
XELJANZ XR
TOFACITINIB
Tabletas recubiertas de liberación prolongada
1 Frasco, 7 Tabletas, 11 mg
1 Frasco, 30 Tabletas, 11 mg
1 Frasco, 90 Tabletas, 11 mg
1 Frasco, 14 Tabletas, 22 mg
1 Frasco, 30 Tabletas, 22 mg
1 Frasco, 90 Tabletas, 22 mg
FORMA FARMACÉUTICA Y FORMULACIÓN:
Cada TABLETA RECUBIERTA contiene:
Citrato de tofacitinib equivalente a 11 mg de tofacitinib
Excipiente cbp 1 tableta
Cada TABLETA RECUBIERTA contiene:
Citrato de tofacitinib equivalente a 22 mg de tofacitinib
Excipiente cbp 1 tableta
INDICACIONES TERAPÉUTICAS:
Artritis reumatoide: XELJANZ® XR (tofacitinib) está indicado para el tratamiento de pacientes adultos con artritis reumatoide (AR) activa de moderada a severa y que han tenido una respuesta inadecuada a uno o más fármacos antirreumáticos modificadores de la enfermedad (FARME).
Artritis psoriásica: XELJANZ® XR (tofacitinib) está indicado para el tratamiento de pacientes adultos con artritis psoriásica activa.
Colitis ulcerosa: XELJANZ® XR (tofacitinib) está indicado para la inducción y el mantenimiento del tratamiento en pacientes adultos con colitis ulcerosa (CU) activa de moderada a grave con una respuesta inadecuada, pérdida de respuesta o intolerancia a corticoides, azatioprina (AZA), 6-mercaptopurina (6-MP) o antagonistas del factor de necrosis tumoral (TNF, por sus siglas en inglés).
FARMACOCINÉTICA Y FARMACODINAMIA:
Propiedades farmacocinéticas:
Código ATC: L04AF01.
XELJANZ® XR: Tras la administración oral de XELJANZ® XR, las concentraciones plasmáticas máximas se logran a las 4 horas y la vida media es de aproximadamente 6 a 8 horas. Las concentraciones en estado de equilibrio se alcanzan en el plazo de 48 horas y la acumulación es insignificante tras la administración una vez al día. El ABC y la Cmáx de tofacitinib para XELJANZ® XR 11 mg administrado una vez al día son bioequivalentes a aquellas de Xeljanz® 5 mg administrado dos veces al día. El ABC y la Cmáx de tofacitinib para XELJANZ® XR 22 mg administrado una vez al día son equivalentes a aquellos de Xeljanz® 10 mg administrado dos veces al día.
Absorción y distribución: Tofacitinib se absorbe bien y presenta una biodisponibilidad oral del 74% tras la administración de Xeljanz®. La coadministración de Xeljanz® con una comida rica en grasas no causó cambios en el ABC, mientras que la Cmáx se redujo un 32%. En ensayos clínicos, tofacitinib se administró sin tener en cuenta la comida.
Después de la administración intravenosa, el volumen de distribución es de 87 L. Aproximadamente el 40% de tofacitinib circulante está ligado a proteínas. Tofacitinib se une predominantemente a la albúmina y, aparentemente, no se une a la α1-glucoproteína ácida. Tofacitinib se distribuye equitativamente entre los glóbulos rojos y el plasma.
XELJANZ® XR: La coadministración de XELJANZ® XR 11 mg y 22 mg con una comida rica en grasas no causó cambios en el ABC, mientras que la Cmáx aumentó un 27% y un 19%, respectivamente. El Tmáx se extendió aproximadamente 1 hora para XELJANZ® XR 11 mg y 22 mg.
Metabolismo y eliminación: Los mecanismos de depuración de tofacitinib son el metabolismo hepático en un 70% y la excreción renal del medicamento primario en un 30%, aproximadamente. El metabolismo de tofacitinib está mediado primariamente por CYP3A4, con una contribución menor de CYP2C19. En un estudio de radiomarcado humano, más del 65% de la radiactividad circulante total estuvo representada por medicamento no modificado y el 35% se atribuyó a 8 metabolitos, cada uno de los cuales fue responsable de menos del 8% de la radiactividad total. Se han observado todos los metabolitos en especies animales y se predice que tienen ≤ 10% de la potencia de tofacitinib para la inhibición de JAK1/3. No se detectó evidencia de estereoconversión en las muestras humanas. La actividad farmacológica de tofacitinib se atribuye a la molécula primaria. In vitro, tofacitinib es un sustrato para la resistencia a múltiples medicamentos (MDR, por sus siglas en inglés) 1, pero no para la proteína de resistencia al cáncer de mama (BCRP, por sus siglas en inglés), el polipéptido transportador de aniones orgánicos (OATP, por sus siglas en inglés) 1B1/1B3 o el transportador de cationes orgánicos (OCT, por sus siglas en inglés) ½, y no es un inhibidor de la MDR1, el OAT P1B1/1B3, la OCT2, el transportador de aniones orgánicos (OAT, por sus siglas en inglés) 1/3 o la proteína asociada a la resistencia a varios medicamentos (MRP, por sus siglas en inglés) en concentraciones clínicamente significativas.
En la Figura 1 se muestran los datos farmacocinéticos y las recomendaciones de dosificación para poblaciones especiales e interacciones medicamentosas.
Las modificaciones necesarias para las poblaciones especiales se describen en la sección Dosis y vía de administración.
Farmacocinética en pacientes con AR: El análisis de la farmacocinética poblacional en pacientes con artritis reumatoide indicó que la exposición sistémica (ABC) de tofacitinib en los extremos de peso corporal (40 kg, 140 kg) fue similar a la de un paciente de 70 kg. Se calculó que los pacientes de edad avanzada de 80 años tuvieron un ABC < 5% más alta en comparación con la edad media de 55 años. Se estimó que las mujeres tienen un ABC un 7% más bajo que la de los varones. Los datos disponibles también mostraron que no existen diferencias importantes en el ABC de tofacitinib entre los pacientes blancos, negros y asiáticos. Se observó una relación lineal aproximada entre el peso corporal y el volumen de distribución, que provocó concentraciones máximas (Cmáx) más altas y concentraciones mínimas (Cmín) más bajas en pacientes de menos peso. Sin embargo, esta diferencia no se considera importante desde el punto de vista clínico. Se estima que la variabilidad entre pacientes (coeficiente de variación porcentual) en el ABC de Xeljanz® es del 27% aproximadamente.
Farmacocinética en pacientes con artritis psoriásica activa: El análisis de farmacocinética poblacional en pacientes con artritis psoriásica activa indicó que la exposición sistémica (ABC) del tofacitinib en los extremos del peso corporal (61 kg, 109 kg [percentil 10 y 90 en el conjunto de datos poblacionales]) era similar a aquella de un paciente de 83.3 kg. Se estimó que los pacientes de edad avanzada de 80 años tenían un ABC un 10% mayor en relación con la media de edad de 50 años. Se estimó que las mujeres tenían un 5% menos de ABC en comparación con los hombres. Los datos disponibles también han demostrado que no existen diferencias importantes en el ABC del tofacitinib entre los pacientes de raza blanca, negra y asiática. Se estima que la variabilidad entre sujetos (coeficiente de variación porcentual) en el ABC de Xeljanz® es de aproximadamente un 32%.
Farmacocinética en pacientes con colitis ulcerosa activa: El análisis de la farmacocinética poblacional en pacientes con colitis ulcerosa indicó que no había diferencias clínicamente relevantes en la exposición al tofacitinib (ABC), en función de la edad, el peso, el sexo y la raza. La exposición en las mujeres fue un 15% mayor que en los hombres, y los pacientes asiáticos tuvieron una exposición un 7.3% mayor que los no asiáticos. El volumen de distribución aumentó con el peso corporal, lo que tuvo como resultado concentraciones máximas (Cmáx) más altas y concentraciones mínimas (Cmín) más bajas en los pacientes de menor peso corporal. Sin embargo, esta diferencia no se considera clínicamente relevante. Se estima que la variabilidad entre sujetos (% del coeficiente de variación) del ABC del tofacitinib es de aproximadamente un 23% y un 25% con las dosis de 5 mg dos veces al día, y de 10 mg dos veces al día, respectivamente, en pacientes con colitis ulcerosa.
Insuficiencia renal: Los pacientes con insuficiencia renal leve, moderada y grave tuvieron un ABC un 37%, 43% y 123% mayor, respectivamente, en comparación con los pacientes sanos (ver sección Dosis y vía de administración). En pacientes con enfermedad renal en estado terminal, la contribución de la diálisis a la depuración total de tofacitinib fue relativamente pequeña.
Insuficiencia hepática: Los pacientes con insuficiencia hepática leve y moderada tenían un ABC un 3% y 65% mayor, respectivamente, en comparación con los pacientes sanos. No se estudiaron pacientes con insuficiencia hepática grave (ver sección Dosis y vía de administración).
Población pediátrica: No se establecieron la farmacocinética, la seguridad ni la eficacia de tofacitinib en pacientes pediátricos.
Figura 1. Recomendación de dosificación de XELJANZ® XR basada en los datos farmacocinéticos*
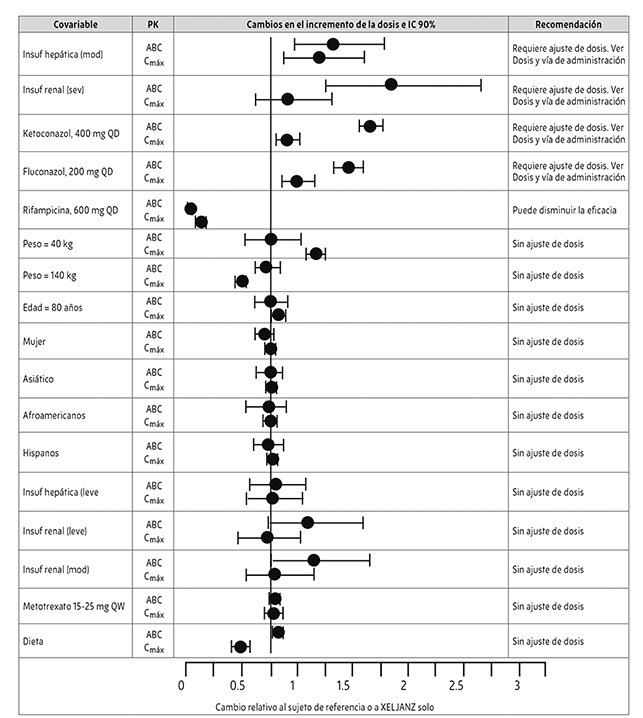
Las comparaciones de peso, edad, sexo y raza se basan en los datos de pacientes con AR, con referencia a valores de 70 kg, 55 años, masculino y blanco, respectivamente.
NOTA: Los grupos de referencia de los datos de insuficiencia renal y hepática corresponden a sujetos con funciones renales o hepáticas normales, respectivamente; el grupo de referencia de la interacción medicamentosa y de los estudios de efecto de los alimentos corresponde a la administración de Xeljanz® solo; Mod. = moderado; Sev. = severo; Insuf. = Insuficiencia.
* El ajuste de la dosis requerido para las poblaciones especiales se describe en Dosis y vía de administración.
Propiedades farmacodinámicas:
Mecanismo de acción: Tofacitinib es un inhibidor potente y selectivo de la familia de JAK con un alto grado de selectividad en comparación con otras cinasas en el genoma humano. En los ensayos de cinasa, tofacitinib inhibe JAK1, JAK2, JAK3 y, en menor medida, TyK2. En entornos celulares en los que las cinasas JAK señalan en pares, tofacitinib inhibe, preferentemente, la señalización mediante receptores heterodiméricos asociados con JAK3 y/o JAK1 con una selectividad funcional por sobre los receptores que señalizan a través de pares de JAK2. La inhibición de JAK1 y JAK3 mediante tofacitinib bloquea la señalización a través de los receptores que contienen cadenas gamma comunes para diversas citocinas, que incluyen IL-2, -4, -7, -9, -15 y -21. Estas citocinas son esenciales para la activación, la proliferación y el funcionamiento de los linfocitos, y la inhibición de su señalización puede, por lo tanto, ocasionar la modulación de varios aspectos de la respuesta inmune. Adicionalmente, la inhibición de JAK1 puede resultar en la disminución de la señalización por citocinas proinflamatorias adicionales, tales como IL-6 e interferones del tipo I. A exposiciones más altas, la inhibición de la señalización de eritropoyetina podría ocurrir a través de la inhibición de la señalización de JAK2.
Efecto farmacodinámico: En pacientes con artritis reumatoide, el tratamiento de hasta 6 meses con Xeljanz® se asoció con reducciones dependientes de la dosis de células asesinas naturales (NK) CD16/56+ circulantes, con reducciones máximas estimadas que se produjeron aproximadamente entre 8 y 10 semanas después de iniciar el tratamiento. En general, estos cambios se resolvieron en el plazo de 2 a 6 semanas después de la interrupción del tratamiento. El tratamiento con Xeljanz® se asoció con aumentos dependientes de la dosis en los recuentos de linfocitos B. Los cambios en los recuentos de linfocitos T circulantes y en los subgrupos de linfocitos T (CD3+, CD4+ y CD8+) fueron pequeños e inconsistentes.
Después del tratamiento a largo plazo (la duración mediana del tratamiento con Xeljanz® de aproximadamente 5 años), los recuentos de CD4+ y CD8+ mostraron reducciones medianas del 28% y 27%, respectivamente, en relación con el valor inicial. En contraste con la reducción observada después de la dosificación a corto plazo, los recuentos de células asesinas naturales CD16/56+ mostraron un aumento mediano del 73% con respecto al valor inicial. Los recuentos de linfocitos B CD19+ no mostraron otros aumentos después del tratamiento con Xeljanz® a largo plazo. Estos cambios volvieron a los valores iniciales después de la interrupción temporal del tratamiento. No se observaron indicios de un mayor riesgo de infecciones graves u oportunistas, ni de herpes zóster con valores bajos de recuentos de linfocitos CD4+, CD8+ o NK, o recuentos elevados de linfocitos B.
Los cambios en los niveles séricos totales de IgG, IgM e IgA después de 6 meses de administración de Xeljanz® en pacientes con artritis reumatoide fueron pequeños, no dependieron de la dosis y fueron similares a los observados con el placebo.
Después del tratamiento con Xeljanz® en pacientes con artritis reumatoide, se observaron rápidas disminuciones de la proteína C reactiva (PCR) sérica y se mantuvieron durante toda la dosificación. Los cambios observados en la PCR durante el tratamiento con Xeljanz® no se revierten completamente en el plazo de 2 semanas después de la interrupción, lo que indica una duración mayor de la actividad farmacodinámica en comparación con la vida media.
En pacientes con artritis psoriásica activa se han observado cambios similares en los linfocitos T, los linfocitos B y la PCR sérica, aunque no se evaluó su reversibilidad. No se evaluaron las inmunoglobulinas séricas totales en pacientes con artritis psoriásica activa.
Seguridad clínica: En un gran estudio de seguridad posterior a la autorización (PASS, por sus siglas en inglés) aleatorizado y abierto realizado en pacientes con AR de 50 años y mayores, con al menos un factor de riesgo cardiovascular adicional y con una dosis estable de metotrexato, los pacientes se trataron con tofacitinib 5 mg dos veces al día, tofacitinib 10 mg dos veces al día o un inhibidor del TNF. En particular, en febrero de 2019, la dosis del tofacitinib en el grupo del estudio con 10 mg dos veces al día se redujo a 5 mg dos veces al día después de que se determinara que la frecuencia de embolia pulmonar aumentaba en el grupo de tratamiento con tofacitinib 10 mg dos veces al día frente al inhibidor del TNF. Además, la mortalidad por todas las causas aumentó en el grupo de tratamiento con tofacitinib 10 mg dos veces al día frente a los grupos de tratamiento con inhibidores del TNF y tofacitinib 5 mg dos veces al día. En los datos finales del estudio, los pacientes del grupo de tratamiento con tofacitinib 10 mg dos veces al día se analizaron en su grupo de tratamiento originalmente aleatorizado. A continuación, se presentan los resultados de los datos finales de seguridad del estudio para determinados eventos.
Mortalidad: Las tasas de incidencia (IR, por sus siglas en inglés), (IC del 95%), de la mortalidad por todas las causas para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento con 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.50 (0.33, 0.74), 0.80 (0.57, 1.09), 0.65 (0.50, 0.82) y 0.34 (0.20, 0.54) eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR, por sus siglas en inglés), (IC del 95%), para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.49 (0.81, 2.74), 2.37 (1.34, 4.18) y 1.91 (1.12, 3.27), respectivamente.
Las IR (IC del 95%) de las muertes asociadas a la infección para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día), y el inhibidor del TNF fueron de 0.08 (0.02, 0.20), 0.18 (0.08, 0.35), 0.13 (0.07, 0.22), y 0.06 (0.01, 0.17) eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR), (IC del 95%), para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.30 (0.29, 5.79), 3.10 (0.84, 11.45) y 2.17 (0.62, 7.62), respectivamente.
Las IR (IC del 95%) de las muertes asociadas a eventos cardiovasculares para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.25 (0.13, 0.43), 0.41 (0.25, 0.63), 0.33 (0.23, 0.46) y 0.20 (0.10, 0.36) eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR), (IC del 95%), para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.26 (0.55, 2.88), 2.05 (0.96, 4.39) y 1.65 (0.81, 3.34), respectivamente.
Las IR (IC del 95%) de las muertes asociadas a neoplasias malignas para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.10 (0.03, 0.23), 0.00 (0.00, 0.08), 0.05 (0.02, 0.12) y 0.02 (0.00, 0.11) eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR) (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 4.88 (0.57, 41.74), 0 (0.00, Inf) y 2.53 (0.30, 21.64), respectivamente.
Las IR (IC del 95%) de las muertes asociadas a otras causas (excluyendo infecciones, eventos cardiovasculares, neoplasias malignas) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.08 (0.02, 0.20), 0.21 (0.10, 0.38), 0.14 (0.08, 0.23) y 0.06 (0.01, 0.17) eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR) (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.30 (0.29, 5.81), 3.45 (0.95, 12.54) y 2.34 (0.67, 8.16), respectivamente.
En los estudios clínicos de Xeljanz® que incluyeron 10 mg dos veces al día, las tasas de incidencia de mortalidad por todas las causas en pacientes tratados con Xeljanz® 10 mg dos veces al día no han sido mayores que las tasas en pacientes tratados con Xeljanz® 5 mg dos veces al día. Las tasas de mortalidad en pacientes tratados con Xeljanz® son similares a aquellas informadas en pacientes con AR, PsA y CU tratados con terapias biológicas.
Infecciones: Las IR (IC del 95%) de todas las infecciones para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día), y el inhibidor del TNF fueron de 41.74 (39.21, 44.39), 48.73 (45.82, 51.77), 45.02 (43.10, 47.01) y 34.24 (32.07, 36.53) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR), (IC del 95%), para Xeljanz® 5 mg dos veces al día, Xeljanz 10 mg dos veces al día y todo Xeljanz® fue de 1.20 (1.10, 1.31), 1.36 (1.24, 1.49) y 1.28 (1.18, 1.38), respectivamente.
Las IR (IC del 95%) de las infecciones graves para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 2.86 (2.41, 3.37), 3.64 (3.11, 4.23), 3.24 (2.89, 3.62) y 2.44 (2.02, 2.92) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR) (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.17 (0.92, 1.50), 1.48 (1.17, 1.87) y 1.32 (1.07, 1.63), respectivamente.
Las IR (IC del 95%) de las infecciones oportunistas para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.76 (0.54, 1.04), 0.91 (0.66, 1.22), 0.84 (0.67, 1.04) y 0.42 (0.26, 0.64) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.82 (1.07, 3.09), 2.17 (1.29, 3.66) y 1.99 (1.23, 3.22), respectivamente. La mayoría de las infecciones oportunistas en los grupos de tratamiento con Xeljanz® fueron infecciones oportunistas por herpes zóster; también se informó una cantidad limitada de eventos con tuberculosis. Excluyendo las infecciones oportunistas por herpes zóster y tuberculosis, las IR (IC del 95%) de todas las demás infecciones oportunistas para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.08 (0.02, 0.20), 0.14 (0.06, 0.30), 0.11 (0.05, 0.20) y 0.06 (0.01, 0.17) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el cociente de riesgos (HR), (IC del 95%), para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.30 (0.29, 5.82), 2.40 (0.62, 9.29) y 1.84 (0.51, 6.59), respectivamente.
Las IR (IC del 95%) del herpes zóster (incluye todos los eventos de herpes zóster) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 3.75 (3.22, 4.34), 3.94 (3.38, 4.57), 3.84 (3.45, 4.26) y 1.18 (0.90, 1.52) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el HR (IC del 95%) para el herpes zóster con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 3.17 (2.36, 4.27), 3.33 (2.48, 4.48) y 3.25 (2.46, 4.29), respectivamente.
Infecciones graves en un estudio de seguridad observacional posautorización: Los datos de un estudio de seguridad observacional posautorización que evaluó tofacitinib en pacientes con AR procedentes de un registro (US Corrona) mostraron una tasa de incidencia numéricamente más alta de infecciones graves con la tableta de liberación prolongada de 11 mg administrada una vez al día que con la tableta recubierta de 5 mg administrada dos veces al día. Las tasas brutas de incidencia (IC del 95%), (es decir, sin ajustar por edad o sexo), de la disponibilidad de cada formulación a los 12 meses después del inicio del tratamiento fueron 3.45 (1.93, 5.69) y 2.78 (1.74, 4.21), y a los 36 meses fueron 4.71 (3.08, 6.91) y 2.79 (2.01, 3.77) pacientes con eventos por 100 pacientes-año en los grupos de tableta de liberación prolongada de 11 mg una vez al día y tableta recubierta de 5 mg dos veces al día, respectivamente. El HR no ajustado fue 1.30 (IC del 95%: 0.67, 2.50) a los 12 meses y 1.93 (IC del 95%: 1.15, 3.24) a los 36 meses para la dosis de 11 mg de liberación prolongada una vez al día en comparación con la dosis de tableta recubierta de 5 mg dos veces al día. Los datos se basan en un pequeño número de pacientes con eventos observados con intervalos de confianza relativamente grandes y un tiempo de seguimiento limitado disponible en el grupo de dosis de liberación prolongada una vez al día de 11 mg después de 24 meses.
Tromboembolismo:
Tromboembolismo venoso: Las IR (IC del 95%) del tromboembolismo venoso (TEV) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.33 (0.19, 0.53), 0.70 (0.49, 0.99), 0.51 (0.38, 0.67) y 0.20 (0.10, 0.37) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el HR (IC del 95%) para el TEV con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.66 (0.76, 3.63), 3.52 (1.74, 7.12) y 2.56 (1.30, 5.05), respectivamente.
Las IR (IC del 95%) de la embolia pulmonar (EP) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.17 (0.08, 0.33), 0.50 (0.32, 0.74), 0.33 (0.23, 0.46) y 0.06 (0.01, 0.17) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el HR (IC del 95%) para EP con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 2.93 (0.79, 10.83), 8.26 (2.49, 27.43) y 5.53 (1.70, 18.02), respectivamente. En los pacientes tratados con tofacitinib en donde se observó EP, la mayoría (97%) tenían factores de riesgo de TEV).
Las IR (IC del 95%) de la trombosis venosa profunda (TVP) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.21 (0.11, 0.38), 0.31 (0.17, 0.51), 0.26 (0.17, 0.38) y 0.14 (0.06, 0.29) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el HR (IC del 95%) para la TVP con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.54 (0.60, 3.97), 2.21 (0.90, 5.43) y 1.87 (0.81, 4.30), respectivamente.
En un análisis exploratorio post hoc de marcadores biológicos dentro de un gran estudio PASS aleatorizado realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional, se observó la aparición de TEV posterior con mayor frecuencia en los pacientes tratados con tofacitinib con un nivel de dímero D ≥ 2 × LSN a los 12 meses de tratamiento, frente a aquellos con un nivel de dímero D < 2 × LSN. Esta observación no se identificó en los pacientes tratados con TNFi. La interpretación está limitada por la baja cantidad de eventos de TEV y la disponibilidad restringida de la prueba de dímero D (solo se evaluó en el periodo inicial, en el mes 12 y al final del estudio). En los pacientes que no tuvieron un TEV durante el estudio, los niveles medios de dímero D se redujeron significativamente en el mes 12 en relación con el periodo inicial en todos los grupos de tratamiento. Sin embargo, se observaron niveles de dímero D ≥ 2 × LSN en el mes 12 en aproximadamente el 30% de los pacientes sin eventos posteriores de TEV, lo que indica una especificidad limitada de las pruebas de dímero D en este estudio. Teniendo en cuenta los datos y las limitaciones generales de este análisis exploratorio post hoc de marcadores biológicos, la utilidad de realizar el monitoreo del dímero D en el contexto de la mitigación del riesgo de eventos de TEV es limitada.
Tromboembolismo arterial: Las IR (IC del 95%) del tromboembolismo arterial (TEA) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.92 (0.68, 1.22), 0.94 (0.68, 1.25), 0.93 (0.75, 1.14) y 0.82 (0.59, 1.12) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, el HR (IC del 95%) de TEA con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fue de 1.12 (0.74, 1.70), 1.14 (0.75, 1.74) y 1.13 (0.78, 1.63), respectivamente.
Eventos cardiovasculares adversos importantes (MACE), incluido el infarto de miocardio: Los eventos cardiovasculares adversos importantes (MACE, por sus siglas en inglés) incluyen el infarto de miocardio no mortal, la apoplejía no mortal y las muertes cardiovasculares, excluida la embolia pulmonar mortal. Las IR (IC del 95%) de MACE para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.91 (0.67, 1.21), 1.05 (0.78, 1.38), 0.98 (0.79, 1.19) y 0.73 (0.52, 1.01) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.24 (0.81, 1.91), 1.43 (0.94, 2.18) y 1.33 (0.91, 1.94), respectivamente.
En los grupos de tratamiento con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® y TNFi, hubo un total de 19, 19, 38 y 11 pacientes con eventos de IM, respectivamente. De estos totales, la cantidad de pacientes con IM mortal fue de 0, 3, 3 y 3, respectivamente, mientras que la cantidad de pacientes con IM no mortal fue de 19, 16, 35 y 8, respectivamente. Por lo tanto, las IR que siguen son para el IM no mortal. Las IR (IC del 95%) del IM no mortal para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.37 (0.22, 0.57), 0.33 (0.19, 0.53), 0.35 (0.24, 0.48) y 0.16 (0.07, 0.31) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 2.32 (1.02, 5.30), 2.08 (0.89, 4.86) y 2.20 (1.02, 4.75), respectivamente.
Neoplasias malignas, excluido el NMSC: Las IR (IC del 95%) de las neoplasias malignas, excluido el cáncer de piel no melanoma (NMSC, por sus siglas en inglés), para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 1.13 (0.87, 1.45), 1.13 (0.86, 1.45), 1.13 (0.94, 1.35) y 0.77 (0.55, 1.04) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.47 (1.00, 2.18), 1.48 (1.00, 2.19) y 1.48 (1.04, 2.09), respectivamente.
Las IR (IC del 95%) del linfoma para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día), y el inhibidor del TNF fueron de 0.07 (0.02, 0.18), 0.11 (0.04, 0.24), 0.09 (0.04, 0.17), y 0.02 (0.00, 0.10) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 3.99 (0.45, 35.70), 6.24 (0.75, 51.86) y 5.09 (0.65, 39.78), respectivamente.
Las IR (IC del 95%) del cáncer de pulmón para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día), y el inhibidor del TNF fueron de 0.23 (0.12, 0.40), 0.32 (0.18, 0.51), 0.28 (0.19, 0.39) y 0.13 (0.05, 0.26) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.84 (0.74, 4.62), 2.50 (1.04, 6.02) y 2.17 (0.95, 4.93), respectivamente.
NMSC: Las IR (IC del 95%) del NMSC para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día), y el inhibidor del TNF fueron de 0.61 (0.41, 0.86), 0.69 (0.47, 0.96), 0.64 (0.50, 0.82) y 0.32 (0.18, 0.52) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.90 (1.04, 3.47), 2.16 (1.19, 3.92) y 2.02 (1.17, 3.50), respectivamente.
Las IR (IC del 95%) del carcinoma de células basales para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.37 (0.22, 0.58), 0.33 (0.19, 0.54), 0.35 (0.24, 0.49) y 0.26 (0.14, 0.44) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.43 (0.71, 2.90), 1.28 (0.61, 2.66) y 1.36 (0.72, 2.56), respectivamente.
Las IR (IC del 95%) del carcinoma cutáneo de células escamosas para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día), y el inhibidor del TNF fueron de 0.29 (0.16, 0.48), 0.45 (0.29, 0.69), 0.37 (0.26, 0.51), y 0.16 (0.07, 0.31) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.82 (0.77, 4.30), 2.86 (1.27, 6.43) y 2.32 (1.08, 4.99), respectivamente.
Perforaciones gastrointestinales: Las IR (IC del 95%) de perforaciones gastrointestinales para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 0.17 (0.08, 0.33), 0.10 (0.03, 0.24), 0.14 (0.08, 0.23) y 0.08 (0.02, 0.20) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el inhibidor del TNF, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 2.20 (0.68, 7.15), 1.29 (0.35, 4.80) y 1.76 (0.58, 5.34), respectivamente.
Fracturas: Las IR (IC del 95%) de las fracturas para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, todo Xeljanz® (combina los grupos de tratamiento de 5 mg dos veces al día y 10 mg dos veces al día) y el inhibidor del TNF fueron de 2.79 (2.34, 3.30), 2.87 (2.40, 3.40), 2.83 (2.50, 3.19) y 2.27 (1.87, 2.74) pacientes con eventos por cada 100 pacientes-año, respectivamente. En comparación con el TNFi, los HR (IC del 95%) para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y todo Xeljanz® fueron de 1.23 (0.96, 1.58) 1.26 (0.97, 1.62) y 1.24 (0.99, 1.55), respectivamente.
Pruebas de laboratorio:
Pruebas de enzimas hepáticas: Los porcentajes de pacientes con al menos un aumento de ALT > 1 vez el LSN, 3 veces el LSN y 5 veces el LSN posterior al periodo inicial para el grupo de tratamiento con Xeljanz® 5 mg dos veces al día fueron de 52.83, 6.01 y 1.68, respectivamente. Los porcentajes para el grupo de tratamiento de Xeljanz® 10 mg dos veces al día fueron de 54.46, 6.54 y 1.97, respectivamente. Los porcentajes para todo Xeljanz® (combina Xeljanz® 5 mg dos veces al día y Xeljanz® 10 mg dos veces al día) fueron de 53.64, 6.27 y 1.82, respectivamente. Los porcentajes para el grupo de tratamiento con inhibidores del TNF fueron de 43.33, 3.77 y 1.12, respectivamente.
Los porcentajes de pacientes con al menos un aumento de AST > 1 vez el LSN, 3 veces el LSN y 5 veces el LSN posterior al periodo inicial para el grupo de tratamiento con Xeljanz® 5 mg dos veces al día fueron de 45.84, 3.21 y 0.98, respectivamente. Los porcentajes para el grupo de tratamiento de Xeljanz® 10 mg dos veces al día fueron de 51.58, 4.57 y 1.62, respectivamente. Los porcentajes para todo Xeljanz® (combina Xeljanz® 5 mg dos veces al día y Xeljanz® 10 mg dos veces al día) fueron de 48.70, 3.89 y 1.30, respectivamente. Los porcentajes para el grupo de tratamiento con inhibidores del TNF fueron de 37.18, 2.38 y 0.70, respectivamente.
Lípidos: A los 12 meses, en los grupos de tratamiento con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día e inhibidor del TNF, el aumento porcentual medio del colesterol LDL fue de 13.80, 17.04 y 5.50, respectivamente. A los 24 meses, el aumento porcentual medio fue de 12.71, 18.14 y 3.64, respectivamente.
A los 12 meses, en los grupos de tratamiento con Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día e inhibidor del TNF, el aumento porcentual medio del colesterol HDL fue de 11.71, 13.63 y 2.82, respectivamente. A los 24 meses, el aumento porcentual medio fue de 11.58, 13.54 y 1.42, respectivamente.
Eficacia clínica:
Artritis reumatoide: XELJANZ® XR 11 mg una vez al día ha demostrado equivalencia farmacocinética (ABC y Cmáx) con Xeljanz® 5 mg dos veces al día. La dosis recomendada de XELJANZ® XR es de 11 mg una vez al día. Toda la información provista en esta sección es aplicable para XELJANZ® XR.
La eficacia y la seguridad de Xeljanz® se evaluaron en seis estudios aleatorizados, doble ciego, controlados y multicéntricos efectuados en pacientes > 18 años con artritis reumatoide activa, diagnosticada según los criterios del American College of Rheumatology (Colegio Americano de Reumatología [ACR, por sus siglas en inglés]). Los pacientes tenían, como mínimo, 6 articulaciones sensibles al tacto y 6 inflamadas al momento de la aleatorización (4 articulaciones inflamadas y sensibles al tacto en el Estudio II). Se administró Xeljanz® 5 o 10 mg dos veces al día como monoterapia (Estudio I) combinado con FARME (Estudio II) en pacientes que tuvieron una respuesta inadecuada a esos medicamentos, y combinado con MTX en pacientes que tuvieron una respuesta inadecuada a MTX (Estudio III y Estudio IV) o en los que la eficacia fue inadecuada o que no tuvieron tolerancia, como mínimo, a un agente biológico inhibidor del TNF aprobado (Estudio V).
El Estudio I fue un estudio de monoterapia de 6 meses, en el cual 610 pacientes con artritis reumatoide activa de moderada a severa que presentaron una respuesta inadecuada a los FARME (no biológicos o biológicos) recibieron Xeljanz® 5 o 10 mg dos veces al día o placebo. En la visita del mes 3, todos los pacientes aleatorizados al tratamiento con placebo avanzaron de modo ciego a un segundo tratamiento predeterminado con Xeljanz® 5 mg o 10 mg administrado dos veces al día. Los criterios primarios de valoración en el mes 3 consistieron en la proporción de pacientes que lograron una respuesta ACR20, cambios en el Cuestionario de evaluación de salud - Índice de incapacidad (HAQ-DI, por sus siglas en inglés) y las Tasas de puntuación de la actividad de la enfermedad DAS28-4(ESR) < 2.6.
El Estudio II fue un estudio de 12 meses en el cual 792 pacientes con artritis reumatoide activa de moderada a severa que presentaron una respuesta inadecuada a los FARME no biológicos recibieron Xeljanz® 5 o 10 mg dos veces al día o placebo agregado a un tratamiento de FARME de fondo (excluyendo los tratamientos con inmunosupresores potentes como azatioprina y la ciclosporina). En la visita del mes 3, todos los pacientes aleatorizados que no respondieron al tratamiento con placebo avanzaron de modo ciego a un segundo tratamiento predeterminado con Xeljanz® 5 o 10 mg administrado dos veces al día. Al final del mes 6, todos los pacientes tratados con placebo avanzaron al segundo tratamiento predeterminado de manera ciega. Los criterios primarios de valoración consistieron en la proporción de pacientes que lograron una respuesta ACR20 en el mes 6, cambios en el cuestionario HAQ-DI en el mes 3 y tasas de DAS28-4(ESR) < 2.6 en el mes 6.
El Estudio III fue un estudio de 12 meses en el que participaron 717 pacientes con artritis reumatoide activa de moderada a severa que habían presentado una respuesta inadecuada a MTX. Los pacientes recibieron Xeljanz® 5 o 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea cada dos semanas o placebo agregado a MTX de fondo. Los pacientes en el grupo placebo avanzaron al igual que en el Estudio II. Los criterios primarios de valoración fueron la proporción de pacientes que lograron una respuesta ACR20 en el mes 6, HAQ-DI en el Mes 3 y la DAS28-4(ESR) < 2.6 en el mes 6.
El Estudio IV fue un estudio de 2 años con un análisis planificado para el primer año en el cual 797 pacientes con artritis reumatoide activa de moderada a severa que presentaron una respuesta inadecuada a MTX recibieron Xeljanz® 5 o 10 mg dos veces al día o placebo agregado a MTX de fondo. Los pacientes en el grupo placebo avanzaron al igual que en el Estudio II. Los criterios primarios de valoración fueron la proporción de pacientes que lograron una respuesta ACR20 en el mes 6, un cambio medio desde el periodo inicial en el puntaje total del método Sharp (mTSS) modificado por Van der Heijde en el mes 6, el HAQ-DI en el mes 3 y un DAS28-4(ESR) <2.6 en el mes 6.
El Estudio V fue un estudio de 6 meses en el cual 399 pacientes con artritis reumatoide activa de moderada a severa que presentaron una respuesta inadecuada a al menos un agente inhibidor biológico de TNF aprobado, recibieron Xeljanz® 5 o 10 mg dos veces al día o placebo agregado a MTX de fondo. En la visita del mes 3, todos los pacientes aleatorizados al tratamiento con placebo avanzaron de manera ciega a un segundo tratamiento predeterminado con Xeljanz® 5 o 10 mg administrado dos veces al día. Los criterios primarios de valoración del mes 3 fueron la proporción de pacientes que lograron una respuesta ACR20, el cuestionario HAQ-DI y un DAS28-4(ESR) < 2.6.
El Estudio VI fue un estudio de monoterapia de 2 años con un análisis planificado en el primer año en el que 952 pacientes no tratados previamente con MTX que tenían artritis reumatoide activa de moderada a severa recibieron Xeljanz® 5 o 10 mg dos veces al día o MTX titulados por dosis por más de 8 semanas, de 10 a 20 mg por semana. Los criterios primarios de valoración fueron el cambio medio respecto del periodo inicial en el mTSS modificado por van der Heijde en el mes 6 y la proporción de pacientes que tuvieron una respuesta ACR70 en el mes 6.
Respuesta clínica:
Respuesta de ACR: En la Tabla 1, se muestran los porcentajes de pacientes tratados con Xeljanz® que lograron respuestas ACR20, ACR50 y ACR70 en los Estudios I, II, IV, V y VI. En todos los estudios, los pacientes tratados con 5 o 10 mg de Xeljanz® dos veces al día tuvieron tasas de respuestas ACR20, ACR50 y ACR70 estadísticamente significativas en el mes 3 y en el mes 6 en comparación con los pacientes tratados con placebo (o en comparación con los tratados con MTX en el Estudio VI).
En el Estudio IV, las tasas de respuesta ACR20/50/70 en el mes 12 se mantuvieron hasta el mes 24.
En el Estudio VI (Tabla 1), la diferencia entre MTX y ambos grupos de tofacitinib para obtener tasas de respuesta ACR20, ACR50 y ACR70 fue de importancia estadística en todos los puntos temporales (p ≤ 0.0001). La administración de tofacitinib como monoterapia a pacientes no tratados previamente con MTX mejoró significativamente los signos y síntomas de la AR en comparación con MTX. La eficacia observada con tofacitinib se mantuvo hasta el mes 24.
En los Estudios I, II y V, se observó una mejora en la tasa de respuesta ACR20 en comparación con el placebo en el plazo de 2 semanas.
Durante las fases controladas de 3 meses (Estudios I y V) y de 6 meses (Estudios II, III y IV) de los estudios, los pacientes tratados con Xeljanz® con dosis de 10 mg dos veces al día, por lo general, presentaron tasas de respuesta mayores que los pacientes tratados con Xeljanz® 5 mg dos veces al día. En el Estudio III, los criterios primarios de valoración fueron la proporción que logró una respuesta ACR20 en el mes 6, un cambio en el HAQ-DI en el mes 3 y un DAS28-4(ESR) < 2.6 en el mes 6. Los datos de estos resultados primarios fueron 51.5%, 52.6%, 47.2% y 28.3%; -0.55%, -0.61%, -0.49% y -0.24%; y 6.2%, 12.5%, 6.7% y 1.1% para la dosis de Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab de 40 mg por vía subcutánea cada dos semanas y los grupos tratados con placebo, respectivamente. En el caso de un criterio secundario de valoración especificado previamente, las tasas de respuesta ACR70 en el mes 6 para los grupos de Xeljanz® 5 mg administrado dos veces al día y de 10 mg administrado dos veces al día fueron considerablemente mayores que las de adalimumab (19.9%, 21.9% y 9.1%, respectivamente).
El efecto del tratamiento fue similar en los pacientes independientemente del estado del factor reumatoide, la edad, el sexo, la raza o el estado de la enfermedad. El tiempo de inicio fue rápido (tan pronto como en la semana 2 en los Estudios I, II y V) y la magnitud de respuesta continuó mejorando a medida que avanzaba el tratamiento. Tal como ocurrió con la respuesta ACR general en los pacientes tratados con Xeljanz® 5 o 10 mg dos veces al día, cada uno de los componentes de la respuesta ACR mejoró de forma consistente a partir del periodo inicial, entre los cuales se incluyen los recuentos de articulaciones inflamadas y sensibles al tacto; la evaluación general del paciente y el médico; los puntajes del índice de discapacidad; la evaluación del dolor y el PCR, en comparación con los pacientes que recibieron placebo más MTX u otros FARME en todos los estudios.
Respuesta DAS28-4 (ESR): Los pacientes de los estudios en Fase 3 tuvieron un Puntaje de actividad de la enfermedad (DAS28-4[ESR]) medio de 6.1 a 6.7 en el periodo inicial. Se observaron reducciones importantes en el DAS28-4(ESR) desde el periodo inicial (mejoría media) de 1.8 a 2.0 y de 1.9 a 2.2 en los pacientes tratados con Xeljanz® 5 y 10 mg, respectivamente, en comparación con los pacientes tratados con placebo (0.7 a 1.1) a los 3 meses. En el mes 6, la proporción de pacientes que logró una remisión clínica de DAS28 (DAS28-4(ESR) < 2.6) en los Estudios II, III y IV fue significativamente mayor en los pacientes que recibieron Xeljanz® 5 o 10 mg (del 6 al 9% y del 13 al 16%, respectivamente) en comparación con los pacientes tratados con placebo entre los cuales de un 1 a un 3% lograron una remisión clínica. En el Estudio III, los porcentajes observados de pacientes que lograron DAS28-4(ESR) < 2.6 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y adalimumab en el mes 6 fueron 6.2%, 12.5% y 6.7%, respectivamente.
En un análisis combinado de los estudios de Fase 3, la dosis de 10 mg administrada dos veces al día proporcionó un beneficio mayor que la dosis de 5 mg administrada dos veces al día en varias mediciones de signos y síntomas: mejoría respecto del valor inicial (tasas de respuesta ACR20, ACR50 y ACR70) y el logro del estado de actividad de la enfermedad objetivo (ya sea DAS28-4[ESR] < 2.6 o DAS28-4[ESR] ≤ 3.2). Se mostraron mayores beneficios con los 10 mg en comparación con los 5 mg en las mediciones más estrictas (es decir, las tasas de respuesta ACR70 y a DAS28-4[ESR] < 2.6).
Tabla 1. Proporción de pacientes con respuesta ACR
|
Estudio I: Pacientes con respuesta inadecuada a los FARME |
||||
|
Índice de respuesta (%) |
Tiempo |
Placebo |
Monoterapia con Xeljanz® 5 mg dos veces al día |
Monoterapia con Xeljanz® 10 mg dos veces al día |
|
N = 120 |
N = 241 |
N = 242 |
||
|
ACR20 |
Mes 3 |
27 |
60 |
66 |
|
Mes 6 |
NA |
69 |
71 |
|
|
Mes 12 |
NA |
NA |
NA |
|
|
Mes 24 |
NA |
NA |
NA |
|
|
ACR50 |
Mes 3 |
13 |
31 |
37 |
|
Mes 6 |
NA |
42 |
47 |
|
|
Mes 12 |
NA |
NA |
NA |
|
|
Mes 24 |
NA |
NA |
NA |
|
|
ACR70 |
Mes 3 |
6 |
15 |
20 |
|
Mes 6 |
NA |
22 |
29 |
|
|
Mes 12 |
NA |
NA |
NA |
|
|
Mes 24 |
NA |
NA |
NA |
|
|
Estudio II: Pacientes con respuesta inadecuada a los FARME, por lo general MTX |
||||
|
Índice de respuesta (%) |
Tiempo |
Placebo |
FARME |
FARME |
|
N = 157 |
N = 311 |
N = 309 |
||
|
ACR20 |
Mes 3 |
27 |
56 |
65 |
|
Mes 6 |
31 |
53 |
58 |
|
|
Mes 12 |
NA |
51 |
57 |
|
|
ACR50 |
Mes 3 |
10 |
27 |
34 |
|
Mes 6 |
13 |
34 |
37 |
|
|
Mes 12 |
NA |
33 |
43 |
|
|
ACR70 |
Mes 3 |
2 |
8 |
14 |
|
Mes 6 |
3 |
13 |
16 |
|
|
Mes 12 |
NA |
19 |
26 |
|
|
Estudio IV: Pacientes con respuesta inadecuada a MTX |
||||
|
Índice de respuesta (%) |
Tiempo |
Placebo + MTX |
Xeljanz® 5 mg dos veces al día + MTX |
Xeljanz® 10 mg dos veces al día + MTX |
|
N = 154 |
N = 309 |
N = 309 |
||
|
ACR20 |
Mes 3 |
27 |
56 |
66 |
|
Mes 6 |
25 |
51 |
62 |
|
|
Mes 12 |
NA |
49 |
56 |
|
|
Mes 24 |
NA |
41 |
50 |
|
|
ACR50 |
Mes 3 |
8 |
29 |
36 |
|
Mes 6 |
8 |
32 |
44 |
|
|
Mes 12 |
NA |
32 |
39 |
|
|
Mes 24 |
NA |
29 |
40 |
|
|
ACR70 |
Mes 3 |
3 |
11 |
17 |
|
Mes 6 |
1 |
15 |
22 |
|
|
Mes 12 |
NA |
19 |
27 |
|
|
Mes 24 |
NA |
17 |
26 |
|
|
Estudio V: Pacientes con respuesta inadecuada a inhibidores del TNF |
||||
|
Índice de respuesta (%) |
Tiempo |
Placebo + MTX |
Xeljanz® 5 mg dos veces al día + MTX |
Xeljanz® 10 mg dos veces al día + MTX |
|
N = 131 |
N = 132 |
N = 133 |
||
|
ACR20 |
Mes 3 |
24 |
42 |
48 |
|
Mes 6 |
NA |
52 |
55 |
|
|
Mes 12 |
NA |
NA |
NA |
|
|
Mes 24 |
NA |
NA |
NA |
|
|
ACR50 |
Mes 3 |
8 |
27 |
28 |
|
Mes 6 |
NA |
37 |
30 |
|
|
Mes 12 |
NA |
NA |
NA |
|
|
Mes 24 |
NA |
NA |
NA |
|
|
ACR70 |
Mes 3 |
2 |
14 |
11 |
|
Mes 6 |
NA |
16 |
16 |
|
|
Mes 12 |
NA |
NA |
NA |
|
|
Mes 24 |
NA |
NA |
NA |
|
|
Estudio VI: No tratados previamente con MTX |
||||
|
Índice de respuesta (%) |
Tiempo |
MTX |
Monoterapia con Xeljanz® 5 mg dos veces al día |
Monoterapia con Xeljanz® 10 mg dos veces al día |
|
N = 184 |
N = 369 |
N = 394 |
||
|
ACR20 |
Mes 3 |
52 |
70 |
78 |
|
Mes 6 |
51 |
71 |
76 |
|
|
Mes 12 |
51 |
68 |
72 |
|
|
Mes 24 |
42 |
64 |
64 |
|
|
ACR50 |
Mes 3 |
20 |
40 |
50 |
|
Mes 6 |
27 |
47 |
56 |
|
|
Mes 12 |
34 |
50 |
56 |
|
|
Mes 24 |
28 |
49 |
49 |
|
|
ACR70 |
Mes 3 |
5 |
20 |
27 |
|
Mes 6 |
12 |
25 |
38 |
|
|
Mes 12 |
15 |
29 |
38 |
|
|
Mes 24 |
15 |
34 |
38 |
|
En la Tabla 1, se presentan los resultados de la proporción de pacientes con respuesta ACR en los Estudios I, II, IV, V y VI. Se observaron resultados similares en el Estudio III.
En la Tabla 2, se presentan los resultados de los componentes de los criterios de respuesta ACR de los Estudios IV y V. Se observaron resultados similares en los Estudios I, II y III.
Tabla 2. Componentes de la respuesta ACR en el mes 3 en los estudios IV y V
|
Estudio IV: Pacientes con respuesta inadecuada a MTX |
||||
|
Componente |
Tiempo |
Placebo + MTX |
Xeljanz® 5 mg dos veces al día + MTX |
Xeljanz® 10 mg dos veces al día + MTX |
|
N = 156 |
N = 316 |
N = 309 |
||
|
Cantidad de articulaciones sensibles al tacto (0-68) |
Periodo inicial |
23 |
24 |
23 |
|
Mes 3 |
18 |
13 |
10 |
|
|
Cantidad de articulaciones inflamadas (0-66) |
Periodo inicial |
14 |
14 |
14 |
|
Mes 3 |
10 |
6 |
6 |
|
|
Dolora |
Periodo inicial |
55 |
58 |
58 |
|
Mes 3 |
47 |
35 |
29 |
|
|
Evaluación global del pacientea |
Periodo inicial |
54 |
58 |
57 |
|
Mes 3 |
47 |
35 |
29 |
|
|
Índice de discapacidad (HAQ-DI)b |
Periodo inicial |
1.31 |
1.41 |
1.39 |
|
Mes 3 |
1.19 |
1.00 |
0.84 |
|
|
Evaluación global del médicoa |
Periodo inicial |
56 |
59 |
58 |
|
Mes 3 |
43 |
30 |
25 |
|
|
PCR (mg/L) |
Periodo inicial |
13.7 |
15.5 |
17.0 |
|
Mes 3 |
14.6 |
6.9 |
4.4 |
|
|
Estudio V: Pacientes con respuesta inadecuada a inhibidores del TNF |
||||
|
Componente |
Tiempo |
Placebo + MTX |
Xeljanz® 5 mg dos veces al día + MTX |
Xeljanz® 10 mg dos veces al día + MTX |
|
N = 132 |
N = 133 |
N = 134 |
||
|
Cantidad de articulaciones sensibles al tacto (0-68) |
Periodo inicial |
28 |
28 |
28 |
|
Mes 3 |
21 |
16 |
13 |
|
|
Cantidad de articulaciones inflamadas (0-66) |
Periodo inicial |
17 |
16 |
17 |
|
Mes 3 |
12 |
8 |
7 |
|
|
Dolora |
Periodo inicial |
61 |
66 |
60 |
|
Mes 3 |
53 |
39 |
38 |
|
|
Evaluación global del pacientea |
Periodo inicial |
62 |
65 |
59 |
|
Mes 3 |
53 |
41 |
37 |
|
|
Índice de discapacidad (HAQ-DI)b |
Periodo inicial |
1.63 |
1.60 |
1.50 |
|
Mes 3 |
1.44 |
1.20 |
1.10 |
|
|
Evaluación global del médicoa |
Periodo inicial |
64 |
65 |
59 |
|
Mes 3 |
44 |
35 |
31 |
|
|
PCR (mg/L) |
Periodo inicial |
16.7 |
19.3 |
15.7 |
|
Mes 3 |
18.2 |
6.2 |
4.8 |
|
a Escala análoga visual: 0 = mejor, 100 = peor.
b Índice de discapacidad del cuestionario de evaluación de salud: 0 = mejor, 3 = peor; 20 preguntas; categorías: vestimenta y aseo, capacidad de levantarse, alimentarse, caminar, higiene, alcance, agarre y realizar actividades.
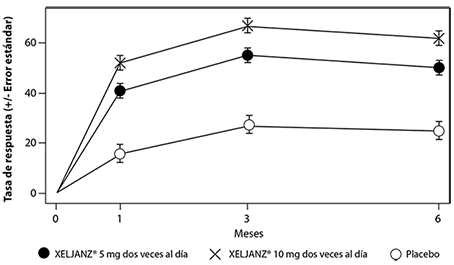
El porcentaje de pacientes con respuesta ACR20 por visita en el Estudio IV se muestra en la Figura 2. Se observaron respuestas similares en los Estudios I, II, III y V.
Figura 2. Porcentaje por visita de pacientes con respuesta ACR20 en el Estudio IV
Respuesta radiográfica: Se efectuaron dos estudios para evaluar el efecto de Xeljanz® con respecto al daño estructural de las articulaciones. En el Estudio IV y en el Estudio VI se evaluó, de manera radiográfica, la inhibición de la progresión del daño estructural de las articulaciones, y ésta se expresó como el cambio medio desde el periodo inicial en mTSS y sus componentes, el puntaje de erosión y el puntaje del estrechamiento del espacio articular (JSN), en los meses 6 y 12. También se evaluó la proporción de pacientes sin progresión radiográfica (cambio de mTSS menor o igual que 0.5).
En el Estudio IV, Xeljanz® 10 mg administrados dos veces al día con MTX de fondo generó una inhibición significativamente mayor de la progresión del daño estructural, en comparación con el placebo más MTX, en los meses 6 y 12. Cuando se administró a una dosis de 5 mg dos veces al día, Xeljanz® más MTX mostraron efectos similares en la progresión media del daño estructural (sin significancia estadística). El análisis de la erosión y el puntaje de JSN fueron consistentes con los resultados generales. Estos resultados se muestran en la Tabla 3.
En el grupo de placebo con MTX, el 78% de los pacientes no presentó progresión radiográfica en el mes 6, en comparación con el 89% y el 87% de los pacientes tratados con Xeljanz® 5 o 10 mg dos veces al día, respectivamente, más MTX. Ambos son importantes en comparación con el placebo más MTX.
Tabla 3. Cambios radiográficos en los meses 6 y 12
|
Estudio IV |
|||||
|
Placebo + MTX N = 139 Media (DE)a |
Xeljanz® 5 mg dos veces al día + MTX N = 277 Media (DE)a |
Xeljanz® 5 mg dos veces al día + MTX diferencia media respecto del placebob (IC) |
Xeljanz® 10 mg dos veces al día + MTX N = 290 Media (DE)a |
Xeljanz® 10 mg dos veces al día + MTX diferencia media respecto del placebob (IC) |
|
|
mTSSc Periodo inicial Mes 6 Mes 12 |
33 (42) 0.5 (2.0) 1.0 (3.9) |
31 (48) 0.1 (1.7) 0.3 (3.0) |
- -0.3 (-0.7, 0.0) -0.6 (-1.3, 0.0) |
37 (54) 0.1 (2.0) 0.1 (2.9) |
- -0.4 (-0.8, 0.0) -0.9 (-1.5, 0.2) |
|
Puntaje de erosiónc Periodo inicial Mes 6 Mes 12 |
14 (19) 0.1 (1.0) 0.3 (2.0) |
14 (24) 0.1 (1.0) 0.2 (1.7) |
- -0.1 (-0.3, 0.1) -0.1 (-0.4, 0.2) |
18 (28) 0.0 (0.7) 0.0 (1.1) |
- -0.1 (-0.3, 0.1) -0.3 (-0.6, 0.0) |
|
Puntaje de JSNc Periodo inicial Mes 6 Mes 12 |
18 (24) 0.3 (1.5) 0.7 (2.9) |
17 (25) 0.1 (1.1) 0.1 (1.9) |
- -0.3 (-0.6, 0.1) -0.5 (-1.0, 0.0) |
20 (28) 0.1 (1.8) 0.1 (2.6) |
- -0.3 (-0.6, 0.0) -0.6 (-1.1, -0.1) |
a DE = desviación estándar.
b Diferencia entre la medias de los mínimos cuadrados de Xeljanz® menos placebo (IC del 95% = intervalo de confianza del 95%).
c Los datos del mes 6 y del mes 12 corresponden a cambios medios con respecto al periodo inicial.
Como se muestra en la Tabla 4, en el Estudio VI la monoterapia con Xeljanz® provocó una inhibición significativamente mayor de la progresión del daño estructural en comparación con MTX en los meses 6 y 12, que también se mantuvo en el mes 24. El análisis de la erosión y los puntajes de JSN fueron consistentes con los resultados generales.
En el grupo de MTX, el 70% de los pacientes no presentaron progresión radiográfica en el mes 6, en comparación con el 84% y el 90% de los pacientes tratados con Xeljanz® 5 mg o Xeljanz® 10 mg dos veces al día, respectivamente, ambos significativos en comparación con el MTX.
Tabla 4. Cambios radiográficos en los meses 6 y 12
|
Estudio VI |
|||||
|
MTX N = 166 Media (DE)a |
Xeljanz® 5 mg dos veces al día N = 346 Media (DE)a |
Xeljanz® 5 mg dos veces al día diferencia media respecto de MTXb (IC) |
Xeljanz® 10 mg dos veces al día N = 369 Media (DE)a |
Xeljanz® 10 mg dos veces al día diferencia media respecto de MTXb (IC) |
|
|
mTSSc Periodo inicial Mes 6 Mes 12 |
17 (29) 0.8 (2.7) 1.3 (3.7) |
20 (40) 0.2 (2.3) 0.4 (3.0) |
- -0.7 (-1.0, -0.3) -0.9 (-1.4, -0.4) |
19 (39) 0.0 (1.2) 0.0 (1.5) |
- -0.8 (-1.2, -0.4) -1.3 (-1.8, -0.8) |
|
Puntaje de erosiónc Periodo inicial Mes 6 Mes 12 |
8 (15) 0.5 (1.9) 0.6 (2.2) |
10 (21) 0.1 (1.4) 0.1 (1.6) |
- -0.4 (-0.7, -0.2) -0.6 (-0.8, -0.3) |
9 (20) 0.0 (0.7) 0.0 (1.0) |
- -0.5 (-0.7, -0.3) -0.7 (-0.9, -0.4) |
|
Puntaje de JSNc Periodo inicial Mes 6 Mes 12 |
8 (16) 0.4 (1.3) 0.6 (2.1) |
11 (21) 0.1 (1.4) 0.3 (2.1) |
- -0.2 (-0.5, 0.0) -0.4 (-0.7, 0.0) |
9 (20) 0.1 (0.9) 0.0 (0.9) |
- -0.3 (-0.5, -0.1) -0.6 (-0.9, -0.3) |
a DE = desviación estándar.
b Diferencia entre la medias de los mínimos cuadrados de Xeljanz® menos MTX (IC del 95% = intervalo de confianza del 95%).
c Los datos del mes 6 y del mes 12 corresponden a cambios medios con respecto al periodo inicial.
Respuesta de la función física y resultados relacionados con la salud: La mejoría del funcionamiento físico se midió con el HAQ-DI. En el mes 3 (Estudios I, II, III y V) y el mes 6 (Estudios II y III), los pacientes que recibieron Xeljanz® 5 mg y 10 mg dos veces al día demostraron mejoras significativamente mayores con respecto al periodo inicial en las funciones físicas en comparación con los pacientes que recibieron placebo. Tan pronto como en la semana 2 de los Estudios I y II, los pacientes tratados con Xeljanz® 5 o 10 mg dos veces al día exhibieron una mejora significativamente mayor en sus funciones físicas en comparación con los pacientes que recibieron placebo. En el Estudio III, las mejoras medias en el HAQ-DI se mantuvieron hasta 12 meses en los pacientes tratados con Xeljanz®. Las mejoras medias del HAQ-DI se mantuvieron por 36 meses en los estudios de extensión abiertos y en curso. En comparación con los pacientes tratados con adalimumab, en el mes 3, los pacientes tratados con Xeljanz® 5 mg dos veces al día tuvieron disminuciones similares desde el periodo inicial en los valores del HAQ-DI y los pacientes tratados con 10 mg dos veces al día tuvieron disminuciones del HAQ-DI considerablemente mayores. En la Tabla 5, se muestra el cambio medio respecto del periodo inicial en los valores del HAQ-DI en el mes 3 en los Estudios I a VI.
Tabla 5. Cambio medio respecto de los valores iniciales en el HAQ-DI
|
Estudio I: Pacientes con respuesta inadecuada a los FARME |
||||
|
Tiempo |
Placebo |
Monoterapia con Xeljanz® 5 mg |
Monoterapia con Xeljanz® 10 mg dos veces al día |
|
|
Cambio medio de los LS en el HAQ-DI en el mes 3a |
-0.19 |
-0.50** |
-0.57** |
|
|
Estudio II: Pacientes con respuesta inadecuada a los FARME |
||||
|
Placebo + FARME(s) |
Xeljanz® 5 mg dos |
Xeljanz® 10 mg dos veces al día + FARME(s) |
||
|
Cambio medio de los LS en el HAQ-DI en el mes 3a |
-0.21 |
-0.46** |
-0.56** |
|
|
Estudio III: Pacientes con respuesta inadecuada a MTX |
||||
|
Placebo + MTX |
Xeljanz® 5 mg |
Xeljanz® 10 mg |
Adalimumab 40 mg |
|
|
Cambio medio de los LS en el HAQ-DI en el mes 3a |
-0.24 |
-0.55** |
-0.61** |
-0.49** |
|
Estudio IV: Pacientes con respuesta inadecuada a MTX |
||||
|
Placebo + MTX |
Xeljanz® 5 mg |
Xeljanz® 10 mg dos veces al día + MTX |
||
|
Cambio medio de los LS en el HAQ-DI en el mes 3a |
-0.15 |
-0.40b |
-0.54 |
|
|
Estudio V: Pacientes con respuesta inadecuada a inhibidores del TNF |
||||
|
Placebo N = 118 |
Xeljanz® 5 mg dos veces al día + MTX N = 117 |
Xeljanz® 10 mg dos veces al día + MTX N = 125 |
||
|
Cambio medio de los LS en el HAQ-DI en el mes 3a |
-0.18 |
-0.43** |
-0.46** |
|
|
Estudio VI: |
||||
|
Placebo + MTX N = 171 |
Monoterapia con Xeljanz® 5 mg dos veces al día N = 355 |
Monoterapia con Xeljanz® 10 mg dos veces al día N = 381 |
||
|
Cambio medio de los LS en el HAQ-DI en el mes 3a |
-0.47 |
-0.75** |
-0.85** |
|
a Punto temporal de eficacia primaria.
b La importancia estadística no se pudo declarar en el Estudio IV debido a un procedimiento de reducción.
** p < 0.0001, Xeljanz® en comparación con placebo + MTX/FARME.
Los resultados se obtuvieron a partir de un modelo lineal longitudinal con el cambio respecto del periodo inicial como variable dependiente y el tratamiento, el periodo inicial, la visita y la región como efectos fijos, y el paciente como efecto aleatorio.
IC = intervalo de confianza, LS = mínimos cuadrados, N = cantidad de pacientes, MTX = metotrexato, QOW = cada dos semanas, HAQ-DI = Cuestionario de evaluación de la salud - Índice de discapacidad
En los 5 estudios, la calidad de vida relacionada con la salud se evaluó mediante el Cuestionario de salud en formulario abreviado (SF-36). En estos estudios, en el mes 3, los pacientes que recibieron Xeljanz® 10 mg dos veces al día demostraron mejoras significativamente mayores con respecto al periodo inicial, en comparación con los pacientes tratados con placebo en los 8 dominios del SF-36, así como también en el Resumen del componente físico (PCS, por sus siglas en inglés) y en el Resumen del componente mental (MCS, por sus siglas en inglés). En el mes 3, ambos grupos tratados con Xeljanz® mostraron mejoras significativamente mayores con respecto al periodo inicial en comparación con los grupos tratados con placebo en los 8 dominios, así como también en el PCS y el MCS de los Estudios I, IV y V. En los Estudios III y IV, las mejoras medias de SF-36 se mantuvieron hasta 12 meses en los pacientes tratados con Xeljanz®.
En el mes 3, la mejora en la fatiga se evaluó mediante la escala de Evaluación funcional de enfermedades crónicas - Fatiga (FACIT-F) en todos los estudios. En los 5 estudios, los pacientes que recibieron Xeljanz® 5 o 10 mg dos veces al día mostraron mejoras significativamente mayores con respecto al periodo inicial en la fatiga en comparación con los pacientes que recibieron placebo. En los Estudios III y IV, las mejoras medias en la FACIT-F se mantuvieron hasta 12 meses en los pacientes tratados con Xeljanz®.
En el mes 3, la mejora del sueño se evaluó mediante las escalas de resumen del Índice de problemas del sueño I y II del Estudio de resultados médicos del sueño (MOS-Sleep, en inglés) en todos los estudios. En los Estudios II, III y IV, los pacientes que recibieron Xeljanz® 5 o 10 mg dos veces al día presentaron mejoras significativamente mayores con respecto al periodo inicial en ambas escalas en comparación con los pacientes tratados con placebo. En los Estudios III y IV, las mejoras medias en ambas escalas se mantuvieron hasta 12 meses en los pacientes tratados con Xeljanz®.
El aumento en la productividad se evaluó utilizando la escala del Cuestionario de limitaciones del trabajo (WLQ, por sus siglas en inglés) en el mes 3 en todos los estudios. Los pacientes que recibieron Xeljanz® 10 mg dos veces al día demostraron una mejora significativamente mayor con respecto a los valores iniciales en la Escala resumen de resultados globales en comparación con el placebo en los Estudios III, IV y V. En los Estudios III y IV, las mejoras en los resultados globales se mantuvieron hasta 12 meses en los pacientes tratados con Xeljanz® 10 mg dos veces al día.
Durabilidad de las respuestas clínicas: La durabilidad del efecto se evaluó mediante las tasas de respuesta ACR20, ACR50, ACR70, HAQ-DI media y DAS28-4(ESR) media en los tres estudios de IR de FARME de fase 3 con una duración de al menos un año. La eficacia en todos los grupos de tratamiento con tofacitinib se mantuvo hasta el final de los estudios. También se proporciona evidencia de persistencia de la eficacia con el tratamiento con tofacitinib hasta 6 años a partir de los datos de un gran estudio PASS aleatorizado realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo CV adicional, así como los estudios de seguimiento a largo plazo y abiertos de hasta 8 años ya finalizados.
Artritis psoriásica: XELJANZ® XR 11 mg una vez al día ha demostrado una exposición equivalente (ABC y Cmáx) a la de Xeljanz® 5 mg dos veces al día. La dosis recomendada de XELJANZ® XR es de 11 mg una vez al día.
El programa de desarrollo clínico de Xeljanz® para evaluar la eficacia y la seguridad en pacientes con artritis psoriásica incluyó 2 ensayos de confirmación multicéntricos, aleatorizados, doble ciego y controlados con placebo realizados en 816 pacientes de 18 años y mayores. Todos los pacientes tenían artritis psoriásica activa desde hace al menos 6 meses según los Criterios de clasificación de la artritis psoriásica (CASPAR, por sus siglas en inglés), al menos 3 articulaciones sensibles al tacto/con dolor y al menos 3 articulaciones inflamadas, y psoriasis en placa activa. En los dos ensayos clínicos se incluyeron pacientes con diferentes subtipos de artritis psoriásica (no excluyentes entre sí), incluyendo < 5 articulaciones o afectación asimétrica (21%), ≥ 5 articulaciones afectadas (90%), afectación de la articulación interfalángica distal (DIP, por sus siglas en inglés) (61%), artritis mutilante (8%) y espondilitis (19%). Los pacientes de estos ensayos clínicos tuvieron un diagnóstico de artritis psoriásica durante una mediana de 5.5 años (rango de 3.0 a 6.0 años). En el periodo inicial, el 80%, el 53% y el 69% de los pacientes presentó entesitis, dactilitis y un área de superficie corporal (ASC) psoriásica total ≥ 3%, respectivamente. Todos los pacientes debían recibir tratamiento con una dosis estable de FARME sintéticos convencionales (FARMEsc; el 79% recibió metotrexato, el 13% sulfasalazina, el 7% leflunomida, el 1% otros FARMEsc) y se les permitió recibir una dosis baja estable de corticosteroides orales (el 21% recibió el equivalente a ≤ 10 mg/día de prednisona) y/o antiinflamatorios no esteroideos (AINE; el 57% los recibió). En ambos ensayos clínicos, los criterios de valoración primarios fueron la respuesta ACR20 y el cambio en el HAQ-DI en el mes 3.
El Estudio PsA-I fue un ensayo clínico de 12 meses de duración realizado en 422 pacientes que tuvieron una respuesta inadecuada a un FARMEsc (el 67% y el 33% eran pacientes con una respuesta inadecuada a 1 FARMEsc y ≥ 2 FARMEsc, respectivamente) y que no habían recibido tratamiento con un FARME biológico inhibidor del TNF (TNFi). Los pacientes se aleatorizaron en una proporción de 2:2:2:1:1 para recibir Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea una vez cada 2 semanas, placebo a la secuencia de tratamiento con Xeljanz® 5 mg dos veces al día, o placebo a la secuencia de tratamiento con Xeljanz® 10 mg dos veces al día, respectivamente; el medicamento del estudio se añadió al tratamiento de fondo con FARMEsc. En la visita del mes 3, todos los pacientes aleatorizados al tratamiento con placebo avanzaron de forma ciega a una dosis predeterminada de Xeljanz® 5 o 10 mg dos veces al día. El Estudio PsA-I no se diseñó para demostrar la no inferioridad o superioridad con respecto al adalimumab.
El Estudio PsA-II fue un ensayo clínico de 6 meses de duración realizado en 394 pacientes que tuvieron una respuesta inadecuada al menos a 1 TNFi aprobado (el 66%, el 19% y el 15% eran pacientes con una respuesta inadecuada a 1 TNFi, 2 TNFi y ≥ 3 TNFi, respectivamente). Los pacientes se aleatorizaron en una proporción de 2:2:1:1 para recibir Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, placebo a la secuencia de tratamiento con Xeljanz® 5 mg dos veces al día, o placebo a la secuencia de tratamiento con Xeljanz® 10 mg dos veces al día, respectivamente; el medicamento del estudio se añadió al tratamiento de fondo con FARMEsc. En la visita del mes 3, los pacientes asignados al placebo avanzaron de forma ciega a una dosis predeterminada de Xeljanz® 5 o 10 mg dos veces al día como en el Estudio PsA-I.
Respuesta clínica:
Signos y síntomas: En el mes 3, los pacientes tratados con Xeljanz® 5 o 10 mg dos veces al día tuvieron tasas de respuesta más altas (p ≤ 0.05) frente al placebo para ACR20, ACR50 y ACR70 en el Estudio PsA-I y para ACR20 y ACR50 en el Estudio PsA-II; las tasas de respuesta ACR70 también fueron más altas tanto para Xeljanz® 5 mg dos veces al día como para Xeljanz® 10 mg dos veces al día frente al placebo en el Estudio PsA-II, aunque las diferencias frente al placebo no fueron estadísticamente significativas (p > 0.05) (Tabla 6). El examen de la edad, el sexo, la raza, la actividad inicial de la enfermedad y el subtipo de artritis psoriásica no identificó diferencias en la respuesta a Xeljanz®. La cantidad de pacientes con artritis mutilante fue demasiado baja para permitir una evaluación significativa.
Tabla 6. Proporción de pacientes con respuesta ACR en los Estudios PsA-I y PsA-II
|
Porcentaje de pacientes |
|||||||
|
Pacientes con una respuesta inadecuada a FARME sintéticos convencionalesc (sin tratamiento previo con TNFi) |
Pacientes con una respuesta inadecuada a TNFid |
||||||
|
Estudio PsA-I |
Estudio PsA-II |
||||||
|
Grupo de tratamiento |
Placebo |
Xeljanz® 5 mg dos veces al día |
Xeljanz® |
Adalimumab |
Placebo |
Xeljanz® 5 mg dos veces al día |
Xeljanz® 10 mg dos veces al día |
|
Na |
105 |
107 |
104 |
106 |
131 |
131 |
132 |
|
ACR20 |
|||||||
|
Mes 3 |
33% |
50%* |
61%* |
52%* |
24% |
50%* |
47%* |
|
Mes 6 |
NAb |
59% |
67% |
64% |
NA |
60% |
49% |
|
Mes 12 |
NA |
68% |
70% |
60% |
-e |
- |
- |
|
ACR50 |
|||||||
|
Mes 3 |
10% |
28%* |
40%* |
33%* |
15% |
30%* |
28%* |
|
Mes 6 |
NA |
38% |
46% |
42% |
NA |
38% |
30% |
|
Mes 12 |
NA |
45% |
48% |
41% |
- |
- |
- |
|
ACR70 |
|||||||
|
Mes 3 |
5% |
17%* |
14%* |
19%* |
10% |
17% |
14% |
|
Mes 6 |
NA |
18% |
32% |
30% |
NA |
21% |
14% |
|
Mes 12 |
NA |
23% |
31% |
29% |
- |
- |
- |
a N es la cantidad de pacientes aleatorizados y tratados.
b NA = no aplicable, ya que los datos del tratamiento con placebo no están disponibles más allá del mes 3 debido al avance del placebo.
c Respuesta inadecuada al menos a un FARMEsc por falta de eficacia y/o intolerabilidad.
d Respuesta inadecuada al menos a un inhibidor del TNF (TNFi) por falta de eficacia y/o intolerabilidad.
e El Estudio PsA-II tuvo una duración de 6 meses.
* p ≤ 0.05 para el tratamiento activo frente al placebo.
Al igual que con las respuestas ACR, en los pacientes tratados con Xeljanz® 5 o 10 mg dos veces al día en los Estudios PsA-I y PsA-II, cada uno de los componentes de la respuesta ACR mejoró de forma coherente con respecto al periodo inicial en el mes 3, incluidos los recuentos de articulaciones sensibles al tacto/con dolor e inflamadas, la evaluación del dolor de la artritis por parte del paciente, la evaluación global de la artritis por parte del paciente y del médico, el HAQ-DI y la PCR, en comparación con los pacientes que recibieron el placebo (Tabla 7).
Tabla 7. Componentes de la respuesta ACR en el periodo inicial y el mes 3 en los Estudios de PsA-I y PsA-II
|
Pacientes con respuesta inadecuada a FARME sintético convencional (sin tratamiento previo con TNFi) |
Pacientes con respuesta inadecuada a TNFi |
||||||
|---|---|---|---|---|---|---|---|
|
Estudio de PsA-I |
Estudio de PsA-II |
||||||
|
Grupo de tratamiento |
Placebo |
Xeljanz® 5 mg dos veces al día |
Xeljanz® 10 mg dos veces al día |
Adalimumab 40 mg SC c/2 sem |
Placebo |
Xeljanz® 5 mg dos veces al día |
Xeljanz® 10 mg dos veces al día |
|
N al periodo inicial |
105 |
107 |
104 |
106 |
131 |
131 |
132 |
|
Componente de ACRa |
|||||||
|
Cantidad de articulaciones sensibles al tacto/dolorosas (0-68) Periodo inicial Mes 3 |
20.6 14.6 |
20.5 12.2 |
20.3 9.9 |
17.1 10.8 |
19.8 15.1 |
20.5 11.5 |
25.5 14.5 |
|
Cantidad de articulaciones inflamadas (0-66) Periodo inicial Mes 3 |
11.5 7.1 |
12.9 6.3 |
11.7 4.3 |
9.8 4.0 |
10.5 7.7 |
12.1 4.8 |
12.8 6.1 |
|
Evaluación del paciente sobre el dolor de artritisb Periodo inicial Mes 3 |
53.2 44.7 |
55.7 34.7 |
54.4 28.5 |
50.7 32.5 |
54.9 48.0 |
56.4 36.1 |
59.5 38.1 |
|
Evaluación global del paciente sobre la artritisb Periodo inicial Mes 3 |
53.9 44.4 |
54.7 35.5 |
53.6 29.8 |
50.6 32.9 |
55.8 49.2 |
57.4 36.9 |
58.5 38.8 |
|
HAQ-DIc Periodo inicial Mes 3 |
1.11 0.95 |
1.16 0.81 |
1.08 0.71 |
1.10 0.75 |
1.25 1.09 |
1.26 0.88 |
1.37 1.03 |
|
Evaluación global del médico sobre la artritisb Periodo inicial Mes 3 |
53.8 35.4 |
54.6 29.5 |
55.2 23.6 |
50.5 26.3 |
53.7 36.4 |
53.5 27.0 |
55.8 25.6 |
|
PCR (mg/L) Periodo inicial Mes 3 |
10.4 8.60 |
10.5 4.02 |
8.1 2.68 |
14.3 3.10 |
12.1 11.44 |
13.8 7.72 |
15.0 7.25 |
a Los datos mostrados son los valores medios en el periodo inicial y en el mes 3.
b Escala analógica visual (VAS por sus siglas en inglés): 0 = mejor, 100 = peor.
c HAQ-DI = Índice de discapacidad del cuestionario de evaluación de la salud (por sus siglas en inglés): 0 = mejor, 3 = peor; 20 preguntas; categorías: vestirse y aseo personal, levantarse, comer, caminar, higiene, alcance, agarre y actividades.
En la Figura 3 se muestra el porcentaje de pacientes con respuesta ACR20 por visita en los Estudios PsA-I y PsA-II. En los pacientes tratados con Xeljanz® en los Estudios PsA-I y PsA-II, se observaron tasas de respuesta ACR20 significativamente mayores en el plazo de 2 semanas, en comparación con el placebo (Figura 3). Después del mes 3, las tasas de respuesta ACR se mantuvieron o mejoraron hasta el mes 6 (Estudios PsA-I y PsA-II) y el mes 12 (Estudio PsA-I).
Figura 3. Porcentaje de pacientes con respuesta ACR20 por visita
a) Hasta el mes 12 en el Estudio PsA-I
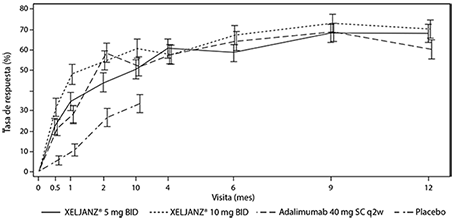
b) Hasta el mes 6 en el Estudio PsA-IIa
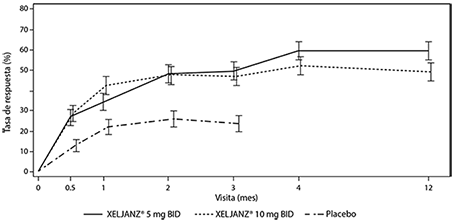
En los Estudios PsA-I y PsA-II, la comparación de Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y adalimumab (solo en el Estudio PsA-I) con el placebo fue significativa (valor p ≤ 0.05) en los meses 0.5, 1, 2 y 3.
BID = dos veces al día; c/2 sem = por vía subcutánea una vez cada 2 semanas.
Los pacientes aleatorizados al tratamiento con placebo avanzaron a Xeljanz® 5 o 10 mg dos veces al día de forma ciega en el mes 3; los resultados de la fase con Xeljanz® de la secuencia de tratamiento placebo → Xeljanz® (es decir, después del mes 3) no se incluyen en la figura para mejorar la legibilidad.
a El Estudio PsA-II tuvo una duración de 6 meses.
En los pacientes con entesitis en el periodo inicial, se observó un beneficio en la entesitis con el tratamiento con Xeljanz®. En el Estudio PsA-I, el cambio con respecto al periodo inicial en la puntuación del Índice de entesitis de Leeds fue de -0.8, -1.5, -1.1 y -0.4 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea una vez cada dos semanas y el placebo, respectivamente, en el mes 3; y -1.7, -1.6 y -1.6 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y adalimumab 40 mg por vía subcutánea una vez cada 2 semanas, respectivamente, en el mes 12. En el Estudio PsA-II, el cambio con respecto al periodo inicial en la puntuación del Índice de entesitis de Leeds fue de -1.3, -1.3 y -0.5 para XELJANZ 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y el placebo, respectivamente, en el mes 3; y de -1.5 y -1.6 para Xeljanz® 5 mg dos veces al día y Xeljanz® 10 mg dos veces al día, respectivamente, en el mes 6.
En el Estudio PsA-I, la resolución de la entesitis en el mes 3 se produjo en el 33.3%, 40.6%, 47.4% y 21.5% de los pacientes que recibieron Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea una vez cada dos semanas y el placebo, respectivamente. En el Estudio PsA-II, la entesitis se resolvió en el mes 3 en el 39.8%, 32.3% y 21.5% de los pacientes que recibieron Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y el placebo, respectivamente.
En los pacientes con dactilitis en el periodo inicial, se observó evidencia de beneficio en la dactilitis con el tratamiento con Xeljanz®. En el Estudio PsA-I, el cambio con respecto al periodo inicial en la Puntuación de severidad de la dactilitis fue de -3.5, -5.5, -4.0 y -2.0 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea una vez cada dos semanas y el placebo, respectivamente, en el mes 3; y de -7.4, -7.5 y -6.1 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y adalimumab 40 mg por vía subcutánea una vez cada 2 semanas, respectivamente, en el mes 12. En el Estudio PsA-II, el cambio con respecto al periodo inicial en la Puntuación de severidad de la dactilitis fue de -5.2, -5.4 y -1.9 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y placebo, respectivamente, en el mes 3; y de -6.0 y -6.0 para Xeljanz® 5 mg dos veces al día y Xeljanz® 10 mg dos veces al día, respectivamente, en el mes 6.
En el Estudio PsA-I, la resolución de la dactilitis en el mes 3 se produjo en el 34.4%, 60.0%, 46.6% y 32.8% de los pacientes que recibieron Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea una vez cada dos semanas y el placebo, respectivamente. En el Estudio PsA-II, la dactilitis ocurrió en el mes 3 en el 51.5%, 50.8% y 28.6% de los pacientes que recibieron Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y placebo, respectivamente.
La evidencia del beneficio en las manifestaciones cutáneas de la artritis psoriásica se evaluó mediante el PASI75 (mejora de ≥ 75% desde el periodo inicial en el Índice de área y severidad de la psoriasis), en pacientes con artritis psoriásica activa que tenían un ASC psoriásica total ≥ 3%. En el Estudio PsA-I, las tasas de respuesta del PASI75 en el mes 3 fueron mayores para Xeljanz® 5 mg dos veces al día y Xeljanz® 10 mg dos veces al día frente al placebo. En el Estudio PsA-II, las tasas de respuesta del PASI75 en el mes 3 fueron mayores (sin importancia estadística) para Xeljanz® 5 mg dos veces al día y mayores para Xeljanz® 10 mg dos veces al día frente al placebo. Después del mes 3, el beneficio en este dominio se mantuvo o mejoró hasta el mes 6 (Estudios PsA-I y PsA-II) y el mes 12 (Estudio PsA-I).
La actividad de la enfermedad de la artritis psoriásica también se midió mediante la Actividad mínima de la Enfermedad (MDA, por sus siglas en inglés) y la Puntuación de la actividad de la enfermedad de la artritis psoriásica (PASDAS, por sus siglas en inglés). En el Estudio PsA-I, las tasas de MDA en el mes 3 fueron del 26%, 26%, 25% y 7% en los pacientes que recibieron Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea una vez cada dos semanas y el placebo, respectivamente. En el Estudio PsA-II, las tasas de MDA en el mes 3 fueron del 23%, 21% y 15% en los pacientes que recibieron Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y el placebo, respectivamente. Después del mes 3 en los pacientes tratados con Xeljanz®, las tasas de MDA se mantuvieron o mejoraron hasta el mes 6 (Estudios PsA-I y PsA-II) y el mes 12 (Estudio PsA I).
En el Estudio PsA-I, el cambio en el PASDAS con respecto al periodo inicial en el mes 3 fue de -2.0, -2.4, -2.2 y -1.2 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día, adalimumab 40 mg por vía subcutánea una vez cada dos semanas y el placebo, respectivamente. En el Estudio PsA-II, el cambio en el PASDAS con respecto al periodo inicial en el mes 3 fue de -1.9, -2.1 y -0.8 para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y placebo, respectivamente. Después del mes 3 en los pacientes tratados con Xeljanz®, el cambio en el PASDAS con respecto al periodo inicial se mantuvo o mejoró hasta el mes 6 (Estudios PsA-I y PsA-II) y el mes 12 (Estudio PsA-I).
Función física: La mejora del funcionamiento físico se midió mediante el HAQ-DI. Los pacientes que recibieron Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día demostraron una mayor mejoría (p ≤ 0.05) desde el periodo inicial en el funcionamiento físico, en comparación con el placebo en el mes 3 (Tabla 8). La mejoría del HAQ-DI con respecto al periodo inicial en los pacientes tratados con Xeljanz® se mantuvo o mejoró hasta el mes 6 (Estudios PsA-I y PsA-II) y el mes 12 (Estudio PsA-I).
Tabla 8. Cambio con respecto al periodo Inicial en el HAQ-DI en los Estudios PsA-I y PsA-II
|
Cambio medio de los mínimos cuadrados con respecto al periodo inicial en el HAQ-DI: diferencia con respecto al placebo |
|||||
|
Pacientes con una respuesta inadecuada a FARME sintéticos convencionalesb (sin TNFi) |
Pacientes con una respuesta inadecuada a TNFic |
||||
|
Estudio PsA-I |
Estudio PsA-II |
||||
|
Grupo de tratamiento |
Xeljanz® 5 mg dos veces al día |
Xeljanz® 10 mg dos veces al día |
Adalimumab 40 mg SC c/2 sem |
Xeljanz® 5 mg dos veces al día |
Xeljanz® 10 mg dos veces al día |
|
Na |
107 |
104 |
106 |
129 |
132 |
|
Mes 3 |
-0.17* |
-0.22* |
-0.20* |
-0.25* |
-0.22* |
a N es la cantidad total de pacientes en el análisis estadístico.
b Respuesta inadecuada al menos a un FARME sintético convencional (FARMEsc) por falta de eficacia y/o intolerabilidad.
c Respuesta inadecuada al menos a un inhibidor del TNF (TNFi) por falta de eficacia y/o intolerabilidad.
* p ≤ 0.05 para el tratamiento activo frente al placebo.
La tasa de respuesta al HAQ-DI (definida como una disminución de ≥ 0.35 con respecto al periodo inicial) en el mes 3 en los Estudios PsA-I y PsA-II fue del 53% y 50%, respectivamente, en los pacientes que recibieron Xeljanz® 5 mg dos veces al día, del 55% y 41%, respectivamente, en los pacientes que recibieron Xeljanz® 10 mg dos veces al día, del 31% y 28%, respectivamente, en los pacientes que recibieron el placebo, y del 53% en los pacientes que recibieron adalimumab 40 mg por vía subcutánea una vez cada dos semanas (solamente Estudio PsA-I).
Otros resultados relacionados con la salud: El estado de salud general se evaluó mediante la encuesta de salud de Formulario corto (SF-36). En los Estudios PsA-I y PsA-II, los pacientes que recibieron Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día tuvieron una mayor mejora con respecto al periodo inicial en comparación con el placebo en la puntuación del Resumen del componente físico (PCS) y en la puntuación del dominio de funcionamiento físico, y no empeoraron en la puntuación del Resumen del componente mental (MCS, por sus siglas en inglés) en el mes 3. Las mejoras en el SF-36 se mantuvieron hasta el mes 6 (Estudios PsA-I y PsA-II) y hasta el mes 12 (Estudio PsA-I).
Los resultados de salud relacionados con la fatiga se evaluaron mediante la Evaluación funcional de la terapia de la Enfermedad crónica - Fatiga (FACIT-F, por sus siglas en inglés). En los Estudios PsA-I y PsA-II, los pacientes que recibieron Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día presentaron una mayor mejoría con respecto al periodo inicial en comparación con el placebo en la puntuación total de la FACIT-F, la puntuación del dominio de experiencia y la puntuación del dominio de impacto en el mes 3.
Las mejoras en la FACIT-F se mantuvieron hasta el mes 6 (Estudios PsA-I y PsA II) y hasta el mes 12 (Estudio PsA-I).
Respuesta radiográfica: En el Estudio PsA-I, la progresión del daño estructural de la articulación se evaluó radiográficamente utilizando la Puntuación total de Sharp modificada (mTSS, por sus siglas en inglés) de Van der Heijde y se evaluó la proporción de pacientes con progresión radiográfica (aumento de la mTSS con respecto al periodo inicial superior a 0.5) en el mes 12. En el mes 12, el 96%, 95% y 98% de los pacientes que recibieron Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y adalimumab 40 mg SC cada 2 semanas, respectivamente, no presentaron progresión radiográfica (aumento de la mTSS con respecto al periodo inicial menor o igual a 0.5).
Colitis ulcerosa: El programa de desarrollo clínico de Fase 3 de Xeljanz® para la indicación de colitis ulcerosa incluyó 3 estudios de confirmación (Estudio UC-I, Estudio UC-II y Estudio UC-III) y un estudio de extensión a largo plazo abierto (Estudio UC-IV).
Estudios de confirmación: La seguridad y la eficacia de Xeljanz® para inducir y mantener la remisión se evaluaron en 3 estudios multicéntricos, doble ciego, aleatorizados y controlados con placebo: 2 estudios de inducción idénticos (8 semanas de duración; Estudio UC-I y Estudio UC-II) y 1 estudio de mantenimiento (52 semanas de duración; Estudio UC-III). Estos estudios pivotales incluyeron a pacientes adultos con colitis ulcerosa activa de moderada a severa (puntuación total de la Clínica Mayo de 6 a 12, con una subpuntuación de endoscopía de al menos 2, y una subpuntuación de sangrado rectal de al menos 1) y que tuvieron un fracaso con al menos 1 de los siguientes tratamientos o eran intolerantes a ellos: corticosteroides orales o intravenosos, azatioprina, 6-MP o inhibidor del TNF. La actividad de la enfermedad se evaluó mediante el índice de puntuación de Mayo (de 0 a 12), que constaba de 4 subpuntuaciones (de 0 a 3 para cada subpuntuación): frecuencia de las deposiciones, sangrado rectal, hallazgos en la endoscopía y evaluación global del médico. Un eritema marcado, ausencia de patrón vascular, friabilidad y erosiones se definió como subpuntuación de 2 en la endoscopía; un sangrado espontáneo y ulceración se definió como una subpuntuación de 3 en la endoscopía. Los pacientes que completaron el Estudio UC-I o el Estudio UC-II y lograron una respuesta clínica fueron elegibles para ser realeatorizados al Estudio UC-III.
Durante los estudios, se permitió que los pacientes utilizaran dosis estables de aminosalicilatos orales y corticosteroides (dosis diaria equivalente de prednisolona de hasta 25 mg). La reducción de los corticosteroides fue necesaria en el ingreso al Estudio UC-III. Xeljanz® se administró como monoterapia (es decir, sin administración concomitante de productos biológicos ni inmunosupresores) para la colitis ulcerosa durante los estudios.
Además de los estudios anteriores, también se evaluó la seguridad y la eficacia de Xeljanz® en un estudio de extensión a largo plazo abierto (Estudio UC-IV). Los pacientes que completaron 1 de los estudios de inducción (Estudio UC-I o Estudio UC-II), pero que no lograron una respuesta clínica, o los pacientes que completaron o se retiraron anticipadamente debido al fracaso del tratamiento en el estudio de mantenimiento (Estudio UC-III) fueron elegibles para el Estudio UC-IV. Los pacientes del Estudio UC-I o del Estudio UC-II que no lograron una respuesta clínica después de 8 semanas en el Estudio UC-IV fueron excluidos del Estudio UC-IV. Se requirió la reducción de los corticosteroides luego del ingreso al Estudio UC-IV.
Estudios de inducción (Estudio UC-I y Estudio UC-II): En el Estudio UC-I y el Estudio UC-II, 1139 pacientes se aleatorizaron (598 y 541 pacientes respectivamente) a Xeljanz® 10 mg dos veces al día o al placebo con una proporción de asignación de tratamiento de 4:1. En el Estudio UC-I y el Estudio UC-II, el 51.7%, 73.2% y 71.9% de los pacientes tuvieron un fracaso anterior o eran intolerantes a los inhibidores del TNF (51.3% en el Estudio UC-I y 52.1% en el Estudio UC-II), a los corticosteroides (74.9% en el Estudio UC-I y 71.3% en el Estudio UC-II), y/o a los inmunosupresores (74.1% en el Estudio UC-I y 69.5% en el Estudio UC-II), respectivamente. En el periodo inicial, el 46.1% de los pacientes recibía corticoides orales como tratamiento concomitante para la colitis ulcerosa (45.5% en el Estudio UC-I y 46.8% en el Estudio UC-II). Las características clínicas iniciales fueron en general similares entre los pacientes tratados con Xeljanz® y los que recibieron el placebo.
El criterio primario de valoración del Estudio UC-I y del Estudio UC-II fue la proporción de pacientes en remisión en la semana 8, y el criterio secundario de valoración clave fue la proporción de pacientes con mejora del aspecto endoscópico de la mucosa en la Semana 8. La remisión se definió como una remisión clínica (una puntuación total de Mayo ≤ 2 sin ninguna subpuntuación individual > 1) y una subpuntuación de sangrado rectal de 0. La mejora del aspecto endoscópico de la mucosa se definió como una subpuntuación de endoscopía de 0 o 1.
En la Tabla 9 se muestran los resultados de eficacia del Estudio UC-I y del Estudio UC-II con base en los resultados de la endoscopía leída centralmente. Una proporción significativamente mayor de pacientes tratados con Xeljanz® 10 mg dos veces al día alcanzó la remisión, la mejora del aspecto endoscópico de la mucosa y la respuesta clínica en la semana 8, en comparación con el placebo en ambos estudios.
Tabla 9. Proporción de pacientes que cumplieron con los criterios de valoración de la eficacia en la semana 8
(Estudio de inducción UC-I y Estudio UC-II, Endoscopía leída centralmente)
|
Estudio UC-I |
||||
|
Criterio de valoración |
Placebo |
Xeljanz® 10 mg |
Diferencia del tratamiento (IC del 95%) |
Valor p |
|
N = 122 |
N = 476 |
|||
|
Remisión en la semana 8a |
8.2% |
18.5% |
10.3% (4.3, 16.3) |
0.0070 |
|
Mejora del aspecto endoscópico de la mucosa en la semana 8b |
15.6% |
31.3% |
15.7% (8.1, 23.4) |
0.0005 |
|
Normalización del aspecto endoscópico de la mucosa en la semana 8c |
1.6% |
6.7% |
5.1% (1.9, 8.3) |
0.0345 |
|
Respuesta clínica en la semana 8d |
32.8% |
59.9% |
27.1% (17.7, 36.5) |
< 0.0001 |
|
Estudio UC-II |
||||
|
Criterio de valoración |
Placebo |
Xeljanz® 10 mg |
Diferencia del tratamiento (IC del 95%) |
Valor p |
|
N = 112 |
N = 429 |
|||
|
Remisión en la semana 8a |
3.6% |
16.6% |
13.0% (8.1, 17.9) |
0.0005 |
|
Mejora del aspecto endoscópico de la mucosa en la semana 8b |
11.6% |
28.4% |
16.8% (9.5, 24.1) |
0.0002 |
|
Normalización del aspecto endoscópico de la mucosa en la semana 8c |
1.8% |
7.0% |
5.2% (1.8, 8.6) |
0.0425 |
|
Respuesta clínica en la semana 8d |
28.6% |
55.0% |
26.4% (16.8, 36.0) |
< 0.0001 |
IC = intervalo de confianza; N = cantidad de pacientes en el conjunto de análisis.
a La remisión se definió como la remisión clínica (una puntuación de Mayo ≤ 2 sin ninguna subpuntuación individual > 1) y una subpuntuación de sangrado rectal de 0.
b La mejora del aspecto endoscópico de la mucosa se definió como una subpuntuación de la endoscopía de Mayo de 0 (enfermedad normal o inactiva) o 1 (eritema, patrón vascular disminuido).
c La normalización del aspecto endoscópico de la mucosa se definió como una subpuntuación endoscópica de Mayo de 0.
d La respuesta clínica se definió como una disminución con respecto al valor inicial de la puntuación Mayo de ≥ 3 puntos y ≥ 30%, con una disminución concomitante de la subpuntuación del sangrado rectal de ≥ 1 punto o una subpuntuación absoluta de sangrado rectal de 0 o 1.
Los resultados de eficacia con base en las lecturas endoscópicas en los centros del estudio fueron coherentes con los resultados con base en las endoscopías leídas centralmente.
Los resultados de los estudios individuales, Estudio UC-I y Estudio UC-II, fueron similares. Los datos agrupados proporcionan una estimación más precisa de la diferencia de tratamiento entre Xeljanz® 10 mg dos veces al día y el placebo en la semana 8: el 11.6% (IC del 95%, 7.7% - 15.5%) en remisión y el 16.3% (IC del 95%, 11.0% - 21.6%) con mejora del aspecto endoscópico de la mucosa.
En ambos subgrupos de pacientes con o sin fracaso previo de un inhibidor del TNF, una mayor proporción de pacientes tratados con Xeljanz® 10 mg dos veces al día alcanzó la remisión y la mejora del aspecto endoscópico de la mucosa en la semana 8 en comparación con el placebo. Esta diferencia de tratamiento fue coherente entre los 2 subgrupos (Tabla 10).
Tabla 10. Proporción de pacientes que cumplieron con los criterios primarios y secundarios de valoración de la eficacia clave en la semana 8 por subgrupos de tratamiento con inhibidores del TNF (Estudio de inducción UC-I y Estudio UC-II, Endoscopías leídas centralmente)
|
Estudio UC-I |
|||
|
Criterio de valoración |
Placebo N = 122 |
Xeljanz® 10 mg Dos veces al día |
Diferencia del Tratamiento (IC del 95%) |
|
Remisión en la semana 8a |
|||
|
Con fracaso previo de un inhibidor del TNF |
1.6% |
11.1% |
9.5% |
|
Sin fracaso previo de un inhibidor del TNFb |
15.5% |
26.2% |
10.7% |
|
Mejora del aspecto endoscópico de la mucosa en la semana 8c |
|||
|
Con fracaso previo de un inhibidor del TNF |
6.3% |
22.6% |
16.4% |
|
Sin fracaso previo de un inhibidor del TNFb |
25.9% |
40.3% |
14.5% |
|
Estudio UC-II |
|||
|
Criterio de valoración |
Placebo |
Xeljanz® 10 mg |
Diferencia del tratamiento (IC del 95%) |
|
Remisión en la semana 8a |
|||
|
Con fracaso previo de un inhibidor del TNF |
0.0% |
11.7% |
11.7% |
|
Sin fracaso previo de un inhibidor del TNFb |
7.7% |
21.7% |
14.0% |
|
Mejora del aspecto endoscópico de la mucosa en la semana 8c |
|||
|
Con fracaso previo de un inhibidor del TNF |
6.7% |
21.6% |
15.0% |
|
Sin fracaso previo de un inhibidor del TNFb |
17.3% |
35.7% |
18.4% |
IC = Intervalo de confianza; N = Cantidad de pacientes en el conjunto de análisis; TNF = Factor de necrosis tumoral.
a La remisión se definió como la remisión clínica (una puntuación de Mayo ≤ 2 sin ninguna subpuntuación individual > 1) y una subpuntuación de sangrado rectal de 0.
b Se incluyó a pacientes que no habían recibido inhibidores del TNF.
c La mejora del aspecto endoscópico de la mucosa se definió como una subpuntuación de la endoscopía de Mayo de 0 (enfermedad normal o inactiva) o 1 (eritema, patrón vascular disminuido).
Los cambios con respecto al periodo inicial en las subpuntuaciones de sangrado rectal y frecuencia de las deposiciones se evaluaron en cada visita del Estudio UC-I y del Estudio UC-II, y se muestran en la Figura 4. Se observaron mejoras significativas con respecto al periodo inicial en el sangrado rectal y la frecuencia de las deposiciones en los pacientes tratados con Xeljanz®, en comparación con el placebo. Las diferencias de tratamiento en el sangrado rectal y la frecuencia de las deposiciones entre Xeljanz® 10 mg dos veces al día y el placebo fueron significativas tan pronto como en la semana 2, la primera visita programada del estudio, y en cada visita posterior.
Figura 4. Cambios con respecto al periodo inicial en las subpuntuaciones del sangrado rectal y la frecuencia de las deposiciones (Estudio UC-I y Estudio UC-II)
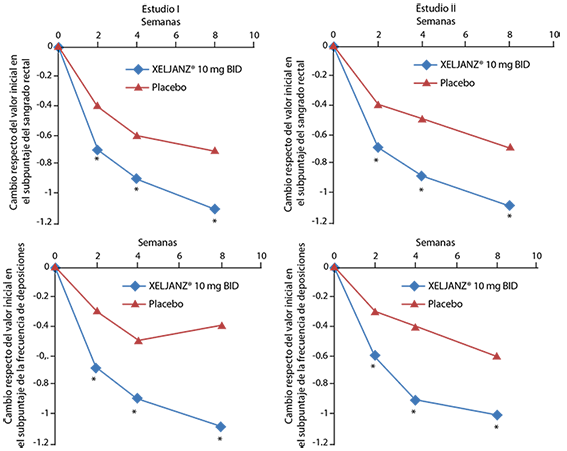
BID = dos veces al día.
* p < 0.001 para Xeljanz® 10 mg dos veces al día en comparación con el placebo.
En la Figura 5 se muestran los cambios en la puntuación parcial de Mayo con respecto al periodo inicial, a partir de los datos agrupados del Estudio UC-I y del Estudio UC-II. Se observaron mejoras significativas con respecto al periodo inicial en la puntuación parcial de Mayo en los pacientes tratados con Xeljanz® 10 mg dos veces al día, en comparación con el placebo en cada visita del estudio. Las diferencias del tratamiento en la puntuación parcial de Mayo entre Xeljanz® 10 mg dos veces al día y el placebo fueron significativas tan pronto como en la semana 2, de forma similar al sangrado rectal y la frecuencia de las deposiciones observadas en los estudios de inducción individuales.
Figura 5. Cambio con respecto al periodo inicial en la puntuación parcial de Mayo
(Datos agrupados de los Estudios UC-I y UC-II)
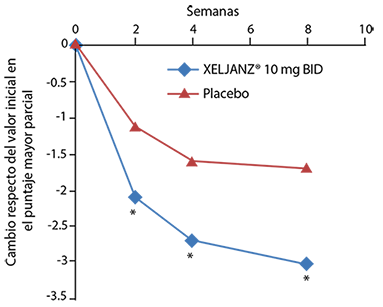
BID = dos veces al día.
* p < 0.001 para Xeljanz® 10 mg dos veces al día, en comparación con el placebo.
Estudio de mantenimiento (Estudio UC-III): Un total de 593 pacientes que completaron 8 semanas en uno de los estudios de inducción y que lograron una respuesta clínica entraron en el Estudio UC-III; 179 (30.2%) pacientes estaban en remisión en el periodo inicial del Estudio UC-III. Los pacientes se realeatorizaron a Xeljanz® 5 mg dos veces al día, a Xeljanz® 10 mg dos veces al día o al placebo durante 52 semanas con una proporción de asignación de tratamiento de 1:1:1. La reducción de los corticosteroides fue obligatoria para los pacientes que recibían corticosteroides en el periodo inicial.
En el periodo inicial del Estudio UC-III, 289 (48.7%) pacientes recibían corticosteroides orales; 265 (44.7%), 445 (75.0%) y 413 (69.6%) pacientes habían tenido un fracaso previo del tratamiento con inhibidores del TNF, corticosteroides e inmunosupresores, respectivamente, o eran intolerantes a ellos.
El criterio primario de valoración del Estudio UC-III fue la proporción de pacientes en remisión en la semana 52. Hubo dos criterios secundarios de valoración clave: la proporción de pacientes con mejora del aspecto endoscópico en la semana 52 y la proporción de pacientes con remisión sostenida sin corticosteroides tanto en la semana 24 como en la semana 52 entre los pacientes en remisión en el periodo inicial del Estudio UC-III.
En la Tabla 11 se resumen los resultados de eficacia del Estudio UC-III con base en los resultados de la endoscopía leída centralmente. Una proporción significativamente mayor de pacientes en los grupos de tratamiento con Xeljanz® 5 mg dos veces al día y Xeljanz® 10 mg dos veces al día alcanzó los siguientes criterios de valoración en la semana 52, en comparación con el placebo: remisión, mejora del aspecto endoscópico de la mucosa, normalización del aspecto endoscópico de la mucosa, mantenimiento de la respuesta clínica, remisión entre los pacientes en remisión en el periodo inicial, y remisión sostenida sin corticosteroides tanto en la semana 24 como en la semana 52 entre los pacientes en remisión en el periodo inicial.
Tabla 11. Proporción de pacientes que cumplieron con los criterios de valoración de la eficacia en el estudio de mantenimiento UC-III (lectura de endoscopía central)
|
Diferencia del Tratamiento frente al Placebo (IC del 95%) |
|||||
|
Criterio de valoración |
Placebo |
Xeljanz® 5 mg |
Xeljanz® |
Xeljanz® 5 mg |
Xeljanz® 10 mg dos veces al día |
|
N = 198 |
N = 198 |
N = 197 |
|||
|
Remisión en la semana 52a |
11.1% |
34.3% |
40.6% |
23.2%* |
29.5%* |
|
Mejora del aspecto endoscópico de la mucosa en la semana 52b |
13.1% |
37.4% |
45.7% |
24.2%* |
32.6%* |
|
Normalización del aspecto endoscópico de la mucosa en la semana 52c |
4.0% |
14.6% |
16.8% |
10.6%** |
12.7%* |
|
Mantenimiento de la respuesta clínica en la semana 52d |
20.2% |
51.5% |
61.9% |
31.3%* |
41.7%* |
|
Remisión en la semana 52 entre los pacientes en remisión en el periodo iniciala,f |
10.2% |
46.2% |
56.4% |
36.0%* |
46.2%* |
|
Remisión sostenida sin corticosteroides tanto en la semana 24 como en la semana 52 entre los pacientes en remisión en el periodo iniciale,f |
5.1% |
35.4% |
47.3% |
30.3%* |
42.2%* |
|
Criterio de valoración |
Placebo |
Xeljanz® 5 mg |
Xeljanz® 10 mg |
Xeljanz® 5 mg |
Xeljanz® 10 mg |
|
N = 101 |
N = 101 |
N = 87 |
|||
|
Remisión sin corticosteroides en la semana 52 entre los que tomaban corticosteroides en el periodo inicial |
10.9% |
27.7% |
27.6% |
16.8% |
16.7% |
* p < 0.0001.
** p < 0.001, para Xeljanz® frente al placebo.
IC = intervalo de confianza; N = cantidad de pacientes en el conjunto de análisis.
a La remisión se definió como la remisión clínica (una puntuación de Mayo ≤ 2 sin ninguna subpuntuación individual > 1) y una subpuntuación de sangrado rectal de 0.
b La mejora del aspecto endoscópico de la mucosa se definió como una subpuntuación de la endoscopía de Mayo de 0 (enfermedad normal o inactiva) o 1 (eritema, patrón vascular disminuido).
c La normalización del aspecto endoscópico de la mucosa se definió como una subpuntuación endoscópica de Mayo de 0.
d El mantenimiento de la respuesta clínica se definió por una disminución de la puntuación Mayo inicial del estudio de inducción (UC-I, UC-II) de ≥ 3 puntos y ≥ 30%, con una disminución concomitante de la subpuntuación de sangrado rectal de ≥ 1 punto o de la subpuntuación de sangrado rectal de 0 o 1. Los pacientes debían estar en respuesta clínica en el periodo inicial del Estudio UC-III de mantenimiento.
e La remisión sostenida sin corticosteroides se definió como estar en remisión y no tomar corticosteroides durante al menos 4 semanas antes de la visita, tanto en la semana 24 como en la semana 52.
f N = 59 para el placebo, N = 65 para Xeljanz® 5 mg dos veces al día, N = 55 para Xeljanz® 10 mg dos veces al día.
En ambos subgrupos de pacientes con o sin fracaso previo de un inhibidor del TNF, una mayor proporción de pacientes tratados con Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día alcanzaron los siguientes criterios de valoración en la semana 52 del Estudio UC-III, en comparación con el placebo: remisión, mejora del aspecto endoscópico de la mucosa, o remisión sostenida sin corticosteroides tanto en la semana 24 como en la semana 52 entre los pacientes en remisión en el periodo inicial (Tabla 12). Esta diferencia de tratamiento con respecto al placebo fue similar entre Xeljanz® 5 mg dos veces al día y Xeljanz® 10 mg dos veces al día en el subgrupo de pacientes sin fracaso previo de un inhibidor del TNF. En el subgrupo de pacientes con fracaso previo del inhibidor del TNF, la diferencia de tratamiento observada con respecto al placebo fue numéricamente mayor para Xeljanz® 10 mg dos veces al día que para Xeljanz® 5 mg dos veces al día, entre 9.7 y 16.7 puntos porcentuales en los criterios primarios y secundarios de valoración clave.
Tabla 12. Proporción de pacientes que cumplieron con los criterios primarios y secundarios clave de valoración de la eficacia en el Estudio UC-III de mantenimiento por subgrupo de tratamiento con inhibidores del TNF (lectura de endoscopía central)
|
Diferencia del tratamiento frente al placebo (IC del 95%) |
|||||
|
Criterio de valoración |
Placebo |
Xeljanz® 5 mg |
Xeljanz® 10 mg dos veces al día |
Xeljanz® 5 mg |
Xeljanz® 10 mg |
|
N = 198 |
N = 198 |
N = 197 |
|||
|
Remisión en la semana 52a |
|||||
|
Con fracaso previo de un inhibidor del TNF |
10/89 |
20/83 |
34/93 |
12.9% |
25.3% |
|
Sin fracaso previo de un inhibidor del TNFb |
12/109 |
48/115 |
46/104 |
30.7% |
33.2% |
|
Mejora del aspecto endoscópico de la mucosa en la semana 52c |
|||||
|
Con fracaso previo de un inhibidor del TNF |
11/89 |
25/83 |
37/93 |
17.8% |
27.4% |
|
Sin fracaso previo de un inhibidor del TNFb |
15/109 |
49/115 |
53/104 |
28.8% |
37.2% |
|
Remisión sostenida sin corticosteroides tanto en la semana 24 como en la semana 52 entre los pacientes en remisión en el periodo iniciald |
|||||
|
Con fracaso previo de un inhibidor del TNF |
1/21 |
4/18 |
7/18 |
17.5% |
34.1% |
|
Sin fracaso previo de un inhibidor del TNFb |
2/38 |
19/47 |
19/37 |
35.2% |
46.1% |
IC = intervalo de confianza; N = cantidad de pacientes en el conjunto de análisis; TNF = factor de necrosis tumoral.
a La remisión se definió como la remisión clínica (una puntuación de Mayo ≤ 2 sin ninguna subpuntuación individual > 1) y una subpuntuación de sangrado rectal de 0.
b Se incluyó a pacientes que no habían recibido inhibidores del TNF.
c La mejora del aspecto endoscópico de la mucosa se definió como una subpuntuación de la endoscopía de Mayo de 0 (enfermedad normal o inactiva) o 1 (eritema, patrón vascular disminuido).
d La remisión sostenida sin corticosteroides se definió como estar en remisión y no tomar corticosteroides durante al menos 4 semanas antes de la visita, tanto en la semana 24 como en la semana 52.
En la Figura 6, se muestra la proporción de pacientes que cumplieron con los criterios de fracaso del tratamiento en el tiempo en el Estudio UC-III de mantenimiento. El fracaso del tratamiento se definió como un aumento de la puntuación de Mayo de al menos 3 puntos con respecto al periodo inicial del estudio de mantenimiento, acompañado de un aumento de la subpuntuación de sangrado rectal de al menos 1 punto, y un aumento de la subpuntuación endoscópica de al menos 1 punto, lo que supone una subpuntuación endoscópica absoluta de al menos 2 tras un tratamiento mínimo de 8 semanas en el estudio. La subpuntuación endoscópica leída centralmente se utilizó para determinar el fracaso del tratamiento.
La proporción de pacientes de ambos grupos de Xeljanz® que presentaron un fracaso del tratamiento fue menor, en comparación con el placebo, en cada punto temporal tan pronto como en la semana 8, el primer punto temporal en el que se evaluó el fracaso del tratamiento.
Figura 6. Tiempo hasta el fracaso del tratamiento del Estudio UC-III de mantenimiento (curvas de Kaplan-Meier)
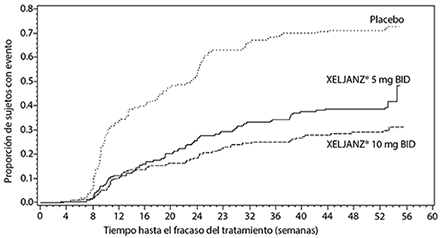
BID = dos veces al día.
P < 0.0001 para Xeljanz® 5 mg dos veces al día frente al placebo.
P < 0.0001 para Xeljanz® 10 mg dos veces al día frente al placebo.
Otros resultados relacionados con la salud: El estado de salud general se evaluó mediante la encuesta de salud de formulario corto (SF-36). En el Estudio UC-I de inducción y el Estudio UC-II, los pacientes que recibieron Xeljanz® 10 mg dos veces al día demostraron una mayor mejora con respecto al periodo inicial, en comparación con el placebo, en las puntuaciones del resumen del componente físico (PCS) y del resumen del componente mental (MCS, por sus siglas en inglés), y en los 8 dominios del SF-36. En el Estudio UC-III de mantenimiento, los pacientes que recibieron Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día demostraron un mayor mantenimiento de la mejora, en comparación con el placebo, en las puntuaciones del PCS y el MCS, y en los 8 dominios del SF-36 en la semana 24 y en la semana 52.
El estado de salud específico de la enfermedad se evaluó con el Cuestionario de la enfermedad inflamatoria intestinal (IBDQ, por sus siglas en inglés) en pacientes con colitis ulcerosa. En el Estudio UC-I de inducción y en el Estudio UC-II, los pacientes que recibieron Xeljanz® 10 mg dos veces al día demostraron una mayor mejoría con respecto al periodo inicial, en comparación con el placebo, en las puntuaciones totales y de los 4 dominios del IBDQ en la semana 8. En el Estudio UC-III de mantenimiento, los pacientes que recibieron Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día demostraron un mayor mantenimiento de la mejora, en comparación con el placebo, en las puntuaciones totales y de los 4 dominios del IBDQ en la semana 24 y en la semana 52.
Las utilidades del estado de salud se evaluaron con el Cuestionario EuroQoL de 5 dimensiones (EQ-5D) en pacientes con colitis ulcerosa. En el Estudio UC-I de inducción, los pacientes que recibieron Xeljanz® 10 mg dos veces al día demostraron una mayor mejoría con respecto al periodo inicial, en comparación con el placebo, en la puntuación de utilidad en la semana 8; en el Estudio UC-II de inducción, los pacientes que recibieron Xeljanz® 10 mg dos veces al día no demostraron una mayor mejoría con respecto al periodo inicial, en comparación con el placebo, en la puntuación de utilidad en la Semana 8. En el estudio UC-III de mantenimiento, los pacientes que recibieron Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día demostraron un mayor mantenimiento de la mejora con respecto al periodo inicial, en comparación con el placebo, en las puntuaciones de utilidad del estado de salud del EQ-5D en la semana 24 y en la semana 52.
La productividad laboral y el deterioro de la actividad (WPAI, por sus siglas en inglés) se evaluaron con el Cuestionario WPAI en la colitis ulcerosa (WPAI-CU) en pacientes con colitis ulcerosa. En el Estudio UC-I de inducción, los pacientes que recibieron Xeljanz® 10 mg dos veces al día demostraron una mayor mejoría con respecto al periodo inicial, en comparación con el placebo, en los dominios de presentismo, pérdida de productividad laboral y deterioro de la actividad no laboral, pero no en el dominio de absentismo en la semana 8; en el Estudio UC-II de inducción, los pacientes que recibieron Xeljanz® 10 mg dos veces al día demostraron una mayor mejoría con respecto al periodo inicial, en comparación con el placebo, en el dominio de deterioro de la actividad no laboral, pero no en los dominios de absentismo, presentismo o pérdida de productividad laboral en la semana 8. En el Estudio UC-III de mantenimiento, los pacientes que recibieron Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día demostraron un mayor mantenimiento de la mejoría con respecto al periodo inicial, en comparación con el placebo, en las puntuaciones de los dominios de presentismo y deterioro de la actividad no laboral, pero no en los dominios de absentismo o pérdida de productividad laboral en la semana 52.
Estudio de extensión abierto (Estudio UC-IV): En el Estudio UC-IV, se inscribieron un total de 944 pacientes, que se asignaron a Xeljanz® 5 mg dos veces al día (para los pacientes en remisión en el periodo inicial del Estudio UC-IV) o a Xeljanz® 10 mg dos veces al día (para todos los demás pacientes que entraron en el Estudio UC-IV).
Entre los pacientes que participaron en el Estudio UC-IV, 295 recibieron Xeljanz® 10 mg dos veces al día y no lograron una respuesta clínica en 1 de los estudios de inducción (Estudio UC-I o Estudio UC-II) y, luego, continuaron recibiendo Xeljanz® 10 mg dos veces al día en el Estudio UC-IV. Tras 8 semanas adicionales de tratamiento con Xeljanz® 10 mg dos veces al día (16 semanas en total), 154/293 (52.6%) pacientes lograron una respuesta clínica y 42/293 (14.3%) pacientes alcanzaron la remisión.
Además, 58 pacientes recibieron Xeljanz® 10 mg dos veces al día en el Estudio UC-I o en el Estudio UC-II y lograron una respuesta clínica, se les redujo la dosis a Xeljanz® 5 mg dos veces al día en el Estudio UC-III y presentaron un fracaso del tratamiento y, luego, se les aumentó la dosis a Xeljanz® 10 mg dos veces al día en el Estudio UC-IV. Después de 2 meses de tratamiento con Xeljanz® 10 mg dos veces al día en el Estudio UC-IV, se logró la remisión y la curación de la mucosa en 20/58 (34.5%) y 24/58 (41.4%) pacientes, respectivamente. En el mes 12 del Estudio UC-IV, 25/48 (52.1%) y 29/48 (60.4%) de estos pacientes alcanzaron la remisión y la curación de la mucosa, respectivamente. Además, hubo 65 pacientes inscritos durante al menos 1 año antes de la fecha de corte de los datos, el 8 de julio de 2016, en el Estudio UC-IV después de alcanzar la remisión al final del Estudio UC-III mientras recibían Xeljanz® 5 mg dos veces al día o Xeljanz® 10 mg dos veces al día. En el mes 12 del Estudio UC-IV, 48/65 (73.8%) de estos pacientes seguían en remisión mientras recibían Xeljanz® 5 mg dos veces al día.
CONTRAINDICACIONES:
Se recomienda no iniciar el tratamiento con XELJANZ® XR en pacientes con:
• Hipersensibilidad conocida al medicamento o a cualquier excipiente de la fórmula.
• Clara evidencia o resultados de laboratorio que indiquen síndromes de inmunodeficiencia.
• Infecciones activas severas.
• Insuficiencia hepática grave.
RESTRICCIONES DE USO DURANTE EL EMBARAZO Y LA LACTANCIA:
No existen estudios adecuados y bien controlados sobre la administración de XELJANZ® XR en mujeres embarazadas. Tofacitinib ha demostrado ser teratogénico en ratas y conejos, y tiene efectos sobre la fertilidad en ratas hembra, el parto y el desarrollo peri y posnatal (ver sección Precauciones en relación con efectos de carcinogénesis, mutagénesis, teratogénesis y sobre fertilidad). No debe administrarse XELJANZ® XR durante el embarazo a menos que sea claramente necesario.
Se recomienda a las mujeres con capacidad reproductiva que empleen un método anticonceptivo efectivo durante el tratamiento con XELJANZ® XR y durante al menos 4 semanas después de la última dosis.
Según los datos publicados, tofacitinib está presente en la leche materna. La información sobre los efectos de tofacitinib en el lactante a partir de la literatura publicada y los datos posteriores a la comercialización se limita a un pequeño número de casos sin efectos adversos relacionados causalmente. No existen datos sobre los efectos en la producción de leche. Las mujeres no deben amamantar durante el tratamiento con XELJANZ® XR.
REACCIONES SECUNDARIAS Y ADVERSAS:
Artritis reumatoide: Los siguientes datos incluyen 6 estudios doble ciego, controlados, multicéntricos de distintas duraciones de entre 6 y 24 meses (Estudios I a VI; ver sección Farmacocinética y farmacodinamia). En estos estudios, se asignó aleatoriamente y se trató a 3200 pacientes con monoterapia de Xeljanz® 5 mg dos veces por día (616 pacientes) o 10 mg dos veces por día (642 pacientes) y con Xeljanz® 5 mg dos veces al día (973 pacientes) o 10 mg dos veces al día (969 pacientes) en combinación con FARME (incluyendo, metotrexato).
Todos los pacientes en estos estudios tenían artritis reumatoide de moderada a severa. La población del estudio con Xeljanz® tenía una edad media de 52.1 años y un 83.2% eran mujeres.
La población de seguridad a largo plazo incluye todos los pacientes que participaron en un estudio doble ciego controlado (incluidos los estudios en fase de desarrollo anteriores) y, luego, participaron en uno de los dos estudios de seguridad a largo plazo.
Se trató un total de 6194 pacientes (estudios de fase 1, 2, 3 y de extensión a largo plazo) con cualquier dosis de Xeljanz®, con una duración media de 3.13 años, y se acumularon 19405.8 años-paciente de exposición total al medicamento con base en más de 8 años de exposición continua a Xeljanz®.
También se incluye información sobre la seguridad de un estudio (N = 4362) de seguridad posterior a la autorización aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (factores de riesgo CV definidos como: tabaquismo actual, diagnóstico de hipertensión, diabetes mellitus, antecedentes familiares de enfermedad coronaria prematura, antecedentes de enfermedad arterial coronaria, incluidos los antecedentes de procedimiento de revascularización, injerto de derivación arterial coronaria, infarto de miocardio, paro cardiaco, angina inestable, síndrome coronario agudo y presencia de enfermedad extraarticular asociada a la AR, p. ej., nódulos, síndrome de Sjögren, anemia de la enfermedad crónica, manifestaciones pulmonares), y que estaban con una dosis de fondo estable de metotrexato. La mayoría (más del 90%) de los pacientes con tofacitinib que eran fumadores actuales o exfumadores tenían una duración de tabaquismo de más de 10 años y una mediana de 35.0 y 39.0 años de tabaquismo, respectivamente.
Los pacientes se aleatorizaron de forma abierta a tofacitinib 10 mg dos veces al día, tofacitinib 5 mg dos veces al día, o a un inhibidor del TNF (el inhibidor del TNF era etanercept 50 mg una vez a la semana o adalimumab 40 mg cada dos semanas) en una proporción de 1:1:1. Los criterios coprimarios de valoración son la neoplasia maligna adjudicada (excluido el NMSC) y los MACE adjudicados; la incidencia acumulada y la evaluación estadística de los criterios de valoración eran ciegas. El estudio es uno impulsado por eventos que también requiere el seguimiento de al menos 1500 pacientes durante 3 años. Se interrumpió el tratamiento del estudio con tofacitinib 10 mg dos veces al día y se cambió a los pacientes a 5 mg dos veces al día debido a una señal de EP dependiente de la dosis.
Artritis psoriásica: Xeljanz® 5 mg dos veces al día y 10 mg dos veces al día se estudiaron en 2 ensayos clínicos de fase 3 doble ciego realizados en pacientes con artritis psoriásica activa (PsA por sus siglas en inglés).
El Estudio PsA-I tuvo una duración de 12 meses e incluyó a 422 pacientes con una respuesta inadecuada a un FARMEsc y que no habían recibido tratamiento con un FARME biológico inhibidor del TNF (TNFi). El Estudio PsA-I incluyó un periodo controlado con placebo de 3 meses y también incluyó adalimumab 40 mg por vía subcutánea una vez cada 2 semanas durante 12 meses. El Estudio PsA-II tuvo una duración de 6 meses e incluyó a 394 pacientes con una respuesta inadecuada a al menos un TNFi aprobado. El Estudio PsA-II incluyó un periodo de 3 meses controlado con placebo. Todos los pacientes de los ensayos clínicos debían recibir tratamiento con una dosis estable de un FARMEsc (la mayoría recibió metotrexato [78.2%]). En los ensayos clínicos de fase 3, los pacientes se aleatorizaron y trataron con Xeljanz® 5 mg dos veces al día (238 pacientes) o Xeljanz® 10 mg dos veces al día (236 pacientes). La población del estudio aleatorizada y tratada con Xeljanz® (474 pacientes) incluía 45 (9.5%) pacientes de 65 años o más y 66 (13.9%) pacientes con diabetes en el periodo inicial.
Se llevó a cabo un ensayo clínico abierto adicional a largo plazo que incluyó a 680 pacientes con artritis psoriásica que participaron originalmente en cualquiera de los 2 ensayos clínicos controlados doble ciego. Los pacientes que participaron en este ensayo clínico abierto se trataron inicialmente con Xeljanz® 5 mg dos veces al día. A partir del mes 1, se permitió el aumento escalonado de la dosis a Xeljanz® 10 mg dos veces al día a discreción del investigador; también se permitió la reducción posterior de la dosis a 5 mg dos veces al día. Esto limita la interpretación de los datos de seguridad a largo plazo con respecto a la dosis.
De los 783 pacientes (desde el 10 de mayo de 2016) que recibieron dosis de Xeljanz® de 5 mg dos veces al día o de 10 mg dos veces al día en los ensayos clínicos de artritis psoriásica, 665 recibieron tratamiento durante 6 meses o más, de los cuales 437 recibieron tratamiento durante un año o más, de los cuales 44 recibieron tratamiento durante 24 meses o más.
Colitis ulcerosa: Los siguientes datos de seguridad se basaron en 4 estudios aleatorios, doble ciego, controlados con placebo: 2 estudios de inducción de fase 3 de diseño idéntico (UC-I y UC-II), un estudio de mantenimiento de fase 3 (UC-III) y un estudio de inducción fase 2 de una dosis (UC-V). Los pacientes con colitis ulcerosa de actividad moderada a grave se inscribieron en los estudios de inducción de fase 2 y fase 3. En los estudios de inducción, los pacientes aleatorizados recibieron tratamiento con Xeljanz® 10 mg dos veces al día (938 pacientes combinados) o placebo (282 pacientes combinados) hasta por 8 semanas. Los pacientes que completaron el Estudio UC-I o el Estudio UC-II y lograron una respuesta clínica ingresaron al Estudio UC-III. En el Estudio UC-III, los pacientes fueron realeatorizados, de modo que 198 pacientes recibieron Xeljanz® 5 mg dos veces al día, 196 pacientes recibieron Xeljanz® 10 mg dos veces al día, y 198 pacientes recibieron placebo hasta por 52 semanas. Se prohibió el uso concomitante de inmunosupresores o productos biológicos durante estos estudios. Se permitieron dosis estables concomitantes de corticosteroides orales en los estudios de inducción, con disminución gradual de corticosteroides hasta la interrupción obligatoria dentro de las 15 semanas de haber ingresado al estudio de mantenimiento. Además de los estudios de inducción y mantenimiento, la seguridad a largo plazo fue evaluada en un estudio abierto de extensión a largo plazo (Estudio UC-IV).
Experiencia en ensayos clínicos: Las reacciones adversas graves más comunes en la artritis reumatoide y artritis psoriásica fueron infecciones graves (ver sección Precauciones generales).
En estudios de inducción y mantenimiento, en todos los grupos de tratamiento, las categorías más comunes de reacciones adversas graves en pacientes con colitis ulcerosa fueron infecciones y desórdenes intestinales.
Artritis reumatoide: En el caso de la artritis reumatoide, las reacciones adversas informadas con mayor frecuencia durante los primeros 3 meses en los ensayos clínicos controlados con placebo (que se produjeron en ≥ 2% de los pacientes tratados con Xeljanz® como monoterapia o en combinación con FARME) fueron dolor de cabeza, infecciones de las vías respiratorias superiores, nasofaringitis, hipertensión, náuseas y diarrea.
La proporción de pacientes que interrumpieron el tratamiento debido a cualquier tipo de reacción adversa durante los primeros 3 meses de los estudios doble ciego, controlados con placebo o con metotrexato fue del 3.8% en los pacientes a quienes se administró Xeljanz® y del 3.2% en los pacientes tratados con placebo. Las infecciones más comunes que causaron la interrupción del tratamiento fueron el herpes zóster y la neumonía.
Artritis psoriásica: En la artritis psoriásica activa, las reacciones adversas informadas con mayor frecuencia durante las primeras 12 semanas en los ensayos clínicos controlados con placebo (ocurrieron en ≥ 2% de los pacientes tratados con Xeljanz® y al menos 1% más que la tasa observada en pacientes con placebo) fueron bronquitis, diarrea, dispepsia, fatiga, dolor de cabeza, nasofaringitis, faringitis.
La proporción de pacientes que interrumpieron el tratamiento debido a alguna reacción adversa durante las primeras 12 semanas de los estudios doble ciego controlados con placebo fue de 3.2% para pacientes tratados con Xeljanz® y de 2.5% para pacientes tratados con placebo. La infección más común que dio como resultado la interrupción del tratamiento fue sinusitis.
En general, el perfil de seguridad observado en pacientes con artritis psoriásica activa tratados con Xeljanz® fue coherente con el perfil de seguridad en pacientes con artritis reumatoide.
Colitis ulcerosa: Las reacciones adversas que ocurrieron en por lo menos el 2% de los pacientes que recibían Xeljanz® 10 mg dos veces al día y por lo menos 1% más grande que las observadas en pacientes recibiendo placebo en los estudios de inducción (Estudio UC-I, Estudio UC-II y Estudio UC-V) fueron: incremento de la creatina fosfocinasa sérica, nasofaringitis, pirexia y dolor de cabeza.
En los estudios de inducción y mantenimiento, en todos los grupos de tratamiento, las categorías más comunes de reacciones adversas graves fueron desordenes gastrointestinales e infecciones, y la reacción adversa grave más común fue el empeoramiento de la colitis ulcerosa.
En los estudios clínicos controlados para la colitis ulcerosa, 1 caso de cáncer de mama fue reportado en un paciente tratado con placebo y no se observaron casos de neoplasias sólidas o linfomas en los pacientes tratados con Xeljanz®. También se observaron neoplasias malignas en el estudio de extensión a largo plazo en pacientes con colitis ulcerosa tratados con Xeljanz®, incluyendo cánceres sólidos y linfomas.
En los estudios de inducción y mantenimiento, la razón más frecuente para la discontinuación del estudio fue el empeoramiento de la colitis ulcerosa. Excluyendo las discontinuaciones debidas al empeoramiento de la colitis ulcerosa, la proporción de pacientes que discontinuaron el tratamiento debido a las reacciones adversas fue menor del 5% en cualquiera de los grupos de tratamiento de Xeljanz® o placebo en estos estudios.
En general, el perfil de seguridad observado en pacientes con colitis ulcerosa tratados con Xeljanz® fue consistente con el perfil de seguridad de Xeljanz® para las demás indicaciones.
Las reacciones adversas al medicamento (RAM) enumeradas en la siguiente tabla se presentan por clasificación por órganos y sistemas (SOC, por sus siglas en inglés); se presentan los efectos no deseados en orden decreciente de gravedad.
Tabla 13. Reacciones adversas al medicamento por clasificación por órganos y sistemas, y proporción de incidencia (AR, PsA y CU)a
|
Clasificación por órganos y sistemas |
Reacciones adversas |
Proporción de incidencia en AR (%) |
Proporción de incidencia en PsA (%) |
Proporción de incidencia en CU (%) |
|---|---|---|---|---|
|
Infecciones e infestaciones |
Nasofaringitis |
7.1 |
8.0 |
11.7 |
|
Infección del tracto urinario |
4.9 |
2.6 |
2.8 |
|
|
Bronquitis |
4.4 |
3.6 |
2.8 |
|
|
Herpes zóster |
3.3 |
1.0 |
3.1 |
|
|
Influenza |
2.2 |
1.2 |
2.8 |
|
|
Sinusitis |
2.1 |
2.2 |
2.0 |
|
|
Faringitis |
2.0 |
2.6 |
1.8 |
|
|
Neumonía |
1.4 |
0.6 |
0.3 |
|
|
Infección viral |
0.6 |
0.7 |
0.8 |
|
|
Herpes simple |
0.7 |
0.7 |
0.5 |
|
|
Gastroenteritis viral |
0.2 |
0.6 |
0.8 |
|
|
Celulitis |
0.8 |
0.3 |
– |
|
|
Pielonefritis |
0.2 |
0.1 |
0.3 |
|
|
Diverticulitis |
0.2 |
– |
– |
|
|
Tuberculosis |
0.2 |
– |
– |
|
|
Sepsis |
0.09 |
– |
– |
|
|
Artritis bacterianab |
0.07 |
– |
– |
|
|
Meningitis criptocócicac |
– |
– |
– |
|
|
Infección micobacteriana atípicac |
– |
– |
– |
|
|
Infección compleja por Mycobacterium aviumc |
– |
– |
– |
|
|
Tuberculosis de sistema nervioso centralc |
– |
– |
– |
|
|
Encefalitisc |
– |
– |
– |
|
|
Fascitis necrotizantec |
– |
– |
– |
|
|
Bacteriemiac |
– |
– |
– |
|
|
Bacteriemia estafilocócicac |
– |
– |
– |
|
|
Neumonía |
– |
– |
– |
|
|
Urosepsis |
0.02 |
– |
– |
|
|
Tuberculosis diseminada |
0.02 |
– |
– |
|
|
Neumonía Pneumocystis jirovecii |
0.02 |
– |
– |
|
|
Neumonía bacteriana |
0.02 |
– |
– |
|
|
Infección por Cytomegalovirus |
– |
– |
– |
|
|
Neoplasia benigna, maligna y no especificada (incluidos quistes y pólipos) |
Cánceres de piel no melanoma |
0.2 |
0.1 |
0.5 (Poco común) |
|
Trastornos hematológicos y del sistema linfático |
Anemia |
2.7 |
0.7 |
3.1 |
|
Leucopenia |
0.8 |
0.6 |
0.3 |
|
|
Neutropenia |
0.6 |
0.6 |
0.3 |
|
|
Linfopenia |
0.4 |
– |
– |
|
|
Trastornos del sistema inmunológico |
Hipersensibilidad al fármacoe |
0.9 |
0.9 |
1.0 |
|
Trastornos del metabolismo y nutrición |
Hiperlipidemia |
0.8 |
0.6 |
0.8 |
|
Dislipidemia |
1.1 |
0.9 |
0.5 |
|
|
Deshidratación |
0.2 |
0.3 |
– |
|
|
Trastornos psiquiátricos |
Insomnio |
1.0 |
0.7 |
1.0 |
|
Trastornos del sistema nervioso |
Dolor de cabeza |
5.3 |
5.8 |
5.8 |
|
Parestesia |
0.6 |
1.2 |
0.5 |
|
|
Trastornos vasculares |
Hipertensión |
4.7 |
2.6 |
2.0 |
|
Tromboembolismo venosof |
0.3 |
0.1 |
– |
|
|
Trastornos respiratorios, torácicos y mediastínicos |
Tos |
2.6 |
1.2 |
2.8 |
|
Disnea |
0.7 |
1.0 |
0.5 |
|
|
Congestión nasal |
0.5 |
– |
0.3 |
|
|
Trastornos gastrointestinales |
Diarrea |
4.3 |
4.1 |
3.1 |
|
Náusea |
4.1 |
3.2 |
2.3 |
|
|
Dispepsia |
2.7 |
1.7 |
1.3 |
|
|
Vómito |
2.1 |
0.4 |
2.3 |
|
|
Dolor abdominal |
1.7 |
1.9 |
3.1 |
|
|
Gastritis |
1.8 |
1.2 |
0.3 |
|
|
Trastornos hepatobiliares |
Esteatosis hepática |
0.6 |
0.4 |
– |
|
Trastornos de la piel y del tejido subcutáneo |
Erupción |
1.9 |
1.3 |
4.3 |
|
Acné |
0.7 |
1.1 |
1.5 |
|
|
Prurito |
0.5 |
0.3 |
0.5 |
|
|
Eritema |
0.3 |
0.9 |
– |
|
|
Trastornos musculoesqueléticos y del tejido conectivo |
Artralgia |
2.9 |
1.2 |
8.6 |
|
Inflamación articular |
0.5 |
0.4 |
0.3 |
|
|
Tendinitis |
0.4 |
– |
– |
|
|
Dolor musculoesquelético |
0.8 |
0.6 |
0.8 |
|
|
Trastornos generales y reacciones en el sitio de administración |
Edema periférico |
2.4 |
1.2 |
0.5 |
|
Fatiga |
1.1 |
1.7 |
3.1 |
|
|
Pirexia |
1.4 |
0.6 |
2.3 |
|
|
Investigaciones |
Aumento de creatina fosfocinasa sanguínea |
3.5 |
3.9 |
4.8 |
|
Aumento de peso |
2.2 |
1.2 |
1.8 |
|
|
Aumento de la gama-glutamil transferasa |
1.4 |
1.0 |
1.0 |
|
|
Aumento de colesterol en sangre |
0.7 |
0.3 |
1.0 |
|
|
Aumento de lipoproteínas de baja densidad |
0.3 |
– |
0.3 |
|
|
Aumento de enzimas hepáticas |
0.9 |
– |
– |
|
|
Aumento de creatinina en sangre |
0.7 |
0.1 |
– |
|
|
Aumento en las transaminasas |
0.6 |
0.1 |
– |
|
|
Prueba de función hepática anormal |
0.2 |
0.1 |
– |
|
|
Lesión, envenenamiento y complicaciones del procedimiento |
Esguince de ligamento |
0.6 |
0.7 |
0.8 |
|
Contractura muscular |
0.4 |
0.3 |
0.3 |
RAM = reacción adversa al medicamento; CPNM = cáncer de piel no melanoma; PsA = artritis psoriásica; PT = término preferido; AR = artritis reumatoide; CU = colitis ulcerosa.
a Las frecuencias se basan en datos agrupados de ensayos clínicos aleatorizados de fase 3 (excluyendo el Estudio A3921133).
b La frecuencia de la artritis bacteriana se determina mediante las frecuencias combinadas de los PT de artritis bacteriana y artritis infecciosa.
c Las RAM solo se informaron en estudios abiertos de extensión a largo plazo; por lo tanto, se estimó la frecuencia de estas RAM en los ensayos aleatorizados de fase 3.
d El CPNM identificado como RAM en 2013; CPNM no es un PT: la frecuencia se determina combinando frecuencias para PT de cáncer de células basales y cáncer de piel de células escamosas.
e Datos de informes espontáneos (se han observado eventos como angioedema y urticaria). También se observaron algunos eventos en los ensayos clínicos.
f Tromboembolia venosa (p. ej., embolia pulmonar y trombosis venosa profunda, trombosis venosa retinal).
* Para AR y PsA, tofacitinib 5 mg (10 mg) 2 veces al día incluye sujetos que fueron aleatorizados a tofacitinib 5 mg (10 mg) 2 veces al día y placebo cambiados al grupo de tofacitinib 5 mg (10 mg) 2 veces al día. Para los que cambiaron del grupo del placebo, el recuento de eventos comienza desde el día 1 de la dosis de tofacitinib. Los sujetos que fueron aleatorizados para recibir placebo cambiaron al grupo de tofacitinib 5 mg (10 mg) dos veces al día, interrumpieron el estudio durante el periodo de tratamiento con placebo y no recibieron tratamiento con tofacitinib, fueron excluidos del análisis.
– No se observó ningún evento.
Infecciones en general:
Artritis reumatoide: En los estudios clínicos controlados de fase 3 de 6 y 24 meses, las tasas de infecciones en el grupo de monoterapia con Xeljanz® 5 mg dos veces al día (en total, 616 pacientes) y de 10 mg dos veces al día (en total, 642 pacientes) fueron del 16.2% (100 pacientes) y del 17.9% (115 pacientes), respectivamente, en comparación con 18.9% (23 pacientes) en el grupo de placebo (en total, 122 pacientes). En los estudios de 6, 12 o 24 meses de duración con FARME como tratamiento de fondo, las tasas de infecciones en el grupo de 5 mg dos veces al día (en total, 973 pacientes) y 10 mg dos veces al día (en total, 969 pacientes) en el grupo de Xeljanz® más FARME fueron del 21.3% (207 pacientes) y del 21.8% (211 pacientes), respectivamente, en comparación con 18.4% (103 pacientes) en el grupo de placebo más FARME (en total, 559 pacientes).
Las infecciones informadas con mayor frecuencia fueron infecciones de las vías respiratorias superiores y nasofaringitis (3.7% y 3.2%, respectivamente).
La tasa total de infecciones con Xeljanz® en la población de seguridad a largo plazo a toda exposición (total de 4867 pacientes) fue de 46.1 pacientes con eventos cada 100 años-paciente (43.8 y 47.2 pacientes con eventos para 5 mg y 10 mg dos veces al día, respectivamente). En el caso de pacientes tratados con monoterapia (en total, 1750), las tasas fueron 48.9 y 41.9 pacientes con eventos cada 100 años-paciente para 5 mg y 10 mg dos veces al día, respectivamente. En el caso de los pacientes tratados con FARME como tratamiento de fondo (en total, 3117), las tasas fueron 41.0 y 50.3 pacientes con eventos cada 100 años-paciente para 5 mg y 10 mg dos veces al día, respectivamente.
También se notificaron infecciones en un PASS aleatorizado de gran tamaño en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia).
Artritis psoriásica: En los estudios controlados de fase 3 de 6 y hasta 12 meses, la frecuencia de infecciones en los grupos con Xeljanz® 5 mg dos veces al día (238 pacientes) y Xeljanz® 10 mg dos veces al día (236 pacientes) fue de 37.8% y 44.5%, respectivamente. La frecuencia de las infecciones en el periodo controlado con placebo de 3 meses fue de 23.5% para el grupo con Xeljanz® 5 mg dos veces al día (238 pacientes), 28.8% para el grupo con Xeljanz® 10 mg dos veces al día (236 pacientes) y 15.7% para el grupo con placebo (236 pacientes).
Las infecciones informadas con mayor frecuencia en el periodo controlado con placebo de 3 meses fueron nasofaringitis (5.9% y 5.5% en los grupos de dosis de 5 mg dos veces al día y 10 mg dos veces al día, respectivamente) e infecciones de las vías respiratorias superiores (5.0% y 4.7% en los grupos de dosis de 5 mg dos veces al día y 10 mg dos veces al día, respectivamente).
A partir de mayo de 2016, la tasa general de infecciones con Xeljanz® en la población de seguridad a largo plazo para dosis combinadas fue de 63.5 pacientes con eventos por cada 100 pacientes-año.
Colitis ulcerosa: En los estudios de inducción aleatorizados de fase 2/3 de 8 semanas, las proporciones de pacientes con infecciones fueron 21.1% para Xeljanz® 10 mg dos veces al día comparado con 15.2% para el placebo. En el estudio de mantenimiento aleatorizado fase 3 de 52 semanas, la proporción de pacientes con infecciones fue 35.9% para Xeljanz® 5 mg dos veces al día, 39.8% para Xeljanz® 10 mg dos veces al día y 24.2% para placebo. En la experiencia de tratamiento completo con Xeljanz® en el programa de colitis ulcerosa, la incidencia general de infección fue 65.7 eventos por 100 pacientes-año (involucrando a 47.9% de los pacientes). La infección más común fue nasofaringitis, ocurriendo en 16.8% de los pacientes.
Infecciones graves:
Artritis reumatoide: En estudios clínicos controlados de 6 y 24 meses, la tasa de infecciones graves en el grupo de monoterapia con Xeljanz® 5 mg dos veces al día fue de 1.7 pacientes con eventos cada 100 años-paciente. En el grupo de monoterapia con Xeljanz® 10 mg dos veces al día, la tasa fue de 1.6 pacientes con eventos cada 100 años-paciente, y la tasa para el grupo de placebo fue de 0 eventos cada 100 años-paciente y la tasa para el grupo de metotrexato fue de 1.9 pacientes con eventos cada 100 años-paciente.
En estudios con una duración de 6, 12 o 24 meses, las tasas de infecciones graves en los grupos de Xeljanz® 5 mg y 10 mg dos veces al día más FARME fueron de 3.6 y 3.4 pacientes con eventos cada 100 años-paciente, respectivamente, en comparación con 1.7 pacientes con eventos cada 100 años-paciente en el grupo de placebo más FARME.
En la población expuesta de seguridad a largo plazo compuesto por ensayos clínicos de fase 2 y fase 3 y estudios clínicos de extensión a largo plazo, las tasas globales de infecciones graves fueron de 2.4 y 3.0 pacientes con eventos por 100 pacientes-año para los grupos de Xeljanz® de 5 mg y 10 mg dos veces al día, respectivamente. Las infecciones graves más comúnmente reportadas con Xeljanz® incluyen neumonía, herpes zóster, infección de vías urinarias, celulitis, gastroenteritis y diverticulitis. Se han reportado casos de infecciones oportunistas (ver la sección Precauciones generales).
De los 4271 pacientes que se inscribieron en los Estudios I a VI, un total de 608 pacientes con artritis reumatoide de 65 años y mayores, incluidos 85 pacientes de 75 años y mayores. La frecuencia de infección grave entre los pacientes tratados con Xeljanz® de 65 años y mayores fue mayor que la frecuencia entre los pacientes menores de 65 años. Debido a que hay una mayor incidencia de infecciones en pacientes de edad avanzada en general, se debe tener precaución cuando se trate a pacientes de edad avanzada.
Las infecciones graves también se reportaron en un PASS aleatorizado de gran tamaño en pacientes con AR que tenían 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia).
Artritis psoriásica: En los estudios de fase 3 de 6 y 12 meses, la tasa de infecciones graves en el grupo con Xeljanz® 5 mg dos veces al día fue de 1.30 pacientes con eventos por cada 100 pacientes-año. En el grupo de Xeljanz® 10 mg dos veces al día, la tasa fue de 2.0 pacientes con eventos por cada 100 pacientes-año.
En la población de seguridad a largo plazo, la tasa de infecciones graves fue de 1.4 pacientes con eventos por cada 100 pacientes-año para pacientes tratados con Xeljanz®. La infección sería más común informada con Xeljanz® fue la neumonía.
Colitis ulcerosa: En los estudios de inducción aleatorios de Fase 2/3 de 8 semanas, la proporción de pacientes con infecciones graves en pacientes tratados con Xeljanz® 10 mg dos veces al día fue del 0.9% (8 pacientes) en comparación con el 0.0% en pacientes tratados con placebo. En el estudio de mantenimiento, aleatorizado de 52 semanas fase 3, las tasas de incidencia de infecciones graves en pacientes tratados con Xeljanz® 5 mg dos veces al día (1.35 eventos por 100 pacientes-año) y en pacientes tratados con Xeljanz® 10 mg dos veces al día (0.64 eventos por 100 pacientes-año) no fueron más altas en comparación con el placebo (1.94 eventos por 100 pacientes-año). La tasa de incidencia de infecciones graves en toda la experiencia de tratamiento con Xeljanz® en pacientes con colitis ulcerosa fue de 2.05 eventos por cada 100 pacientes-año. No hubo agrupación aparente en tipos específicos de infecciones graves.
Reactivación viral: En los estudios clínicos de Xeljanz®, los pacientes japoneses y coreanos parecen tener una mayor tasa de herpes zóster que aquella observada en otras poblaciones. Se informaron eventos de herpes zóster en un PASS aleatorizado de gran tamaño en pacientes con AR que tenían 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia).
Tromboembolismo venoso:
Artritis reumatoide: Se informaron eventos de EP y TVP en un PASS aleatorizado de gran tamaño en pacientes con AR que tenían 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver la sección Farmacocinética y farmacodinamia).
Estudios finalizados de artritis reumatoide: En el periodo de 4 a 12 semanas con placebo de los estudios aleatorizados y controlados de 4 semanas a 24 meses de duración, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día, tofacitinib 10 mg dos veces al día y placebo fueron de 0.00 (0.00, 0.57), 0.00 (0.00, 0.77) y 0.40 (0.01, 2.22) pacientes con eventos por cada 100 pacientes-año, respectivamente; las IR (IC del 95%) de la TVP fueron 0.00 (0.00, 0.57), 0.21 (0.01, 1.16) y 0.40 (0.01, 2.22) pacientes con eventos por cada 100 pacientes-año, respectivamente.
En el periodo con aleatorización completa de los estudios controlados de 4 semanas a 24 meses de duración, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día y tofacitinib 10 mg dos veces al día fueron de 0.12 (0.02, 0.34) y 0.15 (0.03, 0.44) pacientes con eventos por cada 100 pacientes-año, respectivamente; las IR (IC del 95%) de la TVP fueron de 0.15 (0.04, 0.40) y 0.10 (0.01, 0.36) pacientes con eventos por cada 100 pacientes-año, respectivamente.
En la población de seguridad a largo plazo, que incluye la exposición durante los estudios aleatorizados y controlados finalizados y los estudios de extensión abiertos a largo plazo, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día y tofacitinib 10 mg dos veces al día fueron de 0.12 (0.06, 0.22) y 0.13 (0.08, 0.21) pacientes con eventos por cada 100 pacientes-año, respectivamente; las IR (IC del 95%) de la TVP fueron de 0.17 (0.09, 0.27) y 0.15 (0.09, 0.22) pacientes con eventos por cada 100 pacientes-año, respectivamente.
Artritis psoriásica: En el periodo de 3 meses con placebo de los estudios aleatorizados y controlados finalizados de 6 a 12 meses de duración, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día, tofacitinib 10 mg dos veces al día y placebo fueron de 0.00 (0.00, 6.75), 0.00 (0.00, 6.78) y 0.00 (0.00, 6.87) pacientes con eventos por cada 100 pacientes-año, respectivamente; las IR (IC del 95%) de la TVP fueron de 0.00 (0.00, 6.75), 0.00 (0.00, 6.78) y 0.00 (0.00, 6.87) pacientes con eventos por cada 100 pacientes-año, respectivamente.
En el periodo con aleatorización completa de los estudios controlados de 6 a 12 meses finalizados, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día y tofacitinib 10 mg dos veces al día fueron de 0.00 (0.00, 1.83) y 0.00 (0.00, 1.87) pacientes con eventos por cada 100 pacientes-año, respectivamente; las IR (IC del 95%) de la TVP fueron de 0.00 (0.00, 1.83) y 0.51 (0.01, 2.83) pacientes con eventos por cada 100 pacientes-año, respectivamente.
En la población de seguridad a largo plazo, que incluye la exposición durante los estudios aleatorizados y controlados finalizados y el estudio de extensión abierto a largo plazo en curso, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día y tofacitinib 10 mg dos veces al día fueron de 0.11 (0.00, 0.60) y 0.00 (0.00, 0.58) pacientes con eventos por cada 100 pacientes-año, respectivamente; las IR (IC del 95%) de la TVP fueron de 0.00 (0.00, 0.40) y 0.16 (0.00, 0.87) pacientes con eventos por cada 100 pacientes-año, respectivamente.
Colitis ulcerosa: En los estudios de inducción aleatorizados y controlados con placebo de 8 semanas de duración finalizados, la IR (IC del 95%) de la EP para tofacitinib 10 mg dos veces al día y placebo fueron de 0.00 (0.00, 2.22) y 1.98 (0.05, 11.04) pacientes con eventos por cada 100 pacientes-año; la IR (IC del 95%) de la TVP fueron de 0.00 (0.00, 2.22) y 1.99 (0.05, 11.07) pacientes con eventos por cada 100 pacientes-año, respectivamente.
En el estudio de mantenimiento aleatorizado finalizado de 52 semanas de duración, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día y tofacitinib 10 mg dos veces al día fueron de 0.00 (0.00, 2.48) y 0.00 (0.00, 2.35) pacientes con eventos por cada 100 pacientes-año, respectivamente; las IR (IC del 95%) de la TVP fueron de 0.00 (0.00, 2.48) y 0.00 (0.00, 2.35) pacientes con eventos por cada 100 pacientes-año, respectivamente.
En la población de seguridad a largo plazo, que incluye la exposición durante los estudios aleatorizados y controlados finalizados y el estudio de extensión abierto a largo plazo en curso, las IR (IC del 95%) de la EP para tofacitinib 5 mg dos veces al día y tofacitinib 10 mg dos veces al día fueron de 0.00 (0.00, 0.54) y 0.20 (0.05, 0.52) pacientes con eventos por cada 100 pacientes-año, respectivamente, las IR (IC del 95%) de la TVP fueron de 0.00 (0.00, 0.54) y 0.05 (0.00, 0.28) pacientes con eventos por cada 100 pacientes-año, respectivamente.
Experiencia clínica en pacientes con artritis reumatoide no tratados previamente con metotrexato: El Estudio VI fue un estudio clínico controlado por principio activo en pacientes con AR no tratados previamente con metotrexato (ver sección Farmacocinética y farmacodinamia). La experiencia en seguridad en estos pacientes fue consistente con los Estudios I-V.
Pruebas de laboratorio: En los ensayos clínicos en artritis psoriásica y colitis ulcerosa, los cambios en los linfocitos, neutrófilos y lípidos observados con el tratamiento con Xeljanz® fueron similares a los cambios observados en los ensayos clínicos en artritis reumatoide.
En los ensayos clínicos en artritis psoriásica y colitis ulcerosa, los cambios en las pruebas de enzimas hepáticas observados con el tratamiento de Xeljanz® fueron similares a los cambios observados en los ensayos clínicos en artritis reumatoide donde los pacientes recibieron FARME de fondo.
Artritis reumatoide:
Linfocitos: En los estudios clínicos controlados sobre artritis reumatoide, se produjeron disminuciones confirmadas en los recuentos de linfocitos por debajo de 500 células/mm3 en el 0.23% de los pacientes con dosis combinadas de 5 mg dos veces al día y 10 mg dos veces al día.
En la población de seguridad a largo plazo con artritis reumatoide, se produjeron disminuciones confirmadas en los recuentos de linfocitos por debajo de 500 células/mm3 en el 1.3% de los pacientes con dosis combinadas de 5 mg y 10 mg dos veces al día.
Los recuentos confirmados de linfocitos < 500 células/mm3 se asociaron con un aumento de la incidencia de infecciones tratadas y graves (ver sección Precauciones generales).
Neutrófilos: En los estudios clínicos controlados sobre artritis reumatoide, se produjeron disminuciones confirmadas en el ANC por debajo de 1000 células/mm3 en el 0.08% de los pacientes con dosis combinadas de 5 mg y 10 mg dos veces al día. No se observaron disminuciones confirmadas de ANC por debajo de 500 células/mm3 en ningún grupo de tratamiento. No hubo una relación clara entre la neutropenia y la aparición de infecciones graves.
En la población de seguridad a largo plazo, el patrón y la incidencia de los descensos confirmados en ANC se mantuvieron consistentes con lo observado en los ensayos clínicos controlados (ver sección Precauciones generales).
Pruebas de enzimas hepáticas:
Artritis reumatoide: Se observaron con poca frecuencia aumentos confirmados en las enzimas hepáticas > 3 veces el límite superior normal (3 × LSN). En pacientes que presentaron aumento de las enzimas hepáticas, la modificación del régimen de tratamiento, tal como la reducción en la dosis del FARME concomitante, la interrupción del tratamiento con Xeljanz® o la reducción de la dosis de Xeljanz® dio lugar a la disminución o la normalización de las enzimas hepáticas.
En la parte controlada del estudio de monoterapia de fase 3 (0 a 3 meses), (Estudio I, ver sección Farmacocinética y farmacodinamia), se observaron aumentos de alanina aminotransferasa (ALT) > 3 × LSN en un 1.65%, 0.41% y 0% de los pacientes que recibieron placebo, Xeljanz® 5 mg y 10 mg dos veces al día, respectivamente. En este estudio, se observaron aumentos de AST > 3 × LSN en 1.65%, 0.41% y 0% de los pacientes que recibieron placebo, Xeljanz® 5 mg y 10 mg dos veces al día, respectivamente.
En el estudio de monoterapia de fase 3 (0 a 24 meses), (Estudio VI, ver sección Farmacocinética y farmacodinamia), se observaron aumentos de ALT > 3 × LSN en 7.1%, 3.0%, y 3.0% de los pacientes a quienes se les administró metotrexato, Xeljanz® 5 mg y 10 mg dos veces al día, respectivamente. En este estudio, se observaron aumentos de AST > 3 × LSN en 3.3%, 1.6% y 1.5% de los pacientes que recibieron metotrexato, Xeljanz® 5 mg y 10 mg dos veces al día, respectivamente.
En la parte controlada de los estudios de Fase 3 con FARME de fondo (0 a 3 meses), (Estudios II a V; ver sección Farmacocinética y Farmacodinamia), se observaron aumentos de ALT > 3 × LSN en 0.9%, 1.24% y 1.14% de los pacientes que recibieron placebo, Xeljanz® 5 mg y 10 mg dos veces al día, respectivamente. En estos estudios, se observaron aumentos de AST > 3 × LSN en 0.72%, 0.50% y 0.31% de los pacientes que recibieron placebo, Xeljanz® 5 mg y 10 mg dos veces al día, respectivamente.
Se informaron elevaciones de ALT y AST en un PASS aleatorizado de gran tamaño en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia).
Lípidos: Los aumentos de los parámetros lipídicos (colesterol total, colesterol LDL, colesterol HDL, triglicéridos) se evaluaron por primera vez un mes después del inicio del tratamiento con Xeljanz® en los ensayos clínicos controlados doble ciego de artritis reumatoide. Los aumentos se observaron en este punto temporal y se mantuvieron estables a partir de entonces.
Artritis reumatoide: Los cambios en los parámetros lipídicos con respecto a los valores iniciales hasta el final del estudio (6 a 24 meses) en los estudios clínicos controlados de artritis reumatoide se resumen a continuación:
• La media del colesterol LDL aumentó un 15% en el grupo tratado con Xeljanz® 5 mg dos veces al día y un 20% en el grupo tratado con Xeljanz® 10 mg dos veces al día al mes 12, y aumentó un 16% en el grupo tratado con Xeljanz® 5 mg dos veces al día y el 19% en el grupo tratado con Xeljanz® 10 mg dos veces al día al mes 24.
• La media del colesterol HDL aumentó un 17% en el grupo de Xeljanz® 5 mg dos veces al día y un 18% en el grupo de Xeljanz® 10 mg dos veces al día en el mes 12, y aumentó un 19% en el grupo de Xeljanz® 5 mg dos veces al día y un 20% en el grupo de Xeljanz® 10 mg dos veces al día en el mes 24.
Se notificaron elevaciones del colesterol LDL y del colesterol HDL en un PASS aleatorizado de gran tamaño en pacientes con AR de 50 años o mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia).
Al momento del abandono del tratamiento con tofacitinib, los niveles de lípidos volvieron al valor inicial.
En artritis reumatoide, las proporciones medias de colesterol LDL/HDL y las proporciones de apolipoproteína B (ApoB)/ApoA1 se mantuvieron esencialmente sin cambios en los pacientes tratados con XELJANZ®.
En un estudio clínico controlado, los incrementos del colesterol LDL y de la ApoB descendieron a niveles pretratamiento en respuesta a la terapia con estatinas.
En la vigilancia de población de seguridad a largo plazo, los incrementos en el perfil lipídico fueron consistentes con lo que se observó en los estudios clínicos controlados.
PRECAUCIONES EN RELACIÓN CON EFECTOS DE CARCINOGÉNESIS, MUTAGÉNESIS, TERATOGÉNESIS Y SOBRE LA FERTILIDAD:
En estudios no clínicos se observaron efectos sobre los sistemas inmunitarios y hematopoyético que se atribuyeron a las propiedades farmacológicas (inhibición de JAK) de tofacitinib. En dosis de importancia clínica, se observaron efectos secundarios de inmunosupresión, como infecciones bacterianas, virales y linfoma. Otros hallazgos con dosis muy por encima de las exposiciones en seres humanos incluyeron efectos en el hígado, los pulmones y el aparato gastrointestinal.
Se observaron linfomas en 3 de 8 monos adultos y 0 de 14 monos jóvenes tratados con una dosis de tofacitinib de 5 mg/kg dos veces al día. La dosis máxima sin efecto adverso observado (NOAEL) para los linfomas fue de 1 mg/kg dos veces al día. El ABC no unida con 1 mg/kg dos veces al día fue de 341 ng• h/mL, que es aproximadamente la mitad del ABC no unida con 10 mg dos veces al día y similar al ABC no unida con 5 mg dos veces al día en humanos.
Tofacitinib no es mutagénico ni genotóxico en función de los resultados de una serie de pruebas in vitro e in vivo sobre mutaciones genéticas y aberraciones cromosómicas.
La capacidad carcinogénica de tofacitinib se evaluó con estudios de carcinogenicidad en ratones transgénicos rasH2 de 6 meses y estudios de carcinogenicidad en ratas de 2 años. Tofacitinib no fue carcinogénico en ratones en una dosis alta de como máximo 200 mg/kg/día (ABC del medicamento no unido de ~19 veces el ABC humano con 10 mg dos veces al día). Se observaron tumores benignos con células de Leydig en ratas: los tumores benignos con células de Leydig en ratas no se asocian con un riesgo de tumores con células de Leydig en seres humanos. Se observaron hibernomas (neoplasia maligna del tejido adiposo pardo) en ratas hembra con dosis ≥ 30 mg/kg/día (ABC del medicamento no unido de ~41 veces el ABC humano con 10 mg dos veces al día). Se observaron timomas benignos en ratas hembra, que recibieron dosis solamente de 100 mg/kg/día reducidas a 75 mg/kg/día (ABC del medicamento no unido de ~94 veces el ABC humano con 10 mg dos veces al día).
Se demostró que tofacitinib es teratógeno en ratas y conejos y que, en las ratas, produce efectos sobre la fertilidad de la hembra, el parto y el desarrollo peri/posnatal. Tofacitinib no tuvo efectos en la fertilidad de los machos, la motilidad ni la concentración del esperma. Tofacitinib se secreta en la leche de ratas lactantes. En estudios realizados en ratas y monos jóvenes, los efectos relacionados con tofacitinib en el sistema inmunitario fueron similares a los de los animales adultos. No hubo efectos relacionados con el tofacitinib en el sistema reproductivo o el desarrollo óseo en machos y hembras.
INTERACCIONES MEDICAMENTOSAS Y DE OTRO GÉNERO:
Interacciones que afectan la administración de XELJANZ® XR: Dado que tofacitinib se metaboliza mediante la CYP3A4, es posible la interacción con medicamentos que inhiben o inducen la CYP3A4. La exposición de tofacitinib se ve aumentada cuando se coadministra con inhibidores potentes del citocromo P450 (CYP) 3A4 (p. ej., ketoconazol) o cuando la administración de uno o más medicamentos concomitantes tiene como consecuencia tanto la inhibición moderada de CYP3A4 y la inhibición potente del CYP2C19 (p. ej., fluconazol) (ver sección Dosis y vía de administración).
La exposición a tofacitinib se ve disminuida cuando se coadministra con inductores potentes de CYP (p. ej., rifampicina). Es poco probable que los inhibidores de CYP2C19 solos o la glucoproteína P alteren de manera importante la PK de tofacitinib.
La administración concomitante con metotrexato (15 mg a 25 mg de MTX una vez a la semana) no tuvo ningún efecto en la farmacocinética de tofacitinib. La coadministración de ketoconazol, un fuerte inhibidor de CYP3A4, con una sola dosis de tofacitinib aumentó el ABC y la Cmáx en un 103% y un 16%, respectivamente. La coadministración de fluconazol, un inhibidor moderado de CYP3A4 y un inhibidor fuerte de CYP2C19, aumentó el ABC y la Cmáx de tofacitinib en un 79% y un 27%, respectivamente. La administración concomitante de tacrólimus (Tac), un inhibidor leve de CYP3A4, aumentó el ABC de tofacitinib en un 21% y disminuyó la Cmáx de tofacitinib en un 9%. La coadministración de ciclosporina (CsA), un inhibidor moderado de CYP3A4, aumentó el ABC de tofacitinib en un 73% y disminuyó la Cmáx de tofacitinib en un 17%. No se ha estudiado la administración combinada de dosis múltiples de tofacitinib con inmunosupresores potentes para el tratamiento de la artritis reumatoide, artritis psoriásica o colitis ulcerosa. La coadministración de rifampicina, un inductor potente de CYP3A4, disminuyó el ABC y la Cmáx de tofacitinib en un 84% y 74%, respectivamente (ver sección Dosis y vía de administración).
Potencial de XELJANZ® XR de influir en la farmacocinética de otros medicamentos: Los estudios in vitro indican que tofacitinib no inhibe ni induce significativamente la actividad de las más importantes CYP metabolizantes de medicamentos en seres humanos (CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6 y CYP3A4) en concentraciones superiores a 80 veces la Cmáx total en estado estacionario en dosis de 5 mg y 10 mg dos veces al día en pacientes con artritis reumatoide, artritis psoriásica y colitis ulcerosa. Estos resultados in vitro fueron confirmados por un estudio de interacciones medicamentosas en seres humanos que demuestra que no se producen cambios en la farmacocinética de midazolam, un sustrato altamente sensible de CYP3A4, al coadministrarse con tofacitinib.
Los estudios in vitro indican que tofacitinib no inhibe significativamente la actividad de las más importantes uridin 5-difosfo-glucuronosiltransferasas (UGT) metabolizantes de medicamentos en seres humanos, (UGT1A1, UGT1A4, UGT1A6, UGT1A9 y UGT2B7) en concentraciones superiores a 250 veces la Cmáx total en estado estacionario en dosis de 5 mg y 10 mg dos veces al día en pacientes con artritis reumatoide, artritis psoriásica y colitis ulcerosa.
Los datos in vitro indican que también es bajo el potencial de tofacitinib para inhibir transportadores, como glucoproteína P, polipéptidos transportadores de aniones orgánicos, transportadores aniónicos o catiónicos orgánicos en concentraciones terapéuticas.
La coadministración de tofacitinib no tuvo ningún efecto en la farmacocinética de anticonceptivos orales, levonorgestrel y etinilestradiol, en voluntarias sanas.
La coadministración de tofacitinib con metotrexato 15 a 25 mg una vez a la semana disminuyó el ABC y la Cmáx de metotrexato en un 10% y un 13%, respectivamente. El grado de disminución de la exposición al metotrexato no garantiza que haya modificaciones en la dosificación individualizada con metotrexato.
La coadministración de tofacitinib no tuvo un efecto sobre la farmacocinética de metformina, lo que indica que tofacitinib no interfiere con el transportador catiónico orgánico (OCT2) en voluntarios sanos.
En los pacientes con artritis reumatoide y colitis ulcerosa, la depuración oral de tofacitinib no varía con el tiempo, lo que indica que tofacitinib no normaliza la actividad enzimática de CYP en estos pacientes. Por lo tanto, no se espera que la coadministración con tofacitinib cause aumentos de importancia clínica en el metabolismo de los sustratos de CYP en pacientes con AR y colitis ulcerosa.
Población pediátrica: Solo se han realizado estudios de interacción medicamentosa en adultos.
ALTERACIONES EN LOS RESULTADOS DE PRUEBAS DE LABORATORIO:
Parámetros de laboratorio:
Linfocitos: Los recuentos de linfocitos de < 500 células/mm3 se asociaron con una mayor incidencia de infecciones tratadas y graves. No se recomienda iniciar el tratamiento con XELJANZ® XR en pacientes con un recuento de linfocitos bajo (es decir, < 500 células/mm3). No se recomienda el tratamiento con XELJANZ® XR en pacientes que desarrollaron un conteo absoluto de linfocitos confirmado < 500 células/mm3 Los linfocitos se deben monitorear en el periodo inicial y cada 3 meses a partir de entonces. Para ver las modificaciones recomendadas basadas en el recuento de linfocitos, ver sección Dosis y vía de administración.
Neutrófilos: El tratamiento con Xeljanz® estuvo relacionado con un aumento de la incidencia de neutropenia (< 2000 células/mm3) en comparación con el placebo. No se recomienda iniciar el tratamiento con XELJANZ® XR en pacientes con un recuento bajo de neutrófilos (es decir, ANC < 1000 células/mm3). Para pacientes que toman XELJANZ® XR 22 mg una vez al día que desarrollan un ANC persistente de 500-1000 células/mm3, reduzca la dosis de Xeljanz® a XELJANZ® XR 11 mg una vez al día hasta que el ANC sea > 1000 células/mm3. Para pacientes que toman XELJANZ® XR 11 mg una vez al día que desarrollan un ANC persistente de 500-1000 células/mm3, interrumpa XELJANZ® XR hasta que el ANC sea > 1000 células/mm3. No se recomienda el tratamiento con XELJANZ® XR en pacientes que desarrollan un recuento absoluto de neutrófilos confirmado de < 500 células/mm3. Deben monitorearse los neutrófilos en el periodo inicial y después de 4 a 8 semanas de tratamiento y cada 3 meses a partir de entonces (ver secciones Dosis y vía de administración, y Reacciones secundarias y adversas).
Hemoglobina: No se recomienda iniciar el tratamiento con XELJANZ® XR en pacientes con valores bajos de hemoglobina (es decir, < 9 g/dL). El tratamiento con XELJANZ® XR debe interrumpirse en pacientes que presenten niveles de hemoglobina < 8 g/dL o cuyo nivel de hemoglobina disminuya en > 2 g/dL con el tratamiento. Debe monitorearse la hemoglobina en el periodo inicial y después de 4 a 8 semanas de tratamiento y cada 3 meses a partir de entonces (ver secciones Dosis y vía de Administración, y Reacciones secundarias y Adversas).
Lípidos: El tratamiento con Xeljanz® se ha asociado con aumentos en los parámetros lipídicos, tales como el colesterol total, el colesterol LDL y el colesterol de lipoproteína de alta densidad (HDL). Los efectos máximos se observaron generalmente en el plazo de 6 semanas. También se informaron aumentos de colesterol total, colesterol LDL y colesterol HDL en un PASS aleatorizado de gran tamaño en pacientes con AR que tenían 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia).
La evaluación de los parámetros lipídicos se debe realizar aproximadamente 4 a 8 semanas después del inicio del tratamiento con XELJANZ® XR. Se debe controlar a los pacientes de acuerdo con las directrices clínicas (p. ej., Programa nacional educativo sobre el colesterol [National Cholesterol Educational Program]) para el manejo de la hiperlipidemia. Los aumentos en el colesterol total y LDL asociados con Xeljanz® pueden disminuir a los niveles previos al tratamiento con la administración de estatinas.
PRECAUCIONES GENERALES:
Infecciones graves: Se informaron infecciones graves y, en algunas ocasiones, mortales que se deben a patógenos bacterianos, micobacterianos, fúngicos invasivos, virales u otros patógenos oportunistas en pacientes que reciben agentes inmunomoduladores, incluidos los FARME biológicos y Xeljanz®. Las infecciones graves informadas con mayor frecuencia con Xeljanz® incluyeron neumonía, infección de vías urinarias, celulitis, herpes zóster, bronquitis, choque séptico, diverticulitis, gastroenteritis, apendicitis y sepsis. Entre las infecciones oportunistas informadas con el tratamiento con Xeljanz® se hallaban: tuberculosis y otras infecciones micobacterianas, criptococo, histoplasmosis, candidiasis esofágica, herpes zóster multidermatomal, infección por citomegalovirus e infecciones por el virus BK y listeriosis. Algunos pacientes presentaron enfermedad diseminada en lugar de enfermedad localizada y, con frecuencia, los pacientes con artritis reumatoide tomaban inmunomoduladores concomitantes, como metotrexato o corticosteroides que, además de la artritis reumatoide, pueden predisponer a las infecciones. También se pueden producir otras infecciones graves que no se informaron en los estudios clínicos (p. ej., coccidioidomicosis).
En un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional, se observó un aumento dependiente de la dosis de infecciones graves en pacientes tratados con el tofacitinib, en comparación con el inhibidor del TNF (ver sección Farmacocinética y farmacodinamia). Algunas de estas infecciones graves tuvieron como resultado la muerte. También se informaron infecciones oportunistas en el estudio.
No se debe iniciar tratamiento con XELJANZ® XR en pacientes con una infección activa, incluidas las infecciones localizadas. Los riesgos y beneficios del tratamiento deben tomarse en consideración antes de iniciar el tratamiento con XELJANZ® XR en pacientes con infecciones crónicas o recurrentes, o en aquellos que han estado expuestos a la tuberculosis, cuentan con antecedentes de una infección grave u oportunista, han residido o viajado a lugares de tuberculosis endémica o micosis endémicas, o presentan afecciones subyacentes que puedan predisponer a la infección.
Se debe monitorear a los pacientes detenidamente a fin de determinar el desarrollo de signos y síntomas de infección durante y después del tratamiento con XELJANZ® XR. Debe interrumpirse la administración de XELJANZ® XR si un paciente desarrolla una infección grave, una infección oportunista o sepsis. Un paciente que desarrolla una nueva infección durante el tratamiento con XELJANZ® XR debe someterse a una evaluación diagnóstica inmediata y completa apropiada para un paciente inmunodeprimido, se debe iniciar el tratamiento antimicrobiano apropiado y se debe monitorear de cerca al paciente.
Debido a que existe una mayor incidencia de infecciones en las poblaciones de pacientes de edad avanzada y diabéticos en general, se debe tener precaución al tratar a los pacientes de edad avanzada y a los pacientes con diabetes (ver sección Reacciones secundarias y adversas). También se recomienda tener precaución en el caso de pacientes con antecedentes de enfermedad pulmonar crónica, ya que podrían ser más propensos a contraer infecciones. Se han informado eventos de enfermedad pulmonar intersticial (algunos con desenlace mortal) en pacientes tratados con XELJANZ® XR, un inhibidor de la cinasa Jano (JAK, por sus siglas en inglés), en ensayos clínicos y en el entorno de poscomercialización, aunque se desconoce la función de la inhibición de la JAK en estos eventos.
El riesgo de contraer infecciones podría ser mayor con grados mayores de linfopenia y se debe considerar el recuento de linfocitos al evaluar el riesgo de infección individual del paciente. Los criterios relacionados con la interrupción del tratamiento y el monitoreo de la linfopenia se analizan en la sección Dosis y vía de administración.
Tuberculosis: Se debe evaluar a los pacientes y realizarles pruebas para detectar infecciones latentes o activas antes y durante la administración de XELJANZ® XR, de acuerdo con las directrices aplicables.
Los pacientes con tuberculosis latente deben recibir tratamiento con la terapia antimicobacteriana estándar antes de la administración de XELJANZ® XR.
El tratamiento antituberculoso también debe considerarse antes de la administración de XELJANZ® XR en pacientes con antecedentes de tuberculosis latente o activa en los que no puede confirmarse un curso adecuado de tratamiento y para pacientes con un resultado negativo para la prueba de tuberculosis latente, pero que presentan factores de riesgo para la infección tuberculosa. Se recomienda consultar a un profesional de la salud con experiencia en el tratamiento contra la tuberculosis para ayudar en la decisión acerca de si resulta apropiado iniciar el tratamiento antituberculoso en un paciente en particular.
Los pacientes deben ser monitoreados detenidamente para determinar el desarrollo de signos y síntomas de tuberculosis, incluidos los pacientes que tuvieron resultados negativos en las pruebas para la infección tuberculosa latente antes de iniciar el tratamiento.
Reactivación viral: La reactivación viral se ha informado con el tratamiento con FARME, y se han observado casos de reactivación del virus del herpes (p. ej., herpes zóster) en los estudios clínicos con XELJANZ® XR. En un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional, se observó un aumento de los casos de herpes zóster en pacientes tratados con tofacitinib, en comparación con el inhibidor del TNF (ver sección Farmacocinética y farmacodinamia). Se han informado casos de reactivación de la hepatitis B después de la comercialización en pacientes tratados con Xeljanz®. Se desconoce el efecto de XELJANZ® XR en la reactivación de la hepatitis viral crónica. Los pacientes con resultados positivos para hepatitis B o C durante las pruebas de detección se excluyeron de los ensayos clínicos. La detección de la hepatitis viral se debe realizar según los lineamientos clínicos antes de comenzar el tratamiento con tofacitinib.
El riesgo de herpes zóster parece ser mayor en pacientes japoneses y coreanos tratados con Xeljanz®.
Tromboembolismo venoso: Se ha observado TEV en pacientes que toman XELJANZ® XR en ensayos clínicos y en informes posteriores a la comercialización. En un estudio PASS aleatorizado de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional, los pacientes se trataron con tofacitinib 5 mg dos veces al día, tofacitinib 10 mg dos veces al día o un inhibidor del TNF. Se observó un aumento dependiente de la dosis de eventos de embolia pulmonar (EP) en los pacientes tratados con tofacitinib, en comparación con el inhibidor del TNF (ver sección Farmacocinética y farmacodinamia). Muchos de estos eventos de EP fueron graves y algunos tuvieron como resultado la muerte. Los eventos de EP se informaron con mayor frecuencia en este estudio en los pacientes que tomaban tofacitinib en relación con otros estudios en todo el programa de tofacitinib (ver secciones Farmacocinética y farmacodinamia, y Reacciones secundarias y adversas).
Se observaron eventos de TVP en los tres grupos de tratamiento en este estudio (ver sección Farmacocinética y farmacodinamia).
Evalúe a los pacientes para detectar factores de riesgo de TEV antes de iniciar el tratamiento y periódicamente durante el mismo. Administre XELJANZ® XR con precaución en pacientes de 65 años y mayores, y en pacientes en los que se identifican otros factores de riesgo de TEV (p. ej., antecedentes de trombosis). Evalúe urgentemente a los pacientes con signos y síntomas de TEV. Suspenda el tofacitinib mientras se evalúa la sospecha de TEV, independientemente de la dosis o la indicación.
Eventos cardiovasculares adversos importantes (incluido el infarto de miocardio): En un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional, los pacientes se trataron con tofacitinib 5 mg dos veces al día, tofacitinib 10 mg dos veces al día o un inhibidor del TNF. Se observaron MACE, incluidos eventos de infarto de miocardio, en los tres grupos de tratamiento en este estudio. Se observó un aumento de infartos de miocardio no mortales en los pacientes tratados con tofacitinib, en comparación con el inhibidor del TNF (ver sección Farmacocinética y farmacodinamia). Los MACE, incluidos los eventos de infarto de miocardio, fueron más comunes en pacientes de 65 años y mayores, y en pacientes que son o fueron por largo tiempo fumadores, y en pacientes con antecedentes de enfermedad cardiovascular aterosclerótica (ECVA). Se debe tener precaución en el tratamiento de pacientes de 65 años y mayores, pacientes que son o fueron, por largo tiempo fumadores y pacientes con otros factores de riesgo cardiovascular (p. ej., antecedentes de ECVA). En pacientes con estos factores de riesgo, se debe completar una evaluación individualizada de riesgo-beneficio antes de tomar una decisión sobre el inicio o la continuación del tratamiento (ver sección Farmacocinética y farmacodinamia).
Neoplasia maligna y trastorno linfoproliferativo (excluido el cáncer cutáneo no melanoma [NMSC, por sus siglas en inglés]): Considere los riesgos y beneficios del tratamiento con XELJANZ® XR antes de iniciar el tratamiento en pacientes con neoplasia maligna actual o antecedentes de la misma distinta del cáncer de piel no melanoma (NMSC) tratado satisfactoriamente o cuando se considere continuar el tratamiento con XELJANZ® XR en pacientes que desarrollen una neoplasia maligna. Existe la posibilidad de que XELJANZ® XR afecte las defensas del huésped contra las neoplasias malignas.
Se observó un aumento de neoplasias malignas en pacientes tratados con tofacitinib en comparación con un inhibidor del TNF en un PASS aleatorizado de gran tamaño en pacientes de 50 años y mayores con AR con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia). Las neoplasias malignas, excluyendo el NMSC, fueron más comunes en pacientes de 65 años y mayores, y en pacientes que eran fumadores, actuales o pasados, desde hace mucho tiempo. Se debe tener precaución al tratar a pacientes de 65 años y mayores, a pacientes, actuales o pasados, fumadores desde hace mucho tiempo, y pacientes con otros factores de riesgo de malignidad (p. ej., malignidad actual o antecedentes de malignidad). En pacientes con estos factores de riesgo, se debe completar una evaluación individualizada de riesgo-beneficio antes de tomar una decisión sobre el inicio o la continuación del tratamiento (ver sección Farmacocinética y farmacodinamia).
Se han observado linfomas en pacientes tratados con XELJANZ® XR y en pacientes tratados con Xeljanz® en un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia). Los pacientes con artritis reumatoide, en particular aquellos que tienen una enfermedad muy activa, y los pacientes con psoriasis pueden tener un riesgo mayor (hasta varias veces más) que la población general de desarrollar un linfoma. El papel de XELJANZ® XR en el desarrollo de linfomas es incierto.
Se ha observado cáncer de pulmón en pacientes tratados con XELJANZ® XR. También se observaron cánceres de pulmón en pacientes tratados con Xeljanz® en un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional; se observó un aumento en los pacientes tratados con Xeljanz® 10 mg dos veces al día, en comparación con el inhibidor del TNF (ver sección Farmacocinética y farmacodinamia). De los 30 cánceres de pulmón informados en el estudio realizado en pacientes que tomaban tofacitinib, todos menos 2 fueron en pacientes que son o fueron. Los pacientes con artritis reumatoide pueden tener un mayor riesgo que la población general de desarrollar cáncer de pulmón. El papel de XELJANZ® XR en el desarrollo del cáncer de pulmón es incierto.
Se observaron otras neoplasias malignas en estudios clínicos y en el marco posterior a la comercialización incluyendo, pero no limitado, cáncer de mama, melanoma, cáncer de próstata y cáncer pancreático.
Se desconoce el papel del tratamiento con XELJANZ® XR en el desarrollo y la evolución de las neoplasias malignas.
Las recomendaciones para cáncer de piel no melanoma se presentan abajo.
Artritis reumatoide: En estudios clínicos controlados de fase 3 en pacientes con artritis reumatoide se diagnosticaron 26 neoplasias malignas (excluidos los NMSC), entre ellas 5 linfomas en 26 pacientes que recibían Xeljanz®/Xeljanz® más FARME, en comparación con 0 neoplasias malignas (excluidos los NMSC) en pacientes del grupo que recibió placebo/placebo más FARME y 2 en 2 pacientes del grupo que recibió adalimumab, 1 en 1 paciente del grupo que recibió metotrexato. Se trataron 3800 pacientes (3942 pacientes-año de observación) con Xeljanz® por periodos de 2 años como máximo, mientras que 681 pacientes (203 pacientes-año de observación) recibieron tratamiento con placebo durante 6 meses como máximo y 204 pacientes (179 pacientes-año de observación) recibieron tratamiento con adalimumab durante 12 meses. La tasa de incidencia ajustada a la exposición de neoplasias malignas y linfoma fue de 0.66 y 0.13 eventos por cada 100 pacientes-año, respectivamente, en los grupos que recibieron Xeljanz®.
En la población de seguridad a largo plazo (4867 pacientes), en los estudios de artritis reumatoide, la tasa de neoplasias (excluidos los NMSC) y linfoma fue de 0.97 y 0.09 eventos cada 100 pacientes-año, respectivamente, lo que es consistente con la tasa observada en el periodo controlado.
En un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional, se observó un aumento de las neoplasias malignas (excluido el NMSC) en los pacientes tratados con Xeljanz®, en comparación con el inhibidor del TNF (ver sección Farmacocinética y farmacodinamia). Las neoplasias malignas (excluido el NMSC) fueron más frecuentes en pacientes de 65 años y mayores, y en pacientes que son o fueron por largo tiempo fumadores.
Artritis psoriásica: En 2 ensayos clínicos controlados de fase 3 realizados en pacientes con artritis psoriásica activa, se produjeron 3 neoplasias malignas (excluido el NMSC) en 474 pacientes (298 años-paciente de observación) que recibieron Xeljanz® más FARMEsc (6 a 12 meses de exposición), en comparación con 0 neoplasias malignas en 236 pacientes (52.3 años-paciente) en el grupo con placebo más FARMEsc (3 meses de exposición) y 0 neoplasias malignas en 106 pacientes (91 años-paciente) en el grupo con adalimumab más FARMEsc (12 meses de exposición). No se informaron linfomas. La tasa de incidencia ajustada por exposición de neoplasias malignas (excluido el NMSC) fue de 1.95 pacientes con eventos y 0 pacientes con eventos por cada 100 años-paciente en los grupos de Xeljanz® que recibieron 5 mg dos veces al día y 10 mg dos veces al día, respectivamente.
En la población de seguridad compuesta por los 2 ensayos clínicos controlados de fase 3 y el ensayo de extensión a largo plazo (783 pacientes), la tasa de neoplasias malignas (excluido el NMSC) fue de 0.63 pacientes con acontecimientos por cada 100 años-paciente.
Colitis ulcerosa: En los estudios de inducción y mantenimiento controlados con placebo para la colitis ulcerosa no se produjeron neoplasias malignas (excluido el NMSC) en ningún grupo de Xeljanz®. En toda la experiencia de tratamiento con Xeljanz® para la colitis ulcerosa, se han informado neoplasias malignas (excluido el NMSC) con una tasa de incidencia global de 0.5 eventos por cada 100 años-paciente.
Cáncer de piel no melanoma: Se han informado casos de cáncer de piel no melanoma (NMSC) en pacientes tratados con XELJANZ® XR. También se informaron NMSC en un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores, y al menos un factor de riesgo cardiovascular adicional. En este estudio se observó un aumento del NMSC en general, incluidos los carcinomas cutáneos de células escamosas, en los pacientes tratados con tofacitinib, en comparación con el inhibidor del TNF (ver sección Farmacocinética y farmacodinamia). Dado que existe una mayor incidencia de NMSC en los pacientes de edad avanzada y en los pacientes con antecedentes de NMSC, se debe tener precaución al tratar a este tipo de pacientes. Se recomienda el examen periódico de la piel en los pacientes con mayor riesgo de padecer cáncer de piel (ver la Tabla 13 de la sección Reacciones secundarias y adversas).
Perforaciones gastrointestinales: Se han informado eventos de perforación gastrointestinal en ensayos clínicos, incluido un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional (ver sección Farmacocinética y farmacodinamia). Se desconoce el papel de la inhibición de la JAK en estos eventos. Los eventos se informaron principalmente como perforación diverticular, peritonitis, absceso abdominal y apendicitis. En los ensayos clínicos sobre artritis reumatoide, la tasa de incidencia de perforación gastrointestinal en todos los estudios (fase 1, fase 2, fase 3 y extensión a largo plazo) de todas las dosis de todos los grupos de tratamientos fue de 0.11 eventos por 100 pacientes-año con el tratamiento con Xeljanz®. Todos los pacientes con artritis reumatoide que desarrollaron perforaciones gastrointestinales tomaban medicamentos antiinflamatorios no esteroideos (AINE) y/o corticosteroides de manera concomitante. Se desconoce la contribución relativa de estos medicamentos concomitantes en comparación con Xeljanz® para el desarrollo de perforaciones gastrointestinales. La tasa de incidencia en los ensayos clínicos de artritis psoriásica (fase 3 y extensión a largo plazo) fue de 0.13 pacientes con eventos por cada 100 años-paciente con el tratamiento con Xeljanz®.
En los estudios de inducción controlados con placebo para la colitis ulcerosa, se produjo una perforación gastrointestinal (todos los casos) en 2 (0.2%) pacientes tratados con Xeljanz® 10 mg dos veces al día y en 2 (0.9%) pacientes que recibieron el placebo. En el estudio de mantenimiento de fase 3 para la colitis ulcerosa, no se informó ninguna perforación gastrointestinal (todos los casos) en los pacientes tratados con Xeljanz® y sí en 1 paciente tratado con el placebo.
XELJANZ® XR se debe administrar con precaución en pacientes que puedan estar en mayor riesgo de perforación gastrointestinal (p. ej., pacientes con antecedentes de diverticulitis). Los pacientes que presentan síntomas abdominales de nueva aparición deben ser evaluados de inmediato para la identificación temprana de perforación gastrointestinal.
Fracturas: Se han observado fracturas en pacientes tratados con XELJANZ® XR en estudios clínicos y en el entorno posterior a la comercialización.
En estudios clínicos controlados de fase 3 realizados en pacientes con AR, durante los 0 a 3 meses de exposición, las tasas de incidencia de fracturas para Xeljanz® 5 mg dos veces al día, Xeljanz® 10 mg dos veces al día y el placebo fueron de 2.11, 2.56 y 4.43 pacientes con eventos por cada 100 años-paciente, respectivamente.
En un estudio PASS aleatorizado y de gran tamaño realizado en pacientes con AR de 50 años y mayores con al menos un factor de riesgo cardiovascular adicional, se observaron fracturas en todos los grupos de tratamiento con Xeljanz® y con inhibidores del TNF (ver sección Farmacocinética y farmacodinamia).
Se debe tener precaución en los pacientes con factores de riesgo de fracturas conocidos, como los pacientes de edad avanzada, las mujeres y los pacientes con uso de corticosteroides.
Hipersensibilidad: Se han observado reacciones como angioedema y urticaria que pueden reflejar hipersensibilidad al medicamento en pacientes que reciben XELJANZ® XR. Algunos eventos fueron graves. Muchos de estos eventos se produjeron en pacientes con antecedentes de alergias múltiples. Si se produce una reacción de hipersensibilidad grave, suspenda rápidamente la administración del tofacitinib mientras se evalúa la causa o causas posibles de la reacción.
Hipoglucemia en pacientes tratados por diabetes: Se han notificado casos de hipoglucemia tras el inicio del tratamiento con XELJANZ® en pacientes que reciben medicación para la diabetes. Puede ser necesario ajustar la dosis de la medicación antidiabética en caso de que se produzca hipoglucemia.
Vacunas: No se dispone de datos sobre la transmisión secundaria de infección por vacunas de microorganismos vivos atenuados a pacientes que reciben XELJANZ® XR. Se recomienda no administrar vacunas de microorganismos vivos atenuados concurrentemente con XELJANZ® XR. Se recomienda que todos los pacientes actualicen su programa de vacunación de acuerdo con las directrices actuales de vacunación vigentes antes de iniciar el tratamiento con XELJANZ® XR. El intervalo comprendido entre las vacunas de microorganismos vivos y el comienzo del tratamiento con tofacitinib debe coincidir con los lineamientos actuales de vacunación relacionados con agentes inmunomoduladores. De manera coherente con estos lineamientos, si se administra la vacuna contra el herpes zóster, solamente se podrá administrar a pacientes con antecedentes conocidos de varicela o que sean seropositivos para el virus zóster de varicela. La vacunación se debe realizar por lo menos 2 semanas antes, preferentemente 4 semanas previas, al inicio de agentes inmunosupresores tales como tofacitinib.
En un ensayo clínico controlado, se evaluó la respuesta humoral a la vacunación concurrente con vacunas polisacáridas antineumocócicas y antiinfluenza en pacientes con artritis reumatoide que iniciaban el tratamiento con tofacitinib 10 mg dos veces al día o con placebo. Un porcentaje similar de pacientes alcanzó una respuesta humoral satisfactoria a la vacuna para la influenza (≥ 4 veces el aumento en ≥ 2 de 3 antígenos) en los grupos de tratamiento con tofacitinib (57%) y placebo (62%). Se observó una modesta reducción en el porcentaje de pacientes que alcanzaron una respuesta humoral satisfactoria a la vacuna polisacárida antineumocócica (≥ 2 veces el aumento en ≥ 6 de 12 serotipos) en pacientes que recibían monoterapia con tofacitinib (62%) y monoterapia con metotrexato (62%), en comparación con el placebo (77%), con una mayor reducción en la tasa de respuesta de pacientes que recibieron tofacitinib y metotrexato (32%). Se desconoce su importancia clínica.
Un estudio de vacunas por separado evaluó la respuesta humoral a la vacunación concurrente con vacunas polisacáridas antineumocócicas y antiinfluenza en pacientes que recibieron tofacitinib 10 mg dos veces al día durante una mediana de aproximadamente 22 meses. Más del 60% de los pacientes tratados con tofacitinib (con o sin metotrexato) presentaron respuestas satisfactorias a las vacunas polisacáridas antineumocócicas y antiinfluenza. De forma coherente con el ensayo controlado, los pacientes que recibieron tofacitinib y MTX presentaron una tasa de respuesta inferior a la vacuna polisacárida antineumocócica, en comparación con la monoterapia con tofacitinib (66% frente a 89%).
En un estudio controlado en pacientes con artritis reumatoide sobre la administración de metotrexato de fondo, se evaluaron las respuestas humoral y mediada por células a la inmunización con una vacuna de virus vivos atenuados (Zostavax) indicada para la prevención del herpes zóster. La inmunización se realizó entre 2 y 3 semanas antes de iniciar un tratamiento de 12 semanas con tofacitinib 5 mg dos veces al día o con placebo. Seis semanas después de la inmunización con la vacuna contra el herpes zóster, los receptores de tofacitinib y placebo mostraron similares respuestas humorales y mediadas por células (cambio medio de los anticuerpos IgG VZV de 2.11 en tofacitinib 5 mg dos veces al día y de 1.74 en placebo dos veces al día; aumento de IgG VZV ≥ 1.5 en el 57% de quienes recibieron tofacitinib y en el 43% de quienes recibieron placebo; cambio medio de células formadoras de manchas ELISPOT células T VZV de 1.5 mg en tofacitinib 5 mg dos veces al día y 1.29 en placebo dos veces al día). Estas respuestas fueron similares a las observadas en voluntarios sanos de 50 años y mayores.
En este estudio, un paciente presentó diseminación de la cepa de vacunas del virus zóster de varicela 16 días después de la vacunación. El paciente no había tenido contacto previo con el virus de la varicela, según lo evidencia la ausencia de antecedentes de infección por varicela y la falta de anticuerpos contra la varicela en el periodo inicial. Se interrumpió el tratamiento con tofacitinib y el sujeto se recuperó después del tratamiento con dosis estándar de medicamento antiviral. Los análisis posteriores mostraron que este paciente presentó respuestas robustas de células T antivaricela y anticuerpos a la vacuna aproximadamente 6 semanas después de la vacunación, pero no 2 semanas después de la vacunación, tal como se espera para una infección primaria.
Pacientes con insuficiencia renal: No se requieren ajustes de la dosis en pacientes con insuficiencia renal leve o moderada. La dosis de XELJANZ® XR no debe exceder los 11 mg una vez al día en pacientes que sufren insuficiencia renal severa. Para conocer las recomendaciones específicas de ajuste de dosis para cada indicación, ver sección Dosis y vía de administración.
En los ensayos clínicos no se evaluó XELJANZ® XR en pacientes con valores iniciales de depuración de creatinina (estimados por la ecuación de Cockroft-Gault) < 40 mL/min (ver secciones Farmacocinética y farmacodinamia, y Dosis y vía de administración).
Pacientes con insuficiencia hepática: No se requiere ajuste de la dosis en pacientes con insuficiencia hepática leve. La dosis de XELJANZ® XR no debe exceder los 11 mg una vez al día en pacientes con insuficiencia hepática moderada. Para las recomendaciones de ajuste de dosis específicas para cada indicación, ver sección Dosis y vía de administración.
XELJANZ® XR no se debe administrar a pacientes con insuficiencia hepática severa (ver sección Dosis y vía de administración). En los ensayos clínicos no se evaluó XELJANZ® XR en pacientes con insuficiencia hepática severa ni en pacientes con serología positiva para VHB o VHC.
Combinación con otros tratamientos:
Artritis reumatoide: No se ha estudiado y se debe evitar la administración de XELJANZ® XR en pacientes con AR en combinación con FARME biológicos tales como los antagonistas del TNF, antagonistas de IL-1R, antagonistas de IL-6R, anticuerpos monoclonales anti-CD20 y moduladores de coestimulación selectiva e inmunosupresores potentes tales como azatioprina y ciclosporina debido a la posibilidad de aumento de inmunosupresión y aumento del riesgo de infección.
Artritis psoriásica: No se ha estudiado y se debe evitar la administración de XELJANZ® XR en pacientes con artritis psoriásica activa en combinación con FARME biológicos tales como antagonistas del TNF, antagonistas de IL-17, y antagonistas de IL-12/IL-23, e inmunosupresores potentes tales como la azatioprina y la ciclosporina, debido a la posibilidad de un aumento de la inmunosupresión y un mayor riesgo de infección.
No se ha estudiado la administración de XELJANZ® XR en combinación con inhibidores de la fosfodiesterasa 4 en los ensayos clínicos de XELJANZ® XR.
Colitis ulcerosa: No se ha estudiado y se debe evitar la administración de XELJANZ® XR en pacientes con colitis ulcerosa en combinación con agentes biológicos tales como antagonistas del TNF y vedolizumab, e/o inmunosupresores potentes tales como azatioprina, 6-mercaptopurina, tacrólimus y ciclosporina, debido a la posibilidad de un aumento de la inmunosupresión y un mayor riesgo de infección.
General: Como con cualquier otro material no deformable, se debería tener precaución cuando se administre XELJANZ® XR a pacientes con estrechamiento gastrointestinal preexistente severo (patológico o iatrogénico). Han habido reportes raros de síntomas obstructivos en pacientes con estenosis conocida en asociación con la ingestión de medicamentos que utilizan una formulación de liberación modificada no deformable.
Efectos sobre la capacidad para conducir y operar maquinarias: No se realizaron estudios formales sobre los efectos en la capacidad para conducir y utilizar máquinas.
DOSIS Y VÍA DE ADMINISTRACIÓN:
XELJANZ® XR no ha sido estudiado y debe evitarse su uso en combinación con los antagonistas del TNF, antagonistas del IL-1R, antagonistas del IL-6R, anticuerpos monoclonales anti-CD20, antagonistas IL-17, antagonistas IL-12/IL-23, anti-integrinas, moduladores selectivos de la coestimulación e inmunosupresores potentes como azatioprina, ciclosporina y tacrólimus, debido a la posibilidad de una mayor inmunosupresión y al mayor riesgo de infecciones.
El tratamiento con XELJANZ® XR debe interrumpirse si el paciente desarrolla una infección grave hasta que se controle dicha infección.
Método de administración: XELJANZ® XR se administra vía oral con o sin alimentos.
Trague las tabletas de XELJANZ® XR completas e intactas. No las triture, divida o mastique.
Posología para la artritis reumatoide: XELJANZ® XR se puede administrar como monoterapia o en combinación con metotrexato (MTX) u otros FARME no biológicos.
La dosis recomendada de XELJANZ® XR es de 11 mg una vez al día.
XELJANZ® XR 11 mg una vez al día ha demostrado equivalencia farmacocinética (ABC y Cmáx) con Xeljanz® 5 mg dos veces al día. Toda la información provista en esta sección para la indicación de artritis reumatoide es aplicable a Xeljanz® 5 mg dos veces al día y XELJANZ® XR 11 mg una vez al día ya que contienen el mismo principio activo (tofacitinib).
Cambio de Xeljanz® tabletas a XELJANZ® XR tabletas para la posología de artritis reumatoide: Puede cambiarse a los pacientes tratados con Xeljanz® 5 mg dos veces al día a XELJANZ® XR 11 mg una vez al día el día posterior a la última dosis de Xeljanz® 5 mg.
Posología para la artritis psoriásica: La dosis recomendada de XELJANZ® XR es de 11 mg administrados una vez al día en combinación con FARMEsc.
XELJANZ® XR 11 mg una vez al día ha demostrado una exposición equivalente (ABC y Cmáx) a la de Xeljanz® 5 mg dos veces al día. Toda la información proporcionada en esta sección para la indicación de artritis psoriásica es aplicable a Xeljanz® 5 mg dos veces al día y a XELJANZ® XR 11 mg una vez al día, ya que contienen el mismo principio activo (tofacitinib).
Cambio de Xeljanz® tabletas a XELJANZ® XR tabletas para la posología para la artritis psoriásica: Los pacientes tratados con Xeljanz® 5 mg dos veces al día pueden cambiar a XELJANZ® XR 11 mg una vez al día, al día siguiente de la última dosis de Xeljanz® 5 mg.
Posología para la colitis ulcerosa:
XELJANZ® XR: La dosis recomendada de XELJANZ® XR es de 22 mg una vez al día durante al menos 8 semanas; seguido por XELJANZ® XR 11 mg o 22 mg una vez al día dependiendo de la respuesta terapéutica.
Suspenda el tratamiento de inducción con XELJANZ® XR en los pacientes que no muestren evidencia de beneficio terapéutico en la semana 16.
En el caso de los pacientes refractarios, como los que han tenido un fracaso en el tratamiento previo con antagonistas del TNF, debe considerarse la continuación de la dosis de mantenimiento con Xeljanz® 10 mg dos veces al día o XELJANZ® XR 22 mg una vez al día.
Los pacientes que no consiguen mantener el beneficio terapéutico con Xeljanz® 5 mg dos veces al día pueden beneficiarse de un aumento a Xeljanz® 10 mg administrado dos veces al día.
Los pacientes que no consiguen mantener el beneficio terapéutico con XELJANZ® XR 11 mg una vez al día pueden beneficiarse de un aumento a XELJANZ® XR 22 mg administrados una vez al día.
En general, administre la dosis efectiva más baja para mantener el beneficio terapéutico.
Dosis de mantenimiento: Xeljanz® 5 mg dos veces al día o XELJANZ® XR 11 mg una vez al día. Para pacientes con pérdida de respuesta durante el tratamiento de mantenimiento, se puede considerar Xeljanz® 10 mg dos veces al día o XELJANZ® XR 22 mg una vez al día y limitarse a la duración más corta, con una cuidadosa consideración de los riesgos y beneficios para los pacientes individuales. Use la dosis efectiva más baja necesaria para mantener la respuesta.
Cambio de Xeljanz® tabletas a XELJANZ® XR tabletas: Los pacientes tratados con Xeljanz® 5 mg dos veces al día pueden cambiar a XELJANZ® XR 11 mg una vez al día, al día después de la última dosis de Xeljanz® 5 mg.
Los pacientes tratados con Xeljanz® 10 mg dos veces al día pueden cambiar a XELJANZ® XR 22 mg una vez al día, al día siguiente de la última dosis de Xeljanz® 10 mg.
Ajustes de dosis debidos a alteraciones en los resultados de laboratorio (ver sección Precauciones generales): Los ajustes de dosis o la interrupción de la dosis pueden ser necesarios para el manejo de alteraciones en los resultados de laboratorio relacionadas con la dosis, que incluyen linfopenia, neutropenia y anemia, tal como se describe en las Tablas 14, 15 y 16 a continuación.
No se recomienda iniciar el tratamiento con XELJANZ® XR en pacientes con un recuento de linfocitos menor que 500 células/mm3.
Tabla 14. Ajustes de dosis por linfopenia
|
Recuento bajo de linfocitos (ver sección Precauciones generales) |
|
|
Valor de laboratorio (células/mm3) |
Recomendación |
|
Recuento de linfocitos ≥ 500 |
Mantenga la dosis. |
|
Recuento de linfocitos < 500 (confirmado por análisis repetidos) |
Interrumpa la administración de XELJANZ® XR. |
No se recomienda iniciar el tratamiento con XELJANZ® XR en pacientes con un recuento absoluto de neutrófilos (ANC, por sus siglas en inglés) menor que < 1000 células/mm3.
Tabla 15. Ajustes de dosis por neutropenia
|
Recuento absoluto de neutrófilos (ANC) bajo |
|
|
Valor de laboratorio (células/mm3) |
Recomendación |
|
ANC > 1000 |
Mantenga la dosis. |
|
ANC 500-1000 |
En el caso de reducciones persistentes en este rango, interrumpa la administración de XELJANZ® XR hasta que el ANC sea > 1000. Para los pacientes que reciben XELJANZ® XR 11 mg una vez al día, interrumpa la administración de XELJANZ® XR. Cuando el ANC sea > 1000, reanude el tratamiento con XELJANZ® XR 11 mg una vez al día. Para los pacientes que reciben XELJANZ® XR 22 mg una vez al día, reduzca la dosis a XELJANZ® XR 11 mg una vez al día. Cuando el ANC sea >1000, reanude XELJANZ® XR 22 mg una vez al día en función de la respuesta clínica. |
|
ANC < 500 (confirmado por análisis repetidos) |
Interrumpa la administración de XELJANZ® XR. |
Se recomienda no iniciar el tratamiento con XELJANZ® XR en pacientes con hemoglobina < 9 g/dL.
Tabla 16. Ajustes de dosis por anemia
|
Valor bajo de hemoglobina (ver sección Precauciones generales) |
|
|
Valor de laboratorio |
Recomendación |
|
≤ 2 g/dL de disminución y ≥ 9.0 g/dL |
Mantener la dosis. |
|
> 2 g/dL de disminución o menos que 8.0 g/dL (confirmado por análisis repetidos) |
Interrumpa la administración de XELJANZ® XR hasta que los valores de hemoglobina se hayan normalizado. |
Poblaciones especiales:
Insuficiencia renal: Si la dosificación es XELJANZ® XR 11 mg una vez al día, la dosis recomendada en pacientes con insuficiencia renal grave es una tableta de XELJANZ® XR 11 mg en días alternos (ver secciones Farmacocinética y farmacodinamia y Precauciones Generales).
A continuación, se proporcionan recomendaciones específicas para cada indicación.
Si la dosificación es XELJANZ® XR 22 mg una vez al día, la dosis recomendada en pacientes con insuficiencia renal grave es una tableta de XELJANZ® XR 11 mg una vez al día (ver secciones Farmacocinética y farmacodinamia y Precauciones Generales).
A continuación, se proporcionan recomendaciones específicas para cada indicación.
Artritis reumatoide: No se requiere ajustar la dosis en pacientes con insuficiencia renal leve o moderada. La dosificación de XELJANZ® XR no debe exceder los 11 mg una vez al día en pacientes que sufren insuficiencia renal grave (incluidos, entre otros, aquellos sometidos a hemodiálisis) (ver secciones Farmacocinética y farmacodinamia y Precauciones generales).
Artritis psoriásica: No se requiere ajustar la dosis en pacientes con insuficiencia renal leve o moderada. La dosis recomendada de XELJANZ® XR es de 11 mg en días alternos, en pacientes con insuficiencia renal grave (incluidos, entre otros, aquellos sometidos a hemodiálisis) (ver secciones Farmacocinética y farmacodinamia y Precauciones generales).
Colitis ulcerosa: No se requiere ajustar la dosis en pacientes con insuficiencia renal leve o moderada. En pacientes con insuficiencia renal grave (incluidos, entre otros, aquellos sometidos a hemodiálisis), la dosis recomendada de XELJANZ® XR es de 11 mg en días alternos, si la dosis en presencia de una función renal normal es de 11 mg una vez al día (ver secciones Farmacocinética y farmacodinamia y Precauciones generales).
En los pacientes con insuficiencia renal grave (incluidos, entre otros, aquellos sometidos a hemodiálisis), la dosis recomendada de XELJANZ® XR es de 11 mg una vez al día si la dosis en presencia de una función renal normal es de 22 mg una vez al día (ver secciones Farmacocinética y farmacodinamia y Precauciones generales).
Insuficiencia hepática: No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve. Si la dosificación es XELJANZ® XR 11 mg una vez al día, la dosis recomendada en pacientes con insuficiencia hepática moderada es una tableta de XELJANZ® XR 11 mg en días alternos.
Si la dosificación es XELJANZ® XR 22 mg una vez al día, la dosis recomendada en pacientes con insuficiencia hepática moderada es una tableta de XELJANZ® XR 11 mg una vez al día.
A continuación, se proporcionan recomendaciones específicas para cada indicación.
Artritis reumatoide: No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve. XELJANZ® XR no se debe administrar a pacientes con insuficiencia hepática grave. La dosificación de XELJANZ® XR no debe exceder los 11 mg una vez al día en pacientes con insuficiencia hepática moderada (ver secciones Farmacocinética y farmacodinamia y Precauciones generales).
Artritis psoriásica: No se requiere ajuste de dosis en pacientes con insuficiencia hepática leve. XELJANZ® XR no se debe administrar a pacientes con insuficiencia hepática grave. La dosificación recomendada de XELJANZ® XR es de 11 mg en días alternos, en pacientes con insuficiencia hepática moderada (ver secciones Farmacocinética y farmacodinamia y Precauciones generales).
Colitis ulcerosa: No es necesario ajustar la dosis en pacientes con insuficiencia hepática leve. XELJANZ® XR no debe administrarse en pacientes con insuficiencia hepática grave.
En pacientes con insuficiencia hepática moderada, la dosis recomendada de XELJANZ® XR es de 11 mg una vez al día si la dosis en presencia de una función hepática normal es de 22 mg una vez al día, y la dosis recomendada de XELJANZ® XR es de 11 mg en días alternos. La dosis en presencia de una función hepática normal es de 11 mg una vez al día (ver secciones Farmacocinética y farmacodinamia y Precauciones generales).
Pacientes con artritis reumatoide, artritis psoriásica y colitis ulcerosa que reciben inhibidores del citocromo P450 y 2C19 (CYP3A4 y CYP2C19): Para indicaciones con una dosis máxima recomendada de XELJANZ® XR 11 mg una vez al día, en pacientes que reciben inhibidores potentes de CYP3A4 (p. ej., ketoconazol), o uno o más medicamentos concomitantes que dan como resultado una inhibición moderada de CYP3A4 y una inhibición potente de CYP2C19 (p. ej., fluconazol), la dosificación recomendada es XELJANZ® XR 11 mg en días alternos.
A continuación, se proporcionan recomendaciones específicas para cada indicación.
Artritis reumatoide: La dosificación de XELJANZ® XR no debe exceder 11 mg una vez al día en pacientes que reciben inhibidores potentes de CYP3A4 (p. ej., ketoconazol). La dosificación de XELJANZ® XR no debe exceder 11 mg una vez al día en pacientes que recibieron uno o más medicamentos concomitantes que causen tanto una inhibición moderada de CYP3A4 y una inhibición potente de CYP2C19 (p. ej., fluconazol).
Artritis psoriásica: La dosis recomendada de XELJANZ® XR es de 11 mg en días alternos en pacientes que reciben inhibidores potentes del CYP3A4 (p. ej., ketoconazol). La dosis recomendada de XELJANZ® XR es de 11 mg en días alternos en pacientes que reciben uno o más medicamentos concomitantes que dan como resultado tanto una inhibición moderada del CYP3A4 y una inhibición potente del CYP2C19 (p. ej., fluconazol).
Colitis ulcerosa: En pacientes que reciben inhibidores potentes de CYP3A4 (p. ej., ketoconazol), o uno o más medicamentos concomitantes que producen tanto una inhibición moderada de CYP3A4 y una inhibición potente de CYP2C19 (p. ej., fluconazol), la dosis de XELJANZ® XR debe reducirse a 11 mg una vez al día si el paciente toma 22 mg una vez al día, y la dosis de XELJANZ® XR debe reducirse a 11 mg en días alternos si el paciente toma 11 mg una vez al día.
Pacientes con artritis reumatoide, artritis psoriásica y colitis ulcerosa que reciben inductores del citocromo P450 (CYP3A4 y CYP2C19): La coadministración de XELJANZ® XR con inductores potentes de CYP (p. ej. rifampicina), puede provocar la pérdida de respuesta clínica o una respuesta clínica reducida (ver sección Interacciones medicamentosas y de otro género). No se recomienda la coadministración de inductores potentes de CYP3A4 con XELJANZ® XR.
Pacientes de edad avanzada (≥ 65 años): No se requiere un ajuste de la dosis en pacientes de 65 años y mayores.
Pacientes pediátricos: No se ha establecido aún la seguridad y eficacia de XELJANZ® XR en niños neonatos a < 18 años.
MANIFESTACIONES Y MANEJO DE LA SOBREDOSIFICACIÓN O INGESTA ACCIDENTAL:
No existe experiencia de sobredosis con XELJANZ® XR. No existe un antídoto específico para la sobredosis de XELJANZ® XR. El tratamiento debe ser sintomático y de apoyo. En caso de sobredosis, se recomienda monitorear al paciente para detectar signos y síntomas de reacciones adversas. Los pacientes que desarrollen reacciones adversas deben recibir un tratamiento adecuado.
Los datos farmacocinéticos incluyen hasta una dosis única de 100 mg en voluntarios sanos indican que se espera que más del 95% de la dosis administrada se elimine en el plazo de 24 horas.
PRESENTACIONES:
Frasco con 7, 30 o 90 tabletas de 11 mg y un desecante.
Frasco con 14, 30 o 90 tabletas de 22 mg y dos desecantes.
RECOMENDACIONES SOBRE ALMACENAMIENTO:
Consérvese a no más de 30 °C.
Consérvese el frasco bien cerrado.
Vida útil: 24 meses.
LEYENDAS DE PROTECCIÓN:
Su venta requiere receta médica. Información exclusiva para médicos. No se deje al alcance de los niños. No se use en el embarazo ni lactancia. Contiene un desecante NO INGERIBLE, consérvese dentro del envase.
Reporte las sospechas de reacción adversa a los correos: farmacovigilancia@cofepris.gob.mx y MEX.AEReporting@pfizer.com
o a la línea de Pfizer: 800 401 2002.
PFIZER, S.A. de C.V.
Km. 63 Carretera México-Toluca,
Zona Industrial, C.P. 50140,
Toluca, México, México.
Reg. Núm. 055M2021 SSA


