual la neurotoxina botulínica tipo A ejerce sus efectos sobre las terminaciones nerviosas colinérgicas se puede describir a través de cuatro pasos, que incluyen:
a) Unión: La cadena pesada de la neurotoxina botulínica tipo A se une con una selectividad y afinidad excepcionalmente altas a los receptores encontrados solamente en las terminales colinérgicas.
b) lnternalización: Constricción de la membrana de la terminal nerviosa y absorción de la toxina dentro de la terminal nerviosa (endocitosis).
c) Translocación: El segmento amino-terminal de la cadena pesada de la neurotoxina forma un poro en la membrana de la vesícula, el puente disulfuro se rompe y la cadena ligera de la neurotoxina pasa a través del poro hacia el citosol.
d) Proteólisis: Después de que la cadena ligera es liberada, rompe específicamente una proteína blanco (SNAP25), la cual es esencial para la liberación de acetilcolina. Se inhibe la contracción muscular si no hay liberación de acetilcolina (parálisis).
La recuperación completa de la función/transmisión del impulso de la placa terminal se restablece hasta 4 meses después por brotes terminales nerviosos y reconexión con la placa terminal.
Propiedades farmacocinéticas:
a) Características generales de la sustancia activa: No es viable la realización de estudios clásicos de cinética y distribución debido a que la sustancia activa debe aplicarse en cantidades muy pequeñas (picogramos por inyección) y a causa de que se une muy rápida e irreversiblemente a las terminales colinérgicas.
La toxina botulínica nativa es un complejo de alto peso molecular, que además de la neurotoxina (150 kD) contiene otras proteínas bacterianas no tóxicas, como hemaglutininas y no hemaglutininas. En contraste con otras preparaciones, XEOMEEN® contiene neurotoxina pura (150 kD) y está libre de complejo proteínico, por lo tanto, tiene una carga de proteínas extrañas baja. La carga administrada de proteínas extrañas se considera uno de los factores de falla terapéutica secundaria, relacionada a NAF (formación de anticuerpos neutralizantes).
Al igual que muchas otras proteínas de su tamaño, se ha demostrado que la neurotoxina botulínica tipo A experimenta transporte axonal retrógrado después de la inyección intramuscular. Sin embargo, no se ha encontrado el pasaje trans-sináptico retrógrado de la neurotoxina botulínica tipo A activa hacia el sistema nervioso central.
La neurotoxina botulínica tipo A unida al receptor sufre un proceso de endocitosis antes de alcanzar su objetivo (SNAP-25) y eventualmente es degradada de manera intracelular. La molécula de la neurotoxina botulínica tipo A circulante que no se ha unido a los receptores de la terminal colinérgica presináptica es fagocitada o pinocitada, sufriendo de esta forma degradación como cualquier proteína circulante.
Distribución de la sustancia activa en los pacientes: No se han realizado estudios farmacocinéticos de XEOMEEN® en humanos por las razones detalladas con anterioridad.
Datos de seguridad preclínica: Los datos preclínicos no muestran riesgos especiales para los seres humanos de acuerdo con los estudios convencionales farmacológicos de seguridad cardiovascular.
Los hallazgos en los estudios de toxicidad a dosis repetidas realizados con XEOMEEN® se relacionaron principalmente con su acción farmacodinámica.
No se observaron signos de intolerancia local.
Los estudios de toxicidad reproductiva con XEOMEEN® no mostraron efectos adversos sobre la fertilidad masculina o femenina en conejos ni efectos directos sobre el desarrollo embriofetal o pre y posnatal en ratas y/o conejos. Sin embargo, la administración de XEOMEEN® en estudios de embriotoxicidad a niveles de dosis que exhiben toxicidad materna aumentó el número de abortos en conejos y disminuyó ligeramente el peso corporal fetal en ratas.
No existe información acerca de los efectos potenciales de XEOMEEN® sobre genotoxicidad o carcinogénesis.
Resultados de estudios clínicos: Uso terapéutico.
La equivalencia terapéutica de XEOMEEN® en comparación con un producto que contiene la toxina botulínica A de tipo convencional toxina onabotulínica (900 kD) se muestra en dos estudios comparativos Fase III de dosis única, uno en pacientes con blefaroespasmo (estudio MRZ 60201-0003, n = 300) y uno en pacientes con distonía cervical (estudio MRZ 60201-0013, n = 463). Los resultados de los estudios también sugieren que XEOMEEN® y este producto de comparación tienen eficacia y perfil de seguridad similares en pacientes con blefaroespasmo o cuando se utiliza la conversión de dosis con proporción 1:1. También son similares en el inicio de acción (4 días en blefaroespasmo y 7 días en distonía cervical) y en la duración del efecto (110 días).
En el estudio pivote (estudio doble ciego, controlado con placebo, multicéntrico, EudraCT Número 2005-003951-11) realizado en pacientes con espasticidad post-EVC del miembro superior, 148 pacientes fueron aleatorizados para recibir XEOMEEN® (N = 73) o placebo (N = 75). La dosis acumulada después de hasta 6 tratamientos repetidos en un ensayo clínico fue en promedio de 1,333 unidades (un máximo de 2,395 unidades) durante un periodo de hasta 89 semanas. Ningún paciente desarrolló anticuerpos neutralizantes.
Según lo determinado como parámetro primario de eficacia (tasas de respuesta para los flexores de la muñeca, puntuación de la Escala de Ashworth en la semana 4, la respuesta definida como mejoría de al menos 1 punto en la escala de Ashworth de 5 puntos), los pacientes tratados con XEOMEEN® (tasa de respuesta: 68.5%) tuvieron una probabilidad 3.97 veces mayor de ser respondedores con respecto a los pacientes tratados con placebo (tasa de respuesta: 37.3%, IC 95%: 1.90 a 8.30, p<0.001, población con intención a tratar-ITT).
La tasa de respuesta fue similar en los hombres en comparación con las mujeres en el periodo de extensión abierta del estudio pivote (dosis flexibles eran posibles en este periodo de prueba), en la que se reclutaron 145 pacientes y se realizaron hasta 5 ciclos de inyección, así como en el estudio ciego (EudraCT Número 2006-003036-30) en el que la eficacia y la seguridad de XEOMEEN® fueron evaluadas en dos diluciones diferentes, en 192 pacientes con espasticidad del miembro superior de diversa etiología.
Resultados de estudios clínicos en Líneas Glabelares:
La eficacia y duración de XEOMEEN® en líneas glabelares se respalda en los siguientes estudios:
En un estudio con 180 pacientes con edades entre 20-60 años con líneas glabelares moderadas a graves, se evaluó el tiempo para el inicio de acción y la duración del efecto del tratamiento con XEOMEEN® comparado con la toxina onabotulínica tipo A y la toxina abobotulínica tipo A para el tratamiento de las líneas glabelares. La mediana de tiempo para el inicio de acción fue de 3.02 días para XEOMEEN® comparado con 5.29 y 5.32 días para la toxina onabotulínica y abobotulínica, respectivamente. En los pacientes masculinos el inicio de acción fue de 3.36 días con XEOMEEN® comparado con 5.89 y 5.93 días para las toxinas onabotulínica y abobotulínica, respectivamente. La duración del efecto del tratamiento fue mayor para los pacientes tratados con XEOMEEN® con 146.12 días (4.8 meses), comparado con las toxinas onabotulínica y abobotulínica (140 y 139 días, respectivamente). Para todos los productos, la duración del efecto del tratamiento fue mayor en mujeres que en hombres. Resultó interesante que 4 pacientes del grupo tratado con XEOMEEN® continuaron mostrando el efecto terapéutico hasta el día 180 (6 meses) posteriores a la aplicación. (Rappl T. et al., 2013).
En otro estudio, de un brazo, prospectivo, prueba de concepto con 23 pacientes, se evaluó la eficacia, inicio de acción y duración del efecto de XEOMEEN® para el tratamiento de las Líneas glabelares. Los pacientes fueron tratados con 25 U de XEOMEEN®, en cinco puntos distintos en la glabela de manera equitativa en ambos lados.
El porcentaje de respondedores al realizar la contracción máxima de la glabela en los días 2-4 fue de 95.2% y 85%, de acuerdo a la mejoría de 1 punto y 2 puntos en la escala de 5 puntos, respectivamente. Al momento de esta evaluación, 84% del máximo efecto había ocurrido. En todas las evaluaciones, el cambio desde la evaluación basal en el promedio del puntaje de las líneas glabelares con la contracción máxima fue estadísticamente significativo, con un promedio de al menos 1 punto de mejoría desde la evaluación inicial hasta 5 meses después del tratamiento. La tasa de respuesta a la contracción máxima de la glabela, de acuerdo a las dos definiciones, fue de 100% para las siguientes 2 evaluaciones (día 8±1 y día 14±12). (Prager W, et al 2013).
La eficacia y seguridad de XEOMEEN® en líneas glabelares también ha sido demostrada en los estudios MRZ60201-0520, MRZ60201-0527, MRZ60201-0724, MRZ60201-0741 (para más de 2 ciclos de inyecciones) y en dosis repetidas y a largo plazo, en el estudio MRZ60201-0609 (para más de 8 ciclos de inyecciones). La evidencia de la eficacia y seguridad en la indicación en líneas perioculares y frontales (líneas faciales superiores) está basada en los resultados de los estudios Fase III MRZ60201-0617 y MRZ60201_3076_1, respectivamente.
CONTRAINDICACIONES:
• Hipersensibilidad a la sustancia activa o a cualquiera de los excipientes.
• Trastornos generalizados de actividad muscular (p. ej. miastenia grave, síndrome Lambert-Eaton).
• Infección o inflamación en el sitio de inyección propuesto.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
Embarazo: No hay datos adecuados acerca del uso de XEOMEEN® en mujeres embarazadas. Los estudios en animales han demostrado toxicidad reproductiva. Se desconoce el riesgo potencial en humanos. En consecuencia, XEOMEEN® no debe usarse durante el embarazo, sólo debe usarse en casos donde sea claramente necesario durante el embarazo y si el beneficio potencial justifica el riesgo.
Lactancia: No hay información disponible sobre si XEOMEEN® se excreta a través de la leche materna humana. Por tal motivo, no se recomienda el uso de XEOMEEN® en mujeres durante el periodo de lactancia.
Fertilidad: No hay información clínica disponible del uso de la toxina botulínica tipo A. No se detectaron reacciones adversas sobre la fertilidad en conejos machos y hembras.
REACCIONES SECUNDARIAS Y ADVERSAS: Pueden ocurrir eventos adversos debido a una mala aplicación de las inyecciones de la toxina botulínica tipo A, mismas que temporalmente pueden paralizar grupos de músculos cercanos. Los eventos adversos generalmente se observan durante la primera semana después del tratamiento, pero siempre son de naturaleza temporal.
La debilidad muscular localizada es un efecto farmacológico esperado de la toxina botulínica tipo A.
Como se espera para cualquier procedimiento de inyección, el dolor localizado, inflamación, parestesias, hipoestesia, sensibilidad, hinchazón, edema, eritema, prurito, sangrado, infección localizada y/o hematomas pueden estar asociados con la inyección.
El dolor y/o ansiedad relacionada con la aguja puede generar respuestas vasovagales, incluyendo hipotensión sintomática transitoria, náusea, tinnitus y síncope.
Se han comunicado casos de síntomas similares a los de la gripe y reacciones de hipersensibilidad incluyendo hinchazón, edema (también, lejos del lugar de la inyección), eritema, prurito, rash (local y general) y disnea.
Cuando se tratan condiciones neurológicas, los efectos colaterales relacionados con la difusión de la toxina hacia lugares distantes del sitio de inyección han sido reportados muy raramente (debilidad muscular exagerada, disfagia y neumonitis por aspiración, con resultado fatal en algunos casos). La aparición de eventos adversos como éstos, no pueden ser descartados con el uso de XEOMEEN® en indicaciones estéticas.
Reacciones de hipersensibilidades graves y/o inmediatas incluso anafilaxia, enfermedad del suero, urticaria, edema de los tejidos blandos y disnea se han reportado en raras ocasiones. Algunas de estas reacciones se han reportado después del uso de la toxina botulínica tipo A convencional con complejo proteínico, ya sea sola o en combinación con otros agentes conocidos por causar reacciones similares.
Frecuencia de acuerdo con diferentes indicaciones: Con base en la experiencia clínica, a continuación se ofrece información sobre la frecuencia de los eventos adversos para las distintas indicaciones. Las categorías de la frecuencia son: muy comunes (>1/10); comunes (>1/100, <1/10); poco comunes (>1/1,000, <1/100), raros (>1/10,000, <1/1,000); muy raros (<1/10,000).
• Blefaroespasmo:
Tabla 1. Eventos adversos basados en la experiencia clínica en blefaroespasmo
|
Sistema |
Reacción adversa |
|
|
Trastornos del Sistema Nervioso Central |
Poco comunes |
Cefalea, Parálisis facial |
|
Trastornos oculares |
Muy comunes |
Ptosis palpebral, ojos secos, visión borrosa |
|
Comunes |
Discapacidad visual |
|
|
Poco comunes |
Diplopía, incremento del lagrimeo |
|
|
Alteraciones gastrointestinales |
Comunes |
Boca seca |
|
Poco comunes |
Disfagia |
|
|
Trastornos generales y condiciones del sitio de administración |
Comunes |
Dolor en el sitio de inyección |
|
Poco comunes |
Fatiga |
|
|
Alteraciones musculo-esqueléticas y del tejido conectivo |
Poco comunes |
Debilidad muscular |
|
Trastornos de la piel y tejido subcutáneo |
Poco comunes |
Erupción cutánea o rash |
• Distonía cervical:
Tabla 2. Eventos adversos basados en la experiencia clínica en Distonía cervical
|
Sistema |
Reacción Adversa |
|
|
Alteraciones gastrointestinales |
Muy comunes |
Disfagia |
|
Comunes |
Boca seca, náusea |
|
|
Trastornos generales y condiciones del sitio de administración |
Comunes |
Dolor en el sitio de inyección, Astenia |
|
Alteraciones musculo-esqueléticas y del tejido conectivo |
Comunes |
Dolor en el cuello, debilidad muscular, dolor, rigidez muscular y espasmos musculares |
|
Trastornos del Sistema Nervioso |
Comunes |
Cefalea, Presíncope, mareo |
|
Poco comunes |
Alteraciones del habla |
|
|
Infecciones e infestaciones |
Comunes |
Infección del tracto respiratorio superior |
|
Trastornos respiratorios, torácicos y mediastinales |
Poco comunes |
Disfonía, disnea |
|
Trastornos de la piel y del tejido subcutáneo |
Comunes |
Hiperhidrosis |
|
Poco comunes |
Erupción cutánea o rash |
|
El tratamiento de la distonía cervical puede causar disfagia en diferentes niveles de gravedad con la posibilidad de aspiración que puede requerir intervención médica. La disfagia puede persistir durante dos a tres semanas después de la inyección, se ha informado de un caso con persistencia de cinco meses.
La disfagia podría ser dosis-dependiente.
• Espasticidad:
Tabla 3. Eventos adversos basados en la experiencia clínica en Espasticidad de miembro superior
|
Sistema |
Reacción Adversa |
|
|
Alteraciones gastrointestinales |
Comunes |
Boca seca |
|
Poco comunes |
Disfagia, náusea |
|
|
Trastornos generales y condiciones del sitio de administración |
Poco comunes |
Astenia |
|
Alteraciones musculo-esqueléticas y del tejido conectivo |
Poco comunes |
Debilidad muscular, Dolor en la extremidad, Mialgia |
|
Trastornos del Sistema Nervioso Central |
Poco comunes |
Cefalea, hipoestesia |
* Algunos de estos eventos adversos pueden estar relacionados con la enfermedad.
• Líneas glabelares:
Tabla 4. Eventos adversos basados en la experiencia clínica con líneas glabelares
|
Sistema |
Reacción Adversa |
|
|
Trastornos generales y condiciones del sitio de administración |
Poco comunes |
Hematoma en el sitio de inyección, enfermedad parecida a la influenza, sensibilidad (local), fatiga, dolor en el sitio de inyección, incomodidad (sensación de pesadez de párpados/cejas) |
|
Alteraciones musculo-esqueléticas y del tejido conectivo |
Comunes |
Signo de Mefisto |
|
Poco comunes |
Asimetría facial (asimetría de cejas), espasmos musculares (sobre las cejas) |
|
|
Trastornos del Sistema Nervioso |
Comunes |
Cefalea |
|
Trastornos oculares |
Poco comunes |
Edema de párpado, visión borrosa, ptosis de párpado |
|
Trastornos de la piel y del tejido subcutáneo |
Poco comunes |
Prurito, ptosis de ceja |
|
Infecciones e infestaciones |
Poco comunes |
Nasofaringitis |
|
Trastornos vasculares |
Poco comunes |
Hematomas |
• Líneas perioculares:
Tabla 5. Eventos adversos basados en la experiencia clínica en líneas perioculares
|
Sistema |
Reacción Adversa |
|
|
Trastornos generales y condiciones del sitio de administración |
Comunes |
Hematoma en el sitio de inyección |
|
Alteraciones oculares |
Comunes |
Edema del párpado, ojo seco |
• Líneas faciales superiores:
Tabla 6. Eventos adversos basados en la experiencia clínica en Líneas faciales superiores
|
Sistema |
Reacción Adversa |
|
|
Trastornos generales y condiciones del sitio de administración |
Comunes |
Hematoma en el sitio de inyección, dolor en el sitio de inyección, eritema en el sitio de inyección, incomodidad (sensación de pesadez de área frontal) |
|
Alteraciones oculares |
Comunes |
Ptosis de párpado, ojo seco |
|
Trastornos del Sistema Nervioso |
Muy común |
Cefalea |
|
Comunes |
Hipostesia |
|
|
Trastornos en tejido subcutáneo y en la piel |
Comunes |
Ptosis de ceja |
|
Trastornos del tejido músculo esquelético y conectivo |
Comunes |
Asimetría Facial Signo Mefisto |
|
Trastorno gastrointestinales |
Comunes |
Náusea |
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD: No se han realizado estudios sobre genotoxicidad, carcinogenicidad o estudios de desarrollo pre y postnatales con XEOMEEN®.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO: No se han realizado estudios de interacción medicamentosa. Teóricamente, el efecto de la toxina botulínica tipo A se puede potenciar por antibióticos aminoglucósidos u otros medicamentos que interfieren con la transmisión neuromuscular, por ejemplo, relajantes musculares del tipo de la tubocurarina. En consecuencia, el uso concomitante de XEOMEEN® con aminoglucósidos o espectinomicina requiere cuidado especial. Los relajantes musculares de acción periférica se deben usar con precaución; de ser necesario, reduciendo la dosis inicial del relajante o usando una sustancia de acción intermedia, como vecuronio o atracurio, en lugar de sustancias con efectos más duraderos. Las 4-aminoquinolinas pueden reducir el efecto de XEOMEEN®.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO: No se han reportado alteraciones en las pruebas de laboratorio.
PRECAUCIONES GENERALES: Pacientes con enfermedades neuromusculares pueden tener un riesgo mayor de presentar debilidad muscular exagerada. El uso de productos con toxina botulínica tipo A en este tipo de pacientes debe ser bajo supervisión de un especialista y sólo deberán ser utilizados si el beneficio del tratamiento es considerado mayor al riesgo. Pacientes con historia de disfagia y bronco-aspiración deben ser tratados con extrema precaución cuando son tratados por alguna indicación neurológica.
Antes de administrar XEOMEEN®, es necesario conocer la anatomía del sitio a inyectar, así como cualquier variación o alteración causada por procedimientos quirúrgicos previos. Para el tratamiento de distonía cervical y espasticidad, XEOMEEN® debe ser administrado cuidadosamente cuando los sitios de inyección se encuentren cercanos a estructuras susceptibles, tales como la arteria carótida, ápices pulmonares y esófago.
Si la inyección de toxina botulínica tipo A es realizada en un lugar inadecuado, pueden ocurrir efectos indeseables como la parálisis temporal de grupos musculares adyacentes.
XEOMEEN® debe ser usado con precaución:
• En presencia de trastornos de la coagulación de cualquier tipo.
• En pacientes bajo tratamiento con anticoagulantes o bajo otros tratamientos.
• En pacientes con alteraciones neuromusculares, tales como Esclerosis Lateral Amiotrófica.
• En músculos específicos que muestran debilidad o atrofia pronunciada.
• En niños y adolescentes (por ejemplo con parálisis cerebral, hasta que los datos de los estudios clínicos controlados con XEOMEEN® estén disponibles (ver sección de Población pediátrica).
Existen reportes de eventos adversos que pueden estar relacionados con la difusión de la toxina a sitios lejanos al lugar de aplicación. En el tratamiento de indicaciones neurológicas algunos de estos efectos pueden ser potencialmente mortales. Se han reportado muy raros casos fatales. Los efectos indeseables como estos no pueden descartarse por completo con el uso de XEOMEEN® en indicaciones estéticas.
Pacientes tratados con dosis terapéuticas pueden desarrollar debilidad muscular exagerada. Los pacientes y sus cuidadores deben ser advertidos de buscar atención médica inmediata si se producen alteraciones en la deglución, el habla o trastornos respiratorios.
El tratamiento para indicaciones estéticas con XEOMEEN® no está recomendado para pacientes con un historial de disfagia y bronco-aspiración.
Se han reportado reacciones de hipersensibilidad con productos con toxina botulínica tipo A. Si se producen reacciones graves (por ejemplo, reacciones anafilácticas) y/o reacciones de hipersensibilidad inmediata, el tratamiento médico adecuado debe ser instituido.
Los efectos terapéuticos de la toxina botulínica tipo A pueden ser incrementados o disminuidos a través de la aplicación de dosis repetidas. Posibles razones de esto incluyen: diferentes técnicas de reconstitución utilizadas, intervalos de tratamiento utilizados, músculos inyectados, variación marginal de la actividad de la toxina, el resultado del procedimiento utilizado para la prueba biológica o la falta de respuesta secundaria o formación de anticuerpos. Las dosis demasiado frecuentes pueden aumentar el riesgo de formación de anticuerpos, lo que puede dar como resultado un fracaso del tratamiento.
A los pacientes previamente acinéticos se les debe advertir que reanudan la actividad gradualmente después de la inyección de XEOMEEN®.
Se debe tener cuidado para asegurar que XEOMEEN® no se inyecte en un vaso sanguíneo.
• Blefaroespasmo: Debido al efecto anticolinérgico de la toxina botulínica tipo A, XEOMEEN® debe usarse con precaución en los pacientes con riesgo de desarrollar glaucoma de ángulo cerrado.
A fin de evitar un ectropión, deberá evitarse su aplicación en el área inferior del párpado; de ser necesario deberá emplearse un tratamiento vigoroso para cualquier defecto epitelial. Esto puede requerir el uso de gotas protectoras, ungüento, lentes de contacto protectores suaves o el cierre del ojo mediante parche u otros medios.
El evitar las inyecciones cerca del músculo elevador superior del párpado puede reducir la complicación de ptosis. Puede desarrollarse diplopía como resultado de la difusión de la toxina botulínica tipo A al oblicuo inferior. Evitar las inyecciones en el párpado inferior puede reducir esta complicación.
La reducción del parpadeo después de la inyección de XEOMEEN® en el músculo orbicular puede conducir a exposición de la córnea, defectos epiteliales persistentes y ulceración corneal, especialmente en los pacientes con trastornos del nervio craneal VII (nervio facial). Deberá realizarse un examen cuidadoso de la sensibilidad corneal en los pacientes que se hayan sometido previamente a operaciones oculares.
Puede ocurrir equimosis ligera en el tejido blando del párpado. Una presión suave e inmediata en el sitio de la inyección puede reducir al límite dicho riesgo.
• Distonía cervical: Los pacientes con distonía cervical deben ser informados sobre la posibilidad de experimentar disfagia de leve a grave, que puede conducir a aspiración, disnea y la necesidad de un tubo de alimentación gástrica. Limitar la dosis inyectada en el músculo esternocleidomastoideo a menos de 100 U puede reducir la aparición de disfagia. Los reportes señalan que los pacientes con una menor masa muscular en el cuello o que requieren inyecciones bilaterales en el músculo esternocleidomastoideo tienen un mayor riesgo de disfagia. La disfagia es atribuible a la difusión de la toxina botulínica tipo A hacia la musculatura esofágica.
• Espasticidad: XEOMEEN® como tratamiento para la espasticidad focal ha sido estudiado en asociación con regímenes de cuidado estándar y no tiene como objetivo reemplazar estas modalidades de tratamiento. Es probable que XEOMEEN® no sea eficaz alrededor de una contractura articular fija.
Población pediátrica: Para el tratamiento de la espasticidad en niños, XEOMEEN® solo debe usarse en niños/adolescentes de 2 años de edad o más. Se han reportado muy raramente informes espontáneos de posible diseminación a distancia de la toxina para preparaciones de toxina botulínica tipo A en pacientes pediátricos con comorbilidades, predominantemente con parálisis cerebral. En general, la dosis utilizada en estos casos fue superior a la recomendada para estos productos.
Se han reportado raros eventos espontáneos de muerte asociados a veces con neumonía por aspiración en niños con parálisis cerebral grave después del tratamiento con productos con toxina botulínica, incluido el uso no indicado en la etiqueta (por ejemplo, el área del cuello). Se debe tener mucho cuidado al tratar a pacientes pediátricos que tienen debilidad neurológica significativa, disfagia o antecedentes recientes de neumonía por aspiración o enfermedad pulmonar. El tratamiento en pacientes con un estado de salud subyacente deficiente debe administrarse solo si se considera que el beneficio potencial para el paciente individual supera los riesgos.
Efectos sobre la capacidad para conducir y usar maquinaria: XEOMEEN® tiene una influencia menor o moderada sobre la capacidad para conducir y usar maquinaria. Se debe informar a los pacientes que, si se produce pérdida de fuerza, debilidad muscular, visión borrosa, cansancio, mareo o ptosis, deben evitar conducir un coche o participar en otras actividades potencialmente peligrosas.
DOSIS Y VÍA DE ADMINISTRACIÓN: La dosis en unidades recomendada para XEOMEEN® no se puede intercambiar con otras preparaciones de toxina botulínica tipo A. Varios estudios han demostrado eficacia, seguridad e inicio de acción similares entre XEOMEEN® y la toxina onabotulínica tipo A de 900 kD con una tasa de conversión 1:1 (Benecke, 2005; Roggenkämper 2006; Jost W, 2005) y con una tasa de conversión de 2-6:1 entre la toxina Abobotulínica tipo A y toxina onabotulínica tipo A/XEOMEEN® (Dressier D, 2014; Bentivoglio AR, et al., 2012; Jimenez-Shahed, 2012; Marchetti, A, et al., 2005; Chen JJ, et al., 2013).
Instrucciones para la Reconstitución de XEOMEEN®: Un vial de XEOMEEN® 100 o 50 unidades de toxina botulínica tipo A (150 kD), libre de complejo proteínico. En el vial debe existir vacío debido a su proceso de manufactura.
La apariencia del contenido del vial de XEOMEEN® es un polvo blanco, liofilizado. XEOMEEN® debe ser reconstituido antes de la inyección con solución de cloruro de sodio al 0.9%. La cantidad de solución salina utilizada en la reconstitución determina el número de unidades de XEOMEEN® por ml de solución lista para utilizarse. La siguiente tabla muestra las posibles diluciones de acuerdo al volumen de solución salina utilizado:
Tabla de Dilución para vial de 100 U
|
Cantidad de diluyente añadido (cloruro de sodio a 0.9% para inyección) |
Dosis resultante en Unidades por 0.1 ml |
|
0.5 ml |
20 U |
|
1.0 ml |
10 U |
|
1.25 ml |
8 U |
|
2.0 ml |
5 U |
|
2.5 ml |
4 U |
|
4.0 ml |
2.5 U |
|
5.0 ml |
2 U |
Tabla de Dilución para vial de 50 U
|
Cantidad de diluyente añadido (cloruro de sodio al 0.9% para inyección) |
Dosis resultante en Unidades por 0.1 ml |
|
0.5 ml |
10 U |
|
1.0 ml |
5 U |
|
1.25 ml |
4 U |
|
2.0 ml |
2.5 U |
|
2.5 ml |
2 U |
|
4.0 ml |
1.25 U |
|
5.0 ml |
1 U |
Nota: Si entra aire al vial sin abrir, por ejemplo, por insertar una jeringa vacía, el vacío provocara la distribución del polvo (liofilizado) en el vial. Se asentará sobre el vidrio y el tapón de hule. Esto puede provocar la reconstitución incompleta del polvo y reducir la actividad biológica. Cuando se extrae la solución lista para usarse, no toque la pared del vial con la aguja porque puede disminuir su filo y así provocar una inyección más dolorosa.
Pasos para la Reconstitución de un vial de XEOMEEN®:
Paso 1: Limpiar el tapón de hule del vial de XEOMEEN® con alcohol (70%). Llenar la jeringa adecuada con el volumen de solución salina (NaCI) al 0.9% deseado (ver las tablas de dilución mostradas en la sección anterior). Una aguja corta de 20-27 gauge es recomendada para la reconstitución de XEOMEEN®.
Después de la inserción vertical de la aguja a través del tapón de hule, el solvente es inyectado de manera suave dentro del vial, con la finalidad de evitar la formación de espuma. Debido al sistema de vacío cerrado de XEOMEEN®, la solución salina deberé ser aspirada dentro del vial.
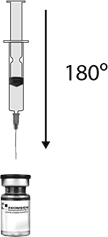
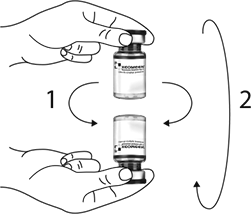
Paso 2: Retire la jeringa del vial y mezcle XEOMEEN® con el solvente girando lentamente e invirtiendo el vial cuidadosamente (no lo agite con fuerza). Asegurando que la solución toque todas las paredes del vial, incluyendo el tapón de hule.
Paso 3: Llene una jeringa adecuada con la solución lista para usarse. Las jeringas más utilizadas para las indicaciones estéticas son 1.0, 0.5 o 0.3 ml con una aguja de 30 o 31 gauge. Para las indicaciones neurológicas, las jeringas más utilizadas comúnmente son: Para espasticidad y distonía cervical: 27 gauge x 1"½; para estrabismo; 27 gauge x ½"; para blefaroespasmo: 30 gauge x ½".
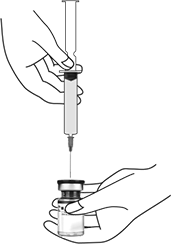
La solución lista para usarse debe ser clara, incolora y libre de partículas. XEOMEEN® no debe ser utilizado si la solución reconstituida tiene una apariencia opaca o contiene material floculado o en partículas.
La solución reconstituida de XEOMEEN® debe ser utilizada para inyección intramuscular y puede ser almacenado en refrigeración por 24 h a una temperatura de 2°C a 8ºC.
Para desechar los residuos con seguridad, los viales caducos sin usar deberán reconstituirse con una pequeña cantidad de agua y después introducirse al autoclave. Los viales y jeringas usadas, así como los derrames ocurridos, deberán introducirse a la autoclave. Los residuos de XEOMEEN® deberán de inactivarse usando una de las siguientes soluciones: Etanol al 70%, isopropanol al 50% solución diluida de hidróxido de sodio (NaOH 0.1 N) o solución diluida de hipoclorito de sodio (al menos al 0.1% NaOCI). Los viales usados, las jeringas y otros materiales empleados deben ser eliminados en contenedores apropiados.
XEOMEEN® sólo deberá ser administrado por médicos calificados, con acreditación y experiencia comprobada en la aplicación de toxina botulínica tipo A.
La dosis óptima y el número de sitios de inyección en el músculo tratado deberán ser elegidos de manera individual por el médico. Debe realizarse una titulación de la dosis.
Una disminución o aumento de la dosis XEOMEEN® es posible mediante la administración de un volumen de inyección menor o mayor. Cuanto más pequeño sea el volumen de la inyección, se producirá una menor sensación de presión y una menor propagación de la toxina botulínica tipo A en el músculo inyectado. Esto puede reducir los efectos sobre los músculos cercanos cuando se inyectan pequeños grupos musculares.
• Blefaroespasmo: Después de su reconstitución, XEOMEEN® se inyecta usando una aguja estéril, con calibre 27-30/0.30-0.40 mm. No es necesario emplear guía electromiográfica. La dosis inicial recomendada es de 1.25-2.5 U (Volumen de 0.05-0.1 mL) en cada sitio, inyectada en los músculos orbiculares en las zonas medial y lateral del párpado superior y zona lateral del párpado inferior.
La inyección también se puede aplicar en sitios adicionales, como el área de la ceja, en la zona lateral del orbicular y en el área facial superior en caso de que los espasmos aquí interfieran con la visión. Evitar las inyecciones cerca del músculo elevador superior del párpado puede reducir las complicaciones de ptosis. Puede desarrollarse diplopía como resultado de la difusión de la toxina botulínica tipo A al Oblicuo Inferior.
El promedio de inicio del efecto se observa en los cuatro días siguientes a la inyección. El efecto de cada tratamiento dura aproximadamente de 3 a 4 meses; sin embargo, éste puede ser significativamente más largo o corto. El procedimiento puede repetirse de ser necesario.
Debido al efecto anticolinérgico de la toxina botulínica tipo A, XEOMEEN® debe usarse con precaución en los pacientes con riesgo de desarrollar glaucoma de ángulo cerrado.
La reducción del parpadeo después de la inyección de XEOMEEN® en el músculo orbicular puede conducir a exposición de la córnea, defectos epiteliales persistentes y ulceración corneal.
Se define como respuesta insuficiente al efecto que no dura más de dos meses. En las sesiones subsecuentes de tratamiento, la dosis se puede incrementar hasta el doble si la respuesta al tratamiento inicial se considera insuficiente; generalmente esto se define como un efecto que no dura más de dos meses. Sin embargo, al parecer es poco el beneficio que se obtiene de inyectar más de 5.0 U por sitio. La dosis inicial no debe exceder de 25 U por ojo.
Es raro que el efecto sea permanente después de una inyección única. En el manejo del blefaroespasmo, la dosis total no debe rebasar las 100 U cada 12 semanas. Si el paciente requiere administración adicional de XEOMEEN®, ésta puede ser inyectada con intervalos flexibles de mínimo 6 semanas.
Dosis y puntos de inyección por músculo en blefaroespasmo:
|
Músculo |
Unidades (rango) |
Número de Sitios de Inyección |
|
Orbicular |
1.25-5 U Por sitio de inyección |
3-5 |
• Dosis y puntos de Inyección por músculo en estrabismo:
|
Músculo |
Unidades (rango) |
Número de Sitios de Inyección |
|
Recto superior, inferior, lateral y medial |
1.25-7.5 U Por músculo y de acuerdo al tipo de estrabismo |
1 |
• Distonía cervical: Para los músculos superficiales se puede usar una aguja desechable estéril de calibre 25-30/0.30-0.50 mm; mientras que para la musculatura más profunda se puede usar una aguja de calibre 22/0.70 mm.
En la práctica, la dosis total máxima por sesión de tratamiento generalmente no es mayor a 200 U. Se pueden administrar dosis hasta de 300 U. No deben administrarse más de 50 U en un mismo sitio de inyección.
El tratamiento de la distonía cervical típicamente puede incluir inyección de XEOMEEN® en los músculos esternocleidomastoideo, elevador de la escápula, escaleno, esplenio y/o trapecio. Esta lista no es exhaustiva ya que cualquiera de los músculos responsables de la posición de la cabeza puede requerir tratamiento.
En el tratamiento de la distonía cervical con XEOMEEN®, la dosis se debe ajustar para cada paciente de manera individual, con base en la posición de la cabeza y cuello del paciente, la ubicación del dolor, la hipertrofia del músculo, el peso corporal del paciente y la respuesta terapéutica.
En el tratamiento de la distonía cervical, la localización de los músculos involucrados con guía electromiográfica puede ser útil para aislar músculos únicos. La masa del músculo y el grado de hipertrofia o atrofia son factores que deben tomarse en cuenta al momento de elegir la dosis adecuada.
El empleo de sitios de inyección múltiples permite a XEOMEEN® una cobertura más uniforme de las áreas de inervación del músculo distónico y es especialmente útil en músculos grandes. El número óptimo de sitios de inyección depende del tamaño del músculo a ser químicamente desnervado.
El músculo esternocleidomastoideo no debe inyectarse bilateralmente, pues existe un riesgo elevado de efectos adversos (disfagia en particular) cuando se aplican a este músculo inyecciones o dosis bilaterales de más de 100 U.
La mediana de inicio del efecto se observa en los siete días posteriores a la inyección. El efecto de cada tratamiento dura aproximadamente 4 meses; sin embargo, éste puede tener una duración significativamente más larga o corta.
Si el paciente requiere administración adicional de XEOMEEN® ésta puede ser inyectada con intervalos flexibles de mínimo 6 semanas.
|
Músculo |
Unidades (Rango) |
Número de sitios de Inyección |
|
Esternocleidomastoideo |
5-50 U |
1-3 |
|
Elevador de la escápula |
5-60 U |
1-2 |
|
Escaleno anterior, medio y/o posterior |
5-25 U por Músculo |
1-3 |
|
Esplenio de la cabeza |
10-100 U |
1-3 |
|
Trapecio ascendente, transverso y/o descendente |
5-20 U por Sitio de inyección |
2-4 |
Fuente: Jost W. Pictorial Atlas of Botulium Toxin Injection Quintessence Publishing 2012.
• Espasticidad: Después de su reconstitución, XEOMEEN® se inyecta usando una aguja estéril adecuada. La dosis exacta y el número de sitios a inyectar deberán ser determinados en cada paciente, con base en el tamaño, número y localización de los músculos a tratar, la gravedad de la espasticidad y la presencia de debilidad muscular local.
Puede ser necesario localizar los músculos involucrados con guía electromiográfica o técnicas de estimulación nerviosa. El empleo de sitios de inyección múltiples permite a XEOMEEN® tener un contacto más uniforme con las áreas de inervación del músculo y es especialmente útil cuando se inyectan músculos grandes.
En el tratamiento de espasticidad de la extremidad superior, los rangos de dosis recomendadas por músculo para adultos se proveen en la siguiente tabla:
• Miembro superior:
|
Patrón clínico Músculo |
Unidades (Rango) |
Número de sitios de inyección por músculo |
|
Muñeca flexionada |
||
|
Flexor carpi radialis |
25-100 U |
1-2 |
|
Flexor carpi ulnaris |
20-100 U |
1-2 |
|
Puño cerrado |
||
|
Flexor digitorum superficialis |
40-100 U |
2 |
|
Flexor digitorum profundus |
40-100 U |
2 |
|
Codo flexionado |
||
|
Brachioradialis |
25-100 U |
1-3 |
|
Biceps |
75-200 U |
2-4 |
|
Brachialis |
40-150 U |
1-2 |
|
Antebrazo pronado |
||
|
Pronator quadratus |
10-50 U |
1 |
|
Pronator teres |
25-75 U |
1-2 |
|
Pulgar en palma |
||
|
Flexor pollicis longus |
10-50 U |
1 |
|
Adductor pollicis |
5-30 U |
1 |
|
Flexor pollicis brevis/opponens |
5-30 U |
1 |
Fuente: Kanovsky P, et al. J Rehabil Med 2011;43:486-92.
• Miembro inferior*:
|
Patrón clínico Músculo |
Unidades (Rango) |
Número de sitios de inyección por músculo |
|
Aducción de muslos |
||
|
Adductor longus |
20-100 U |
1-3 |
|
Adductor magnus |
30-150 U |
1-3 |
|
Adductor brevis |
20-80 U |
1-2 |
|
Gracilis |
20-60 U |
1-3 |
|
Iliopsoas (psoas minor + psoas major + iliacus) |
25-200 U |
1-3 |
|
Pectineus |
20-50 U |
1 |
|
Rodilla flexionada |
||
|
Biceps femoris |
40-140 U |
1-3 |
|
Semimembranosus |
20-100 U |
1-3 |
|
Semitendinosus |
20-80 U |
1-3 |
|
Gastrocnemius |
20-100 U por cabeza |
1-3 |
|
Rodilla extendida |
||
|
Rectus femoris (quadriceps femoris) |
20-100 U |
1-3 |
|
Vastus medialis (quadriceps femoris) |
20-80 U |
1-3 |
|
Vastus intermedius (quadriceps femoris) |
20-80 U |
1-2 |
|
Vastus lateralis (quadriceps femoris) |
20-80 U |
1-3 |
|
Pie equinovaro |
||
|
Gastrocnemius medial |
20-100 U |
1-3 |
|
Gastrocnemius lateral |
20-100 U |
1-3 |
|
Soleus |
20-80 U |
2-4 |
|
Tibialis posterior |
20-100 U |
1-3 |
|
Tibialis anterior |
20-80 U |
1-3 |
|
Pulgar en Extensión |
||
|
Extensor hallucis longus |
20-40 U |
1-2 |
|
Dedos en garra |
||
|
Flexor digitorum longus |
10-40 U |
1-2 |
|
Quadratus plantae |
5-20 U |
1-2 |
* Fuente: Grupo de trabajo de la Sociedad Española de Rehabilitación y Medicina Física (SERMEF). Guía de Práctica Clínica para el Tratamiento de la Espasticidad con Toxine Botulínica. Sociedad Española de Rehabilitación y Medicina Física; 2010. Jost W. Pictorial Atlas of Botulium Toxin Injection Quintessence Publishing 2012.
Se han reportado crisis convulsivas de nueva aparición o recurrentes, típicamente en pacientes que presentan esta predisposición. La relación exacta de las crisis convulsivas a la inyección de la neurotoxina botulínica no ha sido establecida. La dosis de los tratamientos subsecuentes debe ser adaptada de acuerdo a la necesidad individual del paciente. La dosis total no debe ser mayor a 400 unidades por sesión de tratamiento.
El periodo entre cada sesión de tratamiento debe ser de 12 semanas como mínimo. La mediana de tiempo para el inicio del efecto se observa dentro de cuatro días después de la inyección. El efecto máximo como una mejoría del tono muscular se ve usualmente en 4 semanas. El efecto de cada tratamiento puede durar aproximadamente 4 meses.
• Indicaciones Estéticas (Líneas Faciales Hiperquinéticas): El tiempo promedio para el inicio del efecto con XEOMEEN® se ha observado dentro de los 2-3 días después de la inyección en las líneas glabelares (Rappl T, 2013). El máximo efecto fue percibido dentro de las primeras 4 semanas. En general, el efecto del tratamiento tiene una duración de más de 150 días (Rappl T, 2013; Prager W, 2013; Oliveira de Morais O, 2012); sin embargo, la duración puede ser significativamente más larga o más corta.
• Líneas Faciales Hiperquinéticas:
Fuente: Carruthers J, 2004; Carruthers J, 2013.
• Líneas Glabelares:
|
Músculos a Inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Corrugador, procerus, Depresor superciliar, orbicular de los párpados, Occipito-frontal (vientre frontal) |
Mujeres: 10 a 30 U |
5 |
|
Hombres: 20 a 40 U |
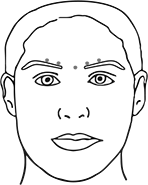
• Líneas Frontales: Para reducir el riesgo de blefaroptosis, deben de evitarse las inyecciones cerca del levator palpebrae superioris y en la porción craneal del orbicularis oculis.
|
Músculos a inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Occipito-frontal (vientre frontal), considerando interacciones con procerus, corrugadores y orbicular de los párpados |
5-15 U |
4 a 10 |

• Líneas Periorbitales:
|
Músculos a Inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Porciones laterales del orbicular de los párpados |
Mujeres: 5-15 U Por lado |
3 a 4 por lado |
|
Hombres: 10-15 U por lado |

Las inyecciones muy cercanas al músculo cigomático mayor deben ser evitadas para prevenir la ptosis de labios.
• Líneas nasales transversales:
|
Músculos a Inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Nasal |
1-2.5 U por lado |
1 por lado |
|
Procerus |
1 U, en caso necesario |
1 en la línea media |

• Levantamiento de Cejas:
|
Músculos a Inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Depresor superciliar central |
3-7 U en la cola de la ceja, más inactivación de los depresores centrales |
3 |

• Líneas Periorales: Las inyecciones muy cercanas al músculo cigomático mayor deben ser evitadas para prevenir la ptosis de labios.
|
Músculos a inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Orbicular de los labios |
2 a 6 U divididas igualmente en los sitios de inyección |
2-7 |

• Líneas Mentonianas:
|
Músculos a Inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Mentoniano |
5-10 U |
1 sitio |

• Área del Músculo Depresor del Ángulo de los Labios:
|
Músculos a Inyectar |
Unidades (Rango) |
No. de puntos habituales para inyección |
|
Depresor del ángulo de los labios |
1-7.5 U |
1 por lado |

Los intervalos entre tratamientos no deberán ser menores a 3 meses. Antes y durante la inyección, el dedo pulgar y el dedo índice deben ser usados para aplicar presión firme debajo del borde de la órbita, con la finalidad de evitar la difusión de la solución en esta región. Se debe mantener una alineación superior y medial de la aguja durante la inyección.
Para reducir el riesgo de ptosis palpebral, se deberán evitar las inyecciones cerca del músculo elevador del párpado superior y dentro de la porción craneal del músculo orbicular de los párpados. Las inyecciones dentro del músculo corrugador deben ser realizadas en la porción medial del músculo y en la porción central del vientre del músculo, al menos, 1 cm por encima del borde óseo de la órbita.
En todas las Indicaciones: Si el efecto del tratamiento no es observado dentro del mes posterior a la inyección inicial, las siguientes medidas deberán realizarse:
1. Verificación del efecto de la toxina botulínica tipo A en el músculo inyectado: por ejemplo, una electromiografía en un centro especializado.
2. Análisis de las razones de no respuesta; por ejemplo, inadecuada identificación de los músculos a ser inyectados, uso de muy baja dosis, técnica inadecuada de inyección, existencia de contracturas fijas, presencia de músculos antagonistas débiles, posible desarrollo de anticuerpos.
3. Revisar si el tratamiento con toxina botulínica tipo A es un tratamiento adecuado para ese caso.
4. Si no se han presentado reacciones adversas durante el tratamiento inicial, un curso adicional de tratamiento puede ser realizado bajo las siguientes condiciones: a) Ajuste de la dosis de acuerdo al análisis de la reciente falla terapéutica; b) Realizar la aplicación guiada con EMG; c) Apegarse a los intervalos de aplicación mínimos recomendados entre el tratamiento inicial y los tratamientos repetidos.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
Síntomas de la Sobredosis: Dosis altas de toxina botulínica tipo A pueden dar como resultado parálisis neuromuscular pronunciada distinta del sitio de inyección con una variedad de síntomas. Los síntomas incluyen debilidad general, ptosis, diplopía, dificultades para respirar, hablar, parálisis de los músculos respiratorios o dificultades para deglutir dando por resultado neumonía por aspiración.
Medidas en caso de sobredosis: En caso de sobredosis, el paciente debe ser controlado médicamente para los síntomas de debilidad muscular excesiva o parálisis muscular. Puede ser necesario un tratamiento sintomático. Apoyo respiratorio puede ser necesario si se produce una parálisis de los músculos respiratorios.
PRESENTACIONES:
Caja de cartón con 1 frasco ámpula de 100 U de Toxina Botulínica.
Caja de cartón con 1 frasco ámpula de 50 U de Toxina Botulínica.
RECOMENDACIONES SOBRE ALMACENAMIENTO: Consérvese a no más de 25ºC.
El vial reconstituido se puede almacenar por hasta 24 horas en refrigeración, a 2-8ºC, siempre y cuando la reconstitución se haya llevado a cabo bajo condiciones controladas y asépticas. Desde un punto de vista microbiológico, el producto debe ser utilizado inmediatamente.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. No se deje al alcance de los niños. Literatura exclusiva para médicos. No se administre durante el embarazo ni en la lactancia.
Fabricado en Alemania por:
Merz Pharma GmbH & Co. KGaA
o
IDT Biologika GmbH
Para:
Merz Pharma GmbH & Co. KGaA
Ludwigstraβe 22, 64354 Reinheim, Alemania
Acondicionado por:
Merz Pharma GmbH & Co. KGaA
Ludwigstraβe 22, 64354 Reinheim, Alemania
Importado, Distribuido y Representante legal por:
MERZ PHARMA, S.A. de C.V.
Parque Industrial Prologis, Av. Del Pozo S/N Nave 5C-2,
Col. Recursos Hidráulicos, C.P. 54913,
Tultitlán, México, México
Reg. Núm. 321M2006 SSA IV
®Marca Registrada


